

Abstract
Gliomas account for approximately 80% of primary malignant tumors in the central nervous system. Despite aggressive therapy, the prognosis of patients remains extremely poor. Glioma stem cells (GSCs), considered a potential target of therapy for their crucial role in therapeutic resistance and tumor recurrence, are believed to be key factors in the disappointing outcome. Here, we took advantage of GSCs as the cell model to perform high-throughput drug screening, and the old antibiotic clofoctol was identified as the most effective compound, showing reduction of colony formation and induction of apoptosis of GSCs. Moreover, growth of tumors was obviously inhibited in vivo after clofoctol treatment especially in primary patient-derived xenografts and transgenic xenografts. The anticancer mechanisms demonstrated by analysis of related downstream genes and discovery of the targeted binding protein revealed that clofoctol exhibited the inhibition of GSCs by upregulation of Krüppel-like factor 13 (KLF13), a tumor suppressor gene, through clofoctol’s targeted binding protein, Upstream of N-ras (UNR). Collectively, these data demonstrate that induction of KLF13 expression suppressed growth of gliomas and provide a potential therapy for gliomas targeting GSCs. Importantly, our results also identify the RNA-binding protein UNR as a drug target.
Keywords: Oncology, Therapeutics
Keywords: Brain cancer, Drug screens, Drug therapy

Introduction
Gliomas are the most common type of primary malignant tumor in the central nervous system. Among these, glioblastoma, or WHO grade IV glioma, accounts for 55% of all cases and is the most aggressive and lethal type (1). Despite advances in conventional treatment, the prognosis of treated glioma currently remains very poor (2). The outlook for this devastating disease remains bleak, and the reasons for this are diverse. It is partly because of the diffuse and infiltrative nature of the tumor, which limits the feasibility of surgical removal (3), the complex heterogeneity at the genomic and molecular levels (4), and genomic instability (5), which results in difficulty in choosing targeted molecular therapies. It is also partly because of the existence of glioma stem cells (GSCs), which are believed to be responsible for the rapid tumor recurrence after surgery, radiation, and chemotherapy (6).
GSCs were first reported in 2003 (7) and were further confirmed in 2012 (8). They are small populations of cells in glioma tumor samples that possess characteristics associated with stem/progenitor cells, such as the capacity for self-renewal, the expression of stem cell–related markers, and multilineage differentiation capacity, indicating that GSCs can give rise to proliferating progenitor-like and more differentiated tumor cells found in the tumor mass (9). Thus, GSCs have been considered the cellular origin of gliomas. Moreover, researchers found that the growth of glioblastoma was consistent with a markedly neutral process referring to a conserved proliferative hierarchy rooted in GSCs (10). As related research has progressed, it has been demonstrated that GSCs also play important roles in angiogenesis, hypoxia response, invasion, and, especially, therapeutic resistance of glioma after standard radiotherapy and chemotherapy treatment, suggesting that GSCs escape elimination and become the basis for rapid tumor recurrence (11). Temozolomide (TMZ), an alkylating agent regarded as the standard treatment since its discovery (12), can trigger DNA damage mainly by methylating the O6 position of guanine through its intermediate product, 5,3-(methyl)-1-(triazen-1-yl) imidazole-4-carboxamide (MTIC) (13). This represents an ideal target for the most common DNA repair pathways. Controversially, higher expression levels of O6-methyl-guanine-DNA-methyltransferase (MGMT), the key repair enzyme for TMZ-induced DNA damage, have increasingly been reported in GSCs compared with non-stem cells. That allows GSCs to benefit from enhanced DNA repair based on MGMT expression. High activity of DNA damage responses in GSCs is proposed to underlie persistence in tumors and make GSCs a distinct population that causes relapse and metastasis by giving rise to new tumors (14, 15). Therefore, development of specific therapies targeted at GSCs holds promise for improvement of survival and quality of life in glioma patients. Researchers have already made efforts to identify these kinds of compounds, particularly with old drugs used clinically for other diseases, e.g., metformin (16) and niclosamide (17). Small-molecule compounds, such as LLP-3 (18), and inhibitors of dopamine receptor D4, such as PNU 96145E and L-741,742 (19), have also been reported to affect GSCs’ growth. However, to date, an optimal drug that can be used to clinically target GSCs has not yet been approved.
In this study, using unbiased drug screening, we have identified an old antibiotic, clofoctol, as a potential anticancer drug targeting GSCs in vitro and in vivo. Moreover, gene expression changes after drug exposure and the identification of a targeted binding protein by drug affinity responsive target stability (DARTS) revealed that the impairment of GSCs was associated with upregulation of Krüppel-like factor 13 (KLF13) mediated by its binding protein, Upstream of N-ras (UNR).
Results
A high-throughput screening identified clofoctol as an inhibitor of GSCs.
Isocitrate dehydrogenase (IDH) mutation status is a strong prognostic marker of gliomas. It was originally discovered that IDH1 and IDH2 mutation is present in about 80% of low-grade gliomas, while the wild type of IDH is more frequent in high-grade gliomas and is believed to be associated with poor patient survival (20). In this study, for improvement of therapy of gliomas, we first used the 4 previously established and identified GSCs (GSC2, GSC5, U87MG SLC, and U251 SLC; refs. 21, 22) with wild-type status of IDH1 and IDH2 as the cell model for primary drug screening. Genotype analysis of the 4 GSCs showed that they have almost the same status of other mutations commonly seen in human gliomas, such as TP53, EGFR, α-thalassemia/mental retardation syndrome X-linked (ATRX), and promoter of the telomerase reverse transcriptase (TERT) (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI124979DS1).
For screening, we tested 1920 compounds from the MicroSource Spectrum Collection, comprising 1163 drugs used clinically, 448 compounds with undetermined activity, 287 compounds in experimental stages, and 22 compounds with agricultural uses (Figure 1A). The screening was performed using MTS assay to determine relative cell viability 48 hours after treatment with 10-μM concentrations of a single application in the 4 GSCs. Every compound that reduced the mean cell viability of the 4 GSCs to less than 70% of control levels was considered as a “hit.” Seventy-one compounds fulfilled this criterion (Figure 1B). Since efficacy and safety are the basis for transformation of compounds to clinical drugs, we then measured compounds’ efficacy by treating GSCs at a lower concentration (1 μM) and evaluated the relative safety of compounds by measuring the relative cell viability in 3 normal human cell lines (293ET, ReNcell-CX, and H1-NSC) at 10 μM. Thirteen compounds were ultimately selected that showed potential GSC-specific inhibition without similarly affecting normal control cells (Figure 1, B and C, and Supplemental Table 2).
Figure 1. High-throughput screening to identify inhibitors of GSCs.

(A) Compound classification of the MicroSource Spectrum Collection. (B) Schematic representation of the screening process. (C) All 13 compounds used to treat the 4 GSCs for 48 hours with a relative cell viability (vs. control) less than 70%. Data are presented as the mean ± SEM, and samples were assayed in triplicate. (D) Clofoctol concentration in mouse brain following the last treatment of vehicle and clofoctol (10 mg/kg, daily) i.v. for 13 days. Control, n = 4; 5 minutes, n = 3; 15 minutes, n = 4; 45 minutes, n = 4; 90 minutes, n = 4. Data are presented as the mean ± SEM. (E) Selection criteria for compound 12 (clofoctol) as the primary lead compound. In the “IC50” row, “+” indicates that the IC50 values in the 4 GSCs were less than those in normal human cells; “–” indicates that they were not. In “Coculture assay,” “++” and “+” indicate that compounds could selectively inhibit GSC2-GFP and U87MG SLC–GFP cells; “–” indicates not. In “Safety in zebrafish,” “+++,” “++,” and “+” indicate that compounds had almost no, lower, and minor toxicity; “–” indicates that compounds had strong toxicity. In “In vivo effects in zebrafish,” “+” indicates that with compound treatment, the tumors in the zebrafish xenograft model decreased; “–” indicates that this did not occur. In “BBB,” “+” indicates that compounds were predicted to pass through the BBB; “–” indicates that they were not; and “U” indicates undetermined.
