

Abstract
An electrokinetically pumped sheath-flow nanoelectrospray interface provides an efficient means of transferring ions from a capillary electrophoresis system into a mass spectrometer. To characterize its performance, we analyzed angiotensin solutions prepared in a background of 0.25 mg/mL of a BSA tryptic digest. Calibration curves were prepared from 10 zeptomole (10−21 mol) to 10 femtomole (10−14 mol) of angiotensin injected into the capillary. A parallel reaction monitoring approach was used; MS1 was set to m/z = 523.8 ± 2, and fragment ion signals at 263.1389 (y2+) and 784.4095 (b6+) were used to generate selected ion electropherograms. Calibration curves based on peak areas were linear (log-log slope of 0.94 for the y2+ fragment and 0.98 for the b6+ fragment). We then injected 1-zmol (600 copies) of angiotensin in the BSA background using a 10-μm ID separation capillary. Triplicate analyses consistently produced co-migrating peaks for the two fragment ions. The b6+ electropherogram showed no background signal, whereas the y2+ electropherogram showed a few noise spikes that were smaller than the peak maximum. Bienayme-Tchebycheff inequality was used to estimate detection limits of 230 ymol (140 ions) injected into the separation capillary. To the best of our knowledge, these electropherograms present the smallest number of molecules detected using mass spectrometry coupled with a separation.
Keywords: Electrospray ionization, capillary zone electrophoresis, zeptomole detection, sub-zeptomole detection limit
Graphical Abstract
1. Introduction
Laser-induced fluorescence produces extraordinarily low detection limits for solution-phase analyte [1]. Incorporation of fluorescence detection for separations produced zeptomole detection in the 1980s and single molecule detection in the 1990s [2,3]. However, fluorescence is a low-information content detector, and mass spectrometry tends to be much more useful detection method for complex samples.
There have been a handful of reports of zeptomole detection limits using electrospray ionization-mass spectrometry. In 1994, Andren and colleagues reported low attomole detection limits for neurotensin using a Vestec electrospray source coupled to a Finnigan MAT TSQ 700 mass spectrometer [4]. Belov and colleagues reported 30 zmol detection limits for 8 kDa proteins using a 3.5 tesla FTICR mass spectrometer in 2000 [5]. Four years later, the same group improved these results by coupling 15-μm ID HPLC with a 11.4 T FTICR mass spectrometer; five peptides were detected from a 75 zmole aliquot of a BSA tryptic digest, and detection limits were extrapolated to 10 zmol [6]. Also in 2004, Syka and colleagues reported a novel linear-quadrupole/FT mass spectrometer with low zeptomole detection limits for tryptic peptides [7]. In 2015, Li and colleagues coupled an open-tubular liquid chromatography system with a Q-Exactive mass spectrometer and reported 10–50 zmol detection limits for tryptic peptides [8]. Last year, Lipponen and colleagues reported a microcapillary electrospray ionization platform with an extrapolated detection limit of 60 zmol [9]. Also last year, the Nemes group reported a limit of detection of 260 zmole and a 4-log-order dynamic range for angiotensin II using a CE-nanoESI-MS system and parallel reaction monitoring [10]. The capillary in this case had an ID of 20 μm, and the interface was coupled to a Qq-TOF Impact HD from Bruker. Finally, single +20 ions of myoglobin have been detected in an LTQ Orbitrap XL, in a direct infusion experiment; this experiment suggested the noise floor of the Orbitrap electronics is six elementary charges [11].
We developed a high-sensitivity sheath-flow electrospray interface for CZE. There have been three generations of this interface, optimizing the distance between the tip of the capillary and the emitter orifice and the outer diameter of the emitter tip [12–14]. The second generation interface etched the distal tip of the capillary to a 60 μm OD [13]. By etching the separation capillary, the distance between the end of the capillary and the opening of the emitter tip was reduced to ~200 μm. This reduction led to a dramatic increase in sensitivity, and reproducible separations of an E. coli protein digest were demonstrated using loading amounts from 400 fg to 84 pg on a 10 μm ID/150 μm OD separation capillary. The most abundant peptide in E. coli, the elongation factor Tu, was used to estimate the 3 σ LOD as approximately 1 zmole (~600 molecules). It is important to note that this detection limit is an estimate based on a three-order of magnitude extrapolation from the injected amount.
