

Abstract
While it is known that cancer cell interactions with vascular endothelial cells (ECs) drive metastatic cancer cell extravasation from blood vessels into secondary tumor sites, the mechanisms of action are still poorly understood. Here, we tested the hypothesis that neuraminidase‐induced degradation of EC surface glycocalyx (GCX), particularly the sialic acid (SA) residue components of the GCX, will substantially increase metastatic cancer cell attachment to ECs. To our knowledge, our study is the first to isolate the role of GCX SA residues in cancer cell attachment to the endothelium, which were found to be differentially affected by the presence of neuraminidase and to indeed regulate metastatic cancer cell homing to ECs. We hope that this work will eventually translate to identification of EC GCX‐based cancer markers that can be therapeutically targeted to hinder the progression of metastasis.
Keywords: endothelial glycocalyx, metastatic cancer cells, secondary tumor, sialic acid
Abbreviations
- 4T1
Stage IV metastatic breast cancer cells
- CS
chondroitin sulfate
- DAPI
4′6‐diamidino‐2‐phenylindole
- DMEM
Dulbecco's modified Eagle medium
- EC
endothelial cell
- FBS
fetal bovine serum
- GCX
glycocalyx
- HA
hyaluronic acid
- HS
heparan sulfate
- HUVEC
human umbilical vein endothelial cells
- Lu‐ECAM
lung EC adhesion molecule‐1
- MAL II
Maakia amurensis lectin II
- Neur
neuraminidase
- PS
penicillin–streptomycin
- RFPEC
rat fat pad endothelial cell
- SA
sialic acid
- SNA
Sambucus nigra (elderberry bark) lectin
- WGA
wheat germ agglutinin lectin
1. INTRODUCTION
Cancer metastasis is one of the major causes of cancer‐related deaths.1, 2 During metastasis, primary tumor cells migrate from the parent tumor into neighboring tissues to form secondary tumors.3 This occurs when cells from the primary tumor migrate to and intravasate nearby blood vessels, travel through these vessels, and eventually extravasate the vessels at distant tissues where secondary tumors can form.4 The migration of cancer cells out of blood vessels, requires initial homing to and crossing of the adhesive endothelium, and it has been established that this occurs due to dysfunction of the endothelium's glycocalyx (GCX) coating (Figure 1a).5, 6
Figure 1.
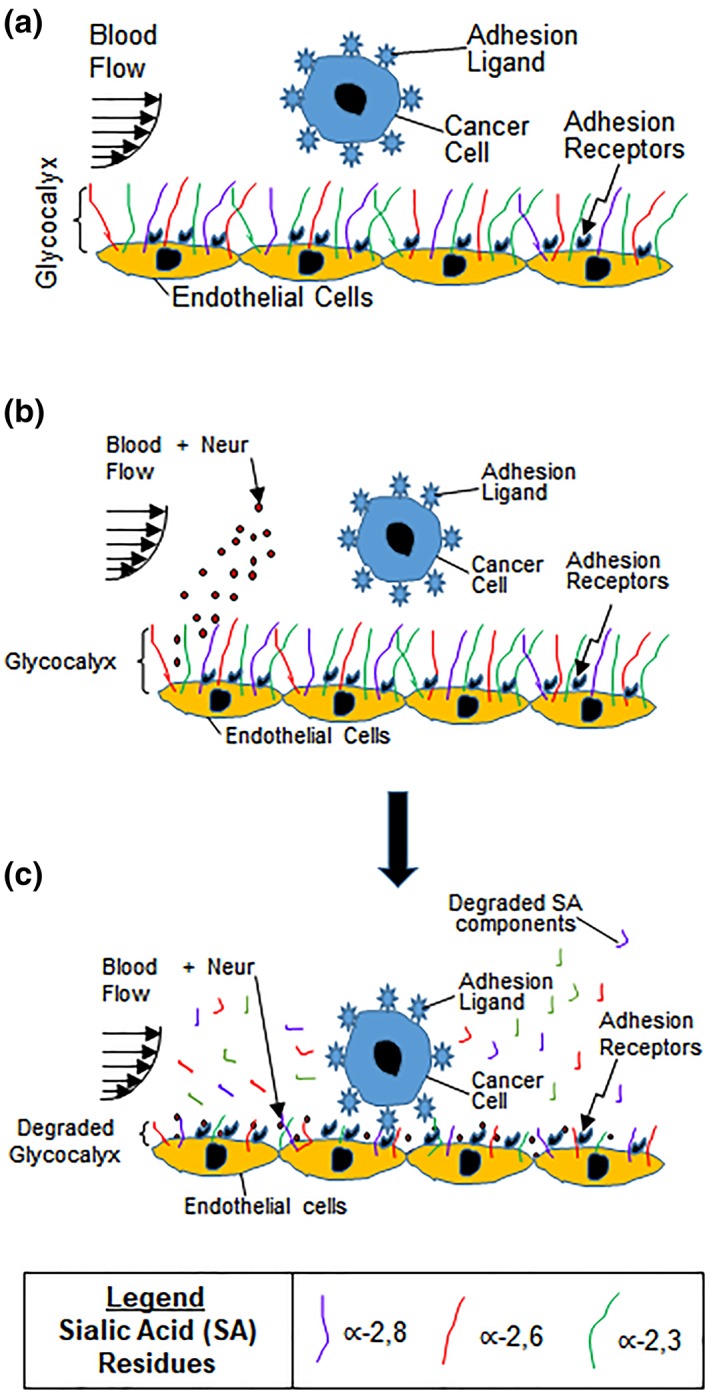
(a) Drawing shows endothelium with intact GCX. As metastatic cancer cells move with blood flow, the healthy GCX blocks the adhesion ligands on the cancer cells from attaching to the adhesion receptors on the endothelium lining of the blood vessel wall. We hypothesize that cancer cell attachment to the endothelium is caused by endothelial GCX degradation. (b, c) These drawings illustrate the conclusions of the findings reported herein. Our observations provide evidence that the systemic increase in Neur (b), which coincides with metastatic cancer conditions, degrades the GCX as a whole and as applied to its∝‐2,6‐linked and ∝‐2,3‐linked SA residues (c). This GCX degradation leads to increased cancer cell attachment to ECs, and we speculate that this is mediated by exposure of adhesion receptors on the endothelium which become accessible to adhesion ligands on cancer cells (c). ECs, endothelial cells; GCX, glycocalyx; SA, sialic acid
GCX covers most mammalian cells. For endothelial cells (ECs), a healthy GCX forms a selective barrier between ECs and their neighboring environment, by blocking adhesion receptors on the endothelium from binding to ligands on cells and certain molecules from the environment (Figure 1a).7, 8, 9, 10, 11, 12 The EC GCX is composed of a variety of sugar chains, glycoproteins, and soluble proteins. For example, the GCX includes core proteins on which heparan sulfate (HS), chondroitin sulfate (CS), and hyaluronic acid (HA) glycosaminoglycan sugar chains are attached.9 HS is the most abundant GCX component. The core proteins are also attached to sialic acid (SA) on their terminal ends.13 Degradation of these key components of GCX is associated with certain conditions and diseases including inflammation, sepsis, atherosclerosis, and cancer.14, 15, 16, 17, 18
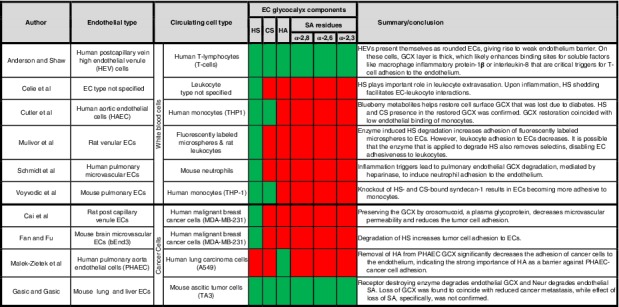
To date, only a few cell mechanism studies have explored how HS, CS, HA, and SA regulate homing of cells, particularly white blood cells and cancer cells, from vessel circulation to the endothelium surface.6, 19, 20, 21, 22, 23, 24, 25, 26, 27 Selected reports are summarized in Table 1, which shows that the majority of investigations have been performed in the context of white blood cells and inflammation,19, 20, 21, 22, 24, 28, 29, 30 and more work is required in the cancer milieu to address metastasis.6, 25, 26, 27 The white blood cell studies inform our understanding of GCX‐mediated EC adhesiveness to cancer cells, because cancer cells utilize inflammation mechanisms to metastasize18, 31, 32, 33 and inflammatory agonists are known to cause endothelial GCX shedding34 that can expose endothelium to circulating cell adhesion. With respect to GCX component‐specific shedding, research has largely been focused on studying the role of HS loss in cancer and inflammatory cell adhesion, due to its abundance in the GCX (Table 1).6, 20, 22, 23, 35 Some work has gone into investigating how losses in HS and CS, combined, contribute to endothelium adhesiveness.21, 24 Experiments conducted to isolate and elucidate the mechanisms of EC adhesiveness involving HA are rare,26 and studies isolating SA‐mediated EC adhesiveness have not been performed at all, to our knowledge (Table 1).
