

Summary
The identity and heterogeneity of glial progenitors and their contributions to brain tumor malignancy remain elusive. By applying lineage-targeted single-cell transcriptomics, we uncover an unanticipated diversity of glial progenitor pools with unique molecular identities in developing brain. Our analysis identifies distinct transitional intermediate states and their divergent developmental trajectories in astroglial and oligodendroglial lineages. Moreover, intersectional analysis uncovers analogous intermediate progenitors during brain tumorigenesis, wherein oligodendrocyte-progenitor intermediates are abundant, hyper-proliferative and progressively reprogrammed towards a stem-like state susceptible to further malignant transformation. Similar actively cycling intermediate progenitors are prominent components in human gliomas with distinct driver mutations. We further unveil lineage-driving networks underlying glial fate specification and identify Zfp36l1 as necessary for oligodendrocyte-astrocyte lineage transition and glioma growth. Together, our results resolve the dynamic repertoire of common and divergent glial progenitors during development and tumorigenesis and highlight Zfp36l1 as a molecular nexus for balancing glial cell-fate decision and controlling gliomagenesis.
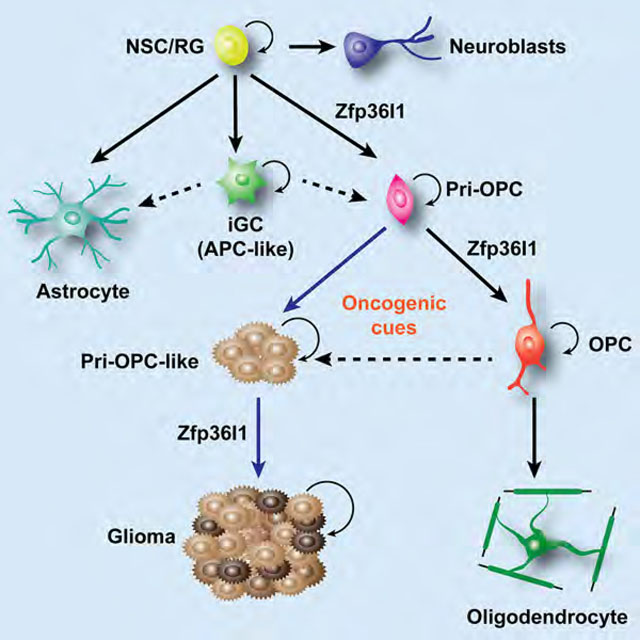
Graphical Abstract
eTOC blurb
By applying lineage-targeted single-cell transcriptomics analysis, Weng and colleagues uncover distinct intermediate glial progenitors in the neonatal brain, and their malignant counterparts in murine and human gliomas. Lineage-driving network analysis further identifies Zfp36l1 as a pivotal regulator for glial fate specification and glioma growth.
Introduction
Abnormal development of glial progenitors, including astrocyte lineage precursors and oligodendrocyte precursor cells (OPCs), contributes to tumorigenesis and various neurological diseases (Gallo and Deneen, 2014; Zong et al., 2015). Although single-cell analysis of human glioma tissues has been reported (Filbin et al., 2018; Patel et al., 2014; Tirosh et al., 2016; Venteicher et al., 2017), the tumorigenic cell of origin and the molecular links between native glial progenitors and pre-cancerous/neoplastic cells during glioma transformation have not been fully defined. Understanding the transformation potential of diverse glial progenitors during brain tumorigenesis should reveal avenues to selectively target transformed cells for cancer therapy.
Until recently, studies of glial cells had largely been limited to the analysis of in vitro cultures or bulk tissues confounded by heterogeneity (Dugas et al., 2006; Zhang et al., 2014). Astrocytes can be derived from radial glia or neural stem cells in the developing CNS (Kriegstein and Alvarez-Buylla, 2009; Molofsky et al., 2012), while the identity of astrocyte lineage precursors and their diversity in the developing cortex remain elusive. Astrocyte heterogeneity has been characterized in different regions of the adult brain based on cell surface markers (Lin et al., 2017), but such population-based approaches have likely failed to resolve the full extent of underlying heterogeneity and progenitor cell identity. Recent single-cell studies indicate that there is regional diversity among oligodendrocyte lineage cells in the murine central nervous system (Marques et al., 2018; Marques et al., 2016), however, whether the OPC pool exhibits diverse states and lineage plasticity at the specific time-window during brain development and malignancy has not been entirely defined. These unresolved issues impelled us to explore lineage-targeted transcriptomics and intersectional analysis of glial progenitors and glioma-forming cells at the single-cell level to identify key cellular components and molecular determinants for brain tumorigenesis.
Here we describe targeted high-throughput single-cell RNA-sequencing (scRNA-seq) on prospective astrocyte lineage cells and OPC populations isolated by fluorescence activated cell sorting (FACS) from neonatal mouse cortices. We found that astrocyte lineage cells are much more dynamic than previously appreciated in the developing cortex and uncovered a transitional progenitor population during astrocyte lineage development. In contrast to the astrocyte lineage, the progenitors of oligodendrocytes exhibited a fate-restricted continuum that encompassed a primitive OPC intermediate population prior to OPC commitment in the neonatal cortex. Application of scRNA-seq to a murine model of glioblastoma (GBM) revealed that primitive OPC intermediates disproportionately contributed to glioma formation. Analyses of different tumorigenic phases suggested that reprogramming of the OPC intermediates into a stem-like state, rather than direct stem-cell proliferation, resulted in malignant transformation. Similar actively cycling oligodendrocyte-progenitor intermediates were prominent components in human gliomas caused by distinct driver mutations. A machine-learning algorithm identified an RNA-binding protein, Zfp36l1, as a critical regulator of glial fate specification and glioma growth, suggesting that this network could be targeted to develop a lineage-specific therapy for malignant glioma.
Results:
Single-cell transcriptomics reveals distinct glial progenitors in developing brain
Human GFAP promoter-driven GFP expression in hGFAP-GFP transgenic brains has been previously shown to mark astrocyte lineage cells (Ge et al., 2012; Zhuo et al., 1997). We performed droplet-based scRNA-seq (Macosko et al., 2015) on FACS-sorted GFP+ cells from the neonatal cortices of hGFAP-GFP animals at P5 and P6, when astrocyte precursors undergo proliferation and differentiation (Ge et al., 2012; Sauvageot and Stiles, 2002) (Figure 1A).
Figure 1. Unsupervised ordering of the hGFAP-GFP-derived cells reveals developmental hierarchy.

(A) Scheme for analysis of hGFAP-GFP+ cells using scRNA-seq from neonatal cortices (n=5 mice).
(B) t-SNE analysis of hGFAP-GFP+ cell clusters.
(C) Heatmap of hGFAP-GFP+ cells ordered as t-SNE (n = 815). Columns, individual cells; rows, genes.
(D) The proportions of distinct clusters among total hGFAP-GFP+ cells.
(E) Dot plot of levels of selected marker genes in subpopulations.
(F-G) t-SNE plots of F) astrocyte (Astro), radial glia (RG), and iGC and G) marker genes.
(H) Comparison of astrocyte and iGC clusters with adult astrocyte populations.
(I-J) t-SNE plot of I) OPC and pri-OPC cells and J) marker genes.
(K) Pseudo-time ordering of pri-OPCs and OPCs in hGFAP-GFP+ dataset. Red line, the predicted trajectory.
(L) Predicted lineage trajectories from RG-like cells in hGFAP-GFP+ cells.
Unsupervised clustering using t-SNE (Macosko et al., 2015) revealed nine clusters with distinct gene expression signatures (Figure 1B–C). Gene ontology analysis (Chen et al., 2009) classified these clusters into discrete subpopulations including radial-glia-like cells, astrocytes, OPCs, neuroblasts, neurons (GABAergic and glutamatergic), and ependymal cells (Figure 1B–C). Clustering was independently verified with BackSPIN (Marques et al., 2016) and PAGODA (Fan et al., 2016) (Figure S1A).
Among hGFAP-GFP+ cells, 14.6% were astrocytes and 5.4% were radial-glia-like (Figure 1D). By subclustering of the astrocyte lineage group, we identified a cell cluster expressing the markers of both astrocytic signature genes (e.g., Slc1a3 and Aldh1l1) and oligodendrocyte lineage genes (e.g., Olig1 and Olig2; Figure 1B–D) (Lu et al., 2000; Zhou et al., 2000), suggesting that these may be transitional intermediate glial progenitor cells (iGCs). A subgroup of iGC population, but not mature astrocytes, that expressed cell-cycle-related genes (e.g., Mki67) were defined as cycling iGCs (Figure 1C). The signature genes are highly cell-type-specific in distinct glial progenitor cells (Figure 1E). Moreover, t-SNE visualization showed that the astrocyte lineage cells were segregated into astrocyte, radial-glia-like, and iGC subpopulations, exhibiting specific signature (Figure 1F,G).
We next compared the neonatal astrocytic populations to five previously identified populations of adult astrocytes. The astrocytes in the neonatal cortex were highly correlated with the adult astrocyte population C (Lin et al., 2017) (Figure 1H). The iGC population resembled postnatal radial glia (Hochgerner et al., 2018) and adult quiescent neural stem cells (NSCs) (Dulken et al., 2017) (Figure S1B), suggesting that iGCs are unique to the immature astrocyte population in the developing cortex.
Unexpectedly, the hGFAP-GFP+ cells also included cells with gene signatures associated with committed OPCs, marked by Pdgfra, and a primitive OPC subpopulation (pri-OPCs), which expressed low levels of Pdgfra and high levels of Olig1/2 (Figure 1B–E). A t-SNE plot based on differential gene expression revealed these two distinct OPC clusters: OPCs (e.g. Pdgfra+ and Cspg4+), and pri-OPCs (e.g. Ppp1r14b+, Ascl1+, Btg2+ and Hes6+) (Figure 1I–J). The pri-OPC population most closely resembled adult activated NSCs (Dulken et al., 2017) (Figure S1B). An unsupervised pseudo-time analysis using Slingshot (Fletcher et al., 2017), was consistent with a developmental trajectory from pri-OPC to OPC (Figure 1K) with increased expression of Pdgfra over the trajectory and high levels of early lineage genes (e.g. Ppp1r14b) in pri-OPCs (Figure S1C). A fraction of pri-OPCs and OPCs expressed cell-cycle genes, indicating that they are proliferating during early oligodendrogenesis (Figure 1C). The other cell clusters expressed the markers of neuronal subpopulations, GABAergic (e.g., Dlx1) and glutamatergic (e.g., Neurod1) neurons, and ependymal cells (e.g., Foxj1 and Wdr52) (Figures 1C, S1D).
