

Abstract
Tonic inhibitory currents, mediated by extrasynaptic GABAA receptors, are elevated at a delay following stroke. Flavonoids minimise the extent of cellular damage following stroke, but little is known about their mode of action. We demonstrate that the flavonoid, 2′-methoxy-6-methylflavone (0.1–10 µM; 2′MeO6MF), increases GABAA receptor tonic currents presumably via δ-containing GABAA receptors. Treatment with 2′MeO6MF 1–6 h post focal ischaemia dose dependently decreases infarct volume and improves functional recovery. The effect of 2′MeO6MF was attenuated in δ−/− mice, indicating that the effects of the flavonoid were mediated via δ-containing GABAA receptors. Further, as flavonoids have been shown to have multiple modes of action, we investigated the anti-inflammatory effects of 2′MeO6MF. Using a macrophage cell line, we show that 2′MeO6MF can dampen an LPS-induced elevation in NFkB activity. Assessment of vehicle-treated stroke animals revealed a significant increase in circulating IL1β, TNFα and IFγ levels. Treatment with 2′MeO6MF dampened the stroke-induced increase in circulating cytokines, which was blocked in the presence of the pan-AKT inhibitor, GSK690693. These studies support the hypothesis that compounds that potentiate tonic inhibition via δ-containing GABAA receptors soon after stroke can afford neuroprotection.
Keywords: Photothrombotic stroke, inflammation, tonic inhibition, motor behaviour, γ-aminobutyric acid
Introduction
Stroke is a leading cause of death and disability, with survivors exhibiting varied levels of functional recovery.1 Stroke-induced cell death occurs due to an excess release of glutamate,2 and as such, research has focused largely on developing neuroprotectants that act to block glutamate-mediated cell death.2,3 In addition, changes in γ-aminobutyric acid (GABA) function following cerebral ischaemia and the protective benefits of GABAergic compounds targeting synaptic GABAA receptors (GABAARs) have been reported.4–7 Despite GABAAR agonists showing great promise in animal models of stroke, they have failed to translate into positive clinical outcomes,8 most likely due to a lack of subtype specificity resulting in unwanted side effects, limiting their therapeutic potential.9–11
Recently we demonstrated marked improvements in post-stroke functional recovery that implicate extrasynaptic GABAARs as novel targets to help stroke sufferers.6 Importantly, dampening tonic inhibitory currents from three days post-stroke leads to improved functional recovery without affecting infarct size.6 However, protective mechanisms after stroke are best achieved soon after stroke. We speculate that the elevation in tonic currents is an endogenous mechanism that is activated, albeit at a delay, to minimise the extent of cell death (see Figure 1(a) and (b) acute period shown in blue).6,7 However, by day 3, the infarct is more or less fully formed, and it is unlikely to respond to any therapeutic interventions to minimise the extent of cell death. Yet as activating extrasynaptic GABAARs, containing either α5 or δ-subunits,12 is known to dampen neuronal excitability, stimulating extrasynaptic GABAARs (within hours after a stroke), could dampen neuronal glutamate-mediated excitability and reduce infarct size. As the activity and function associated with GABAARs varies extensively, the development of ligands that target extrasynaptic GABAARs may therefore lead to more effective treatments for stroke.
Figure 1.

Stroke induces a delayed elevation in tonic GABAAR currents in layer 2/3 pyramidal neurons. Schematic showing early and subacute treatment windows post-stroke (a). Panel (b) shows a flow diagram of the treatment windows along with treatment options using GABA compounds targeting extrasynaptic sites. Early tonic GABA signalling limits the extent of neuronal cell death (shown in blue), whereas tonic GABA signalling during the subacute phase (shown in red) impairs stroke recovery and therefore needs to be dampened. Whole-cell patch-clamp recordings were made from post-stroke brain slices, within 500 µm of infarct, from layer 2/3 pyramidal neurons. Representative traces showing the tonic inhibitory currents in control (n = 19) and peri-infarct neurons at 3 (n = 8), 6 (n = 8), 12 (n = 9) and 24 h (n = 12) and three days (n = 12) post-stroke (c). Tonic currents were revealed by the shift in holding currents after blocking all GABAARs with gabazine (SR95531); > 100 µM). Box plot (whiskers: minimum and maximum; lines: median) showing a small decrease on tonic inhibitory currents in peri-infarct cortical neurons early (3–6 h) after stroke with significance observed at the 3 h time point, while showing a significant elevation tonic inhibition at 24 h onwards (d). Horizontal bars indicate the application of the GABAAR antagonist SR95531. Cells were voltage clamped at + 10 mV. *=P < 0.05, **=P < 0.01, ***=P < 0.001, compared to tonic currents from control animal. GABA: γ-aminobutyric acid.
In addition to changes in glutamate, pro-inflammatory cytokines have also been previously shown to contribute to delayed cell death, oedema and impaired functional recovery. Recent work has shown that cytokines can alter the expression of extrasynaptic GABAAR subunits and impair memory.13 In addition, GABA compounds can modulate pro-inflammatory pathways and protect against damage.14 This indicated that compounds that alter both GABAergic signalling and dampen inflammation would offer a two-pronged approach to affording neuroprotection.
Flavonoids are secondary plant metabolites involved in a variety of biological processes including modulation of GABAAR function,15–17 and modulation of inflammatory pathways.18 In the present study, we report that the synthetic flavonoid, 2′-methoxy-6-methylflavone (2′MeO6MF), increases mouse extrasynaptic GABAAR activity as evidenced by the elevated tonic inhibitory currents in brain slices. We further show that modulation of δ-containing GABAARs within 6 h offers neuroprotection and improved functional recovery and dampens the stroke-induced inflammatory response. Thus, our data support the hypothesis that increasing tonic inhibitory currents early within 6 h following stroke offers a novel therapeutic approach to minimise neuronal damage.
Material and methods
Preparation of brain slices
All experiments were approved by either the Aarhus University Animal Ethics Committee for recordings from hippocampal dentate gyrus granule cells (DGGC), or the University of Otago Animal Ethics Committee for recordings from layer 2/3 pyramidal neurons. Recordings were performed in accordance with the NIH Animal Protection Guidelines for the care and use of animals for scientific purposes and are reported according to the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines. C57BL/6 J male mice were maintained on a 12:12 h light/dark cycle with unrestricted access to food and water. For recordings from hippocampal DGGC, on post-natal day 18–24 (P18–P24) mice were anaesthetised with isoflurane and decapitated in accordance with Danish and European legislation regarding laboratory animals. For whole-cell patch-clamp recordings, slices were perfused with bubbled ACSF (33 ± 1℃) at 2–3 ml/min and neurons were visualised using a custom-built infrared microscope equipped with a 40X water-immersion objective (Olympus, Ballerup, Denmark) and CCD100 camera (DAGE-MTI, Michigan City, IN), as described previously.19 To improve slice quality, 3 mM kynurenic acid, 0.2 mM ascorbic acid and 0.2 mM pyruvic acid were added to the ACSF used for both the slicing and storage incubation solutions.
For recordings from layer 2/3 pyramidal neurons following sham or stroke mice, strokes were induced in 2–3-month-old mice (see below) and then mice were anaesthetised with isoflurane and brains isolated from either sham controls or 3, 6, 12 and 24 h and three days post-stroke to measure tonic currents.6 All recordings were made from peri-infarct cortical layer 2/3 pyramidal neurons within the primary motor cortex, as previously described.6 Neurons were voltage clamped in whole-cell configuration using a MultiClamp-700B amplifier using microelectrodes (3–5 MΩ) filled with a caesium-methyl-sulfonate (CsMeSO4)-based internal pipette solution.
Both inhibitory postsynaptic current (IPSC) amplitude and frequency were analysed offline using MiniAnalysis software. For collecting the events, IPSC amplitude threshold was set to 25 pA and IPSCs > 25 pA were collected and the inter-event intervals were calculated for these IPSCs. A 2 min segment before washing in 2′MeO6MF and a 2 min segment before washing in gabazine were selected to assess the effect of 2′MeO6MF on synaptic currents.
