

SUMMARY
Under-connectivity between cerebral cortical association areas may underlie cognitive deficits in neurodevelopmental disorders including the 22q11.2 Deletion Syndrome (22q11DS). Using the LgDel 22q11DS mouse model, we assessed cellular, molecular and developmental origins of under-connectivity, and its consequences for cognitive function. Diminished 22q11 gene dosage reduces long-distance projections, limits axon and dendrite growth, and disrupts mitochondrial and synaptic integrity in layer 2/3 but not 5/6 projection neurons (PNs). Diminished dosage of Txnrd2, a 22q11 gene essential for reactive oxygen species catabolism in brain mitochondria, recapitulates these deficits in WT layer 2/3 PNs; Txnrd2 re-expression in LgDel layer 2/3 PNs rescues them. Anti-oxidants reverse LgDel or Txnrd2 related layer 2/3 mitochondrial, circuit and cognitive deficits. Accordingly, Txnrd2-mediated oxidative stress reduces layer 2/3 connectivity and impairs cognition in the context of 22q11 deletion. Anti-oxidant restoration of mitochondrial integrity, cortical connectivity and cognitive behavior defines oxidative stress as a therapeutic target in neurodevelopmental disorders.
Keywords: neurodevelopmental disorders, cortical projection neurons, reactive oxygen species, mitochondria, autism spectrum disorders, schizophrenia, under-connectivity
Graphical Abstract
eTOC
Fernandez, Meechan et al. use a mouse model of DiGeorge/22q11 deletion syndrome -a genetic neurodevelopmental disorder- to characterize biological mechanisms of neuronal under-connectivity, establish their role in higher-order behavior and identify an effective pharmacological therapy.
INTRODUCTION
Connections between and within association regions of the cerebral cortex—regions that do not receive inputs from thalamic sensory relays, process primarily one modality, or project to motor neurons—are thought to be the basis for complex behaviors (Bota et al., 2015; Crowe et al., 2013; Fuster, 2009). These connections may also be a pathogenic target in disorders where cognitive capacity is compromised (Geschwind and Levitt, 2007; Kwan, 2013). Indeed, a compelling hypothesis for such dysfunction in several neurodevelopmental disorders, including the genomic deletion syndrome 22q11.2 Deletion Syndrome (22q11DS; also known as DiGeorge or Velo-Cardio-Facial Syndrome/VCFS), proposes that under-connectivity of association cortices underlies behavioral deficits (Just et al., 2004; Schipul et al., 2012). Functional imaging in individuals with 22q11DS (Hall et al., 2013; Kwan, 2013; Padula et al., 2015; Schreiner et al., 2017) and several other neurodevelopmental disorders (Liston et al., 2011; Romme et al., 2017) suggest association cortices are under-connected. Nevertheless, how this focal change in a distinct class of cortical circuits occurs, its quantitative cellular basis, and behavioral consequences remain unknown. We used mouse genetic models of 22q11DS to determine whether association cortical areas are under-connected, whether changes arise during development due to diminished 22q11 gene dosage, and whether such divergence impacts murine cognitive behavior.
Association cortico-cortical connections are made largely by layer 2/3 projection neurons (PNs). Disruptions of these connections, which constitute the majority of afferents and efferents from and to association cortices (Bannister, 2005; Bota et al., 2015; D’Souza and Burkhalter, 2017; Swanson et al., 2017) result in cognitive and social deficits. The molecular, cellular, and microcircuit foundations of association cortico-cortical circuitry underlying complex behaviors—or their disruption in several brain disorders—have proven difficult to analyze in humans or non-human primates; specific mechanisms that mediate circuit development remain largely unknown. Studies in mice have identified general mechanisms for initial differentiation of cortical PN dendrites (Whitford et al., 2002) and major axon pathways that originate from these neurons (Alcamo et al., 2008; Barnes et al., 2007; Britanova et al., 2008; Lodato and Arlotta, 2015; McKenna et al., 2015; Molyneaux et al., 2005). Nevertheless, establishment and maintenance of connections between distinct association areas and differentiation of local microcircuit elements necessary for function have not been explored. We asked whether diminished 22q11 gene dosage selectively and quantitatively regulates these events for a discrete association cortico-cortical circuit critical for cognitive function.
We assessed quantitative regulation of cortico-cortical connections between two association areas: medial frontal association (mFAC) and lateral entorhinal cortices (laEC). We focused on differentiation of layer 2/3 PNs that make these connections, and defined the impact of these mechanisms on cognitive capacity in the LgDel mouse (Merscher et al., 2001), a genomically accurate model of key 22q11DS clinical features (22q11DS; McDonald-McGinn et al., 2015; Meechan et al., 2015a) for a behavior that relies upon this circuit. We found quantitative cortico-cortical under-connectivity in multiple cell biological domains, characterized the role of altered layer 2/3 PN differentiation in this change, defined its consequences for a reversal learning task that depends upon mFAC and laEC connections (Meechan et al., 2015b), and identified a causal molecular mechanism that reflects diminished dosage of a single 22q11 gene—the mitochondrial enzyme Thioredoxin-Reductase 2 (Txnrd2)—essential for catabolism of reactive oxygen species. Anti-oxidant therapy resolves oxidative-stress related cortico-cortical under-connectivity and behavioral deficits in LgDel mice. Our results provide new mechanistic insight and a potential therapeutic approach for disrupted cortical circuit development in 22q11DS.
RESULTS
Local and long distance cortico-cortical connectivity in LgDel
We first assessed local and long-distance projections from projection neurons (PNs) in the medial frontal association cortex (mFAC) in LgDel mice (Meechan et al., 2015b). We retrogradely labeled cortical PNs whose connections to mFAC contribute to several complex behaviors (Sigurdsson and Duvarci, 2015) and quantified their laminar and regional distribution throughout the ipsilateral and contralateral cortical hemispheres (Figure 1A). In WT and LgDel, most cortico-cortical projections originate from layer 2/3 PNs (≥75%, bins 1/2/3; Figure 1A,B). Nevertheless, regional distribution and frequency of LgDel cortico-cortical connections changes (Figure 1C). The frequency of retrogradely labeled layer 2/3 PNs is enhanced (red bars) adjacent to mFAC in LgDel (levels 1–2, prox. to injection, Figure 1C,D) but diminished in distal targets (black bars, levels 3–7, Figure 1C,D). Projections from posterior regions—including lateral entorhinal cortex (laEC), a major source/target of mFAC connections and functional partner implicated in neurodevelopmental disorders (Ellegood et al., 2015; Reep et al., 1987)—are substantially decreased in LgDel (Figure 1D). Apparently, mFAC projections from layer 2/3 PNs are redistributed: proximal inputs increase and distal projections decline, resulting in long-distance under-connectivity of LgDel mouse association cortices.
Figure 1:

Long-distance cortico-cortical connectivity is altered in the LgDel. A) Representative coronal section from a mouse brain injected stereotaxically with Biocytin3Alexafluor488 in medial frontal association cortex (mFAC). Retrogradely labeled cells throughout the anterior-posterior cortical axis were plotted and transformed to polar coordinates to generate raster plots relative to the mid-point of the horizontal midline. B) Laminar distribution of labeled cells in WT and LgDel lateral entorhinal cortex (laEC) is similar; however, their overall density appears diminished in LgDel. Graph represents the mean percentage of cells in each of 5 equidistant bins and error bars SEM. C) Schematic of injection site and location of coronal sections analyzed for retrogradely labeled cortical neurons (left). On this cartoon of a dorsal view of the adult mouse brain, key cortical areas are indicated in the left hemisphere to define the location of labeled neurons in each of the representative sections, and the relationship to the raster plots (right); AC: medial frontal association cortex, SS: somatosensory cortex, Ent: Entorhinal cortex, Vis: Visual cortex, RS: retrosplenial cortex. For each section, the angular location and frequency of all retrograde labeled cortical cells (right) is shown in raster plots for WT (n=3; black) and LgDel (n=3; red) in the ipsilateral (left) and contralateral (right) hemispheres. Colored bars directly beneath raster plots indicate regions of LgDel over-connectivity (red bars; ≥ 5× density v. WT) or under-connectivity (black bars; ≤5× density v. WT). D) Representative ipsilateral and contralateral sections at location 5 (posterior, panel 1C). Reduced labeling is apparent in LgDel laEC (90–120°, arrows) and visual/visual association cortex (~40°, arrowheads). Increased labeling is seen in a small medial dorsal region in the ipsilateral hemisphere (asterisk). Scale bar 500 μm.
Selective changes of neuronal and synaptic integrity in layer 2/3 PNs
We next assessed the cell biological integrity of mFAC layer 2/3 PN microcircuitry in LgDel and WT mice (Figure 2A,B). For maximal resolution, we used quantitative wide-field block face electron microscopic (EM) analysis to evaluate layer 2/3 PN cell bodies, processes, organelles, and synaptic specializations. LgDel layer 2/3 PNs are cytologically compromised; nuclei are electron lucent with shallower nuclear indentations (Figure 2B and inset). The distribution of ER, golgi, ribosomes, as well as tubular and filamentous cytoskeletal elements in layer 2/3 apical dendrites is altered (Figure 2B1). Mitochondria in LgDel layer 2/3 PN cell bodies and apical dendrites are pale, swollen and dysmorphic (compare Figure 2A1,2 with 2B1–4). Because mitochondrial number, structure and function are interdependent (Hung et al., 2018), we compared several morphometric indices of mitochondrial integrity in LgDel and WT layer 2/3 PNs and the surrounding neuropil. Mitochondrion frequency decreases nearly 2× from WT in cell bodies and apical dendrites (cell b.: LgDel<WT, p<0.002, n=10 WT cells, 12 LgDel cells; apical d.: LgDel < WT, p<0.0005, n=12 WT, 12 LgDel; 1-way ANOVA+Holm-Sidak; Figure 2E and Supplemental data (SD) 1). We then determined the aspect ratio of individual mitochondria (the max./min. diameter), an established cell biological measure of mitochondrial oxidative stress (SD2) (Koopman et al., 2005). Values closer to “1” (round, swollen) indicate maximal stress, and substantially greater than “1” (elongated, narrow), minimal stress. In WT layer 2/3 PN cell bodies this ratio is 2.31±0.11 (n=9 cells), and in apical dendrites, 3.77±0.18 (n=12 cells). In LgDel layer 2/3 PNs, these ratios decrease: cell body, 1.56±0.06 (p<0.0001, n=12 cells; 1-way ANOVA+Holm-Sidak); apical dendrite, 2.45±0.17 (LgDel<WT, p<0.0001, n=12). Apparently, there is substantial mitochondrial stress in LgDel layer 2/3 PNs.