Immunostaining also indicated the co-expression of neuronal markers, Sp8 and Dlx2, in a fraction of hGFAP-GFP+ cells in the developing cortex (Figure S1E–G). The presence of neurons within hGFAP-GFP+ populations may result from perdurance of GFP expression from ventricular zone neural progenitor cells. Corroborating Drop-seq data, single-cell sequencing of FACS-sorted hGFAP-GFP+ cells with SMART-seq of 110 sorted hGFAP-GFP+ single cells revealed similar cellular clusters (Figure S1H).
To further investigate cell trajectories during glial cell development, we utilized Slingshot, a statistical framework for inferring branching lineage assignments and developmental distances (Fletcher et al., 2017) and principal component analysis (PCA). The cell lineage development was predicted to start from radial glia passing through iGCs, after which two distinct trajectories were identified that led to either an OPC or an astrocytic fate (Figure 1L). The intermediate iGC populations were located between astrocytes and OPCs, supporting the hypothesis that iGCs are a transitional cell type.
In vivo validation of markers of astrocytes and their lineage precursors
To validate single-cell clustering, we performed immunostaining for the neural cell type-specific markers in the cortex of the hGFAP-GFP mice at P5. The GFP+ cells were detected in astrocytes marked by GFAP and glutamine synthetase (GS) (Figure 2A–B). Consistent with transcriptome analysis, we also detected a population of GFP+ cells expressing both astrocytic markers (e.g., GFAP or Slc1a3) and the oligodendrocyte lineage marker Olig2 (Figure 2C–E), suggesting that these are the transitional iGC population. Furthermore, a proportion of GFP+ cells expressed a radial glia marker Blbp, an OPC marker PDGFRα, and a pri-OPC marker Ppp1r14b (Figure 2F–H).
Figure 2. Identification of hGFAP-GFP classification and astrocyte-enriched transcription factors revealed by scRNA-seq.

(A) Immunolabeling for GFAP and GS in the cortex of P5 hGFAP-GFP mice.
(B) The percentage of indicated cells among hGFAP-GFP+ cells in P5 mouse cortices (n=4 for GFAP; n=3 for GS and PDGFRα).
(C) Immunolabeling for GFAP, Olig2, and Slc1a3 from P5 hGFAP-GFP mice.
(D) Zoom on boxed area in panel C.
(E) The percentage of Olig2+ and Olig2– cells among hGFAP-GFP+GFAP+ (left) or hGFAP-GFP+Slc1a3+ (right) cells in P5 mouse cortices (n=3).
(F) Immunolabeling of Blbp in the cortices from hGFAP-GFP mice at P3.
(G) Expression of PDGFRα in the cortices of P5 hGFAP-GFP mice.
(H) Immunolabeling for Ppp1r14b and Olig2 in the cortices of hGFAP-GFP mice at P3.
(I) Immunolabeling for Olig2 and Ki67 from P5 hGFAP-GFP mice.
(J) Left, enlarged images of panel I show cells co-labeled with Ki67 (arrows) and cells without Ki67 (arrowheads). Right, percentage of Olig2+ and Olig2– cells among Ki67+ hGFAP-GFP+ double positive cells (>300 cell counts from 3 cortices).
(K) Immunolabeling for Olig2 and GFAP or Slc1a3 in adult cortices at P60.
(L) The percentage of GFAP+ or Slc1a3+ among Olig2+ cells at P5 and P60 (>400 cell counts from 3 cortices at each stage).
(M) Immunolabeling for BHLHE40 in P5 hGFAP-GFP cortices.
(N-P) Immunolabeling for N) PRDM16 and GS in P14 mouse spinal cord and O) BHLHE40 and GS in human cortices. DAPI, blue. Arrows; co-labeled cells. P) Percentage of BHLHE40+ or PRDM16+ in GS+ cells (>300 cell counts from 3 samples).
Data are presented as means ± SEM. Scale bars, 50 μm in A, C, G, I, K, M; 20 μm in F, N, O; 10 μm in D, H, J and G (inset).
A recent study based on GFP expression in hGFAP-GFP transgenic cortices at P6 suggested that cortical astroglia result from the local proliferation of astrocytes (Ge et al., 2012). We therefore analyzed proliferating cells, in the hGFAP-GFP cortex at P5. Remarkably, among hGFAP-GFP+ cells that were proliferative (Ki67+), we found that 84.2% were positive for Olig2 (Figure 2I–J), indicating that these dividing cells are likely iGC or OPCs, rather than differentiated astrocytes, consistent with the gene expression profile clustering analysis (Figure 1C). In contrast to the neonatal cortex, few iGC-like cells were detected in adulthood (Figure 2K–L).
Single-cell analysis identified a set of regulatory genes enriched in astrocyte clusters (Figure S1I–J) including transcriptional regulators Bhlhe40 and Prdm16 (Zhang et al., 2014). To validate their specific expression in astrocytes, we performed immunostaining in the developing cortex and spinal cord. The majority of hGFAP-GFP+ cells in the mouse cortex at P5 (71.3%) were co-immunostained with BHLHE40 (Figure 2M). Expression of PRDM16 and BHLHE40 was also detected in GS+ astrocyte in mouse spinal cord and human cortical sections, respectively (Figure 2N–P). These observations suggest that PRDM16 and BHLHE40 are useful markers of mouse and human astrocytes.
Single-cell analysis reveals distinct oligodendroglial progenitor states and a restricted lineage trajectory
To distinguish between two possible trajectories in early glial cell development – that iGCs transition into pri-OPCs or vice versa – we utilized scRNA-seq to characterize cellular heterogeneity of PDGFRα-expressing OPCs in the early neonatal cortex. PDGFRα expression has been shown to mark OPCs in the brain (Woodruff et al., 2001), so we isolated nuclear-GFP+ presumptive OPCs from the cortices of PDGFRα-H2bGFP mice (Klinghoffer et al., 2002) at P1 and P3, when most of the oligodendrocyte lineage cells are at the immature precursor stage (Figure 3A).
Figure 3. Identification of the developmental hierarchy of PDGFRα-GFP-derived cells.

(A) t-SNE plot of PDGFRα-GFP+ populations (n=5 per timepoint).
(B) Heatmap of PDGFRα-GFP+ cells ordered as t-SNE.
(C) The proportions of distinct clusters among total PDGFRα-GFP+ cells.
(D) Dot plot of the expression level of selected marker genes in subpopulations.
(E-F) t-SNE plot of E) OPC lineage cells and F) marker genes.
(G) Pseudo-time ordering of pri-OPCs, OPCs, and iOLs in the PDGFRα-GFP+ dataset.
(H) pri-OPC, OPC, and iOL-specific genes along the pseudo-timeline.
(I) Heatmap of expression dynamics among pri-OPCs, OPCs, and iOLs.
(J) Comparison of clusters in PDGFRα-GFP+ profiles with the expression signatures of neural and oligodendrocyte lineage progenitors.
(K) PCA comparison of iOL, OPC, and pri-OPC clusters in PDGFRα-GFP+ cortex with pre-OPC and OPC populations in P0 and P5 hippocampi.
(L) Correlations based on scaled expression values for comparisons shown in panel K.
Unsupervised clustering based on gene expression patterns (Macosko et al., 2015) and gene ontology enrichment analyses (Chen et al., 2009) revealed eight distinct groups: OPCs, pri-OPCs, cycling OPCs with a cell-mitotic expression signature, immature pre-myelinating oligodendrocytes (iOLs), neuroblasts, cycling neuroblasts, astrocytes, and pericytes (Figure 3A–B, S2A–B). Their gene expression profiles were correlated to previously identified neural cell types (Zhang et al., 2014) (Figure S2C). The most abundant cell populations were OPCs, pri-OPCs, and cycling OPCs; only about 1% were iOLs (Figure 3C). Another rare population (1.4%) had an astrocytic gene signature (Figure 3B–C), consistent with the notion that very few oligodendrocyte progenitors give rise to astrocyte lineage cells during normal development (Kang et al., 2010). Some additional rare cell types were also present including pericytes, likely derived from PDGFRα+ vasculature, and the leptomeninges. Based on incidence and amplitude of signature gene expression each delineated cellular cluster had a distinct regulatory state (Figure 3D). Subclustering analysis further showed that PDGFRα-GFP+ progenitors in the oligodendrocyte lineage were separated into two main clusters of OPCs (Pdgfrahigh or Cspg4high) and pri-OPCs (Olig2+ and Pdgfra low/Cspg4low) (Figures 3E–F and S2D).
Pseudo-time analysis with Slingshot revealed a trajectory from pri-OPCs (e.g., Ascl1, Ppp1r14b, and Btg2) to OPCs (e.g., Pdgfra, Cspg4, and Epn2) to iOLs (e.g., Plp1, Bmp4, and Neu4) (Figure 3G, H), and gene expression dynamics on the pseudo-temporal axis (Figure 3H–I). scRNA-seq of PDGFRa-GFP+ cells using SMART-seq confirmed this trajectory (Figure S2E–F). Cell-cycle gene signatures appeared in both pri-OPCs and OPCs but not in iOLs (Figure 3I). Although genes enriched in OPCs and iOLs were previously reported (Marques et al., 2016; Zhang et al., 2014), our data provide a high-resolution view during early postnatal cortical oligodendrogenesis and defined the pri-OPC population (Figure 3J). The gene signature of the cortical pri-OPC population resembled that of pre-OPCs from the mouse hippocampus at P0 and P5 (Hochgerner et al., 2018; La Manno et al., 2018) (Figure 3K–L), although the cortical pri-OPCs are not exactly the same as the hippocampal pre-OPCs.
Notably, a cluster of PDGFRα-H2bGFP+ cells exhibited gene expression signatures characteristic of neuroblasts. These cells had strong expression of the neuronal marker Sp8 but lacked Olig2 expression (Figure S2G–H). The proportion of Sp8+ cells among PDGFRα-GFP cells was much higher in the embryonic cortex at E14.5 than P3 (Figure S2I–J). Transposase-accessible-chromatin (ATAC-seq) (Buenrostro et al., 2015) of PDGFRα-GFP cells isolated from E14.5 and P5 brains revealed that chromatin of neuronal genes such as Dcx, Dlx1/2 and Stmn2 was more accessible at E14.5 than at P5, whereas stronger ATAC-seq peak signals were detected in OPC/iOL-associated genes (e.g., Olig2, Cnp, Nkx6–2) at P5 (Figure S2K–M). These observations suggest that PDGFRa promoter activity is higher in neuroblasts at early stages than late developmental stages and that GFP+ neuronal cells are likely due to GFP perdurance from PDGFRα-GFP+ neuroblast precursor cells.