Table 1.
Representative reports of the role of the GCX in blocking or enabling EC adhesiveness to cells in the blood circulation, including white blood cells and cancer cells
|
Notes: Green shading indicates that some progress has been made in understanding the role of a specific GCX components in EC interaction with circulating cells, while red shading indicates that little or no progress has been made for some GCX components.
Abbreviations: CS, chondroitin sulfate; ECs, endothelial cells; GCX, glycocalyx; HA, hyaluronic acid; HS, heparan sulfate; SA, sialic acid.
Relevant to endothelium adhesiveness to cells in the blood vessels, SA is of particular interest because it is a major contributor to cellular and molecular recognition.36, 37, 38 This is due to the diversity of SA residues, which are differentiated through post‐transcriptional sialytransferase activity.22, 27, 28 On ECs, several SA residues are expressed, but the most notable include the ∝‐2,6‐linked, ∝‐2,3‐linked, and ∝‐2,8‐linked SA residues. These residues recognize and interact with cells and molecules through lectins, which are proteins that recognize and bind to SA and other sugars.37 In addition, SA residues are uniquely composed of amino acids and carboxyl groups that make them negatively charged, further enhancing SA's ability to facilitate recognition and binding or repulsion to other cells or molecules. SA's terminal location, diversity, composition, and charge, taken together, are important determinants of EC GCX‐mediated endothelial barrier function. Yet, while the role of SA in the cancer cell GCX has been extensively studied and found as a marker of oncogenesis and tumor survival,39, 40, 41 the role of SA in the endothelial GCX during oncogenesis has yet to be studied.
Another reason for our interest in SA stems from reports that the SA‐degrading enzyme, neuraminidase (Neur),42 is strongly associated with cancer metastasis and other pathologies. Specifically, Neur upregulation has been reported in hepatocellular carcinoma and ovarian cancer, and Neur has been noted to be an oncogene that enhances proliferation and migration of metastatic cancer cells.35, 43 Based on these reports, it is surprising that not much effort has been made to elucidate the impact of excessive Neur on ECs, which are likely to respond by shedding their SA residues, leading to cancer cell access to the endothelium.
In the present study, we aimed to test the hypothesis that Neur‐induced degradation of EC GCX, particularly the shedding of SA residues, will result in substantial increase in metastatic cancer cell attachment to the endothelium. To test our hypothesis, we first characterized the expression of the GCX, both generally and with a specific focus on a SA residue. We then investigated the effects of Neur on the GCX. Last, we investigated the effect of GCX degradation on the attachment of metastatic cancer cells to the endothelium. Our findings indicate that SA residues are differentially affected by the presence of Neur and confirm that GCX degradation indeed leads to increased cancer cell attachment to the endothelium. We anticipate that this body of work will extend our knowledge on the role played by GCX, specifically regarding SAs, in regulating movement of metastatic cancer cells from blood vessels to secondary metastatic sites. This will lead to future research to identify innovative GCX‐based markers that can be therapeutically targeted to hinder the progression of cancer metastasis.
2. MATERIALS AND METHODS
2.1. Endothelial cell culture
GCX‐rich rat fat pad endothelial cells (RFPECs)44 at passages 24 to 30 were seeded at 15,000–20,000 cells/cm2 onto 12‐mm No. 1 glass coverslips (Fisher Scientific). REFPECs were grown in Dulbecco's modified Eagle medium (DMEM, Invitrogen), 1% penicillin–streptomycin (PS), 10% fetal bovine serum (FBS, Gibco Life Technologies), humidity, 37°C, and 5% CO2, for 3 days until they reached full confluency.
2.2. Neur enzyme degradation of the endothelial GCX
To model endothelial GCX degradation, RFPECs were cultured for 2 hr in DMEM/1% PS/10% FBS with 0 (untreated control), 15, 135, 1,215, or 3,645 mU/mL of the SA‐degrading enzyme, Neur from Clostridium perfringens (Sigma).45 After the 2‐hr period ended, RFPECs were cultured in enzyme‐free culture media before further experimentation.
2.3. Endothelial GCX lectin staining, confocal microscopy, and image analysis
Fluorescent staining was performed to assess GCX expression on untreated or Neur‐treated RFPECs. This was achieved using lectins, proteins that recognize and bind to sugars.37 After the Neur treatment period, RFPEC monolayers, fixed with 2% paraformaldehyde/0.1% glutaraldehyde, were incubated with various GCX labeling‐lectins (Table 1). Secondary labeling was done with Alexa Fluor 488 (AF‐488) conjugated streptavidin (Table 1) before applying Vectashield anti‐fade media containing 4′6‐diamidino‐2‐phenylindole (from Vector Labs; indicated in Table 1) to stain the cell nuclei. Further details are described in Supporting Information (SI) Materials and Methods.
RFPECs were then imaged using a Zeiss Confocal Laser Scanning Microscope 700 with a ×63 magnification objective (oil). Analysis was performed using NIH ImageJ software. For detailed methods, see SI Materials and Methods and our previously published work.46
2.4. Cancer cell culture, attachment assay, fluorescence microscopy, and image analysis
Please refer to SI Materials and Methods for details. In brief, Stage IV metastatic mouse breast cancer cells (4T1) (ATCC) at passages 5 to 10 were cultured in DMEM/1% PS/10% FBS, humidity, 37°C, and 5% CO2. Passaging or experimental usage occurred at 80% confluency. For experiments, 4T1 cells were labeled with CellTracker Red CMTPX Dye (Thermo Fisher Scientific) and 103 cells/mL were coincubated with a confluent monolayer of either untreated or enzyme treated RFPECs. After 30 min of coincubation, nonattached 4T1 cells were removed, leaving attached 4T1 cells behind. Attached 4T1 cells and underlying RFPECs were imaged using a Zeiss Z1 Observer fluorescence microscope at ×10 magnification. A red filter (excitation of 558 nm) was used to distinguish and image CellTracker‐labeled 4T1 cells. RFPECs were visualized by phase contrast. The total number of attached 4T1 cells and the total number of RFPECs in the field of view were counted using the ImageJ cell counting tool. The 4T1 cell count was divided by the RFPEC cell count. This quantity was normalized by the value obtained for 4T1 adhesion to untreated RFPECs (control).
2.5. Statistics
Please refer to SI Materials and Methods for details. In brief, data sets were reduced to means ± SEM. When two treatments were compared, Student's t tests were used to determine statistical significances between groups. For multiple comparisons, one‐way ANOVA analyses and Tukey post hoc tests were used to determine statistical significance. An alpha value of p < .05 was used for both t tests and ANOVA.
3. RESULTS AND DISCUSSION
The endothelial GCX plays a significant role as a barrier between the endothelium and circulating cells present in the blood stream, including cancer cells.6, 19, 20, 21, 22, 23, 24, 25, 26, 27 The importance of the endothelial GCX in enabling EC adhesiveness to cancer and other types of circulating cells, primarily when its HS component is shed or in the presence of heparinase enzyme that specifically degrades HS, has been well established (Table 1).6, 19, 20, 21, 22, 23, 24, 25, 27 Regulation of EC adhesiveness by endothelial GCX components such as CS and HA has also been demonstrated (Table 1).6, 20, 21, 22, 23, 24, 25, 26 Another important endothelial GCX component, namely SA, its residues, and its degrading enzyme, Neur, remains understudied in the context of EC adhesion to circulating cancer cells (Table 1). The present study aimed to fill this gap, by studying the impact of Neur‐induced degradation of EC GCX, particularly the SA component, on metastatic cancer cell attachment to the endothelium.
3.1. Neur elevation in the EC environment destabilizes wheat germ agglutinin‐labeled GCX
RFPECs were selected as an appropriate cell model for this study because of their ability to produce robust GCX even in static conditions, while other cell culture models require shear stress to stimulate the synthesis of GCX.44 Upon the initial inspection of RFPECs treated with Neur, we observed that RFPEC monolayers exposed to Neur had preserved morphology (compared to untreated samples) indicating that only the GCX would be affected by the presence of the enzyme and not the underlying endothelium (Figure 2a–e).
Figure 2.
