

Abstract
Mitochondrial disorders are a group of metabolic conditions caused by impairment of the oxidative phosphorylation system. There is currently no clear evidence supporting any pharmacological interventions for most mitochondrial disorders, except for coenzyme Q10 deficiencies, Leber hereditary optic neuropathy, and mitochondrial neurogastrointestinal encephalomyopathy. Furthermore, some drugs may potentially have detrimental effects on mitochondrial dysfunction. Drugs known to be toxic for mitochondrial functions should be avoided whenever possible. Mitochondrial patients needing one of these treatments should be carefully monitored, clinically and by laboratory exams, including creatine kinase and lactate. In the era of molecular and ‘personalized’ medicine, many different physicians (not only neurologists) should be aware of the basic principles of mitochondrial medicine and its therapeutic implications. Multicenter collaboration is essential for the advancement of therapy for mitochondrial disorders. Whenever possible, randomized clinical trials are necessary to establish efficacy and safety of drugs. In this review we discuss in an accessible way the therapeutic approaches and perspectives in mitochondrial disorders. We will also provide an overview of the drugs that should be used with caution in these patients.
Keywords: coenzyme Q10, drugs, mitochondria, mtDNA, mitochondrial diseases, toxicity
Introduction
Mitochondrial disorders (MD) are an extraordinarily complex group of diseases caused by impairment of mitochondrial functions, especially of the mitochondrial respiratory chain.1 Mitochondria are dynamic organelles whose maintenance requires about 1500 proteins. Mutations in either the mitochondrial (mtDNA) or nuclear (nDNA) genome can disrupt myriads of metabolic and homeostatic functions of the cell.2
For this reason, from a genetic perspective, there are two major groups of MD: the diseases due to defects in mtDNA and those due to mutations in nDNA. Only the last ones follow the Mendelian laws of inheritance. MtDNA large-scale single deletions are sporadic and not inheritable. Inherited MtDNA point mutations follow the rules of mitochondrial inheritance (maternal transmission, mitotic segregation, heteroplasmy, and the threshold effect).1
Cells contain multiple copies of mtDNA (polyplasmy), which, in usual conditions, are identical to one another (homoplasmy). Heteroplasmy refers to the coexistence of two populations of mitochondria: normal and mutated. Mutated mtDNA has to reach a certain critical ‘threshold’ in a tissue to cause dysfunction (threshold effect). Because of the mitotic segregation (which is the random assignment of mitochondria between the daughter cells), the mutation load can change from one cell generation to the next, and therefore it can either surpass or fall below the pathogenic threshold (Figure 1).1
Figure 1.
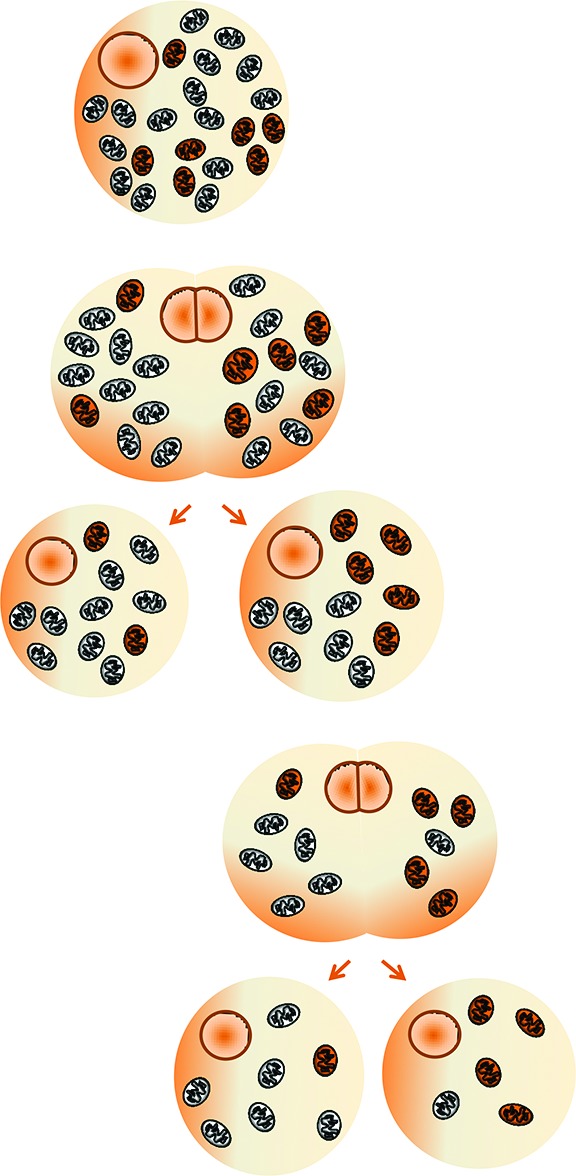
Heteroplasmy and mitotic segregation: schematic representation. Because of the mitotic segregation (random share-out of mutated and nonmutated mitochondria between the daughter cells), the mutation load can change from one cell generation to the next, and with time, it can either surpass or fall below the pathogenic threshold.
It has been observed that cells attempt to maintain wild-type mtDNA density through cell volume reduction, and thus power demand reduction, until a minimum volume. These studies provide evidence for the physiological significance of mtDNA density and emphasize the need for performing single-cell volume measurements jointly with mtDNA quantification.3 This new concept of ‘mtDNA density’ may have implications for the development of new therapies for MD due to mtDNA mutations.3
Recently, the paradigm of maternal inheritance of mtDNA has been questioned.4 Biparental mtDNA transmission was reported in three unrelated multigeneration families, with an autosomal dominant inheritance mode.4 In our opinion, these exceptional families where paternal mtDNA could be passed to the offspring do not invalidate the central dogma of maternal inheritance of mtDNA.4 However, we agree that there is a strong need for further study aimed at elucidating the molecular mechanisms and the biomedical implications of this unusual mode of inheritance.4
The effects of mutations (in both nuclear and mitochondrial genomes) affecting the respiratory chain may be variable, heterogeneous, and multisystemic, with possible involvement of nearly all tissues and organs, including visual and auditory pathways, heart, central nervous system (CNS), and skeletal muscle (Tables 1 and 2). Some of the ‘red flags’ for MD are exercise intolerance and myopathy, ophthalmoparesis and/or eyelid ptosis, sensorineural hearing loss, axonal neuropathy, hypertrophic cardiomyopathy, optic neuropathy, pigmentary retinopathy, diabetes, short stature, migraine, and lactic acidosis.1
Table 1.
Some of the most frequent clinical features of mitochondrial diseases.
| Central nervous system | Migraine, myoclonus, cognitive impairment, stroke-like episodes, seizures, ataxia, dystonia, parkinsonism, tremor, psychiatric involvement |
| Peripheral nervous system | Axonal multifocal neuropathy |
| Skeletal muscle | Weakness, ophthalmoparesis, eyelid ptosis, exercise intolerance, myoglobinuria, respiratory impairment, hypotonia |
| Visual system | Pigmentary retinopathy, cataract, optic neuropathy |
| Acoustic system | Sensorineural hearing loss |
| Digestive system | Malabsorbition, intestinal pseudo-obstruction |
| Kidney | Tubulopathy, Fanconi syndrome |
| Metabolic/endocrine apparatus | Lactic acidosis, multiple lipomatosis, short stature, diabetes, hypothyroidism, hypoparathyroidism |
| Heart | Cardiomyopathy, conduction system defects, Wolff–Parkinson–White syndrome |
| Hematopoietic system | Sideroblastic anemia |
Table 2.
Mitochondrial syndromes. Selected, well-established mitochondrial syndromes are shown. However, many patients do not show these specific clinical pictures and are affected by a variety of complex syndromes (myopathies, neuropathies, cardiomyopathies, encephalomyopathies, multisystemic diseases, etc.) or partial pictures.
| Main feature(s) | Associated feature(s) | Inheritance | Common genetic findings | Treatment of choice | |
|---|---|---|---|---|---|
| Alpers syndrome | Childhood myocerebrohepatopathy | Autosomal recessive | POLG mutations | Symptomatic (avoid valproate) | |
| Autosomal dominant optic atrophy (ADOA) | Optic neuropathy (blindness) | Autosomal dominant | OPA1 mutations | Symptomatic | |
| Coenzyme Q10 deficiency | Ataxia or myopathy or multisystem disease | Autosomal recessive | Different genes | Coenzyme Q10 | |
| Kearns–Sayre Syndrome (KSS) | Ocular myopathy (ptosis, ophthalmoparesis) | Ataxia, cardiac conduction defects | Sporadic | Single large-scale deletion of mtDNA | Symptomatic |
| Leber hereditary optic neuropathy (LHON) | Optic neuropathy (blindness) | Maternal (low penetrance, higher in male smokers) | Different mtDNA mutations | Idebenone | |
| Leigh syndrome | Severe pediatric encephalopathy | Autosomal recessive or maternal | Different nuclear genes or mtDNA (e.g. m.8993T>G) | Symptomatic | |
| Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) | Stroke-like episodes | Hearing loss, diabetes | Maternal | m.3243A>G | Symptomatic |
| Myoclonic epilepsy with ragged red fiber (MERRF) | Myoclonus | Ataxia, myopathy | Maternal | m.8344A>G | Symptomatic (e.g. Levetiracetam) |
| Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE) | Gastrointestinal dysmotility | Leukodystrophy, ocular myopathy, peripheral neuropathy | Autosomal recessive | TYMP mutations | Allogeneic hematopoietic stem cell transplantation |
| Neuropathy, Ataxia, Retinitis Pigmentosa (NARP) | Ataxia | Neuropathy, Retinitis Pigmentosa | Maternal | m.8993T>G | Symptomatic |
| Non syndromic hearing loss (NSHL) | Hearing loss | Maternal | m.1555A>G | Symptomatic (avoid aminoglycosides) | |
| Progressive external ophthalmoplegia (PEO) | Ocular myopathy | Myopathy | Autosomal dominant, recessive, maternal, or sporadic | Different nuclear genes, different mtDNA mutations, mtDNA single deletion | Symptomatic |
mtDNA, mitochondrial DNA.