Figure 2:

Layer 2/3 projection neuron (PN) cytological and synaptic integrity is compromised in LgDel frontal cortex. A) Block-face SEM image of a WT layer 2/3 PN at low magnification showing the cell body, the nucleus with deep nuclear indentations (inset), and a well-defined apical dendrite with high density of ribosome rosettes, elongated mitochondria, filamentous and tubular cytoskeletal elements. A1) Higher magnification images of WT mitochondria (m), ribosomes (arrowheads), neurofilaments (arrow) and microtubules (arrow) in a layer 2/3 apical dendrite. A2–4) WT mitochondria and synapses (asterisks), in and around an apical dendrite in the adjacent neuropil and WT layer 2/3 neuropil synapses. The dendrite and adjacent neuropil elements have healthy mitochondria (A2,3), typical pre- and post-synaptic specializations, and a high frequency of presynaptic vesicles (A3,4). B) Block-face SEM image of LgDel layer 2/3 PN. The nucleus is pale and labeled chromatin is sparse, the nuclear envelope smooth with few, shallow, nuclear indentations (inset). B1) The apical dendrite cytoplasm has fewer, shorter mitochondria, diminished ribosomes, as well as fewer filamentous and tubular cytoskeletal elements. B2–4) Mitochondria and synapses in and around the apical dendrite of a layer 2/3 PN. The mitochondria are swollen and round rather than elongated; their cristae are distended and disordered. B5,6) Layer 2/3 pre-synaptic terminals (asterisks) are dilated, and have few organelles or synaptic vesicles as well as diminished presynaptic densities. Postsynaptic profiles (ps) are similarly organelle-sparse. C) WT and D) LgDel layer 5/6 PNs are indistinguishable. Insets Apical dendrites have substantial density of ribosome rosettes, robust neurofilament and microtubules networks, and deep nuclear indentations. C1–3, D1–3) WT (C1–3) and LgDel (D1–3) layer 5/6 PN mitochondria do not differ in morphology or apparent distribution. C4–6, D4–6) Synaptic vesicle density in WT (C4–6) and LgDel (D4–6) layer 5/6 PN-adjacent neuropil appears equivalent. E) Left to right: Quantification of WT and LgDel PN cell body, dendrite, and neuropil mitochondria, pre-synaptic frequency, and synaptic vesicle density in layer 2/3 and 5/6. (*p<0.0021, 0.00057, 0.0001, 0.008, 0.011, respectively).
Data are represented as Mean +SEM Scale bar 5 μm.
Synaptic integrity and efficiency is sensitive to mitochondrial dysfunction (Devine and Kittler, 2018); thus we assessed the cytological properties of synapses made on layer 2/3 PNs as well as in the surrounding neuropil. LgDel synaptic mitochondria in neuropil immediately adjacent to layer 2/3 PNs are far less frequent (neuropil: LgDel<WT; p<0.0001; n=12 LgDel, 12 WT; ANOVA+Holm-Sidak; Figure 2E) and appear swollen and rounder, with distorted cristae (Figure 2B1–4). LgDel pre-synaptic profiles are vacuolated with apparently fewer synaptic vesicles and diminished pre-synaptic densities (compare Figure 2A3,4 with 2B5,6). Apical dendrite pre-synaptic terminals decline by 25% (p<0.008, n=12 WT, 12 LgDel, 1-way ANOVA+Holm-Sidak; Figure 2E); however, there are no significant differences in cell body or neuropil terminals. Finally, synaptic vesicle density is reduced by 25% in LgDel layer 2/3 neuropil (p<0.011, n=12 LgDel, 12 WT, 1-way ANOVA+Holm-Sidak; Figure 2E). These layer 2/3 PN changes indicate cortico-cortical microcircuit under-connectivity in LgDel.
In contrast with LgDel layer 2/3 PN anomalies, LgDel and WT layer 5/6 PNs are indistinguishable. The apical dendritic as well as perikaryal cytoplasm have comparable densities of organelles, microtubular and neurofibrillary elements. Mitochondria throughout the cell bodies and apical dendrites of layer 5/6 LgDel and WT PNs are neither dysmorphic nor diminished in frequency (Figure 2C,D). Mitochondrial frequencies in WT versus LgDel mFAC layer 5/6 PN cell bodies, apical dendrites, and adjacent neuropil are indistinguishable (cell b.: LgDel ≅ WT, p<0.60; apical d.: LgDel ≅ WT, p<0.60; neuropil: LgDel ≅ WT, p<0.82; n= 8 WT, 8 LgDel, Student’s T-test; Figure 2E). Apical dendrite pre-synaptic terminals are equally frequent in the 2 genotypes (LgDel pre-synaptic WT, p≤0.32, n=8 LgDel, 8 WT, Student’s T-test; Figure 2E). Finally, there is no difference in synaptic vesicle density (LgDel ≅WT, n=8 LgDel, 8 WT, p<0.94, Student’s T-test; Figure 2E). Thus, cytological and synaptic indicators of cortico-cortical under-connectivity due to heterozygous 22q11 gene deletion are limited to LgDel layer 2/3 PNs that make a majority of association cortico-cortical projections.
Axon and dendrite development in LgDel layer 2/3 PNs
We next asked whether diminished 22q11 gene dosage selectively compromises initial growth or differentiation of individual LgDel versus WT layer 2/3 or 5/6 PNs using a primary cortical culture assay (Ahlemeyer and Baumgart-Vogt, 2005). Effectively 100% of these cells, harvested either from E16.5 or E14.5 cortices, are neurons; cultures from E16.5 fetal cortices are 98% Cux2-positive apparent layer 2/3 PNs in vitro (layer 2/3 PNivt: 98±1.3%Cux2-labeled; n=35 neurons/3 experiments, p<4×109, 1-way ANOVA), while Ctip2-positive cells predominate in E14.5 cultures (Figure 3B–E). Additional quantitative expression profiling (data not shown) confirms mRNA profiles consistent with layer 2/3 PN identities for E16.5 (layer 2/3 PNivt), and layer 5/6 PN identity for E14.5 (layer 5/6 PNivt) cultures.
Figure 3:

Diminished growth of LgDel layer 2/3 PNs in vitro (PNivt). A) The experimental paradigm used to establish layer 2/3 versus 5/6 PN cultures. B) These cultures (neurons for analysis identified via electroporation of membrane bound Enhanced GFP (mbEGFP), as shown in panel A) yield nearly all neurons (NeuN; red nuclear labeling), with few if any glial cells (GFAP; no labeling apparent in this panel). C) Cultured neurons harvested from E16.5 cerebral cortices (2/3 PNivt; mbEGFP) uniformly express Cux2 (blue), a selective marker layer for layer 2/3 PNs in vivo. D) Cultured neurons harvested from E14.5 cerebral cortices (5/6 PNivt) more frequently express Ctip2 (red), a layer 5/63selective marker. E) mbEGFP labeled neurons in E16.5 (layer 2/3 PNivt) cultures have been quantified for co-expression of NeuN (neuronal marker), Cux2, and Ctip2 frequency. F) A WT layer 2/3 PNivt with highly branched dendrites (brackets, inset) and long branching axon. G) A LgDel layer 2/3 PNivt; its dendrites appear shorter and less complex (brackets, inset), and the axon appears shorter. H) Quantitative comparisons of WT and LgDel layer 2/3 axon length (p<0.0272), branching (p<4×106), and branch order (* indicates p<0.011 and 0.0001 respectively). I) Quantitative comparisons of WT and LgDel layer 2/3 dendrite length (p<0.0013), branching (p<0.00003), and branch order (* indicates 0.013, 0.002, 0.012, 0.013 respectively). J,K) WT (J) and LgDel (K) layer 5/6 PNivt (identity confirmed as +Ctip2; not shown) do not have apparent differences in growth or differentiation. Axons and dendrites of layer 5/6 PNivt in both genotypes, however, are shorter and less branched than those of layer 2/3 PNs (brackets, inset). L) Quantitative comparisons of WT and LgDel layer 5/6 PN axon and dendrite length and branching; all measures in the two genotypes are statistically indistinguishable (p>0.3).
All graphs represent mean values +SEM. Scale bar 100 μm for 3B, C, F, G and 25 μm for 3D, J, and K.
If diminished long-distance connections in vivo reflect disrupted axon growth, which likely contributes to in vivo long distance under-connectivity, LgDel layer 2/3 PNivt axon lengths should be less than WT. LgDel layer 2/3 PNivt axons are 23% shorter than WT counterparts (LgDel<WT; n=50 WT + 52 LgDel neurons/10 brains/genotype/5 litters, p≤0.0272, 1-way ANOVA+Holm-Sidak; Figure 3F,G,H, and SD3), and branching declines by 45% (LgDel<WT, p<4.0×106; Figure 3H) with 36% fewer 1° and 76% fewer 2° branches (n=50 neurons/10 brains/genotype/5 litters, p<0.011, 0.0001, respectively, 1-way ANOVA+Holm-Sidak; Figure 3H), consistent with reduced long distance cortico-cortical projections made by LgDel layer 2/3 PNs in vivo. We next asked if LgDel layer 2/3 PNivt dendrite growth or differentiation is altered. LgDel layer 2/3 PNivt dendrite length decreases by 22% (LgDel<WT: n=50 WT + 52 LgDel neurons/10 brains/genotype/5 litters, p≤0.0013, 1-way ANOVA+Holm-Sidak; Figure 3I, and SD3), and branching declines (LgDel<WT: p<0.00003; Figure 3I), with 26% 1°, 29% 2°, 33% 3° and 62% fewer 4° dendrites (p≤0.013, 0.002, 0.012, and 0.013, respectively, 1-way ANOVA+Holm-Sidak; Figure 3I). The decline in initial dendrite and axon growth and branching in LgDel layer 2/3 PNsivt suggests a developmental origin for LgDel layer 2/3 PN under-connectivity.
To assess whether these deficits are selective for layer 2/3 PNs—consistent with our in vivo EM observations—we analyzed layer 5/6 PNivt axon and dendrite growth. In parallel with our ultrastructural observations, LgDel and WT layer 5/6 PNsivt are indistinguishable from one another. LgDel layer 5/6 PNivt axon length and branching is equivalent to WT, as is dendrite growth and branching (LgDel ≅ WT; n=20 cells/4 brains/genotype/2 litters; Figure 3J,K,L). Accordingly, LgDel layer 2/3—but not layer 5/6—PNs have growth and differentiation deficits in vitro consistent with a developmental basis for association cortico-cortical under-connectivity due to diminished 22q11 gene dosage.
