

Abstract
Protein–protein interaction (PPI) plays an extremely remarkable role in the growth, reproduction, and metabolism of all lives. A thorough investigation of PPI can uncover the mechanism of how proteins express their functions. In this study, we used gene ontology (GO) terms and biological pathways to study an extended version of PPI (protein–protein functional associations) and subsequently identify some essential GO terms and pathways that can indicate the difference between two proteins with and without functional associations. The protein–protein functional associations validated by experiments were retrieved from STRING, a well-known database on collected associations between proteins from multiple sources, and they were termed as positive samples. The negative samples were constructed by randomly pairing two proteins. Each sample was represented by several features based on GO and KEGG pathway information of two proteins. Then, the mutual information was adopted to evaluate the importance of all features and some important ones could be accessed, from which a number of essential GO terms or KEGG pathways were identified. The final analysis of some important GO terms and one KEGG pathway can partly uncover the difference between proteins with and without functional associations.
1. Introduction
Protein is the material foundation of all living things [1]. Protein–protein interaction (PPI) plays an extremely significant role in the growth, reproduction, and metabolism of any life, even in a single cell [2, 3]. Proteins can be easily clustered in three methods: (I) homology of protein subunits, (II) stability of interactions, and (III) combination mode of subunits [4–6]. By connecting related proteins, PPI initiates the action of various functional or structural proteins in every single cell [4]. Given that proteins influence different biological processes, even in single cells, conducting a study on PPI to further determine protein functions and life activities is a relevant endeavor.
PPI has been thoroughly studied both in experimental and computing scenarios. To study PPI via experiments, coimmunoprecipitation, Western blot, and yeast two-hybrid systems are generally adopted [7, 8]. As for computational methods, several algorithms have been developed to identify PPI, and the two main ones are the topology-free approaches and the graph-based approaches, which are based on distances between proteins and specialized clustering techniques, respectively [9, 10]. Some other computational methods predict PPIs from protein sequences using machine learning. Jansen et al. developed a Bayesian network to integrate multiple genomic features to predict PPIs [11]. Shen et al. trained a support vector machine classifier using conjoint triad features derived from sequences [12]. Pan et al. first used latent Dirichlet allocation model to extract latent topic features from the conjoint triad features, then the learned topic features were fed into a random forest classifier to predict PPIs [13]. Hashemifar et al. trained a deep learning model to predict PPIs using evolutionary information with random projection and data augmentation [14]. In addition, with the development and innovation of computational technologies, the use of updated algorithms has allowed researchers to predict and study PPIs conveniently and accurately alongside the utilization of different databases and methods.
Gene ontology (GO) is a bioinformatic concept that was originally proposed to unify the representation of genes and gene products of many species [15, 16]. The ontology covers three main domains, namely, (I) cellular component, (II) molecular function, and (III) biological process, which can easily cluster all genes and gene products with a directed acyclic graph (DAG) [16]. For convenience, the ontological domains are widely used in computational biology to avoid redundancy of different annotations of a single functional or structural gene [17, 18]. GO terms, which have been updated given the development of biological science, can summarize the specific role of genes and their products in living cells, and they are regarded as powerful tools in computational biology science [16]. Different kinds of PPIs are also included in the various terms of GO annotations. The specific locations or functions of PPIs in cells have been investigated to easily describe and distinguish the several kinds of GO terms. The GO annotations contain informative signals for PPIs. For example, Patil and Nakamura trained a machine learning classifier to infer PPIs using features derived from sequence similarity, shared GO terms and domains [19]. Ben-Hur et al. used a kernel method to integrate sequences, GO annotations, local network properties and homologous interactions for predicting PPIs [20]. Stefan et al. generated features for proteins from GO DAG; then, the extracted features were fed into a random forest classifier to predict PPIs [21]. However, these studies only adopted the GO annotations to construct the model for predicting PPIs. They did not analyze which GO annotations were highly related to the determination of PPIs. In addition, genes can be clustered into several biological pathways. Some essential pathways may be highly related to PPIs.
In this study, we investigated an extended version of PPI (protein–protein function associations) by using GO terms and KEGG pathways. Considering the fact that few PPI studies with computational methods investigated which GO terms were highly related to the determination of PPIs, the purpose of this study was to identify key GO terms or KEGG pathways that can indicate the difference between two proteins with and without functional associations. We first extracted protein–protein functional associations with experiment validations reported in Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) [22, 23], a well-known database on collected associations between proteins, as the positive samples, and then we randomly selected proteins to constitute the negative samples. Considering that the random selection of negative samples may influence the results, 10 sets of negative samples were constructed, thereby constituting 10 datasets, each of which contained the same positive samples. Each protein–protein functional association was encoded into a vector by using the GO terms and KEGG pathways. Then, mutual information was adopted to evaluate the importance of all features in each dataset. From the feature lists, in which features were ranked in the decreasing order of their importance, some important features were identified, and their corresponding GO terms or KEGG pathways were obtainable. Finally, we analyzed some most important GO terms and one KEGG pathway to partly uncover the difference between proteins with and without functional associations.
2. Materials and Methods
2.1. Materials
All human protein–protein functional associations used in this study were retrieved from STRING (http://www.string-db.org/, version 9.1) [22, 23], a well-known public database on several collected associations between proteins from various organisms. These associations have been derived from the following four sources: (I) genomic context, (II) high-throughput experiments, (III) (conserved) coexpression, and (IV) previous knowledge. To obtain the human protein–protein functional associations in this database, we downloaded a file named “protein.links.detailed.v9.1.txt.gz” and then extracted lines starting with “9606” (i.e., the code of human in STRING). A total of 2,425,314 human protein–protein functional associations involving 20,770 proteins were accessed. The purpose of this study is to identify some important GO terms or KEGG pathways that can indicate the difference between two proteins with and without functional associations. Thus, we refined the 20,770 proteins as follows: (1) utilize CD-HIT [24] to discard similar proteins such that the similarity between any two remaining proteins was less than 0.25 and (2) exclude proteins whose GO term or KEGG pathway information was not available, from which we obtained 8,916 proteins. The derived proteins can comprise 588,154 human protein–protein functional associations. Furthermore, we selected 70,392 human protein–protein functional associations among the above-mentioned associations. The “Experimental” scores of these associations are larger than zero, meaning that they are validated by solid experiments. These associations involved 6,623 human proteins. For convenience, these associations were termed positive associations in this study and are provided in Supplementary Material S1.