Slice electrophysiology
For whole-cell patch-clamp recordings, slices were perfused with bubbled ACSF (33 ± 1℃) at 2–3 ml/min and neurons were visualised, as described previously.19 Patch electrodes were prepared on a DMZ-universal puller (Zeitz Instruments GmbH, Munich, Germany) from thick-walled borosilicate glass (Garner Glass Co., Claremont, CA, USA) with an open tip resistance (3–5 MΩ) when filled with intracellular solution containing (in mM): 140 CsCl, 2 MgCl2, 0.05 EGTA and 10 HEPES, adjusted to pH 7.2 with CsOH (280–290 mosmol/kg).
Recordings were carried out using a MultiClamp 700B amplifier (Molecular Devices, Union City, CA, USA). Voltage-clamp recordings were carried out at a holding potential (Vhold) of −70 mV. During voltage-clamp recordings, whole-cell capacitances and series resistances were noted and resistances were compensated by about 70% (lag 10 µs). Recordings were discontinued if resistances increased by > 50% or exceeded 20 MΩ. 2′MeO6MF was perfused for 5–10 min before tonic currents were assessed.
All recordings were low-pass filtered (8-pole Bessel) at 3 kHz, digitised at 20 kHz, and acquired using Digidata 1440 A and pCLAMP10 for recording from layer 2/3 pyramidal cells or using a BNC-2110 D/A converter and a PCI-6014 board (National Instruments, Austin, TX, USA) and custom-written LabVIEW 6.1-based software (EVAN v. 1.4, courtesy of Istvan Mody, CA, USA) for recordings from DGGCs. Since a large difference in the size of granule cells is often seen, tonic currents were normalised to the cell capacitance in histograms to report current density. Unpaired Student’s t-test was used to compare means with two-tailed P < 0.05 as the significance level. Data are presented as means ± SD, with N indicating the number of neurons.
In vivo stroke assessments and brain uptake studies
Chemicals and reagents
Triethylamine was purchased from Sigma-Aldrich (St Louis, MO). Acetonitrile and methanol of HPLC grade were obtained from Merck KGaA (Darmstadt, Germany), and water was obtained from a Millipore purification system (Billerica, MA). 2′MeO6MF was synthesised in house as previously described.16
Mouse brain uptake study
All animal experiments were approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee and were performed in accordance with the Australian National Health and Medical Research Council guidelines for the care and use of animals for scientific purposes. Male BALB/cJ Asmu mice (6–8 weeks of age; 25–30 g) were used in brain uptake studies, except to assess brain uptake after stroke where male C57BL/6 J (2–3 months old) mice were used (approved by the University of Otago, animal ethics committee). Mice had free access to food and water during all experimental periods. An aliquot (200 µl) of a solution of 2′MeO6MF equivalent to 30 mg/kg of body weight in a vehicle consisting of dimethyl sulfoxide (DMSO) (5% v/v), Tween 80 (1% v/v) and saline (94% v/v) was administered to mice by intraperitoneal (i.p.) injection. At 0.2, 0.5, 1, 1.5 and 2 h post-dose, mice (n = 4 at each time point) were anaesthetised with an i.p. dose of ketamine (133 mg/kg) and xylazine (10 mg/kg), blood was collected by cardiac puncture and the whole brain was removed following cervical dislocation. To assess drug uptake after stroke, sham or stroke mice were given a single i.p. dose of vehicle or 2′MeO6MF (30 mg/kg) and sacrificed 0.5 h post-dose (n = 4 mice/group). Plasma and brain samples were stored at −20℃ until analysis by HPLC. The brain homogenate concentrations of 2′MeO6MF were corrected for the amount of compound remaining in the brain microvasculature, which was calculated using a brain microvascular plasma volume of 0.039 ml/g (determined following intravenous administration of the BBB impermeable marker14C-sucrose).
Photothrombosis model of focal ischaemia
All experiments were approved by the University of Otago Animal Ethics Committee and were performed in accordance with the NIH Animal Protection Guidelines for the care and use of animals for scientific purposes and are reported according to the ARRIVE guidelines. Focal stroke was induced in the left hemisphere using the photothrombosis method in adult (2–3 months old) male C57BL/6 J mice weighing 25–27 g.6,20–22 Briefly, under isoflurane anaesthesia (2–2.5% in O2) mice were placed in a stereotactic apparatus, the skull exposed through a midline incision, cleared of connective tissue and dried. A cold light source (KL1500 LCD, Zeiss) attached to a 40X objective giving a 2 mm diameter illumination was positioned 1.5 mm lateral from Bregma and 0.2 ml of Rose Bengal solution (Sigma; 10 g/l in normal saline, i.p.) was administered. After 5 min the brain was illuminated through the intact skull for 15 min. Mice were housed under a 12 h light/dark cycle with ad libitum access to food and water.
2′MeO6MF (0.1–30 mg/kg) was dissolved in 10% DMSO and then diluted in 0.9% saline. 2′MeO6MF was given i.p. 1 h after stroke, with a second dose given at 24 h by an independent experimenter not undertaking any of the histological, biochemical or behavioural assessments to randomise the animals and minimise bias. All animals were randomly assigned to a treatment group post-stroke by a person not undertaking any of the post-stroke assessments, to ensure that all animals in any given cage received a different treatment and to maintain blindness throughout the study. In one experiment, 2′MeO6MF (30 mg/kg) was dosed at either 1, 3 or 6 h after stroke. To assess whether 2′MeO6MF neuroprotection was occurring via the AKT signalling pathway, the pan-AKT inhibitor, GSK690693 (30 mg/kg i.p), was given 1 h after stroke at the same time as 2′MeO6MF.
Behavioural assessment
Animals were tested once on both the gridwalking and cylinder tasks, one week prior to surgery to establish baseline performance levels. For all of the studies, animals were tested one week post-stroke at approximately the same time each day at the end of their dark cycle. Behaviours were scored by observers who were blind to the treatment group of the animals in the study as previously described.6,20,23
Infarct size
At seven days post-stroke animals were anaesthetised, transcardially perfused with 4% paraformaldehyde and brains extracted and processed histologically using cresyl violet staining in order to quantify infarct volume as previously described.6,20,23
Assessment of circulating pro-inflammatory cytokine levels
Blood samples were collected into EDTA-treated tubes three days post-stroke and plasma collected following centrifugation at 1000 × g for 10 min, 4℃. Plasma was then stored at −80℃ until required for analysis of circulating interleukin (IL)-1β, tumour necrosis factor (TNF)-α and IFN-γ levels.
Multiplex protein analysis
Multiplex suspension bead array immunoassay was performed using the Luminex 100™ analyser (Luminex Corporation, Austin, TX, USA) to identify a panel of cytokines (IL-1β, TNF-α and IFN-γ) using the Bio-Plex mouse cytokine assay kit (BioRad Laboratories Inc., NZ), according to the manufacturer’s instructions and as previously described.14 This is a multiplexed, particle-based, flow cytometric assay that utilises anti-cytokine monoclonal antibodies linked to microspheres incorporating distinct proportions of two fluorescent dyes.
Fifty microlitres of each plasma sample or cytokine standard (diluted 1:2 in serum diluent) were pipetted into the accompanying microtitre plate (MultiScreen 96-well filter plate), containing diluted antibody-coated bead complexes and incubation buffer. Samples were incubated for 30 min at room temperature (in the dark). After filtering and washing with assay diluent (100 µl/well, three times) using a vacuum manifold, 25 µl Bio-Plex detection antibody was added to all wells, incubated for 30 min at room temperature (in the dark) and washed as previously described. Fifty microlitres of streptavidin–phycoerythrin was added to all wells and incubated for 10 min at room temperature (in the dark). All microtitre wells received a final wash, 125 µl of Bio-Plex assay buffer added to each well and analysed. A minimum of 100 events (beads) was collected for each of the cytokines and median fluorescence intensities were obtained. Cytokine concentrations were automatically calculated based on standard curve data using Bio-Plex Manager™ software with a detectable range of 2–32,000 pg/ml. Calculated concentrations were exported to an Excel spread sheet for analysis.