Disrupted layer 2/3 PN differentiation in the LgDel cerebral cortex
To determine whether diminished layer 2/3 PN growth and differentiation in vivo underlies cortico-cortical under-connectivity due to diminished 22q11 gene dosage, we labeled individual layer 2/3 PNs selectively using sparse recombination of an eYFPfl reporter via Cux2:CreERT (Figure 4A) and assessed dendritic growth and differentiation at postnatal (P) day 21. P21 LgDel layer 2/3 PN apical dendrite lengths are 50% of WT (LgDel<WT, n=20 neurons/4 brains/genotype/4 litters, p<0.0001, 1-way ANOVA+Holm-Sidak; Figure 4B–D, and SD4). LgDel layer 2/3 PN apical dendrite branching is also diminished; branch points decline by 70% (LgDel<WT: p<0.0002, 1-way ANOVA+Holm-Sidak; Figure 4D, and SD5), and branch order declines by 56% (LgDel<WT: p<0.0042, 1-way ANOVA+Holm-Sidak; Figure 4D, and SD5). Basal dendrite length and branching points also decline; however, these changes do not reach statistical significance (Figure 4D, SD4, and SD5). Nevertheless, basal dendrite branch order is significantly reduced (LgDel<WT: p<0.026, 1-way ANOVA+Holm-Sidak; Figure 4D, and SD5). Finally, apical dendrite retraction, an indicator of neuronal metabolic stress (Shimada et al., 2006), is seen in 70% of LgDel versus 0% WT layer 2/3 PNs (Figure 4C, arrows). Thus, parallel to our in vitro observations, LgDel layer 2/3 PN dendrite growth and branching, critical for circuit development (Purves et al., 1986) is diminished in vivo.
Figure 4:

LgDel layer 2/3 PN growth and differentiation is disrupted in vivo. A) A schematic of the genetic strategy used to selectively and sparsely label WT and LgDel layer 2/3 PNs in vivo. Low doses of tamoxifen were given to P8 pups to elicit recombination of the floxed eGFP allele in a small subset of layer 2/3 PNs. B) Selectively genetically labeled individual WT layer 2/3 PNs in 2D projections of confocal 3D image sets. Dendritic branching is frequent, and arborization is extensive. C) Selectively genetically labeled LgDel layer 2/3 PNs in 2D projections of confocal 3D image sets. Dendritic branching, especially for the apical dendrite, appears diminished, and the terminal portion of some apical dendrites (large arrows) are beaded, a sign of apical dendritic retraction (inset). D) Quantitative comparison between WT and LgDel layer 2/3 PNs of apical dendrite length (*p<0.0001), and branching (*p<0.0002), as well as basal dendritic length (n.s.; no significant differences) and branching (n.s.). Maximum branch order was significantly reduced for LgDel apical dendrites (*p<0.0042) as well as basal dendrites (*p<0.0264).
All graphs represent mean value + SEM. Scale bar 25 μm.
Mitochondrial oxidative stress disrupts layer 2/3 PN growth
In a wide range of cells and tissues, aberrant nuclear and cytoskeletal morphology, distended mitochondria, and disrupted growth and differentiation are associated with oxidative stress due to mitochondrial dysfunction (Sasaki and Yoshida, 2015; Wai and Langer, 2016). Accordingly, the selective changes in LgDel layer 2/3 PNs we identified in vivo and in vitro may reflect mitochondrial dysfunction due to diminished 22q11 gene dosage. Thus, we compared reactive oxygen species (ROS) levels, an established indicator of mitochondrial integrity (Valko et al., 2007), in LgDel and WT layer 2/3 and 5/6 PNivt. LgDel mitochondrial ROS in layer 2/3 PNivt, measured in mitochondria found in processes as well as cell bodies using the superoxide indicator mitoSOX in individual living neurons, increases 45±5% above WT levels (n=50 cells/4 brains/genotype/4 litters, p<0.0001; 1-way ANOVA+Holm-Sidak; Figure 5A, and SD6), and cytosolic ROS, measured with cellROX, increases by 37±7% above WT (n=50 cells/4 brains/genotype/4 litters, p<0.0001; 1-way ANOVA+Holm-Sidak; Figure 5B, and SD6). Apparently, ROS-dependent anti-oxidant defense (Birben et al., 2012; Dey et al., 2016) essential for WT mitochondrial and cytosolic homeostasis operates inefficiently in LgDel layer 2/3 PNs. To determine whether this change is selective for layer 2/3, we also measured mitochondrial ROS levels in layer 5/6 PNsivt. Mitochondrial ROS levels are equivalent in layer 5/6 PNivt from the two genotypes (LgDel mitoSOX: 94% of WT, i.e. 100%, ±7%SEM; LgDel: n=35 cells/6 brains/2 litters, WT: n= 25 cells/4 brains/2 litters, p=0.57; Figure 5C). Thus, ROS metabolism is selectively disrupted in layer 2/3 but not 5/6PNsivt, consistent with selective mitochondrial oxidative stress in layer 2/3 but not 5/6 PNs in vivo.
Figure 5:

Increased Reactive Oxygen Species (ROS) underlies LgDel layer 2/3 PN growth deficits. A) Mitochondrial (Mito) ROS levels, detected with the ROS specific probe mitoSOX, are elevated in LgDel layer 2/3 PNivt. Mitochondria in LgDel 2/3 PNivt are more intensely and frequently labeled. Densitometric quantification in spinning-disk confocal images of individual live WT and LgDel layer 2/3 PNivt (right) confirms apparent differences in ROS levels in the two genotypes (*p<0.0001). B) Cytosolic (Cell) ROS, detected with the cellROX probe, shows enhancement of ROS levels in cell bodies and processes of LgDel layer 2/3 PNivt, similar to the change in mitochondrial ROS (*p< 0.0001). C) mitoSOX levels do not differ significantly (ns) in E14.5 (5/6 PNivt) cortical cultures between genotypes. D) Schematic of anti-oxidant activities of Pyruvate (Pyr), and N-acetylcysteine (NAC) used to assess mitochondrial dysregulation in LgDel PNivt. Pyr acts as a metabolic feedstock (1) by generating AcetylCoA that can directly enter the TCA Cycle. Furthermore, Pyr can directly reduce ROS formed during oxidative phosphorylation (2). NAC directly reduces ROS (3) while also providing cysteine required in the synthesis of Glutathione (GSH), which reduces hydrogen peroxides via the TRX/PRX pathway (4). E) Pyr restores mitoSOX in LgDel layer 2/3 PNivt to WT levels, confirmed by densitometric measurement in live cells (right; *p<0.046). F) NAC restores mitoSOX labeling in LgDel layer 2/3 PNs to WT levels, (right; *p<0.0001). Images in (E) and (F) were collected and processed identically. Dotted lines (right panels) in (E) and (F) indicate outlines of imaged layer 2/3 PNivt where mitoSOX labeling has diminished to low levels compared to untreated LgDel layer 2/3 PNivt. G) Axon and dendrite differentiation in WT+Pyr and LgDel+Pyr treated layer 2/3 PNivt are indistinguishable. H) WT+NAC and LgDel+NAC treated layer 2/3 PNivt are indistinguishable. I) Pyr and NAC restore LgDel layer 2/3 PNivt axon and dendrite length to WT levels. Quantification of total axon and dendritic length in untreated WT, as well as Pyr or NAC treated WT and LgDel layer 2/3 PNivt (LgDel & LgDel+Pyr: *p<0.0004, axons; *p<0.0001, dendrites; LgDel & LgDel+NAC: *p<0.0047, axons; *p<0.0461, dendrites).
All graphs represent mean values +SEM. Scale bar 5 μm for 5A, B, E, and F. 10 μm for 5C. 50 μm for 5G, and H.
If elevated ROS underlies LgDel layer 2/3 PN growth deficits, reducing mitochondrial and cytoplasmic ROS to WT levels should restore LgDel layer 2/3 PNivt growth. We treated LgDel and WT layer 2/3 PNs in vitro with two distinct mitochondrial metabolic support agents (Figure 5D). Sodium Pyruvate (Pyr; Wang et al., 2007) is a pre-metabolite that increases the production of NADH for enhanced efficiency of glycolysis and the TCA cycle as well as acting directly as a ROS scavenger (Figure 5D, left). N-acetyl Cysteine (NAC; Berk et al., 2013) increases Glutathione stores to enhance the efficiency of Peroxiredoxin (PRX)/Thioredoxin (TRX) ROS clearance as well as chemically reducing ROS (Figure 5D, right; Aruoma et al., 1989). In response to the two anti-oxidants (LgDel+Pyr, LgDel+NAC, WT+NAC) mitochondrial ROS levels in LgDel layer 2/3 PNsivt return to WT levels (Figure 5E,F). Pyr treatment reduces LgDel mitoSOX levels by 29.51% (Pyr: n=20 LgDel cells, and 24 LgDel+Pyr cells/4 brains/genotype/2 litters, p≤0.046; Student’s T-test; Figure 5E) and by 27.57% in LgDel- NAC treated cells (NAC: n=23 LgDel cells + 24 LgDel +NAC cells/4 brains/genotype/2 litters, p≤0.0001; Student’s T-test; Figure 5F). In parallel, Pyr and NAC restore LgDel layer 2/3 PNivt axon and dendrite growth to WT levels (Pyr, axons: LgDel+Pyr>LgDel, p<0.0004; dendrites: LgDel+Pyr>LgDel, p<0.0001, n=33 cells /3 brains/condition/3 litters, 1-way ANOVA+Holm-Sidak; NAC, axons: LgDel+NAC>LgDel, p<0.0047; dendrites: LgDel+NAC>LgDel, p<0.0048, n=50 cells/5 brains/condition/5 litters, 1-way ANOVA+Holm-Sidak; Figure 5G–I, and SD3). Thus, diminishing ROS to WT levels using two distinct anti-oxidants is sufficient to restore axon and dendrite growth in LgDel layer 2/3 PNsivt.