Detection limit is defined as the amount (or concentration) of analyte that generates a signal three times larger than the noise in the baseline. There are two issues in this definition of detection limit. First, it assumes a linear calibration curve. This assumption may not hold for low amounts of peptides, which can be lost to the surface of plasticware used to hold samples. This loss will only be observed when a calibration curve extends to close to the detection limit. Most earlier studies estimated detection limits by linear extrapolation of calibration curves over several orders of magnitude, and would be blind to loss of zeptomole amounts of sample.
Second, this definition of detection limit is typically based on the assumption of Gaussian distributed background signal. As we will show, such an assumption is not valid for measurements made near the detection limit of Orbitrap mass spectrometer.
In this manuscript we demonstrate the reproducible detection of two fragment ions from injection of 1 zmol (600 ions) of angiotensin onto a CZE system coupled to a Q-Exactive HF using our electrokinetically-pumped nanoelectrospray interface. To obtain the detection of a peptide at this level, it was necessary to prepare the sample in a much higher concentration of a BSA tryptic digest; this matrix apparently saturated active binding sites on plasticware, allowing analysis of this minute amount of angiotensin. Based on our 1-zmol electropherogram, we extrapolate a 240 ymol detection limit using a method based on the Bienayme-Tchebycheff inequality, which provides a robust estimate of detection limit for non-Gaussian background signals.
Experimental
Materials and Reagents
Formic acid (FA) and hydrofluoric acid (HF) were purchased from Fisher Scientific (Pittsburgh, PA, USA). LC-MS grade water and methanol were from Honeywell Burdick & Jackson (Wicklow, Ireland). Fused silica capillary (10 μm ID/150 μm OD; 20 μm ID/150 μm OD; 10 μm ID/360μm OD) was purchased from Polymicro Technologies (Phoenix, AZ, USA).
Sample Preparation
Bovine serum albumin (BSA) was dissolved in 100 mM NH4HCO3 (pH 8.0) containing 8 M urea, and denatured at 37° C for 30 minutes. A standard reduction and alkylation was performed using dithiothreitol (DTT) and iodoacetic acid (IAA). The solution was diluted to a urea concentration of less than 2 M, and the proteins were digested at 37°C with trypsin at a trypsin/protein ratio of 1/30 (w/w) for 8 hrs. The digestion was ended by acidification with FA, and the protein digest was desalted using a C18-SepPak column (Waters, Milford, MA). The solution was then lyophilized with a vacuum concentrator (Thermo Fisher Scientific, Marietta, OH), and the dried protein pellet was stored at −80 °C.
Stock solutions of angiotensin II were prepared in MS-Grade water, and aliquots were stored at −80° C. Solutions were created at angiotensin II concentrations shown in Table 1, both with and without BSA digest. These solutions were made fresh daily in 0.05% FA and stored at 4° C.
Table 1 –
Sample concentration and injection volume
| Injection Volume | Concentration | Amount |
|---|---|---|
| Detection limit - 50 pL | 20 pM | 1 zmol – 600 copies |
| Calibration curve - 1 nL | 10 pM | 10 zmol – 6 × 103 copies |
| 100 pM | 100 zmol – 6 × 104 copies | |
| 1 nM | 1 amol – 6 × 105 copies | |
| 10 nM | 10 amol – 6 × 106 copies | |
| 100 nM | 100 amol – 6 × 107 copies | |
| 1 μM | 1 fmol – 6 × 108 copies | |
| 10 μM | 10 fmol – 6 × 109 copies |
All samples were diluted in a sample buffer of 0.05% formic acid. The angiotensin II solutions were diluted to a concentration 10X the concentration desired for the injection, and the last dilution was into a 0.25 mg/mL BSA digest. The solutions for injections of 1 zeptomole loading amount were diluted into a 1 mg/mL BSA digest. Any samples within the picomolar range were remade for each injection to ensure accuracy.