Assessment of LPS-induced NFkB activity in RAWblue™ macrophage cells
RAWblue™ macrophages were grown in DMEM supplemented with 10% heat-inactivated FBS and 200 µg/ml Zeocin™ (selection agent). These cells allow for a colorimetric assessment of NFkB activity upon lipopolysaccharide (LPS) stimulation as determined by quantifying the level of secreted embryonic alkaline phosphatase (SEAP). For experiments, RAWblue™ macrophages were seeded to a flat-bottom 96-well plate at a density of 5 × 104 cells/ml (in the absence of Zeocin) and left untreated for 16 h. RAWblue™ macrophages were then pre-incubated with 2′MeO6MF (10−4–10−3 M) for 30 min prior to adding in LPS (2.5 ng/ml). The supernatant was then collected 6 h after LPS stimulation and the levels of SEAP were determined using the Quantiblue™ assay. Therefore, 30 µl of the supernatant and 80 µl of the Quantiblue™ solution were mixed, incubated at 37℃ for 3 h and the absorbance read at 620 nm. Data are from three independent experiments performed in triplicate.
GABA subunit expression in RAWblue™ macrophage cells
GABA subunit (α1, α2, α5, β1, β2 and δ) mRNA levels were assessed by quantitative real-time polymerase chain reaction (qPCR). Total RNA from RAWblue™ macrophage cultures were extracted using the Qiagen RNeasy kit and genomic DNA was removed using DNAse (Ambion) following the manufacturer’s protocols. The purity (RNA with ratio of absorbance at 260 and 280 nm ≥ 2) and amount of the RNA was measured spectrophotometrically (NanoDrop 2000, Thermo Scientific, USA). Total RNA (750 ng) was used to synthesis the first-strand complementary DNA (cDNA) using Super Script III (Life Technologies) following the manufacturer’s protocol. After reverse transcription, the cDNA was amplified by qPCR using SyBr green master mix (Applied Biosystems, Foster City, CA) and each of the following primer (250 nM) sets; α1: forward – CATGACAGTGCTCCGGCTAA, reverse – GCCATACTCTGTGATACGCA; α2: forward – TTCAAAGCCACTGGAGGAAAAC, reverse – GCAGCAGAGACCATACATTGC; α5: forward – GCTGACCCATCCTCCAAACA, reverse – TGGAGACTGTGGGTGCATTC; β1: forward – ATCGAGAGAGTTTGGGGCTTC, reverse – GCTGGGTTCATTGGAGCTGT; β2: forward – TAGTGGGCACGAGGGTTAGA, reverse – ATGACGATCCACCACAGCAG; δ: forward – AGGAACGCCATCGTCCTTTT, reverse – CTTGACGACGGGAGATAGCC; SDHA: forward – GCCCATGCCAGGGAAGATTA, reverse – TGTTCCCCAAACGGCTTCTT. qPCR was performed on a Roche Lightcycler (Roche, Minneapolis, MN) with the following cycling parameters: 40 cycles of 95℃, 15 s; 60℃, 30 s; 72℃, 40 s. After amplification, a denaturing curve was performed to ensure the presence of unique amplification products. All reactions were performed in triplicate. Expression of mRNA was assessed by evaluating threshold cycle (CT) values. The CT values were normalised against the expression level of the housekeeping gene SDHA.
Statistical analysis
All data are expressed as mean ± SD and plotted as box and whisker graphs. Electrophysiological data were analysed using Mann–Whitney non-parametric tests for comparisons between groups. To compare the cumulative probabilities of IPSC inter-event intervals and amplitudes, significance was determined using the two-tailed Kolmogorov–Smirnov test using MiniAnalysis software. For histological, biochemical and behavioural assessments post-stroke, one-way analysis of variances and Newman–Keuls’ multiple pair-wise comparisons for post hoc comparisons were used. An n = 5 was used per treatment group for histological and biochemical assessments and an n = 7 per group for behavioural assessments. The level of significance was set at P < 0.05.
Power analysis
For behavioural experiments, six animals per group are required to achieve > 80% power (86% calculated), considering the following parameters: α = 0.05; with an effect size = 1.5. For histological experiments, five animals per group are required to achieve > 80% power (91% calculated), considering the following parameters: α = 0.05; effect size 1; three concentrations; two groups, and correlation between measures = 0.5. Parameters were determined from our prior work, in which we have demonstrated significant behavioural effects,6,21,22,24 and on the assumption that variance was about 25%. It should be noted that more conservative effect sizes were used for those experiments, as it is harder to assess recovery over time between groups than looking at the effects of drug treatments on stroke size. Prior studies have also determined that an N = 4–5 for BBB permeability and biochemical assessments is sufficient. Where possible an n-value greater than the calculated values was used. Power calculations were performed with G Power Software (version 3.1.5).
No deaths were reported during these studies. Two mice were excluded from analysis, one from the stroke group and one from the stroke + treatment group due to the lack of stroke being detected. This is most likely due to experimenter error with the Rose Bengal most likely being injected into the bladder. Additional animals were set-up to replace these two mice.
Results
Tonic inhibitory GABAAR currents are altered at a delay after stroke
We have previously reported that tonic inhibitory currents are elevated three days post-stroke.6 As we have not established when these currents begin to increase, we examined tonic inhibitory currents in the peri-infarct cortex of mice after a photothrombotic stroke to the forelimb motor cortex. Layer 2/3 pyramidal neuron whole-cell voltage-clamp recordings were obtained from brain slices generated ex vivo 3, 6, 12 and 24 h after stroke and compared to recordings obtained three days post-stroke and sham controls (Figure 1(c)). Recordings revealed a significant treatment effect (F(5,50) = 12.77, P < 0.0001), with recordings obtained at either 3 or 6 h after stroke showing a small decrease in GABAA-receptor-mediated tonic inhibition (33% decrease in Itonic; P = 0.0281 at 3 h, n = 8; 21% decrease in Itonic; P = 0.2761 at 6 h, n = 8), compared to neurons from sham controls (n = 19: Figure 1(d)). In contrast, recordings 12 h after stroke showed no differences (3% increase in Itonic; P = 0.7383 at 12 h, n = 9), whilst recordings obtained at either 24 h or three days after stroke showed a significant increase in tonic inhibitory currents compared to controls (57% increase in Itonic; P = 0.009 at 24 h, n = 12; 136% increase in Itonic; P = 0.0001 at three days, n = 12, Figure 1(d)). These findings infer that tonic inhibitory currents increase at a substantial delay after stroke.
2′MeO6MF increases tonic inhibitory currents in granule cells
2′MeO6MF was applied to acute hippocampal slices, to measure tonic currents in DGGCs, which predominantly express δ-containing GABAARs.25 Recordings revealed a significant treatment effect (F(3,20) = 5.37, P < 0.0071), in the presence of 2′MeO6MF. In control slices, application of the GABAAR antagonist, SR95531, revealed a tonic current of 3.6 ± 0.7 pA (current density 0.72 ± 0..43 pA/pF, N = 7; Figure 2(a) and (b)). While a low concentration of 2′MeO6MF (1 µM) did not affect the SR95531-sensitive current (3.9 ± 0.9 pA, current density 0.67 ± 0.15 pA/pF, N = 5; Figure 2(a) and (b)), higher concentrations of 2′MeO6MF (3–10 µM) induced a concentration-dependent increase in tonic currents (3 µM: 8.5 ±1.5 pA, 1.11 ± 0.39 pA/pF, N = 5, P < 0.05; 10 µM: 14.5 ± 4.1 pA, 2.56 ± 1.72 pA/pF, N = 7, P < 0.05; Figure 2(a) and (b)).
Figure 2.

2′MeO6MF acts on δ-containing GABAARs and increases tonic GABAAR currents in DGGCs. The effect of 2′MeO6MF on GABAAR-mediated inhibitory currents was assessed using DGGCs. Panel (a) shows representative holding current traces across time that were used to estimate the tonic currents from control and 2′MeO6MF-treated (1–10 µM) granule cells. 2′MeO6MF alone induces a small increase in tonic inhibition in granule cells. Horizontal bars indicate the application of the GABAAR antagonist SR95531 (>100 µM). 2′MeO6MF (1 and 3 µM) induced little or no increase in tonic current, while 10 µM 2′MeO6MF was able to induce a clear increase in tonic current in granule cells. Panel (b) box plot (whiskers: minimum and maximum; lines: median) showing the average (mean ± SD) GABAAR-mediated current density in granule cells in control (n = 7) and after application of 1 µM (n = 5), 3 µM (n = 5) or 10 µM (n = 7) 2′MeO6MF. Panel (c) and (d) show that 2′MeO6MF enhances phasic inhibition by increasing mIPSC amplitude at all three doses tested (1–10 µM). Panel (c) shows representative average mIPSC (n > 50 traces) in control conditions and after application of 2′MeO6MF (1–10 µM). Panel (d) shows an overlay of both the control mIPSC trace (black) and the 10 µM 2′MeO6MF mIPSC trace (red), highlighting the increase in amplitude upon application of 2′MeO6MF. **=P < 0.05, compared to the control tonic current. 2′MeO6MF: 2′-methoxy-6-methylflavone.