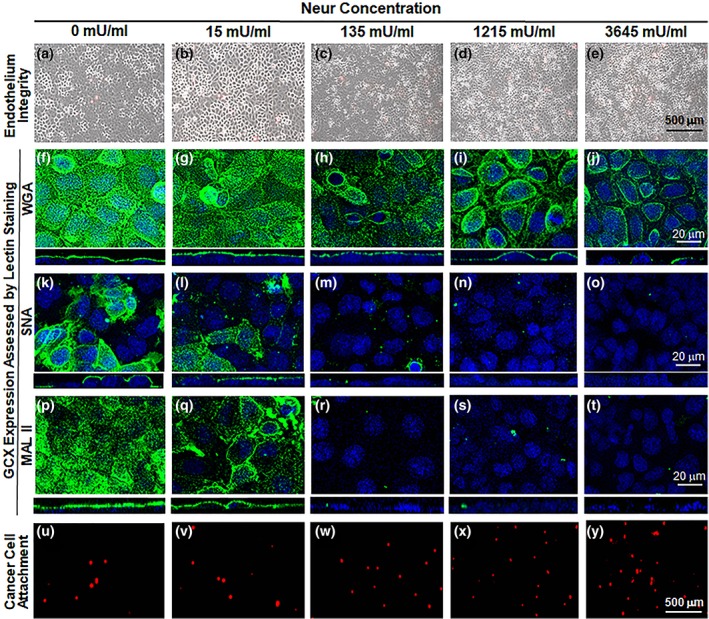
Images show the effect of exposure to various concentrations of Neur enzyme on the GCX, endothelium integrity, and cancer cell attachment to ECs. As shown, Neur concentrations include 0, 15, 135, 1,215, and 3,645 mU/mL. (a–e) ×5 magnification phase contrast images merged with red fluorescence micrographs, to confirm integrity of the EC layer in all conditions and show cancer cells attached to the endothelium. Scale bar equal to 500 μm is shown. (f–t) ×63 magnification confocal micrographs of GCX labeled with green fluorescence conjugated to the following lectins: WGA (f–j), SNA (k–o), and MAL II (p–t). The blue is DAPI, which labels EC nuclei. Scale bar equal to 20 μm is shown. (u–y) ×5 magnification red fluorescence micrographs clarify the presence of CellTracker Red labeled cancer cells. Scale bar equal to 500 μm is shown. DAPI, 4′6‐diamidino‐2‐phenylindole; ECs, endothelial cells; GCX, glycocalyx; MAL II, Maakia amurensis lectin II; SNA, Sambucus nigra (elderberry bark) lectin; WGA, wheat germ agglutinin
To determine the effects of Neur on GCX integrity, RFPECs were first labeled with wheat germ agglutinin (WGA) (Figure 2f–i). WGA is reported to delineate the overall GCX structure better than other lectins,47 and binds to a number of SA residues along with a few other GCX components (Table 2). We discovered that at baseline conditions (i.e., in the absence of Neur), WGA‐labeled GCX covered 73.8 ± 7.4% of RFPEC monolayers (Figure 2f), represented by a normalized value of 1.00 ± 0.07 (Figures 3a and 4a). WGA‐labeled GCX thickness was 1.46 ± 0.02 μm, a normalized value of 1.00 ± 0.02 (Figures 3b and 4b). Previously, Reitsma et al reported a WGA‐labeled GCX layer to cover about 90% of the endothelium and to be 2.3 ± 0.1 μm thick.49 The coverage and thickness of the WGA‐labeled GCX in our study was less, which could be attributed to differential GCX expression in varying vascular tissue beds.7, 50, 51 Additionally, our in vitro GCX is expected to be thinner than that found in vivo due to the absence of bovine serum albumin in the experimental media and also the collapse of the GCX that occurs outside of the in vivo environment.52
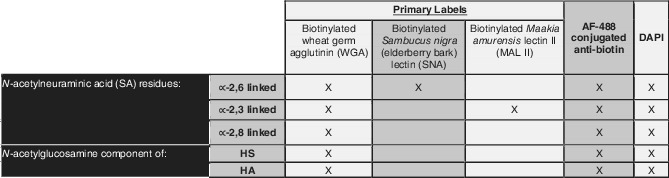
Table 2.
This table indicates the lectins (purchased from Vector Labs) that were used in labeling GCX
|
Notes: As shown, WGA is a general lectin for GCX, labeling three SA residues along with N‐acetylglucosamine, a component of HS and HA. SNA and MAL II are specific to only ∝‐2,6 linked and ∝‐2,6 linked residues of SA, respectively.23, 48 All lectins were biotinylated, enabling fluorescence labeling with biotin antibody conjugated to AF‐488. Once lectins were applied, followed by the secondary antibody, cell nuclei were all labeled with DAPI.
Abbreviations: DAPI, 4′6‐diamidino‐2‐phenylindole; GCX, glycocalyx; HA, hyaluronic acid; HS, heparan sulfate; SA, sialic acid.
Figure 3.
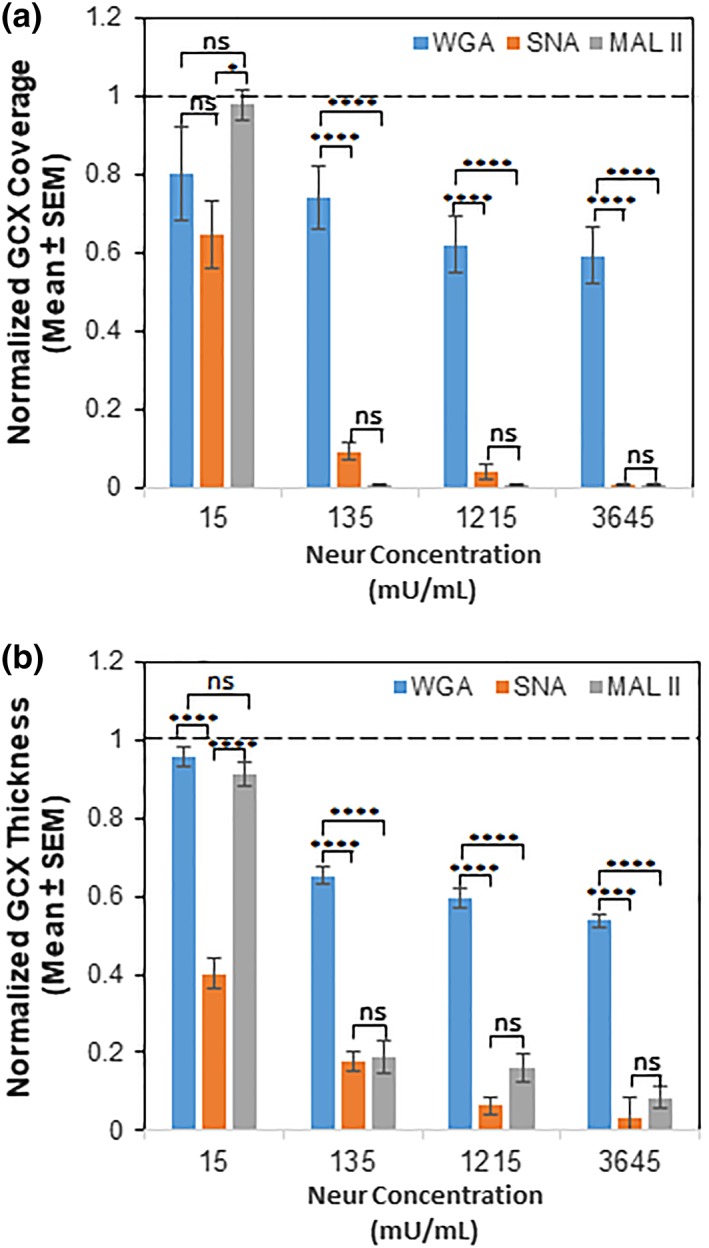
Comparing the coverage (a) and thickness (b) of GCX components that bind WGA, SNA, and MAL II at various Neur concentrations. Compared to untreated baseline conditions as indicated by the dashed line (‐ ‐ ‐), Neur concentration of 15 mU/mL slightly reduced thickness of and coverage of the endothelium by WGA‐labeled GCX, SNA‐labeled ∝‐2,6‐linked SA residue, and MAL II‐labeled ∝‐2,3‐linked SA residue. SNA‐labeled ∝‐2,6‐linked SA residue was reduced most statistically significantly. 135 mU/mL of Neur slightly reduced WGA‐labeled GCX but statistically significantly reduced SNA‐labeled ∝‐2,6‐linked SA residue and MAL II‐labeled ∝‐2,3‐linked SA residue. 1,215 mU/mL and 3,654 mU/mL of Neur further reduced WGA‐labeled GCX, SNA‐labeled ∝‐2,6‐linked SA residue, and MAL II‐labeled ∝‐2,3‐linked SA residue. Results are normalized based on 0 mU/mL conditions. Significance differences between groups are denoted as *****p < .0001, and “ns” denotes nonsignificance. GCX, glycocalyx; MAL II, Maakia amurensis lectin II; SA, sialic acid; SNA, Sambucus nigra (elderberry bark) lectin; WGA, wheat germ agglutinin
Figure 4.