CZE-ESI-MS/MS Analysis
A capillary zone electrophoresis system was constructed from a sample injection block that is similar to ref 15, the third-generation electrokinetically-pumped nanoelectrospray interface [14], and a Q-Exactive HF mass spectrometer (Thermo).
The calibration curve from 10 zmol to 10 fmol was generated with a 36-cm long, 20 μm ID capillary. The capillaries were initially conditioned by flushing with 1 M NaOH followed by background electrolyte (0.5% FA or 0.1% FA) for 10 minutes at 40 PSI. The capillary was flushed with water (MS-Grade) between separations.
1 zmol injections were run to estimate the detection limit. The 1-zmol separations were generated with a 42-cm long, 10-μm ID capillary, pretreated as described for the calibration curve. The narrow ID capillary tended to plug, which discouraged attempts to construct a calibration curve at higher concentrations.
For the calibration curve, the samples were injected by applying 8 PSI for 2 seconds, after which the injection end of the capillary was dipped in MS-Grade water to remove contaminating sample from the exterior of the capillary before being placed in background electrolyte for electrophoresis. Injection volume was calculated using Poiseuille’s law. Electrophoresis was performed at a separation voltage of 22 kV, with the electrospray voltage set at 1.7 kV.
For the 1-zmol data, samples were injected by applying 6 PSI for 2 seconds. The capillary tip was washed as described above. Electrophoresis was performed at a separation voltage of 25.2 kV. The electrospray voltage set at 2.2 kV.
A third-generation electrokinetically pumped nanospray interface was used to couple the separation capillary and mass spectrometer. The distal tip of the separation capillary was etched to ~ 30 μm diameter by hydrofluoric acid (appropriate safety protocols must be employed while handling HF). Emitter tips were pulled from borosilicate capillaries (1 mm OD, 0.75 mm ID) to an opening diameter of 15 μm for the calibration curve, and 40 μm for the 1 zmol experiment using a Sutter puller (P-1000). The puller used a temperature setting from 460 – 470° C and a pull velocity of ~20. The electrospray sheath electrolyte was 0.1% FA, 10% methanol in water.
The separation capillaries were coupled to the Q-Exactive HF mass spectrmeter, operated in positive ion mode. Data were collected using a parallel reaction monitoring (PRM) method with the parameters shown in Table 2. The data files for the calibration curve were collected in Profile mode, while the data files for the 1 zmol runs were collected in Full Profile mode. The calibration curve data files were converted from RAW files to mzXML files using MSConvert [16], and imported into MATLAB for analysis. Selected ion electropherograms were generated by summing the intensity across a 0.075 AMU window around the target m/z.
Table 2 -.
Method Parameters for PRM
| Scan Range | 350 – 1800 m/z |
| Inclusion | On; 10.0 PPM (Mass List: 523.77340, +2) |
| Resolution | 30,000 |
| AGC Target | 2e5 |
| Max Injection Time | 200 ms |
| Isolation Window | 2.0 m/z |
| NCE | 25 |
Results and Discussion
Calibration Curve
We prepared samples for a calibration curve by doping minute amounts of angiotensin into a 0.25 g/L solution of BSA tryptic digest. We discovered that the BSA background was necessary to detect low concentrations of angiotensin; no signal was observed without the added BSA for all but the largest angiotensin injection amounts. We presume that angiotensin is adsorbed on active sites on the plasticware used to prepare the sample, and that the BSA digest blocks those high-activity sites.
The Q-Exactive HF was programmed in PRM mode. The parent ion was set at m/z 523.77340 (+2) corresponding to angiotensin II (human, Asp-Arg-Val-Tyr-Ile-His-Pro-Phe). The two most intense fragment ions (b6+ and y2+) of angiotensin II were extracted with 0.075 Da mass tolerance to construct a calibration curve. Data were collected in triplicate.