We previously reported that 2′MeO6MF can have some effects on synaptic GABAARs.16 To assess whether 2′MeO6MF might affect GABAAR-mediated post-synaptic currents, we carried out experiments in the presence of TTX to isolate miniature IPSCs (mIPSCs). mIPSC amplitudes in the presence of 1, 3 and 10 µM 2′MeO6MF increased from 24.7 ± 2.3 pA (N = 5) to 29.8 ± 4.2 pA (N = 6, P < 0.05), 31.7 ± 6.0 (N = 6, P < 0.05) and 29.9 ± 3.3 (N = 6, P < 0.05), respectively (Figure 2(c) and (d)). No changes in the rise time, frequency and decay of mIPSCs were observed (Figure 2(c) and (d)). These findings are very similar with our previous publication.26 These observations suggest that 2′MeO6MF exerts a strong potentiation of δ-containing GABAARs, and a potential, weak positive modulatory effect of the flavonoid on synaptic GABAARs.
2′MeO6MF increases tonic currents in layer 2/3 pyramidal neurons after stroke
We next assessed whether 2′MeO6MF could also increase tonic inhibitory currents after stroke in the peri-infarct region, specifically in layer 2/3 pyramidal neurons of the motor cortex. We show that stroke induces a delayed increase in Itonic within the peri-infarct region that could offer an extended therapeutic window to minimise the extent of cell death (Figure 1). To assess the effects of 2′MeO6MF on tonic currents after stroke, 2′MeO6MF (30 mg/kg i.p.) was administered to mice 1 h after stroke and then slices generated 2 h later, 3 h post-stroke. Based on our recordings from acute slices generated from stroked mice treated with 2′MeO6MF (30 mg/kg i.p.) 3 h after stroke, we report a 36.81 ± 19.42% (n = 11) increase in Itonic compared to vehicle-treated stroked mice ( − 5.45 ± 12.75%, n = 10; P < 0.0001: Figure 3(a) and (b)). To assess the effects of 2′MeO6MF on phasic inhibitory currents we analysed frequency and amplitude of IPSCs with and without 2′MeO6MF. No difference in either amplitude or frequency (inter-event intervals) of IPSCs was observed following application of 2′MeO6MF (Figure 3(c) and (d), respectively). These data confirm our hypothesis that systemic administration of 2′MeO6MF can increase tonic inhibitory currents, with minimal effects observed on phasic inhibitory currents and is therefore a viable treatment option for further investigation as a neuroprotectant.
Figure 3.
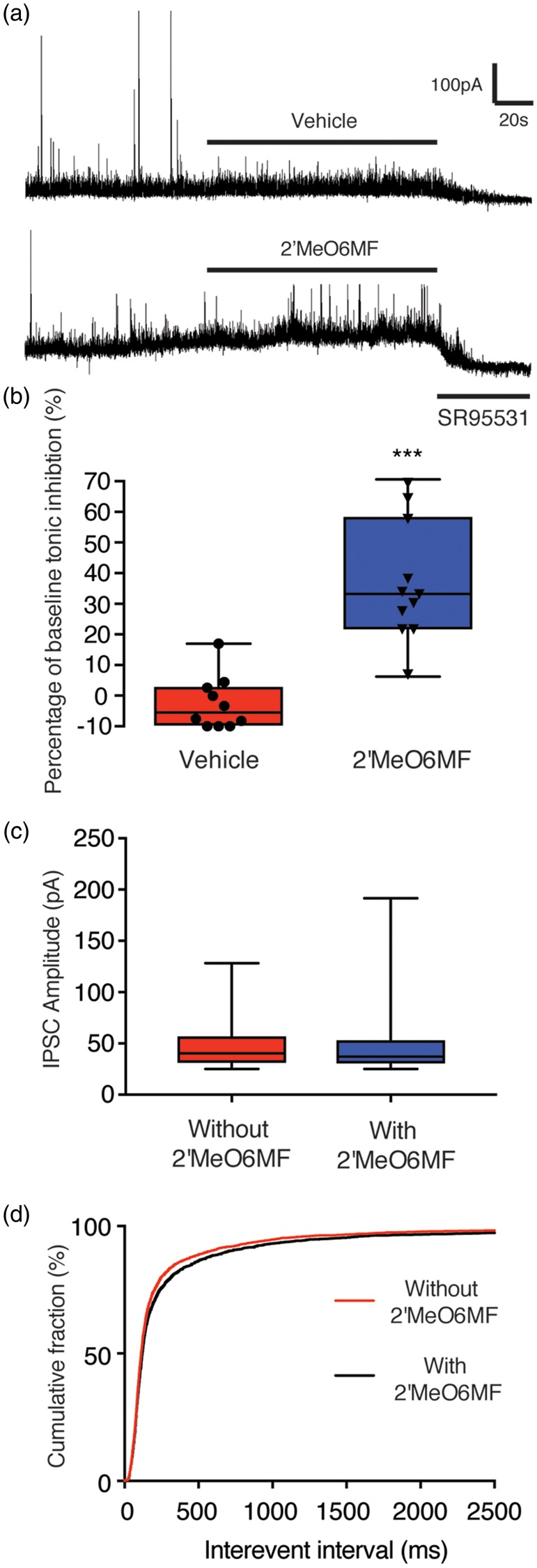
2′MeO6MF increases tonic, but not phasic GABAAR currents in layer 2/3 pyramidal neurons after stroke. Recordings from layer 2/3 pyramidal neurons 3 h after stroke in the presence of 2′MeO6MF revealed a significant increase in tonic inhibitory currents in mice that received an initial 30 mg/kg i.p. dose of 2′MeO6MF (n = 11) 1 h after stroke, when compared to vehicle-treated controls (n = 10 (a)). Horizontal bars indicate the application of either 2′MeO6MF or vehicle, or the GABAAR antagonist SR95531. Cells were voltage clamped at + 10 mV. Data are presented as box plot (whiskers: minimum and maximum; lines: median) for mean ± SD (b). No change in phasic inhibitory currents was observed in mice treated with 30 mg/kg i.p. dose of 2′MeO6MF 1 h after stroke, when compared to vehicle-treated controls ((c) and (d)). Median IPSC amplitudes were not different (without 2′MeO6MF: 46.39 ± 19.9 pA; with 2′MeO6MF: 45.48 ± 21.79 pA; data collected from 2700 events: (c)). Cumulative probability curves reveal no difference in median inter-event intervals, i.e. no difference in frequency of IPSCs – without 2′MeO6MF (112 ms), with 2′MeO6MF (119.6 ms (d)). ***=P < 0.001, compared with stroke + vehicle-treated controls. IPSC: ; 2′MeO6MF: 2′-methoxy-6-methylflavone.
2′MeO6MF crosses the BBB following systemic administration
To enable correlation of in vitro and in vivo data, we next evaluated whether 2′MeO6MF crosses the mouse BBB. We report that administration of a single i.p. dose of 2′MeO6MF (30 mg/kg) to naïve (Figure 4(a1)) and stroked (Figure 4(a2)) mice resulted in robust plasma as well as brain exposure with peak levels observed at 0.5 h following dosing, and with concentrations declining over the next hours. At 2 h following dosing the concentrations were below the limit of quantification suggesting efficient clearance (Figure 4(a)). The amount of 2′MeO6MF in the brain at the peak level equates to a total brain homogenate concentration of 3–10µM, a concentration that increases tonic inhibition in vitro.
Figure 4.