Identification of transcriptional regulatory networks that drive glial lineage specification
To identify the transcriptional regulators (TRs) that drive oligodendrocyte or astrocyte lineage commitment, we developed a machine-learning algorithm that interrogates cell-lineage-driving TRs based on differentially expressed genes, target binding potential, and cellular cluster relationships (Figure 4A). This analysis revealed sets of significantly enriched TRs in OPC and astrocyte populations (Figure 4B–C; Table S1). For OPC-driving TR sets, we detected previously known transcriptional regulators of oligodendrocyte fate commitment and differentiation including Olig1/2, Sox10, and Nkx2–2 (Dugas et al., 2006), which validated our approach. We also identified factors not previously shown to function in oligodendrocyte lineage development (e.g., Ppp1r14b, Zfp36l1, Ostf1; Figures 4B and S3A). Among the candidate driving factors for astrocyte lineage development were astrocyte differentiation associated genes (e.g., Id3, Nfia, Sox9), Notch signaling effectors (e.g., Hes5, Hey2, Hes1), astrocyte-enriched Prdm16 and Bhlhe40, and potential developmental regulators (e.g., Rfx4, Trps1, Gli3) (Figures 4C and S3B).
Figure 4. Regulatory networks underling glial lineage specification.

(A) Workflow to identify driver genes.
(B-C) The top representative TRs ranked by Z-score in B) OPC and C) astrocyte clusters.
(D) Intersection of OPC and astrocyte TRs. Relative Z-scores were normalized to the average of individual Z-scores of all genes in each gene list.
(E) Zfp36l1 in situ hybridization combined with Olig2 and Ascl1 immunostaining at P7.
(F) Diagram depicting Nestin-Cre-mediated excision of Zfp36l1 floxed alleles.
(G) In situ hybridization analysis of Zfp36l1 mRNA in the cortical ventricular zone region of P1 brain from control and Zfp36l1-cKO mice.
(H) H&E-stained brain sections from control and Zfp36l1 iKO mice at P14.
(I) Immunolabeling of OLIG2, PDGFRα, and MBP from control and Zfp36l1-cKO cortices at P7.
(J-K) The numbers of J) Olig2+ (left) or PDGFRα+ (right) cells and K) Ppp1r14b+/Olig2+ pri-OPC-like cells in P7 control and Zfp36l1-cKO cortices.
(L) Immunolabeling for GFAP and GS in control and Zfp36l1-cKO cortices at P7.
(M-N) The numbers of M) GS+ cells and N) GFAP+/Olig2+ iGC-like cells in the cortices of P7 control and Zfp36l1-cKO mice.
(O) Immunostaining for GFAP and GS with tdTomato in Zfp36l1-cKO;tdTomato cortices at P7. Arrows indicate colabeled cells.
(P) qRT-PCR analysis of indicated gene expression from rat OPCs after Ppp1r14b depletion.
(Q-R) Rat OPCs treated with control and Ppp1r14b siRNAs were Q) immunostained for MBP and Olig2 and R) percentage of MBP+ OLs were determined. Cells were differentiated with or without T3 after PDGF-AA withdrawal for 72 hours.
Data are presented as means ± SEM; n=3 independent experiments or animals/genotype; *p<0.05; **p < 0.01; ***p < 0.001; n.s., not significant. Scale bars, 1 mm in H; 100 μm in G, Q; 50 μm in E, I, L and O.
To identify the potential regulators of glial lineage choice, we focused on a subset of candidate lineage-driving transcriptional regulators that are shared for both OPC and astrocyte lineages (Figures 4D and S3C). Among them, Zfp36l1, encoding an RNA-binding zinc-finger protein of the C3H type (Stumpo et al., 2004), had a much higher lineage-driving potential in OPCs than astrocytes (Figure 4D). Zfp36l1 is mainly expressed in oligodendrocyte progenitors (Figure S3D) (Marques et al., 2016). Although a suitable Zfp36l1 antibody for immunohistochemistry is not available, mRNA in situ hybridization indicates that Zfp36l1 expression is expressed in subventricular zone (SVZ) progenitors during early developmental stages (Figure S3E). Zfp36l1 expression was detected in Ascl1+ SVZ progenitors and Olig2+ oligodendrocyte lineage cells at the edge of the SVZ and the boundary of the corpus callosum at P7 (Figure 4E), suggesting a potential role of Zfp36l1 in regulating oligodendroglial cell fate commitment.
Lineage-driving factor Zfp36l1 controls oligodendrocyte-astrocyte lineage transition
To determine the role of Zfp36l1 in glial fate specification in the developing brain, we selectively ablated Zfp36l1 floxed alleles in a neural progenitors and radial glia in a Nestin-Cre line (Zhuo et al., 2001) to generate Zfp36l1fl/fl; Nestin-Cre+/− mice (Zfp36l1-cKO; Figure 4F). mRNA in situ hybridization revealed substantial reduction in Zfp36l1 in neural progenitors in the SVZ and the cortical region at P1 in Zfp36l1-cKO mice compared to controls (Figure 4G). Zfp36l1-cKO animals were born at the expected Mendelian frequency; however, Zfp36l1-cKO mice exhibited substantially enlarged lateral ventricles (Figure 4H).
Immunostaining indicated that numbers of Olig2+ and PDGFRα+-OPCs and pri-OPCs were reduced in the cortices of the Zfp36l1-cKO mice at P7 compared to controls (Figure 4I–K). Expression of the myelin protein MBP was also diminished in both the corpus callosum and cortical regions (Figure 4I). Zfp36l1-cKO mice exhibited generalized tremors, likely due to the myelination deficiency. Expression of astrocytic markers GFAP and GS were increased in Zfp36l1-cKO cortices compared to controls (Figure 4L–M), although the numbers of iGCs were comparable (Figure 4N). To confirm that GS+ and GFAP+ cells were derived from the Cre-mediated Zfp36lfl/fl recombined cells, we bred mice bearing Zfp36l1fl/fl and Nestin-Cre with a tdTomato Cre reporter line. The immunostaining results showed that the GFAP- and GS-positive cells were co-labeled with tdTomato+ cells (Figure 4O), indicating that these ectopic astrocytes were descended from the Zfp36l1-deleted progenitors at the expense of OPCs. These results suggest that Zfp36l1 controls oligodendrocyte-astrocyte fate transition in the developing brain.
To further validate the machine learning approach, we examined the function of Ppp1r14b, which exhibits a high potential for driving OPC lineage progression (Figure 4B). We found that knockdown of Ppp1r14b in primary OPCs inhibited expression of myelin genes as well as OPC differentiation into mature oligodendrocytes (Figure 4P–R), suggesting that Ppp1r14b regulates OPC lineage progression.
Heterogeneous glial progenitors in glioma revealed by single-cell RNA-seq
To investigate the cellular diversity in glioma and their relationship with native glial progenitors, we established an animal model of malignant glioma induced by a DNp53-PDGFB retrovirus expressing dominant-negative p53 (DN-p53) and PDGFB, potent inducers of proneural GBM formation (Lei et al., 2011; Lu et al., 2016). Brain tumors formed around three weeks after stereotaxic delivery of DN-p53-PDGFB retroviruses into the cortical white matter at a gliogenic stage P2 (Figure 5A). Tumor tissues harvested 35 days post injection (dpi), in the aggressive tumorigenic phase, were dissociated into single-cell suspensions and analyzed by scRNA-seq.
Figure 5. Cellular heterogeneity of murine malignant gliomas revealed by scRNA-seq.

(A) H&E-staining showing the malignant glioblastoma tissue (arrow) at dpi 35. Scale bar: 1 mm.
(B) t-SNE analysis of single cells from glioma core tissues at dpi 35.
(C) Left, comparison of OPC-like cells with human GBM subtypes based on TCGA datasets. Right, distributions of subtype scores.
(D) Heatmap of mouse glioma cell clusters (excluding immune and endothelial cells). Selected marker genes are displayed on the right.
(E) Pearson’s correlation coefficient between normal and tumor cell populations.
(F) The proportions of distinct identified clusters in single-cell data from glioblastoma.
(G) Distributions of the pri-OPC scores across pri-OPC-like cells from tumors and OPC populations in normal cells.
(H) The ratio of OPC/COP-like to pri-OPC-like cells in PDGFRα, hGFAP-GFP+, and tumor scRNA-seq pools.
(I) Venn diagram depicting the intersection of top 100 marker genes (ranked by p-value) in pri-OPC-like subpopulations between PDGFRα-GFP and tumor datasets.
(J) Gene ontology analysis of unique genes showing p-value (-log10) and representative terms.
***p < 0.001; one-way ANOVA with post hoc Tukey’s test.
Unsupervised clustering analysis identified eight different clusters with distinct gene expression signatures characteristic of pri-OPC-like and iGC-like populations, committed OPCs (COP), and immune cell populations, but with low abundance of astrocytes, radial glia, mature oligodendrocytes, neuronal cells and endothelial cells (Figures 5B, S4A–B). The pri-OPC-like cells scored highly for the proneural GBM gene signature, but low for neural, classical, and mesenchymal signatures (Verhaak et al., 2010) (Figure 5C). The subclustering for neural cell types identified cellular characteristics that parallel that of the normal neonatal cortex, including astrocyte-like, iGC-like, COP-like, pri-OPC-like, cycling OPC (G1/S and G2/M), ependymal cells, and cell populations unique to tumor cells with stress and hypoxia signatures (Figure 5D). We next compared the gene expression signatures of glial lineage cells in normal and malignant brain tumor tissues. A population of cells in the tumor tissues exhibited a stronger similarity of expression patterns to pri-OPCs than normal OPCs or iOLs in the developing cortex (Figure 5E). These “pri-OPC-like” cells had a partial OPC signature including PDGFRα expression (Figure 5E). In addition, a cell population appeared to be correlated to both OPC and iOL (Figure 5E) as COP-like cells. The gene expression profiles of iGC-like precursors and astrocytes resembled those of the native developmental counterparts (Figure 5E).
Strikingly, pri-OPC-like cells (e.g. Olig1/2, Ascl1, and Ppp1r14b) and their mitotic cells in G1/S and G2/M phases were present in the highest abundance in neural cell types in the tumor tissues (72.6 %; Figure 5F). In addition, the pri-OPC signature score and their proportion were higher in tumors than in neonatal brains (Figure 5G,H), suggesting the expansion and amplification of pri-OPC-like populations during tumorigenesis. The unique gene sets in PDGFRα-GFP cell populations showed gene signatures enriched in gliogenesis and oligodendrocyte development (Figure 5I–J). In contrast, the unique gene sets in tumors exhibited pathway components enriched in GBM, astrocytoma, and stem-cell-related genes (Figure 5J), suggesting that pri-OPC analogs in the proneural-like glioma tissues contribute to malignant transformation during tumorigenesis.
pri-OPC-like intermediates exhibit a transit-amplifying property during gliomagenesis
t-SNE visualization of glioma cell populations showed that a large majority of the cells exhibited expression of the signature genes for pri-OPCs (Figures 6A–B, S4C). To further explore the identity of pri-OPC-like cells in tumor tissues, we evaluated expression of stemness signature gene sets related to glioma formation (Tirosh et al., 2016) in our dataset. Strikingly, we found that most pri-OPC-like cells expressed stemness signature markers such as Sox2, Ccnd2, Sox11, and Chd7, and exhibited a higher stemness score than committed OPCs, iGC-like cells, and astrocytes (Figure 6C–D). In addition, gene expression of pri-OPC-like cells in tumors correlated most strongly with the adult activated NSC signature (Figure S4D) and exhibited the highest stemness score (Figure S4E). These observations suggest that the pri-OPC-like cells are amplified in the tumor tissues and acquire stem cell-like properties.