To extract the difference between positive associations and any two proteins without functional associations, some negative associations are necessary. Given that negative associations are substantially more than the positive ones, we constructed 211,176 differing pairs of proteins, which were thrice as many as positive associations, and each of them was produced as follows: (1) random selection of two different proteins from 6,623 proteins, and (II) these two proteins cannot comprise an association reported in STRING. The obtained negative and positive associations constituted a dataset. Considering that the produced negative associations may influence the results, we randomly produced 10 sets of negative associations. Each of the sets, together with the positive associations, constituted a dataset, thus producing 10 datasets, which were denoted as DS1, DS2,…, DS10. By analyzing these datasets, some essential information for protein–protein functional associations can be discovered. The whole procedures are illustrated in Figure 1.
Figure 1.

The whole procedures for analyzing protein–protein functional associations based on gene ontology (GO) and KEGG pathways. The raw 2,425,314 human PPIs were retrieved from STRING and refined by excluding similar proteins and selecting those validated by experiments, resulting in 70,392 PPIs. 6,623 proteins were involved in investigated PPIs and used to construct ten sets of protein pairs, each of which combined with 70,392 PPIs to constitute ten datasets. Each sample was represented by GO and KEGG features, which were evaluated by mutual information, producing ten feature lists, from which we extracted most important features, corresponding to 134 GO terms and one KEGG pathway.
2.2. Representation of Protein–Protein Function Associations
GO terms [16] and KEGG pathways [25] are always used to elucidate and describe molecular functions, cellular components, and biological and signal processes of genes. From Gene Ontology Consortium [16], 17,916 GO terms were retrieved. Accordingly, a protein p can be encoded as
| (1) |
where
| (2) |
For two proteins p1 and p2 that comprised either a positive association or a negative association P = (p1, p2), because there was no order information in P, i.e., (p1, p2) was identical to (p2, p1), it was not appropriate to simply combine the features of p1 and p2. To exclude the order information of P, we adopted the following scheme that has been used in some studies [26, 27]. For P = (p1, p2), it was encoded into a vector by using vGO(p1) and vGO(p2) as follows:
| (3) |
Moreover, according to KEGG [25], there were 279 pathways, based on which the protein p can be represented by
| (4) |
where
| (5) |
Similarly, P = (p1, p2) can be encoded into
| (6) |
By integrating the GO term and KEGG pathway information of proteins into P = (p1, p2), each association can be finally encoded as
| (7) |
A total of 36,390 features were used to represent each positive association or negative association. The information of each GO term or KEGG pathway was contained by these two features.
2.3. Feature Evaluation with Mutual Information
As mentioned in Section 2.2, several features were used to represent each protein–protein functional association. However, not all are highly related to sufficiently determine the differences between positive and negative associations, i.e., not all GO terms and KEGG pathways can be used to mark the associations. Here, we adopted the mutual information (MI) of each feature and target (class labels of samples) to evaluate the importance of each feature. The evaluations use the following equation to access the relationship between the two variables of x and y:
| (8) |
where p(x) and p(y) are the marginal probabilistic density of variables x and y, while p(x, y) is their joint probabilistic density.
Given a dataset in which each sample is represented by N features, after the MI values of all features were calculated, features were sorted by their MI values in decreasing order, thereby producing a feature list named MaxRel feature list, which is formulated as
| (9) |
where fi represents a feature in the dataset.
To quickly implement the program of MI, we adopted the program of minimum redundancy maximum relevance (mRMR) method [28], which integrates the MI program. This program has been applied in solving several complicated biological problems [26, 29–45].
3. Results
3.1. Results of the Feature Evaluation
As mentioned in Section 2.2, each association in the 10 datasets was represented by 36,390 features. We calculated the MI value of each feature in each of the datasets DS1, DS2,…, DS10. Subsequently, ten MaxRel feature lists could be accessed. A part of these 10 lists is provided in Supplementary Material S2.
3.2. Extracting Important GO Terms and KEGG Pathways
Features with high ranks (large MI values) in the MaxRel feature list are more important than those with low ranks (small MI values). For the MI value, we set 0.01 as the threshold to select important features in each MaxRel feature list, thus producing 10 feature sets denoted as F1, F2,…, F10. The numbers of selected features in these sets are listed in Table 1. In the tabulation, the sizes of the 10 feature sets are nearly the same. After the features in these 10 sets were combined, 158 features were obtained (Supplementary Material S3). The obtained number (i.e., 158) did not differ much from the size of each feature set, which indicates that the majority of the 158 features were included in each set. In particular, among the 158 features, 146 features were included in all 10 feature sets, while 2, 1, 2, 3, and 4 feature/s were included in nine, eight, seven, six, and less than six feature sets (Figure 2), respectively. Considering that the negative associations in each of the 10 datasets somewhat differed, we predicted that the random selection of negative PPIs will not have a strong influence on the selection of the 158 features; i.e., the features can effectively determine the difference between positive and negative associations. Figure 3 shows a heat map of MI values of the 158 features in the 10 datasets. In the figure, the MI values of each of the 158 features in the 10 datasets are nearly the same. Similarly, the distributions of the MI values of the 158 features in the 10 datasets are nearly the same, which validates the above-mentioned results. Subsequently, an extensive investigation to further uncover the mechanism of proteins with function associations was conducted.
Table 1.
Number of selected features in each MaxRel feature list.
| Dataset | Number of selected features |
|---|---|
| DS 1 | 154 |
| DS 2 | 154 |
| DS 3 | 153 |
| DS 4 | 155 |
| DS 5 | 149 |
| DS 6 | 150 |
| DS 7 | 155 |
| DS 8 | 152 |
| DS 9 | 153 |
| DS 10 | 153 |
Figure 2.