2′MeO6MF crosses the BBB, is neuroprotective and enhances functional recovery after focal cerebral ischaemia. Plasma (•, µg/ml) and brain (○, µg/g) concentrations of 2′MeO6MF were assessed following a single 30 mg/kg i.p. dose to BALB/cJ Asmu mice (n = 4/group: (a1)). Brain concentrations (left ipsilateral hemisphere (ipsi) and right contralateral hemisphere (contra) of 2′MeO6MF were assessed following a single 30 mg/kg i.p. dose to both sham and stroke C57BL/6 J mice 30 min post-stroke (n = 4: (a2)). Stroke induces a slight increase in brain concentrations at the site of stroke only (ipsi – red solid bar) compared to sham controls (blue bars) and the non-stroke hemisphere (contra – red chequered bar). Assessment of infarct volume was carried out by quantifying cresyl violet stained sections generated seven days post-stroke revealed that treatment with 2′MeO6MF 1 h post-stroke with a second dose given 24 h later resulted in a dose-dependent decrease in infarct volume (0.1 mg/kg–30 mg/kg: n = 5/treatment group (b)). Representative cresyl violet-stained sections from vehicle-treated stroke controls and stroke + 2′MeO6MF treatment are shown in (b1) with the quantifications shown in (b2). Behavioural measurements of functional recovery, gridwalking function for forelimb function and cylinder task for forelimb asymmetry are shown in (c) and (d), respectively. On both tasks, similar to the histological data, 2′MeO6MF resulted in a decrease in the number of foot faults on the gridwalking task and an improvement in forelimb asymmetry in the cylinder task. The therapeutic window for 2′MeO6MF (30 mg/kg dose) was assessed by starting the dose at either 1 -, 3 - or 6 h with a second dose given 24 h post-stroke and quantifying the volume of infarction seven days later (e). Representative cresyl violet-stained sections from vehicle-treated stroke controls and stroke + 2′MeO6MF treatment are shown in (e1) with the quantifications shown in (e2). Assessment of infarct volumes following dosing of 2′MeO6MF at 1 -, 3 - and 6 h showed a significant decrease in infarction compared to stroke + vehicle controls. Data are presented as mean ± SD. * = P < 0.05, **=P < 0.01 and ***=P < 0.01 compared with stroke + vehicle-treated controls. 2′MeO6MF: 2′-methoxy-6-methylflavone.
2′MeO6MF is neuroprotective and enhances functional recovery after focal cerebral ischaemia
As not all strokes have a reperfusion component, we investigated the protective effects of 2′MeO6MF in the focal photothrombosis model of stroke. This model has minimal reperfusion and it is in general harder to protect against cell death. Mice were treated with an initial i.p. dose of 2′MeO6MF (0.1–30 mg/kg) or vehicle 1 h post-stroke, with a second dose given at 24 h. These doses had no effect on body temperature (data not shown) nor did they induce sedation. Treatment with 2′MeO6MF resulted in a dose-dependent decrease in infarct volume (F(3,16) = 8.435; P < 0.0014: vehicle, 2.23 ± 0.26 mm3 versus 2′MeO6MF (0.1 mg/kg), 1.69 ± 0.12 mm3, P < 0.05; 2′MeO6MF (5 mg/kg), 1.14 ± 0.73 mm3, P < 0.05, and 2′MeO6MF (30 mg/kg) 0.89 ± 0.54 mm3, P < 0.01: n = 5 per group; Figure 4(b1) and (b2)) when assessed one week post-stroke.
We next tested the mice behaviourally on both the gridwalking (forelimb function) and cylinder (forelimb asymmetry) tasks. Behavioural assessments revealed an increase in the number of foot faults on the gridwalking test (n = 7 per group; Figure 4(c)) and an increase in spontaneous forelimb asymmetry in the cylinder task (n = 7 per group; Figure 4(d)) one week post-stroke.6,21 Treatment with 2′MeO6MF resulted in a dose-dependent decrease in the number of foot faults on the gridwalking task (F(3,32) = 11.27, P < 0.001) and an improvement in forelimb asymmetry in the cylinder task F(3,32) = 3.081, P < 0.413).
To establish the therapeutic window for 2′MeO6MF, we treated cohorts of mice with 30 mg/kg 2′MeO6MF starting 1 -, 3 - or 6 h post-stroke (Figure 4(e1 and 2)). Assessment of infarct volume one week post-stroke revealed a significant treatment (F(3,24) = 4.38, P < 0.0113), compared to vehicle-treated stroke controls (Figure 4(e1)). Treatment with 2′MeO6MF resulted in a significant decrease in infarct volume at all three time points (vehicle: 2.33 ± 0.47 mm3, n = 7 versus 30 mg/kg 2′MeO6MF at 1 h: 1.45 ± 0.71 mm3, n = 7, P < 0.001; 3 h: 1.89 ± 0.26 mm3, n = 7, P < 0.05; and 6 h: 1.79 ± 0.62 mm3, n = 7, P < 0.05; Figure 4(e2)).
2′MeO6MF is not neuroprotective following focal cerebral ischaemia in δ−/− mice
To assess whether 2′MeO6MF was affording protection via δ-containing GABAARs, we induced stroke and behaviourally tested wild-type (WT) and δ-subunit knockout mice (δ−/−)27 in the presence and absence of 2′MeO6MF (n = 6 per group). Assessment of infarct volume seven days post-stroke revealed a significant treatment effect (F(3,17) = 3.714, P < 0.032) with extensive damage to the motor cortex in vehicle-treated WT and δ−/− mice (1.69 ± 0.08 mm3, 1.44 ± 0.27 mm3; respectively: Figure 5(a) and (b)). Treatment with 2′MeO6MF (30 mg/kg i.p.) resulted in a significant decrease in infarct volume in WT (0.9 ± 0.3 mm3, P < 0.05; Figure 5(a) and (b)) but not in δ−/− (1.46 ± 0.24 mm3, P > 0.05) mice.
Figure 5.

2′MeO6MF does not afford neuroprotection or enhance functional recovery in δ−/− mice after stroke. WT or δ−/− mice were dosed with 2′MeO6MF (30 mg/kg i.p.) or vehicle at 1 h after stroke with a second dose given 24 h later. Assessment of infarct volume using cresyl violet staining was carried out on brain sections generated seven days after stroke. Treatment with 2′MeO6MF revealed a significant decrease in infarct volume in WT but not in δ−/− mice ((a) and (b)) when compared to vehicle-treated controls. Behavioural measurements of functional recovery, gridwalking function for forelimb function and cylinder task for forelimb asymmetry are shown in (c) and (d), respectively. On both tasks, similar to the histological data, 2′MeO6MF resulted in a decrease in the number of foot faults on the gridwalking task and an improvement in forelimb asymmetry in the cylinder task in WT but not in δ−/− mice compared to vehicle-treated stroke controls. All data are presented as mean ± SD for an n = 6 per treatment group. *=P < 0.05 compared with stroke + vehicle-treated WT controls. ** = P<0.01. KO: d-/- knockout; 2′MeO6MF: 2′-methoxy-6-methylflavone; WT: wild type.
Behavioural assessments revealed an increase in the number of foot faults on the gridwalking test (Figure 5(c)) and an increase in spontaneous forelimb asymmetry in the cylinder task (Figure 5(d)) one week post-stroke in both WT and δ−/− mice. Assessment of functional recovery revealed a significant treatment effect on both the gridwalk (F(3,47) = 5.27, P < 0.0032) and cylinder tasks (F(3,47) = 3.10, P < 0.0355). Administration of 2′MeO6MF to WT mice resulted in a significant improvement in motor function on both the gridwalk (P < 0.01) and cylinder tasks (P < 0.05). However, administration of 2′MeO6MF to δ−/− mice failed to show any improvement in motor function (P > 0.05). These data suggest that 2′MeO6MF is mediating its neuroprotective effects via δ-containing GABAARs.
2′MeO6MF modulates inflammatory pathways
Inflammatory processes play an important role in the modulation and progression of stroke-induced brain damage. The initial inflammatory response is detrimental due to the induction of apoptotic processes,27 and stimulation of tonic GABA responses,13 which hinders regaining function after stroke.6
To first confirm that immune cells express GABA subunit and in turn could respond directly to a 2′MeO6MF, we performed qPCR on macrophage cells for α1, α2, α5, β1, β2 and δ-subunits. Assessment of subunit expression levels revealed that the macrophage cells expressed α5, β1 and δ-subunits, had low levels of α1 and β2-subunit expression and the α2-subunit was not detectable (Figure 6(a)). We next looked at the efficacy of 2′MeO6MF to dampen an LPS-medicated increase in NFkB activity, which is a hallmark sign of inflammation. RAWblue™ macrophage cells in the presence of LPS (2.5 ng/ml) show a marked increase in NFkB activity (P < 0.001; Figure 6(b)), which in the presence of the vehicle (2.5% DMSO) shows no further change. LPS-stimulated cells treated with 2′MeO6MF show a marked decrease in NFkB activity (P < 0.001; Figure 6(b)) with levels not being significantly different to control unstimulated LPS controls.