Figure 6. pri-OPCs are a transit-amplifying tumorigenic population during tumorigenesis.

(A-C) t-SNE analysis of A) glia-related cells, B) pri-OPC marker genes, and C) stemness-related genes from 35 dpi glioma dataset.
(D) Distribution of stemness genes across major tumor subpopulations.
(E) Left, expression of selected cell cycle genes (rows) in individual tumor cells (columns). Cells were ordered by cell-cycle score. Right, t-SNE plot of G1/S or G2/M marker genes.
(F) Percentage of cycling OPCs in PDGFRα-GFP, hGFAP-GFP, and tumor cell datasets.
(G) Percentage of proliferating (Ki67+) cells that were Olig2+ (left) and Sox2+ (right) in PDGFRα-GFP and tumor scRNA-seq pools.
(H) Proportions of immune-related cells in single-cell profiles from dpi 25 and 35 mouse gliomas.
(I) Major cell populations (dots) from dpi 25 and 35 tumors scored for the pri-OPC and stemness signatures. Correlation values are in the top right quadrant.
(J) The percentage of actively cycling cells among pri-OPC-like cells in dpi 25 and 35 tumors.
(K) The relative cycling scores of pri-OPC-like cells from dpi 25 and 35 gliomas.
(L) Ratios of CNVs in pri-OPCs normalized against the “normal” cluster of macrophage/microglia at dpi 25 (above) and dpi 35 (below).
(M-N) t-SNE plot of M) glia-related cells and N) pri-OPC-like and stemness marker genes after excluding immune cells in human IDH-O dataset.
(O) Percentage of proliferating cells in pri-OPC-like and other neural cell populations in human glioma datasets.
***p < 0.001; one-way ANOVA with post hoc Tukey’s test in D, Student’s t test in K.
Further subclustering analysis based on cell-cycle markers indicated that the majority of pri-OPCs also expressing cell-cycle genes in G1/S and G2/M phases (Figure 6E). The fraction of cycling OPCs in G1/S or G2/M phases (34.4%) was substantially higher than that in PDGFRα-GFP+ or hGFAP-GFP+ OPC populations from normal neonatal brains (Figure 6F). This increase in mitotic pri-OPC-like cells in tumors suggests that pri-OPC-like cells are the transit-amplifying cell population that fuels tumor growth.
In contrast to native glial populations isolated from normal developing cortex, in tumors we detected cells expressing stress-associated signature genes (Figures 6A, S5A–B), and hypoxia-associated genes (Figures 6A, S5C–D). These signatures were present in a subpopulation of OPC-like cells (Figure S5E) consistent with observations in human gliomas and other tumor tissues (Patel et al., 2014; Puram et al., 2017). Among glial progenitors, the frequency of Mki67+ cells among Olig2+ or Sox2+ OPC-like intermediates was higher in tumor tissues compared with frequencies in the developing cortex (Figure 6G), consistent with an expansion of the pri-OPC progenitor population during tumorigenesis.
Similar to the heterogeneity of astroglial cells in the developing cortex, we also identified both astrocyte-like cells (expressing Gja1 and Aqp4) and iGC-like populations (expressing Olig2 and astrocyte markers Slc1a2 and Slc1a3) (Figures S4 and 5F–G). The iGC-like cluster was present at a higher proportion in tumor tissues than normal developing cortex (Figure S5H). Intriguingly, radial glia markers were hardly detectable in the tumor tissues (Figure S5I), suggesting that radial glia/NSC-like cells do not actively divide during tumorigenesis in the malignant glioma model.
Reprogramming of oligodendrocyte-progenitor-like intermediates towards a tumorigenic phenotype during tumorigenesis
To better understand the progression of distinct cells during different phases of gliomagenesis, we examined cellular compositions of tumor tissues at an early stage of tumorigenesis at dpi 25 in the animal model by scRNA-seq. The clusters of tumor cells at dpi 25 were similar to those at dpi 35 (Figure S5J–L), whereas immune cells (mainly, microglia or macrophages) were more abundant at dpi 35 than dpi 25 (Figure 6H), suggesting that the complexity of the tumor microenvironment is higher at the late stage of tumorigenesis. Interestingly, although the percentage of pri-OPC populations among neural cell groups was comparable (Figure S5M), the pri-OPC-like cells at the late phase at dpi 35 had a higher correlation coefficient score with respect to stemness signature genes than at dpi 25 (Figure 6I). In addition, the ratio of actively cycling cells among pri-OPC-like cells was significantly higher at dpi 35 than dpi 25, indicating that at the later stage pri-OPC-like cells have higher proliferative capacity (Figure 6J–K). These data suggested that pri-OPC-like cells undergo reprogramming into a more stem-like state during the progression of tumorigenesis.
Because glioma cells frequently harbor large-scale chromosomal alterations, we estimated copy number variations (CNVs) from the average expression of genes in individual large chromosomal regions within each cell (Patel et al., 2014). Compared with microglia/macrophages within the tumor lesion, which are non-malignant cells of a distinct lineage and presumably have ‘normal’ copy numbers, the majority of pri-OPC-like cells at dpi 35 had extensive copy number aberrations (Figure 6L). Alterations included copy number gains at loci such as Ccna2, Ccne2, and Mcm2, genetic alterations frequently seen in human gliomas (Brennan et al., 2013). In contrast, at dpi 25, few CNVs were detected in pri-OPC populations (Figure 6L), suggesting that oligodendrocyte progenitor-like cells are progressively reprogrammed towards a tumorigenic phenotype due to an increase in genomic instability.
To investigate whether the observations in the mouse glioma model reflect the tumorigenesis process in human brain tumors, we analyzed the single-cell transcriptomic signatures of different human gliomas with distinct driver mutations including oligodendrogliomas (IDH-O), astrocytoma (IDH-A), GBM, and diffuse midline gliomas (Filbin et al., 2018; Patel et al., 2014; Tirosh et al., 2016; Venteicher et al., 2017). We observed a prominent pri-OPC-like progenitor population expressing pri-OPC markers ASCL1, PDGFRA, BTG2, and OLIG2 in the human gliomas compared to committed OPCs and oligodendrocytes (Figures 6M–N, S6). Markedly, the pri-OPC-like populations from distinct human gliomas exhibited a stemness-associated signature (Figures 6N, S6) and were the predominant mitotic cell proportion among neural cells in tumor tissues (Figure 6O). These observations suggest reprogramming of pri-OPCs to a primitive and mitotic state endowed with stemness properties during human glioma tumorigenesis, similar to that observed in the animal model. Further, this suggests that these human gliomas may originate from the same intermediate glial progenitors, particularly pri-OPC-like intermediates. Our observations suggest that tumorigenesis influences the abundances of the glial progenitor populations with substantial reprogramming of OPC-like cells to adopt a ‘stemness’ program during tumorigenic progression.
Targeting lineage-driving determinant Zfp36l1 inhibits the initiation and growth of glioma
Given its requirement for OPC fate specification in the developing cortex (Figure 4), we hypothesized that Zfp36l1 is critical for the growth of gliomas such as proneural GBMs, which display strong OPC signatures (Verhaak et al., 2010). Zfp36l1 was enriched in pri-OPC-like and cycling pri-OPC-like cells in our murine proneural GBM model (Lu et al., 2016) (Figure 7A). We inhibited expression of Zfp36l1 in tumor cells isolated from the mouse glioma tissues using siRNA. Zfp36l1 deficiency significantly decreased the rate of glioma cell proliferation (Figure 7B–D). In addition, when tumor cells were transduced with Zfp36l1 shRNA lentiviral vectors, OPC-associated genes were downregulated, whereas astrocyte-associated signature genes were upregulated (Figures 7E–F, S7A).
Figure 7. Zfp36l1 is critical for the initiation and growth of glioma.

(A) Expression of Zfp36l1 in seven clusters from mouse gliomas.
(B) RT-qPCR quantification of Zfp36l1 in mouse gliomas cells.
(C) BrdU labeling in control and si-Zfp36l1-treated tumor cells.
(D) Percentages of BrdU+ cells in control and si-Zfp36l1-treated tumor cells.
(E-F) qRT-PCR analysis of E) proneural and F) classical tumor-associated genes in primary tumor cells infected by control or Zfp36l1 shRNA.
(G) H&E-staining of brain sections of Zfp36l1fl/+ or Zfp36l1fl/fl mice at 30, 60, and 100 and dpi. Arrows indicate tumor regions.
(H) Kaplan-Meier survival analysis of control Zfp36l1fl/+ (n=13) and Zfp36l1fl/fl (n=15) mice after injection of retrovirus. *** p < 0.001 (log-rank test).
(I) Immunostaining of Ki67 within the tumor regions from control and Zfp36l1-iKO mice.
(J) Percentages of Ki67+ cells in control (n=3) and Zfp36l1-iKO (n=2) tumors.
(K) Levels of ZFP36L1 in normal brain (n=207) and primary GBM (n=163) from the TCGA and the GTEx datasets.
(L-M) Survival probability of L) GMB patients and M) low-grade glioma patients with high and low/medium expression of ZFP36L1.
(N-P) Relative qPCR expression of N) ZFP36L1 in human GBM cells (GBM3264) infected with control or Zfp36l1 shRNA lentivirus for 8 days were evaluated N) by qPCR for expression of ZFP36L1, O) for sphere formation, and P) sphere number.
(Q-R) Relative qPCR expression of Q) cell cycle and proneural genes and R) proneural and classical tumor genes in primary proneural GBM TS543 and GBM3264 with ZFP36L1 knockdown over control.
Data are presented as means ± SEM; n=3; ***p < 0.001; **p < 0.01; *p<0.05; Student’s t test. Scale bars, 1 mm in G; 100 μm in O; 10 μm in C and I.
See also Figure S7.
