

Abstract
Stroke-prone spontaneously hypertensive rats (SHRSP) on high-salt diet are characterized by extremely high arterial pressures, and have been endorsed as a model for hypertensive small vessel disease and vascular cognitive impairment. However, rapidly developing malignant hypertension is a well-known cause of posterior reversible encephalopathy syndrome (PRES) in humans, associated with acute neurological deficits, seizures, vasogenic cerebral edema and microhemorrhages. In this study, we aimed to examine the overlap between human PRES and SHRSP on high-salt diet. In SHRSP, arterial blood pressure progressively increased after the onset of high-salt diet and seizure-like signs emerged within three to five weeks. MRI revealed progressive T2-hyperintense lesions suggestive of vasogenic edema predominantly in the cortical watershed and white matter regions. Histopathology confirmed severe blood–brain barrier disruption, white matter vacuolization and microbleeds that were more severe posteriorly. Hematological data suggested a thrombotic microangiopathy as a potential underlying mechanism. Unilateral common carotid artery occlusion protected the ipsilateral hemisphere from neuropathological abnormalities. Notably, all MRI and histopathological abnormalities were acutely reversible upon switching to regular diet and starting antihypertensive treatment. Altogether our data suggest that SHRSP on high-salt diet recapitulates the neurological, histopathological and imaging features of human PRES rather than chronic progressive small vessel disease.
Keywords: Animal models, hypertension, posterior cerebral reversible encephalopathy, cerebral blood flow, white matter
Introduction
Since its description in 1975, the stroke-prone spontaneously hypertensive rat (SHRSP) has been extensively used as a model of complicated chronic hypertension.1–3 In addition to nephroangiosclerosis and cardiac hypertrophy, cerebral lesions form innately over time in this strain.4,5 Pathological characterization of SHRSP has revealed arteriolar fibrinoid necrosis in brain and kidney.6,7 The onset and progression of the vascular disease are accelerated by high-salt Japanese permissive diet.8 Imaging studies suggested that cerebral lesions in SHRSP on high-salt diet developed rather acutely and resembled vasogenic edema.9,10
The SHRSP on high-salt diet has also been proposed as a model of vascular cognitive impairment,11–16 a chronic brain disease linked to age, hypertension and diabetes.17,18 In humans, MRI findings of acute and chronic hypertension differ significantly. Typically, chronic hypertension manifests as progressive periventricular white matter T2 hyperintensities in the form of leukoaraiosis.19 Acutely, severe hypertension can result in posterior reversible encephalopathy syndrome (PRES), presenting with seizures, focal neurological signs and symptoms, hemorrhages and vasogenic edema.20 In contrast to the T2 hyperintensities in chronic hypertension, T2 hyperintensities in PRES are reversible on imaging, and usually predominate in the posterior areas of the brain. To resolve whether SHRSP on high-salt diet models chronic hypertensive small vessel disease and vascular cognitive impairment or PRES, we undertook a longitudinal multimodal investigation of neurological, histopathological and imaging signs and their reversibility in this experimental paradigm.
Materials and methods
Animals
All experimental procedures were carried out in accordance with the ARRIVE guidelines, and the Guide for Care and Use of Laboratory Animals (NIH Publication No. 85-23, 1996), and were approved by the institutional review board (MGH Subcommittee on Research Animal Care [SRAC]). A total of 53 SHRSP (A3 subline) or spontaneously hypertensive rats (SHRs) were purchased from Charles River laboratories, MA, USA. We used 12-week-old male rats for all experiments. Two animals were lost due to premature death of unexplained cause, and a spinal cord hemorrhage.
Experimental groups and timelines
We maintained SHRSP on regular or high-salt diet (randomly picked up from the cages), and SHR on high-salt diet, and sacrificed the animals at different time points for neuropathological analyses. To test reversibility, a subset of animals was switched from high-salt to regular diet at or two days after the onset of neurological signs (SxO), and started on antihypertensive treatment. In order to further examine whether elevated perfusion pressures contribute to neuropathological changes, we performed permanent unilateral common carotid artery ligation in a subset of rats. Experimental groups included: (i) SHRSP on regular diet followed for two (n = 2) or four to five (n = 6) weeks; (ii) SHRSP on high-salt diet (Japanese permissive diet) for two (n = 7) or four to five (n = 13) weeks; (iii) SHRSP with right common carotid artery occlusion (RCCAO) on high-salt diet for five weeks (n = 9); (iv) SHRSP on high-salt diet until the onset of neurological signs, followed by regular diet and antihypertensive treatment for seven days for reversal (REV; n = 5); (v) SHRSP on high-salt diet until two-days after SxO followed by REV for 10 days (n = 4); and (vi) SHR with RCCAO on high-salt diet for five weeks (n = 5). Because SHRSP on regular diet for two or four to five weeks did not yield any abnormalities in any of the endpoints, all SHRSP on regular diet were pooled for all analyses. In addition, groups (iv) and (v) were analyzed separately only for MRI, and otherwise pooled for all other analyses. Two different experimental protocols were implemented in SHRSPs (Figure 1(a)). In protocol 1, rats were started on high-salt diet and sacrificed either at two weeks (n = 7), or at the onset of neurological signs (SxO) or at four to five weeks if animals remained normal (n = 22). In protocol 2, after a baseline MRI, rats were started on high-salt diet until the onset of neurological signs, and another MRI was performed. A subset of the rats was then switched to REV, followed by another MRI before sacrifice seven days later (n = 5). One animal was excluded in this subset due to a large developmental cyst. In another subset, rats were kept on high-salt diet for another two days to examine progression, and MRI was repeated; this group was then switched to REV, followed by an MRI before sacrifice 10 days later (n = 4). Control (naïve) SHRSPs were simply followed on regular diet for two (n = 2) or four to five weeks (n = 6) and sacrificed for histopathology. Animals were monitored twice a day for neurological signs including altered level of consciousness, motor slowing, seizure-like limb shaking, and forelimb paralysis (clasp reflex). Barnes maze was performed at three weeks. One animal was excluded from all the analyses due to a spinal cord hemorrhage, and one animal with RCCAO died unexpectedly two weeks after high-salt diet onset.
Figure 1.

Experimental timelines, deficit-free survival, and neurological signs. (a) Naïve SHRSPs were maintained on regular diet (n = 8). A subset was sacrificed at two weeks to examine early histopathological changes (n = 2). Remaining animals were examined using Barnes maze (BM) at three weeks, followed for a total of 35 days for neurological signs, and then sacrificed for histopathological examination. In protocol 1, 29 SHRSPs were assigned to control (n = 20) or right common carotid artery occlusion (RCCAO, n = 9) groups, and then started on high-salt Japanese permissive diet (JPD). A subset of the control group was sacrificed at two weeks to examine early histopathological changes (n = 7). Remaining animals were examined using BM at three weeks, and followed until the onset of neurological signs (SxO). At SxO, all animals were sacrificed for histopathological studies. Protocol 2 was designed to test reversibility followed by MRI (n = 9). Animals in protocol 2 had a baseline MRI and started on JPD. A second MRI was performed at SxO. One animal was excluded due to a developmental cyst. In the immediate reversal experiment, MRI was repeated seven days after SxO, and animals were sacrificed for histopathology (n = 4). In the delayed reversal experiment, MRI was repeated 2 days after SxO while still on JPD and 10 days after reversal, after which animals were sacrificed for histopathology (n = 4). (b) Neurological signs started during the third week on high-salt Japanese permissive diet (JPD) and progressed throughout the five-week follow-up in SHRSP. Right common carotid artery ligation (RCCAO) on JPD did not significantly differ from JPD alone. Amongst the 37 animals (representing 100% of the pie chart), most common initial neurological sign was seizure-like activity, whereas motor slowing and paralysis, and frank barrel rolling seizures were less common. *p < 0.05 vs. RD, log-rank (Mantel–Cox) test. The initial sample sizes are shown on the figure; numbers of animals at risk at 10, 20 and 30 days were 22, 22 and 19 for JPD, and 9, 9 and 6 for RCCAO + JPD groups, respectively. The pie chart shows the % of all animals that displayed a specific sign. (c) Barnes maze test performed before the emergence of neurological signs showed spatial learning deficits in SHRSP on JPD. *p < 0.05 vs. RD; one-way ANOVA followed by Sidak's multiple comparisons test (whiskers, full range; boxes, interquartile range; horizontal line, median; + , mean).