The heterogeneity is further increased by other not-modifiable factors, including gender5 and nuclear and mtDNA genetic background,6 and by potentially modifiable gene–environment interactions (i.e. smoke5 and drugs).7
Because of the multisystem involvement, a wide range of medical specialists (general practitioners, internists, pediatricians, cardiologists, endocrinologist, audiologists, ophthalmologists, etc.) may first encounter these patients, and the clinical approach may become a ‘diagnostic odyssey’.8 The prevalence of MD is 1–2/10,000, and in fact they represent one of the most frequently encountered metabolic conditions.9 Therefore, we believe that in the era of molecular and ‘personalized’ medicine, physicians (not only neurologists) should know the basic principles of mitochondrial medicine and its therapeutic implications.
This review will focus on the ‘properly called’ MD (diseases of the mitochondrial respiratory chain). Other potentially treatable metabolic and neurodegenerative conditions ‘indirectly’ linked to mitochondrial dysfunction will not be discussed here. These diseases, which are not directly caused by dysfunction of the electron transport chain, include Friedreich ataxia,10 ethylmalonic encephalopathy (treated with metronidazole plus N-acetylcysteine),11 acyl-CoA dehydrogenation deficiency (treated with riboflavin),12 primary carnitine deficiency, vitamin E deficiency, and secondary coenzyme Q10 deficiencies (treated with administration of the defective metabolite).7
Here, we will discuss the therapeutic approaches and perspectives in MD. We will also provide an overview of the drugs that should be used with caution in these patients. We searched PubMed in May 2019 for all articles about ‘mitochondrial disorder* OR mitochondrial disease* OR mitochondrial *myopath*’, and we reviewed the abstracts to identify relevant publications.
Etiopathogenetic approaches
Can we treat mitochondrial dysfunction?
Despite great progress in the molecular pathogenesis of MD, the treatment possibilities are still very limited.7 Apart from symptomatic treatments, therapies that have been attempted include antioxidants, respiratory chain cofactors, and other metabolites. However, there is no clear evidence supporting the use of any of these interventions in most MD,13 and further research is needed. There have been very few randomized controlled clinical trials, the majority of which were short and involved few participants with heterogeneous phenotypes.13
Coenzyme Q10, or ubiquinone, is widely used for MD, because of the generally positive anecdotal data together with the lack of major toxicity.7 This cofactor is located on the inner mitochondrial membrane and shuttles electrons from complexes I and II and from the oxidation of fatty acids and branched chain aminoacids to complex III of the mitochondrial respiratory chain. Reduced coenzyme Q10 has also antioxidant properties.7 A randomized, double-blind crossover trial with coenzyme Q10 was performed on eight patients with MD.14 Several measures showed a trend toward improvement on treatment.14 In another more recent crossover trial, 30 patients with MD received coenzyme Q10 (1200 mg/day) for 60 days.15 The treatment had some effects on aerobic capacity and postexercise lactate, without affecting other relevant variables.15
Unfortunately, crystalline CoQ10 is lipophilic, water insoluble, and poorly absorbed in the gut.16 Therefore, there is a strong need for using solubilized formulations with certified bioavailability. Coenzyme Q10 analogs that target mitochondria, such as ‘MitoQ’, have been also developed and widely used as antioxidants,17 but they have not been evaluated yet in patients with MD.
Regarding other metabolites and cofactors, carnitine (up to 3 g/day) resulted in some improvement in isolated cases, but objective evidence is unavailable.7 However, it has been observed that chronic oral L-carnitine supplementation to MD patients increases a plasma metabolite (trimethylamine oxide) that has been linked to cardiovascular disease.18 Therefore, further studies to evaluate both the efficacy and long-term safety are warranted before routine carnitine supplementation.18 A randomized, controlled trial with creatine on six MD patients showed some improvement of high-intensity activities.19 Another controlled crossover trial studied the effects of creatine in 16 participants with mitochondrial myopathy.20 No significant effects of treatment were noted.20 More recently, a randomized, double-blind, placebo-controlled, crossover study with a combination therapy (creatine, coenzyme Q10, and lipoic acid) was performed on 17 patients with heterogeneous MD.21 The combination therapy could lower lactate and oxidative stress biomarkers.21 Riboflavin is a cofactor for the respiratory chain that was reported to elicit some positive effects in children with complex II deficiency22 and other rare patients with specific biochemical defects, such as the myopathic form of coenzyme Q10 deficiency due to mutations in the electron-transferring flavoprotein dehydrogenase (ETFDH) gene.23
A high fat (or ketogenic)24 diet can be beneficial for some MD patients with complex I deficiency, but further clinical studies are needed.25 However, to date, no specific dietary manipulation has shown consistent benefit.
Looking forward to an effective treatment, one of the choice therapies, well tolerated and safe, is coenzyme Q10, possibly in its reduced form (ubiquinol, at least 300 mg a day), alone or combined with other metabolites (i.e. riboflavin, α-lipoic acid, or creatine).7
Can we treat oxidative stress?
A plausible pathogenic mechanism in MD is oxidative stress.7 Protein oxidative damage is increased in patients with mitochondrial myopathy, especially in the more severely affected ones, supporting the tight links between mitochondrial dysfunction and oxidative stress.26
There has been interest in cysteine donors. Glutathione deficiency has been reported in MD, and the biosynthesis of glutathione depends on cysteine availability. Our group performed a crossover double-blind trial to evaluate if 30-day supplementation with a whey-based cysteine donor could modify oxidative stress markers.26 Clinical scores and lactate at rest and after exercise were not modified by the 30-day supplementation, but the cysteine donor markedly improved oxidative stress markers as well as total glutathione levels.26
Cysteine is also required for the 2-thiomodification of mitochondrial tRNAs.27 Interestingly, supplementation with L-cysteine could partially rescue the mitochondrial translation defect in fibroblasts carrying the m.8344A>G or the m.3243A>G mutation.27 N-acetyl-cysteine had some beneficial effects on mitochondrial translation in fibroblasts from patients with autosomal recessive mutations in genes affecting mitochondrial translation.27 Further studies are needed to explore the full potential of cysteine supplementation as a treatment for patients with MD.
A recent preclinical study evaluated the therapeutic potential of cysteamine bitartrate in three MD models spanning three evolutionarily distinct species (Caenorhabditis elegans, zebrafish, human cells).28 Cysteamine bitartrate is an approved therapy for nephropathic cystinosis, potentially able to enhance glutathione biosynthesis.28 It could improve mitochondrial function in these models, even if not through modulation of total glutathione levels. Careful consideration is required to evaluate safe and effective dosages of cysteamine bitartrate for clinical trials.28
Furthermore, treatment of complex I deficient cells with JP4-039, a novel mitochondrion-targeted reactive oxygen species (ROS) scavenger, could decrease levels of superoxide and increase basal and maximal respiratory rate.29
Another study showed that the mitochondrial ROS-redox modulator KH176 could have some beneficial effects in a mammalian model of Leigh disease.30 Leigh disease is a devastating pediatric neurometabolic disorder, for which an effective treatment is not available. It has also been suggested that the antioxidant EPI-743 may have some beneficial effects in MD, especially in Leigh disease. This molecule is supposed to enhance the biosynthesis of glutathione.31 Definitive results of well-conducted clinical trials are not available yet.32
Interestingly, exercise may also protect mitochondria against oxidative damage, at least in animal models.33 Chronic normobaric hypoxic conditions were also able to prevent the development of the disease in a mouse model of MD (Ndufs4 KO).34 Median life span was remarkably increased from 58 to 270 days, whereas hyperoxia had the opposite effect. The detailed mechanisms are still unclear, but oxidative stress is likely involved.34
It must be noted that mitochondrial ROS production at physiological concentrations appears to be integral to cell signaling and the modulation of gene expression.2 Further studies are strongly needed to clarify if oxidative stress may represent a therapeutic target. In the meanwhile, antioxidants should be used with caution in MD as well as in other diseases linked to mitochondrial dysfunction.
The example of primary coenzyme Q10 deficiency
Primary coenzyme Q10 deficiency is a very rare autosomal recessive MD.35 It is due to mutations in coenzyme Q10 biosynthetic genes, and it has been mainly associated with the infantile multisystemic and cerebellar ataxic phenotypes.7
Coenzyme Q10 deficiency is a treatable condition; therefore it is important to consider this diagnostic possibility.7 An early supplementation with high-dose coenzyme Q10 could radically change the natural history. Patients with all phenotypes of coenzyme Q10 deficiency have shown improvement with oral coenzyme Q10 administration. CNS symptoms are only partially ameliorated, likely because of irreversible neural damage before treatment and because of poor penetration of coenzyme Q10 across the blood–brain barrier.7
Primary coenzyme Q10 deficiency should be treated with coenzyme Q10 supplementation and not with short-tail Q10 analogs, such as idebenone, because these analogs cannot replace coenzyme Q10 in the respiratory chain.7 Patients were given various doses of coenzyme Q10 ranging from 90 to 2000 mg daily. The beneficial effects of exogenous coenzyme Q10 require high doses and long-term administration.7
A therapeutic attempt with coenzyme Q10 should be considered in every MD patient with myopathy and/or ataxia in whom coenzyme Q10 deficiency has not been excluded.7
Although some human coenzyme Q10 deficiencies respond to dietary supplementation with coenzyme Q10, in general the uptake and assimilation of this very hydrophobic lipid is inefficient.36 Simple coenzyme Q10 derivatives may act to enhance biosynthesis of coenzyme Q10 or may bypass certain deficient steps in the coenzyme Q10 biosynthetic pathway.36 For instance, β-resorcylic acid was reported to be more effective than coenzyme Q10 in a mouse model of coenzyme Q10 deficiency with an encephalopathic phenotype.37 β-resorcylic acid is an analog of the coenzyme Q10 precursor 4-hydroxybenzoic acid.37 Further studies are needed to evaluate if its use could be considered in human coenzyme Q10 deficiencies.