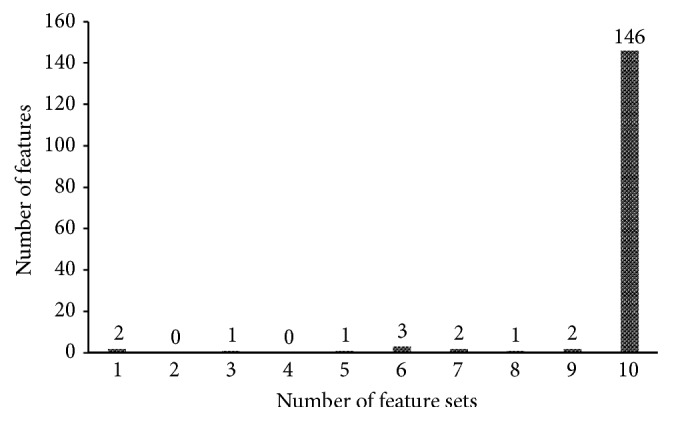
Distribution of 158 selected features: 146, 2, 1, 2, 3, and 4 feature/s in 10, 9, 8, 7, 6, and less than 6 feature sets derived from 10 datasets, respectively.
Figure 3.
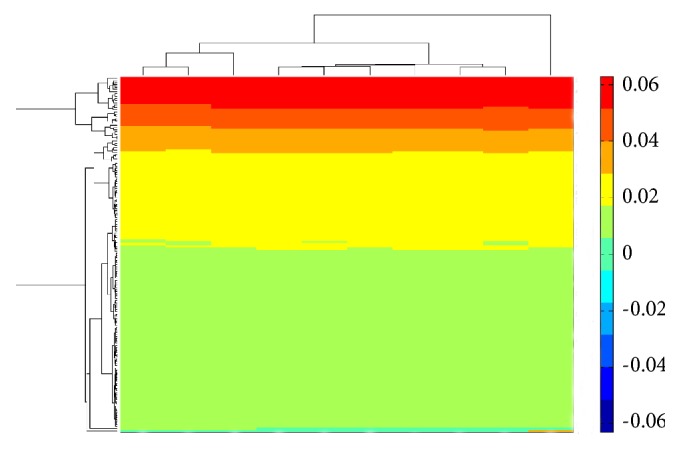
Heat map of MI values of 158 features in the 10 datasets. X-axis represents ten datasets; Y-axis represents 158 features.
A careful checking showed that the important 158 features were derived from 134 GO terms and one KEGG pathway (Supplementary Material S4). To further evaluate their importance, we adopted a calculation technique called rating score measurement for each GO term. In this paper, the rating score is expressed as the sum of MI values of the related features in the 10 MaxRel feature lists. The scores are also provided in Supplementary Material S4. The rating score for the KEGG pathway (hsa03010) was 0.107, while the distribution of rating scores for 134 GO terms is illustrated in Figure 4.
Figure 4.
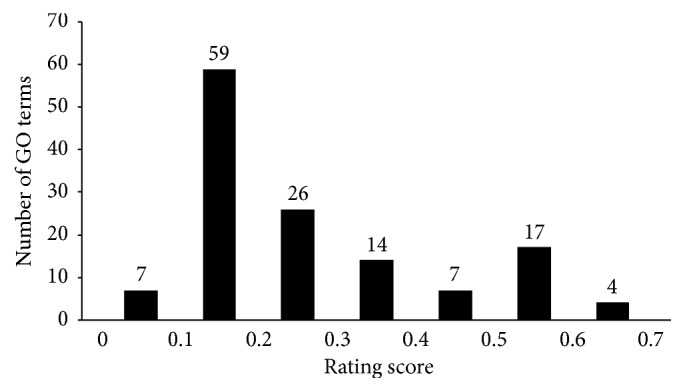
The distribution of the rating scores of 134 selected GO terms.
3.3. Analysis of the Importance of Selected Features
As mentioned in Section 3.2, we finally selected 158 features that were deemed to be highly related to PPIs. To confirm such conclusion, we did the following test. For each of ten datasets mentioned in Section 2.1, each sample in the dataset was represented by these 158 features. And we also randomly constructed 100 feature sets, each of which consisted of 158 features. Samples in DS1 were represented by features in each of these feature sets to comprise 100 datasets. The classic classification algorithm, random forest (RF) [44, 46–50], was performed on all above-mentioned datasets, evaluated by tenfold cross-validation. The predicted results were counted as Matthews correlation coefficient (MCC) [40, 44, 47, 51–53], which are shown in Figure 5. It can be observed that the RF with selected 158 features yielded the MCCs between 0.55 and 0.60, while the RF with randomly selected 158 features generated the MCCs around 0.23. Clearly, the selected 158 features can capture the essential properties of PPIs, thereby providing more powerful distinguishing ability. Investigation on these features can help uncover the mechanisms of PPIs.
Figure 5.