To further assess the tumorigenic function of Zfp36l1 in vivo, we performed microinjection of retrovirus carrying DN-P53;PDGFB-Cre into the cortical white matter of control and Zfp36l1fl/fl mice. In the Zfp36l1fl/fl mice the Cre recombinase converted Zfp36l1-floxed alleles to complete knockout alleles. Histological analysis revealed that the Zfp36l1-iKO had no detectable tumor mass at dpi 30 or 60, when all the virus-injected control mice had developed tumors with full penetrance (Figure 7G). Most control mice died before dpi 60 due to the extensive growth of tumors; the Zfp36l1-iKO mice had a significantly extended survival curve (Figure 7H). Although tumors were detected in 2 of 15 Zfp36l1-iKO mice at 100 dpi (Figure 7G), immunostaining showed greatly reduced Ki67+ proliferative cells in these tumors (Figure 7I–J). In addition, there were higher frequencies of GFAP+ astrocyte-like cells and iGC-like cells and lower frequencies of pri-OPC-like cells in the Zfp36l1-iKO tumor tissues than in control tumors (Figure S7B–C), consistent with the role of Zfp36l1 in regulating oligodendroglial-astroglial fate switch in the developing brain.
Analysis of TCGA datasets of human gliomas showed that ZFP36L1 was expressed at higher levels in human GBMs than normal brain (Figure 7K). Importantly, patients with GBM and low-grade gliomas with high levels of ZFP36L1 expression exhibited a significantly lower survival probability than those with low expression levels (Figure 7L–M), indicating that ZFP36L1 may have a pro-oncogenic role. To examine the effects of ZFP36L1 depletion on human GBM cell growth, we transduced lentiviral vectors delivering ZFP36L1 shRNA into patient-derived proneural GBM cells TS543 and GBM3264 (Lu et al., 2016) to knockdown ZFP36L1 (Figure 7N). The size and number of spheres formed in sh-ZFP36L1-transduced tumor cells were substantially diminished compared to controls (Figure 7O–P). In addition, cell-cycle-related genes and OPC-associated proneural genes characteristic of GBM were downregulated and expression of astrocyte-associated signature genes were increased upon ZFP36L1 depletion (Figure 7Q–R). These observations indicate that ZFP36L1 is critical for murine and human glioma cell growth and tumor cell fate switch and suggest that a convergent mechanism controls normal gliogenesis and glioma tumorigenesis.
Discussion
Lineage-targeted single-cell analysis uncovered common and divergent molecular and cellular dynamics of glial progenitors and malignant counterparts
Malignant glioma is notoriously heterogeneous at the cellular level, and tumors consist of a substantial proportion of glial progenitor-like cells (Tirosh et al., 2016). The single-cell transcriptomic analyses of targeted lineage precursor populations presented here revealed glial progenitor heterogeneity and two previously uncharacterized intermediate progenitor cells in the neonatal brain, namely, pri-OPCs and iGCs. Our unbiased sorting approaches are more likely to capture the complete cell lineage heterogeneity as compared to cell type assignment based on known cell markers. Despite the distinct transcriptome profiling among these glial progenitor populations, it is possible that these progenitors could be transient states of a more limited set of glia in a stage-dependent manner, representing a developmental continuum along the lineage.
Despite the dissimilarity of cell compositions between normal brain and tumor tissues, we detected pri-OPC-like and iGC-like cells in tumor tissues, which paralleled their counterparts observed in the normal developing brain, suggesting that tumorigenesis mirrors ontogeny. Importantly, we found that actively cycling pri-OPC intermediate progenitors are predominant cellular components in human gliomas caused by distinct genetic mutations, indicating common molecular and cellular networks that link normal glial progenitors and their malignant counterparts. Given that gliomas can occur in different regions (e.g., hemispheric and midline structures) (MacDonald et al., 2011; Monje et al., 2011), it remains to be defined whether specific glial precursor cells in different brain regions correlate with the patterns of gliomagenesis during childhood and adolescence.
scRNA-seq revealed divergent developmental trajectories and proliferation potentials of glial progenitor intermediates
In the neonatal cortex, hGFAP-GFP marks astrocyte lineage cells (Ge et al., 2012). In contrast, hGFAP-GFP mainly labels adult neural stem cells in subependymal zone, SVZ, and dentate gyrus regions of adult mice (Beckervordersandforth et al., 2010; Dulken et al., 2017; Hochgerner et al., 2018). Our single-cell analysis of neonatal cortices reveals that GFP+ cell populations include astrocytes, OPCs, and neuroblasts, suggesting that a population of GFP+ cells in the neonatal cortex is derived from hGFAP-GFP-labeled neural stem cells. Strikingly, we identified an unexpected iGC population related to the well-known astroglial and oligodendroglial cells in the neonatal brain. Cell trajectory analyses indicated that these astroglial/oligodendroglial lineage “double-positive cells” are most likely immature transitional cells. A recent study suggested a local generation of astrocytes through proliferation in the developing cortex (Ge et al., 2012). However, our single-cell data indicate that an Olig2+ iGC subpopulation, but not astrocytes, expresses cell-cycle-related genes, suggesting that the proliferating astrocytic progenitors are a population of transitional iGCs, rather than committed or differentiated astrocytes. Our pseudo-timeline analysis predicted that the intermediate iGCs might lead to astrocytic and oligodendrocytic developmental trajectories; thus, the Olig1/2-expressing iGCs might behave like bipotential glial progenitors.
PDGFRα-GFP-derived progenitors are mainly confined to oligodendrocyte lineage cells, suggesting a developmental fate-restricted continuum along the oligodendrocyte lineage. Lineage trajectory analysis identified a previously uncharacterized progenitor population as pri-OPCs in the neonatal cortex. These early pri-OPCs might serve as early oligodendrocyte progenitors with lineage plasticity (Cai et al., 2007; Zhu et al., 2012). They represent a small population in the adult brain (Figure S7D–S7E), and likely reflect a subset of Sox2+/Olig2+ oligodendroglial precursor cells (Figure S7F) in adulthood (Gibson et al., 2014). It is possible that pri-OPCs are not fate-restricted in certain contexts, and there may be additional layers of heterogeneity not been revealed by these analyses, although pri-OPCs do not appear to produce astrocyte lineage cells.
FACS isolation of PDGFRα-GFP+ cells from the neonatal cortex yielded a number of neuronal populations; this was not the case in this population of cells isolated from juvenile and adult brains (Marques et al., 2016). The presence of neuronal markers diminishes over the course of development, suggesting that PDGFRα-GFPlow cells are neuronal lineage cells driven by transient PDGFRα promoter activity (Kang et al., 2010). Alternatively, this could be the result of perdurance of GFP expression from a common precursor of OPC and neuroblasts expressing PDGFRα (Rivers et al., 2008), expression of which is turned off in neuroblasts, even though the GFP expression remains.
Lineage-driving determinant Zfp36l1 is critical for oligodendrocyte-astrocyte lineage transition and gliomagenesis
We developed a machine-learning algorithm to identify the regulatory networks that drive the specification of distinct glial cell fates. We observed a potential bifurcation of glial sublineage states that enabled us to identify putative regulators of glial fate specification. Among the lineage-driving regulators expressed by both oligodendrocyte and astrocyte lineages, we identified an RNA-binding protein Zfp36l1 that appears to regulate oligodendrocyte fate specification. Although Zfp36l1 is largely restricted to OPCs within the oligodendrocyte lineage, it is detected in other lineages, suggesting that Zfp36l1 might have a function in other cell systems (Hodson et al., 2010; Nasir et al., 2012).
We also demonstrated a critical role of Zfp36l1 for tumor cell growth in the murine glioma model and patient-derived GBM cells. Deletion or downregulation of Zfp36l1 increased astrocytic gene expression and astrocyte differentiation, suggesting that Zfp36l1 inhibition may divert the fate of the proliferating tumor cells with OPC characteristics to astrocyte-like cells and maintain them in a postmitotic state. Thus, our data suggest a commonality of gene regulation between gliogenesis and tumorigenesis and indicate that targeting the lineage-driving determinant Zfp36l1 may inhibit glioma cell growth. Future studies of how the lineage-specific regulatory networks regulate glial lineage trajectories and brain tumorigenesis may reveal additional selective treatment vulnerabilities for malignant gliomas.
Reprogramming of pri-OPC intermediates results in the stem-like phenotype of glioma
Although de-differentiation of mature neural cell types could potentially induce glioma tumorigenesis (Friedmann-Morvinski et al., 2012), our data suggest that pri-OPC intermediates undergo a phenotypic shift through reprogramming into a stem-like state susceptible to further tumor transformation in a self-reinforcing loop. We observed that the actively cycling cells were highly overrepresented among these pri-OPC progenitor intermediates in gliomas. This is in contrast to the traditional cancer stem cell model, which posits stem cells as a minority of malignant cells. The increase of the intermediate iGC population in tumor tissues compared to normal developing cortices suggests that nascent astrocyte precursors like iGCs could be poised to transition into a pri-OPC state resulting in hyper-proliferation. Thus, distinct cellular niches might undergo dynamic transitions over the course of tumorigenesis and contribute to different tumor phenotypes.
Notably, the pri-OPC-like cells were the predominant cycling cell population in the human gliomas evaluated, resembling the tumorigenesis process in the animal model. Our observations suggest that reprogramming and amplification of pri-OPCs to a cancerous, stem-like phenotype, rather than direct proliferation of neural stem cells, results in brain tumor pathogenesis and progression. This is consistent with previous observations that OPCs are a cell of origin in anatomically distinct gliomas (Liu et al., 2011; Monje et al., 2011) and with our recent observation that elimination of Olig2+ mitotic OPC-like progenitors abrogates tumor growth in a GBM animal model (Lu et al., 2016). Nonetheless, we cannot conclude that gliomagenesis has a single cell of origin as the actual mutation even in cases where these cells account for the bulk of the proliferation could be in upstream progenitors.
The single-cell transcriptome data reported here will serve as a resource for understanding the heterogeneity and identity of distinct glial progenitors and their contributions to brain tumor formation. Our findings demonstrate the importance of single-cell mapping and reconstructing fundamental processes of progenitor dynamics and plasticity during development and tumorigenesis. Identification of lineage-specific vulnerabilities for targeting malignant gliomas is an essential step toward a glioma treatment avenue.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Qing Richard Lu (Richard.Lu@cchmc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
PDGFRα-H2BGFP knock-in male mice (the Jackson Laboratory, catalog no. 007669) and hGFAP-GFP mice (the Jackson Laboratory, catalog no. 003257) are purchased from Jackson Laboratory. Mice homozygous for floxed alleles of Zfp36l1 f/f (Stumpo et al., 2004) were crossed with mice carrying Cre recombinase driven by the nestin promoter (Nestin-Cre+/−) to generate Zfp36l1 cKO (Zfp36l1f/f; Nestin-Cre+/−) and heterozygous control (Zfp36l1f/+; Nestin-Cre+/−) mice. Animals of either sex were used in the study and littermates were used as controls unless otherwise indicated. The mouse strains used in this study were generated and maintained on a mixed C57Bl/6;129Sv;CD-1 background and housed (four or less animals per cage) in a vivarium with a 12-hour light/dark cycle. All animal studies were approved by the Institutional Animal Care and Use Committees of the Cincinnati Children’s Hospital Medical Center, USA.