Figure 6.
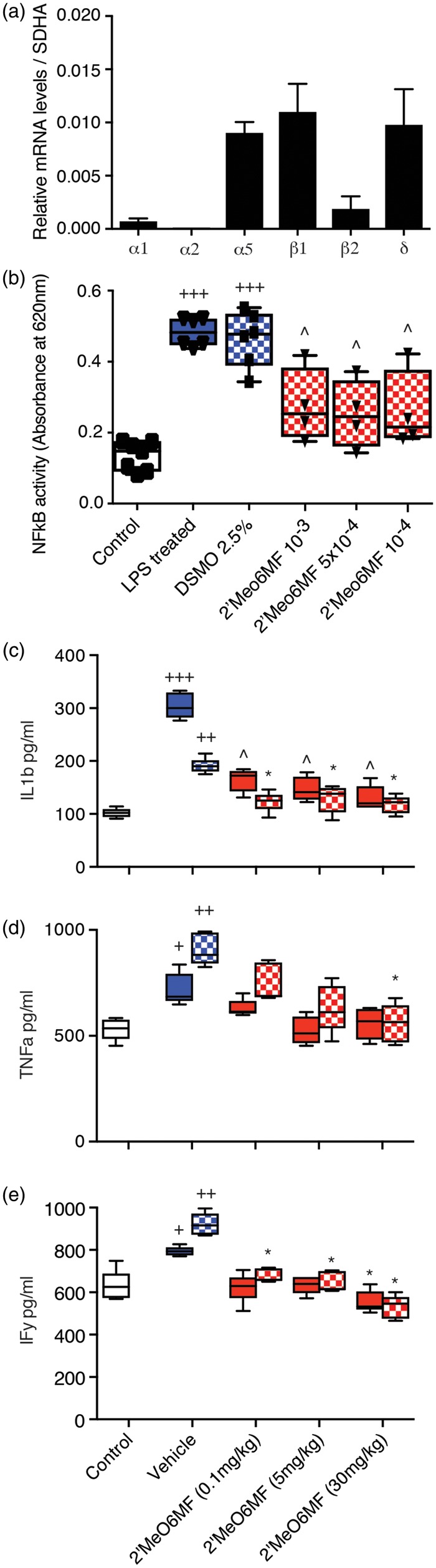
2′MeO6MF decreases NFkB activity and circulating pro-inflammatory cytokines. The level of expression for various GABA subunits was assessed in the RAWblue™ macrophage cell line. Macrophage cells were shown to express α5, β1 and δ, with very low levels of α1, α2 and β2 being detected (a). Upon stimulation of macrophage with LPS (2.5 ng/ml) a significant increase in NFkB activity was detected (b). Addition of the 2′MeO6MF (10−3–10−4), but not the vehicle (2.5% DMSO), to the media resulted in a significant decrease in NFkB activity (b). Data are presented as mean ± SD. +++=P < 0.001 compared with controls; ^=P < 0.01 compared to LPS or LPS + DMSO-treated controls. 2′MeO6MF: 2′-methoxy-6-methylflavone. The effects of sham-operated controls (white bars), stroke + vehicle treatment (blue bars) and stroke + 2′MeO6MF treatment (red bars) on circulating IL-1β (c), TNF-α (d) and IFN-γ levels (e) were assessed in plasma collected 1 - (solid bars) and three days (chequered bars) post-stroke. Stroke induces a significant elevation in circulating pro-inflammatory IL-1β (c), TNF-α (d) and IFN-γ (e) levels. Treatment with 2′MeO6MF dosing at 1 h with a second dose at 24 h resulted in a significant decrease in circulating IL-1β and IFN-γ ((c) and (e), respectively) levels at 0.1, 5 and 30 mg/kg. Treatment with 2′MeO6MF also resulted in a significant decrease in circulating TNF-α (d) levels three days following dosing at 30 mg/kg, but not on day 1, nor following dosing at the either 0.1 or 5 mg/kg. All data are presented as mean ± SD. +=P < 0.05, ++=P < 0.01 and +++=P < 0.001 compared with sham-operated controls; *=P<0.05, ^=P < 0.01 compared with stroke + vehicle-treated controls. DMSO: ; IF: interferon; LPS: ; TNF: tumour necrosis factor.
To confirm our in vitro findings, we next assessed the effects of 2′MeO6MF on the inflammatory response after stroke in vivo. In corroboration with prior studies, we demonstrate that cerebral ischaemia results in a significant increase in circulating pro-inflammatory cytokines, IL-1β (sham control: 105.81 ± 8.45 pg/ml, versus stroke + vehicle: 190.75 ± 16.82 pg/ml; P < 0.01), TNF-α (sham control: 624.56 ± 63.42 pg/ml versus stroke + vehicle: 920.88 ± 88.01 pg/ml; P < 0.05), and interferon (IFN)-γ (sham control: 624.75 ± 49.75 pg/ml versus stroke + vehicle: 866.13 ± 67.05 pg/ml; P < 0.05, Figure 6(c) to (e)).
Treatment with 2′MeO6MF resulted in a significant decrease in circulating IL-1β and IFN-γ levels (Figure 6(c): P < 0.05 for all doses; and Figure 6(e): P < 0.05 for 0.1 and 5 mg/kg and P < 0.01 for 30 mg/kg, respectively) one and three days post-stroke. Assessment of TNF-α revealed a dose-dependent decrease in circulating levels that was significant for the high dose of 2′MeO6MF only (30 mg/kg: P < 0.05, Figure 6(d)). These data suggest that 2′MeO6MF has anti-inflammatory actions that are mediated via the AKT signalling pathway.
Co-administration with the pan-AKT inhibitor GSK690693 blocked the neuroprotection afforded by 2′MeO6MF
Pro-inflammatory cytokines have been previously shown to contribute to delayed cell death, oedema and impaired functional recovery. Recent work has shown that cytokines can alter the expression of extrasynaptic GABAAR subunits and impair memory.13 Activation of GABAARs can signal through multiple pathways including the AKT signalling pathway.25 As the AKT signalling pathway has been shown to play a major role in neuroprotection, we tested the hypothesis that inhibition of AKT could prevent 2′MeO6MF-induced neuroprotection.28
Assessment of infarct volume seven days post-stroke revealed a significant treatment effect (F(3,31) = 5.49, P < 0.0038), with the pan-AKT inhibitor, GSK690693 (30 mg/kg i.p) given 1 h post-stroke, resulted in an increase in infarct volume (stroke + AKT: 2.58 ± 0.58 mm3, n = 7) compared to vehicle-treated controls (stroke + vehicle: 2.33 ± 0.47 mm3, n = 10; Figure 7(a)), and when administered at the same time as 2′MeO6MF completely blocked 2′MeO6MF protective effects (stroke + 2′MeO6MF + AKT: 2.52 ± 0.99 mm3, n = 7: versus stroke + 2′MeO6MF: 1.45 ± 0.71 mm3, n = 7; P < 0.05). In addition, assessment of functional recovery revealed a significant treatment effect on both the gridwalk (F(3,62) = 14.11, P < 0.0001) and cylinder tasks (F(3,62) = 16.57, P < 0.0001). Co-administration of GSK690693 with 2′MeO6MF blocked the 2′MeO6MF-mediated functional recovery on both the gridwalking (P < 0.001, Figure 7(b)) and cylinder (P < 0.05, Figure 7(c)) tasks, whilst animals treated with GSK690693 alone performed significantly worse compared to vehicle-treated stroke controls. Furthermore, GSK690693 completely blocked the effects of 2′MeO6MF on circulating cytokine levels, and when administered alone resulted in a significant increase in circulating IL-1β (Figure 7(d)) and TNF-α (Figure 7(e)) levels, one and three days post-stroke.
Figure 7.