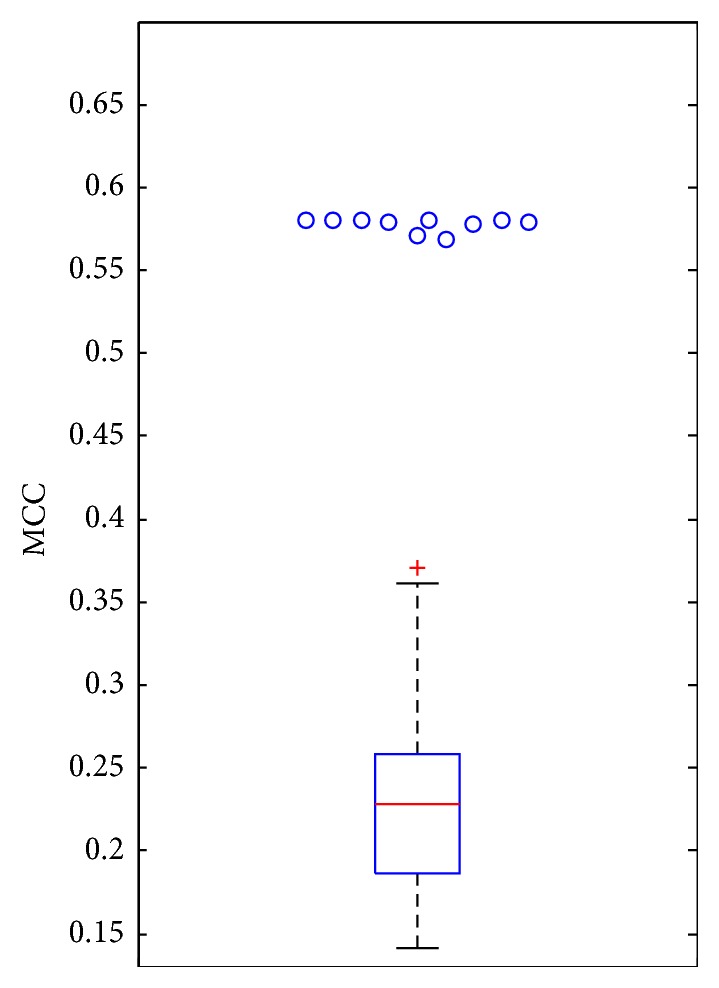
The performance of the random forest (RF) on ten datasets, in which samples were represented by selected 158 features or randomly selected 158 features, evaluated by tenfold cross-validation. The box plot indicates the distribution of MCCs yielded by RF with randomly selected 158 features and the circles represent the MCCs yielded by RF with selected 158 features on ten datasets. It is clear that based on selected 158 selected features, RF produced much better performance, implying the strong associations between these features and PPIs.
4. Discussion
As mentioned in Section 3.2, 134 GO terms and one KEGG pathway were regarded important in determining the difference between positive and negative associations. This section gave a detailed analysis on them.
4.1. Analysis of Key GO Terms
Analyzing above-mentioned 134 GO terms one by one is difficult. Here, we selected the most important 21 GO terms with rating scores larger than 0.5 for detailed analysis, which are listed in Table 2. 21 GO terms can be clustered into three groups: cellular component, molecular function, and biological process [15]. The distribution of the aforementioned 21 GO terms on these three groups is shown in Figure 6. Eleven GO terms are clustered into cellular component, three terms into molecular function, and seven terms into biological process. All these GO terms can be proven or inferred as associated with PPIs in published literature, as to be discussed below.
Table 2.
Information of most important 21 GO terms.
| GO term ID | GO term | Rating score | Group |
|---|---|---|---|
| GO:0044260 | cellular macromolecule metabolic process | 0.688 | Biological process |
| GO:0043170 | macromolecule metabolic process | 0.640 | Biological process |
| GO:0044428 | nuclear part | 0.618 | Cellular component |
| GO:1901363 | heterocyclic compound binding | 0.600 | Molecular function |
| GO:0032991 | protein-containing complex | 0.593 | Cellular component |
| GO:0097159 | organic cyclic compound binding | 0.591 | Molecular function |
| GO:0031981 | nuclear lumen | 0.590 | Cellular component |
| GO:0044238 | primary metabolic process | 0.589 | Biological process |
| GO:0003676 | nucleic acid binding | 0.583 | Molecular function |
| GO:0090304 | nucleic acid metabolic process | 0.569 | Biological process |
| GO:0071704 | organic substance metabolic process | 0.556 | Biological process |
| GO:0044237 | cellular metabolic process | 0.552 | Biological process |
| GO:0005634 | nucleus | 0.549 | Cellular component |
| GO:0044446 | intracellular organelle part | 0.547 | Cellular component |
| GO:0044424 | intracellular part | 0.537 | Cellular component |
| GO:0044422 | organelle part | 0.536 | Cellular component |
| GO:0070013 | intracellular organelle lumen | 0.529 | Cellular component |
| GO:0005622 | intracellular | 0.523 | Cellular component |
| GO:0043233 | organelle lumen | 0.521 | Cellular component |
| GO:0031974 | membrane-enclosed lumen | 0.514 | Cellular component |
| GO:0006139 | nucleobase-containing compound metabolic process | 0.506 | Biological process |
Figure 6.
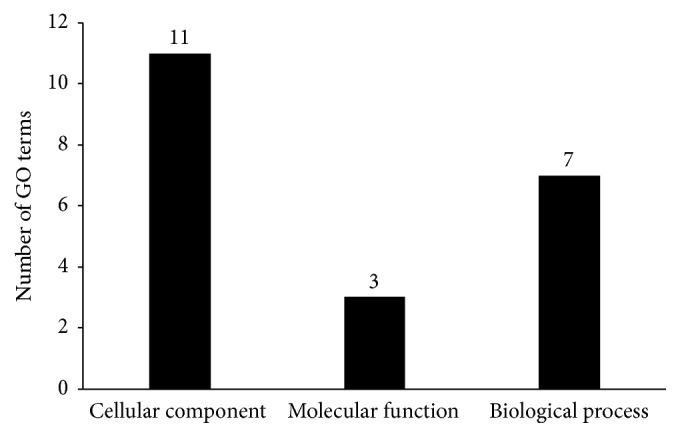
Distribution of 21 GO terms on three groups: cellular component, molecular function, and biological process.
Cellular Component GO Terms. As described above, eleven of the 21 GO terms clustered as cellular components refer to the part of a single cell and its specific extracellular environment, taking account for more than 52% of selected GO terms [16]. Comparing with molecular function and biological process as other two GO categories, which mostly reflect the indirect and functional relationships between different proteins, cellular component reflects the direct interactive relationships. Thus, the enrichment of functional clustered GO terms in such GO category indicated that subcellular localization and regional protein distribution may contribute more to the distinction of positive and negative associations. Direct PPIs which take the majority of all PPIs relied on the direct molecular interactions between proteins. The participants of most positive associations must share similar physical subcellular localizations, while those of the negative ones do not have to. Therefore, comparing to molecular function and biological process, it is quite reasonable for the cellular component category of GO terms to take the majority of all the enriched biological processes contributing to the recognition of positive PPIs.