Primary OPC and Culture
Primary rat OPCs were isolated and cultured as described previously with slight modifications (Chen et al., 2007). Briefly, mixed glial cells were initially cultured in DMEM-F12 medium supplied with 15% FBS, then switched to B104 conditioned medium for 2 days before isolating OPCs by mechanical detachment in an orbital shaker. Isolated rat OPCs were grown in Sato growth medium supplemented with mitogens 10 ng/ml PDGF-AA and 20 ng/ml bFGF, and differentiated in OL Differentiation Medium (Sato medium supplemented with 15 nM T3 and 10 ng/ml ciliary neurotrophic factor).
Mouse and Human GBM Cell Culture
Mouse, human tumor cell (GBM3264) and sphere cultures were established and maintained in serum-free DMEM/F12 medium (Life Technologies), containing B27 (without vitamin A, Invitrogen, Carlsbad, CA), epidermal growth factor (20 ng/mL, Peprotech), and basic fibroblast growth factor (20 ng/mL; Peprotech). The human GBM proneural cell lines (TS543) were maintained in Neural Stem Cell (NSC) Basal Medium with NSC proliferation supplements, 10 ng/ml EGF, 20 ng/ml basic-FGF and 2 μg/ml Heparin (Stem Cell Technologies, Vancouver, Canada) as previously described (Lu et al., 2016).
Human Glioma Single Cell RNA-seq Datasets
We analyzed the single cell RNA-seq expression profiles of human GBM, IDH mutant oligodendroglioma, IDH mutant astrocytoma and H3K27M diffuse midline glioma from the GEO public resource (http://www.ncbi.nlm.nih.gov/geo/) and the accession numbers are GSE82211 (Patel et al., 2014), GSE70630 (Tirosh et al., 2016), GSE89567 (Venteicher et al., 2017) and GSE102130 (Filbin et al., 2018). We performed clustering by R package Seurat (https://satijalab.org/seurat/). To exclude the batch effect of distinct patients, we used unique marker genes for variable genes analysis, they were used for principle component analysis (PCA). The statistically significant PCs were used for two-dimension t-distributed stochastic neighbor embedding (t-SNE).
Human glioma patient survival and gene expression data were analyzed from TCGA (https://tcga-data.nci.nih.gov/docs/publications/tcga/), GTEx projects (https://gtexportal.org/), as well as http://gepia.cancer-pku.cn/ and http://ualcan.path.uab.edu/.
METHOD DETAILS
Single-cell Isolation and Library Preparation
Neonatal pups are euthanized on the ice, decapitated, and the brain was immediately removed and submerged in fresh ice-cold HBSS-HEPES (15 mM HEPES, Gibco 15630–060 and 15 mM Glucose, Sigma G8769 dissolved in HBSS Sigma H6648), cortices were carefully dissected and minced by blade. The tissue pieces were incubated with 3 ml of TrypLE 5× solution (dilute in HBSS-HEPES by 10× TrypLE, Gibco A12177) for 10 min in a 37°C-incubator following by gentle trituration through Pasteur pipettes with polished tip for ten times, filtered the suspension by 40mm strainer, centrifuge at 1300 rpm to get the single cell pellet.
Collect the GFP+ alive cell by excluding the population of GFP- and 7-AAD+ (Stemcell 75001) death cells performed on MoFloXDP (Beckman Coulter). For droplet sequencing (Drop-seq), we followed the procedure as described by Macosko et al. (Macosko et al., 2015). The cDNA libraries were purified, quantified, and then sequenced on the Illumina Hiseq 2500. In addition, GFP+ single cells were prepared using the C1 Single-Cell Auto Prep System (Fluidigm) according to the manufacturer’s instructions. For single cell RNA-seq data from Drop-seq and Fluidigm C1 platforms, we discarded low quality cells which obviously deviate from the major cell population. We performed two quality measurements: the number of genes and the average expression level (log2(TPM+1)) of a curated list of housekeeping genes. In PDGFRα-GFP populations, we excluded the cells with either fewer than 500 detected genes or an average housekeeping expression level below 0.35 (recovered 100% cells). In the hGFAP-GFP cells, we excluded those cells with either fewer than 900 detected genes or an average housekeeping expression level below 1.6 (recovered 41% cells). On average, we recovered 2280, 4581 transcripts per cell, which represented 1301, and 2096 unique genes expressed per cell, respectively.
For single cell RNA-seq of tumor cells, the tumor tissues were digested by TryplE with collagenase Ⅰ, and single cell suspension was treated by Red Blood Cell Lysis Buffer (Sigma, 11814389001). Cells from tumor tissues at dpi 25 and dpi 35 were sequenced by drop-seq or 10× genomics, respectively. Chromium Single Cell 3’ Library & Gel Bead Kit v2 (120237), Chromium Single Cell A Chip Kit (120236) and Chromium i7 Multiplex Kit (120262) were used along with a 10× GemCode Single Cell Instrument, per the manufacturer’s manuals (document CG00052; Rev A). Base on the distribution of cells ordered by percentage of mitochondrial genes, housekeeping genes and detected gene numbers, we excluded those cells with either less than 600 detected genes or an average housekeeping expression level below 1.3 in dpi 35 (recovered 100% cells), as well as excluded those cells with either more than 2800 detected genes or less than 700, and an average mitochondrial expression level more than 0.2 in dpi 25 (recovered 73% cells). For each set of drop-seq, isolated cells at each stage were pooled together and processed to reduce the risk of batch effects. The histograms of UMIs were shown in figshare (https://figshare.com/s/439405353ba2c51b5d51).
Cell Clustering and Visualization
We performed unsupervised clustering by R package Seurat (Macosko et al., 2015). The highly variable genes were identified from these cells using Seurat with the default setting for mouse datasets, they were used for principle component analysis (PCA). The statistically significant PCs were used for two-dimension t-distributed stochastic neighbor embedding (tSNE). Differentially expressed gene (adjusted p-value<0.05 and more than 1.5-fold change or p-value<0.05) for scRNA-seq data were shown as Table S2. The clustered cell matrix for hGFAP-GFP, PDGFRα-GFP and mouse glioma was shown in figshare (https://figshare.com/s/439405353ba2c51b5d51). We verified the clustering by two other different methods: Backspin and PAGODA (Pathway and Geneset Overdispersion Analysis). The hGFAP-GFP and PDGFRa-GFP datasets were clustered using the BackSPIN algorithm as previously described (Marques et al., 2016). In short, the BackSPIN algorithm executed a bi-clustering by sorting the cells and genes into a one-dimensional ordering where a binary split is performed based on the distribution of genes within each ordering. The algorithm repeatedly performs feature selection and subsequent splits until an appropriate threshold is achieved. For clustering the hGFAP-GFP and PDGFRa-GFP datasets using PAGODA (SCDE R-package) (Fan et al., 2016), First, the drop-out rate is determined and the amplification noise is estimated to fit the error models for each single cell. Poor cells with abnormal fit will be removed. PAGODA estimates the overdispersion in genes and gene sets for cell clustering analysis. The overdispersion in gene sets is defined as the amount of variance explained by the first principal component exceed the background expectation. Gene annotation from GO terms are used to facilitate the process by finding pathways with significantly excess of coordinated variability. Finally, cells are clustered according to their patterns in all significant aspects.
For the dataset from Fluidigm C1 cells, we performed unsupervised clustering by ICGS in AltAnalyze (http://www.AltAnalyze.org). All the ICGS, Marker Finder and PCA plot were following the guidance of toolkit AltAnalyze (http://www.AltAnalyze.org). For further subclassification, Marker Finder was performed. After classification, PCA was conducted by Altanalyze using the clustered matrix. Individual gene plot was executed by input distinct genes. For the minimum Pearson correlation cutoff, we used 0.4 for Fluidigm C1.
Analysis of Cell Lineages Trajectory
We used a recently developed cell lineage inference algorithm, Slingshot (Version 0.0.0.9005, https://github.com/kstreet13/slingshot), to predict lineage trajectories and bifurcations by ordering cells along trajectories. Slingshot takes as input a matrix of reduced dimension normalized expression measures using PCA and cell clustering assignments. Lineages are defined by ordered sets of clusters beginning with the root node and terminating in the most distal cluster(s) with only one connection. Potential fitting curves are drawn to the subsets of cells that potentially make up each lineage. The ordering provided by Slingshot, analogous to pseudo developmental time points, is referred to herein as developmental order. The cluster representing RG was chosen as the starting root node.
To analyze the timing of oligodendrocyte development, we extracted cells from OPC and immature oligodendrocyte clusters from the original cell dataset. The subpopulations were pooled for analysis. The most variable genes among all the single cells were identified by Seurat. A pseudo developmental timeline of single cells was then calculated with the package Slingshot, using the most variable genes as time ordering genes. Based on the established differentiation direction of oligodendrocyte (from pri-OPC to OPC to iOL), the direction of pseudotime axis was determined. OPC and pri-OPC in hGFAP-GFP dataset were explored as the same method.
Identification of Lineage-driving Transcriptional Regulators
1) Overview of the driving transcriptional regulator (TR) detection method: we call a set of TRs the driving TRs of a cell group if they activate and regulate the cell group’s differentially expressed genes (DEGs). Because of the regulation relationships with the DEGs, the driving TRs are expected to bind these genes more frequently than random bindings. Thus, in order to decide whether a TR is a driving TR, we compare its binding frequency (to the DEGs) with the binding frequency due to randomness. To do this, for each given cell group, we first detect a set of DEGs, then estimate the TR-DEG binding state matrix (binding or not binding) and the TR-DEG binding probability matrix (due to randomness). 2) Differentially expressed genes: we identify DEGs of each cell group using EdgeR. 3) TR-gene binding state matrix and TR-DEG binding state matrix: In order to compute the binding frequency of each TR, we first infer the TR-gene binding state (whether a TR is binding to a gene or not) based on association of TR-gene expression. Since TR-gene regulation is tissue-specific, we consider the problem within the scope of a certain tissue. We assume that, in a specific tissue, a gene should have similar expression pattern with a TR that regulates (binds) it. Pearson correlation coefficient is employed to assess the association between expression of a TR and a gene, and Student’s t-test of correlation coefficient is performed to decide if the TR-gene pair is statistically significantly associated, namely, if the gene is bound by the TR. The inferred binding state of all TR-gene pairs are organized into a TR-gene binding state matrix, and that of all TR-DEG pairs are organized into a TR-DEG binding state matrix. 4) TR-gene binding probability matrix and TR-DEG binding probability matrix: The binding probability, or the frequency of random bindings, between a gene (or its enhancer regions) and a TR can be impacted by two factors: 1) the affinity of a gene to TRs (the tendency that this gene is bound by any TR); and 2) the affinity of a TR to genes (the tendency that such TR binds any gene). Genes may have different affinities to TRs because of their different properties such as gene length and GC content. Similarly, TRs have different affinity to genes due to differences in features such as motif characteristics. TR with short motifs are more likely to bind a gene by chance. However, all these causing factors can be hardly given as priori knowledge. We estimate the effects of all these factors from the data. Regardless of all complex causes, the affinity of a gene to TRs can be captured by totally how many TRs can bind the gene, and the affinity of a TR to genes can be indicated by totally how many genes it binds.