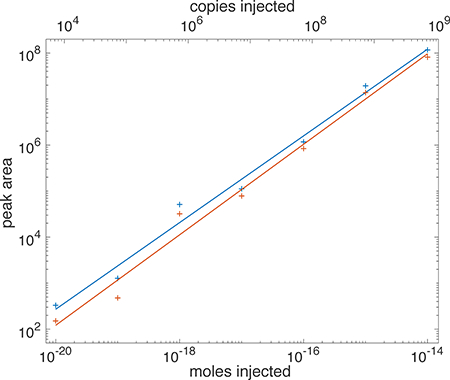
Figure 1 presents a log-log calibration curve. The fragment ion signal increased linearly with angiotensin loading amount for five orders of magnitude (b6+ log-log slope = 0.930, r = 0.990; y2+ log-log slope = 0.987, R2 = 0.988). Higher loading amounts were not used to minimize contamination of the system.
Figure 1 –

log-log calibration curve of PRM analysis of angiotensin. Moles injected are shown on the bottom X-axis, and copies of angiotensin injected are shown on the top axis. Peaks were fit with a Gaussian function; peak area is the product of the height and width of the fitted function. Several 10 zmol injections did not generate peaks for mz 784; a value of zero area was used in calculating that average.
High sensitivity analysis: 1 zmol injection
We modified the instrument to increase sensitivity. We used a 10-μm ID separation capillary, which is a four-fold decrease in capillary cross-section compared to the calibration curve, which produces a proportional increase in analyte concentration for the same loading amount. A slightly longer capillary was also used, which modestly reduced band broadening due to injection volume. Finally, a larger emitter orifice was used, which allowed placing the tip of the etched capillary within the orifice, further improving sensitivity.
Figure 2 presents three successive electropherograms generated from 1-zmol injections (600 copies). The data are presented as butterfly plots, with the m/z 263 data plotted as positive numbers and the 784 data as negative. The two fragments comigrate, which provides high confidence for the detection of angiotensin at this minute loading amount. The mean migration time was 148 ± 6 seconds, and the variation in time is likely due to a drift in room temperature.
Figure 2.

PRM electropherograms generated from injection of 1 zmol (600 molecules) of angiotensin. Data are presented as butterfly plots, with the data at m/z = 263 plotted as positive numbers and the data at m/z = 784 plotted as negative numbers.
Detection limit
The data presented in Figure 2 are intriguing. With the exception of one spike, the background generated at m/z784 equals zero. The background for m/z = 263 consists of a set of spikes with width equal to one sampling interval and with similar peak heights. Run1 generated 16 spikes with intensity = 734 ± 80 (mean ± standard deviation), Run2 generated 12 spikes with intensity = 690 ± 66, and Run3 generated 19 spikes with intensity = 665 ± 62. The decrease in average spike intensity is statistically significant and presumably due to instrument drift.
We interpret this data as arising from a thresholding algorithm used by the QExactive HF to truncate noise below some level, and that the spikes are produced in the m/z 263 by noise that occasionally exceed the threshold. Without details of the algorithm used by the Q-Exactive HF to threshold low-intensity signal, any extrapolation of this data to lower injection amounts is problematic. However, the consistent observation of co-migrating fragment ions in the three electropherograms provides confidence in an estimate of the detection limit as less than 1 zmol injected into the capillary.
There are several approaches to calculate detection limits from this data. The classic definition is
| (1) |
where signal is the peak height, stdbackground is the standard deviation of the background, and the injection amount is 1 zmol. In this case, we define the background as the baseline before 125 seconds, which corresponds to a mass LOD of 80 ymole (~50 copies). However, the classic definition of detection limit is based on normally-distributed background signal which is inappropriate for this non-Gaussian background noise.