The cellular component GO term with the highest rating score was GO: 0044428, describing the nuclear part of the eukaryotic cells, involving in chromosomes housing and replicating. Such processes involve multiple effective PPIs, like Esc2 and Rad51 [54, 55]. Therefore, the functional enrichment of genes involved in such cellular component may be more probable to participate in an actual PPI, contributing to the recognition of positive PPIs. Similarly, GO: 0031981, describing the nuclear lumen region, and GO: 0005634, describing a more general region of the cell, nucleus, may also involve in multiple PPIs. It has been widely reported that the nucleus region involves multiple subgroup of PPIs, regulating the expression and replication of genes [56–58]. Therefore, having nucleus as one of the busiest regions in cells, genes identified in such region may actually tend to be participating in certain PPIs.
Apart from the nucleus region of the cell, according to our results, we also identified that cellular regions associated with functional organelles may also be related to PPIs. GO: 0044422, describing the organelle part of cells, GO: 0070013, describing intracellular organelle lumen, GO:0044446, describing the intracellular organelle part, and GO:0043233, describing organelle lumen, have all been screened out as the potential cellular components that may be associated with positive PPIs [59]. Similar with the nucleus regions, comparing to extracellular matrix and other intracellular regions, the organelles and its related biochemical reactions space involve in more actually interacting PPIs [59–61]. Therefore, PPIs that locate in such region tend out to actually happen, indicating that these GO terms contribute to describing an effective gene cellular component features of genes that actually participate in PPIs.
Apart from such specific GO terms, we also identified some more general ones, like GO: 0032991 (protein-containing complex), GO: 0044424 (intracellular part), GO: 0005622 (intracellular), and GO: 0031974 (membrane-enclosed lumen). They all describe the regions that enrich significant biological processes of the cells. Therefore, actual PPIs tend to enrich in such region, revealing the specific PPI distribution pattern in the eukaryotic cells.
Molecular Function GO Terms. Three molecular function associated GO terms were extracted. The top GO term was GO: 1901363, describing heterocyclic compound binding. According to recent publications, various PPIs can actually be functional enriched in the heterocyclic compound binding, like the interactions between PDK1 and AKT in the eukaryotic cells [62, 63]. Therefore, genes that participate in such molecular function may tend to be more probable to actually contribute to PPIs. Similarly, the other molecular function GO term, named GO: 0097159, which describes organic cyclic compound binding, also involves various PPIs, like interactions among TBK1, PDPK1, and AURKA [64]. As for the last term, GO: 0003676, it describes the nucleic acid binding. As analyzed above, the nucleus region, where nucleic acid binding processes mostly occur, can distinguish the positive and negative PPIs due to its relative high interaction frequency [56–58]. Therefore, it is quite reasonable to speculate that such molecular function GO term may also be related to PPIs.
Biological Process GO Terms. Apart from above-mentioned cellular component and molecular function associated GO terms, we also identified a group of functional enrichment results that can be clustered into the biological processes cluster. All these GO terms describe effective metabolic processes in the cells. GO: 0044260 and GO: 0043170 describe the macromolecule metabolic processes. According to recent publications, such metabolic processes involve various PPIs, like the interactions in mTOR signaling pathways [65]. Apart from that, GO: 0044238, describing the primary metabolic process, has also been confirmed to contribute to PPIs. Considering the normal anabolic and catabolic processes, all involving functional PPIs [66–68], it is quite reasonable for genes participating in such biological processes to also participate in effective PPIs. The following three GO terms, GO: 0090304 (nucleic acid metabolic process), GO: 0006139 (nucleobase-containing compound metabolic process), and GO: 0071704 (organic substance metabolic process), may also contribute to PPIs, considering that the nucleus region has been discussed to be quite significant for PPIs [56–58] and proteins turn out to be one of the major subgroups of organic substance in eukaryotic cells; these three GO terms may also actually contribute to the identification of PPIs. As for the remaining GO term, GO: 0044237, it describes a general concept of all the cellular metabolic processes. Considering the analyses listed above, metabolic processes in the cells enrich various actual PPIs and are reasonable to be predicted and screened out as a potential identifier for positive PPIs.
On the basis of the analyses, all 21 GO terms are involved in different aspects of PPI, and they can be used to mark proteins with functional associations. For the remaining GO terms shown in Supplementary Material S4, it is anticipated that they also have associations with PPIs.
4.2. Analysis of Other GO Terms and KEGG Pathways
As for other GO terms extracted in this study, although not so relevant with PPIs as such GO terms described in Section 4.1, some of them have also been reported to be functionally related to certain PPIs. For instance, GO: 0006807, describing nitrogen compound metabolism, has been widely reported to be functionally related to compound-protein interactions but not protein–protein interactions [69, 70]. However, when extensively studying biological processes of such GO term, we found out that various specific PPIs are just like the interactions between the protein products of TIMP1 and MMP2 [71]. Therefore, in this study, some identified GO terms have not been directly reported to contribute to the PPIs. However, by digging deep into the actual biological processes, molecular functions and cellular components of them, we actually found that various novel identified PPIs are associated with these GO terms.
Furthermore, one KEGG pathway hsa03010 was obtained in our study. It describes the ribosome associated pathway. Considering that genes/proteins that participate in such pathway may interact with each other, forming the complex of ribosome, such KEGG pathway, may also contribute to the distinction of positive and negative PPIs.
5. Conclusions
This study investigated protein–protein functional associations based on GO terms and KEGG pathways. By using mutual information, we identified important GO terms and KEGG pathways that can describe the difference between actual associations and pairs of proteins without associations and help understand the mechanisms of protein interactions. A possible future research direction is to further use these GO terms and KEGG pathways to build a computational method for inferring novel associations between proteins, enriching the biological functional annotation of proteins.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (31701151), Natural Science Foundation of Shanghai (17ZR1412500), National Key R&D Program of China (2018YFC0910403), Shanghai Sailing Program (16YF1413800), The Youth Innovation Promotion Association of Chinese Academy of Sciences (CAS) (2016245), the fund of the Key Laboratory of Stem Cell Biology of Chinese Academy of Sciences (201703), and Science and Technology Commission of Shanghai Municipality (STCSM) (18dz2271000).