Importantly, a variety of strategies could be used to prioritize hit compounds for follow-up experiments. Here, we opted for an array involving 5 criteria. IC50 values detected in different cell lines confirmed that, with the exception of compounds 8 and 10, the selected compounds could specifically inhibit the viability of GSCs (Supplemental Table 2). GSCs and a control normal human astrocyte cell line, HA, labeled with GFP or RFP by lentivirus infection, respectively, and selected by FACS of cocultures, were further used to determine and confirm the compound selectivity. Compounds 7, 8, 10, 12, and 13 showed specificity in 2 coculture models, GSC2-GFP+HA-RFP and U87MG SLC–GFP+HA-RFP, which indicated that these compounds inhibit cell viability of GSCs in association with relatively lower toxicity in normal cells (Supplemental Figure 1, A and B). To further measure the toxicity, 13 hits were administered to egg water of zebrafish embryos at the 1- to 2-cell stage and 4 days after fertilization; only compounds 6 and 12 showed relatively lower toxicities and had no impact on zebrafish development, embryonic death, malformation, or yolk edema (Supplemental Figure 2A). We then examined the in vivo efficacy of the compounds in a zebrafish glioma model, and found that compounds 5, 7, 8, 10, 12, and 13 could markedly inhibit tumor growth (Figure Supplemental 2B). As gliomas are intracranial, the blood-brain barrier (BBB) constitutes a specific hurdle for drug delivery. The ability to cross this barrier was predicted by ADMET (absorption, distribution, metabolism, excretion, and toxicity) in silico (Supplemental Table 3). Only compound 7 (ivermectin) was excluded based on its nearly total inability to penetrate the BBB. Permeability of compounds 10, 12, and 13 cannot be defined by ADMET. Since compounds 10 and13 were excluded by high toxicities in normal human cells or in zebrafish development, we then only tested compound 12 concentration in mouse brains and found 152–187 ng/g of tissue weight at 5–90 minutes after the last i.v. administration of 10 mg/kg body weight of compound 12 (Figure 1D). This shows that compound 12 is permeable in crossing the BBB. Based on the collective analysis of the above experiments, further studies focused only on compound 12 (clofoctol) (Figure 1E).
As previously reported, clofoctol is an antibiotic drug that has been used for the treatment of upper respiratory tract infections in France and Italy for decades (23). The proposed mechanism of its antibacterial activity is the alteration of bacterial membrane permeability, owing to its hydrophobic nature (24). More recently, it was also reported that clofoctol inhibited protein translation via active unfolded protein response pathways, leading to the induction of G1 cell cycle arrest in prostate cancer. Thus, clofoctol could potentially serve as an anticancer drug (25). Here, we report its anticancer activity in gliomas for the first time to our knowledge.
Clofoctol can inhibit GSCs in vitro and in vivo.
To validate the selective inhibition of GSCs, pharmacodynamic analysis of clofoctol was performed. Four GSC cell lines and 3 normal human cell lines — H1-NSC (26), 293ET, and HA — were investigated. The obtained dose-response curves showed consistent courses for all cell lines, and respective IC50 values are listed. We found that, compared with normal human cells, clofoctol could selectively inhibit GSCs (Figure 2A). The selective inhibition was also demonstrated by recording of the cell death process in a coculture model, GSC2-GFP+HA-RFP treated with clofoctol. The coculture model was treated with DMSO or clofoctol at 3 μM. With clofoctol treatment, the intensity of GFP fluorescence declined rapidly and drastically, whereas the RFP fluorescence did not (Supplemental Video 1). Furthermore, continuous monitoring and endpoint cell viability analysis after exposure to the compound and TMZ revealed that clofoctol could inhibit GSCs, whereas GSCs were TMZ-resistant (Figure 2B). We then tested the therapeutic outcome of clofoctol combined with TMZ in GSCs, and measurement of combination index (CI) values (CI < 1) indicated the synergistic effect between these 2 drugs (Figure 2C). The results revealed the potential of clofoctol to improve chemotherapy for patients with gliomas. Meanwhile, we found that after treatment with clofoctol the related number of tumorspheres was also obviously decreased (Figure 2D). Moreover, to further estimate the potential alterations in colony formation that clofoctol induced in GSCs, we pretreated GSCs with clofoctol for 6 hours. Then, the same number of living cells was collected for assessment of the inhibition of colony formation ability of GSCs after withdrawal of clofoctol. The results showed that the clonogenic potential was decreased; strikingly, tumorsphere formation was completely abolished after pretreatment with 3 μM and 10 μM clofoctol, and no living cells could be collected in GSC2 and GSC5 (Figure 2, E and F, and Supplemental Figure 3, A and C). To further evaluate the effects of clofoctol on GSCs, tertiary tumorsphere assays were then used after dissociation of secondary tumorspheres. Quantification of related tumorspheres indicated that a single application of the compound could still reduce the frequencies of colony formation even after a second passage (Figure 2G and Supplemental Figure 3, B and D).
Figure 2. Clofoctol inhibits GSCs in vitro and in vivo.

(A) Clofoctol was tested for effects on the cell viability of GSCs (red) and normal human cells (black). Data are representative of 3 wells for more than 3 times. (B) Time course of clofoctol and TMZ treatments in GSCs. (C) Sequential treatment of clofoctol plus TMZ in GSCs (48 hours, n = 3). CI was calculated by CalcuSyn demo version 2.0 software (ComboSyn, Inc). (D) Analysis of tumorsphere formation of GSC2 after clofoctol treatment. (E and F) Limiting dilution assay (E) (n = 10 for each group) and secondary tumorsphere assay (F) of GSC2 cells after pretreatment with clofoctol for 6 hours. (G) Third tumorsphere assay of GSC2 cells collected from second tumorspheres after pretreatment with clofoctol for 6 hours. (H) GSC2 cells were implanted s.c. into nude mice. The mice were treated with vehicle (n = 8) or clofoctol (20 mg/kg) (n = 7) once daily via i.p. injection for 11 days. The tumor volume was calculated using a modified ellipsoid formula. (I) In vivo bioluminescent image (left) and quantitative analysis (right) of GSC2-derived xenografts in mice treated with vehicle and clofoctol (10 mg/kg, daily) i.v. for 13 days from day 7. (J) GSC2 cells were implanted intracranially into nude mice. Mice were treated with vehicle or 10 mg/kg clofoctol i.v. daily for 13 days, and the relative survival curves (control, n = 8; clofoctol-treated, n = 8) are shown. Data are presented as the mean ± SEM. For A, B, D, and F–H, samples were assayed in triplicate. Data in D, F, and G were analyzed by ANOVA. Two-tailed Student’s t test was used in H and I. Mantel-Cox test was used in J. *P < 0.05, **P < 0.01, ***P < 0.001.
Xenograft models of gliomas were used to further determine the in vivo anticancer activity of clofoctol. For the subcutaneous xenograft, GSC2 cells were implanted s.c. into nude mice. Then, mice were treated with vehicle or clofoctol by i.p. administration for 11 days after the tumor volume reached approximately 100 mm3. Tumor volume was measured during the course of the treatment. As a result, tumor growth was significantly inhibited by clofoctol (Figure 2H). The ability of clofoctol to attenuate tumor progression was next examined in an intracranial xenograft model. Here, nude mice received intracranial injections of Luc-GSC2 stable cells, and the resulting tumor was allowed to develop for 7 days before clofoctol or vehicle was administered intravenously. Quantitation of the results from 3 mice in each group confirmed that clofoctol markedly suppressed tumor growth by bioluminescence imaging (Figure 2I) and the related survival of mice with clofoctol treatment was also extended (Figure 2J). Moreover, we established primary patient-derived xenografts (PDXs) by transplanting tumor specimens (Supplemental Table 4) subcutaneously into nude mice and treated them with clofoctol. Consistently, we observed that clofoctol treatment exhibited significant reduction in tumor growth (Figure 3A). Further, transgenic mouse glioblastoma models created by constitutive expression of active H-RasV12 and silencing of p53, previously reported by Niola and colleagues (27, 28), were used to evaluate antitumor effects of clofoctol. After 12 days of injection, MRI analysis was used for grouping, and then mice were treated with clofoctol (10 mg/kg, i.v.) for 10 days. Similarly, clofoctol obviously inhibited growth of glioblastoma (Figure 3, B and C). Immunohistochemical staining of representative tumor sections from the clofoctol-treated group expressed less of the GSC marker CD133 both in PDXs (Supplemental Figure 4, left) and in transgenic mouse models (Figure 3D), indicating that clofoctol inhibited GSCs in vivo. Thus, by effect evaluation by multiple xenograft models, clofoctol appeared to be a promising candidate for glioma therapy.
Figure 3. Clofoctol inhibits growth of gliomas in vivo in PDXs and transgenic xenografts.