In this case, it is instead appropriate to use a method reported by Knoll that is based on the Bienayme-Tchebycheff inequality, which provides a robust estimate for non-Gaussian background noise [17]. This method estimates detection limit as
| (2) |
where Klod is a constant based on the measurement window divided by the peak width, hn is the height of the largest noise peak, Minjected is the sample mass injected, and hs is the height of the signal. The measurement window for the background is 125 s and the peak width at half-height is ~2s, so that the window is roughly 50 times the peak width, which corresponds to a Klod = 0.92. As noted above, hn/hs ~0.25, and detection limit is estimated to be 230 ymol or 140 ions injected into the separation capillary, which is a factor of four less than our injected amount.
Conclusions
We draw several conclusions from these data.
First, the electrokinetically pumped nanoelectrospray interface is remarkably efficient in introducing ions into the mass spectrometer. Although this interface uses sheath flow, that flow does not appear to interfere with the transfer of ions from the separation capillary into the mass spectrometer.
Second, the Orbitrap Q-Exactive HF mass spectrometer has ion optics that are very efficient in transferring precursor ions to the collision cell, and then transferring fragment ions to the Orbitrap mass analyzer. Assuming that the noise of the Orbitrap is roughly equivalent to six elementary charges [11], then roughly 20 ions are detected in our 1-zmol injection, which corresponds to an overall ion transmission from injection onto the separation capillary to detection in the Orbitrap mass analyzer of 3%.
Third, care is required to block active sites on vessels that contact the sample to minimize loss of minute concentrations of analyte. We employed a large excess of a BSA tryptic digest in our experiments to saturate those active sites. Alternatives should be investigated.
Fourth, in favorable cases, it is possible to detect injections of 1 zmol of peptide in capillary zone electrophoresis with an electrokinetically-pumped nanoelectrospray interface.
Finally, capillary electrophoresis coupled with the Orbitrap mass spectrometer produces six order of magnitude dynamic range measurements. That dynamic range approaches the performance of laser-induced fluorescence, which providing a much higher information content signal for compound identification.18−19
Highlights.
We report the detection of 1 zmol amounts of a peptide injected onto a capillary, separated by zone electrophoresis, and detected with an Orbitrap mass spectrometer. These detection limits required careful consideration of the sample’s matrix to minimize peptide loss.
Potential Reviewers.
Chris Le – University of Alberta - Xc.LE@UAlberta.CA
Mingliang Ye – Dalian Institute of Chemical Physics - mingliang@dicp.ac.cn
Huwei Liu – Peking University - hwliu@pku.edu.cn
Philip Britz-McKibbin – McMaster University - britz@mcmaster.ca
Michael Breadmore – University of Tasmania - mcb@utas.edu.au
Acknowledgements
We thank Drs. Matthew Champion and William Boggess in the Notre Dame Mass Spectrometry and Proteomics Facility for their help with this project. This work was funded by the National Institutes of Health (Grant R01GM096767).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dovichi NJ, Martin JC, Jett JH, Keller RA, Attogram detection limit for aqueous dye samples by laser-induced fluorescence. Science 219 (1983) 845–847. [DOI] [PubMed] [Google Scholar]
- 2.Cheng YF, Dovichi NJ, Subattomole amino acid analysis by capillary zone electrophoresis and laser-induced fluorescence. Science 242 (1988) 562–564. [DOI] [PubMed] [Google Scholar]
- 3.Chen DY, Dovichi NJ, Single-molecule detection in capillary electrophoresis: Molecular shot noise as a fundamental limit to chemical analysis. Anal. Chem 68 (1996) 690–696. [Google Scholar]