The pan-AKT inhibitor prevents 2′MeO6MF from affording protection and functional recovery. Assessment of infarct volume seven days post-stroke revealed that treatment with 2MeO6MF (30 mg/kg i.p.) resulted in a significant decrease in infarction (n = 7) compared to stroke + vehicle (n = 7 (a)). Treatment with the pan-AKT inhibitor, GSK690693 (30 mg/kg i.p) given 1 h post-stroke with a second dose at 24 h, resulted in a small increase in infarct volume (n = 7) compared to vehicle-treated controls, and when administered at the same time as 2′MeO6MF completely blocked 2′MeO6MF protective effects (n = 7). Functional recovery was assessed using behavioural measurements of forelimb functions (gridwalking (b)) and forelimb asymmetry (cylinder task (c)). Treatment with 2′MeO6MF resulted in a significant improvement in functional recovery on the gridwalking task (b) and in the cylinder task (c). Co-administration of GSK690693 with 2′MeO6MF blocked the 2′MeO6MF-mediated functional recovery on both the gridwalking (b) and cylinder (c) tasks. Animals treated with GSK690693 alone performed significantly worse on both the gridwalking (b) and cylinder (c) tasks compared to stroke + vehicle controls. Assessment of circulating cytokine levels revealed that treatment with 2′MeO6MF decreased both IL-1β (d) and TNF-α (e) levels, one day (solid bars) and three days (chequered bars) post-stroke. Treatment with GSK690693 completely blocked the effects of 2′MeO6MF and when administered alone resulted in a significant increase in circulating IL-1β (d) and TNF-α (e) levels, one and three days post-stroke. All data are presented as mean ± SD. + =P < 0.05, ++=P < 0.01 and +++=P < 0.001 compared with sham + vehicle-treated controls; *=P < 0.05, **=P < 0.01 and ***=P < 0.001 compared with stroke + vehicle-treated controls; ^=P<0.05 and $=P < 0.001 compared with time-matched stroke + 2′MeO6MF treatment. 2′MeO6MF: 2′-methoxy-6-methylflavone.
Discussion
The mechanisms whereby GABAergic compounds achieve neuroprotection after stroke have not been fully elucidated. One strategy to improve the performance of GABAergic compounds is to target specific GABAAR subtypes. Subtype-specific GABAAR compounds are more likely to show an effect and are already being trialled for conditions such as Down’s syndrome, affective disorders, schizophrenia and autism.10,11 The need to assess subtype-specific GABA compounds will enable us to better understand the GABAergic processes that occur after stroke. This is further highlighted with clinical reports showing that zolpidem can result in a transient improvement in aphasia.29 Furthermore, we have previously shown that negative allosteric modulators for α5-containing GABAARs can enhance functional recovery when started > three days after stroke.6 It is important to note that by day 3 after stroke, the infarct is fully formed, and drug treatments designed to increase cortical excitability at or beyond this time point do not affect the infarct size per se.6,20
GABAARs mediate both phasic and tonic inhibitory signals.30 Tonic GABAARs in the hippocampus, cortex and thalamus contain either α5 - or δ-subunits.31,32 Antagonism of α5 - or δ-subunits enhances pyramidal neuron firing to afferent inputs19,31 and enhances neuronal network excitability.12 Given the primary role that extrasynaptic GABAARs play in modulating neuronal excitability, modulation of either α5- or δ-subunits early would offer a viable approach for dampening the glutamate-mediated hyperexcitability.2 This is confirmed in the current set of experiments where we show that 2′MeO6MF can increase tonic inhibitory currents and minimise the extent of cell death after stroke.
Flavonoids can modulate GABAergic function,15–17 with recent evidence showing that some flavonoids can modulate tonic inhibition.33 In corroboration with these findings, we demonstrate that the synthetic flavonoid, 2′MeO6MF, potentiates δ-containing GABAARs to modulate tonic currents in brain slice recordings. As the subunit compositions of δ-containing GABAARs in the DGGC are predominantly α4β2δ,34 and 2′MeO6MF (10 µM) results in a significant increase in tonic GABA currents, our observations suggest that 2′MeO6MF exerts a strong action most likely on δ-containing GABAARs, and a potential, weak positive modulatory action of the flavonoid on synaptic GABAARs. This is further supported by our previous data showing that 2′MeO6MF (1–300 µM) neither activates or enhances the response to GABA EC10 at recombinant α5β1–3γ2 L receptors.16 In addition, we show that the neuroprotective effects of 2′MeO6MF were absent in the δ−/− mice. The lack of an increase in infarct volume in the δ−/− mice is consistent with our previously published data, where we report that genetic deletion of α5 - or δ-GABAARs does not affect infarct size or neuronal number in peri-infarct cortex.6 Unlike pharmacological antagonism genetic deletion of α5 - or δ-GABAARs most likely triggers a compensatory upregulation of the other receptor35 and therefore obscures their roles in neuroprotection immediately following stroke.
We also demonstrate that increasing tonic inhibitory currents early after stroke (within 6 h) by means of treatment with 2′MeO6MF offers a potential new therapeutic approach for minimising stroke damage. Despite the efficient clearance of 2′MeO6MF from plasma, it is important to note that the neuroprotective effects of this flavonoid are still observed following systemic administration to mice. Given the clearance profile of 2′MeO6MF, one may speculate that giving multiple doses or intravenous infusion of 2′MeO6MF to ensure brain concentrations are maintained at therapeutic levels for a prolonged period of time could lead to further improvements. Future studies will be required to ascertain what are safe levels of change in tonic currents. The rational here is that too much tonic inhibition could be deleterious and result in either receptor desensitisation,36 or increased seizure activity, which has been reported to occur in both humans37 and animals.38
Astrocytes play a significant role in modulating network excitability via changes in tonic inhibition.39 Recently, it was shown that GABA could be released from glial cells via the anion channel Bestrophin 1,40 which could account for the increase in extracellular GABA observed during various pathological states, including stroke.6,7 In addition to modulating neurotransmitter levels, astrocytes and infiltrating immune cells (macrophage, lymphocytes, etc.) release cytokines.41 Circulating pro-inflammatory cytokines such as IL-1β and TNF-α contribute to delayed cell death and oedema. GABA compounds can modulate pro-inflammatory pathways and protect against mitochondrial damage.14 We show here that treatment with 2′MeO6MF can attenuate circulating pro-inflammatory cytokine levels in an AKT-dependent manner. Recent work demonstrated that IL-1β can change the surface expression of α5-containing GABAARs and is associated with impaired memory.13 We show here using 2′MeO6MF that a reciprocal relationship also exists between pro-inflammatory cytokine levels and δ-containing GABAARs. Elevated levels of cytokines could be associated with the delayed increase in tonic inhibition after stroke.6 What we demonstrate is that increasing tonic inhibitory currents early affords significant protection, dampens an LPS-mediated increase in NFkB activity and also dampens circulating cytokines levels in an AKT-dependent manner. Even though the relationship between circulating cytokine levels and changes in tonic inhibitory currents was first documented by Wang et al.,13 the mechanisms linking the two remain poorly understood. What we hypothesis is happening is that the infiltration of immune cells triggers a local release cytokines in peri-infarct regions, which in turn contributes to an increase in tonic inhibitory currents. A change in tonic inhibitory current does not itself directly affect cytokine levels unless the compound being used acts directly on immune cells to prevent the release of cytokines. We show that 2′MeO6MF not only increases cortical pyramidal neuron tonic inhibitory currents, but also acts on immune cells to dampen NFkB activity and release of cytokines. Increasing tonic inhibitory currents does not increase inflammation or cytokine levels, which is why such compounds can be used early after stroke to prevent damage; however, an increase in circulating cytokines can lead to an increase in tonic inhibitory currents.