With the affinities of different genes and of different TRs taken into account, we assume that TR-gene pairs’ random binding states follow a 2D Fisher’s noncentral hypergeometric distribution. Then the probability of a TR-gene pair’s random binding can be estimated as:
where Pij is the probability that the ith TR binds the jth gene. ni is conceptually the number of genes that the ith TR binds and computationally the sum of the ith row in TR-gene binding state matrix. Similarly, mj is the number of TRs that bind to the jth gene i.e., the sum of the jth column in TR-gene binding state matrix. {ri} and {wj} are variables reflecting the ith TR’ affinity to genes and the jth gene’s affinity to TRs, respectively. Pijs can be obtained by solving the equation system. It can be proved that the solution for Pijs is unique. In practice, Pijs are obtained by first initializing {ri}, then updating {wj} and {ri} iteratively. All Pijs together form a TR-gene binding probability matrix, from which Pijs of only DEGs are selected to compose a TR-DEG binding probability matrix.
5) Driving TR detection: Having obtained the TR-DEG binding state matrix and TR-DEG random binding probability matrix, we compare them to detect driving TRs of each given cell group. For each TR, we decide it as a driving TR if its binding frequency to the cell group’s DEGs is statistically significantly higher than its random binding probability to these DEGs.
The test statistics, which indicates how higher the observed binding frequency is than the null random binding probability, is computed as following:
where k is the index of DEG; Yik is the binding state (1 for binding and 0 for non-binding) of the ith TR and kth DEG. Pik is the null random binding probability of the ith TR and kth DEG. Yik and Pik are respectively from the TR-DEG binding state matrix and TR-DEG binding probability matrix. Finally, to facilitate the evaluation of statistical significance, we transform the score[Si] into a Z-score of the ith TR:
which approximately follows a standard Gaussian distribution and from which we can get p-value for the TR.
Active cycling cell analysis
Gene sets representing five phases of the cell cycle (G1/S, S, G2/M, M and M/G1) were refined as previously described (Tirosh et al., 2016). In short, we extracted cycling genes by examining the correlation between the expression pattern of each gene and the average expression pattern of all genes in either dpi25 or dpi35 tumor dataset, and excluding genes with a low correlation (R<0.25). This step removed genes that were identified as phase-specific in HeLa cells but did not correlate with that phase in our single-cell data.
Then the putative actively cycling cells were identified as previous method (Tirosh et al., 2016). Basically, the cells were defined as actively cycling by at least a twofold upregulation and a t-test P value < 0.01 for either the G1/S or the G2/M gene set compared to the average of all cells. Relative proliferating score in both putative actively cycling cells and leftover pri-OPC-like cells were calculating, the average actively cycling score in non-actively cycling cells were normalized to 1.
Stemness score and module calculation
To explore the stemness signature of pri-OPC-like cells in tumor tissues, we applied stemness signature gene sets related to glioma formation (Tirosh et al., 2016) to our dataset, and calculated the average expression of stemness genes in each cell, which represented the stemness score. The stemness score of each group was then calculated. To compare the stemness score in tumor tissues and normal neonatal glial cells, we employed all cells in hGFAP-GFP and PDGFRa-GFP dataset, and the stemness score of each cell in either dataset was calculated and normalized to the average of stemness gene expression of all tumor cells. The stemness score of each group was then calculated.
For pri-OPC score in Figure 5G, we selected all the OPC-lineage cells from PDGFRα-GFP dataset and employed Seurat to get the variable genes. We excluded all the merged genes between pri-OPC and OPC from pri-OPC differential expressed genes (Table S3), the pri-OPC score of each cell in either dataset was calculated and normalized to the average of pri-OPC gene expression of all tumor cells. The pri-OPC score of each group was then calculated. For violin plots, the gene list for each referred gene module were shown in Table S3.
Correlation analysis between cortical pri-OPC and hippocampal pre-OPC
To compare cortical pri-OPC/OPC/iOL with pre-OPC/OPC from the mouse hippocampus at P0 and P5, we firstly extracted gene expression data of pre-OPC and OPC from previously published datasets (Hochgerner et al., 2018; La Manno et al., 2018), and used the Seurat program to combine the datasets of cortical pri-OPC/OPC/iOL with hippocampal pre-OPC/OPC, normalized the expression values, and conducted PCA. The data were then scaled by Seurat and regress to “nUMI” and “Mt- genes”, the correlation coefficient values were calculated by “cor.test” function in R.
Tissue Processing and In Situ Hybridization
Mice at various developmental stages were anesthetized with ketamine/xylazine and perfused with PBS followed by 4% paraformaldehyde (PFA). Brain tissues were dissected and fixed in 4% PFA overnight. dehydrated in 20% sucrose at 4°C, embedded in OCT and cryosectioned at 16 μm or paraffin-embedded for sectioning at 10 μm. For immunostaining of cryosections, tissues were shortly fixed in 4% PFA for 2–4 hours. For vibratome sectioning, tissues were fixed in 4% PFA overnight and embedded by 4% agarose in PBS and sectioned at 50 μm. In situ hybridization was performed as previously described (Lu et al., 2002). Digoxigenin-labeled riboprobes used in the study was murine Zfp36l1.
Immunofluorescence Staining and Immunohistochemistry
Cryosections (16-μm thick) or vibratome sections (50-μm thick) were permeabilized and blocked in blocking buffer (0.3% Triton X-100 and 5% normal donkey serum in PBS) for 1 h at room temperature and overlaid with primary antibodies overnight at 4 °C. For human brain paraffin section staining, we performed antigen retrieval before permeabilization. Antibodies used in the study were: Olig2 (Rabbit, Millipore, AB9610, RRID:AB_10141047; Mouse, Millipore, MABN50, RRID:AB_10807410), PDGFRα (Rat; BD Bioscience, 558774, RRID:AB_397117), APC (Mouse; CC1, Oncogene Research, OP80, RRID:AB_2057371), MBP (Goat; Santa Cruz Biotechnology, sc-13914, RRID:AB_648798), Sp8 (Goat; Santa Cruz Biotechnology, sc-104661, RRID:AB_2194626), Dlx2 (Rabbit; Abcam, ab30339, RRID:AB_731969), Glutamine Synthetase (Mouse; Millipore, MAB302, RRID:AB_2110656), Sharp2 (Rabbit, Abcam, ab97525, RRID:AB_10680936); BHLHE40 (Rabbit, Sigma, HPA028922, RRID:AB_2672828), PRDM16 (Rabbit; LifeSpan BioSciences, LS-B4625, RRID:AB_10797227), GFAP (Mouse; Sigma, G3893, RRID:AB_477010), Ki67 (Rabbit; Thermo Fisher Scientific, RM-9106, RRID:AB_2335745), BrdU (Mouse; BD Pharmingen, 555627, RRID:AB_395993), GFP (Goat, Novus Biologicals, NB100–1770, RRID:AB_10128178; Rabbit, Thermo Sci, A11122, RRID:AB_221569), Slc1a3 (Rabbit; Novus Biologicals, NB100–1869, RRID:AB_2190597), PHI-1 (Mouse; Santa Cruz Biotechnology, sc-514759), Ascl1 (Rabbit; Abcam, ab74065, RRID:AB_1859937), BLBP (Rabbit; Abcam, ab32423, RRID: AB_880078), Sox2 (Goat; Santa Cruz Biotechnology, sc-17320, RRID: AB_2286684). After washing with 0.3% Triton X-100 in PBS, cells or sections were incubated with secondary antibodies conjugated to Cy2, Cy3 or Cy5 (Jackson ImmunoResearch Laboratories) for 2 h at room temperature, stained in DAPI for 10 min, washed in PBS and mounted with Fluoromount-G (SouthernBiotech). Cell images were quantified in a blinded manner. For BrdU staining, cells or tissue sections were denatured with 1 N HCl for 1 hr at 37°C. Sections were neutralized with 0.1 M Borax pH 8.5 (Sigma) for 10 min, washed with PBS and blocked with 5% normal donkey serum (Sigma, Inc.) for 1 hr at room temperature. All immunofluorescence-labeled images were acquired using a Nikon C2+ confocal microscope. For Zfp36l1 in situ combined with immunolabeling, we developed in BM Purple (Roche-11442074001) for 3 days to enhance the signal.
Assay for Transposase-accessible Chromatin Using Sequencing (ATAC-Seq)
ATAC-seq assays were performed as previously described (Buenrostro et al., 2015). Briefly, we isolated nuclei of ~50,000 FACS-sorted PDGFRa-GFP+ cells from the cortices of a pool of 3 individual animals at each E14.5 and P5 stage in a cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630) by exclude GFP− and 7-AAD+ cells. After spinning down at 500 × g for 10 min at 4 °C, nuclei were resuspended in transposition mix containing TD (2× reaction buffer), TDE1 (Nextera Tn5 Transposase) at 37°C for 30 min. The samples were purified using a Qiagen MinElute kit. Transposed DNA fragments were subsequently amplified and purified using Qiagen MinElute PCR Purification Kit. Libraries were generated using the Ad1_noMX and Ad2.1–2.4 barcoded primers and were amplified for 11 total cycles. Libraries were purified with AMPure beads (Agencourt) to remove contaminating primer dimers. All libraries were sequenced on the Illumina HiSeq 2500 with 75 bp single-end reads.
Reads of ATAC-seq data were aligned to mm10 genome using Bowtie with the following options: --best --chunkmbs 200 (http://bowtie-bio.sourceforge.net). Peak calling was performed using Model-based analysis of MACS version 2.12 (https://github.com/taoliu/MACS) with specific parameters without the prebuilt model: --shift −75 --extsize 150 --nomodel --call-summits --nolambda --keep-dup all -p 0.01, to call peaks, which extend and shift the fragments to get the region cut by the Tn5 sites. We calculated the peak_RPKM, then GSEA (v2.2.0) was used to analyze the enrichment of signature gene sets from different cell types in E14.5 and P5 PDGFRα-GFP cells. Homer (http://homer.ucsd.edu/homer/) was used to generate the normalized UCSC bedgraph files (the total number of tags is normalized to 10 million) to show the genome browser tracks.
siRNA Knockdown, Transduction and Sphere Formation Assays
For siRNA knockdown by negative control or Ppp1r14b siRNA (Sigma, SASI_Rn01_00039926, SASI_Rn01_00039931) in rat OPCs, we used Lipofectamine® RNAiMAX Transfection Reagent (ThermoFisher Scientific, 13778150) according to the manufacturer’s protocol. Cells were harvested for immunocytochemistry or qRT-PCR analysis.