(A) Tumor volumes of PDXs over time. The mice were treated with 20 mg/kg clofoctol or vehicle by i.p. administration. PDX1, clofoctol (n = 5), vehicle (n = 5); PDX2, clofoctol (n = 9), vehicle (n = 9); PDX3, clofoctol (n = 9), vehicle (n = 9). Data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control by 2-tailed Student’s t test at each time point. (B and C) Glioblastomas were induced by injection with pTomo-Ras-sip53 lentivirus in C57BL/6 mice. MRI analysis after 12 days of injection was used for grouping (C, left), and then drug effects were valued after treatment with vehicle or 10 mg/kg clofoctol i.v. daily for 10 days (C, right), and MRI graphs of 3 samples for each group are shown (B). *P < 0.05 compared with control by 2-tailed Student’s t test. (D) Immunohistochemical analysis of sections from tumors induced by injection with pTomo-Ras-sip53 lentivirus after clofoctol treatment stained with antibody against CD133. Three individual samples were analyzed. Scale bars: 100 μm.
Gene expression profiling predicts a role for KLF13 in clofoctol-mediated inhibition.
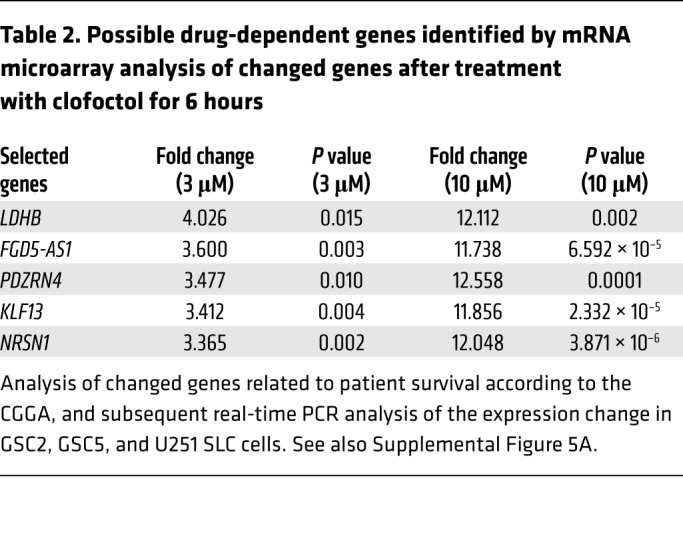
To address the mechanisms by which clofoctol suppresses growth of GSCs, we performed gene expression profiling using mRNA microarray analysis. GSC2 cells were incubated with 1% DMSO or clofoctol at 3 μM and 10 μM for 6 hours before RNA was isolated. Unsupervised hierarchical clustering revealed a high level of consistency across biological replicates (Figure 4A). To gain insight into the pathways regulated by clofoctol, we investigated the changes in genes treated with 10 μM clofoctol versus control using Ingenuity Pathway Analysis (IPA; QIAGEN Inc.; www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/). Diseases and biofunction analysis suggested that clofoctol altered the expression of genes primarily associated with cell apoptosis (Table 1). To confirm this, GSCs were treated with clofoctol or DMSO, and the percentages of annexin V–positive and of annexin V–positive, propidium iodide–positive (PI-positive) apoptotic cells were determined. The results showed that clofoctol indeed induced cell apoptosis (Figure 4, B and C). We further measured increased cleavage of the apoptosis-related proteins PARP and caspase-3 (Figure 4D). Upregulation of cleaved caspase-3 was also detected in the tumor sections from the clofoctol-treated group by immunohistochemical analysis of transgenic mouse models (Figure 4E) and PDXs (Supplemental Figure 4, middle). Then, to find the crucial genes involved in decreasing cellular growth or inducing cell apoptosis after clofoctol treatment, we analyzed differentially expressed genes according to the strategy shown in Supplemental Figure 5A. Seventy-six upregulated genes (P < 0.05, fold change [FC] > 2, FC absolute [10 μM]/FC absolute [3 μM] > 3) and 63 downregulated genes (P < 0.05, fold change > 2) were differentially expressed in both the 3 μM and the 10 μM clofoctol treatment groups. To narrow the range of candidates, we then analyzed the differentially expressed genes that showed a relationship to patient survival, according to the Chinese Glioma Genome Atlas (CGGA) database, followed by real-time PCR analysis of the expression change in 3 glioma stem cell lines, GSC2, GSC5, and U251 SLC (Supplemental Figure 5A). Only 5 upregulated genes were ultimately selected (Table 2). Tumorsphere assays, clofoctol recovery assays, and limited dilution assays were then performed to select and determine the precise functional genes by knockdown of related genes by RNAi. We found that only KLF13 met the criteria (Supplemental Figure 5B).
Figure 4. Expression profiling identifies KLF13 as a target of clofoctol in GSCs.

(A) Unsupervised hierarchical clustering of genes differentially expressed between GSC2 cells treated with 0.1% DMSO and with 3 μM or 10 μM clofoctol for 6 hours (n = 3 for each group). (B and C) Flow cytometry–based quantification of GSC2 (B) and GSC5 (C) cell apoptosis after 6 hours of exposure at different concentrations by annexin V/PI staining. The related quantitative analysis is shown on the right. Data are presented as the mean ± SEM, and samples were assayed in triplicate. ***P < 0.001 compared with the DMSO control by 2-tailed Student’s t test. (D) Western blotting analyzing the cleavage of cell apoptosis proteins (PARP and caspase-3) in GSC2, GSC5, and U251 SLC cells after treatment with clofoctol for 6 hours. Samples were assayed more than 3 times. (E) Immunohistochemical analysis of sections from tumors induced by injection with pTomo-Ras-sip53 lentivirus after clofoctol treatment stained with antibody against cleaved caspase-3. Three individual samples were analyzed. Scale bars: 100 μm.
Table 1. Enriched biofunctions affected by clofoctol treatment by Ingenuity Pathway Analysis.
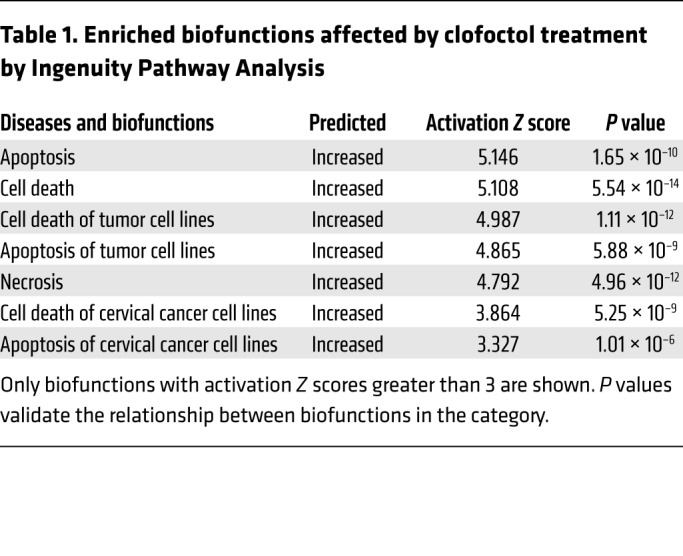
Table 2. Possible drug-dependent genes identified by mRNA microarray analysis of changed genes after treatment with clofoctol for 6 hours.
KLF13 is induced by clofoctol and can inhibit growth of GSCs.