Contributor Information
Fei Yuan, Email: snowhawkyrf@outlook.com.
Yu-Dong Cai, Email: cai_yud@126.com.
Data Availability
The original data used to support the findings of this study are available at STRING dataset and in supplementary information files.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Supplementary Materials
70392 protein–protein functional associations.
A part of MaxRel feature list on ten datasets obtained by mutual information of each feature.
Selected 158 features and their occurrences in the 10 feature sets of the 10 datasets (√: feature is in the feature set; ☓: feature is not included in the feature set).
Extracted important GO terms and their rating scores.
References
- 1.Lodish H. B. A., Matsudaira P., Kaiser C. A., et al. In: Molecular Cell Biology. Freeman W. H., editor. New York, NY, USA: 2004. [Google Scholar]
- 2.Safari-Alighiarloo N., Taghizadeh M., Rezaei-Tavirani M., Goliaei B., Peyvandi A. A. Protein-protein interaction networks (PPI) and complex diseases. Gastroenterology and Hepatology from Bed to Bench. 2014;7(1):17–31. [PMC free article] [PubMed] [Google Scholar]
- 3.Chakraborty C., Priya Doss. C G., Chen L., Zhu H. Evaluating protein-protein interaction (PPI) networks for diseases pathway, target discovery, and drug-design using 'In silico pharmacology'. Current Protein & Peptide Science. 2014;15(6):561–571. doi: 10.2174/1389203715666140724090153. [DOI] [PubMed] [Google Scholar]
- 4.Tepper K., Biernat J., Kumar S., et al. Oligomer formation of tau protein hyperphosphorylated in cells. The Journal of Biological Chemistry. 2014;289(49):34389–34407. doi: 10.1074/jbc.M114.611368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shukla S., Allam U. S., Ahsan A., et al. KRAS protein stability is regulated through SMURF2: UBCH5 complex-mediated β-TrCP1 degradation. Neoplasia (United States) 2014;16(2):115–128. doi: 10.1593/neo.14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zakeri B., Fierer J. O., Celik E., et al. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proceedings of the National Acadamy of Sciences of the United States of America. 2012;109(12):E690–E697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg I. M., editor. Protein Analysis and Purification: Benchtop Techniques. 2nd. 2005. [Google Scholar]
- 8.Snider J., Kittanakom S., Curak J., Stagljar I. Split-ubiquitin based membrane yeast two-hybrid (MYTH) system: a powerful tool for identifying protein-protein interactions. Journal of Visualized Experiments. 2010;(36) doi: 10.3791/1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruckner S., Hüffner F., Karp R. M., Shamir R., Sharan R. Topology-free querying of protein interaction networks. Journal of Computational Biology. 2010;17(3):237–252. doi: 10.1089/cmb.2009.0170. [DOI] [PubMed] [Google Scholar]
- 10.Neyshabur B., Khadem A., Hashemifar S., Arab S. S. NETAL: a new graph-based method for global alignment of protein-protein interaction networks. Bioinformatics. 2013;29(13):1654–1662. doi: 10.1093/bioinformatics/btt202. [DOI] [PubMed] [Google Scholar]
- 11.Jansen R., Yu H., Greenbaum D., et al. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302(5644):449–453. doi: 10.1126/science.1087361. [DOI] [PubMed] [Google Scholar]
- 12.Shen J., Zhang J., Luo X., et al. Predicting protein-protein interactions based only on sequences information. Proceedings of the National Acadamy of Sciences of the United States of America. 2007;104(11):4337–4341. doi: 10.1073/pnas.0607879104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan X.-Y., Zhang Y.-N., Shen H.-B. Large-scale prediction of human protein-protein interactions from amino acid sequence based on latent topic features. Journal of Proteome Research. 2010;9(10):4992–5001. doi: 10.1021/pr100618t. [DOI] [PubMed] [Google Scholar]
- 14.Hashemifar S., Neyshabur B., Khan A. A., Xu J. Predicting protein–protein interactions through sequence-based deep learning. Bioinformatics. 2018;34(17):i802–i810. doi: 10.1093/bioinformatics/bty573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huntley R. P., Sawford T., Mutowo-Meullenet P., et al. The GOA database: gene ontology annotation updates for 2015. Nucleic Acids Research. 2015;43(D1):D1057–D1063. doi: 10.1093/nar/gku1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Gene Ontology Consortium. Gene ontology consortium: going forward. Nucleic Acids Research. 2015;43(D1):D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillis J., Pavlidis P. Assessing identity, redundancy and confounds in gene ontology annotations over time. Bioinformatics. 2013;29(4):476–482. doi: 10.1093/bioinformatics/bts727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jantzen S. G., Sutherland B. J., Minkley D. R., Koop B. F. GO trimming: systematically reducing redundancy in large gene ontology datasets. BMC Research Notes. 2011;4:p. 267. doi: 10.1186/1756-0500-4-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patil A., Nakamura H. Filtering high-throughput protein-protein interaction data using a combination of genomic features. BMC Bioinformatics. 2005;6:p. 100. doi: 10.1186/1471-2105-6-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ben-Hur A., Noble W. S. Kernel methods for predicting protein-protein interactions. Bioinformatics. 2005;21(supplement 1):i38–i46. doi: 10.1093/bioinformatics/bti1016. [DOI] [PubMed] [Google Scholar]
- 21.Maetschke S. R., Simonsen M., Davis M. J., Ragan M. A. Gene ontology-driven inference of protein-protein interactions using inducers. Bioinformatics. 2012;28(1):69–75. doi: 10.1093/bioinformatics/btr610. [DOI] [PubMed] [Google Scholar]
- 22.Von Mering C., Jensen L. J., Snel B., et al. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Research. 2005;33(supplement 1):D433–D437. doi: 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franceschini A., Szklarczyk D., Frankild S., et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Research. 2013;41(1):D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L., Chen L., Zhang Y.-H., et al. Analysis and prediction of drug-drug interaction by minimum redundancy maximum relevance and incremental feature selection. Journal of Biomolecular Structure and Dynamics. 2017;35(2):312–329. doi: 10.1080/07391102.2016.1138142. [DOI] [PubMed] [Google Scholar]