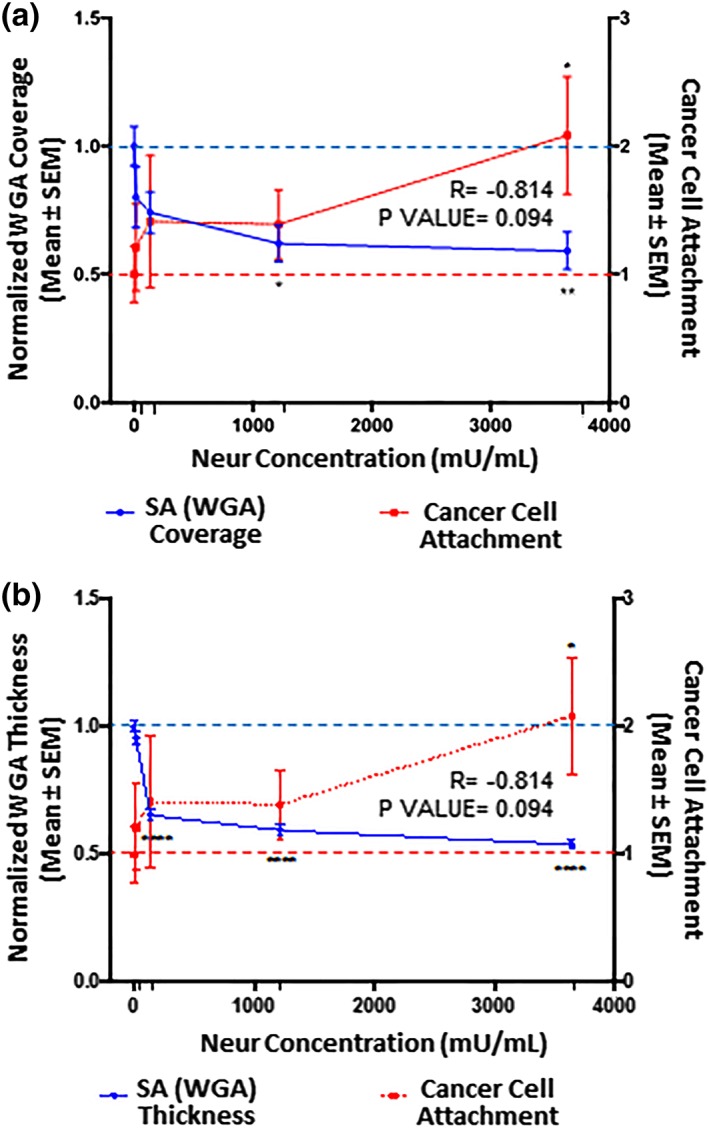
The extent of EC coverage by GCX and the thickness of GCX, as assessed by quantifying WGA labeled GCX, are inversely proportional to the number of cancer cells that attach to endothelium. Results are normalized to 0 mU/mL baseline conditions, which are indicated by the dashed lines (‐ ‐ ‐). Significance is denoted as *p < .05, **p < .01, and ****p < .0001. (a) Compared to 0 mU/mL Neur conditions, WGA‐labeled GCX coverage of ECs only becomes statistically low at high Neur doses of 1,215 and 3,645 mU/mL. N = 3, and representative en face images are shown in Figure 2a–j. (b) Compared to 0 mU/mL Neur conditions, WGA‐labeled GCX thickness is statistically significantly affected by Neur doses of 135, 1,215, and 3,645 mU/mL. N = 3, and representative cross‐section images used for this data are shown in Figure 2a–j. (a, b) Exponential increase in cancer attachment was observed with the increasing Neur concentration. At 0 mU/mL, N = 9; at 15 mU/mL, N = 8; at 135 mU/mL, N = 8; at 1,215 mU/mL, N = 9; and at 3,645 mU/mL, N = 9, and representative images used for this data are shown in Figure 2u–y. EC, endothelial cell; GCX, glycocalyx; WGA, wheat germ agglutinin
After 2‐hr treatment of RFPEC monolayers with 15, 135, 1,215, and 3,645 mU/mL of Neur, the normalized WGA‐labeled GCX coverage of ECs decreased to 0.8 ± 0.1, 0.74 ± 0.08, 0.62 ± 0.07, and 0.59 ± 0.07, respectively (Figures 3a and 4a). Normalized thicknesses of WGA‐labeled GCX for the same sequence of enzyme treatments decreased to 0.96 ± 0.03, 0.65 ± 0.02, 0.59 ± 0.02, and 0.54 ± 0.02, respectively (Figures 3b and 4b).
In summary, WGA‐labeled GCX was resistant to the initial enzyme dose of 15 mU/mL. However, as WGA‐labeled GCX degraded, statistically (p‐value) significant reduction in thickness, but not in coverage, was observed starting at an enzyme concentration of 135 mU/mL. Statistically (p‐value) significant reductions in both coverage and thickness required an enzyme concentration of 1,215 mU/mL. At this 1,215 mU/mL concentration, in the orthogonal views we also started to see discontinuity in the overall appearance of the GCX layer (Figure 2i, orthogonal view), which may expose the underlying endothelial cell membranes. With the highest dose of 3,645 mU/mL, we noticed an ~50% decrease in both the coverage and thickness of WGA‐labeled GCX. This reduction in coverage and thickness matches pathological conditions, such as sepsis and ischemia/reperfusion and some cancers, in the degree of GCX degradation.15, 53
3.2. Increased Neur in the EC environment leads to degradation of ∝‐2,6‐linked SA residue
∝‐2,6‐linked SA residues exhibited a patchy morphology when compared to WGA‐linked GCX (Figure 2k). The discontinuity in expression of ∝‐2,6‐linked SA residue was confirmed using orthogonal views of RFPEC monolayers (Figure 2k) and has also been reported by other studies.54 When we quantified our observation, coverage of the ECs by the ∝‐2,6‐linked SA residue at baseline conditions (i.e., in the absence of enzyme treatment) was 25.8 ± 5.9% of the RFPEC surface (Figure 2k), represented by a normalized value of 1.0 ± 0.23 (Figures 3a and 5a). SNA‐labeled SA thickness was 1.19 ± 0.04 μm, a normalized value of 1.0 ± 0.1 (Figures 3b and 5b).
Figure 5.
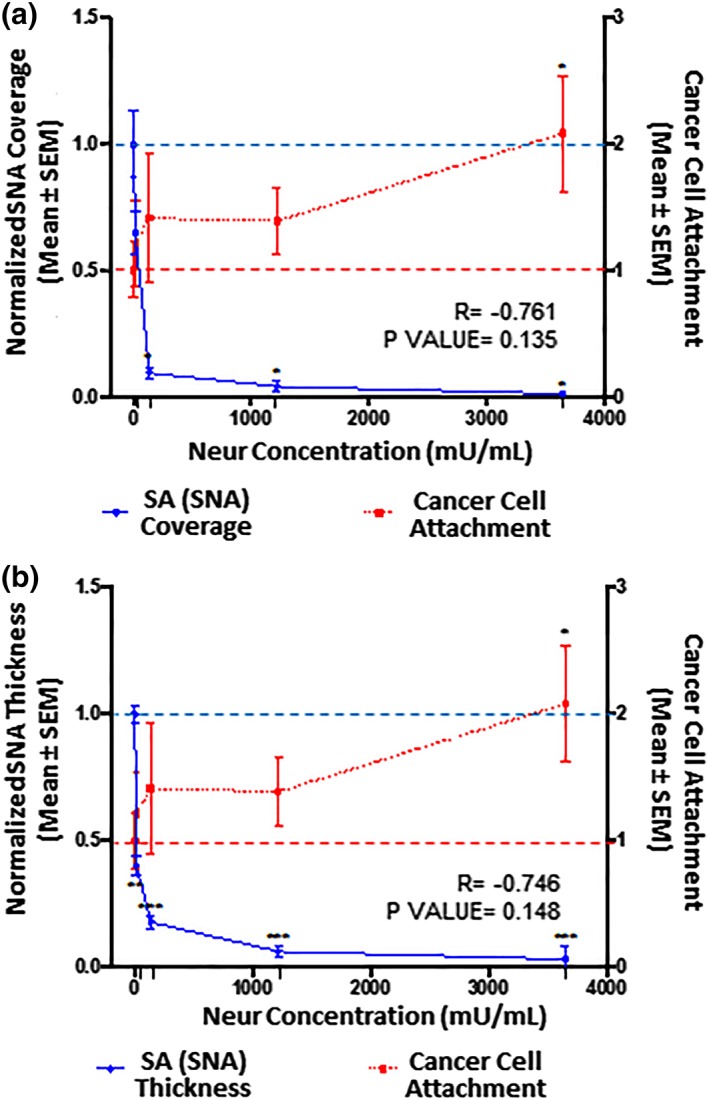
The extent of EC coverage by SA and the thickness of SA, as assessed by quantifying SNA‐labeled ∝‐2,6‐linked SA residue, are compared to the number of cancer cells that attach to endothelium. 0 mU/mL baseline conditions are indicated by the dashed lines (‐ ‐ ‐). Significance is denoted as *p < .05, **p < .01, and ***p < .001. (a) Compared to 0 mU/mL Neur conditions, SNA‐labeled ∝‐2,6‐linked SA residue coverage of ECs becomes statistically low at Neur doses of 135, 1,215, and 3,645 mU/mL. N = 3, and representative en face images are shown in Figure 2k–o. (b) Compared to 0 mU/mL Neur conditions, SNA‐labeled ∝‐2,6‐linked SA residue thickness is statistically significantly affected by Neur doses of 15, 135, 1,215, and 3,645 mU/mL. N = 3, and representative cross‐section images used for this data are shown in Figure 2k–o. (a, b) The observed Neur‐induced increase in cancer attachment as shown in Figure 3 is shown again, for comparison to expression of SNA‐labeled ∝‐2,6‐linked SA residue. At 0 mU/mL, N = 9; at 15 mU/mL, N = 8; at 135 mU/mL, N = 8; at 1,215 mU/mL, N = 9; and at 3,645 mU/mL, N = 9, and representative images used for this data are shown in Figure 2u–y. EC, endothelial cell; SA, sialic acid; SNA, Sambucus nigra (elderberry bark) lectin
We found that 15 mU/mL of Neur enzyme did not significantly degrade the coverage of RFPECs by ∝‐2,6 SA residues (Figures 3a and 5a). Conversely, at the same enzyme concentration ∝‐2,6‐linked SA exhibited a significant decrease in normalized thickness from 1.0 ± 0.2 to 0.40 ± 0.08 (Figures 3b and 5b), representing an ~60% decrease from baseline conditions. Recall, at the same enzyme concentration of 15 mU/mL, WGA‐linked GCX did not exhibit significant decrease in thickness (Figures 3b and 4b). Taken together, these results suggest that ∝‐2,6 SA residues could possibly be the first GCX component degraded by Neur.