Blood pressure measurements
Blood pressure (BP) was measured using the tail cuff method (CODA system, Kent scientific, CT, USA) at baseline, once a week over the first three weeks of high-salt diet, and one week after REV. Measurements were always taken in the morning. Rats were familiarized to the restrainer during three sessions over the 10 days preceding the experiment. For each BP recording session, between three and seven measurements were performed, depending on the tolerance of the animal, and averaged.
High-salt diet (Japanese permissive diet)
After two to three weeks of acclimation, rats were placed on high-salt diet consisting of 1% salt in drinking water in addition to high-salt food (stroke prone rodent diet, 0.39% salt, 0.55% potassium, 16% protein; Zeigler bros Inc., PA, USA). Control animals were fed standard lab chow (ProLab Isopro RMH 3000, irradiated, 0.23% salt, 0.98% potassium, 22.5% protein) and no salt was added to drinking water.
Right common carotid artery ligation
Surgery was performed under 1.5–2% isoflurane anesthesia, temperature monitoring (FHC, ME, USA), and analgesia (buprenorphine 0.08 mg/kg SC). After a midline cervical incision, the right carotid bifurcation was dissected and the common carotid ligated using a 4–0 silk suture. High-salt diet was started the same day.
BP lowering therapy
In the reversal group, we replaced high-salt diet with standard lab chow and drinking water, and started two antihypertensive drugs, selected based on previous reports21 with some modifications. An Alzet osmotic mini-pump (2ML2) filled with nicardipine with a daily delivery rate of 1 mg/kg was implanted under isoflurane anesthesia. Osmotic mini-pumps were primed in saline for at least 24 h before implantation. Buprenorphine (0.08 mg/kg, subcutaneous) was given before and 12 h after the procedure. In addition, enalaprilat (0.9 mg/kg/day, subcutaneous) was administered in two doses every 12 h.
Magnetic resonance imaging
Rats were anesthetized with isoflurane (1%). Heart rate, respiratory rate, and transcutaneous oxygen saturation were monitored to ensure normal physiological state and eliminate potential confounders. Imaging was carried out using a Bruker 4.7-T small-bore scanner (Bruker Biospec, Billerica, MA). Multi-parametric MRI was obtained with single-shot echo planar imaging (EPI) readout (5 slices, slice thickness = 2 mm, field of view = 20 × 20 mm2, image matrix = 48 × 48, bandwidth = 227 kHz). Diffusion-weighted MRI was acquired with two b-values of 250 and 1000 s/mm2 (repetition time (TR)/echo time (TE) = 3000/48 ms, number of average (NA) = 16). Two T2-weigthed images were obtained for T2 mapping with TEs of 30 and 100 ms (TR = 3000 ms, NA = 16). Cerebral blood flow was acquired with amplitude modulated arterial spin labeling MRI (TR/TE = 6500/10.2 ms, NA = 32). Images were processed using Matlab (Mathworks, Natick, MA). Apparent diffusion coefficient (ADC) was derived from S(b) = S(0)*exp(-b*ADC) using b = 250 and 1000 s/mm2. T2 map was calculated as T2 = (TE2-TE1)/(ln(I(TE1))-ln(I(TE2))), where I(TE1,2) are T2-weighted signals obtained at echo times of 30 and 100 ms. Cerebral blood flow (CBF) maps were reconstructed using CBF = λ(Iref–Ilabel)/(2α*Iref*T1app), in which we assumed λ = 0.9 ml/g, α = 0.63 and T1app = 0.84 s.22 Specific values of T2 and ADC parameters were extracted using Image J software. Each lesion was manually outlined and the ROI recorded. Baseline numbers for each lesion were extracted from corresponding images, when available, by ROI transposition. One SHRSP on high-salt diet was excluded from analysis when it showed a large cerebral cyst at baseline.
Barnes maze
Spatial memory retention was assessed using the Barnes maze three weeks after high-salt or regular diet onset in rats without overt neurological signs. Test was carried out using a 122 cm diameter platform with 18 evenly spaced holes near the periphery, and 4 distant spatial cues, as previously described.23 We released the rats from a high edge open box placed at the center of the platform, and used noise as an aversive stimulus to motivate them to the escape (i.e. target) hole. The test consisted of four learning trials per day 10 min apart, for four consecutive days, followed by a probe (retention) test on the fifth day. Each trial lasted 3 min during which the rat would try to find the escape hole. As soon as the escape hole was reached, the noise was stopped and the rat was allowed to rest for one minute before being returned to its cage. The platform was rotated and cleaned with 70% ethanol between each trial. For the probe trial, the escape box was removed from underneath the escape hole. All sessions were video recorded, and time spent to reach the target hole during learning phase, and time spent in the correct quadrant during the probe trial were quantified.
Blood sampling and analysis
Blood was drawn from the tail vein at baseline and every week under brief isoflurane anesthesia. All tests were carried out by the Center for Comparative Medicine laboratories at the Massachusetts General Hospital. Hematological assays (platelets, hemoglobin, reticulocytes) and plasma chemistry (Na+, K+, creatinine) were studied using automated systems (HemaTrue analyzer and Dri-Chem 7000, respectively; Heska Corporation, CO, USA). Blood smears were prepared and interpreted by independent technicians blinded to study groups.
Red blood cell deformability
Deformability of red blood cells (RBCs) from 8 SHRSP on high-salt diet was assessed by the average velocity of cells traversing through microfluidic channels.24 The microfluidic device was casted in polydimethylsiloxane with a microfabricated positive silicon master and bonded to a glass slide after air plasma treatment. It consisted of parallel microchannels (2 µm wide, 5 µm deep, 30 µm long) in the center of a main channel (1 mm wide, 5 µm deep and 5 mm long) and periodic supporting pillars (12 µm diameter, 30 µm apart) away from the constrictions. Hydrostatic pressure-driven flow in the microfluidic device was established by a difference in water column height (5 ml) in two 60 ml Terumo plastic syringe tubes, connected to the reservoirs at each end of the main channel with microbore tubing (0.02 inch internal diameter). A volume of 0.5 ml blood sample was drawn and tested on the same day for each animal. RBCs were washed in PBS at 2000 r/min for 5 min at room temperature and diluted for 200 times in PBS solution containing 1% w/v Bovine Serum Albumin. Cell suspension was injected into the microfluidic device. Movement of RBCs in the pressure-driven flow was recorded at 30 frames per second on an inverted microscope (Zeiss Axiovert 200) equipped with a CCD camera (Hitachi KP-D20AU). Microscopic images of RBCs traversing through the microchannels were processed using Image J.25 Cellular velocity was averaged across 100 ± 20 cells per sample, and normalized by mean corpuscular volume.