- 27.Chen L., Li B.-Q., Zheng M.-Y., Zhang J., Feng K.-Y., Cai Y.-D. Prediction of effective drug combinations by chemical interaction, protein interaction and target enrichment of KEGG pathways. BioMed Research International. 2013;2013:10. doi: 10.1155/2013/723780.723780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng H., Long F., Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Transactions on Pattern Analysis and Machine Intelligence. 2005;27(8):1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 29.Chen L., Zhang Y.-H., Lu G., Huang T., Cai Y.-D. Analysis of cancer-related lncRNAs using gene ontology and KEGG pathways. Artificial Intelligence in Medicine. 2017;76:27–36. doi: 10.1016/j.artmed.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Li B. Q., Zheng L.-L., Feng K.-Y., Hu L.-L., Huang G.-H., Chen L. Prediction of linear B-ceel epitopes with mRMR feature selection and analysis. Current Bioinformatics. 2016;11(1):22–31. doi: 10.2174/1574893611666151119215131. [DOI] [Google Scholar]
- 31.Chen L., Pan X., Hu X., et al. Gene expression differences among different MSI statuses in colorectal cancer. International Journal of Cancer. 2018;143(7):1731–1740. doi: 10.1002/ijc.31554. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y., Ding C., Li T. Gene selection algorithm by combining reliefF and mRMR. BMC Genomics. 2008;9(supplement 2):p. S27. doi: 10.1186/1471-2164-9-S2-S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni Q., Chen L. A feature and algorithm selection method for improving the prediction of protein structural class. Combinatorial chemistry & high throughput screening. 2017;20(7):612–621. doi: 10.2174/1386207320666170314103147. [DOI] [PubMed] [Google Scholar]
- 34.Li J., Lu L., Zhang Y.-H., et al. Identification of synthetic lethality based on a functional network by using machine learning algorithms. Journal of Cellular Biochemistry. 2019;120(1):405–416. doi: 10.1002/jcb.27395. [DOI] [PubMed] [Google Scholar]
- 35.Chen L., Zhang Y., Zheng M., Huang T., Cai Y. Identification of compound–protein interactions through the analysis of gene ontology, KEGG enrichment for proteins and molecular fragments of compounds. Molecular Genetics and Genomics. 2016;291(6):2065–2079. doi: 10.1007/s00438-016-1240-x. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Y., Zhang N., Li B.-Q., Huang T., Cai Y.-D., Kong X.-Y. A method to distinguish between lysine acetylation and lysine ubiquitination with feature selection and analysis. Journal of Biomolecular Structure and Dynamics. 2015;33(11):2479–2490. doi: 10.1080/07391102.2014.1001793. [DOI] [PubMed] [Google Scholar]
- 37.Chen L., Zhang Y., Zou Q., Chu C., Ji Z. Analysis of the chemical toxicity effects using the enrichment of Gene Ontology terms and KEGG pathways. Biochimica et Biophysica Acta (BBA) - General Subjects. 2016;1860(11, Part B):2619–2626. doi: 10.1016/j.bbagen.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 38.Ma X., Guo J., Sun X. Sequence-based prediction of RNA-binding proteins using random forest with minimum redundancy maximum relevance feature selection. BioMed Research International. 2015;2015:10. doi: 10.1155/2015/425810.425810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li F., Li C., Wang M., et al. GlycoMine: a machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics. 2015;31(9):1411–1419. doi: 10.1093/bioinformatics/btu852. [DOI] [PubMed] [Google Scholar]
- 40.Chen L., Wang S., Zhang Y., et al. Identify key sequence features to improve CRISPR sgRNA efficacy. IEEE Access. 2017;5:26582–26590. doi: 10.1109/ACCESS.2017.2775703. [DOI] [Google Scholar]
- 41.Radovic M., Ghalwash M., Filipovic N., Obradovic Z. Minimum redundancy maximum relevance feature selection approach for temporal gene expression data. BMC Bioinformatics. 2017;18(1):p. 9. doi: 10.1186/s12859-016-1423-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niu S., Hu L.-L., Zheng L.-L., et al. Predicting protein oxidation sites with feature selection and analysis approach. Journal of Biomolecular Structure and Dynamics. 2012;29(6):650–658. doi: 10.1080/07391102.2011.672629. [DOI] [PubMed] [Google Scholar]
- 43.Chen L., Wang S., Zhang Y.-H., et al. Prediction of nitrated tyrosine residues in protein sequences by extreme learning machine and feature selection methods. Combinatorial Chemistry & High Throughput Screening. 2018;21(6):393–402. doi: 10.2174/1386207321666180531091619. [DOI] [PubMed] [Google Scholar]
- 44.Zhao X., Chen L., Lu J. A similarity-based method for prediction of drug side effects with heterogeneous information. Mathematical Biosciences. 2018;306:136–144. doi: 10.1016/j.mbs.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 45.Chen L., Zhang Y.-H., Pan X., et al. Tissue expression difference between mRNAs and lncRNAs. International Journal of Molecular Sciences. 2018;19(11):p. 3416. doi: 10.3390/ijms19113416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Breiman L. Random forests. Machine Learning. 2001;45(1):5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 47.Zhao X., Chen L., Guo Z.-H., Liu T. Predicting drug side effects with compact integration of heterogeneous networks. Current Bioinformatics. 2019 [Google Scholar]
- 48.Wang S., Wang D., Li J., Huang T., Cai Y. Identification and analysis of the cleavage site in a signal peptide using SMOTE, dagging, and feature selection methods. Molecular Omics. 2018;14(1):64–73. doi: 10.1039/C7MO00030H. [DOI] [PubMed] [Google Scholar]