Idebenone in Leber disease
Idebenone is a coenzyme Q10 short-tail analog with a more favorable pharmacokinetic profile.7 In recent years, it has been largely used in Leber hereditary optic neuropathy. Leber disease is a mtDNA-related MD mainly involving the optic nerves. It may lead to blindess, especially in male smokers.5
Idebenone could increase ATP production and reduce ROS levels in Leber fibroblasts, but the effect was cell specific and partial. Other Q10 analogs had more variable effects.38
A 24-week multicenter, double-blind, randomized, placebo-controlled trial with idebenone was performed on 85 patients with Leber hereditary optic neuropathy.39 No difference in visual recovery was reported, but post hoc analysis suggested that patients with discordant visual acuities were the most likely to benefit from this treatment.39
A large retrospective study has also suggested that idebenone treatment could significantly improve the frequency of visual recovery in Leber patients, possibly changing the natural history of this disease.40
Further studies are still needed to definitively confirm these preliminary observations.
Other medications, including elamipretide (a small molecule that binds to cardiolipin and stabilizes the inner mitochondrial membrane) and curcumin, are currently under study for Leber disease.31 Furthermore, a number of clinical trials are investigating the efficacy of viral-based gene therapy for Leber patients harboring the m.11778G>A mtDNA mutation,41 but definitive results are not available to date.
The particular case of mitochondrial neurogastrointestinal encephalomyopathy
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an autosomal recessive disease that usually leads to death in early adulthood.42 It is caused by mutations of thymidine phosphorylase (TYMP) gene that lead to deoxynucleoside accumulation in plasma and tissues and subsequently to mitochondrial dysfunction. MNGIE is characterized by gastrointestinal dysmotility with cachexia, leukodystrophy, ocular myopathy, and peripheral neuropathy.42
Therapeutic options aimed at counterbalance the nucleoside accumulation are available, and in fact MNGIE is one of the few MD susceptible to effective treatment.42 Attempted strategies include platelets infusion to restore TYMP activity, continuous ambulatory peritoneal dialysis, hemodialysis, enzyme replacement therapy using recombinant Escherichia coli thymidine phosphorylase entrapped in encapsulated autologous erythrocytes (EE-TP), and liver transplantation.43–49
To date, the treatment of choice is allogeneic hematopoietic stem cell transplantation (HSCT). HSCT is available only for very selected patients50 and may restore the thymidine phosphorylase enzyme function and improve the clinical picture, even if the complication rate is very high in these patients. Hematopoietic gene therapy may represent a promising permanent approach for MNGIE in the future.42
Unfortunately, these approaches are not useful for most MDs that are not caused by the accumulation of noxious metabolites.
Innovative metabolic strategies
Targeting mitochondrial biogenesis is a possibility for treating MD.51 Medications able to enhance mitochondrial biogenesis, and therefore oxidative metabolism, have been defined exercise-mimetic drugs,51 because exercise has been shown to promote mitochondrial biogenesis. In fact, exercise may provide a safe therapeutic option to patients with mitochondrial myopathy, benefiting the biochemical and clinical endpoints.51
Fibrates, which are peroxisomal proliferator-activated receptors (PPAR) pan-agonists, have been suggested as a potential therapeutic agents to correct oxidative phosphorylation in MD.52 Bezafibrate was able to delay the accumulation of cytochrome c (COX)-negative fibers and multiple mtDNA deletions in Twinkle-mutated mice.53
More recently, these positive preliminary findings on fibrates were not confirmed in three recombinant mouse models characterized by defective COX activity.54 Furthermore, bezafibrate showed adverse effects in these models (myopathy, marked hepatomegaly, and body weight loss).54 Another strategy appeared more promising. Two principal systems, PPARs and PGC-1α (PPAR-γ coactivator-1α), are supposed to increase biogenesis of mitochondria and thus may enhance aerobic metabolism acting on partially overlapping sets of genes. AMP-dependent kinase (AMPK) is a key regulator of PGC-1α. Double-recombinant animal models overexpressing PGC-1α in skeletal muscle on a Surf1 knockout background showed marked induction of mitochondrial biogenesis and increased mitochondrial respiratory activities, including COX.54 Interestingly, treatment with the AMPK agonist AICAR (5-aminoimidazole-4-carboxamide ribonucleoside) led to partial correction of COX deficiency in COX-deficient models, with significant motor improvement in the knockout/knockin Sco2 mouse. Therefore, pharmacological activation of mitochondrial biogenesis may be a potentially useful approach.54
Another recent study screened 10 different compounds using fibroblasts derived from seven patients with complex I deficiency, harboring different mutations. AICAR was found to be the most effective molecule, improving growth and ATP content and decreasing oxidative stress; AICAR also activated AMPK, thus increasing mitochondrial biogenesis.55 Therefore, further studies on this drug are warranted. As AICAR may have effects on different homeostatic and regulatory pathways,56 further studies are needed to establish its safety and translatability to humans.55
A study recently appeared in Nature57 showed that de novo nicotinamide adenine dinucleotide (NAD+) synthesis enhanced mitochondrial function. NAD+ production could be pharmacologically increased, ultimately improving mitochondrial function.57
Nicotinamide riboside, a form of vitamin B3 and a natural precursor of NAD+, may be a promising treatment strategy for MD.58 It could prevent both the development and progression of mitochondrial myopathy in a mouse model.58 The treatment also resulted in a significant induction of mitochondrial biogenesis and oxidative metabolism.58 A NAD+ precursor compound named acipimox may also be tested, but data on MD are not available yet.59
Very recently, the administration of febuxostat and inosine was suggested as another potential strategy to enhance cellular ATP levels in MD patients, but further studies are needed.60
Nucleoside supplementation may be beneficial in mtDNA depletion syndromes by substrate enhancement of the purine nucleoside salvage pathway, thus improving liver pathology in these young patients. Studies are also needed to confirm this hypothesis.61
Deoxynucleoside substrate enhancement is a novel therapy, which may ameliorate thymidine kinase 2 (TK2) deficiency in patients.62 TK2 is required for mtDNA maintenance, and autosomal recessive TK2 gene mutations cause depletion and multiple deletions of mtDNA that manifest predominantly as a progressive myopathy, which usually begins in childhood.63 A recent open-label clinical study investigated deoxynucleoside administration to 16 TK2-deficient patients. This study indicated favorable side-effect profiles and clinical efficacy of deoxynucleoside therapies for TK2 deficiency.63
Another recent suggestion is the potential possibility of bypassing blockade of the complex III–IV segment of the respiratory chain by alternative oxidase (AOX). AOX is a nonmammalian enzyme that shunts electrons from quinols directly to oxygen, thus restoring electron flow upstream of complex III.64 AOX transgene could prevent lethal mitochondrial cardiomyopathy in a mouse model of complex III deficiency,64 and it may hypothetically represent a potential therapeutic strategy for MD patients with this specific biochemical defect. A similar study in another MD model (COX15 knockout mouse)65 led to decreased life span and a substantial worsening of the myopathy. Decreased ROS production led to impaired AMPK/PGC-1α signaling and compromised the compensatory responses.65 Noticeably, the antioxidant N-acetylcysteine had a similar effect, decreasing the life span in these mice.65 This study highlighted the benefits of ROS signaling and the potential hazards of indiscriminate antioxidant treatment. However, the idea of bypassing blockade of the respiratory chain is intriguing and further studies may be warranted.
Furthermore, the bromodomain inhibitor I-BET525762A could remodel the mitochondrial proteome, thus increasing levels and activity of the respiratory chain and leading to rescue of the bioenergetic defects and cell death caused by blockade of Complex I.66
A recent pilot study reported that the bacterial protein CNF1 could boost the mitochondrial ATP production in cells derived from a patient with the m.8344A>G mitochondrial mutation, likely acting on the actin cytoskeleton.67 Further studies are needed to clarify if the cytoskeleton may represent a pharmacological target against MD.
Targeting the regulation of lipid dynamics may represent another possibility. For instance the drug, elamipretide, targets the lipid cardiolipin and may stabilize the mitochondrial lipid structure.68 This drug has been assessed in mitochondrial myopathy patients and could improve exercise intolerance and walking distance when administered at the higher dose.68 Further studies are ongoing.
Mitochondria are dynamic organelles undergoing frequent fission and fusion cycles that result in major morphological changes. OPA1 protein is involved in these modifications, and therefore autosomal dominant optic atrophy (ADOA), which is due to OPA1 mutations, has been defined as a disorder of mitochondrial fusion. Active fusion is important to maintain mitochondrial integrity, to reduce ROS production and to activate mtDNA replication to elevate mtDNA copy number. Pharmacological modulation of mitochondrial fusion (e.g. by the hydroxylamine derivative BGP-15) may represent a potential therapeutic strategy for ADOA and possibly other MD, but further studies are needed.69
Mitochonic acid (MA-5) could increase ATP and improve mitochondrial function in MD fibroblasts, and it prolonged the life span of the disease model ‘Mitomouse’. MA-5 was reported to facilitate mitochondrial ATP production and to modulate oxidative stress.70 Furthermore, it may reduce mitochondrial fragmentation and restore crista shape and dynamics.70 Further studies are needed to clarify if MA-5 may be useful in MD patients.