A molecular mechanism for ROS-related aberrant layer 2/3 PN differentiation
Six of the 28 contiguous murine human 22q11 gene orthologues encode mitochondrial proteins that are expressed in cortical neurons (Maynard et al., 2008) and are therefore prime candidates for mitochondrial dysfunction underlying layer 2/3 PN under-connectivity. We electroporated shRNAs targeting Mrpl40, Txnrd2, Slc25a1, Prodh, Tango2 and Zdhhc8—each expressed in layer 2/3 PNs in vitro (qPCR; data not shown)—into WT layer 2/3 PNs and compared LgDel and WT axon and dendrite growth (Figure 6A). Diminished dosage of only one 22q11 mitochondrial gene, Txnrd2, recapitulates LgDel layer 2/3 PN growth deficits in otherwise WT layer 2/3 PNsivt. Txnrd2 encodes the key mitochondrial enzyme for H2O2 clearance via the PRX/TRX pathway (Ren et al., 2017; see Figure 5D), and is expressed in a punctate pattern coincident with the mitochondrial marker apoptosis inhibitory factor (AIF), in layer 2/3 PNsivt (Figure 6B). Following Txnrd2 depletion by shRNA, axon length declines by 29% and dendrite length by 23% (axons: p≤0.0084; dendrites: p≤0.0095; n=50 cells/6 animals/6 litters 1-way ANOVA+Holm-Sidak; Figure 6A,C, and SD7). In parallel, Txnrd2 depletion increases mitochondrial ROS by 27±7.01% and cytosolic ROS by 26.1±10% over NS-WT controls (mito: n=51 cells/4 brains/condition/4 litters, p<0.0058, cyto: n=41 cells/4 brains/condition/4 litters, p<0.042, Student’s T-test; Figure 6D,E). Thus, diminishing Txnrd2 decreases axon and dendrite growth and elevates ROS in WT layer 2/3 PNsivt, reproducing key changes seen selectively in LgDel layer 2/3 PNsivt. To assess the relationship between Txnrd2 expression, layer 2/3 PN growth and ROS, we asked whether the ROS scavenger NAC rescues axon and dendrite growth in shTxnrd2-electroporated neurons as in LgDel (Figure 6F). Axon growth deficits are resolved and total dendrite length increases to WT levels (axons: shRNA-Txnrd2<shRNA-Txnrd2+NAC, p<0.0084; shRNA-Txnrd2+NAC≈nS control, p>0.99; dendrites: shRNA-Txnrd2+NAC> shRNA-Txnrd2, p<0.034; shRNA-Txnrd2+NAC≈NS control, p>0.84; n=50 cells/5 brains/condition/5 litters, 1-way ANOVA+Holm-Sidak; Figure 6F, and SD7).
Figure 6:

Txnrd2 regulates ROS levels, axonal and dendritic growth in layer 2/3 PNivt. A) shRNA-mediated diminished expression (knock-down) of six mitochondrial localized 22q11 genes (Maynard et al., 2008) in LgDel in WT layer 2/3 PNivt. Knock-down of only one gene, Txnrd2, recapitulates LgDel axon and dendrite growth deficits (*p<0.0084), and does so at similar magnitudes seen in LgDel. The mitochondrial ribosomal protein Mrpl40, when depleted, results in a significant increase of axon, but not dendrite length in WT layer 2/3 PNs (*p<0.0049). B) Txnrd2 protein (red) is expressed in a pattern consistent with mitochondrial localization in layer 2/3 PNivt based upon substantial co-localization with the mitochondrial marker Apoptosis Inhibitory Factor (AIF; green) in the cell body and initial segment of neurites. Inset single channel, higher magnification images of Txnrd2 (red) and AIF (green) showing punctate, presumed mitochondrial labeling from a proximal neurite of the layer 2/3 PNivt shown at low power in (B). C) Differences in axon and dendrite growth in non-sense shRNA control (NS-shRNA) and sh-Txnrd2 electroporated layer 2/3 PNs. D) Enhanced mitoSox labeling of mitochondrial ROS in sh-Txnrd2 electroporated WT layer 2/3 PNs (*p,0.0058). E) Enhanced cellROX labeling of cytosolic ROS in sh-Txnrd2 electroporated WT layer 2/3 PNs (*p,0.042). F) NAC-mediated rescue of diminished axon and dendrite growth of sh-Txnrd2 electroporated WT layer 2/3 PNs. NAC restores sh-Txnrd2 layer 2/3 PN axon and dendrite lengths to WT levels, dotted lines, no significant difference (ns). As for non-electroporated WT layer 2/3 PNs, NAC treatment decreases NS-shRNA axon and dendrite growth. G) Txnrd2 re-expression restores LgDel layer 2/3 PN axon and dendrite growth. H, I) Txnrd2 re-expression restores mitochondrial and cytosolic ROS to WT levels in LgDel layer 2/3 PNivt. Insets adjacent to (H) Txnrd2-electroporated cells (t; green) have low levels of mitoSOX (red); untransfected cells (u, not green) have high levels of ROS (red). Insets adjacent to (I): Txnrd2-electroporated cells (t; red) have low levels of cellROX (green); untransfected cells (u, not red) have high levels of ROS (green). At right of (H) and (I) quantification confirms restoration of mitochondrial and intracellular ROS by Txnrd2 re-expression (* p<0.0001). J) Txnrd2 protein (red) in cortical PNs is enhanced in layer 2/3 PNs. The insets, imaged at identical gain, show at higher magnification enhanced Txnrd2 (cytoplasmic labeling; red) in Satb2 expressing (nuclear labeling; green) layer 2/3 (arrowhead) versus 5/6 PNs. K) In P21 layer 2/3 PNs, Txnrd2 protein (punctate cytoplasmic labeling; red) co-localizes with Uqcrc1 (green), a mitochondrial marker. At right, higher magnification images of a single layer 2/3 PN shows Txnrd2 (red; top) labeling of a single layer 2/3 PN as well as the coincident Uqcrc1 labeling in the same neuron (green, bottom). Insets Punctate, presumed mitochondrial, labeling for Txnrd2 (red, top) and Uqcr1 (green, bottom). In the middle panel, arrows indicate punctate co-localization of Txnrd2 and Uqcrc1 in presumed individual mitochondria. L) Apical dendrite length and dendritic branching is significantly decreased in sparsely recombined, eYFP, individual Txnrd2+/− layer 2/3 PNs in vivo, in an otherwise P21 WT mouse compared to WT controls. Apical dendrite retraction is not seen (arrows). Quantification confirms that this decline (apical dendrite length *p<0.0077; apical branch points *p<0.0007; apical max branch order *p<0.0225; basal max branch order *p<0.02) is similar to that in LgDel layer 2/3 PNs in vivo. Pink, shaded horizontal bands on each histogram indicate mean LgDel (LD) values (see Figure 4D) ± S.E.M.
All histogram data represent mean values + SEM. Scale bar 10 μm for 6B. 50 μm for 6C, F, and G. 2.5 μm for 6D, and E. 25 μm for 6L.
We next asked if increasing Txnrd2 levels in LgDel layer 2/3 PNivt above the 50% decrement that results from heterozygous 22q11 deletion (Maynard et al., 2008; Meechan et al., 2007) rescues growth and ROS levels in LgDel layer 2/3 PNivt. Txnrd2 re-expression returns LgDel layer 2/3 PNivt axon and dendrite growth to WT levels (axons: LgDel+Txnrd2>LgDel, p≤0.022; n=50 cells/5 animals/5 litters; dendrites: LgDel+Txnrd2>LgDel, p≤0.028; n=50 cells/5 brains/condition/5 litters; Figure 6G, and SD3). It also decreases mitochondrial and cytosolic ROS to WT levels (LgDel+Txnrd2<LgDel: mito p<0.0001, LgDel+Txnrd2<LgDel: cyto p<0.0001; 1-way ANOVA+Holm-Sidak n=50 cells/4 brains/condition/4 litters; Figure 6H,I, and SD6). Apparently, Pyr, NAC, or Txnrd2 re-expression restores LgDel layer 2/3 PNivt growth deficits, while also diminishing ROS to WT levels.
In the cortex in vivo, Txnrd2 (protein) is enhanced in layer 2/3, but barely detectable in layer 5/6 PNs (Figure 6J). In addition, it co-localizes with Uqcrc1, a mitochondrial respiratory/electron transport chain protein, in individual layer 2/3 PNs (Figure 6K). Thus, we asked if cell-autonomous Txnrd2 deletion in layer 2/3 PNs in vivo recapitulates Txnrd2 depletion phenotypes in vitro using Cux2:CreERT conditional recombination of Txnrd2flox/+ (Conrad et al., 2004) in single layer 2/3 PNs of otherwise WT mice (Figure 6L). Txnrd2+/− layer 2/3 PN dendrite length declines by 21% (Txnrd2+/− < WT, p≤0.0077; n=20 neurons/4 brains/genotype; Figure 6L, and SD4) apical and basal arbors decrease by 53% and 35% (Txnrd2+/− apical< WT, p<0.0007 and Txnrd2+/− basal< WT, p<0.0673 - NS trend; Figure 6L, and SD5), and maximum branch order decreases by 34% and 28% (order, Txnrd2+/− < WT, p≤0.0225, p≤0.020; n=20 neurons/4 brains/genotype/4 litters, 1-way ANOVA+Holm-Sidak; Figure 6L, and SD5). Apical dendrite retraction, seen in LgDel (see Figure 4C) is not seen in Txnrd2fl/+ layer 2/3 PNs (Figure 6L, arrows). Apparently diminished Txnrd2 elevates ROS and diminishes layer 2/3 PN growth and differentiation parallel to LgDel layer 2/3 PN phenotypes that accompany association cortical under-connectivity.
ROS-related rescue of 22q11 deletion-dependent under-connectivity
If Txnrd2 dosage-dependent ROS dysregulation in layer 2/3 PNs in the context of 22q11 gene deletion underlies neuronal deficits leading to cortico-cortical under-connectivity, then NAC treatment in vivo should equally rescue Txnrd2fl/+ and LgDel layer 2/3 PN growth deficits. To establish convergence of diminished Txnrd2 dosage and ROS metabolism on layer 2/3 PN differentiation we asked whether NAC rescues dendritic growth in Txnrd2fl/+ and LgDel layer 2/3 PNs as it does in Txnrd2-depleted WT layer 2/3 PNs in vitro. Neonatal Txnrd2fl/+ and LgDel pups (Txnrd2fl/+ or LgDel+NAC) received NAC via their nursing mothers (das Neves Duarte et al., 2012) from the 1st postnatal day until weaning, and thereafter via their own water supply. NAC from birth through P21 restores dendrite growth, branching, and branch order in individual Txnrd2+/− layer 2/3 PNs or LgDel layer 2/3 PNs to WT levels (Figure 7A,B, SD4, and SD5). All apical dendrite measures in Txnrd2+/−+NAC and LgDel+NAC layer 2/3 PNs are significantly increased from those in untreated Txnrd2+/− and LgDel (dendrite length: Txnrd2+/−+NAC>Txnrd2+/−, p<0.034; branching: p<0.0001; branch order: p<0.0001; dendrite length: LgDel+NAC>LgDel, p<0.0001; branching: p<0.0001; branch order: p<0.0001; Figure 7B). Evidently, Txnrd2 dosage-dependent ROS dysregulation—alone or in the context of broader heterozygous 22q11 deletion—underlies a key aspect of layer 2/3 PN differentiation associated with cortico-cortical under-connectivity.