Further increases in enzyme concentration lead to an exponential decrease in ∝‐2,6‐linked SA residue coverage and thickness on RFPECs. After 2‐hr treatment of confluent RFPECs with 15, 135, 1,215, and 3,645 mU/mL of Neur, the resulting coverage of ECs by ∝‐2,6 SA residue decreased to 0.7 ± 0.1, 0.09 ± 0.05, 0.04 ± 0.02, and 0.010 ± 0.001, respectively (Figures 3a and 5a). The corresponding effect on the thickness of ∝‐2,6‐linked sialic acid for the same Neur doses was observed to decrease 0.40 ± 0.08, 0.18 ± 0.002, 0.06 ± 0.01, and 0.03 ± 0.01, respectively (Figures 3b and 5b). Clearly, as Neur concentration increases, the underlying endothelium membrane is exposed.
3.3. Neur increase degrades ∝‐2,3‐linked residue of GCX SA
33.7 ± 2.4% of the untreated RFPEC surface was covered by ∝‐2,3‐linked SA. This result was normalized to 1.00 ± 0.08 (Figure 6a). The corresponding thickness was 1.83 ± 0.06 μm, also normalized to a value of 1.0 ± 0.08 (Figure 6b). Taking these results into consideration clarifies that ∝‐2,3‐linked SA residue provided less baseline coverage than WGA‐labeled GCX and higher baseline RFPEC coverage than ∝‐2,6‐linked SA residue (Figure 2f–t). In addition, comparing ∝‐2,3‐linked SA residue to ∝‐2,6‐linked SA residue, it is worth mentioning that ∝‐2,3‐linked SA residue expression is less patchy and discontinuous than ∝‐2,6‐linked SA residue (Figure 2k–t). These observations reveal differential expression of these SA residues and may also indicate that different ECs could have different presentations of these SA residues.55 In fact, Cioffi et al reported that ∝‐2,3‐linked SA residues, specifically, are more abundantly expressed in microvessels compared to other SA residues.55
Figure 6.
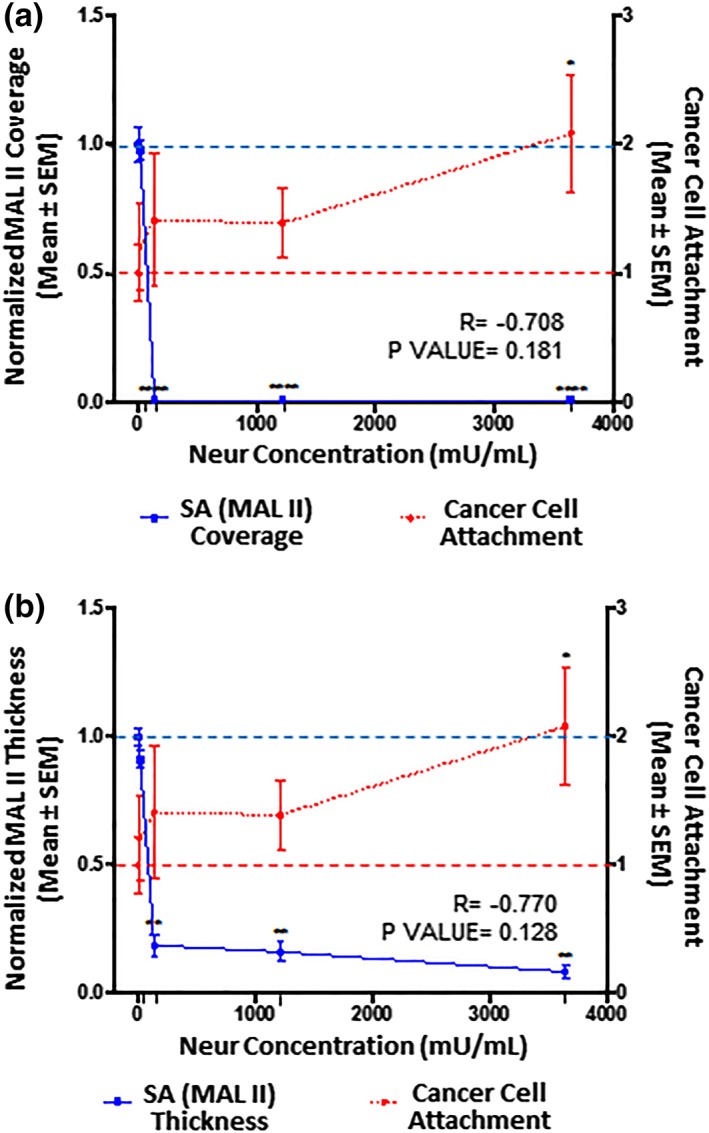
The extent of EC coverage by SA and the thickness of SA, as assessed by quantifying MAL II‐labeled ∝‐2,3‐linked SA residue, are compared to the number of cancer cells that attach to endothelium. Results are normalized to 0 mU/mL baseline conditions, which are indicated by the dashed lines (‐ ‐ ‐). Significance is denoted as *p < .05, **p < .01, and ****p < .0001. (a) Compared to 0 mU/mL Neur conditions, MAL II‐labeled ∝‐2,3‐linked SA residue coverage of ECs becomes statistically low at Neur doses of 135, 1,215, and 3,645 mU/mL. N = 3, and representative en face images are shown in Figure 2p–t. (b) Similarly, compared to 0 mU/mL Neur conditions, SNA‐labeled ∝‐2,6‐linked SA residue thickness is statistically significantly affected by Neur doses of 135, 1,215, and 3,645 mU/mL. N = 3, and representative cross‐section images used for this data are shown in Figure 2p–t. (a, b) The observed Neur‐induced increase in cancer attachment as shown in Figures 3 and 4 are shown again, for comparison to expression of MAL II‐labeled ∝‐2,3‐linked SA residue. At 0 mU/mL, N = 9; at 15 mU/mL, N = 8; at 135 mU/mL, N = 8; at 1,215 mU/mL, N = 9; and at 3,645 mU/mL, N = 9, and representative images used for this data are shown in Figure 2u–y. EC, endothelial cell; MAL II, Maakia amurensis lectin II; SA, sialic acid
RFPEC coverage by ∝‐2,3‐linked SA residue was drastically affected starting from the initial stages of enzymatic treatment (Figures 3a and 6a). After the treatment of RFPECs with 15, 135, 1,215, and 3,645 mU/mL of Neur, the normalized coverage of RFPEC by ∝‐2,3‐linked SA was decreased to 0.98 ± 0.03, 0.007 ± 0.001, 0.008 ± 0.005, and 0.007 ± 0.004, respectively (Figures 3a and 6a). The corresponding normalized thicknesses of ∝‐2,3‐linked SA after enzyme treatment were decreased to 0.9 ± 0.2, 019 ± 0.09, 0.2 ± 0.1, and 0.08 ± 0.03, respectively (Figures 3b and 6b).