Rapamycin is a mTORC1 inhibitor that had beneficial effects in all the models of MD tested, independently of the genetic lesion.71 Given the broad effects of mTOR inhibition, including immunosuppressive action, side effects are a major concern for the use of these compounds. Rapamycin analogs with a better safety profile are being developed. Similarly, rapamycin administration is not a valid therapeutic strategy for every case of MD, and appropriate investigations on patients are warranted.71
Mitochondrial RNA translation may also represent a potential treatment target, but further studies are needed.73
Gene therapy and germline therapy
Future approaches to MD may also include gene therapy and germline therapy. For MD due to mutations in nuclear genes, the problems are not different from those valid for other Mendelian disorders, including choice of appropriate vectors, delivery to the affected tissues, and potential immunological reactions.7 The problems are even more complex for mtDNA-related diseases because of heteroplasmy and polyplasmy.
To achieve mitochondrial gene therapy, developing a mitochondrial transgene expression system that produces therapeutic proteins in mitochondria is essential. For instance, mitochondrial transfection may be achieved using a liposome-based carrier for delivering a cargo to mitochondria via membrane fusion.74
Mitochondria-targeted endonucleases may provide an alternative strategy for treating mtDNA-related MD via targeted destruction of the mutant mtDNA and induction of heteroplasmic shifting.75 This was successfully achieved in cell lines harboring a high proportion of the m.3243A>G mtDNA mutation, suggesting a potential new approach for the treatment of MD as well as the prevention of germline transmission of mutant mtDNA.75 Two recent studies showed the feasibility of this approach in vivo as well.76,77
Interestingly, it has been observed that mtDNA could be efficiently replaced in mature primate oocytes by spindle–chromosomal complex transfer from one egg to an enucleated, mitochondrial-replete egg.78 The reconstructed oocytes with the replaced mtDNA were capable of supporting normal fertilization, embryo development and could produce healthy offspring. Genetic analysis confirmed that nuclear DNA in the infants born so far originated from the spindle donors whereas mtDNA came from the cytoplasm donors.78
In future, similar approaches may offer a reproductive option to prevent mutated mtDNA transmission in MD families. A comprehensive discussion of these options goes beyond the aims of this paper. Recent approaches and advances in the field of mitochondrial replacement therapies are excellently discussed elsewhere.79–81
Treatment of specific clinical features
Stroke-like episodes
‘Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes’ (MELAS) is a mitochondrial phenotype mainly associated with the m.3243A>G mutation of the mtDNA.82 A mitochondrial stroke-like episode is a subacute, evolving brain syndrome linked to seizure activity in genetically determined MD. Unfortunately, experience with the treatment of stroke-like episodes derives only from case report or small series. ‘MELAS’ denote patients with histological, biochemical, and/or molecular evidence of MD who experience stroke-like episodes.82
A double-blind, placebo-controlled, randomized, crossover trial of the lactate-lowering dichloroacetate in MELAS patients harboring the m.3243A>G mutation had to be terminated because of peripheral nerve toxicity, which overshadowed any potential beneficial effect.83
Several evidences point toward a benefit of arginine, a nitric oxide precursor, to both prevent and reduce the severity of stroke-like episodes in MELAS patients.84
Administration of L-arginine, a nitric oxide precursor, to MELAS patients has been suggested to reduce neurological symptoms due to stroke-like episodes and to prevent recurrences.85 Two years of supplementation with oral L-arginine could significantly improve endothelial function.86 L-arginine administration within 30 minutes of a stroke-like episode could decrease frequency and severity of stroke-like episodes.87 L-arginine is usually applied intravenously in a dosage of 0.4–0.5 g/kg.7
Recently, the efficacy and safety of oral and intravenous L-arginine was studied in an open-label trial on pediatric and adult patients with MELAS.88 Oral L-arginine (0.3–0.5 g/kg/day) could extend the interictal phase and decrease the incidence and severity of stroke-like episodes. L-arginine was well tolerated, and no treatment-related adverse events were reported.88 Arginine is safe in pediatric MD patients as well as in adults.89
However, we do not advocate the use of intravenous L-arginine as part of the routine treatment of stroke-like episodes. There is no robust scientific evidence supporting its use. Placebo-controlled studies of oral and intravenous L-arginine in patients with MELAS are strongly needed.
Until specific drugs will be available, the treatment of choice of a stroke-like episode is based on antiepileptic drugs (excluding valproic acid) and other supportive treatments. During stroke-like episodes, steroids may be safely used and could play some benefit.90
Seizures
Treatment of CNS manifestations, including epilepsy and status epilepticus, in patients with MD does not differ from treatment of the same conditions in the general population.91 However, mitochondrion-toxic drugs, such as the antiepileptic drug valproate, mainly in patients with POLG disease, should be avoided.7
Because of its antimitochondrial effects,7 valproate may also have detrimental effects in patients with MELAS92 and in other MD. For instance, children with Alpers syndrome (a severe hepatoencephalopathy due to nuclear mutations in the POLG gene) are at high risk of death from status epilepticus or liver failure, if exposed to valproate.93
Finally, carbonic anhydrase inhibitors topiramate and zonisamide have been reported to cause severe metabolic acidosis in some patients,94–96 and if used in subjects with MD, acidosis may require monitoring.7 Lacosamide can be safe and potentially effective.97
Ketogenic diet may be attempted for treating intractable epilepsy, especially in the treatment of MD due to complex I deficiency.98 Carefully monitoring these patients is critical.
Regarding status epilepticus, attempted treatments include antiepileptic drugs, anesthetic agents, high-dose steroids, and others, with variable success.99 The outcome of mitochondrial status epilepticus is poor.99 Perampanel has been recently suggested as a potential therapeutic alternative in refractory status epilepticus associated with MELAS.100 Intravenous magnesium as anticonvulsant therapy, adapted from practice in eclampsia, was attempted in two unrelated, previously healthy teenage girls who developed refractory status epilepticus.101 In both cases, POLG mutations were found. Magnesium therapy resulted in clinical and neurophysiological improvement and extubation of both patients.101
Myoclonus
Myoclonus is one of the typical features of MERRF syndrome, defined as ‘myoclonic epilepsy with ragged red fibers’, or maybe more correctly ‘myoclonic encephalopathy with ragged red fibers’.102 MERRF is more frequently, but not invariably, associated to the m.8344A>G mtDNA mutation.103
Although valproic acid may be beneficial, it should be used with caution.7 Levetiracetam seems to be a safe and effective alternative104 and may possibly have some mitochondrioprotective effect.105
Other CNS manifestations
As a general rule, treatment of CNS signs and symptoms (e.g. parkinsonism, mood disorders, migraine) in patients with MD does not differ from treatment of the same conditions in general population.91 This is also true for most neuromuscular and internist features such as endocrine abnormalities, cardiomyopathy with heart failure, renal insufficiency, and respiratory insufficiency. Hardly accessible to treatment are cerebellar manifestations (‘mitochondrial ataxias’).106
For instance, mitochondrial parkinsonism does not have distinctive features from idiopathic Parkinson disease.107 Reduced striatal dopamine uptake and good response to levodopa or dopamine agonists have been well documented. Levodopa-induced dyskinesias and motor fluctuations may also occur.107
Regarding antidepressants, an adult MELAS patient with major depressive disorder was reported to show an excellent response to the selective serotonin-norepinephrine reuptake inhibitor medication duloxetine.108
Be careful with these drugs
The potential mitochondrial toxicity of some antiepileptics, with particular reference to valproic acid, has been discussed earlier. Valproate should be avoided, because of the risk of liver failure and status epilepticus, especially in POLG mutation carriers.7 However, for most of the antiepileptic drugs, there is not in vivo evidence of severe mitotoxicity and, therefore, they may be used safely.
Furthermore, because of their prokaryotic origins, mitochondria are susceptible to the antibiotics targeting the bacterial ribosome. This vulnerability is increased by specific mtDNA mutations. Among them, aminoglycosides should be avoided, whenever possible, in patients with known or suspected MD.7 In fact, they may even lead to deafness if administered to patients with some mtDNA mutations, especially the m.1555A>G.109 If aminoglycosides need to be used, it is strongly recommended to screen for mitochondrial DNA mutations before treatment. One MELAS patient developed severe lactic acidosis shortly after the beginning of linezolid treatment.110 Linezolid induces lactic acidosis by inhibiting mitochondrial ribosomal protein synthesis and thus disrupting crucial mitochondrial functions.111 The risk of side effects of use of chloramphenicol is low but serious, namely acute hepatitis or blood dyscrasia. Therefore, preferably alternatives should be prescribed, especially in patients with mitochondrial genetic defects affecting liver and/or bone marrow, such as mtDNA depletion syndromes or Pearson syndrome. MD patients needing one of these antibiotics should be carefully monitored.7
Metformin is an effective agent for reducing hyperglycemia in type 2 diabetes mellitus. Lactic acidosis can develop during metformin intoxication, possibly because of mitochondrial dysfunction.112 Therefore, this drug should be given to patients with MD with monitoring lactate levels.
Statins are currently the most effective medications for reducing low-density lipoprotein (LDL) cholesterol concentrations. The most frequent side effects of statins are myopathic complaints ranging from myalgia to fatal rhabdomyolysis. Statins block production of farnesyl pyrophosphate, an intermediate of coenzyme Q10 biosynthesis.113 Muscle coenzyme Q10 levels and respiratory chain activities were significantly reduced by simvastatin, but not by atorvastatin.114 Simvastatin, but not atorvastatin, reduced muscle mtDNA levels.115 Patients with MD may be prone to develop statin myopathy.116 Statin-induced myopathy was linked to mitochondrial complex III inhibition.117 Therefore, statins should be used with caution and strictly monitoring creatine kinase levels in MD patients.7 Atorvastatin seems to be preferable to simvastatin, but convincing studies are pending.