Histological preparation
Animals were euthanized by transcardiac perfusion with cold PBS and exsanguination under deep anesthesia. Brains were then frozen in 2-methylbutane on dry ice at −35℃. Sets of coronal cryosections were harvested every 500 µm throughout the entire brain. Hematoxylin and eosin staining of 12-µm thick sections was used to seek histopathological evidence of cell death. Histological readouts were examined in the same cohorts on adjacent sections.
Blood–brain barrier
Blood–brain barrier (BBB) integrity was examined by IgG leakage in separate groups of rats at two weeks, at the onset of neurological signs, and between four to five weeks in rats that did not show any neurological signs. Two animals were excluded from the analysis because of poor intracardiac perfusion (one SHR on high-salt diet with RCCAO, and one SHRSP on high-salt diet sacrificed at two weeks). IgG leakage was determined using immunostaining with anti-rat IgG antibody (Jackson, 1/100), for which the sections were dried, post-fixed in 4% paraformaldehyde followed by cold methanol, and incubated at room temperature for 90 min. IgG fluorescence intensity was examined on sections every 1 mm obtained throughout the entire brain (+4 to −8 mm from bregma) for every animal. Sections were scanned at 5 × magnification on a tissueFAXS slide scanner (TissueGnostics, Vienna, Austria) equipped with an Axio observer microscope (Zeiss, Jena, Germany). Each scanning session contained a control animal (SHRSP on regular diet) to serve as internal reference for background calculation. Images were then analyzed using Imaris software (Bitplane, Zurich, Switzerland). Threshold for automatic detection of BBB leakage (IgG positive areas) was calculated based on two regions of interest in cortex and striatum each, at 0 mm and −2 mm from bregma, in controls. Detection threshold was set at the mean optical density (OD) plus 2 standard deviations of the internal control, and applied to all sections in the same scanning session.
Myelin integrity
White matter integrity and myelin were examined using Kluver Barrera (i.e. luxol fast blue) staining of 20 µm-thick post-fixed sections at two coronal section levels aiming +1.0 mm and −3.2 mm from bregma. Sections were scanned at 10× magnification on a tissueFAXS slide scanner (TissueGnostics, Vienna, Austria) equipped with a Zeiss Axio observer microscope (Zeiss, Jena, Germany). Regions of interest (500 × 375 µm) were placed on midline and lateral corpus callosum, external and internal capsules, and fimbria. OD was quantified within each region of interest. In addition, regions of interest were examined by two independent blinded observers, (FH and JM), using a scoring system: 0 (dense myelin), 1 (myelin pallor), 2 (non confluent vacuoles), 3 confluent vacuoles.26 Scores were averaged between the two observers.
Intracerebral hemorrhage detection
Intraparenchymal hemorrhages were examined by diaminobenzidine (DAB) staining, which reacts with erythrocyte peroxidases, unlike other methods that detect iron.27 Therefore, DAB detects bleeding mostly during the acute and subacute stages prior to RBC lysis.28,29 Thirteen serial coronal sections (approximately one every millimeter, between bregma × 9 and + 5 mm) throughout the entire brain were stained with DAB. Sections with bleeding were scanned at 5 × in a tissueFAXS slide scanner (TissueGnostics, Vienna, Austria) equipped with a Zeiss Axio observer microscope (Zeiss, Jena, Germany). The images were then analyzed using the tissueFAXS viewer (TissueGnostics, Vienna, Austria). Each lesion was manually outlined for area measurement, and its anatomical and anteroposterior location noted.
Statistics
This study was designed as exploratory. Data were statistically tested using one- or two-way ANOVA followed by Holm–Sidak’s, Sidak’s, or Dunnett’s multiple comparisons, Kruskal–Wallis followed by Dunn’s multiple comparisons, Student’s t, Mann–Whitney U, log rank (Mantel–Cox), or paired Wilcoxon tests, where appropriate (Prism, GraphPad Software, Inc. La Jolla, CA). All tests were two-sided, and adjusted for multiple comparisons. Lastly, MRI and part of microbleed data set were analyzed using a longitudinal, random intercept, linear mixed effects model (Stata, StataCorp LLC, College Station, TX). Statistical tests and sample sizes for each dataset are stated in the results, figures or figure legends. Significance was set at p < 0.05. Results are expressed as mean ± standard error, or whisker-box plots (whiskers, full range; box, interquartile range; horizontal line, median; cross, mean).
Results
Systemic physiology
Systolic and diastolic BP, and to a lesser extent heart rate (not shown), progressively increased in SHRSP after the onset of high-salt diet compared with regular diet (Supplemental Figure S1). In addition, body weight started to decrease two weeks after the onset of high-salt diet. RCCAO did not influence these changes over time. Reversal by switching to regular diet and antihypertensive treatment was highly effective in reducing the BP to even below baseline levels prior to the onset of high-salt diet, and restored body weight.
Neurological signs
Control SHRSP on regular diet did not develop overt neurological signs at any point during the study (Figure 1(b)). In contrast, SHRSP on high-salt diet developed neurological signs starting as early as 19 days; most rats showed signs by the end of fourth week. Both SHRSP on high-salt diet and SHRSP on high-salt diet after RCCAO showed worse deficit-free survival compared with SHRSP on regular diet (p = 0.003 and 0.038, respectively; Mantel–Cox test). Although the overall deficit-free survival curves did not differ between SHRSP on high-salt diet and SHRSP on high-salt diet after RCCAO (p > 0.05, Mantel–Cox test), a higher proportion of RCCAO plus high-salt diet cohort showed deficit-free survival at the 35-day time point compared to that without RCCAO (1/19 at risk and 3/6 at risk deficit-free in JPD and JPD+RCCAO, respectively; p = 0.009, χ2). Initial neurological signs in most cases included continuous stereotypic and rhythmic, “seizure-like” forelimb contractions, although we did not have electrophysiological confirmation of seizure activity (supplementary video). A smaller proportion showed motor slowing, hindlimb paresis, and frank barrel-rolling seizures as initial signs.
In rats that were free of overt neurological signs at three weeks, we examined spatial learning and memory retention using Barnes maze (Figure 1(c)). Although all groups appeared to acquire spatial memory at approximately the same rate during the learning phase of Barnes Maze (not shown), the time spent in the correct quadrant (i.e. in probe test) was shorter in SHRSP on high-salt diet, compared with SHRSP on regular diet, indicating impaired retention of spatial memory. Notably, SHRSP on high-salt diet after RCCAO did not differ from SHRSP on regular diet, suggesting that RCCAO may ameliorate or delay the cognitive deficits.