In summary, we demonstrate that enhancing tonic inhibition early, within hours of inducing a stroke, affords protection, enhances motor recovery and dampens circulating pro-inflammatory cytokine levels. Importantly, this highlights the need to develop new and better subunit-specific compounds. Together, our results further identify δ-containing GABAARs as novel pharmacological targets for stroke and provide a rational basis for developing future therapies to minimise the extent of cell death and promote recovery after stroke and possibly other brain injuries.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the New Zealand Neurological Foundation, a project grant and a Sir Charles Hercus Fellowship from the Health Research Council of New Zealand (ANC), an Australian National Health and Medical Research Council Grant #1081733 (ANC, JRH, JAN and MC), the Lundbeck Foundation (MMH and KJ), a Lottery Health Postdoctoral Fellowship (SN).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JRH, MC, PKA and NG synthesised and provided the flavonoid. ZD, MMH and KJ performed the recordings from granule cells. EKG and PSN performed the KO animal studies. ANC, LB-B and KP performed the stroke studies, behavioural assessments and measured cytokine levels. SN performed the macrophage cell studies. RYN performed recordings from cortical neurons after stroke. LJ and JAN performed the studies to assess brain and plasma concentrations of the flavonoid. MC and ANC designed the experiments and wrote the paper. All authors reviewed, edited and approved the final version of this manuscript.
References
- 1.Dobkin BH. Training and exercise to drive poststroke recovery. Nat Clin Pract Neurol 2008; 4: 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lipton P. Ischemic cell death in brain neurons. Physiol Rev 1999; 79: 1431–1568. [DOI] [PubMed] [Google Scholar]
- 3.Cook DJ, Teves L, Tymianski M. A translational paradigm for the preclinical evaluation of the stroke neuroprotectant Tat-NR2B9c in gyrencephalic nonhuman primates. Sci Transl Med 2012; 4: 154ra133. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz-Bloom RD, Sah R. Gamma-aminobutyric acid(A) neurotransmission and cerebral ischemia. J Neurochem 2001; 77: 353–371. [DOI] [PubMed] [Google Scholar]
- 5.Clarkson AN, Liu H, Rahman R, et al. Clomethiazole: mechanisms underlying lasting neuroprotection following hypoxia-ischemia. FASEB J 2005; 19: 1036–1038. [DOI] [PubMed] [Google Scholar]
- 6.Clarkson AN, Huang BS, Macisaac SE, et al. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 2010; 468: 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clarkson AN. Perisynaptic GABA receptors the overzealous protector. Adv Pharmacol Sci 2012; 2012: 708428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology 2008; 55: 363–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaulk D, Wells J, Evans S, et al. Long-term effects of clomethiazole in a model of global ischemia. Exp Neurol 2003; 182: 476–482. [DOI] [PubMed] [Google Scholar]
- 10.Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Disc 2011; 10: 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudolph U, Mohler H. GABAA receptor subtypes: therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu Rev Pharmacol Toxicol 2014; 54: 483–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker MC, Semyanov A. Regulation of excitability by extrasynaptic GABA(A) receptors. Results Probl Cell Differ 2008; 44: 29–48. [DOI] [PubMed] [Google Scholar]
- 13.Wang DS, Zurek AA, Lecker I, et al. Memory deficits induced by inflammation are regulated by alpha5-subunit-containing GABAA receptors. Cell Rep 2012; 2: 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarkson AN, Clarkson J, Jackson DM, et al. Mitochondrial involvement in transhemispheric diaschisis following hypoxia-ischemia: clomethiazole-mediated amelioration. Neuroscience 2007; 144: 547–561. [DOI] [PubMed] [Google Scholar]
- 15.Hall BJ, Chebib M, Hanrahan JR, et al. 6-Methylflavanone, a more efficacious positive allosteric modulator of gamma-aminobutyric acid (GABA) action at human recombinant alpha2beta2gamma2L than at alpha1beta2gamma2L and alpha1beta2 GABA(A) receptors expressed in Xenopus oocytes. Eur J Pharmacol 2005; 512: 97–104. [DOI] [PubMed] [Google Scholar]
- 16.Karim N, Curmi J, Gavande N, et al. 2′-Methoxy-6-methylflavone: a novel anxiolytic and sedative with subtype selective activating and modulating actions at GABA(A) receptors. Br J Pharmacol 2012; 165: 880–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanrahan JR, Chebib M, Johnston GA. Flavonoid modulation of GABA(A) receptors. Br J Pharmacol 2011; 163: 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dajas F, Andres AC, Florencia A, et al. Neuroprotective actions of flavones and flavonols: mechanisms and relationship to flavonoid structural features. Cent Nerv Syst Agents Med Chem 2013; 13: 30–35. [DOI] [PubMed] [Google Scholar]
- 19.Drasbek KR, Jensen K. THIP, a hypnotic and antinociceptive drug, enhances an extrasynaptic GABAA receptor-mediated conductance in mouse neocortex. Cereb Cortex 2006; 16: 1134–1141. [DOI] [PubMed] [Google Scholar]
- 20.Clarkson AN, Overman JJ, Zhong S, et al. AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci 2011; 31: 3766–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarkson AN, Lopez-Valdes HE, Overman JJ, et al. Multimodal examination of structural and functional remapping in the mouse photothrombotic stroke model. J Cereb Blood Flow Metab 2013; 33: 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bix GJ, Gowing EK, Clarkson AN. Perlecan domain V is neuroprotective and affords functional improvement in a photothrombotic stroke model in young and aged mice. Transl Stroke Res 2013; 4: 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Overman JJ, Clarkson AN, Wanner IB, et al. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci USA 2012; 109: E2230–E2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez-Valdes HE, Clarkson AN, Ao Y, et al. Memantine enhances recovery from stroke. Stroke 2014; 45: 2093–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glykys J, Mann EO, Mody I. Which GABA(A) receptor subunits are necessary for tonic inhibition in the hippocampus? J Neurosci 2008; 28: 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vardya I, Hoestgaard-Jensen K, Nieto-Gonzalez JL, et al. Positive modulation of delta-subunit containing GABA(A) receptors in mouse neurons. Neuropharmacology 2012; 63: 469–479. [DOI] [PubMed] [Google Scholar]
- 27.Downes CE, Crack PJ. Neural injury following stroke: are Toll-like receptors the link between the immune system and the CNS? Br J Pharmacol 2010; 160: 1872–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bath PM, Gray LJ, Bath AJ, et al. Effects of NXY-059 in experimental stroke: an individual animal meta-analysis. Br J Pharmacol 2009; 157: 1157–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen L, Chaaban B, Habert MO. Transient improvement of aphasia with zolpidem. N Engl J Med 2004; 350: 949–950. [DOI] [PubMed] [Google Scholar]
- 30.Sieghart W. Structure and pharmacology of gamma-aminobutyric acidA receptor subtypes. Pharmacol Rev 1995; 47: 181–234. [PubMed] [Google Scholar]
- 31.Glykys J, Mody I. Activation of GABAA receptors: views from outside the synaptic cleft. Neuron 2007; 56: 763–770. [DOI] [PubMed] [Google Scholar]
- 32.Olsen RW, Sieghart W. GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 2009; 56: 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen Y, Lindemeyer AK, Gonzalez C, et al. Dihydromyricetin as a novel anti-alcohol intoxication medication. J Neurosci 2012; 32: 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herd MB, Haythornthwaite AR, Rosahl TW, et al. The expression of GABAA beta subunit isoforms in synaptic and extrasynaptic receptor populations of mouse dentate gyrus granule cells. J Physiol 2008; 586: 989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glykys J, Mody I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J Neurophysiol 2006; 95: 2796–2807. [DOI] [PubMed] [Google Scholar]
- 36.Bright DP, Renzi M, Bartram J, et al. Profound desensitization by ambient GABA limits activation of delta-containing GABAA receptors during spillover. J Neurosci 2011; 31: 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conrad J, Pawlowski M, Dogan M, et al. Seizures after cerebrovascular events: risk factors and clinical features. Seizure 2013; 22: 275–282. [DOI] [PubMed] [Google Scholar]
- 38.Kelly KM, Kharlamov A, Hentosz TM, et al. Photothrombotic brain infarction results in seizure activity in aging Fischer 344 and Sprague Dawley rats. Epilepsy Res 2001; 47: 189–203. [DOI] [PubMed] [Google Scholar]
- 39.Heja L, Nyitrai G, Kekesi O, et al. Astrocytes convert network excitation to tonic inhibition of neurons. BMC Biol 2012; 10: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee S, Yoon BE, Berglund K, et al. Channel-mediated tonic GABA release from glia. Science 2010; 330: 790–796. [DOI] [PubMed] [Google Scholar]
- 41.Tuttolomondo A, Di Raimondo D, di Sciacca R, et al. Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des 2008; 14: 3574–3589. [DOI] [PubMed] [Google Scholar]