In some instances, β-blockers, including metoprolol, were reported to aggravate an underlying MD.118 However, we do not have enough clinical data to suggest to avoid β-blockers in MD. Other drugs potentially interfering with mitochondrial function, including some antiviral drugs (antiretrovirals and clevudine), are extensively discussed elsewhere.7 Long-term oral antiviral therapy with nucleoside analogs for hepatitis B can induce chronic oxidative damage to mtDNA resulting in qualitative mtDNA abnormalities and toxic myopathy. 119
Many other medications are associated with muscle toxicity (e.g. corticosteroids, amiodarone, chloroquine/hydroxychloroquine, colchicine, vincristine, interferon-α, some neuroleptics, cyclosporine, and other cytotoxic drugs)7 and/or can cause metabolic acidosis (e.g. spironolactone, prostaglandin inhibitors, triamterene, amiloride, trimethoprim, pentamidine).7 Inhibition of the AMPK pathway, and thus of ATP production, is one of the proposed mechanisms possibly mediating mitochondrial toxicity due to chemotherapeutics such as doxorubicin, trastuzumab, and sunitinib.120 Bortezomib was also reported to cause a symptomatic myopathy with storage of lipid droplets together with mitochondrial abnormalities in a patient with multiple myeloma.121 Further studies are also needed to establish the safety of the chemotherapeutics used for HSCT in MNGIE patients, such as ciclosporin and melphalan (see earlier discussion).
Patients with MD needing one of these treatments (Table 3) should be carefully monitored, clinically and by laboratory exams (including creatine kinase and lactate).
Table 3.
Potentially mitochondrion-toxic agents. Mitochondrial patients needing one of these treatments should be carefully monitored, clinically and by laboratory exams, including creatine kinase and lactate (for further details, see text).
| Aminoglycosides |
| Antiretrovirals |
| Clevudine |
| Chloramphenicol |
| Dichloroacetate |
| Isoflurane |
| Linezolid |
| Metformin |
| Propofol |
| Statins |
| Topiramate |
| Valproic acid |
| Zonisamide |
Anesthesia and MD
Patients with MD have received a variety of general and regional anesthetic regimens with no adverse consequences.7 Perioperative complications of lactic acidosis were reported more in inhalation anesthesia than intravenous anesthesia.122 The use of lactate-free intravenous fluids with dextrose should be considered in all MD patients to prevent lactic acidosis. Potentially mitochondrion-toxic agents include isoflurane and propofol.7 Furthermore, children with MD have increased sensitivity to volatile anesthetics.123 MD patients may be at higher risk of developing propofol infusion syndrome.124 Mitochondrial patients are generally considered at increased risk of adverse surgical outcome compared to the general population, but adequate longitudinal studies are still lacking.125 Common malignant hyperthermia precautions are strongly recommend.126 Furthermore, mitochondrial patients should be carefully monitored in the perioperative period.127 Normothermia and normoglycemia should be maintained and metabolic stresses avoided (including prolonged fasting).
Conclusion and perspectives
The symptomatic treatment of the various clinical features (e.g. epilepsy, migraine, parkinsonism, diabetes, heart failure, renal insufficiency, respiratory insufficiency) in subjects with MD does not substantially differ from the treatment of the same conditions in nonmitochondrial patients. However, well-known mitochondrion-toxic drugs such as valproate, aminoglycosides, or linezolid should be used with extreme caution.7
Patient care standards for MD are described in detail elsewhere.128 These include the use of digital hearing aids or cochlear implantation for hearing loss; pacemaker and/or implantable defibrillator for arrhythmias; deep brain stimulation for untreatable movement disorders, etc.128 When needed, renal or cardiac transplantation should be offered after careful multidisciplinary review.128 Furthermore, MD patients should be offered age-appropriate vaccination.128
Appropriate care standards also include enteral nutrition and/or assisted ventilation in MD patients with advanced disease.
When the administration of a mitochondrion-toxic drug is needed, a careful clinical and laboratory follow-up is warranted to precociously recognize and treat possible side effects, such as rhabdomyolysis, lactic acidosis, hepatic failure, and others.7 Furthermore, in MD patients, possible causes of metabolic acidosis should be timely recognized and treated (e.g. sepsis, diabetic ketoacidosis, alcohol intoxications, other intoxications, diarrhea, prolonged fasting, etc.). The physician should not be scared of treating mitochondrial patients when needed. For instance, a not-treated bacterial infection may be more dangerous than the use of appropriate drugs, because infections may precipitate metabolic crises in MD patients.7
Alongside the traditional biochemical and histochemical assays, the molecular diagnosis of MD has been revolutionized by the introduction of next-generation sequencing (NGS). In recent years, high-throughput ‘omics’ techniques capable of detecting differences in a multitude of molecular constituents in organisms (including metabolomics, proteomics, transcriptomics, genomics, and epigenomics) accompanied by sophisticated bioinformatics tools have revealed new details about mitochondrial function and dysfunctions.2
The application of patient-derived induced pluripotent stem cells may allow the development of innovative, more effective cellular models.129 Their use may benefit the search for effective treatments against MD.
Future approaches to these pathologies could include germline therapy and gene therapy.7 Mitochondrial gene therapy seems to be a valuable and promising strategy to treat MD.130 In future, these approaches may offer a reproductive option to prevent mtDNA disease transmission in affected families.
The use of sensitive and valid endpoints is essential to test the effectiveness of potential treatments.131 A set of recommended outcome measures to be implemented in clinical studies has been identified.132 The development of new biomarkers to facilitate clinical development of promising new therapies is a critical issue.133 Furthermore, innovative neuroimaging tecniques may in future serve as surrogate biomarkers in trials investigating therapeutic options in MD.134
Multicenter collaboration is needed for rare diseases, and a cooperative effort of the centers with specific expertise in mitochondrial medicine is essential for the advancement of the treatment options. In some countries, including Italy, this collaboration is an established fact and it appears timely for large, multicenter, well-controlled clinical trials.7
There is still a strong need for more effective therapies, but mitochondrial medicine has entered the evidence-based era.
Acknowledgments
None.
Footnotes
Contributions: All authors contributed to the preparation of this review. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosure and potential conflicts of interest: The authors declare that they have no conflicts of interest. The International Committee of Medical Journal Editors (ICMJE) Potential Conflicts of Interests form for the authors is available for download at http://www.drugsincontext.com/wp-content/uploads/2019/06/dic.212588-COI.pdf
Funding declaration: This work was partially supported by Telethon Grant GUP09004 and Telethon-MITOCON grant GSP16001.
Correct attribution: Copyright © 2019 Orsucci D, Caldarazzo Ienco E, Siciliano G, Mancuso M. Published by Drugs in Context under Creative Commons License Deed CC BY NC ND 4.0.
Article URL: https://www.drugsincontext.com/mitochondrial-disorders-and-drugs:-what-every-physician-should-know/
Provenance: invited; externally peer reviewed.
Drugs in Context is published by BioExcel Publishing Ltd. Registered office: Plaza Building, Lee High Road, London, England, SE13 5PT.
BioExcel Publishing Limited is registered in England Number 10038393. VAT GB 252 7720 07.
For all manuscript and submissions enquiries, contact the Editor-in-Chief gordon.mallarkey@bioexcelpublishing.com
For all permissions, rights and reprints, contact David Hughes david.hughes@bioexcelpublishing.com
Peer review comments to author: 17 May 2019
References
- 1.DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9(8):429–444. doi: 10.1038/nrneurol.2013.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahman J, Rahman S. Mitochondrial medicine in the omics era. Lancet. 2018;391(10139):2560–2574. doi: 10.1016/S0140-6736(18)30727-X. [DOI] [PubMed] [Google Scholar]
- 3.Aryaman J, Johnston IG, Jones NS. Mitochondrial DNA density homeostasis accounts for a threshold effect in a cybrid model of a human mitochondrial disease. Biochem J. 2017;474(23):4019–4034. doi: 10.1042/BCJ20170651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo S, Valencia CA, Zhang J, et al. Biparental inheritance of mitochondrial DNA in humans. Proc Natl Acad Sci U S A. 2018;115(51):13039–13044. doi: 10.1073/pnas.1810946115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirkman MA, Yu-Wai-Man P, Korsten A, et al. Gene-environment interactions in Leber hereditary optic neuropathy. Brain. 2009;132(Pt 9):2317–2326. doi: 10.1093/brain/awp158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orsucci D, Siciliano G, Mancuso M. Revealing the complexity of mitochondrial DNA-related disorders. EBioMedicine. 2018;30:3–4. doi: 10.1016/j.ebiom.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mancuso M, Orsucci D, Filosto M, Simoncini C, Siciliano G. Drugs and mitochondrial diseases: 40 queries and answers. Expert Opin Pharmacother. 2012;13(4):527–543. doi: 10.1517/14656566.2012.657177. [DOI] [PubMed] [Google Scholar]