MRI lesions
Nine SHRSP were longitudinally studied using MRI, in half of which high-salt diet was reversed at SxO, and in the other half two days later (Figure 1(a)); one was excluded from MRI analysis because of a developmental cyst. Seven rats displayed focal seizure-like signs and one rat showed motor slowing; all signs disappeared within 24–48 h after reversal. MRI performed at the onset of neurological signs showed T2 hyperintensities predominantly in the anterior and middle cerebral artery watershed territories as well as deep white matter structures (Figure 2(a) and (b); Supplemental Figure S2). T2 lesion burden progressively increased while on high-salt diet between the onset of neurological signs and 2 days later, mainly due to emergence of new lesions rather than growth of existing lesions (Figure 2(b)). Upon switching to regular diet and starting antihypertensive treatment, T2 lesions diminished within 2 days, and almost completely disappeared within 7–10 days. The T2 signal intensity averaged across the entire cortex or subcortical regions did not show statistically significant changes over time (not shown). Twenty-one T2 lesions were followed over time, 16 of which entirely disappeared after 7–10 days of reversal.
Figure 2.

MRI lesions. (a) Longitudinal MRI showing T2 and cerebral blood flow (CBF, arterial spin labeling) changes at baseline (B), onset of neurological signs (SxO), 2 days after SxO (SxO + 2 days), and 10 days after switching to regular diet and starting antihypertensive treatment (Rev + 10 days) in a representative SHRSP on high-salt Japanese permissive diet. Arrowheads point to the arterial border zone regions that showed a propensity to develop T2 and ADC hyperintensities and mild hypoperfusion. Switching to regular diet and starting antihypertensive treatment reversed T2 and ADC changes and led to relative hyperemia. (b) The time course of lesion morphometry and signal intensity showed a progressive increase in the number and size of T2 lesions in each animal while on high-salt diet (first row). Average T2 and ADC signal intensity within the lesions increased while on high-salt diet, whereas CBF tended to mildly decrease (second row). Switching to regular diet and starting antihypertensive treatment rapidly reversed T2 and ADC changes, and led to significant hyperemia. *p < 0.05 vs. SxO; longitudinal, random intercept, linear mixed effects model, followed by pair-wise multiple comparisons between SxO and each other time point. Connected data points represent a single animal. (c) Images from three representative rats show hypoperfusion within the lesion at SxO, turning into severe hyperemia at Rev + 2 days (arrowheads). Although most severe within the lesion, hyperemia after Rev usually involved the entire brain. Graph on the right shows the time course of CBF changes averaged across the entire cortex. *p < 0.05 vs. SxO; longitudinal, random intercept, linear mixed effects model, followed by pair-wise multiple comparisons between SxO and each other time point. Connected data points represent a single animal.
The average ADC values within the T2 lesions were generally increased suggesting vasogenic edema, with the exception of one lesion that showed decreased ADC at the onset of neurological signs and then normalized (Figure 2(b)). These data suggested predominantly non-ischemic mechanisms of MRI lesions in this model. As with the T2 signal intensity, ADC values rapidly returned to baseline upon high-salt diet reversal. Importantly, we did not observe discrete ADC-dark lesions throughout the cortex or subcortical tissues, with the exception of one animal at the onset of neurological signs (Figure 2(b), blue arrowhead), suggesting that the overall reduced cortical ADC values did not reflect focal ischemic infarcts.
Interestingly, cerebral perfusion within the T2 lesions was variable at the onset of neurological signs, some animals showing up to 50% reduction in CBF, others showing no change. This was likely related to variable rate of lesion progression in different animals, a snapshot of which was captured by each MRI at a fixed time point. However, upon high-salt diet reversal, a marked hyperemia developed throughout the cortex and subcortical tissues that was most marked within the T2-hyperintense lesions (Figure 2(a) and (b)). Some examples of this biphasic CBF pattern are shown in Figure 2(c). The average CBF across the entire cortex (Figure 2(c)) or the subcortical regions (not shown) did not show consistent changes during high-salt diet, but markedly increased after switching to regular diet and starting antihypertensive treatment. In most animals, the imaging signs have returned to normal on the final MRI at 7–10 days after switching to regular diet and starting antihypertensive treatment.
BBB disruption
To determine the histopathological correlates of T2 hyperintensities on MRI, we examined the BBB integrity using IgG leakage (Figure 3). At two weeks after the initiation of high-salt diet in SHRSP, IgG leakage did not differ from SHRSP on regular diet. At the onset of neurological signs, however, there was widespread IgG leakage that showed a predilection for cortical watershed regions corresponding to arterial border zones, and tracked along grossly enlarged white matter bundles, especially the corpus callosum and the external capsule. Leakage did not show a strong anteroposterior trend at the time points studied, but was often asymmetric. RCCAO diminished IgG leakage on the ligated side. IgG leakage completely disappeared within 7–10 days after high-salt diet reversal and antihypertensive treatment.
Figure 3.
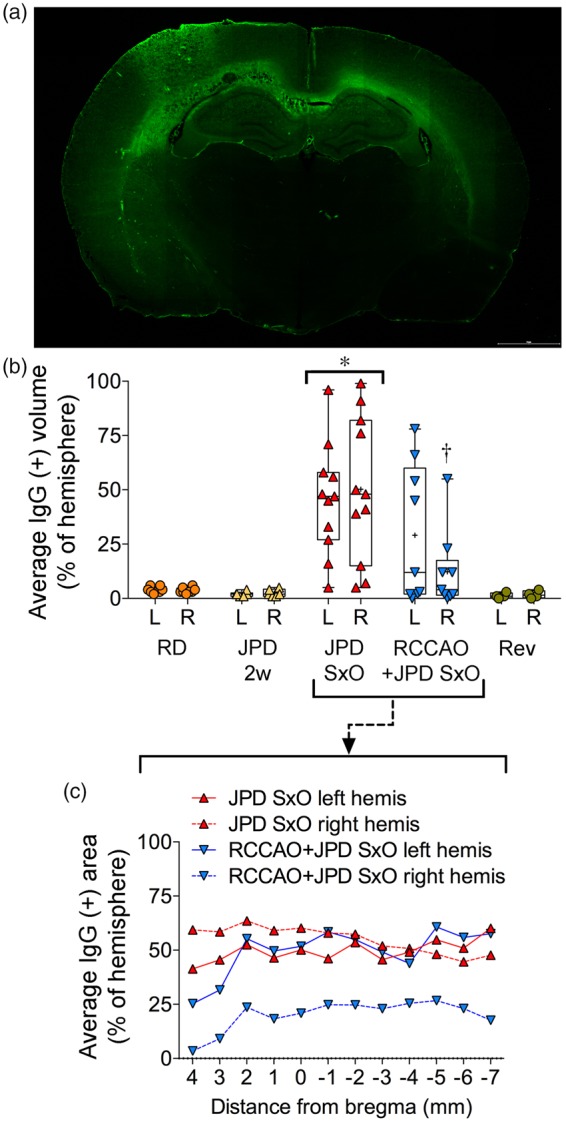
Blood–brain barrier (BBB) disruption. (a) IgG fluorescence shows BBB leakage in corpus callosum (left worse than right) and left anterior and middle cerebral artery border zone in a representative SHRSP on high-salt diet (JPD). Scale bar = 2 mm. (b) Average volume of IgG leakage, expressed as % of each hemisphere shows BBB disruption starts after two weeks on JPD (n = 6), becomes severe at the onset of neurological signs (SxO, n = 11), and completely disappears within 7–10 days after switching to regular diet and starting antihypertensive treatment (Rev, n = 4). Right common carotid artery ligation (RCCAO, n = 9) was associated with significantly less ipsilateral BBB leakage. *p < 0.05 JPD SxO vs. all other groups; †p < 0.05 right (R) vs. left (L) RCCAO+JPD SxO; two-way repeated measures ANOVA followed by Sidak’s (R vs. L) and Holm–Sidak’s (among groups) multiple comparisons tests. Each data point represents a single animal. (c) Average volume of IgG leakage expressed as % of hemisphere in individual section levels from SHRSP on JPD at SxO, with or without BCCAO.