Figure 7:

NAC rescues neuronal and synaptic deficits in Txnrd2+/− and LgDel PNs. A, B) NAC, administered from birth onward, restores dendrite length and branching deficits in sparsely recombined Txnrd2+/− and LgDel layer 2/3 PNs in vivo to WT levels, but does not compromise WT layer 2/3 PN differentiation. The blue, green, and pink shaded horizontal bands indicate mean values, ± S.E.M, for untreated WT (blue), LgDel (LD, pink) and Txnrd2+/− (T2, green)− neurons (see Figure 4D, 6L). C, D) NAC administered from birth onward restores cytological and synaptic integrity of LgDel layer 2/3 PNs. Mitochondrial and cytoskeletal elements (C1,2; D1,2) and synapses (C3,4; D3,4) are indistinguishable in the two genotypes. E) Quantitative analysis of mitochondrial frequency, synapse frequency, and synaptic vesicle density confirms restoration of layer 2/3 PN and neuropil cellular and synaptic integrity. Blue (WT) and pink (LD: LgDel) horizontal bands indicate values for untreated layer 2/3 PNs (see Figure 2E). F) NAC, administered from birth onward, restores deficits in visual reversal learning in LgDel mice to WT levels (*p=0.005; *p=0.0385 2-way ANOVA, reversal session 1–4 scores × genotype, Holm-Sidak). The blue and pink horizontal bars provide a visual indication of the mean sessions to criterion values, ± SEM, for untreated WT and LgDel (LD) mice, respectively.
All histogram data represent mean values + SEM. Scale bar 25 μm for 7A. 5 μm for 7C.
NAC restoration of layer 2/3 PN dendrite growth and branching suggests that layer 2/3 PN cytological and synaptic integrity may also be rescued. We therefore analyzed cellular, dendritic, and synaptic morphology of LgDel and WT layer 2/3 PNs after NAC treatment from birth onward. NAC treatment in WT neurons does not appear to affect cytoskeletal or organelle morphology (Figure 7C). Cytoskeletal elements and organelle integrity in LgDel+NAC layer 2/3 PNs are similar to WT (compare Figure 2A, A1–4 with Figure 7D, D1–4). Mitochondria are no longer dilated and dysmorphic (Figure 7D, D1–4, and SD2; aspect ratios: LgDel+NAC>LgDel; cell body.: 1.91±0.1, n=127 mito/7 neurons/3 animals, p<0.0226; apical dendr.: 3.17±0.24, n=69 mito/8 neurons/3 animals, p<0.0261; 1-way ANOVA+Holm-Sidak). Layer 2/3 apical dendrite pre-synaptic specializations are restored to WT appearance (compare Figure 2B5,6 with Figure 7D3,4). NAC treatment increases LgDel cell body and apical dendrite mitochondrial frequency; however, neither increase reaches statistical significance. In contrast, neuropil mitochondria frequency is significantly greater in NAC-treated LgDel (p<0.005; Figure 7E, and SD1). We note that WT mitochondrial frequency decreases in response to NAC treatment. This change, however, is not accompanied by qualitative or quantitative signs of oxidative stress (see above). Moreover, NAC treatment does not alter WT layer 2/3 PN dendrite growth (see Figure 7A,B). Apical dendrite synapse frequency also increases significantly to WT levels in NAC-treated LgDel (p<0.009; Figure 7E, and SD1). Finally, synaptic vesicle density is significantly greater than untreated LgDel (p<0.008; Figure 7E, and SD1). Apparently, NAC corrects mitochondrial stress3related under-connectivity by restoring cytological, mitochondrial and synaptic integrity of layer 2/3 PNs in LgDel mice.
ROS modulation influences LgDel cognitive behavioral performance
NAC restoration of cytological and synaptic integrity in LgDel layer 2/3 PNs, if relevant to the proposed under-connectivity hypothesis of anomalous function in 22q11DS and other neurodevelopmental disorders, predicts parallel restoration of behavioral capacity. We confirmed a significant deficit for an mFAC-laEC mediated visual reversal-learning task we reported previously for LgDel mice (Meechan et al., 2015b) in a new, larger cohort of young adult LgDel (n=18) and WT (n=13) animals from 10 independent litters (LgDel>WT sessions to criterion, p<0.0051; 2-way ANOVA+Holm-Sidak; Figure 7F). We then did the same behavioral analysis at similar ages in NAC-treated LgDel (n=8) and NAC-treated WT (n=7) counterparts from a total of 4 WT and 6 LgDel litters. In NAC-treated LgDel mice, the number of reversal sessions necessary to learn and perform the task returns to WT untreated levels, significantly improved from that in untreated LgDel mice (p<0.0385; 2-way ANOVA+Holm-Sidak; Figure 7F, and SD8). NAC-treated WT and untreated WT mice are statistically indistinguishable. Thus, NAC resolves cortico-cortical related cognitive deficits in concert with restoring layer 2/3 PN dendritic growth and differentiation as well as neuronal, mitochondrial and synaptic integrity in LgDel mice.
DISCUSSION
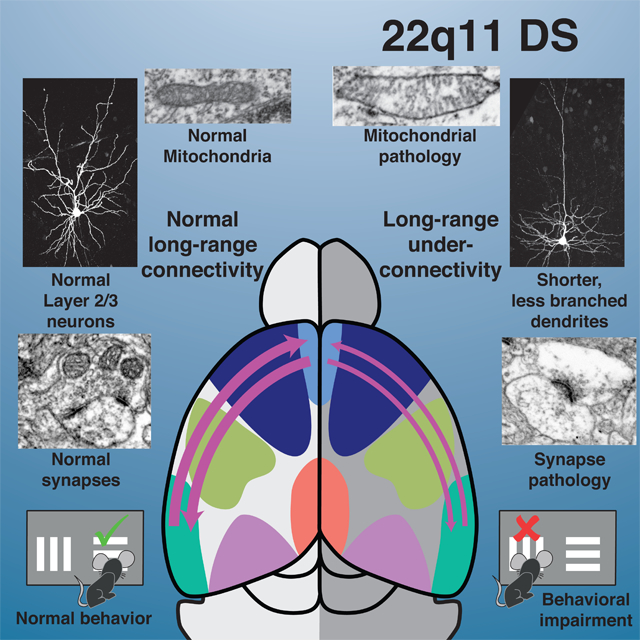
Quantitative under-connectivity of association cortices underlies cognitive deficits in the LgDel mouse model of 22q11DS. This under-connectivity arises due to inefficient reactive oxygen species (ROS) catabolism and mitochondria3associated oxidative stress in layer 2/3, but not 5/6 PNs. The disruption of layer 2/3 PN anti-oxidant defense and its cellular consequences reflect diminished dosage of one 22q11 gene, Txnrd2, a rate-limiting enzyme for ROS clearance in neuronal mitochondria. Anti-oxidants or Txnrd2 re-expression corrects quantitative cellular deficits associated with LgDel layer 2/3 PN-selective under-connectivity. In parallel, anti-oxidant treatment resolves a cognitive deficit associated with cortico-cortical under-connectivity. Thus, we identify a layer 2/3 PN selective developmental mechanism for establishing and maintaining quantitatively appropriate association cortico-cortical connectivity and related behavioral capacity (Figure 8). Based upon these observations, we define a safe, effective therapy that corrects mitochondrial dysfunction, under-connectivity, and cognitive impairment that arises due to diminished gene dosage following heterozygous 22q11 deletion.
Figure 8:

Mitochondrial regulation of cortico-cortical connectivity in developing and adult association cortices. Top Row; left to right: Normal quantitative connectivity is established by layer 2/3 projection neurons that extend axons between distal as well as local association areas. These connections rely critically upon mitochondria-mediated metabolic homeostasis for initial growth and elaboration of axons and dendrites, as well as cytological and synaptic integrity. The mitochondrial enzyme, Thioredoxin-reductase 2, a regulator of reactive oxygen species clearance in the mitochondrial matrix, is key in regulating layer 2/3 projection neuron growth and cortico-cortical connections made by these neurons. Finally, when long distance cortico-cortical connections, in this instance between medial frontal and lateral entorhinal association cortices, are quantitatively sufficient, mice perform optimally on a complex cognitive task, visual reversal learning. Bottom Row: left to right: Disruption of mitochondrial regulation of ROS levels due to diminished Txnrd2 dosage in the LgDel mouse model of 22q11.2 Deletion Syndrome reduces connectivity by selectively compromising layer 2/3 PN growth and synaptic differentiation. These ROS-dependent quantitative changes are rescued by Txnrd2 re-expression in vitro, or by the ROS scavenger N-acetyl cysteine in vitro or in vivo. The restoration of several aspects of mitochondria-regulated layer 2/3 PN cortico-cortical connectivity is accompanied by restored performance on the visual reversal task.
LgDel quantitative under-connectivity is selective for layer 2/3 PNs. Layer 5/6 PNs show no signs of oxidative stress, altered synaptic integrity, or diminished axon and dendrite growth. Of six 22q11 mitochondrial genes—obvious candidates for LgDel layer 2/3 PN ROS dysregulation—only Txnrd2 depletion elevates ROS and diminishes layer 2/3 PN growth similar to full 22q11 deletion. Enhanced Txnrd2 protein levels in layer 2/3, but not layer 5/6, PNs as well as consequences of layer 2/3 PN cell autonomous heterozygous Txnrd2 deletion in vivo suggest Txnrd2-mediated mitochondrial ROS catabolism is necessary to meet bioenergetic demands of developing or mature layer 2/3 PNs (Figure 8). Furthermore, N-acetyl Cysteine (NAC), which selectively enhances ROS catabolism via the TRX/PRX pathway—for which Txnrd2 is rate limiting—rescues Txnrd2-associated layer 2/3 PN deficits in vitro and in vivo. Thus, Txnrd2, independently, or in the context of 22q11 deletion, emerges as a key regulator of mitochondrial metabolism essential to establish and maintain quantitatively appropriate association cortico-cortical connections made by layer 2/3 PNs. Txnrd2, expressed at varying levels, may regulate ROS-dependent growth and homeostasis in several types of CNS projection neurons whose axons extend for distances longer than layer 5/6 PNs but equivalent to or greater than those traversed by layer 2/3 PNs, or in tonically active neurons with enhanced metabolic demands.
LgDel under-connectivity targets development and maintenance of layer 2/3 cortico-cortical circuits in medial frontal association cortex (mFAC), essential for cognitive behaviors (Bicks et al., 2015; Carlen, 2017). Murine association cortices receive little or no innervation from thalamic relay nuclei and send few, if any, efferents to cortico-bulbar or spinal targets (DeNardo et al., 2015); projections are mostly from and to other cortical regions. Thus, diminished layer 2/3 PN connections should disproportionately impact behaviors that rely upon these association cortico-cortical circuits, including those that rely upon integrity of mFAC cortico-cortical circuits. Our results are consistent with this assumption, and associate quantitative change in circuit elements with behavioral change. Elimination of deficits in the reversal-learning task, in parallel with robust restoration of multiple in vivo measures of cortico-cortical under-connectivity, reinforces this assertion. We suggest that developing new approaches to restore quantitative aspects of connectivity—even if earlier developmental disruptions cannot be repaired—can correct or at least ameliorate deficits in complex behaviors that rely critically upon association cortico-cortical connections.