- 4.Andren PE, Emmett MR, Caprioli RM, Micro-Electrospray: Zeptomole/attomole per microliter sensitivity for peptides. J. Am. Soc. Mass Spectrom 5 (1994) 867–869. [DOI] [PubMed] [Google Scholar]
- 5.Belov ME, Gorshkov MV, Udseth HR, Anderson GA, Smith RD, Zeptomole-sensitivity electrospray ionization--Fourier transform ion cyclotron resonance mass spectrometry of proteins. Anal. Chem 72 (2000) 2271–2279. [DOI] [PubMed] [Google Scholar]
- 6.Shen Y, Tolić N, Masselon C, Pasa-Tolić L, Camp DG 2nd, Hixson KK, Zhao R, Anderson GA, Smith RD, Ultrasensitive proteomics using high-efficiency on-line micro-SPE-nanoLC-nanoESI MS and MS/MS. Anal. Chem 76 (2004) 144–154. [DOI] [PubMed] [Google Scholar]
- 7.Syka JE, Marto JA, Bai DL, Horning S, Senko MW, Schwartz JC, Ueberheide B, Garcia B, Busby S, Muratore T, Shabanowitz J, Hunt DF, Novel linear quadrupole ion trap/FT mass spectrometer: performance characterization and use in the comparative analysis of histone H3 post-translational modifications. J. Proteome Res 3 (2004) 621–626. [DOI] [PubMed] [Google Scholar]
- 8.Li S, Plouffe BD, Belov AM, Ray S, Wang X, Murthy SK, Karger BL, Ivanov AR, An Integrated Platform for Isolation, Processing, and Mass Spectrometry-based Proteomic Profiling of Rare Cells in Whole Blood. Mol. Cell Proteomics 14 (2015) 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipponen K, Tähkä S, Sikanen T, Jokinen V, Tatikonda A, Franssila S, Kostiainen R, Kotiaho T, Thiol-ene micropillar array electrospray ionization platform for zeptomole level bioanalysis. Analyst 142 (2017) 2552–2557. [DOI] [PubMed] [Google Scholar]
- 10.Choi CB, Zamarbide M, Manzini MC, Nemes P, Tapered-Tip Capillary Electrophoresis Nano-Electrospray Ionization Mass Spectrometry for Ultrasensitive Proteomics: the Mouse Cortex. J. Am. Soc. Mass Spectrom 28 (2017) 597–607. [DOI] [PubMed] [Google Scholar]
- 11.Makarov A, Denisov E, Dynamics of Ions of Intact Proteins in the Orbitrap Mass Analyzer. J. Am. Soc. Mass Spectrom 20 (2009) 1486–1495. [DOI] [PubMed] [Google Scholar]
- 12.Wojcik R, Dada OO, Sadilek M, Dovichi NJ, Simplified capillary electrophoresis nanospray sheath-flow interface for high efficiency and sensitive peptide analysis. Rapid Commun. Mass Spectrom 24 (2010) 2554–2560. [DOI] [PubMed] [Google Scholar]
- 13.Sun L, Zhu G, Zhao Y, Yan X, Mou S, Dovichi NJ, Ultrasensitive and fast bottom-up analysis of femtogram amounts of complex proteome digests. Angew. Chem. Int. Ed. Engl 52 (2013) 13661–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L, Zhu G, Zhang Z, Mou S, Dovichi NJ, Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. J. Proteome Res 14 (2015) 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krylov SN, Starke DA, Arriaga EA, Zhang Z, Chan NW, Palcic MM, Dovichi NJ, Instrumentation for chemical cytometry. Anal. Chem 72 (2000) 872–877. [DOI] [PubMed] [Google Scholar]
- 16.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick PA, A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol 30 (2012) 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knoll JE, Estimation of the Limit of Detection in Chromatography. J. Chromatogr. Sci 23 (1985) 422–425. [Google Scholar]
- 18.Whitmore CD, Essaka D, Dovichi NJ, Six orders of magnitude dynamic range in capillary electrophoresis with ultrasensitive laser-induced fluorescence detection. Talanta 80 (2009) 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dada OO, Essaka D, Hindsgaul O, Palcic MM, Prendergast J, Schnaar RL, Dovichi NJ, Nine Orders of Magnitude Dynamic Range: Picomolar to Millimolar Concentration Measurement in Capillary Electrophoresis with Laser Induced Fluorescence Detection Employing Cascaded Avalanche Photodiode Photon Counters. Anal. Chem 83 (2011) 2748–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]