Myelin staining
Two weeks after starting high-salt diet myelin appeared normal in all SHRSP. When examined at the onset of neurological signs, there was significant pallor and vacuolization in large white matter tracts (Figure 4). In severe cases, vacuoles became confluent. These abnormalities were most marked in corpus callosum and external capsule, although cortical watershed zones also showed pallor in a subset of SHRSP (Figure 4, see asterisk (*)). When quantified using standardized ROIs placed at two different coronal levels (1.29 ± 0.36 mm anterior and 3.06 ± 0.34 mm posterior to bregma), OD of myelin staining was reduced in central and lateral corpus callosum, the external capsule (Figure 4(b)), fimbria and internal capsule (p < 0.05 vs. high-salt diet two weeks; not shown). Myelin pallor was more severe in the posterior section level compared to the anterior using a grading system (Figure 4(c)). Myelin pallor correlated with IgG leakage volume (Spearman r = 0.64, p < 0.0001). As with BBB disruption, RCCAO reduced the loss of myelin staining in the ipsilateral hemisphere, and all myelin normalized within 7–10 days after switching to regular diet and starting antihypertensive treatment (Figure 4(b)).
Figure 4.
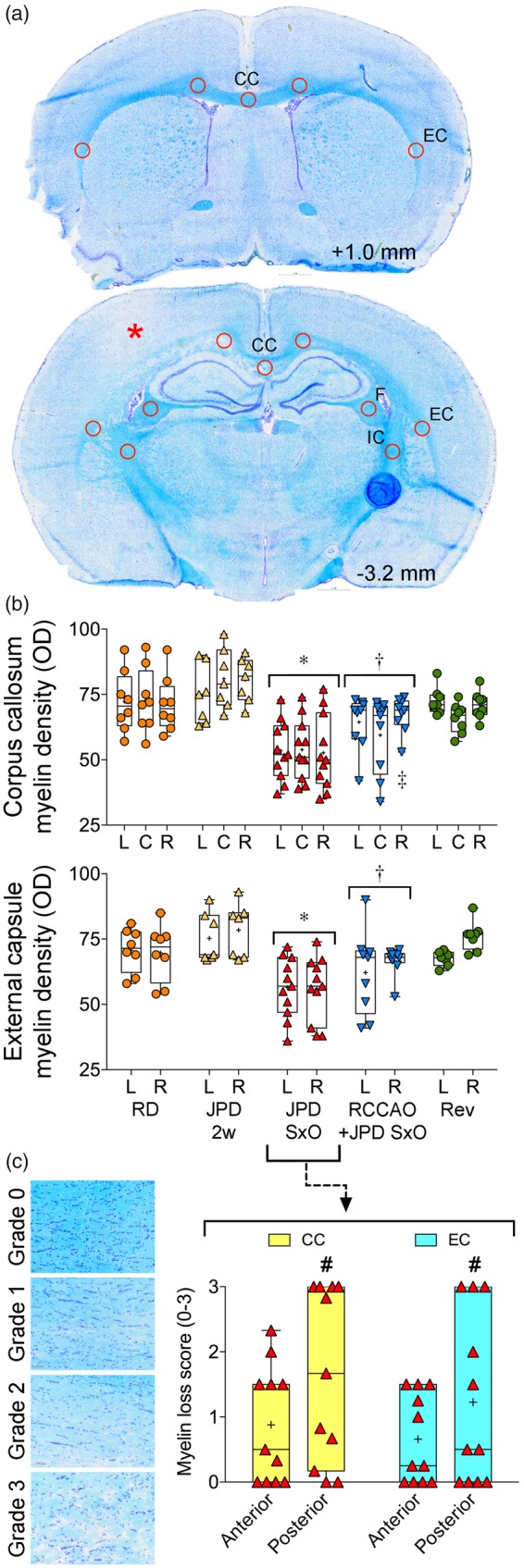
Myelin changes. (a) Coronal sections 1 mm anterior and 3.2 mm posterior to bregma, show normal and abnormal (*) Kluver–Barrera myelin staining, respectively. The corpus callosum (CC), external capsule (EC), fimbria (F) and internal capsule (IC) regions of interest within which the optical density (OD) was quantified and averaged are also shown. (b) Myelin staining (OD) in left (L), center (C) and right (R) CC, and L and R EC, was diminished in SHRSP on high-salt diet (JPD) at the onset of neurological signs (SxO, n = 11), but not at two weeks after JPD onset (2 weeks, n = 7). Staining intensity recovered within 7–10 days after switching to regular diet and starting antihypertensive treatment (Rev, n = 8). Right common carotid artery ligation (RCCAO, n = 9) was associated with stronger myelin staining in the ipsilateral hemisphere. *p < 0.05 vs. RD, JPD 2 weeks and Rev; †p < 0.05 vs. JPD 2 week; ‡p < 0.05 vs. C; two-way repeated measures ANOVA followed by Holm–Sidak's and Sidak’s multiple comparisons tests. Each data point represents a single animal. (c) Myelin grading shows the posterior dominance of staining loss in CC and EC in JPD SxO. #p < 0.05 vs. anterior section level; two-way repeated measures ANOVA followed by Holm–Sidak's multiple comparisons test. Each data point represents a single animal.
Microbleeds
Microbleeds were a conspicuous pathological feature (Figure 5(a)) observed at the onset of neurological signs, but not at two weeks after the onset of high-salt diet (Figure 5(b)). Serial sectioning throughout the entire brain showed that the number of microbleeds was highly variable (0 to > 50) within each hemisphere, and revealed a strong predilection for the posterior brain regions (Figure 5(c)). RCCAO diminished the microbleed counts in the ipsilateral hemisphere. Average microbleed cross sectional area ranged approximately between 0.01 and 0.1 mm2 (i.e. ∼ 100 × 100 to ∼ 300 × 300 µm), and was larger in white matter and cortex (Figure 5(d)). Although the number of microbleeds was highest in the hippocampus, total microbleed burden (i.e. cumulative microbleed area) was higher in white matter than any other brain region by virtue of larger microbleeds (Figure 5(e)). Microbleed burden correlated with the volume of IgG leakage (Spearman r = 0.68, p < 0.0001), some microbleeds showed strong IgG leakage. Except for one animal, microbleeds disappeared within 7–10 days after switching to regular diet and starting antihypertensive treatment (Figure 5(b)).
Figure 5.