3.4. Neur‐induced GCX loss leads to increased cancer cell attachment to ECs
After establishing a degradation profile for GCX and its SA components we coincubated Neur treated and nontreated RFPEC monolayers with labeled 4T1 breast cancer cells to determine the effect of Neur on cancer attachment to the endothelium. Before the addition of the enzyme to ECs, there was a baseline attachment of cancer cells to ECs (Figure 2u), normalized to a value of 1.00 ± 0.22 (Figures 4, 5, 6). This could be attributed to the barrier created by the terminal positions of sugar chains within the GCX matrix.56, 57 These GCX structures found on the apical surface of the GCX structure mediate intercellular recognition and binding.56
Adding 15 mU/mL of Neur resulted in attachment of 1.2 ± 0.3‐fold more 4T1 breast cancer cells to ECs, a 20% increase (Figures 2v, 4, 5, 6). At this enzyme concentration WGA‐labeled GCX and ∝‐2,3‐linked SA decrease in coverage and thickness was not as significant as the decrease in ∝‐2,6‐linked SA (Figures 3, 4, 5, 6). Therefore, 4T1 attachment to ECs could likely be related to the degradation of ∝‐2,6‐linked SA, which may have exposed receptor sites on the surface of the endothelium to facilitate attachment.
Increasing the enzyme dosage to 135 mU/mL resulted in 1.4 ± 0.5‐fold increase in cancer cell attachment to ECs (Figures 2w, 4, 5, 6). Further increasing the enzyme dose to 1,215 mU/mL resulted in a 1.4 ± 0.3‐fold increase in cancer cell attachment compared to control conditions (Figures 2x, 4, 5, 6) a result that was not significantly different from those at the 135 mU/mL concentration. This can be explained by the fact that at both of these Neur concentrations, ∝‐2,6‐linked and ∝‐2,3‐linked SA residues were not visible and WGA‐labeled GCX became clearly discontinuous (Figure 2h,l–n,r,s). These results indicate a point at which loss of both ∝‐2,6‐linked and ∝‐2,3‐linked SA permitted 4T1‐EC attachment. Furthermore, due to statistical insignificance between 4T1 attachment at 135 mU/mL and 1,215 mU/mL, we believe that at the enzyme concentration of 135 mU/mL cancer cell saturation of exposed adhesion receptors occurred. Thus, additional cancer cells were prevented from binding. The reverse of this phenomena was observed by Zhu et al who reported blocked mouse melanoma cell (B16) binding to lung EC adhesion molecule‐1 (Lu‐ECAM‐1) that was competitively bound to monoclonal antibody (6D3) against Lu‐ECAM‐1.58
A final Neur dose of 3,645 mU/mL resulted in a coverage and thickness decrease of WGA‐labeled GCX to 40% and 50%, respectively, in comparison to baseline conditions (Figure 4). The ∝‐2,6‐linked and ∝‐2,3‐linked SA residues were completely removed (Figures 5 and 6). The high level of Neur enzyme concentration and subsequent GCX and SA degradation lead to a twofold increase in attachment of 4T1 breast cancer cells to RFPECs, in comparison with control (Figures 2y, 4, 5, 6). A similar result was reported by Gasic et al, who treated 8‐week old CAF1/J mice with Neur and discovered that there was a significant increase in metastasis formation in mice treated with Neur in comparison with untreated mice.27 Our investigation and the study reported by Gasic27 reveal the important role played by the presence of Neur in the spread of cancer. Recall our finding that intact GCX components extend 1.2–1.8 μm into the extracellular space of the ECs. The shedding of these GCX components presents an opportunity for exposure of EC surface adhesion molecules such as E‐selectin,29 which are membrane‐attached, less than 100 nm in height, and typically covered by the thick GCX. The disparity between GCX thickness and adhesion molecule height could prevent 4T1 breast cancer cells from attaching to the surface of the endothelium36, 59 in conditions where the GCX is not damaged. Conversely, GCX degradation makes the adhesion molecules more accessible to cancer cells, thus facilitating the high level of 4T1‐EC attachment (Figure 1b,c).
4. CONCLUSIONS
The goal of this study was to clarify how the GCX, specifically the understudied SA component of the GCX, contributes to GCX‐mediated protection of the endothelium against cancer cell adhesion. Toward this end, we have confirmed differential expression of WGA‐labeled GCX, ∝‐2,6‐linked SA, and ∝‐2,3‐linked SA on the endothelium. We found that the presence of Neur in the EC environment sheds these components and enables ECs to become adhesive to floating cancer cells (Figure 1b,c). Graded Neur dosing and systematic GCX degradation revealed that there is a necessary threshold of degradation of both GCX coverage and thickness on ECs at which the attachment of 4T1 breast cancer cells to the endothelium is substantially enhanced in a manner that could promote metastasis.
Further understanding of endothelial GCX mechanisms of cancer metastasis is needed. Ongoing work in our lab includes studies of human EC interaction with human cancer cells. We expect the human cell studies will confirm and build upon the rodent cell results reported herein. For example, in critical control experiments, we have confirmed that untreated human umbilical vein endothelial cells (HUVEC; Figure 7a) highly express GCX components such as those targeted by WGA (Figure 7b) (Mensah et al, manuscript in review), similar to what we observed in healthy RFPEC (Figure 7e,f). Other GCX components such as HS show low expression (Figure 7c) in HUVEC, particularly in the absence of physiological flow stimulation, which is usually required for in vitro human EC studies of the GCX.60, 61 This was contrary to what we observed in RFPEC where HS expression was prominent without flow stimulation (Figure 7g). We speculated that the lack of HS is insufficient to enable HUVEC adhesiveness to 4T1 breast cancer cells, because WGA‐labeled GCX is abundant enough to compensate and provide an adequate buffer between the HUVEC and 4T1 cells. Indeed, our control experiments revealed that the level of attachment of 4T1 cells to these untreated HUVEC is quite low and statistically similar to what was observed with RFPEC (Figure 7d,h,i). In additional control experiments, we are incorporating flow perfusion into our EC culture environment (Mensah et al, manuscript in review). This approach will more completely replicate the natural environment in which both the circulating cancer cells and the endothelial cells are conditioned by flow, which tends to affect the morphology, biochemical activities, and other aspects of the cells. For example, a healthy flow environment can move the endothelial GCX scale in the antimetastatic direction while unhealthy flow conditions can have the same effect as GCX‐degrading enzyme and lead the endothelial GCX to become more prometastatic. Finally, in our human cell studies, under flow conditions, we are also comparing the role of SA to the role of EC surface adhesion molecules (Mensah et al, manuscript in review). In the future, we look forward to interrogating additional GCX components (Table 1). The aim is to eventually clarify how the various GCX components and the adhesion molecules synergistically contribute to cancer cell attachment to the endothelium. Studying the role of the EC GCX as compared to the role of the EC adhesion molecules from a perspective that has a human‐context, considers both enzyme and flow conditions, and looks at the multiple GCX components, we will expand upon our pre‐existing knowledge of the relationship between endothelial GCX degradation and cancer attachment. Our long‐term goal is to apply this knowledge to the development of therapeutic means to prevent the endothelial GCX degradation in a hope of hindering or even preventing cancer metastasis.
Figure 7.