KLF13 is reported to be a DNA-binding transcriptional regulator belonging to the Krüppel-like factor family that can specifically bind to GC-rich sequences and related GT and CACCC boxes (29). It has emerged as an essential regulator of cell proliferation, apoptosis, differentiation, and development (30). In apoptosis, KLF13 is considered a negative regulator of the antiapoptotic protein BCLXL (31) or a positive regulator of proapoptotic proteins, such as BIM (32). We found that the expression of KLF13 was markedly increased at the RNA and protein levels in vitro after clofoctol treatment (Figure 5, A and B), and immunohistochemical analysis also showed KLF13 upregulation in gliomas after clofoctol treatment of PDXs (Supplemental Figure 4, right) and transgenic mouse glioblastoma models (Figure 5C). Consistently, the expression of the apoptosis-related downstream gene BIM was increased after clofoctol treatment (Supplemental Figure 8D). To clarify the role of KLF13 in GSCs after clofoctol treatment, we analyzed KLF13 mRNA expression in data from Gene Expression Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn/index.html) (33) and the CGGA database. The results showed that KLF13 mRNA expression was reduced in gliomas (Figure 5D) and, to some degree, the expression decreased as the glioma grade increased (Supplemental Figure 6A). Similarly, analysis of KLF13 protein expression of 12 grade II, 23 grade III, and 46 grade IV glioma tissues, along with 7 normal brain tissues as normal controls, showed decreased KLF13 protein expression in gliomas (Figure 5E). Results indicated that KLF13 expression was significantly lower in high-grade gliomas than in low-grade gliomas (Supplemental Figure 6B). Most importantly, we found that KLF13 mRNA was aberrantlydownexpressed in IDH1 wild-type gliomas compared with the mutated (Figure 5F). Analysis using the CGGA database and the GEPIA website indicated that the expression of higher than median levels of KLF13 mRNA in glioma samples was correlated with greater patient survival (Supplemental Figure 6, C and D). Then, we further analyzed the relationship between KLF13 mRNA expression and survival in tumors of different IDH1 status and found that higher expression of KLF13 mRNA was relevant to better survival in IDH1-wild-type gliomas, while KLF13 mRNA expression was uncorrelated with patient survival in IDH1-mutated gliomas, which have been proven to be associated with significantly better patient survival (Figure 5G). These findings indicate that loss of KLF13 is associated with tumorigenesis, maintenance of gliomas, and poor prognosis, especially in IDH1-wild-type gliomas. Thus, upregulation of KLF13 by clofoctol may explain clofoctol’s inhibition of GSCs.
Figure 5. KLF13 is induced by clofoctol, and aberrant downregulation of KLF13 is connected with tumorigenesis, maintenance of gliomas, and poor prognosis.

(A and B) GSCs were treated with 0.1% DMSO or 3 μM or 10 μM clofoctol for 6 hours. The mRNA expression of KLF13 was determined by quantitative reverse transcriptase PCR in GSC2 (A, n = 3), and KLF13 protein expression was determined by Western blotting in GSC2 and GSC5 (B). ***P < 0.001 compared with control by ANOVA. (C) Immunohistochemical analysis of sections from tumors induced by injection with pTomo-Ras-sip53 lentivirus after clofoctol treatment stained with antibody against KLF13. Three individual samples were analyzed. Scale bars: 100 μm. (D) Expression analysis of KLF13 by the Web-based tool GEPIA. T, tumor sample; N, normal sample; LGG, lower-grade glioma; GBM, glioblastoma multiforme. Number of N is 207; number of LGG is 518; and number of GBM is 163. (E) Relative KLF13 protein levels in 7 control brain tissues and in 81 glioma tissues; β-actin was used as a loading control. See also Supplemental Figure 6B. (F) Expression analysis of KLF13 mRNA in IDH1-mutant (n = 443 in TCGA, n = 152 in CGGA) versus IDH1-wild-type (n = 246 in TCGA, n = 148 in CGGA) tumors using The Cancer Genome Atlas (TCGA) and CGGA databases. (G) Analysis of survival based on KLF13 expression in IDH1-wild-type and IDH1-mutant gliomas by TCGA and CGGA databases. Mantel-Cox test was used. Data are present as the mean ± SEM. Two-tailed Student’s t test was used in D–F. *P <0.05, ***P <0.001.
To further confirm that clofoctol induced inhibition of GSCs by increasing KLF13 expression, we overexpressed KLF13 by infecting GSCs with a related lentivirus (Figure 6A); the cell growth of GSCs was subsequently inhibited (Figure 6B). Meanwhile, the cleavage of PARP and caspase-3 increased compared with a control (Supplemental Figure 7A). Consistently, GSCs overexpressing KLF13 had an inhibition of colony formation (Figure 6, C and D, and Supplemental Figure 7B). MTS assay also showed that KLF13-overexpressing GSCs were more sensitive to clofoctol (Supplemental Figure 7C). KLF13 was then knocked down by RNAi on the basis of the idea that depleting a key factor in the pathway should render GSCs refractive to clofoctol-induced inhibition. Abrogation of KLF13 activity by 2 independent siRNA treatments at the RNA and protein levels (Figure 6E) led to a marked increase in the colony formation ability (Figure 6, F–H, and Supplemental Figure 7, D–F). Importantly, the residual growth of KLF13-knockdown cells was less sensitive to clofoctol treatment than that of control cells, in accord with increase of IC50 value of clofoctol-treated GSC2 cells (Figure 6I) as well as decrease of inhibition of percentage of cell viability in GSC5 and U251 SLC cells (Supplemental Figure 7G). Besides, at a single dose of clofoctol treatment, the relative cell viability of the KLF13-suppressed group was higher than that of a control group at a specific time point (Supplemental Figure 7H). Consistently, this recovery of clofoctol-induced effects by siRNA treatment was also demonstrated by the percentages of annexin V–positive and of annexin V–positive, PI-positive apoptotic cells following treatment at different concentrations (Figure 6, J and K, and Supplemental Figure 7, I and J). Taken together, gain-of-function and loss-of-function experiments indicated that KLF13 was required for the inhibitory effects of clofoctol on GSCs.
Figure 6. KLF13 is induced by clofoctol and can inhibit growth of GSCs.

(A) Western blotting analysis of KLF13 overexpression by related lentivirus. (B) Control [LV5 (EF-1aF/GFP&Puro)] or KLF13-LV-overexpressing [LV5 (EF-1aF/GFP&Puro)-KLF13] GSC2 cells were sorted and plated at 10,000 cells per 100 μL. The relative cell viability was detected at 0, 24, 36, 48, and 72 hours by MTS. Samples were assayed in triplicate. (C) Control-LV or KLF13-LV-overexpressing GSC2 cells were sorted, and 5000 cells per 100 μL were plated. The number of tumorspheres was calculated 5 days later. Data are representative of 3 wells. (D) Limiting dilution tumorsphere assay in control-LV or KLF13-LV-overexpressing GSC2 cells. Ten wells were analyzed for each group. (E) Knockdown of KLF13 was successful at the RNA and protein levels. (F–H) Colony-forming ability was increased by KLF13 knockdown in GSC2 (F), GSC5 (G), and U251 SLC (H) cells. Samples were assayed in triplicate. (I) GSC2 cells transfected with KLF13-si1 and KLF13-si2 were treated with the indicated concentrations of clofoctol for 48 hours, and viability was measured by MTS. IC50 values for each condition are shown, and samples were assayed in triplicate. (J and K) Flow cytometry–based quantification of the apoptosis of GSC2 (J) and GSC5 (K) cells with or without KLF13-siRNA transfection after 6 hours of exposure to different concentrations of clofoctol (0 μM, 3 μM, 10 μM) by annexin V/PI staining. The related quantitative analysis is shown in Supplemental Figure 7, I and J. Data are presented as the mean ± SEM. For B and C, 2-tailed Student’s t test was used. For E–H, ANOVA with Bonferroni multiple-comparisons test was used. *P <0.05, **P < 0.01, ***P <0.001.
Clofoctol increased KLF13 expression via specific binding to UNR.
To further elucidate the mechanism by which clofoctol increased KLF13 expression in GSCs, we took advantage of a newly developed method, termed DARTS (drug affinity responsive target stability), for targeting protein identification without requiring modification or immobilization of a small molecule (34). GSC2 cells were used as the protein source for DARTS, and this analysis revealed a robust, protected band approximately 95 kDa in size in the proteolyzed extracts of clofoctol-treated lysates (Supplemental Figure 8A). The protected band and the matching gel region of the control lane were then examined by mass spectrometry, and we ultimately identified UNR, also called CSDE1 (cold shock domain containing E1), as the primary protein present at higher abundance in the clofoctol-treated samples (Supplemental Table 5). The clofoctol-dependent proteolytic protection of UNR was also observed by immunoblotting of clofoctol-treated lysates (Figure 7A). We then confirmed the binding of UNR protein with clofoctol by isothermal titration calorimetry (ITC) experiments. Calorimetric titration of fusion protein GST-UNR into a solution of clofoctol showed exothermic binding with a dissociation constant (KD) of 69 μM (<100 μM), while there was almost no binding between clofoctol and the control protein GST, with a KD of 282 μM (>100 μM) (Figure 7B and Supplemental Figure 8, B and C).
Figure 7. Clofoctol can specifically bind UNR and regulate KLF13, thus inducing GSC inhibition.