Cerebral microbleeds. (a) Representative coronal sections showing microbleeds of various sizes in SHRSP on high-salt diet (DAB and cresyl violet staining). (b) The number of microbleeds was significantly higher in SHRSP on high-salt diet (JPD) at the onset of neurological signs (SxO, n = 11), but not at two weeks after JPD onset (2 weeks, n = 7). Right common carotid artery ligation (RCCAO, n = 9) reduced the microbleed counts in the ipsilateral hemisphere. Microbleed counts were reduced after switching to regular diet and starting antihypertensive treatment (Rev, n = 8), suggesting cessation of microbleed development. *p < 0.05 vs. RD, JPD 2 weeks and Rev; Kruskal–Wallis followed by Dunn's multiple comparisons test. Each data point represents a single animal. (c) Microbleed counts showed a posterior predominance, and RCCAO diminished microbleeds in the ipsilateral hemisphere. *p < 0.05 vs. anterior half, median [25–75%] of pooled data from JPD and RCCAO+JPD bilaterally; Wilcoxon matched-pairs signed rank test. (d) Microbleed area per lesion in cortex (C), deep grey nuclei (DG), white matter (WM), hippocampus (H), choroid plexus (CP), and septum (S). *p < 0.05 vs. S, p < 0.01 vs. DG, p < 0.001 vs. H; †p < 0.05 vs. DG, p < 0.001 vs. H; general linear mixed effects model with random subject intercepts and variance component error structure to account for the multiple lesions within each ROI and within individual experimental subject (n = 52, 65, 53, 139, 6 and 19 microbleeds in C, DG, WM, H, CP and S, respectively). Please note the log scale. (e) The distribution of microbleed count and total cumulative microbleed area in the same six brain regions.
General histopathology
Routine histology revealed scattered cells with pyknotic nuclei and eosinophilic cytoplasm in cortical regions prone to vasogenic edema (Supplemental Figure S3), which persisted even when other MRI and histopathological lesions disappeared after switching to regular diet and starting antihypertensive treatment (not shown). Nevertheless, we never observed overt microinfarcts suggestive of lacunar strokes due to small vessel disease.
Thrombotic microangiopathy
Serial blood samples revealed a progressive thrombocytopenia that developed within two weeks of high-salt diet, followed one week later by anemia and elevated reticulocyte counts (Figure 6, A and B, left panel), while mean corpuscular volumes and hemoglobin concentrations were normal (data not shown). Resuming regular diet and starting antihypertensive treatment rapidly restored the platelet counts and led to rebound thrombocytosis (Figure 6(a), right panel); however, anemia and elevated reticulocyte counts did not recover within the time frame of our follow up (Figure 6(b) to (d)). Examination of blood smears revealed schistocytes, implicating a thrombotic microangiopathic process with consumptive thrombocytopenia (Supplementary Figure S5). To test whether hemolysis was due to reduced RBC deformability, we measured RBC velocities traversing through microchannels and found no change (Supplementary figure S6). A thrombotic microangiopathic process was further supported by renal failure as evident in elevated plasma creatinine and blood urea nitrogen levels, which only partially normalized upon switching back to regular diet and starting antihypertensive treatment (Supplemental Table 1). Of note, RCCAO did not protect against these hematological abnormalities (not shown).
Figure 6.

Hematological changes. Platelet counts (a), hemoglobin concentration (b), red blood cell (RBC) count (c), reticulocyte percentage at sacrifice time point (d), all suggest a thrombotic microangiopathic process in SHRSP on high-salt diet (JPD). Platelet counts, but not anemia or reticulocyte counts, were rapidly normalized upon resuming regular diet (RD) and starting antihypertensive treatment (Rev). *p < 0.05 JPD vs. RD, and JPD two to four week vs. JPD baseline; †p < 0.05 JPD two to four week and Rev vs. JPD baseline; #p < 0.05 JPD two to four week vs. JPD baseline; ‡p < 0.05 JPD three to four week and Rev vs. JPD baseline; two-way ANOVA followed by Sidak’s and Dunnett's multiple comparisons tests. §p < 0.05 vs. RD; @p < 0.05 vs. all other groups; one-way ANOVA followed by Holm–Sidak's multiple comparisons test. Each data point in C and D represents a single animal.
SHRs
SHR subjected to identical high-salt diet conditions did not develop any of the systemic physiological, neurological, histopathological and hematological abnormalities described above for SHRSP (Supplemental Figure S4, and data not shown).
Discussion
PRES often develops in the setting of malignant hypertension, renal failure or during treatment with certain immunosuppressive drugs, and typically presents with cognitive and visual changes, and seizures.20,30 Here, we show that the neurological and MRI signs, BBB disruption, predominantly posterior white matter changes and microbleeds,1,9,31 and rapid reversal of all pathological findings in SHRSP on high-salt diet recapitulate the clinical hallmarks of human PRES. Thrombocytopenia, regenerative anemia, schistocytes and mild renal failure strongly suggest an acute systemic thrombotic microangiopathic process, likely triggered by malignant hypertension. Although we did not measure the perfusion pressures, the fact that RCCAO ameliorated ipsilateral vasogenic edema (Figure 3), white matter rarefaction (Figure 4) and microbleeds (Figure 5), and preserved neurological function (Figure 1(c)) implicates elevated perfusion pressure as a critical determinant for cerebral pathology, and suggests that RCCAO shields the hemisphere from dangerously elevated perfusion pressures. In support of this, UCCAO has been shown to reduce cerebral perfusion pressure.32 Moreover, unilateral carotid stenosis has been reported to protect the ipsilateral hemisphere from hypertensive vascular changes in a rat model of induced hypertension.33 Altogether, while certain aspects of the pathophysiology may overlap, our data do not support SHRSP on high-salt diet as a translational model for chronic hypertensive small vessel disease or vascular dementia as has been endorsed in the past.11–16,34
The tissue hallmarks of the disease process in SHRSP on high-salt diet were BBB disruption (i.e. IgG leakage, and elevated T2 and ADC on MRI) and microbleeds,35,36 both of which are neuroimaging features of PRES as well.20,37 The severity and spatial distribution of MRI lesions, histopathological abnormalities and neurological signs were closely matched. Morphological changes in white matter and reduced myelin staining intensity in the rat model reflected vasogenic edema rather than true demyelination,30 as they resolved within two days after resuming regular diet and treatment with antihypertensive drugs. Persistent and profound BBB leakage may also lead to extravasation of RBCs as a mechanism of microbleeds. Indeed, BBB leakage has been reported to precede frank intracerebral hemorrhage in SHRSP.38 Spatial distribution of the pathology in the rat also matched human PRES. For example, BBB disruption showed a predilection for the cortical watershed regions, and white matter changes and microbleeds were more severe posteriorly.20,39,40 Although we did not detect a clear posterior dominance of IgG leakage, this was likely due to faster onset and progression of BBB disruption that led to severe and widespread vasogenic edema by the time neurological signs appeared. Hence, examination at earlier time points could have revealed a posterior trend in IgG leakage as well. Even in human PRES, vasogenic edema is not necessarily limited to posterior brain regions, and can involve frontal and temporal regions as well. Indeed, holohemispheric watershed is a major imaging distribution of vasogenic edema in PRES,39 which is exactly what we observed in our study (Figures 2(a), 3(a) and 4(a), Supplemental Figure S2). It is also recognized that T2 hyperintensities of PRES can be asymmetric or even unilateral.20,39 Indeed, ∼ 30% of the SHRSP on high-salt diet in our cohort showed asymmetric neuropathology even in the absence of RCCAO.