- 8.Grier J, Hirano M, Karaa A, Shepard E, Thompson JLP. Diagnostic odyssey of patients with mitochondrial disease: results of a survey. Neurol Genet. 2018;4(2):e230. doi: 10.1212/NXG.0000000000000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaefer AM, McFarland R, Blakely EL, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63(1):35–39. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 10.Mancuso M, Orsucci D, Choub A, Siciliano G. Current and emerging treatment options in the management of friedreich ataxia. Neuropsychiatr Dis Treat. 2010;6:491–499. doi: 10.2147/ndt.s6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Viscomi C, Burlina AB, Dweikat I, et al. Combined treatment with oral metronidazole and n-acetylcysteine is effective in ethylmalonic encephalopathy. Nat Med. 2010;16(8):869–871. doi: 10.1038/nm.2188. [DOI] [PubMed] [Google Scholar]
- 12.Auranen M, Paetau A, Piirila P, et al. Patient with multiple acyl-coa dehydrogenation deficiency disease and flad1 mutations benefits from riboflavin therapy. Neuromuscul Disord. 2017;27(6):581–584. doi: 10.1016/j.nmd.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Chinnery P, Majamaa K, Turnbull D, Thorburn D. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2006;(1):CD004426. doi: 10.1002/14651858.CD004426.pub2. [DOI] [PubMed] [Google Scholar]
- 14.Chen RS, Huang CC, Chu NS. Coenzyme q10 treatment in mitochondrial encephalomyopathies. Short-term double-blind, crossover study. Eur Neurol. 1997;37(4):212–218. doi: 10.1159/000117445. [DOI] [PubMed] [Google Scholar]
- 15.Glover EI, Martin J, Maher A, Thornhill RE, Moran GR, Tarnopolsky MA. A randomized trial of coenzyme q10 in mitochondrial disorders. Muscle Nerve. 2010;42(5):739–748. doi: 10.1002/mus.21758. [DOI] [PubMed] [Google Scholar]
- 16.Masotta NE, Martinefski MR, Lucangioli S, Rojas AM, Tripodi VP. High-dose coenzyme q10-loaded oleogels for oral therapeutic supplementation. Int J Pharm. 2019;556:9–20. doi: 10.1016/j.ijpharm.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Kim YR, Baek JI, Kim SH, et al. Therapeutic potential of the mitochondria-targeted antioxidant mitoq in mitochondrial-ROS induced sensorineural hearing loss caused by idh2 deficiency. Redox Biol. 2019;20:544–555. doi: 10.1016/j.redox.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallance HD, Koochin A, Branov J, et al. Marked elevation in plasma trimethylamine-n-oxide (tmao) in patients with mitochondrial disorders treated with oral l-carnitine. Mol Genet Metab Rep. 2018;15:130–133. doi: 10.1016/j.ymgmr.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarnopolsky MA, Roy BD, MacDonald JR. A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve. 1997;20(12):1502–1509. doi: 10.1002/(SICI)1097-4598(199712)20:12<1502::AID-MUS4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 20.Klopstock T, Querner V, Schmidt F, et al. A placebo-controlled crossover trial of creatine in mitochondrial diseases. Neurology. 2000;55(11):1748–1751. doi: 10.1212/wnl.55.11.1748. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez MC, MacDonald JR, Mahoney DJ, et al. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve. 2007 Feb;35(2):235–242. doi: 10.1002/mus.20688. [DOI] [PubMed] [Google Scholar]
- 22.Bugiani M, Lamantea E, Invernizzi F, et al. Effects of riboflavin in children with complex ii deficiency. Brain Dev. 2006;28(9):576–581. doi: 10.1016/j.braindev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Gempel K, Topaloglu H, Talim B, et al. The myopathic form of coenzyme q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (etfdh) gene. Brain. 2007;130(Pt 8):2037–2044. doi: 10.1093/brain/awm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahola S, Auranen M, Isohanni P, et al. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol Med. 2016;8:1234–1247. doi: 10.15252/emmm.201606592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theunissen TEJ, Gerards M, Hellebrekers D, et al. Selection and characterization of palmitic acid responsive patients with an oxphos complex i defect. Front Mol Neurosci. 2017;10:336. doi: 10.3389/fnmol.2017.00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mancuso M, Orsucci D, Logerfo A, et al. Oxidative stress biomarkers in mitochondrial myopathies, basally and after cysteine donor supplementation. J Neurol. 2010;257(5):774–781. doi: 10.1007/s00415-009-5409-7. [DOI] [PubMed] [Google Scholar]
- 27.Bartsakoulia M, Mupsilonller JS, Gomez-Duran A, Yu-Wai-Man P, Boczonadi V, Horvath R. Cysteine supplementation may be beneficial in a subgroup of mitochondrial translation deficiencies. J Neuromuscul Dis. 2016;3(3):363–379. doi: 10.3233/JND-160178. [DOI] [PubMed] [Google Scholar]
- 28.Guha S, Konkwo C, Lavorato M, et al. Pre-clinical evaluation of cysteamine bitartrate as a therapeutic agent for mitochondrial respiratory chain disease. Hum Mol Genet. 2019 doi: 10.1093/hmg/ddz023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leipnitz G, Mohsen AW, Karunanidhi A, et al. Evaluation of mitochondrial bioenergetics, dynamics, endoplasmic reticulum-mitochondria crosstalk, and reactive oxygen species in fibroblasts from patients with complex i deficiency. Sci Rep. 2018;8(1):1165. doi: 10.1038/s41598-018-19543-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Haas R, Das D, Garanto A, et al. Therapeutic effects of the mitochondrial ros-redox modulator kh176 in a mammalian model of leigh disease. Sci Rep. 2017;7(1):11733. doi: 10.1038/s41598-017-09417-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirano M, Emmanuele V, Quinzii CM. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018;62(3):467–481. doi: 10.1042/EBC20170114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kouga T, Takagi M, Miyauchi A, et al. Japanese leigh syndrome case treated with epi-743. Brain Dev. 2018;40(2):145–149. doi: 10.1016/j.braindev.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Huertas JR, Al Fazazi S, Hidalgo-Gutierrez A, Lopez LC, Casuso RA. Antioxidant effect of exercise: exploring the role of the mitochondrial complex i superassembly. Redox Biol. 2017;13:477–481. doi: 10.1016/j.redox.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain IH, Zazzeron L, Goli R, et al. Hypoxia as a therapy for mitochondrial disease. Science. 2016;352(6281):54–61. doi: 10.1126/science.aad9642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiMauro S, Quinzii CM, Hirano M. Mutations in coenzyme q10 biosynthetic genes. J Clin Invest. 2007;117(3):587–589. doi: 10.1172/JCI31423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Awad AM, Bradley MC, Fernandez-Del-Rio L, Nag A, Tsui HS, Clarke CF. Coenzyme q10 deficiencies: pathways in yeast and humans. Essays Biochem. 2018;62(3):361–376. doi: 10.1042/EBC20170106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hidalgo-Gutierrez A, Barriocanal-Casado E, Bakkali M, et al. Beta-ra reduces dmq/coq ratio and rescues the encephalopathic phenotype in coq9 (r239x) mice. EMBO Mol Med. 2019;11(1) doi: 10.15252/emmm.201809466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu-Wai-Man P, Soiferman D, Moore DG, Burte F, Saada A. Evaluating the therapeutic potential of idebenone and related quinone analogues in Leber hereditary optic neuropathy. Mitochondrion. 2017;36:36–42. doi: 10.1016/j.mito.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011;134(Pt 9):2677–2686. doi: 10.1093/brain/awr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carelli V, La Morgia C, Valentino ML, et al. Idebenone treatment in Leber’s hereditary optic neuropathy. Brain. 2011;134(Pt 9):e188. doi: 10.1093/brain/awr180. [DOI] [PubMed] [Google Scholar]
- 41.Jurkute N, Harvey J, Yu-Wai-Man P. Treatment strategies for Leber hereditary optic neuropathy. Curr Opin Neurol. 2019;32(1):99–104. doi: 10.1097/WCO.0000000000000646. [DOI] [PubMed] [Google Scholar]
- 42.Filosto M, Cotti Piccinelli S, Caria F, et al. Mitochondrial neurogastrointestinal encephalomyopathy (mngie-mtdps1) J Clin Med. 2018;7(11) doi: 10.3390/jcm7110389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spinazzola A, Marti R, Nishino I, et al. Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem. 2002;277:4128–4133. doi: 10.1074/jbc.M111028200. [DOI] [PubMed] [Google Scholar]
- 44.Lara MC, Weiss B, Illa I, et al. Infusion of platelets transiently reduces nucleoside overload in MNGIE. Neurology. 2006;67:1461–1463. doi: 10.1212/01.wnl.0000239824.95411.52. [DOI] [PubMed] [Google Scholar]
- 45.Yavuz H, Ozel A, Christensen M, et al. Treatment of mitochondrial neurogastrointestinal encephalomyopathy with dialysis. Arch Neurol. 2007;64:435–438. doi: 10.1001/archneur.64.3.435. [DOI] [PubMed] [Google Scholar]
- 46.De Giorgio R, Pironi L, Rinaldi R, et al. Liver transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Ann Neurol. 2016;80:448–455. doi: 10.1002/ana.24724. [DOI] [PubMed] [Google Scholar]
- 47.Moran NF, Bain MD, Muqit M, Bax BE. Carrier erythrocyte entrapped thymidine phosphorylase therapy in MNGIE. Neurology. 2008;71:686–688. doi: 10.1212/01.wnl.0000324602.97205.ab. [DOI] [PubMed] [Google Scholar]