Some aspects of layer 2/3 cortico-cortical under-connectivity, however, may be more amenable to adjustment than others. We show that LgDel layer 2/3 PN growth and synaptic integrity can be modified toward WT levels; however, adjusting the overall number of layer 2/3 PNs that project to distal versus proximal association cortical regions may be more challenging. Layer 2/3 PN frequency declines in LgDel association cortices including mFAC due to altered neurogenesis, thus limiting the number of PNs available for cortico-cortical connections (Meechan et al., 2009). This decline also selectively influences cognitive performance (Meechan et al., 2015b), consistent with a role for quantitative connectivity changes in disruption of complex behaviors. Nevertheless, a different subset of 22q11 genes, notably Ranbp1 (Paronett et al., 2015) and Zdhhc8 (Mukai et al., 2015), appears to mediate neurogenesis deficits. Moreover, these dosage-dependent phenotypes—unlike those we associate with postnatal mitochondrial dysfunction—cannot be easily corrected due to limited access (fetal development) and selectivity (non-specific drugs).
We find that these limited numbers of LgDel mFAC layer 2/3 PNs make fewer long distance and more short distance connections, perhaps due to lower bioenergetic demands necessary to generate a local versus long projecting axon. These changes, and parallel mitochondrial deficits, most likely arise shortly after birth—around the time that Txnrd2 reaches an expression peak perinatally (Maynard et al., 2008), in register with final stages of layer 2/3 PN axon, dendrite and synapse differentiation. Cortico-cortical connectivity may be restored by promoting terminal branching of a diminished number of long distance projection axons or adding local as well as intrinsic cortico-cortical target space (dendrites) or synaptic inputs. This scenario is consistent with observations made in children and adults with neurodevelopmental disorder-related behavioral deficits. In these individuals, long-distance cortico-cortical connectivity, inferred from in vivo imaging, is diminished, while short distance or intrinsic cortico-cortical connectivity is enhanced (Barttfeld et al., 2011; Wass, 2011). We show that quantitative restoration of association cortical microcircuit elements from either local (enhanced layer 2/3 PN dendritic growth) or extrinsic sources (additional synapses from additional long distance axon terminal branches or increased short distance inputs) is sufficient to correct behavioral deficits that reflect LgDel layer 2/3 under-connectivity (Figure 8).
We chose N-acetyl cysteine (NAC), a ROS scavenging anti-oxidant, to restore oxidative-stress related under-connectivity in the LgDel mouse because it has no known off-target effects (Atkuri et al., 2007). Moreover, following amidase conversion, it provides cysteine reducing equivalents—presumably replacing those lost due to diminished Txnrd2 dosage—to increase glutathione/glutaredoxin activity (Dringen and Hamprecht, 1999), restoring ROS catabolism and redox homeostasis (Lu and Holmgren, 2014; Zhang et al., 2007). NAC has no detectable impact on WT layer 2/3 PNs, with two exceptions. Axon and dendrite growth of NAC-treated WT layer 2/3 PNs in vitro declines modestly, perhaps due to homeostatic responses to acute changes in ROS levels below baseline or lack of mechanisms to buffer this ROS disequilibrium in the absence of other cell classes, particularly glial cells in vitro. Nevertheless, NAC treatment in vivo does not alter WT layer 2/3 PN dendrite growth. There is, however, a 50% decrease in apical dendrite mitochondria density. These mitochondria, however, are not dysmorphic: aspect ratios are indistinguishable from WT and significantly greater than LgDel. Finally, NAC does not impair WT cognitive behavioral performance. Thus, NAC emerges as a relatively safe and effective therapeutic intervention for oxidative stress-related under-connectivity due to 22q11 gene deletion. Optimal NAC doses, timely diagnosis, and treatment regimens remain to be established, and additional anti-oxidants with equal or increased efficacy and greater specificity remain to be identified.
22q11.2DS has the highest known genetic association with a number of more common neurodevelopmental disorders that fall within broader clinical/diagnostic categories (Marshall et al., 2017; Sanders et al., 2015). Behavioral pathogenesis in these fairly common non-syndromic disorders is thought to reflect altered connections between complex association cortices (Richter et al., 2015; van Erp et al., 2018), parallel to current hypotheses of behavioral pathogenesis in 22q11DS. Our data establish a new framework for considering the origins of selectively disrupted association cortical connections and function, especially in networks defined primarily by association cortico-cortical connections made by layer 2/3 PNs. In many neurodevelopmental disorders beyond 22q11DS, developmental and adult mitochondrial dysfunction and resulting oxidative stress may be a common pathogenic mechanism for association cortical under-connectivity and circuit dysfunction. Such changes provide an accessible, biologically defined target amenable to novel therapies that stabilize or enhance mitochondrial function, restore essential circuit elements, and improve behavior.
STAR METHODS
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Anthony LaMantia (lamantia@gwu.edu)
Animals
The George Washington University IACUC-monitored Animal Resource Facility maintained C57BL/6N (Charles River Laboratories) and LgDel mice (Merscher et al., 2001). Mutant lines were backcrossed for > 10 generations to C57BL/6N for breeding stock. The LgDel mutation (Idd to Hira, mmChr 16) was transmitted paternally. Timed pregnancies were confirmed by vaginal plug (E0.5). Fetuses were genotyped post-hoc for LgDel as well as additional Cre driver, or floxed target alleles using PCR; WT littermates were controls. In vivo experiments used Ai3(RCL-EYFP) designated eYFP (Jackson#007903), Cux2tm2.1(cre)Mull; (Gil-Sanz et al., 2015), designated Cux2:CreERT (U.Mueller) to visualize layer 2/3 PNs and recombine the Txnrd2tm1.1Marc locus (Conrad et al., 2004), designated Txnrd2fl (M. Brielmeier).
Stereotactic injections
Mature male mice were anesthetized with urethane, monitored for continuous anesthesia, injected at 2mm depth with biocytin-conjugated Alexafluor488 (Thermo Fisher Scientific) 1.0 mm forward of bregma, and 0.1 mm lateral to the midline, and after 2 hrs perfused transcardially (4% paraformaldehyde in NaPO4 buffer, ph7.4). Coronal vibratome sections (100 μm) were cut, coverslipped, imaged at high magnification, and composite images generated using a Cell Observer Confocal Microscope (w/Zen software; Zeiss). Labeled cells were plotted and their angle relative to the mid-point of the cortical midline axis was calculated using ImageJ software (see Figure 1) and represented in raster plots from A-P levels standardized based upon independent neuroanatomical landmarks.
In vivo visualization of layer 2/3 PNs
Litters from Cux2:CreERT;eYFP;LgDel × C57BL/6N and Cux2:CreERT;eYFP;Txnrd2fl/+ × C57BL/6N crosses were generated. Animals used for these experiments were not sexed. Recombination was induced in these pups via maternal lactation from nursing dams injected with tamoxifen (dissolved in ethanol and corn oil; final dose, 1mg) once at P1, twice at P2, and once at P3 (Mayes et al., 2011). Dams and pups were undisturbed from P3 until P21, when brains were collected from 4% paraformaldehyde perfused weanlings of both sexes. Thick slices (300μm) were cut and tissue was processed for immunofluorescence. After immunostaining with anti-GFP antibody (chicken, Abcam), slices were dehydrated and cleared with benzyl alcohol: benzyl benzoate (BABB).
Primary cultures, ex vivo electroporations, ROS measurement
Electroporation was performed as previously described with slight modifications (Hand et al., 2005). Dorsal telencephalic progenitors were co-transfected with pCI-mbGFP or pCI-mitoCherry injected into the lateral ventricles of E14.5 or 16.5 embryonic mouse brains; cortices were then dissected, dissociated, and cultured for 5 days (Ahlemeyer and Baumgart-Vogt, 2005). We analyzed 5 days in vitro (5 Divt) cells with at least two visible independent dendrites and an axon at least twice as long as the longest dendrite. Cytosolic and mitochondrial ROS levels were measured at 5 Divt using mitoSOX and cellROX reagents and protocol (Thermo Fisher Scientific). Neurons were traced using imageJ software which calculated average pixel intensity within the outlined cell area. Any nuclear labeling was excluded.
DNA constructs
We identified unique sequences (NCBI nucleotide BLAST) to target shRNA in pSh-MitoGFP/cherry vectors to each 22q11 mitochondria gene. shRNA efficacy was assessed by repression of GFP-fusion constructs expressed in HEK293 cells. A “nonsense” shRNA construct served as a negative control for layer 2/3 PNivt. For overexpression, we sub-cloned the Txnrd2 ORF into pCIG (Megason and McMahon, 2002), with a composite CMV-enhancer/chicken β-actin promoter and EMCV IRES-reporter. The first expression vector (pCI-mbGFP) co-expresses a membrane-targeted GFP (containing the 20 N-terminal amino acids of mouse Gap43, including a fsylation signal sequence), while the second (pCI-mitoGFP/pCI-mitoCherry) co-expresses either a mitochondrially-targeted EGFP or pCI-mitoCherry (utilizing the N-terminal signal peptide of human COX8A for targeting). Derivative versions of this vector were made for shRNA experiments (pSh-MitoGFP/Cherry), by adding a shRNA (short-hairpin) expression cassette obtained from pSilencer 2.1 (Ambion).
Anti-oxidant treatments in vitro and in vivo
Sodium Pyruvate (Pyr) (Gibco) or N-acetyl Cysteine (NAC) (Sigma) was added to LgDel, WT, sh-Txnrd2 or Txnrd2 electroporated layer 2/3 PNivt at 1mM final concentration at plating and media changes at 24 (1 Divt) and 96 hours (4 Divt). LgDel and Txnrd2fl/+ litters received NAC (PharmaNAC) via maternal lactation (900mg/l in maternal drinking water; das Neves Duarte et al., 2012) from P0 through weaning. For weanlings, NAC (PharmaNAC) was provided in drinking water (900 mg/l, fresh solutions 3×/week (Cabungcal et al., 2013) until tissue harvesting and/or completion of behavioral experiments.
Immunocytochemistry
Cortical cultures were fixed in 4% paraformaldehyde at 37° for 15 minutes. P21 Animals were anesthetized and perfused with 4% paraformaldehyde; brains were post-fixed overnight and then processed for histology. The following 1° antibodies were used: NeuN (mouse, Millipore), GFAP (rabbit, Millipore), Cux2 (rabbit, Abcam), Ctip2 (rat, Abcam), GFP (chicken, Abcam), Txnrd2 (rabbit, Abcam), Apoptosis Inhibitory Factor (AIF; mouse, Novus) Satb2 (mouse, Abcam), Uqcrc1 (mouse, Invitrogen); 2° antibodies (Alexa-fluor; various species, Molecular Probes).