As PRES is reversible when treated, histopathological studies in human brain are limited. Edematous white matter and microthrombi have been reported on biopsy or necropsy of PRES patients.41–43 In our study, a subset of animals showed hypoperfusion within the T2 hyperintense lesions in watershed regions at the onset of neurological signs (Figure 2).44 Upon switching to regular diet, however, all T2 hyperintense lesions developed a pronounced hyperemia, although this may have been due to a direct cerebrovascular effect of the antihypertensive drugs. Both hypoperfusion and hyperperfusion have been observed in human PRES as well,37 and poor cerebral autoregulation in border zone regions has been implicated as a mechanism.45 Previous studies have suggested that edema could lead to hypoperfusion and ischemia through capillary compression,44 but we did not observe any overt ischemic infarct by histopathology or MRI during the two-day follow-up after the onset of neurological signs in our study.
Increased RBC fragility has been proposed as a mechanism for hemolytic anemia in SHRSP on high-salt diet.46,47 The small and statistically insignificant drop in RBC deformability three weeks after high-salt diet onset was likely a reflection of reticulocytosis, and thus did not explain the hemolysis (Supplementary Figure S6).48–50 Instead, thrombocytopenia and schistocytes strongly suggested mechanical lysis in the setting of a thrombotic microangiopathy,51 likely related to endothelial dysfunction.52 Mild renal failure was also consistent with such a microangiopathic mechanism. Thrombotic thrombocytopenic purpura has been associated with neuroimaging signs consistent with PRES in many cases,53,54 and malignant hypertension and acute hypertensive encephalopathy have been associated with a thrombotic microangiopathy.55,56
Interestingly, SHR on high-salt diet did not develop any of the systemic, hematological, neurological and histopathological signs that we observed in SHRSP, despite reaching arterial BPs only slightly lower than SHRSP. The reasons for this strain-specific response to high-salt diet are unknown at this time, but presumably relate to genetic differences.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Arthur Sachs fellowship (FH), the Harvard French Scholarship Fund (FH), the Philippe Foundation (FH), NIH/NINDS (K23NS064052, R01NS082285, and R01NS086905 to NSR; NS055104, NS061505 to CA), the Fondation Leducq (CA), the Heitman Foundation (CA), and the Ellison Foundation (CA). This work was conducted with support from Harvard Catalyst, The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health Award UL1 TR001102).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
FH: study design, data generation and interpretation, figures and writing; IZ and PZS: Imaging procedure, post processing and interpretation; JM, AHB, ED and TQ: Data interpretation and technical assistance; ED: red blood cell deformability procedure and data generation; MJC: critical review of manuscript; NR and CA: study design, figures, writing, statistics and critical review of manuscript.
References
- 1.Hazama F, Ooshima A, Tanaka T, et al. Vascular lesions in the various substrains of spontaneously hypertensive rats and the effects of chronic salt ingestion. Jpn Circul J 1975; 39: 7–22. [DOI] [PubMed] [Google Scholar]
- 2.Hazama F, Amano S, Haebara H, et al. Changes in vascular permeability in stroke-prone spontaneously hypertensive rats studied with peroxidase as a tracer. Acta Pathol Japon 1975; 25: 565–574. [DOI] [PubMed] [Google Scholar]
- 3.Yamori Y, Tomimoto K, Ooshima A, et al. Proceedings: developmental course of hypertension in the SHR-substrains susceptible to hypertensive cerebrovascular lesions. Jpn Heart J 1974; 15: 209–210. [DOI] [PubMed] [Google Scholar]
- 4.Engelmann GL, Vitullo JC, Gerrity RG. Morphometric analysis of cardiac hypertrophy during development, maturation, and senescence in spontaneously hypertensive rats. Circ Res 1987; 60: 487–494. [DOI] [PubMed] [Google Scholar]
- 5.Yamori Y, Horie R, Handa H, et al. Pathogenetic similarity of strokes in stroke-prone spontaneously hypertensive rats and humans. Stroke 1976; 7: 46–53. [DOI] [PubMed] [Google Scholar]
- 6.Fredriksson K, Auer RN, Kalimo H, et al. Cerebrovascular lesions in stroke-prone spontaneously hypertensive rats. Acta Neuropathol 1985; 68: 284–294. [DOI] [PubMed] [Google Scholar]
- 7.Ogata J, Fujishima M, Tamaki K, et al. Stroke-prone spontaneously hypertensive rats as an experimental model of malignant hypertension. I. A light- and electron-microscopic study of the brain. Acta Neuropathol 1980; 51: 179–184. [DOI] [PubMed] [Google Scholar]
- 8.Nagaoka A, Iwatsuka H, Suzuki Z, et al. Proceedings: cerebral lesions in the spontaneously hypertensive rats. Effects of NaCl-loading, sex-related difference and genetic analysis. Jpn Heart J 1974; 15: 216–217. [PubMed] [Google Scholar]
- 9.Guerrini U, Sironi L, Tremoli E, et al. New insights into brain damage in stroke-prone rats: a nuclear magnetic imaging study. Stroke 2002; 33: 825–830. [DOI] [PubMed] [Google Scholar]
- 10.Sironi L, Guerrini U, Tremoli E, et al. Analysis of pathological events at the onset of brain damage in stroke-prone rats: a proteomics and magnetic resonance imaging approach. J Neurosci Res 2004; 78: 115–122. [DOI] [PubMed] [Google Scholar]
- 11.Madigan JB, Wilcock DM, Hainsworth AH. Vascular contributions to cognitive impairment and dementia: topical review of animal models. Stroke 2016; 47: 1953–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jalal FY, Yang Y, Thompson J, et al. Myelin loss associated with neuroinflammation in hypertensive rats. Stroke 2012; 43: 1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura-Ohba S, Yang Y, Thompson J, et al. Transient increase of fractional anisotropy in reversible vasogenic edema. J Cereb Blood Flow Metab 2016; 36: 1731–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver J, Jalal FY, Yang Y, et al. Tissue oxygen is reduced in white matter of spontaneously hypertensive-stroke prone rats: a longitudinal study with electron paramagnetic resonance. J Cereb Blood Flow Metab 2014; 34: 890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jalal FY, Yang Y, Thompson JF, et al. Hypoxia-induced neuroinflammatory white-matter injury reduced by minocycline in SHR/SP. J Cereb Blood Flow Metab 2015; 35: 1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Kimura-Ohba S, Thompson J, et al. Rodent models of vascular cognitive impairment. Transl Stroke Res 2016; 7: 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimura S, Saito H, Minami M, et al. Pathogenesis of vascular dementia in stroke-prone spontaneously hypertensive rats. Toxicology 2000; 153: 167–178. [DOI] [PubMed] [Google Scholar]
- 18.Hainsworth AH, Brittain JF, Khatun H. Pre-clinical models of human cerebral small vessel disease: utility for clinical application. J Neurol Sci 2012; 322: 237–240. [DOI] [PubMed] [Google Scholar]
- 19.Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke 1997; 28: 652–659. [DOI] [PubMed] [Google Scholar]
- 20.Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med 1996; 334: 494–500. [DOI] [PubMed] [Google Scholar]