- 48.Bax BE, Bain MD, Scarpelli M, et al. Clinical and biochemical improvements in a patient with MNGIE following enzyme replacement. Neurology. 2013;81:1269–1271. doi: 10.1212/WNL.0b013e3182a6cb4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levene M, Bain MD, Moran NF, et al. Safety and efficacy of erythrocyte encapsulated thymidine phosphorylase in mitochondrial neurogastrointestinal encephalomyopathy. J Clin Med. 2019;8 doi: 10.3390/jcm8040457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halter J, Schüpbach W, Casali C, et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011;46:330–337. doi: 10.1038/bmt.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahmed ST, Craven L, Russell OM, Turnbull DM, Vincent AE. Diagnosis and treatment of mitochondrial myopathies. Neurotherapeutics. 2018;15(4):943–953. doi: 10.1007/s13311-018-00674-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koene S, Smeitink J. Metabolic manipulators: a well founded strategy to combat mitochondrial dysfunction. J Inherit Metab Dis. 2011;34(2):315–325. doi: 10.1007/s10545-010-9162-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yatsuga S, Suomalainen A. Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum Mol Genet. 2012;21(3):526–535. doi: 10.1093/hmg/ddr482. [DOI] [PubMed] [Google Scholar]
- 54.Viscomi C, Bottani E, Civiletto G, et al. In vivo correction of cox deficiency by activation of the ampk/pgc-1alpha axis. Cell Metab. 2011;14(1):80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Golubitzky A, Dan P, Weissman S, Link G, Wikstrom JD, Saada A. Screening for active small molecules in mitochondrial complex I deficient patient’s fibroblasts, reveals aicar as the most beneficial compound. PLoS One. 2011;6(10):e26883. doi: 10.1371/journal.pone.0026883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodyear LJ. The exercise pill — too good to be true? N Engl J Med. 2008;359:1842–1844. doi: 10.1056/NEJMcibr0806723. [DOI] [PubMed] [Google Scholar]
- 57.Katsyuba E, Mottis A, Zietak M, et al. De novo nad(+) synthesis enhances mitochondrial function and improves health. Nature. 2018;563(7731):354–359. doi: 10.1038/s41586-018-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khan NA, Auranen M, Paetau I, et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin b3. EMBO Mol Med. 2014;6(6):721–731. doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van de Weijer T, Phielix E, Bilet L, et al. Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes. 2015;64:1193–1201. doi: 10.2337/db14-0667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kamatani N, Kushiyama A, Toyo-Oka L, Toyo-Oka T. Treatment of two mitochondrial disease patients with a combination of febuxostat and inosine that enhances cellular ATP. J Hum Genet. 2019;64:351–353. doi: 10.1038/s10038-018-0558-0. [DOI] [PubMed] [Google Scholar]
- 61.Munro B, Horvath R, Muller JS. Nucleoside supplementation modulates mitochondrial DNA copy number in the dguok−/− zebrafish. Hum Mol Genet. 2018 doi: 10.1093/hmg/ddy389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lopez-Gomez C, Levy RJ, Sanchez-Quintero MJ, et al. Deoxycytidine and deoxythymidine treatment for thymidine kinase 2 deficiency. Ann Neurol. 2017;81:641–652. doi: 10.1002/ana.24922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Domínguez-González C, Madruga-Garrido M, Mavillard F, et al. Deoxynucleoside therapy for thymidine kinase 2 (TK2) deficient myopathy. Ann Neurol. 2019 doi: 10.1002/ana.25506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajendran J, Purhonen J, Tegelberg S, et al. Alternative oxidase-mediated respiration prevents lethal mitochondrial cardiomyopathy. EMBO Mol Med. 2019;11(1) doi: 10.15252/emmm.201809456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dogan SA, Cerutti R, Beninca C, et al. Perturbed redox signaling exacerbates a mitochondrial myopathy. Cell Metab. 2018;28(5):764–775 e765. doi: 10.1016/j.cmet.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barrow JJ, Balsa E, Verdeguer F, et al. Bromodomain inhibitors correct bioenergetic deficiency caused by mitochondrial disease complex i mutations. Mol Cell. 2016;64(1):163–175. doi: 10.1016/j.molcel.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fabbri A, Travaglione S, Maroccia Z, et al. The bacterial protein cnf1 as a potential therapeutic strategy against mitochondrial diseases: a pilot study. Int J Mol Sci. 2018;19(7) doi: 10.3390/ijms19071825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karaa A, Haas R, Goldstein A, Vockley J, Weaver WD, Cohen BH. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology. 2018;90(14):e1212–e1221. doi: 10.1212/WNL.0000000000005255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szabo A, Sumegi K, Fekete K, et al. Activation of mitochondrial fusion provides a new treatment for mitochondria-related diseases. Biochem Pharmacol. 2018;150:86–96. doi: 10.1016/j.bcp.2018.01.038. [DOI] [PubMed] [Google Scholar]
- 70.Matsuhashi T, Sato T, Kanno SI, et al. Mitochonic acid 5 (ma-5) facilitates atp synthase oligomerization and cell survival in various mitochondrial diseases. EBioMedicine. 2017;20:27–38. doi: 10.1016/j.ebiom.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garone C, Viscomi C. Towards a therapy for mitochondrial disease: an update. Biochem Soc Trans. 2018;46(5):1247–1261. doi: 10.1042/BST20180134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barriocanal-Casado E, Hidalgo-Gutiérrez A, Raimundo N, et al. Rapamycin administration is not a valid therapeutic strategy for every case of mitochondrial disease. EBioMedicine. 2019;42:511–23. doi: 10.1016/j.ebiom.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fakruddin M, Wei FY, Suzuki T, et al. Defective mitochondrial trna taurine modification activates global proteostress and leads to mitochondrial disease. Cell Rep. 2018;22(2):482–496. doi: 10.1016/j.celrep.2017.12.051. [DOI] [PubMed] [Google Scholar]
- 74.Ishikawa T, Somiya K, Munechika R, Harashima H, Yamada Y. Mitochondrial transgene expression via an artificial mitochondrial DNA vector in cells from a patient with a mitochondrial disease. J Control Release. 2018;274:109–117. doi: 10.1016/j.jconrel.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 75.Yang Y, Wu H, Kang X, et al. Targeted elimination of mutant mitochondrial DNA in melas-ipscs by mitotalens. Protein Cell. 2018;9(3):283–297. doi: 10.1007/s13238-017-0499-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gammage PA, Viscomi C, Simard ML, et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med. 2018;24:1691–1695. doi: 10.1038/s41591-018-0165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bacman SR, Kauppila JHK, Pereira CV, et al. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat Med. 2018;24:1940. doi: 10.1038/s41591-018-0166-8. [DOI] [PubMed] [Google Scholar]
- 78.Tachibana M, Sparman M, Sritanaudomchai H, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461(7262):367–372. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Herbert M, Turnbull D. Progress in mitochondrial replacement therapies. Nat Rev Mol Cell Biol. 2018;19(2):71–72. doi: 10.1038/nrm.2018.3. [DOI] [PubMed] [Google Scholar]
- 80.Rai PK, Craven L, Hoogewijs K, Russell OM, Lightowlers RN. Advances in methods for reducing mitochondrial DNA disease by replacing or manipulating the mitochondrial genome. Essays Biochem. 2018;62(3):455–465. doi: 10.1042/EBC20170113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang E, Wu J, Gutierrez NM, et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature. 2016;540(7632):270–275. doi: 10.1038/nature20592. [DOI] [PubMed] [Google Scholar]
- 82.Mancuso M, Orsucci D, Angelini C, et al. The m.3243a>g mitochondrial DNA mutation and related phenotypes. A matter of gender? J Neurol. 2014;261(3):504–510. doi: 10.1007/s00415-013-7225-3. [DOI] [PubMed] [Google Scholar]
- 83.Kaufmann P, Engelstad K, Wei Y, et al. Dichloroacetate causes toxic neuropathy in melas: a randomized, controlled clinical trial. Neurology. 2006;66(3):324–330. doi: 10.1212/01.wnl.0000196641.05913.27. [DOI] [PubMed] [Google Scholar]
- 84.Koenig MK, Emrick L, Karaa A, et al. Recommendations for the management of strokelike episodes in patients with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes. JAMA Neurol. 2016;73(5):591–594. doi: 10.1001/jamaneurol.2015.5072. [DOI] [PubMed] [Google Scholar]
- 85.Yoneda M, Ikawa M, Arakawa K, et al. In vivo functional brain imaging and a therapeutic trial of L-arginine in melas patients. Biochim Biophys Acta. 2012;1820(5):615–618. doi: 10.1016/j.bbagen.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 86.Koga Y, Akita Y, Junko N, et al. Endothelial dysfunction in melas improved by L-arginine supplementation. Neurology. 2006;66(11):1766–1769. doi: 10.1212/01.wnl.0000220197.36849.1e. [DOI] [PubMed] [Google Scholar]
- 87.Koga Y, Akita Y, Nishioka J, et al. L-arginine improves the symptoms of strokelike episodes in melas. Neurology. 2005;64(4):710–712. doi: 10.1212/01.WNL.0000151976.60624.01. [DOI] [PubMed] [Google Scholar]
- 88.Koga Y, Povalko N, Inoue E, et al. Therapeutic regimen of L-arginine for melas: 9-year, prospective, multicenter, clinical research. J Neurol. 2018;265(12):2861–2874. doi: 10.1007/s00415-018-9057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ganetzky RD, Falk MJ. 8-year retrospective analysis of intravenous arginine therapy for acute metabolic strokes in pediatric mitochondrial disease. Mol Genet Metab. 2018;123(3):301–308. doi: 10.1016/j.ymgme.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rossi FH, Okun M, Yachnis A, Quisling R, Triggs WJ. Corticosteroid treatment of mitochondrial encephalomyopathies. Neurologist. 2002;8(5):313–315. doi: 10.1097/01.nrl.0000031010.85777.63. [DOI] [PubMed] [Google Scholar]
- 91.Finsterer J. Treatment of central nervous system manifestations in mitochondrial disorders. Eur J Neurol. 2011;18(1):28–38. doi: 10.1111/j.1468-1331.2010.03086.x. [DOI] [PubMed] [Google Scholar]
- 92.Lin CM, Thajeb P. Valproic acid aggravates epilepsy due to melas in a patient with an a3243g mutation of mitochondrial DNA. Metab Brain Dis. 2007;22(1):105–109. doi: 10.1007/s11011-006-9039-9. [DOI] [PubMed] [Google Scholar]