(A) Western blot showing protection of the UNR protein from pronase digestion upon clofoctol binding in the DARTS assay. Samples of more than 3 individual experiments were assayed. (B) ITC binding curves for GST-UNR with clofoctol. The binding of clofoctol with GST is shown in Supplemental Figure 7D. (C) Knockdown of UNR could decrease the expression of KLF13 at the RNA and protein levels. (D) Limiting dilution tumorsphere assay in control or UNR-knockdown GSC2 cells. Ten wells were analyzed for each group. (E) Percent cell viability inhibition of GSC2 cells transiently transfected with UNR-si3 and UNR-si4 treated with clofoctol for 48 hours. (F) RIP assay analysis of the binding between UNR and KLF13 mRNA. PABPC1 is the positive control. (G) Clofoctol treatment (3 μM) increased the binding between UNR and KLF13 mRNA. (H and I) KLF13 mRNA stability assay in GSC2 cells without (H) or with (I) UNR knockdown. (J and K) Analysis of the expression of KLF13 mRNA (J) and the expression of UNR and KLF13 at the protein level (K) under clofoctol treatment with or without UNR knockdown. Data are presented as the mean ± SEM. For C, E–G, and J, 2-tailed Student’s t test was used. *P < 0.05, **P < 0.01, ***P < 0.001.
UNR is a highly conserved RNA-binding protein that functions in the post-transcriptional control of gene expression by regulating mRNA translation and stability (35, 36). It is essential for mammalian development and is an important regulator of a number of cellular processes, such as differentiation, mitosis, cell cycle, apoptosis, and cancer progression (37–39). Here, we evaluated the potential role of UNR in the regulation of KLF13 as a clofoctol-binding protein.
Four related siRNAs were designed for UNR knockdown, although only siRNA3 and siRNA4 functioned successfully (Figure 7C and Supplemental Figure 8E). We found that knockdown of UNR could markedly downregulate the expression of KLF13 (Figure 7C). Concomitantly, UNR abrogation resulted in an increase in colony formation of GSCs and a rescue of inhibition after treatment with clofoctol at 10 μM, as observed with KLF13 knockdown (Figure 7, D and E). As an RNA-binding protein, the binding motif of UNR has been identified by discriminative regular expression motif elicitation (DREME) analysis of iCLIP enrichment of targets (39). There were 7 possible binding sites of UNR in KLF13 mRNA, indicating the possibility that UNR binds to KLF13 mRNA directly (Supplemental Figure 8F). This was then demonstrated by RNA immunoprecipitation (RIP) analysis (Figure 7F). Moreover, increased binding between UNR and KLF13 mRNA was observed after clofoctol treatment (Figure 7G). We also found that the increased binding between them could lead to an increase of KLF13 mRNA’s stability (Figure 7, H and I). KLF13 mRNA’s half-life analysis showed that clofoctol treatment could decrease the rate of KLF13 mRNA decay. Conversely, the decay rate of KLF13 mRNA was accelerated after UNR knockdown, and, importantly, this kind of acceleration was just reversed slightly by clofoctol treatment. Meanwhile, we found that after knockdown of UNR in GSC2 cells, the expression of KLF13 mRNA had no obvious increase after treatment with clofoctol (Figure 7J). Consistently, results also showed that knockdown of UNR in GSC2 cells could block the upregulation of KLF13 after exposure to clofoctol at the protein level (Figure 7K). We therefore concluded that clofoctol substantially selectively inhibits GSCs, thus impairing glioma progression, by overexpressing the apoptosis-related gene KLF13 through its targeted binding protein UNR.
Discussion
Despite aggressive multimodal therapies, the prognosis of glioma patients remains very poor. This makes identifying novel effective therapies extremely urgent and important. Currently, GSCs have been shown to be significant prognostic factors for shorter overall survival and reduced progression-free survival (40).
In this study, by using IDH1-wild-type GSCs as a cell model, we performed a high-throughput drug screening offering tremendous promise for the rational development of targeted therapies. The results of our study showed that, among the 1920 compounds initially investigated, clofoctol, an old drug for the treatment of mild upper respiratory tract infections, especially in pediatric patients (23, 41, 42), could selectively inhibit the cell viability of GSCs compared with glioma cells and normal human cells. The IC50 value was 4.98–12.17 μM in GSCs, compared with 18.93 μM in normal human 293ET cells. Although Han and colleagues reported that clofoctol reduced ATP concentrations with more potency for NSCs derived from human embryonic stem cells (hESCs) (H9 or I6) than for hESCs (43), pharmacodynamic analysis of clofoctol in our work showed less inhibition of another NSC cell line (H1-derived NSC [H1-NSC]) (26) with an IC50 value of 1084.00 μM (Figure 2A). Clofoctol also selectively targeted GSCs in mixed cocultures with human astrocyte cells. The small molecular weight and hydrophobic nature give clofoctol high permeability to penetrate the BBB. As an antibacterial drug, it is well absorbed after a single rectal dose of 1.5 g, with an average AUC value of 112.78 μmol/L/h in plasma after administration, which is sufficient for clofoctol to inhibit the growth of GSCs (44). Moreover, with clofoctol treatment, growth of gliomas was markedly inhibited in vivo in xenograft models, especially in PDXs and transgenic mouse models. Thus, the anticancer effects of clofoctol that we identified provide promising clues for the development of targeted glioma therapy.
KLF13 was shown to be the crucial functional gene regulated by clofoctol. As reported, KLF13 is a proapoptotic factor. It induces cell apoptosis by acting as a negative regulator for the antiapoptotic protein BCLXL (31) or as a positive regulator for proapoptotic proteins, such as BIM (32). Our studies further indicated that the levels of KLF13 mRNA and protein were markedly increased by clofoctol, and the overexpression of its apoptosis-related downstream gene, BIM, at the RNA level was also observed. Furthermore, we found that knockdown of KLF13 blunted GSC responses to clofoctol, and overexpression of KLF13 inhibited GSCs, much like the effects observed after exposure to clofoctol. These results further highlight the importance of KLF13 in mediating the inhibitory effects of clofoctol. Additional results showed that clofoctol could inhibit the ability of GSCs to undergo tumorsphere formation. Partly, the results reflect that clofoctol might also inhibit the self-renewal ability of GSCs besides induction of apoptosis. These effects might also be suggested by the overexpression of KLF13 after treatment, as KLF13 has reportedly shown activity in the regulation of differentiation of different cells (45, 46).
For further analysis of the mechanism by which clofoctol increased KLF13 expression in GSCs, we used the DARTS method; we found that clofoctol bound to UNR, which could regulate the expression of KLF13. For further confirmation, ITC assay was used, and we found that clofoctol could directly bind UNR protein. UNR has been reported to be a highly conserved RNA-binding protein that functions in post-transcriptional control of gene expression by regulating mRNA translation and stability, which makes it important in many cellular processes. Wurth and colleagues first revealed that it could promote melanoma migration and metastasis. They also found its specific binding motif (39). Moreover, Fishbein and colleagues identified UNR as a somatically mutated driver gene correlating with poor clinical outcomes in neuroendocrine tumors, pheochromocytomas and paragangliomas (47). Here, we reveal the role of UNR in gliomas for the first time to our knowledge. When UNR was knocked down, the expression of KLF13 was markedly decreased. Consistently, UNR knockdown blocked the overexpression of KLF13 after exposure to clofoctol and resulted in recovery from clofoctol drug effects on GSCs, which was similar to the effects of KLF13 knockdown. We performed RIP analysis, and showed that UNR could bind to KLF13 mRNA. Moreover, the binding of UNR and KLF13 mRNA increased after treatment with clofoctol, and the increased binding between them could lead to an increase of KLF13 mRNA’s stability. No doubt the specific mechanism by which UNR regulates KLF13 requires further exploration, which we are analyzing as part of ongoing research. We propose a collective hypothesis regarding the mechanism of clofoctol. Clofoctol can specifically bind the RNA-binding protein UNR, which could increase the binding of UNR and KLF13 mRNA. This enhancement of binding could then result in increased KLF13 expression (at the RNA and protein levels) by increasing the stability of KLF13 mRNA, which induces inhibitory effects in GSCs.
In summary, our results indicate a new therapeutic option, clofoctol, for glioma chemotherapy and reveal its anticancer mechanisms by identifying a targeted binding protein and a related functional gene. These results demonstrate that the activation of KLF13 by related compounds in gliomas is connected to depletion of GSCs, attenuation of disease progression, and prolonged survival in a glioma animal model. This suggests a new strategy of using KLF13 activators for glioma therapy.