- 49.Wang S., Zhang Y.-H., Zhang N., Chen L., Huang T., Cai Y.-D. Recognizing and predicting thioether bridges formed by lanthionine and β-methyllanthionine in lantibiotics using a random forest approach with feature selection. Combinatorial chemistry & high throughput screening. 2017;20(7):582–593. doi: 10.2174/1386207320666170310115754. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Q., Sun X., Feng K., et al. Predicting citrullination sites in protein sequences using mRMR method and random forest algorithm. Comb Chem High Throughput Screen. 2017;20(2):164–173. doi: 10.2174/1386207319666161227124350. [DOI] [PubMed] [Google Scholar]
- 51.Matthews B. W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochimica et Biophysica Acta (BBA) - Protein Structure. 1975;405(2):442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 52.Cui H., Chen L. A binary classifier for the prediction of EC numbers of enzymes. Current Proteomics. 2019;16(5):381–389. [Google Scholar]
- 53.Chen L., Chu C., Zhang Y.-H., et al. Identification of drug-drug interactions using chemical interactions. Current Bioinformatics. 2017;12(6):526–534. [Google Scholar]
- 54.Urulangodi M., Sebesta M., Menolfi D., et al. Local regulation of the Srs2 helicase by the SUMO-like domain protein Esc2 promotes recombination at sites of stalled replication. Genes & Development. 2015;29(19):2067–2080. doi: 10.1101/gad.265629.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aranda S., Rutishauser D., Ernfors P. Identification of a large protein network involved in epigenetic transmission in replicating DNA of embryonic stem cells. Nucleic Acids Research. 2014;42(11):6972–6986. doi: 10.1093/nar/gku374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santucci M., Vignudelli T., Ferrari S., et al. The hippo pathway and YAP/TAZ–TEAD protein–protein interaction as targets for regenerative medicine and cancer treatment. Journal of Medicinal Chemistry. 2015;58(12):4857–4873. doi: 10.1021/jm501615v. [DOI] [PubMed] [Google Scholar]
- 57.Li Y., Collins M., An J., et al. Immunoprecipitation and mass spectrometry defines an extensive RBM45 protein–protein interaction network. Brain Research. 2016;1647:79–93. doi: 10.1016/j.brainres.2016.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCrea P. D., Gottardi C. J. Beyond β-catenin: prospects for a larger catenin network in the nucleus. Nature Reviews Molecular Cell Biology. 2016;17(1):55–64. doi: 10.1038/nrm.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kory N., Farese R. V., Walther T. C. Targeting fat: mechanisms of protein localization to lipid droplets. Trends in Cell Biology. 2016;26(7):535–546. doi: 10.1016/j.tcb.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simmen T., Tagaya M. Organelle communication at membrane contact sites (MCS): from curiosity to center stage in cell biology and biomedical research. Advances in Experimental Medicine and Biology. 2017;997:1–12. doi: 10.1007/978-981-10-4567-7_1. [DOI] [PubMed] [Google Scholar]
- 61.Tagaya M., Arasaki K. Regulation of mitochondrial dynamics and autophagy by the mitochondria-associated membrane. Advances in Experimental Medicine and Biology. 2017;997:33–47. doi: 10.1007/978-981-10-4567-7_3. [DOI] [PubMed] [Google Scholar]
- 62.Abeyrathna P., Su Y. The critical role of Akt in cardiovascular function. Vascular Pharmacology. 2015;74:38–48. doi: 10.1016/j.vph.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rettenmaier T. J., Sadowsky J. D., Thomsen N. D., et al. A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1. Proceedings of the National Acadamy of Sciences of the United States of America. 2014;111(52):18590–18595. doi: 10.1073/pnas.1415365112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ctortecka C., Palve V., Kuenzi B. M., et al. Functional proteomics and deep network interrogation reveal a complex mechanism of action of midostaurin in lung cancer cells. Molecular & Cellular Proteomics. 2018;17(12):2434–2447. doi: 10.1074/mcp.RA118.000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ben-Sahra I., Manning B. D. mTORC1 signaling and the metabolic control of cell growth. Current Opinion in Cell Biology. 2017;45:72–82. doi: 10.1016/j.ceb.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dornelles G. L., Bueno A., de Oliveira J. S., et al. Biochemical and oxidative stress markers in the liver and kidneys of rats submitted to different protocols of anabolic steroids. Molecular and Cellular Biochemistry. 2017;425(1-2):181–189. doi: 10.1007/s11010-016-2872-1. [DOI] [PubMed] [Google Scholar]
- 67.Heiden M. G. V., Cantley L. C., Thompson C. B. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kopitz J. Lipid glycosylation: a primer for histochemists and cell biologists. Histochemistry and Cell Biology. 2017;147(2):175–198. doi: 10.1007/s00418-016-1518-4. [DOI] [PubMed] [Google Scholar]
- 69.Xu J., Zha M., Li Y., et al. The interaction between nitrogen availability and auxin, cytokinin, and strigolactone in the control of shoot branching in rice (Oryza sativa L.) Plant Cell Reports. 2015;34(9):1647–1662. doi: 10.1007/s00299-015-1815-8. [DOI] [PubMed] [Google Scholar]
- 70.Feng X. E., Wang Q. J., Gao J., Ban S. R., Li Q. S. Synthesis of novel nitrogen-containing heterocycle bromophenols and their interaction with Keap1 protein by molecular docking. Molecules. 2017;22(12):p. 2142. doi: 10.3390/molecules22122142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen P., Xuan D., Zhang J. Periodontitis aggravates kidney damage in obese mice by MMP2 regulation. Bratislava Medical Journal. 2018;118(12):740–745. doi: 10.4149/BLL_2017_140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
70392 protein–protein functional associations.
A part of MaxRel feature list on ten datasets obtained by mutual information of each feature.
Selected 158 features and their occurrences in the 10 feature sets of the 10 datasets (√: feature is in the feature set; ☓: feature is not included in the feature set).
Extracted important GO terms and their rating scores.
Data Availability Statement
The original data used to support the findings of this study are available at STRING dataset and in supplementary information files.