- 93.Tzoulis C, Engelsen BA, Telstad W, et al. The spectrum of clinical disease caused by the a467t and w748s polg mutations: a study of 26 cases. Brain. 2006;129(Pt 7):1685–1692. doi: 10.1093/brain/awl097. [DOI] [PubMed] [Google Scholar]
- 94.Mirza NS, Alfirevic A, Jorgensen A, Marson AG, Pirmohamed M. Metabolic acidosis with topiramate and zonisamide: an assessment of its severity and predictors. Pharmacogenet Genomics. 2011;21(5):297–302. doi: 10.1097/FPC.0b013e3283441b95. [DOI] [PubMed] [Google Scholar]
- 95.Belotti EA, Taddeo I, Ragazzi M, et al. Chronic impact of topiramate on acid-base balance and potassium in childhood. Eur J Paediatr Neurol. 2010;14(5):445–448. doi: 10.1016/j.ejpn.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 96.Shiber JR. Severe non-anion gap metabolic acidosis induced by topiramate: a case report. J Emerg Med. 2010;38(4):494–496. doi: 10.1016/j.jemermed.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 97.Primiano G, Vollono C, Dono F, Servidei S. Drug-resistant epilepsy in melas: safety and potential efficacy of lacosamide. Epilepsy Res. 2018;139:135–136. doi: 10.1016/j.eplepsyres.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 98.Paleologou E, Ismayilova N, Kinali M. Use of the ketogenic diet to treat intractable epilepsy in mitochondrial disorders. J Clin Med. 2017;6(6) doi: 10.3390/jcm6060056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rahman S. Mitochondrial diseases and status epilepticus. Epilepsia. 2018;59(Suppl 2):70–77. doi: 10.1111/epi.14485. [DOI] [PubMed] [Google Scholar]
- 100.Santamarina E, Alpuente A, Maisterra O, et al. Perampanel: a therapeutic alternative in refractory status epilepticus associated with MELAS syndrome. Epilepsy Behav Case Rep. 2019;11:92–95. doi: 10.1016/j.ebcr.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Visser NA, Braun KP, Leijten FS, van Nieuwenhuizen O, Wokke JH, van den Bergh WM. Magnesium treatment for patients with refractory status epilepticus due to polg1-mutations. J Neurol. 2011;258(2):218–222. doi: 10.1007/s00415-010-5721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mancuso M, Orsucci D, Angelini C, et al. Myoclonus in mitochondrial disorders. Mov Disord. 2014;29(6):722–728. doi: 10.1002/mds.25839. [DOI] [PubMed] [Google Scholar]
- 103.Mancuso M, Orsucci D, Angelini C, et al. Phenotypic heterogeneity of the 8344a>g mtdna “merrf” mutation. Neurology. 2013;80(22):2049–2054. doi: 10.1212/WNL.0b013e318294b44c. [DOI] [PubMed] [Google Scholar]
- 104.Mancuso M, Galli R, Pizzanelli C, Filosto M, Siciliano G, Murri L. Antimyoclonic effect of levetiracetam in merrf syndrome. J Neurol Sci. 2006;243(1–2):97–99. doi: 10.1016/j.jns.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 105.Gibbs JE, Walker MC, Cock HR. Levetiracetam: antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia. 2006;47(3):469–478. doi: 10.1111/j.1528-1167.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- 106.Mancuso M, Orsucci D, Siciliano G, Bonuccelli U. The genetics of ataxia: through the labyrinth of the Minotaur, looking for Ariadne’s thread. J Neurol. 2014;261(Suppl 2):S528–541. doi: 10.1007/s00415-014-7387-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Orsucci D, Caldarazzo Ienco E, Mancuso M, Siciliano G. Polg1-related and other “mitochondrial parkinsonisms”: an overview. J Mol Neurosci. 2011;44(1):17–24. doi: 10.1007/s12031-010-9488-9. [DOI] [PubMed] [Google Scholar]
- 108.Cozart B, Diaz Vera J, Denham JD, Whiting WL. Treatment of depression with duloxetine in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes. Clin Neuropharmacol. 2018;41(3):103–105. doi: 10.1097/WNF.0000000000000277. [DOI] [PubMed] [Google Scholar]
- 109.Rydzanicz M, Wrobel M, Pollak A, et al. Mutation analysis of mitochondrial 12s rrna gene in polish patients with non-syndromic and aminoglycoside-induced hearing loss. Biochem Biophys Res Commun. 2010;395(1):116–121. doi: 10.1016/j.bbrc.2010.03.149. [DOI] [PubMed] [Google Scholar]
- 110.Cope TE, McFarland R, Schaefer A. Rapid-onset, linezolid-induced lactic acidosis in melas. Mitochondrion. 2011;11(6):992–993. doi: 10.1016/j.mito.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 111.Zuccarini NS, Yousuf T, Wozniczka D, Rauf AA. Lactic acidosis induced by linezolid mimics symptoms of an acute intracranial bleed: a case report and literature review. J Clin Med Res. 2016;8(10):753–756. doi: 10.14740/jocmr2687w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Protti A, Russo R, Tagliabue P, et al. Oxygen consumption is depressed in patients with lactic acidosis due to biguanide intoxication. Crit Care. 2010;14(1):R22. doi: 10.1186/cc8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Marcoff L, Thompson PD. The role of coenzyme q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol. 2007;49(23):2231–2237. doi: 10.1016/j.jacc.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 114.Paiva H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther. 2005;78(1):60–68. doi: 10.1016/j.clpt.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 115.Schick BA, Laaksonen R, Frohlich JJ, et al. Decreased skeletal muscle mitochondrial DNA in patients treated with high-dose simvastatin. Clin Pharmacol Ther. 2007;81(5):650–653. doi: 10.1038/sj.clpt.6100124. [DOI] [PubMed] [Google Scholar]
- 116.Tay SK, Dimauro S, Pang AY, Lai PS, Yap HK. Myotoxicity of lipid-lowering agents in a teenager with melas mutation. Pediatr Neurol. 2008;39(6):426–428. doi: 10.1016/j.pediatrneurol.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 117.Schirris TJ, Renkema GH, Ritschel T, et al. Statin-induced myopathy is associated with mitochondrial complex iii inhibition. Cell Metab. 2015;22(3):399–407. doi: 10.1016/j.cmet.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 118.Samuels C, Koenig MK, Hernandez M, Yadav A, Mosquera RA. Mitochondrial disorder aggravated by metoprolol. Case Rep Pediatr. 2016;2016 doi: 10.1155/2016/7869174. 7869174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fujii T, Takase KI, Honda H, et al. Toxic myopathy with multiple deletions in mitochondrial DNA associated with long-term use of oral anti-viral drugs for hepatitis B: a case study. Neuropathology. 2019;39:162–167. doi: 10.1111/neup.12548. [DOI] [PubMed] [Google Scholar]
- 120.Gorini S, De Angelis A, Berrino L, Malara N, Rosano G, Ferraro E. Chemotherapeutic drugs and mitochondrial dysfunction: focus on doxorubicin, trastuzumab, and sunitinib. Oxid Med Cell Longev. 2018;2018 doi: 10.1155/2018/7582730. 7582730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guglielmi V, Nowis D, Tinelli M, et al. Bortezomib-induced muscle toxicity in multiple myeloma. J Neuropathol Exp Neurol. 2017;76(7):620–630. doi: 10.1093/jnen/nlx043. [DOI] [PubMed] [Google Scholar]
- 122.Miyamoto Y, Miyashita T, Takaki S, Goto T. Perioperative considerations in adult mitochondrial disease: a case series and a review of 111 cases. Mitochondrion. 2016;26:26–32. doi: 10.1016/j.mito.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 123.Morgan PG, Hoppel CL, Sedensky MM. Mitochondrial defects and anesthetic sensitivity. Anesthesiology. 2002;96(5):1268–1270. doi: 10.1097/00000542-200205000-00036. [DOI] [PubMed] [Google Scholar]
- 124.Finsterer J, Frank M. Propofol is mitochondrion-toxic and may unmask a mitochondrial disorder. J Child Neurol. 2016;31(13):1489–1494. doi: 10.1177/0883073816661458. [DOI] [PubMed] [Google Scholar]
- 125.Muravchick S. Clinical implications of mitochondrial disease. Adv Drug Deliv Rev. 2008;60(13–14):1553–1560. doi: 10.1016/j.addr.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 126.Hopkins PM. Malignant hyperthermia: pharmacology of triggering. Br J Anaesth. 2011;107(1):48–56. doi: 10.1093/bja/aer132. [DOI] [PubMed] [Google Scholar]
- 127.Finsterer J, Segall L. Drugs interfering with mitochondrial disorders. Drug Chem Toxicol. 2010;33(2):138–151. doi: 10.3109/01480540903207076. [DOI] [PubMed] [Google Scholar]
- 128.Parikh S, Goldstein A, Karaa A, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017;19 doi: 10.1038/gim.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Inak G, Lorenz C, Lisowski P, Zink A, Mlody B, Prigione A. Concise review: induced pluripotent stem cell-based drug discovery for mitochondrial disease. Stem Cells. 2017;35(7):1655–1662. doi: 10.1002/stem.2637. [DOI] [PubMed] [Google Scholar]
- 130.Coutinho E, Batista C, Sousa F, Queiroz J, Costa D. Mitochondrial gene therapy: advances in mitochondrial gene cloning, plasmid production, and nanosystems targeted to mitochondria. Mol Pharm. 2017;14(3):626–638. doi: 10.1021/acs.molpharmaceut.6b00823. [DOI] [PubMed] [Google Scholar]
- 131.Koene S, van Bon L, Bertini E, et al. Outcome measures for children with mitochondrial disease: consensus recommendations for future studies from a delphi-based international workshop. J Inherit Metab Dis. 2018;41(6):1267–1273. doi: 10.1007/s10545-018-0229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mancuso M, McFarland R, Klopstock T, Hirano M. International workshop: outcome measures and clinical trial readiness in primary mitochondrial myopathies in children and adults. Consensus recommendations. 16–18 November 2016, Rome, Italy. Neuromuscul Disord. 2017;27(12):1126–1137. doi: 10.1016/j.nmd.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Steele HE, Horvath R, Lyon JJ, Chinnery PF. Monitoring clinical progression with mitochondrial disease biomarkers. Brain. 2017;140(10):2530–2540. doi: 10.1093/brain/awx168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mascalchi M, Montomoli M, Guerrini R. Neuroimaging in mitochondrial disorders. Essays Biochem. 2018;62(3):409–421. doi: 10.1042/EBC20170109. [DOI] [PubMed] [Google Scholar]