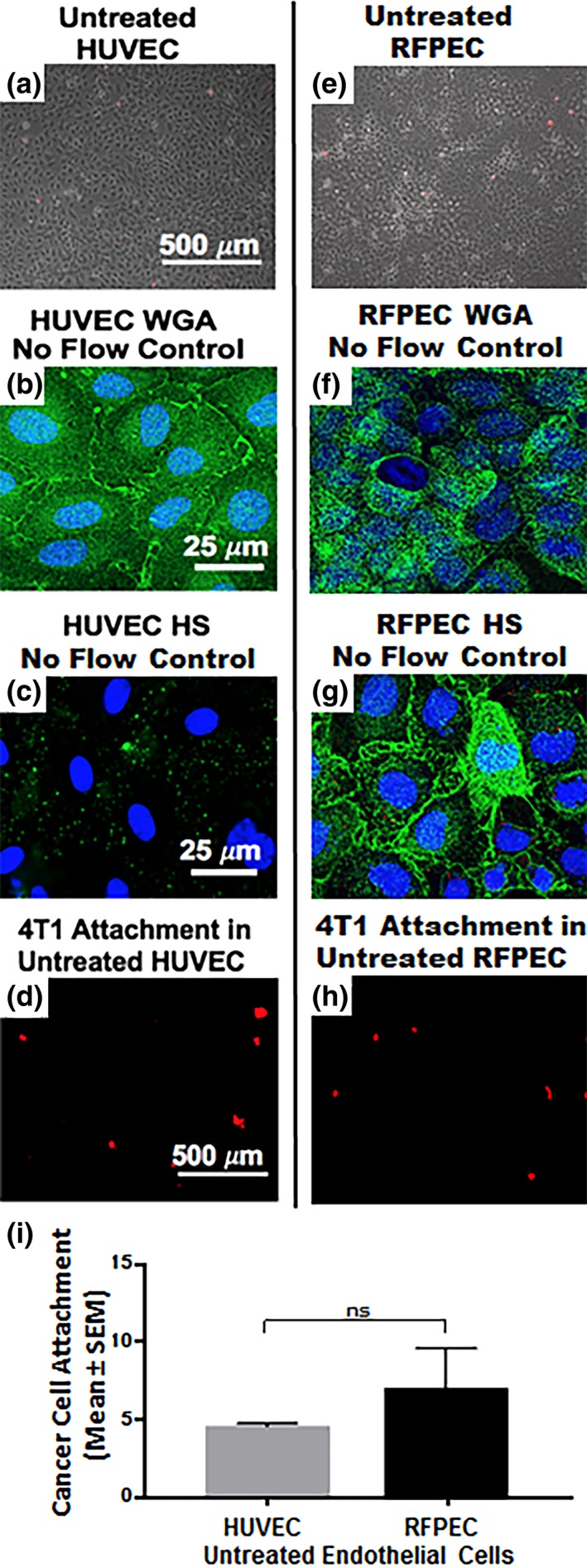
Preliminary data was collected regarding human ECs (HUVEC), their GCX, and the extent of their recruitment of 4T1 breast cancer cells in comparison with RFPEC controls. This human EC data confirms and validates the rat EC data that was the focus of this report. (a) Phase image shows that an untreated HUVEC layer is healthy. (b) WGA‐labeled untreated HUVEC reveals the presence of GCX even in the absence of physiological flow stimulation, which is usually required for in vitro human EC studies. (c) Low expression of HS is observed when these HUVEC, which lack flow stimulation, are labeled with HS antibody. The limited HS is presumably insufficient to expose the EC surface adhesion molecules to 4T1 breast cancer cells, because WGA is abundant enough to compensate and provide adequate coverage. (d) As expected, the level of attachment of 4T1 breast cancer cells to untreated HUVEC is low, similar to what was observed in untreated RFPEC. (e) Phase image shows healthy untreated RFPEC monolayer. (f) Fluorescent image of WGA‐labeled untreated RFPEC monolayer shows intact GCX. (g) Expression of HS is abundant in RFPEC without flow stimulation. (h) Picture shows attachment of 4T1 breast cancer cells to RFPEC monolayers. (i) Plot shows that the difference between RFPEC and HUVEC adhesiveness to 4T1 breast cancer cells is statistically not significant (ns). N = 3 for both cell types that were studied. EC, endothelial cell; GCX, glycocalyx; HS, heparan sulfate; RFPEC, rat fat pad endothelial cell; WGA, wheat germ agglutinin
AUTHOR CONTRIBUTIONS
S.A.M. and E.E.E. designed the experiments. S.A.M., M.Z., and M.P.J. performed the experiments and analyzed the data. S.A.M., I.C.H., V.P.T., M.J.N., and E.E.E. interpreted the results of the experiments. S.A.M., M.Z., and M.P.J. drafted the Figures and manuscript. S.A.M., I.C.M., M.P.J., and E.E.E. edited, revised, and approved the final manuscript. V.P.T., M.J.N., and E.E.E. supervised the project.
Supporting information
Appendix S1 Supporting Information.
ACKNOWLEDGMENTS
We thank Northeastern University Prof. Heather Clark for permitting us to use the confocal microscope in her laboratory. We also thank Jungwoo Lee (University of Massachusetts Amherst) who identified this contribution as the “Best Presentation” in the “Engineering in Cancer Biology and Therapy II” session of the 2016 AIChE Annual Meeting in San Francisco, CA.
Mensah SA, Harding IC, Zhang M, et al. Metastatic cancer cell attachment to endothelium is promoted by endothelial glycocalyx sialic acid degradation. AIChE J. 2019;65:e16634. 10.1002/aic.16634
This presentation was identified by Jungwoo Lee (University of Massachusetts Amherst) as the “Best Presentation” in the “Engineering in Cancer Biology and Therapy II” session of the 2016 AIChE Annual Meeting in San Francisco, CA.
Funding information National Institutes of Health National Heart, Lung, and Blood Institute, Grant/Award Number: K01/HL125499; National Science Foundation Division of Graduate Education, Grant/Award Number: GRFP/DGE‐096843
REFERENCES
- 1. Tian Q, Wang Y, Guo H, et al. Recent perspectives of management of breast cancer metastasis ‐ an update. J BUON. 2017;22(2):295‐300. [PubMed] [Google Scholar]
- 2. Sharma R, Sharma R, Khaket TP, Dutta C, Chakraborty B, Mukherjee TK. Breast cancer metastasis: putative therapeutic role of vascular cell adhesion molecule‐1. Cell Oncol (Dordr). 2017;40(3):199‐208. [DOI] [PubMed] [Google Scholar]
- 3. Li J, King MR. Adhesion receptors as therapeutic targets for circulating tumor cells. Front Oncol. 2012;2:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16(4):201‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reymond N, d'Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13(12):858‐870. [DOI] [PubMed] [Google Scholar]
- 6. Cai B, Fan J, Zeng M, Zhang L, Fu BM. Adhesion of malignant mammary tumor cells MDA‐MB‐231 to microvessel wall increases microvascular permeability via degradation of endothelial surface glycocalyx. J Appl Physiol (1985). 2012;113(7):1141‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Curry FE. Layer upon layer: the functional consequences of disrupting the glycocalyx‐endothelial barrier in vivo and in vitro. Cardiovasc Res. 2017;113(6):559‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghavampour S, Kleefeldt F, Bommel H, et al. Endothelial barrier function is differentially regulated by CEACAM1‐mediated signaling. FASEB J. 2018;32(10):5612‐5625. [DOI] [PubMed] [Google Scholar]
- 9. Mitra R, O'Neil GL, Harding IC, Cheng MJ, Mensah SA, Ebong EE. Glycocalyx in atherosclerosis‐relevant endothelium function and as a therapeutic target. Curr Atheroscler Rep. 2017;19(12):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pillinger NL, Kam P. Endothelial glycocalyx: basic science and clinical implications. Anaesth Intensive Care. 2017;45(3):295‐307. [DOI] [PubMed] [Google Scholar]
- 11. Rienks M, Carai P, van Teeffelen J, et al. SPARC preserves endothelial glycocalyx integrity, and protects against adverse cardiac inflammation and injury during viral myocarditis. Matrix Biol. 2018;74:21‐34. [DOI] [PubMed] [Google Scholar]
- 12. Yen WY, Cai B, Zeng M, Tarbell JM, Fu BM. Quantification of the endothelial surface glycocalyx on rat and mouse blood vessels. Microvasc Res. 2012;83(3):337‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perdicchio M, Ilarregui JM, Verstege MI, et al. Sialic acid‐modified antigens impose tolerance via inhibition of T‐cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci USA. 2016;113(12):3329‐3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cancel LM, Ebong EE, Mensah S, Hirschberg C, Tarbell JM. Endothelial glycocalyx, apoptosis and inflammation in an atherosclerotic mouse model. Atherosclerosis. 2016;252:136‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kataoka H, Ushiyama A, Akimoto Y, Matsubara S, Kawakami H, Iijima T. Structural behavior of the endothelial glycocalyx is associated with pathophysiologic status in septic mice: an integrated approach to analyzing the behavior and function of the glycocalyx using both electron and fluorescence intravital microscopy. Anesth Analg. 2017;125:874‐883. [DOI] [PubMed] [Google Scholar]
- 16. Cooper S, McDonald K, Burkat D, Leask RL. Stenosis hemodynamics disrupt the endothelial cell glycocalyx by MMP activity creating a proinflammatory environment. Ann Biomed Eng. 2017;45:2234‐2243. [DOI] [PubMed] [Google Scholar]
- 17. Ushiyama A, Kataoka H, Iijima T. Glycocalyx and its involvement in clinical pathophysiologies. J Intensive Care. 2016;4(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Becker BF, Jacob M, Leipert S, Salmon AH, Chappell D. Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br J Clin Pharmacol. 2015;80(3):389‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson AO, Shaw S. T cell adhesion to endothelium: the FRC conduit system and other anatomic and molecular features which facilitate the adhesion cascade in lymph node. Semin Immunol. 1993;5(4):271‐282. [DOI] [PubMed] [Google Scholar]
- 20. Celie JW, Beelen RH, van den Born J. Heparan sulfate proteoglycans in extravasation: assisting leukocyte guidance. Front Biosci (Landmark Ed). 2009;14:4932‐4949. [DOI] [PubMed] [Google Scholar]
- 21. Cutler BR, Gholami S, Chua JS, Kuberan B, Anandh Babu PV. Blueberry metabolites restore cell surface glycosaminoglycans and attenuate endothelial inflammation in diabetic human aortic endothelial cells. Int J Cardiol. 2018;261:155‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mulivor AW, Lipowsky HH. Role of glycocalyx in leukocyte‐endothelial cell adhesion. Am J Physiol Heart Circ Physiol. 2002;283(4):H1282‐H1291. [DOI] [PubMed] [Google Scholar]