- 21.Griffin KA, Polichnowski A, Litbarg N, et al. Critical blood pressure threshold dependence of hypertensive injury and repair in a malignant nephrosclerosis model. Hypertension 2014; 64: 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Utting JF, Thomas DL, Gadian DG, et al. Understanding and optimizing the amplitude modulated control for multiple-slice continuous arterial spin labeling. Magn Reson Med 2005; 54: 594–604. [DOI] [PubMed] [Google Scholar]
- 23.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 1979; 93: 74–104. [DOI] [PubMed] [Google Scholar]
- 24.Bow H, Pivkin IV, Diez-Silva M, et al. A microfabricated deformability-based flow cytometer with application to malaria. Lab Chip 2011; 11: 1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods 2012; 9: 671–675. [DOI] [PMC free article] [PubMed]
- 26.Wakita H, Tomimoto H, Akiguchi I, et al. Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: an immunohistochemical study. Acta Neuropathol 1994; 87: 484–492. [DOI] [PubMed] [Google Scholar]
- 27.Campos F, Qin T, Castillo J, et al. Fingolimod reduces hemorrhagic transformation associated with delayed tissue plasminogen activator treatment in a mouse thromboembolic model. Stroke 2013; 44: 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradley WG., Jr MR appearance of hemorrhage in the brain. Radiology 1993; 189: 15–26. [DOI] [PubMed] [Google Scholar]
- 29.Wakisaka Y, Chu Y, Miller JD, et al. Spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. J Cereb Blood Flow Metab 2010; 30: 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. Am J Neuroradiol 2008; 29: 1036–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smeda JS. Hemorrhagic stroke development in spontaneously hypertensive rats fed a North American, Japanese-style diet. Stroke 1989; 20: 1212–1218. [DOI] [PubMed] [Google Scholar]
- 32.Omura-Matsuoka E, Yagita Y, Sasaki T, et al. Hypertension impairs leptomeningeal collateral growth after common carotid artery occlusion: restoration by antihypertensive treatment. J Neurosci Res 2011; 89: 108–116. [DOI] [PubMed] [Google Scholar]
- 33.Giacomelli F, Rooney J, Wiener J. Cerebrovascular ultrastructure and permeability after carotid artery constriction in experimental hypertension. Exp Mol Pathol 1978; 28: 309–321. [DOI] [PubMed] [Google Scholar]
- 34.Bailey EL, McCulloch J, Sudlow C, et al. Potential animal models of lacunar stroke: a systematic review. Stroke 2009; 40: e451–e458. [DOI] [PubMed] [Google Scholar]
- 35.Tamaki K, Sadoshima S, Baumbach GL, et al. Evidence that disruption of the blood-brain barrier precedes reduction in cerebral blood flow in hypertensive encephalopathy. Hypertension 1984; 6(2 Pt 2): I75–181. [DOI] [PubMed] [Google Scholar]
- 36.McKinney AM, Sarikaya B, Gustafson C, et al. Detection of microhemorrhage in posterior reversible encephalopathy syndrome using susceptibility-weighted imaging. Am J Neuroradiol 2012; 33: 896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwartz RB, Jones KM, Kalina P, et al. Hypertensive encephalopathy: findings on CT, MR imaging, and SPECT imaging in 14 cases. Am J Roentgenol 1992; 159: 379–383. [DOI] [PubMed] [Google Scholar]
- 38.Lee JM, Zhai G, Liu Q, et al. Vascular permeability precedes spontaneous intracerebral hemorrhage in stroke-prone spontaneously hypertensive rats. Stroke 2007; 38: 3289–3291. [DOI] [PubMed] [Google Scholar]
- 39.Bartynski WS, Boardman JF. Distinct imaging patterns and lesion distribution in posterior reversible encephalopathy syndrome. Am J Neuroradiol 2007; 28: 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fredriksson K, Kalimo H, Westergren I, et al. Blood-brain barrier leakage and brain edema in stroke-prone spontaneously hypertensive rats. Effect of chronic sympathectomy and low protein/high salt diet. Acta Neuropathol 1987; 74: 259–268. [DOI] [PubMed] [Google Scholar]
- 41.Jacquot C, Glastonbury CM, Tihan T. Is posterior reversible encephalopathy syndrome really reversible? Autopsy findings 4.5 years after radiographic resolution. Clin Neuropathol 2015; 34: 26–33. [DOI] [PubMed] [Google Scholar]
- 42.Lanzino G, Cloft H, Hemstreet MK, et al. Reversible posterior leukoencephalopathy following organ transplantation. Description of two cases. Clin Neurol Neurosurg 1997; 99: 222–226. [DOI] [PubMed] [Google Scholar]
- 43.Horbinski C, Bartynski WS, Carson-Walter E, et al. Reversible encephalopathy after cardiac transplantation: histologic evidence of endothelial activation, T-cell specific trafficking, and vascular endothelial growth factor expression. Am J Neuroradiol 2009; 30: 588–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mies G, Hermann D, Ganten U, et al. Hemodynamics and metabolism in stroke-prone spontaneously hypertensive rats before manifestation of brain infarcts. J Cereb Blood Flow Metab 1999; 19: 1238–1246. [DOI] [PubMed] [Google Scholar]
- 45.Smeda JS, VanVliet BN, King SR. Stroke-prone spontaneously hypertensive rats lose their ability to auto-regulate cerebral blood flow prior to stroke. J Hypertens 1999; 17(12 Pt 1): 1697–1705. [DOI] [PubMed] [Google Scholar]
- 46.Gelosa P, Pignieri A, Gianazza E, et al. Altered iron homeostasis in an animal model of hypertensive nephropathy: stroke-prone rats. J Hypertens 2013; 31: 2259–2569. [DOI] [PubMed] [Google Scholar]
- 47.Suno M, Shibota M, Nagaoka A. Effects of idebenone on lipid peroxidation and hemolysis in erythrocytes of stroke-prone spontaneously hypertensive rats. Arch Gerontol Geriatr 1989; 8: 307–311. [DOI] [PubMed] [Google Scholar]
- 48.Xie L, Jiang Y, Yao W, et al. Studies on the biomechanical properties of maturing reticulocytes. J Biomech 2006; 39: 530–535. [DOI] [PubMed] [Google Scholar]
- 49.Bow H, Pivkin IV, Diez-Silva M, Goldfless SJ, Dao M, Niles JC, et al. A microfabricated deformability-based flow cytometer with application to malaria. Lab on a chip 2011; 11(6): 1065–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohandas N, Johnson A, Wyatt J, et al. Automated quantitation of cell density distribution and hyperdense cell fraction in RBC disorders. Blood 1989; 74: 442–447. [PubMed] [Google Scholar]
- 51.Zheng XL, Sadler JE. Pathogenesis of thrombotic microangiopathies. Annu Rev Pathol 2008; 3: 249–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldberg RJ, Nakagawa T, Johnson RJ, et al. The role of endothelial cell injury in thrombotic microangiopathy. Am J Kidney Dis 2010; 56: 1168–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burrus TM, Mandrekar J, Wijdicks EF, et al. Renal failure and posterior reversible encephalopathy syndrome in patients with thrombotic thrombocytopenic purpura. Arch Neurol 2010; 67: 831–834. [DOI] [PubMed] [Google Scholar]
- 54.Burrus TM, Wijdicks EF, Rabinstein AA. Brain lesions are most often reversible in acute thrombotic thrombocytopenic purpura. Neurology 2009; 73: 66–70. [DOI] [PubMed] [Google Scholar]
- 55.Thachil J. A clue to the pathophysiology of posterior reversible encephalopathy syndrome. Arch Neurol 2010; 67: 1536 ; author reply 1536–1537. [DOI] [PubMed] [Google Scholar]
- 56.Deguchi I, Uchino A, Suzuki H, et al. Malignant hypertension with reversible brainstem hypertensive encephalopathy and thrombotic microangiopathy. J Stroke Cerebrovasc Dis 2012; 21: 915 e17–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.