Methods
Cells and cell culture.
Four GSCs were established and identified by our previous work, and were cultured in serum-free stem cell medium (SFM) (Neurobasal, Gibco, Thermo Fisher Scientific) containing 20 ng/mL basic fibroblast growth factor (bFGF; catalog 100-18B-50UG, PeproTech), 20 ng/mL epidermal growth factor (EGF; catalog AF-100-15-100UG, PeproTech), 10 μg/mL heparin (catalog 9041-08-1, Sigma-Aldrich), 2% B27 (Gibco, Thermo Fihser Scientific), and 2 mmol/L l-glutamine (catalog SH30034.01, HyClone). The induced human neural stem cells, H1-NSCs obtained from D.Q. Pei’s laboratory (Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, China) and ReNcell-CX cells (ATCC), were cultured in medium as for the GSCs with the extra addition of 1% N-2 supplement (Gibco, Thermo Fisher Scientific). We purchased a normal human astrocyte cell line (HA) from ScienCell and cultured it using commercial astrocyte medium (catalog 1801, ScienCell) supplemented with 2% FBS (ScienCell), 1% AGS (ScienCell), and 1% P/S solution (ScienCell).
Coculture assay.
For coculture assay, GSC2-GFP, U87MG SLC–GFP (Hu6-MCS-ubiquitin-Egfp-IRES-puromycin, GeneChem), and the human astrocyte cell HA-RFP (Hu6-MCS-CMV-RFP, GeneChem) were labeled with GFP or RFP by lentivirus infection. Selection of the labeled cells was performed by flow cytometry. GSCs and HA cells were dissociated into single cells, mixed at the ratio of 1000:1000, and then suspended as hanging drops in 50 μL culture media with DMSO or one of the 13 hits at 1 μM and 10 μM. Images were taken at 48 hours.
Xenografts.
AB zebrafish were raised and managed in the Department of Genetics, National Research Institute for Family Planning, according to the guidelines for zebrafish cultivation (48). A zebrafish xenograft glioma model was used to evaluate the effects of compounds in vivo. Anesthetized zebrafish at 48 hours after fertilization were embedded on an agarose platform under anesthesia with tricaine in egg water. Three thousand U87MG SLC cells labeled with GFP by lentivirus infection were injected into the yolks of each zebrafish. After development for 2 days, 3 zebrafish were transferred into each well of 6-well plates containing 5 mL egg water with or without compounds. Drug effects were assessed by the intensity of GFP fluorescence after 48 hours of treatment.
Six- to 8-week-old male BALB/c nude (Weitong Lihua) mice were used to determine the drug effects of clofoctol in vivo. For intracranial xenograft models, 1 × 105 Luc-GSC2 stable cells were collected, resuspended in PBS, and injected into mouse frontal cortexes (coordinates were 1 mm rostral to bregma, 1.5 mm lateral to the midline, and 2.7 mm deep) using a Hamilton microsyringe (10 μl). Tumors were allowed to develop for 7 days before being treated with vehicle or 10 mg/kg clofoctol i.v. daily for 13 days. Tumor size was quantified by bioluminescence imaging, and the survival was recorded and analyzed after all mice died. For subcutaneous xenograft models, GSC2 cells (105 cells per mouse) were implanted s.c. into the left flanks of nude mice. Clofoctol (20 mg/kg daily) in DMSO/physiological saline was given via i.p. injection daily for 11 days after tumors grew to 100 mm3. For PDXs, isometric tumor specimens from patients with Matrigel (catalog 356234, BD Pharmingen) were transplanted s.c. into nude mice and treated. Twenty milligrams per kilogram clofoctol and vehicle were injected by i.p. administration for about 2 weeks. Tumor growth was monitored by measurement of the lengths and widths of tumors. The transgenic glioblastoma mice were purchased from X.D. Zhao’s laboratory (Kunming Institute of Zoology).
Drug permeability assay.
Total brains of mice were collected after the last administration of 13-day clofoctol treatment. The mass spectrometry used a high-resolution accurate mass spectrometer, Thermo Scientific Q Exactive (Thermo Fisher Scientific), coupled with DIONEX UltiMate 3000 UPLC operated in negative ion mode. The capillary temperature was set to 350°C, and capillary voltage was set to 3.5 kV. The sheath and auxiliary gas were set to 40 and 10 units, respectively. The diagnostic product ions were at m/z 363.12879 for clofoctol.
Toxicity analysis by zebrafish.
One- to 2-cell-stage zebrafish embryos were used for analysis of developmental toxicity. Fifty related zebrafish embryos were distributed into each well of 6-well plates in 5 mL of egg water and treated with DMSO as a control or various concentrations of selected compounds for 72 hours with egg water. For toxicity analysis in zebrafish at 96 hours after fertilization, each well of 6-well plates received 8 zebrafish in 5 mL egg water with DMSO or one of the 13 hits at different concentrations. The zebrafish were monitored, and drug effects were recorded every 24 hours in both situations.
Inhibitors.
The MicroSource Spectrum Collection compound library, and clofoctol for further use, were purchased from MicroSource Discovery Inc.
Apoptosis.
Cell apoptosis analysis was performed using a FITC Annexin V Apoptosis Detection Kit I (catalog 55647, BD Pharmingen). GSCs were treated with clofoctol or DMSO. A total of 105 cells were collected after clofoctol exposure and resuspended in 1× binding buffer. Five microliters of FITC Annexin V and 5 μL PI were then added into cell suspension and incubated for 15 minutes at room temperature in the dark. The results were determined by flow cytometry within 1 hour.
Tumorsphere formation assay and limiting dilution assay.
For tumorsphere formation assay, GSCs were dissociated into single cells and diluted to 3000–5000 cells per 100 μL in each well of 96-well plates. The number of tumorspheres with a diameter of at least 50 μm was counted under a microscope after 4–6 days. The limiting dilution assay was performed as described previously (22). In brief, the sphere cells were dissociated into a single-cell suspension and plated in 96-well plates in 100 μL serum-free stem cell medium (SFM) (Neurobasal containing bFGF, EGF, heparin, B27, and l-glutamine). Final cell densities ranged from 200 cells to 1 cell per well in 100-μL volumes. After 10–12 days, the percentage of wells that did not contain spheres (diameter ≤ 50 μm) at each cell plating density was calculated.
Antibodies.
β-Actin (1:3000; catalog A5441, Sigma-Aldrich) was used as internal control. The antibodies used included anti-CD133 (1:1000; catalog PA2049, Boster Bio), anti-KLF13 (1:200; catalog sc-130454, Santa Cruz Biotechnology), anti-UNR (1:1000; catalog A5941, ABclonal), anti–caspase-3 (1:1000; catalog 9665S, Cell Signaling Technology), anti–cleaved caspase-3 (1:500; catalog 9661L, Cell Signaling Technology), and anti-PARP (1:1000; catalog 9542L, Cell Signaling Technology).
mRNA arrays.
Microarray data have been deposited in the Gene Expression Omnibus public database (GEO GSE129047). GSC2 cells were digested with Accutase (catalog A6964, Sigma-Aldrich) and resuspended into 10 mL SFM, with approximately 106 cells per 10-cm2 dish. Cells were treated with 3 μM or 10 μM clofoctol for 6 hours, and collected for mRNA assays in 1 mL TRIzol reagent (Ambion). Total RNA extraction, RNA integrity measurements, total RNA purification, RNA amplification and labeling, and RNA hybridization were performed as previously described (22) by CapitalBio Technology. Expression changes were also analyzed by this company.
DARTS analysis.
The DARTS assay was performed according to the published protocol (49). GSC2 cells were collected, washed with ice-cold PBS, and lysed in M-PER (catalog 78501, Thermo Fisher Scientific) containing 1× protease inhibitor cocktail (catalog 04693124001, Roche) and 1× phosphatase inhibitor cocktail (catalog 04906845001, Roche). The protein concentration of lysate was determined after addition of TNC buffer using the BCA Protein Assay kit (catalog 23227, Pierce). The lysates were distributed into two 1.5-mL tubes and then incubated with DMSO or 100 μM clofoctol for 1 hour at room temperature. After incubation, the mixture was digested using pronase (catalog 10165921001, Roche) at room temperature for 30 minutes. The reaction was stopped by addition of protease inhibitor. Samples were separated by SDS-PAGE and visualized using colloidal Coomassie (catalog 0472, Amresco). A strong protected band with increased staining in the proteolyzed extracts of clofoctol-treated lysates and the related control lane in DMSO lysates were excised and subjected to liquid chromatography–tandem mass spectrometry analysis (Shanghai Applied Protein Technology Inc.). For target verification by Western blotting, DARTS samples were generated as described above, except that the proteolysis was stopped by addition of SDS loading buffer and immediate heating at 95°C for 5 minutes.