Imaging and analysis
All imaging and morphometry was performed blind to genotype. Images were obtained using a Zeiss Cell Observer spinning disk confocal microscope. Isolated, fully labeled PNivt were selected randomly, tiled images obtained with a 63× objective at 512×512 pixels resolution, and maximal projection z-stacks of whole neurons analyzed using SynD automated software (Schmitz et al., 2011) to measure total dendrite or axon length. Sections (300μm) from Cux2:CreERT;eYFP cortices were imaged at 2048×2048 pixels on a Leica TSC SP8MP. Due to sparse labeling, nearly every fully labeled neuron in sections through the relevant cortical area was imaged and analyzed. 3D reconstruction and branch analysis used TREES toolbox automated software (Cuntz et al., 2010). We used 1 and 2-way ANOVA with Holm-Sidak post-hoc tests to evaluate statistical differences.
Electron microscopy
All adult mice tested were male with the exception of 1 LgDel female treated with NAC, and 1 WT female treated with NAC. Mice were anesthetized and perfused transcardially (2.5 % glutaraldehyde, 4% paraformaldehyde in 0.1 M Na-cacodylate buffer). Coronal brain sections (100 μm) were post-fixed in OsO4, infiltrated with aqueous uranyl acetate, dehydrated and flat embedded in resin. Ultrathin (120 nm) sections were cut and placed on a silicon wafer. Images were generated with an FEI Helios FIBSEM equipped with a backscatter (CBS) detector and Helios scanning stage driven by FEI MAPS software. The section was imaged (6003700×) as a low-resolution map, a layer 2/3 or 5/6 region selected, and then high-resolution images captured including entire PNs overlapped 20% with MAPS software to yield high resolution images (1500 pixels resolution, horizontal field; 1 pixel=2.4 nm) for quantitative analysis. These images were then sampled using a rectangular “frame” of standard size that framed the entire neuron, and elements were counted in the cell, and in the surrounding neuropil. A systematic sample was made of pre-and post-synaptic neuropil processes using a standard counting grid, and sampling every other grid square. All EM quantification was performed blind to genotype by at least two independent observers.
Behavioral Testing and Analysis
LgDel and WT mice with or without NAC were trained in an automated touchscreen apparatus, blind to genotype. All mice tested were male with the exception of 1 LgDel female treated with NAC, and 1 WT female treated with NAC. Mice were initially trained to select 1 of 2 visual stimuli for food reward. A daily testing session=20 trials (1st choice trials); however, a correction procedure was used so that following a first incorrect choice the trial was repeated until the animal made the correct response. Once criterion was reached (80% correct over 2 consecutive days) on initial discrimination, reward contingencies were switched and the animal tested until reaching the same criterion (Reversal 1) (Meechan et al., 2015b). The rewarded stimulus was alternated for 3 additional reversals, data are average number of sessions to criterion over the 4 reversals.
Statistics
Comparison between groups was tested by ANOVA with post-hoc tests, or Student’s T-test. Statistical comparisons of data which spanned more than one figure are provided in Supplementary data.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-NeuN | Millipore | Cat# MAB377; RRID: AB_2298772 |
| rabbit anti-GFAP | Millipore | Cat# AB5804; RRID: AB_2109645 |
| rabbit anti-Cux2 | Abcam | Cat# ab130395; RRID: AB_11155898 |
| rat anti-Ctip2 | Abcam | Cat# ab18465; RRID: AB_2064130 |
| chicken anti-GFP | Abcam | Cat# ab13970; RRID: AB_300798 |
| rabbit anti-Txnrd2 | Abcam | Cat# ab1684; RRID: AB_302540 |
| mouse anti-AIF | Novus | Cat# NBP2–37577 |
| mouse anti-Satb2 | Abcam | Cat# ab51502; RRID: AB_882455 |
| mouse anti-Uqcrc1 | Thermo Fisher Scientific | Cat# 459140; RRID:AB_2532227 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sodium Pyruvate | Gibco | Cat# 11360–070 |
| Biocytin Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A12924 |
| CellROX | Thermo Fisher Scientific | Cat# C10444 |
| MitoSOX | Thermo Fisher Scientific | Cat# M36008 |
| Tamoxifen | Enzo Life Sciences | Cat# ALX-550–095-G001 |
| N-Acetyl-L-cysteine | Sigma | Cat# A9165–5G |
| PharmaNAC 900mg tablets | BioAdvantex Pharma | |
| Experimental Models: Organisms/Strains | ||
| Mouse: LgDel : B6.Cg-Del(16Dgcr2-Hira)1Rak/+ | Merscher et al., 2001 | RRID:MGI:5645346 |
| Mouse: Txnrd2fl : B6.129P2/OlaHsd-Txnrd2tm1.1Marc | Conrad et al., 2004 | MGI Cat# 3512409 |
| Mouse: eYFP : B6.Cg-Gt(ROSA)26Sortm3(CAG-EYFP)Hze | Jackson laboratory | RRID:IMSR_JAX:007903 |
| Mouse: Cux2: CreERT : tm3.1(cre/ERT2)Mull | Gil-Sanz et al., 2015 | RRID:MGI:5298040 |
| Oligonucleotides | ||
| shTxnrd2: GATCCCCCAACACAACTGGAAGACAATTCAAGAGATTGTCTTCCAGTTGTGTTGTTTTTCTCGA | This paper | N/A |
| shProdh: GATCCCCGGGCAAAGGATGTTCGGATTCAAGAGATCTCGAACATCCTTTGCCCTTTTTCTCGA | This paper | N/A |
| shZdhhc8: GATCCCCCCACGTCTGATGTGTTAGTTTCAAGAGAACTAACACATCAGACGTGGTTTTTCTCGA | This paper | N/A |
| shTango2: GATCCCCCAGAGGGCCACCTGTATAATTCAAGAGATTATACAGGTGGCCCTCTGTTTTTCTCGA | This paper | N/A |
| shMrpl40: GATCCCCACACACAAGTAGAATTCAATTCAAGAGATTGAATTCTACTTGTGTGTTTTTTCTCGA | This paper | N/A |
| shSlc25a1: GATCCCCCGACAGCAGGAGAGGACTATTCAAGAGATAGTCCTCTCCTGCTGTCGTTTTTCTCGA | This paper | N/A |
| shNon: GATCCCCGGAGATTAGCTATTAGGGATTCAAGAGACCTCTAATCGATAATCCCTTTTTTCTCGA | This paper | N/A |
| Recombinant DNA | ||
| pSH-mitoGFP | This paper | N/A |
| pSH-mitoCherry | This paper | N/A |
| pC-fEGFP | This paper | N/A |
| pC-fEGFP -Txnrd2 | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism5 | https://www.graphpad.com/scientific-software/prism/ | |
| ImageJ | https://imagej.nih.gov/ij/; RRID: SCR_003070 | |
| TreesToolbox | Cuntz et al., 2010 | https://www.treestoolbox.org/ |
| Microsoft Excel | https://products.office.com/en-US/ | |
| Adobe Creative Cloud | https://www.adobe.com/creativecloud.html | |
| Imaris 9.1 | http://www.bitplane.com/imaris/imaris; RRID: SCR_007370 | |
| SynD | Schmitz et al., 2011 | http://www.johanneshjorth.se/SynD/SynD.html |
Acknowledgements:
The National Institute of Child Health and Human Development (HD042182, ASL), Mental Health (F31 MH103021, AF), and the Simons Foundation (SFARI 306796, ASL and SFARI 342005, TM) supported this work. The GW Nanofabrication and Imaging Center is also supported by the NIH National Center for Research Resources (S10RR025565, U54HD090257). We thank Drs. U. Mueller and M. Brielmeier for mouse lines. Cheryl Clarkson-Pardes provided assistance with EM preparation and imaging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: The authors declare no competing interests.
References
- Ahlemeyer B, and Baumgart-Vogt E (2005). Optimized protocols for the simultaneous preparation of primary neuronal cultures of the neocortex, hippocampus and cerebellum from individual newborn (P0.5) C57Bl/6J mice. J Neurosci Methods 149, 110–120. [DOI] [PubMed] [Google Scholar]
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Farinas I, Grosschedl R, and McConnell SK (2008). Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 57, 364–377. [DOI] [PubMed] [Google Scholar]
- Aruoma OI, Halliwell B, Hoey BM, and Butler J (1989). The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free radical biology & medicine 6, 593–597. [DOI] [PubMed] [Google Scholar]
- Atkuri KR, Mantovani JJ, Herzenberg LA, and Herzenberg LA (2007). N-Acetylcysteine:--a safe antidote for cysteine/glutathione deficiency. Current opinion in pharmacology 7, 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AP (2005). Inter- and intra-laminar connections of pyramidal cells in the neocortex. Neuroscience research 53, 95–103. [DOI] [PubMed] [Google Scholar]
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, and Polleux F (2007). LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 129, 549–563. [DOI] [PubMed] [Google Scholar]
- Barttfeld P, Wicker B, Cukier S, Navarta S, Lew S, and Sigman M (2011). A big-world network in ASD: dynamical connectivity analysis reflects a deficit in long-range connections and an excess of short-range connections. Neuropsychologia 49, 254–263. [DOI] [PubMed] [Google Scholar]
- Berk M, Malhi GS, Gray LJ, and Dean OM (2013). The promise of N-acetylcysteine in neuropsychiatry. Trends in pharmacological sciences 34, 167–177. [DOI] [PubMed] [Google Scholar]
- Bicks LK, Koike H, Akbarian S, and Morishita H (2015). Prefrontal Cortex and Social Cognition in Mouse and Man. Frontiers in psychology 6, 1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birben E, Sahiner UM, Sackesen C, Erzurum S, and Kalayci O (2012). Oxidative stress and antioxidant defense. World Allergy Organ J 5, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bota M, Sporns O, and Swanson LW (2015). Architecture of the cerebral cortical association connectome underlying cognition. Proceedings of the National Academy of Sciences of the United States of America 112, E2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, et al. (2008). Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron 57, 378–392. [DOI] [PubMed] [Google Scholar]
- Cabungcal JH, Steullet P, Kraftsik R, Cuenod M, and Do KQ (2013). Early-life insults impair parvalbumin interneurons via oxidative stress: reversal by N-acetylcysteine. Biological psychiatry 73, 574–582. [DOI] [PubMed] [Google Scholar]
- Carlen M (2017). What constitutes the prefrontal cortex? Science 358, 478–482. [DOI] [PubMed] [Google Scholar]
- Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, Beck H, Hatzopoulos AK, Just U, Sinowatz F, et al. (2004). Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Molecular and cellular biology 24, 9414–9423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe DA, Goodwin SJ, Blackman RK, Sakellaridi S, Sponheim SR, MacDonald AW 3rd, and Chafee MV (2013). Prefrontal neurons transmit signals to parietal neurons that reflect executive control of cognition. Nature neuroscience 16, 1484–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuntz H, Forstner F, Borst A, and Hausser M (2010). One rule to grow them all: a general theory of neuronal branching and its practical application. PLoS Comput Biol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza RD, and Burkhalter A (2017). A Laminar Organization for Selective Cortico-Cortical Communication. Frontiers in neuroanatomy 11, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- das Neves Duarte JM, Kulak A, Gholam3Razaee MM, Cuenod M, Gruetter R, and Do KQ (2012). N-acetylcysteine normalizes neurochemical changes in the glutathione-deficient schizophrenia mouse model during development. Biological psychiatry 71, 1006–1014. [DOI] [PubMed] [Google Scholar]
- DeNardo LA, Berns DS, DeLoach K, and Luo L (2015). Connectivity of mouse somatosensory and prefrontal cortex examined with trans3synaptic tracing. Nature neuroscience 18, 1687–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, and Kittler JT (2018). Mitochondria at the neuronal presynapse in health and disease. Nature reviews Neuroscience 19, 63–80. [DOI] [PubMed] [Google Scholar]