- 23. Schmidt EP, Yang Y, Janssen WJ, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217‐1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Voyvodic PL, Min D, Liu R, et al. Loss of syndecan‐1 induces a pro‐inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J Biol Chem. 2014;289(14):9547‐9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fan J, Fu BM. Quantification of malignant breast cancer cell MDA‐MB‐231 transmigration across brain and lung microvascular endothelium. Ann Biomed Eng. 2016;44(7):2189‐2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Malek‐Zietek KE, Targosz‐Korecka M, Szymonski M. The impact of hyperglycemia on adhesion between endothelial and cancer cells revealed by single‐cell force spectroscopy. J Mol Recognit. 2017;30(9):1‐11. [DOI] [PubMed] [Google Scholar]
- 27. Gasic G, Gasic T. Removal of sialic acid from the cell coat in tumor cells and vascular endothelium, and its effects on metastasis. Proc Natl Acad Sci USA. 1962;48:1172‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Constantinescu AA, Vink H, Spaan JA. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol. 2003;23(9):1541‐1547. [DOI] [PubMed] [Google Scholar]
- 29. Lipowsky HH. The endothelial glycocalyx as a barrier to leukocyte adhesion and its mediation by extracellular proteases. Ann Biomed Eng. 2012;40(4):840‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chappell D, Dorfler N, Jacob M, et al. Glycocalyx protection reduces leukocyte adhesion after ischemia/reperfusion. Shock. 2010;34(2):133‐139. [DOI] [PubMed] [Google Scholar]
- 31. Henry CB, Duling BR. TNF‐alpha increases entry of macromolecules into luminal endothelial cell glycocalyx. Am J Physiol Heart Circ Physiol. 2000;279(6):H2815‐H2823. [DOI] [PubMed] [Google Scholar]
- 32. Lipowsky HH, Gao L, Lescanic A. Shedding of the endothelial glycocalyx in arterioles, capillaries, and venules and its effect on capillary hemodynamics during inflammation. Am J Physiol Heart Circ Physiol. 2011;301(6):H2235‐H2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mulivor AW, Lipowsky HH. Inflammation‐ and ischemia‐induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol. 2004;286(5):H1672‐H1680. [DOI] [PubMed] [Google Scholar]
- 34. Lala PK, Orucevic A. Role of nitric oxide in tumor progression: lessons from experimental tumors. Cancer Metastasis Rev. 1998;17(1):91‐106. [DOI] [PubMed] [Google Scholar]
- 35. Ren LR, Zhang LP, Huang SY, et al. Effects of sialidase NEU1 siRNA on proliferation, apoptosis, and invasion in human ovarian cancer. Mol Cell Biochem. 2016;411(1–2):213‐219. [DOI] [PubMed] [Google Scholar]
- 36. Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121‐167. [DOI] [PubMed] [Google Scholar]
- 37. Traving C, Schauer R. Structure, function and metabolism of sialic acids. Cell Mol Life Sci. 1998;54(12):1330‐1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schauer R. Sialic acids as regulators of molecular and cellular interactions. Curr Opin Struct Biol. 2009;19(5):507‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Narayanan S. Sialic acid as a tumor marker. Ann Clin Lab Sci. 1994;24(4):376‐384. [PubMed] [Google Scholar]
- 40. Varki A, Schauer RL, Schauer R. Sialic acids and other nonulosonic acids In: Varki A, Cummings RD, Esko JD, et al., eds. Essentials of Glycobiology. 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2017. [Google Scholar]
- 41. Warren L, Fuhrer JP, Buck CA. Surface glycoproteins of normal and transformed cells: a difference determined by sialic acid and a growth‐dependent sialyl transferase. Proc Natl Acad Sci USA. 1972;69(7):1838‐1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Popoff MR, Dodin A. Survey of neuraminidase production by Clostridium butyricum, Clostridium beijerinckii, and Clostridium difficile strains from clinical and nonclinical sources. J Clin Microbiol. 1985;22(5):873‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hou G, Liu G, Yang Y, et al. Neuraminidase 1 (NEU1) promotes proliferation and migration as a diagnostic and prognostic biomarker of hepatocellular carcinoma. Oncotarget. 2016;7(40):64957‐64966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zeng Y, Ebong EE, Fu BM, Tarbell JM. The structural stability of the endothelial glycocalyx after enzymatic removal of glycosaminoglycans. PLoS One. 2012;7(8):e43168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu W, Wang X, Bai K, Lin M, Sukhorukov G, Wang W. Microcapsules functionalized with neuraminidase can enter vascular endothelial cells in vitro. J R Soc Interface. 2014;11(101):20141027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mensah SA, Cheng MJ, Homayoni H, Plouffe BD, Coury AJ, Ebong EE. Regeneration of glycocalyx by heparan sulfate and sphingosine 1‐phosphate restores inter‐endothelial communication. PLoS One. 2017;12(10):e0186116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kataoka H, Ushiyama A, Kawakami H, Akimoto Y, Matsubara S, Iijima T. Fluorescent imaging of endothelial glycocalyx layer with wheat germ agglutinin using intravital microscopy. Microsc Res Tech. 2016;79(1):31‐37. [DOI] [PubMed] [Google Scholar]
- 48. Sukhikh GT, Ziganshina MM, Nizyaeva NV, et al. Differences of glycocalyx composition in the structural elements of placenta in preeclampsia. Placenta. 2016;43:69‐76. [DOI] [PubMed] [Google Scholar]
- 49. Reitsma S, oude Egbrink MG, Vink H, et al. Endothelial glycocalyx structure in the intact carotid artery: a two‐photon laser scanning microscopy study. J Vasc Res. 2011;48(4):297‐306. [DOI] [PubMed] [Google Scholar]
- 50. Arkill KP, Knupp C, Michel CC, et al. Similar endothelial glycocalyx structures in microvessels from a range of mammalian tissues: evidence for a common filtering mechanism? Biophys J. 2011;101(5):1046‐1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Zandvoort M, Engels W, Douma K, et al. Two‐photon microscopy for imaging of the (atherosclerotic) vascular wall: a proof of concept study. J Vasc Res. 2004;41(1):54‐63. [DOI] [PubMed] [Google Scholar]
- 52. Ebong EE, Macaluso FP, Spray DC, Tarbell JM. Imaging the endothelial glycocalyx in vitro by rapid freezing/freeze substitution transmission electron microscopy. Arterioscler Thromb Vasc Biol. 2011;31(8):1908‐1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Murphy LS, Wickersham N, McNeil JB, et al. Endothelial glycocalyx degradation is more severe in patients with non‐pulmonary sepsis compared to pulmonary sepsis and associates with risk of ARDS and other organ dysfunction. Ann Intensive Care. 2017;7(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Deng X, Zhang J, Liu Y, Chen L, Yu C. TNF‐alpha regulates the proteolytic degradation of ST6Gal‐1 and endothelial cell‐cell junctions through upregulating expression of BACE1. Sci Rep. 2017;7:40256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cioffi DL, Pandey S, Alvarez DF, Cioffi EA. Terminal sialic acids are an important determinant of pulmonary endothelial barrier integrity. Am J Physiol Lung Cell Mol Physiol. 2012;302(10):L1067‐L1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Plattner VE, Ratzinger G, Engleder ET, Gallauner S, Gabor F, Wirth M. Alteration of the glycosylation pattern of monocytic THP‐1 cells upon differentiation and its impact on lectin‐mediated drug delivery. Eur J Pharm Biopharm. 2009;73(3):324‐330. [DOI] [PubMed] [Google Scholar]
- 57. Ghazarian H, Idoni B, Oppenheimer SB. A glycobiology review: carbohydrates, lectins and implications in cancer therapeutics. Acta Histochem. 2011;113(3):236‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhu D, Cheng CF, Pauli BU. Blocking of lung endothelial cell adhesion molecule‐1 (Lu‐ECAM‐1) inhibits murine melanoma lung metastasis. J Clin Invest. 1992;89(6):1718‐1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang J, Chen J, Chesla SE, et al. Quantifying the effects of molecular orientation and length on two‐dimensional receptor‐ligand binding kinetics. J Biol Chem. 2004;279(43):44915‐44923. [DOI] [PubMed] [Google Scholar]
- 60. Cheng MJ, Bal NN, Prabakaran P, et al. Ultrasmall gold nanorods: synthesis and glycocalyx‐related permeability in human endothelial cells. Int J Nanomedicine. 2019;14:319‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yao Y, Rabodzey A, Dewey CF Jr. Glycocalyx modulates the motility and proliferative response of vascular endothelium to fluid shear stress. Am J Physiol Heart Circ Physiol. 2007;293(2):H1023‐H1030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information.