ITC experiments.
For the ITC measurements, the cDNA fragments encoding UNR (forward primer, CGCGGATCCATGAGCTTTGATCCAAACCT; reverse primer, CCGCTCGAGTCAGTCAATGACACCAGCTT) were subcloned into the expression vector pGEX4T-1 (Invitrogen) for a GST-UNR fusion protein. Control GST was individually expressed by vector pGEX4T-1. The proteins were purified in the following manner. First the solution was adsorbed to a Glutathione Sepharose High Performance column (17-5279-01, GE Healthcare). After sufficient adsorption, the column was washed with PBS and eluted with the buffer (50 mM Tris-HCl buffer, pH 8.0, containing 10 mM glutathione). GST-UNR (23 μM) and clofoctol (860 μM) were extensively dialyzed into ITC buffer. The ITC experiments were performed at 25°C using an AutoITC200 microcalorimeter (GE MicroCal Inc.), and the binding isotherms were plotted and analyzed by Origin 8 Software (MicroCal Inc.). The ITC experiments using the control GST were performed analogously to the above-mentioned procedure.
Ribonucleoprotein immunoprecipitation analysis.
RNA immunoprecipitation (RIP) analysis was performed according to the protocol provided by Medical & Biological Laboratories Co. Ltd. (MBL) and using Ribocluster Profiler RIP-Assay kit (catalog RN1001, MBL). Protein A agarose beads (Roche), RNase inhibitor (catalog 2313A, Takara), protease inhibitor, 1 mM DTT (Takara), and TransScript First-Strand cDNA Synthesis Super Mix (catalog AT301-02, TransGen) were also used. Related enrichment of mRNAs was detected by quantitative real-time PCR.
mRNA half-life assay.
GSC2 cells were transfected with the control siRNA and UNR siRNA3 for 48 hours. Actinomycin D (catalog 014-21261, Wako) (5 μg/mL) was added with or without clofoctol treatment, and then the total RNA was harvested at different time points for the quantitative analysis of KLF13 mRNA by quantitative real-time PCR analysis. GAPDH was used as the quantitative control. For each group, the amount at 0 hours was set to 100%.
Viruses and siRNAs.
LV5 (EF-1aF/GFP&Puro) KLF13-overexpressing virus and the control virus were produced by GenePharma; virus Hu6-MCS-CMV-RFP and Hu6-MCS-ubiquitin-Egfp-IRES-puromycin were purchased from GeneChem. Targeting sequences of human KLF13 siRNAs were: si1, CCUCAGGUGUCAAAGUAAA; and si2, GCGAGAAAGUUUACGGGAA. Targeting sequences of human UNR siRNAs were: si1, GGACAGAAAUGGUAAAGAA; si2, GAGAUAAGGUUGAAUUUAG; si3, CCAAUAUAGAAGUUCUGUC; and si4, GAGAUGAUGUUGAAUUUGA.
Statistics.
Data are presented as the mean ± SEM. For experiments with 2 comparisons, 2-tailed Student’s t test was used for statistical analysis. For experiments with multiple comparisons, the level of significance was determined using ANOVA with Bonferroni multiple-comparisons test. Kaplan-Meier survival curves in Figure 2J, Figure 5G, and Supplemental Figure 6, C and D, were statistically analyzed by log-rank (Mantel-Cox) test. All analyses were performed using GraphPad Prism 8 (GraphPad Software). P values less than 0.05 were considered statistically significant. For all figures, *P < 0.05, **P < 0.01, ***P < 0.001.
Study approval.
Fresh glioma samples were obtained from the Beijing Tiantan Hospital and the Beijing Sanbo Brain Hospital. Glioma samples were classified according to Histological Grades of Tumors of the Nervous System (3rd edition, WHO, 2000). Informed consent was obtained, and the study was approved by the Medical Ethics Committee of Beijing Tiantan Hospital. All mouse experiments were carried out in accordance with institutional animal guidelines (21, 22, 50) and abided by the declaration of ethical approval for experiments. All animal studies were approved by the IACUC of the Center for Experimental Animal Research (Beijing, China).
Author contributions
YH designed and performed most of the experiments with assistance from MZ, NT, DL, FW, PH, ZW, and LW. YH analyzed data and wrote the manuscript. ZZ provided the drug library for primary drug screening. TJ provided glioma samples. W Hao, JK, BY, JY, and BQ supervised and analyzed the research data. W Han and XP secured funding for the project, conceived the research study, and contributed to the research design. The manuscript was reviewed and edited by all authors.
Supplementary Material
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2016YFC0902500, 2016YFC0902502, 2016YFA0100702), the National Sciences Foundation of China (31671316, 31670789), the CAMS Innovation Fund for Medical Sciences (2016-I2M-1-001, 2016-I2M-2-001, 2016-I2M-1-004, 2017-I2M-2-004, 2017-I2M-3-010, 2017-I2M-1-004), and the Non-profit Central Research Institute Fund of the Chinese Academy of Medical Sciences (2018RC310014). We are grateful to C.L. Lu and R.Q. Ding of the Department of Genetics, National Research Institute for Family Planning, who helped to design and perform zebrafish-related experiments. We thank A.L. Liu of the Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, for ADMET analysis of the 13 selected compounds. We also thank X.D. Zhao of Kunming Institute of Zoology for providing transgenic mouse glioblastoma models and D.Q. Pei of Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, for providing the NSC cell line H1-NSC. Last but not least, we thank H. Jacquemin-Sablon of INSERM, E362, Bordeaux, France, for useful discussions concerning UNR.
Version 1. 05/21/2019
In-Press Preview
Version 2. 06/24/2019
Electronic publication
Version 3. 07/01/2019
Affiliations corrected for Jingting Kang and Jiangang Yuan.
Version 4. 08/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(8):3072–3085.https://doi.org/10.1172/JCI124979.
Contributor Information
Yan Hu, Email: huyancsu@126.com.
Meilian Zhang, Email: 18811152901@163.com.
Ningyu Tian, Email: tianningyu1204@163.com.
Dengke Li, Email: dengkejuanfang@126.com.
Fan Wu, Email: wufan2014@sina.cn.
Peishan Hu, Email: xiaopei296@sina.com.
Zhixing Wang, Email: 1067836615@qq.com.
Liping Wang, Email: 17839949139@163.com.
Wei Hao, Email: haowei588@126.com.
Jingting Kang, Email: kangjingting@ibms.pumc.edu.cn.
Bin Yin, Email: yinbin_pumc@aliyun.com.
Zhi Zheng, Email: zhizheng100@126.com.
Tao Jiang, Email: taojiang1964@163.com.
Jiangang Yuan, Email: yuanjiangang@pumc.edu.cn.
Boqin Qiang, Email: chiangbq@imicams.ac.cn.
Wei Han, Email: hanwei2012@ibms.pumc.edu.cn.
Xiaozhong Peng, Email: pengxiaozhong@pumc.edu.cn.