- Dey S, Sidor A, and O’Rourke B (2016). Compartment-specific Control of Reactive Oxygen Species Scavenging by Antioxidant Pathway Enzymes. The Journal of biological chemistry 291, 11185–11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, and Hamprecht B (1999). N-acetylcysteine, but not methionine or 2-oxothiazolidine-4-carboxylate, serves as cysteine donor for the synthesis of glutathione in cultured neurons derived from embryonal rat brain. Neuroscience letters 259, 79–82. [DOI] [PubMed] [Google Scholar]
- Ellegood J, Anagnostou E, Babineau BA, Crawley JN, Lin L, Genestine M, DiCicco-Bloom E, Lai JK, Foster JA, Penagarikano O, et al. (2015). Clustering autism: using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Molecular psychiatry 20, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JM (2009). Cortex and memory: emergence of a new paradigm. Journal of cognitive neuroscience 21, 2047–2072. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, and Levitt P (2007). Autism spectrum disorders: developmental disconnection syndromes. Current opinion in neurobiology 17, 103–111. [DOI] [PubMed] [Google Scholar]
- Gil-Sanz C, Espinosa A, Fregoso SP, Bluske KK, Cunningham CL, Martinez-Garay I, Zeng H, Franco SJ, and Muller U (2015). Lineage Tracing Using Cux2-Cre and Cux2-CreERT2 Mice. Neuron 86, 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SS, Jiang H, Reiss AL, and Greicius MD (2013). Identifying large-scale brain networks in fragile X syndrome. JAMA psychiatry 70, 1215–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C, et al. (2005). Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron 48, 45–62. [DOI] [PubMed] [Google Scholar]
- Hung CH, Cheng SS, Cheung YT, Wuwongse S, Zhang NQ, Ho YS, Lee SM, and Chang RC (2018). A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox biology 14, 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just MA, Cherkassky VL, Keller TA, and Minshew NJ (2004). Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain: a journal of neurology 127, 1811–1821. [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JA, and Willems PH (2005). Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. American journal of physiology Cell physiology 289, C881–890. [DOI] [PubMed] [Google Scholar]
- Kwan KY (2013). Transcriptional dysregulation of neocortical circuit assembly in ASD. International review of neurobiology 113, 167–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston C, Malter Cohen M, Teslovich T, Levenson D, and Casey BJ (2011). Atypical prefrontal connectivity in attention-deficit/hyperactivity disorder: pathway to disease or pathological end point? Biological psychiatry 69, 1168–1177. [DOI] [PubMed] [Google Scholar]
- Lodato S, and Arlotta P (2015). Generating neuronal diversity in the mammalian cerebral cortex. Annual review of cell and developmental biology 31, 699–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, and Holmgren A (2014). The thioredoxin antioxidant system. Free radical biology & medicine 66, 75–87. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, et al. (2017). Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nature genetics 49, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayes DA, Rizvi TA, Cancelas JA, Kolasinski NT, Ciraolo GM, Stemmer-Rachamimov AO, and Ratner N (2011). Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer research 71, 4675–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, Sugimoto TJ, Wu Y, Lieberman JA, and Lamantia AS (2008). Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Molecular and cellular neurosciences 39, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, et al. (2015). 22q11.2 deletion syndrome. Nature reviews Disease primers 1, 15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna WL, Ortiz-Londono CF, Mathew TK, Hoang K, Katzman S, and Chen B (2015). Mutual regulation between Satb2 and Fezf2 promotes subcerebral projection neuron identity in the developing cerebral cortex. Proceedings of the National Academy of Sciences of the United States of America 112, 11702–11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Maynard TM, Gopalakrishna D, Wu Y, and LaMantia AS (2007). When half is not enough: gene expression and dosage in the 22q11 deletion syndrome. Gene expression 13, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Maynard TM, Tucker ES, Fernandez A, Karpinski BA, Rothblat LA, and LaMantia AS (2015a). Modeling a model: Mouse genetics, 22q11.2 Deletion Syndrome, and disorders of cortical circuit development. Progress in neurobiology 130, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Rutz HL, Fralish MS, Maynard TM, Rothblat LA, and LaMantia AS (2015b). Cognitive ability is associated with altered medial frontal cortical circuits in the LgDel mouse model of 22q11.2DS. Cerebral cortex 25, 1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Tucker ES, Maynard TM, and LaMantia AS (2009). Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proceedings of the National Academy of Sciences of the United States of America 106, 16434–16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megason SG, and McMahon AP (2002). A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development 129, 2087–2098. [DOI] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. (2001). TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104, 619–629. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, and Macklis JD (2005). Fezl is required for the birth and specification of corticospinal motor neurons. Neuron 47, 817–831. [DOI] [PubMed] [Google Scholar]
- Mukai J, Tamura M, Fenelon K, Rosen AM, Spellman TJ, Kang R, MacDermott AB, Karayiorgou M, Gordon JA, and Gogos JA (2015). Molecular substrates of altered axonal growth and brain connectivity in a mouse model of schizophrenia. Neuron 86, 680–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padula MC, Schaer M, Scariati E, Schneider M, Van De Ville D, Debbane M, and Eliez S (2015). Structural and functional connectivity in the default mode network in 22q11.2 deletion syndrome. Journal of neurodevelopmental disorders 7,23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronett EM, Meechan DW, Karpinski BA, LaMantia AS, and Maynard TM (2015). Ranbp1, Deleted in DiGeorge/22q11.2 Deletion Syndrome, is a Microcephaly Gene That Selectively Disrupts Layer 2/3 Cortical Projection Neuron Generation. Cerebral cortex 25, 3977–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D, Hadley RD, and Voyvodic JT (1986). Dynamic changes in the dendritic geometry of individual neurons visualized over periods of up to three months in the superior cervical ganglion of living mice. J Neurosci 6, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reep RL, Corwin JV, Hashimoto A, and Watson RT (1987). Efferent connections of the rostral portion of medial agranular cortex in rats. Brain research bulletin 19, 203–221. [DOI] [PubMed] [Google Scholar]
- Ren X, Zou L, Zhang X, Branco V, Wang J, Carvalho C, Holmgren A, and Lu J (2017). Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxidants & redox signaling 27, 989–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J, Poustka L, Vomstein K, Haffner J, Parzer P, Stieltjes B, and Henze R (2015). Volumetric alterations in the heteromodal association cortex in children with autism spectrum disorder. European psychiatry : the journal of the Association of European Psychiatrists 30, 214–220. [DOI] [PubMed] [Google Scholar]
- Romme IA, de Reus MA, Ophoff RA, Kahn RS, and van den Heuvel MP (2017). Connectome Disconnectivity and Cortical Gene Expression in Patients With Schizophrenia. Biological psychiatry 81, 495–502. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, and Yoshida H (2015). Organelle autoregulation-stress responses in the ER, Golgi, mitochondria and lysosome. Journal of biochemistry 157, 185–195. [DOI] [PubMed] [Google Scholar]
- Schipul SE, Williams DL, Keller TA, Minshew NJ, and Just MA (2012). Distinctive neural processes during learning in autism. Cerebral cortex 22, 937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz SK, Hjorth JJ, Joemai RM, Wijntjes R, Eijgenraam S, de Bruijn P, Georgiou C, de Jong AP, van Ooyen A, Verhage M, et al. (2011). Automated analysis of neuronal morphology, synapse number and synaptic recruitment. J Neurosci Methods 195, 185–193. [DOI] [PubMed] [Google Scholar]
- Schreiner M, Forsyth JK, Karlsgodt KH, Anderson AE, Hirsh N, Kushan L, Uddin LQ, Mattiacio L, Coman IL, Kates WR, and Bearden CE (2017). Intrinsic Connectivity Network-Based Classification and Detection of Psychotic Symptoms in Youth With 22q11.2 Deletions. Cerebral cortex 27, 3294–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada A, Tsuzuki M, Keino H, Satoh M, Chiba Y, Saitoh Y, and Hosokawa M (2006). Apical vulnerability to dendritic retraction in prefrontal neurones of ageing SAMP10 mouse: a model of cerebral degeneration. Neuropathol Appl Neurobiol 32, 1–14. [DOI] [PubMed] [Google Scholar]
- Sigurdsson T, and Duvarci S (2015). Hippocampal-Prefrontal Interactions in Cognition, Behavior and Psychiatric Disease. Front Syst Neurosci 9, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW, Hahn JD, and Sporns O (2017). Organizing principles for the cerebral cortex network of commissural and association connections. Proceedings of the National Academy of Sciences of the United States of America 114, E9692–E9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, and Telser J (2007). Free radicals and antioxidants in normal physiological functions and human disease. The international journal of biochemistry & cell biology 39, 44–84. [DOI] [PubMed] [Google Scholar]
- van Erp TGM, Walton E, Hibar DP, Schmaal L, Jiang W, Glahn DC, Pearlson GD, Yao N, Fukunaga M, Hashimoto R, et al. (2018). Cortical Brain Abnormalities in 4474 Individuals With Schizophrenia and 5098 Control Subjects via the Enhancing Neuro Imaging Genetics Through Meta Analysis (ENIGMA) Consortium. Biological psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, and Langer T (2016). Mitochondrial Dynamics and Metabolic Regulation. Trends in endocrinology and metabolism: TEM 27, 105–117. [DOI] [PubMed] [Google Scholar]
- Wang X, Perez E, Liu R, Yan LJ, Mallet RT, and Yang SH (2007). Pyruvate protects mitochondria from oxidative stress in human neuroblastoma SK-N-SH cells. Brain research 1132, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wass S (2011). Distortions and disconnections: disrupted brain connectivity in autism. Brain and cognition 75, 18–28. [DOI] [PubMed] [Google Scholar]
- Whitford KL, Dijkhuizen P, Polleux F, and Ghosh A (2002). Molecular control of cortical dendrite development. Annual review of neuroscience 25, 127–149. [DOI] [PubMed] [Google Scholar]
- Zhang H, Go YM, and Jones DP (2007). Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Archives of biochemistry and biophysics 465, 119–126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.