References
- 1.Legler JM, et al. Cancer surveillance series [corrected]: brain and other central nervous system cancers: recent trends in incidence and mortality. J Natl Cancer Inst. 1999;91(16):1382–1390. doi: 10.1093/jnci/91.16.1382. [DOI] [PubMed] [Google Scholar]
- 2.Delgado-López PD, Corrales-García EM. Survival in glioblastoma: a review on the impact of treatment modalities. Clin Transl Oncol. 2016;18(11):1062–1071. doi: 10.1007/s12094-016-1497-x. [DOI] [PubMed] [Google Scholar]
- 3.Wong ET, Lok E, Swanson KD. An evidence-based review of alternating electric fields therapy for malignant gliomas. Curr Treat Options Oncol. 2015;16(8):40. doi: 10.1007/s11864-015-0353-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snuderl M, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Nicholas MK, Lukas RV, Chmura S, Yamini B, Lesniak M, Pytel P. Molecular heterogeneity in glioblastoma: therapeutic opportunities and challenges. Semin Oncol. 2011;38(2):243–253. doi: 10.1053/j.seminoncol.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Auffinger B, Spencer D, Pytel P, Ahmed AU, Lesniak MS. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev Neurother. 2015;15(7):741–752. doi: 10.1586/14737175.2015.1051968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh SK, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. [PubMed] [Google Scholar]
- 8.Chen J, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, et al. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia. 2015;17(3):239–255. doi: 10.1016/j.neo.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan X, et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature. 2017;549(7671):227–232. doi: 10.1038/nature23666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng L, Bao S, Rich JN. Potential therapeutic implications of cancer stem cells in glioblastoma. Biochem Pharmacol. 2010;80(5):654–665. doi: 10.1016/j.bcp.2010.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevens MF, et al. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987;47(22):5846–5852. [PubMed] [Google Scholar]
- 13.Ochs K, Kaina B. Apoptosis induced by DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and Fas/caspase-8 independent. Cancer Res. 2000;60(20):5815–5824. [PubMed] [Google Scholar]
- 14.Yoshimoto K, et al. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front Oncol. 2012;2:186. doi: 10.3389/fonc.2012.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 16.Würth R, et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: a role for metformin-induced inhibition of Akt. Cell Cycle. 2013;12(1):145–156. doi: 10.4161/cc.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wieland A, et al. Anticancer effects of niclosamide in human glioblastoma. Clin Cancer Res. 2013;19(15):4124–4136. doi: 10.1158/1078-0432.CCR-12-2895. [DOI] [PubMed] [Google Scholar]
- 18.Guvenc H, et al. Impairment of glioma stem cell survival and growth by a novel inhibitor for Survivin-Ran protein complex. Clin Cancer Res. 2013;19(3):631–642. doi: 10.1158/1078-0432.CCR-12-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolma S, et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell. 2016;29(6):859–873. doi: 10.1016/j.ccell.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartmann C, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118(4):469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- 21.Wu F, et al. RhoGDIα suppresses self-renewal and tumorigenesis of glioma stem cells. Oncotarget. 2016;7(38):61619–61629. doi: 10.18632/oncotarget.11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu PS, et al. NSPc1 promotes cancer stem cell self-renewal by repressing the synthesis of all-trans retinoic acid via targeting RDH16 in malignant glioma. Oncogene. 2017;36(33):4706–4718. doi: 10.1038/onc.2017.34. [DOI] [PubMed] [Google Scholar]
- 23.Danesi R, Del Tacca M. Clinical study on the efficacy of clofoctol in the treatment of infectious respiratory diseases. Int J Clin Pharmacol Res. 1985;5(3):175–179. [PubMed] [Google Scholar]
- 24.Yablonsky F, Simonnet G. Action of clofoctol on bacterial cell wall synthesis. J Pharmacol. 1982;13(4):515–524. [PubMed] [Google Scholar]
- 25.Wang M, et al. Identification of an old antibiotic clofoctol as a novel activator of unfolded protein response pathways and an inhibitor of prostate cancer. Br J Pharmacol. 2014;171(19):4478–4489. doi: 10.1111/bph.12800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q, et al. A sequential EMT-MET mechanism drives the differentiation of human embryonic stem cells towards hepatocytes. Nat Commun. 2017;8:15166. doi: 10.1038/ncomms15166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niola F, et al. Mesenchymal high-grade glioma is maintained by the ID-RAP1 axis. J Clin Invest. 2013;123(1):405–417. doi: 10.1172/JCI63811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, et al. Cytotoxic indole alkaloid 3α-acetonyltabersonine induces glioblastoma apoptosis via inhibition of DNA damage repair. Toxins (Basel) 2017;9(5):E150. doi: 10.3390/toxins9050150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scohy S, et al. Identification of KLF13 and KLF14 (SP6), novel members of the SP/XKLF transcription factor family. Genomics. 2000;70(1):93–101. doi: 10.1006/geno.2000.6362. [DOI] [PubMed] [Google Scholar]
- 30.Zhang P, et al. A functional screen for Krüppel-like factors that regulate the human gamma-globin gene through the CACCC promoter element. Blood Cells Mol Dis. 2005;35(2):227–235. doi: 10.1016/j.bcmd.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Zhou M, et al. Kruppel-like transcription factor 13 regulates T lymphocyte survival in vivo. J Immunol. 2007;178(9):5496–5504. doi: 10.4049/jimmunol.178.9.5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jing D, et al. Opposing regulation of BIM and BCL2 controls glucocorticoid-induced apoptosis of pediatric acute lymphoblastic leukemia cells. Blood. 2015;125(2):273–283. doi: 10.1182/blood-2014-05-576470. [DOI] [PubMed] [Google Scholar]
- 33.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lomenick B, Jung G, Wohlschlegel JA, Huang J. Target identification using drug affinity responsive target stability (DARTS) Curr Protoc Chem Biol. 2011;3(4):163–180. doi: 10.1002/9780470559277.ch110180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson EC, Catnaigh PÓ. Regulation of the expression and activity of Unr in mammalian cells. Biochem Soc Trans. 2015;43(6):1241–1246. doi: 10.1042/BST20150165. [DOI] [PubMed] [Google Scholar]
- 36.Mihailovich M, Militti C, Gabaldón T, Gebauer F. Eukaryotic cold shock domain proteins: highly versatile regulators of gene expression. Bioessays. 2010;32(2):109–118. doi: 10.1002/bies.200900122. [DOI] [PubMed] [Google Scholar]
- 37.Tinton SA, Schepens B, Bruynooghe Y, Beyaert R, Cornelis S. Regulation of the cell-cycle-dependent internal ribosome entry site of the PITSLRE protein kinase: roles of Unr (upstream of N-ras) protein and phosphorylated translation initiation factor eIF-2alpha. Biochem J. 2005;385(pt 1):155–163. doi: 10.1042/BJ20040963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dormoy-Raclet V, et al. Unr, a cytoplasmic RNA-binding protein with cold-shock domains, is involved in control of apoptosis in ES and HuH7 cells. Oncogene. 2007;26(18):2595–2605. doi: 10.1038/sj.onc.1210068. [DOI] [PubMed] [Google Scholar]
- 39.Wurth L, et al. UNR/CSDE1 drives a post-transcriptional program to promote melanoma invasion and metastasis. Cancer Cell. 2016;30(5):694–707. doi: 10.1016/j.ccell.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Zeppernick F, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14(1):123–129. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- 41.Del Tacca M, Danesi R, Senesi S, Gasperini M, Mussi A, Angeletti CA. Penetration of clofoctol into human lung. J Antimicrob Chemother. 1987;19(5):679–683. doi: 10.1093/jac/19.5.679. [DOI] [PubMed] [Google Scholar]
- 42.Ghilardi PL, Casani A. Treatment of ear, nose and throat infections with clofoctol. Drugs Exp Clin Res. 1985;11(11):815–818. [PubMed] [Google Scholar]
- 43.Han Y, et al. Identification by automated screening of a small molecule that selectively eliminates neural stem cells derived from hESCs but not dopamine neurons. PLoS One. 2009;4(9):e7155. doi: 10.1371/journal.pone.0007155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Danesi R, Gasperini M, Senesi S, Freer G, Angeletti CA, Del Tacca M. A pharmacokinetic study of clofoctol in human plasma and lung tissue by using a microbiological assay. Drugs Exp Clin Res. 1988;14(1):39–43. [PubMed] [Google Scholar]
- 45.Jiang S, et al. KLF13 promotes porcine adipocyte differentiation through PPARγ activation. Cell Biosci. 2015;5:28. doi: 10.1186/s13578-015-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pabona JM, Zeng Z, Simmen FA, Simmen RC. Functional differentiation of uterine stromal cells involves cross-regulation between bone morphogenetic protein 2 and Kruppel-like factor (KLF) family members KLF9 and KLF13. Endocrinology. 2010;151(7):3396–3406. doi: 10.1210/en.2009-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fishbein L, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31(2):181–193. doi: 10.1016/j.ccell.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li D, et al. Developmental mechanisms of arsenite toxicity in zebrafish (Danio rerio) embryos. Aquat Toxicol. 2009;91(3):229–237. doi: 10.1016/j.aquatox.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 49.Pai MY, et al. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol Biol. 2015;1263:287–298. doi: 10.1007/978-1-4939-2269-7_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carlson BL, Pokorny JL, Schroeder MA, Sarkaria JN. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol. 2011;Chapter 14:Unit 14.16. doi: 10.1002/0471141755.ph1416s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.