For siRNA knockdown by negative control or Zfp36l1 siRNA (Sigma, SASI_Mm01_00063508, SASI_Mm01_00063509, SASI_Mm01_00063512) in mouse tumor cells, we use Lipofectamine® RNAiMAX Transfection Reagent (ThermoFisher Scientific, 13778150) according to the manufacturer’s protocol. Cells were harvested after 72 h and performed BrdU staining (pulse-labeled with BrdU for 1 hr before fixation).
For qRT-PCR, mouse and human tumor cells were transduced by non-target control or lentiviral vectors carrying Zfp36l1 shRNA lentivirus (Nasir et al., 2012) for 72 h, the cells were harvested and analyzed by qRT-PCR.
For the human sphere formation assay, the primary spheres were further dissociated into single cells and diluted to a density of 1000 cells/ml. Then the cells were infected by non-target control or lentiviral vectors carrying Zfp36l1 shRNA lentivirus for 48 hours. The 10 μl/well diluted cell suspension was plated to ultralow attachment 96-well plate (Corning Inc., Corning, NY, USA) in serum-free medium. The number of wells with spheres is counted after 8 days induction.
RNA Extraction and qRT-PCR
Analyses were conducted with RNA extracts from cells. Total RNA was extracted per the Trizol (Life Technologies) protocol. cDNA was generated with iScript™ cDNA Synthesis Kit (Bio-Rad). qRT-PCR was performed using the ABI Prism 7700 Sequence Detector System (Perkin-Elmer Applied Biosystems). qRT-PCR analysis is based on the ΔΔCT method with normalization of the raw data to GAPDH genes. For each gene, ΔCT was calculated by subtracting CTGAPDH from CTGENE in either the control or experimental group. We set the average ΔCT of the control as a calibrator, then the 2–ΔΔCT method was used to calculate each relative expression in both control or experimental group. The values in the control were normalized to 1 by dividing each data point with the averaged control value. The primer sequences were included in Table S4.
Copy Number Variation Analysis
CNV analysis in single-cell profiling of tumor tissues was performed as previously described (Patel et al., 2014), with minor changes. Briefly, CNVs were estimated by sorting the analyzed genes by their chromosomal location and applying a moving average to the relative expression values, with a sliding window of 100 genes within each chromosome, then to stabilize the graph, we averaged such these 50 genes again. To normalize these patterns to the reference cluster of “normal cells”, we used the macrophage/microglia cluster with the same tumor tissues in each single-cell profile at different stages of tumorigenesis.
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses were done using Microsoft Excel, GraphPad Prism 6.00 (San Diego California, www.graphpad.com) and RStudio (https://www.rstudio.com/). Data are shown as mean ± SEM or as a Box-and-whisker plot, as a dot plot and as a violin plot. Data distribution was assumed to be normal, but this was not formally tested. Statistical significance was determined using two-tailed Student’s t tests or Wilcoxon rank-sum and signed-rank tests as indicated. One-way ANOVA test was performed by multiple comparisons following Turkey’s ranking tests when comparing multiple groups. Significance was set as * for p < 0.05, ** for p < 0.01, and *** p < 0.001. Correlation significance of distinct groups or scores was assessed by Pearson’s correlation coefficient-test. Values of p<0.05 denoted a statistically significant difference. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally employed in the field. Quantifications were performed from at least three independent experiments. No randomization was used to collect all the data, but they were quantified blindly.
DATA AND SOFTWARE AVAILABILITY
All the scRNA-seq and ATAC-seq data have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE122871.
Supplementary Material
Table S1. Lists of driving transcriptional regulators. Related to Figure 4.
Table S2. Lists of differentially expressed genes for identified clusters. Related to Figures 1, 3 and 5.
Table S4. Primer sequences for q-PCR and genotyping. Related to STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Olig2 | Millipore | Cat#AB9610; RRID:AB_10141047 |
| Mouse anti-Olig2 | Millipore | Cat#MABN50; RRID:AB_10807410 |
| Rat anti-PDGFRα | BD Bioscience | Cat#558774; RRID:AB_397117 |
| Mouse anti-APC (CC1) | Oncogene Research | Cat#OP80; RRID:AB_2057371 |
| Goat anti-MBP | Santa Cruz biotechnology | Cat#sc-13914; RRID:AB_648798 |
| Goat anti-Sp8 | Santa Cruz Biotechnology | Cat#sc-104661; RRID:AB_2194626 |
| Rabbit anti-Dlx2 | Abcam | Cat#ab30339; RRID:AB_731969 |
| Mouse anti-Glutamine Synthetase | Millipore | Cat#MAB302; RRID:AB_2110656 |
| Rabbit anti-Sharp2 | Abcam | Cat#ab97525; RRID:AB_10680936 |
| Rabbit anti-BHLHE40 | Sigma | Cat#HPA028922; RRID:AB_2672828 |
| Rabbit anti-PRDM16 | LifeSpan BioSciences | Cat#LS-B4625; RRID:AB_10797227 |
| Mouse anti-GFAP | Sigma | Cat#G3893; RRID:AB_477010 |
| Rabbit anti-Ki67 | Thermo Fisher Scientific | Cat#RM-9106; RRID:AB_2335745 |
| Mouse anti-BrdU | BD Pharmingen | Cat#555627; RRID:AB_395993 |
| Goat anti-GFP | Novus Biologicals | Cat#NB100–1770; RRID:AB_10128178 |
| Rabbit anti-GFP | Thermo Fisher Scientific | Cat#A11122; RRID:AB_221569 |
| Rabbit anti-Slc1a3 | Novus Biologicals | Cat#NB100–1869; RRID:AB_2190597 |
| Rabbit anti-PHI-1 (Ppp1r14b) | Santa Cruz Biotechnology | Cat#sc-514759 |
| Rabbit anti-Ascl1 | Abcam | Cat#ab74065; RRID:AB_1859937 |
| Rabbit anti-BLBP | Abcam | Cat#ab32423; RRID: AB_880078 |
| Goat anti-Sox2 | Santa Cruz Biotechnology | Cat#sc-17320; RRID: AB_2286684 |
| Biological Samples | ||
| Mouse cortex, spinal cord | This study | N/A |
| Human cortex tissue | CCHMC pathology core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PDGF AA | PeproTech | Cat#100–13A |
| bFGF | PeproTech | Cat#100–18B |
| Insulin from bovine pancreas | Sigma-Aldrich | Cat#I6634 |
| DIG RNA Labeling Mix | Roche | Cat#11277073910 |
| PolyJet | SignaGen Laboratories | Cat#SL100688 |
| MinElute PCR Purification Kit | Qiagen | Cat#28004 |
| Heparin Solution | Stem Cell Technologies | Cat#07980 |
| BCIP®/NBT Alkaline Phosphatase Substrate | Sigma | Cat#B5655 |
| T7 RNA polymerase | Promega | Cat#P207B |
| TRIzol reagent | ThermoFisher Scientific | Cat#15596018 |
| iScript™ cDNA Synthesis Kit, 100 × 20 μl rxns | Bio-Rad | Cat#1708891 |
| Fluoromount-G | SouthernBiotech | Cat#0100–01 |
| RNAiMAX | ThermoFisher Scientific | Cat#13778030 |
| NeuroCult™ NS-A Proliferation Kit (Human) | Stem Cell Technologies | Cat#05751 |
| HBSS | Sigma | Cat#H6648 |
| HEPES | Gibco | Cat#15630–060 |
| Glucose | Sigma | Cat#G8769 |
| TrypLE | Gibco | Cat#A12177 |
| 7-AAD+ | Stem Cell Technologies | Cat#75001 |
| Human CNTF | Peprotech | Cat#450–13 |
| Recombinant Human EGF | Peprotech | Cat#100–15 |
| BM Purple | Roche | Cat#11442074001 |
| Critical Commercial Assays | ||
| BioAnalyzer High Sensitivity Chip | Agilent Technologies | Cat#5067–4626 |
| Hifi HotStart Readymix | Kapa Biosystems | Cat#KK2602 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v2, 16 rxns PN | 10× Genomics | Cat#120237 |
| Chromium Single Cell A Chip Kit, 48 rxns PN | 10× Genomics | Cat#120236 |
| Chromium i7 Multiplex Kit, 96 rxns PN | 10× Genomics | Cat#120262 |
| High Sensitivity DNA Kit | Agilent | Cat#5067–4626 |
| Nextera® XT DNA Library Preparation Kit | Illumina | Cat#FC-131–1024, Cat#FC-131–1096 |
| NEBNext® High-Fidelity 2X PCR Master Mix | NEB | Cat#M0541S |
| 100× SYBR Green I | Invitrogen | Cat#S-7563 |
| Nextera® DNA Sample Preparation Kit (24 Samples) | Illumina | Cat#FC-121–1030 |
| Deposited Data | ||
| Raw and processed data | This paper | GEO: GSE122871 |
| Human GBM of single-cell rna-seq | (Patel et al., 2014) | GEO: GSE82211 |
| IDH mutant oligodendroglioma of single-cell rna-seq | (Tirosh et al., 2016) | GEO: GSE70630 |
| IDH mutant astrocytoma of single-cell rna-seq | (Venteicher et al., 2017) | GEO: GSE89567 |
| H3K27M diffuse midline glioma of single-cell rna-seq | (Filbin et al., 2018) | GEO: GSE102130 |
| P0 and P5 mouse OPC cells | (Hochgerner et al., 2018) | GEO: GSE95753 |
| Experimental Models: Cell Lines | ||
| Human GBM cell line (TS543) | (Lu et al., 2016) | N/A |
| Human GBM cell line (GBM3264) | Jeremy N. Rich | N/A |
| Rat OPC primary culture | This study | N/A |
| Human: HEK 293 cell | ATCC | CRL-11268 |
| Mouse GBM cell line | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| B6.129S4-Pdgfratm11(EGFP)Sor/J | The Jackson Laboratory | Stock No. 007669 |
| FVB/N-Tg(GFAPGFP)14Mes/J | The Jackson Laboratory | Stock No. 003257 |
| Zfp36l1 flox/flox | (Stumpo et al., 2004) | N/A |
| B6.Cg-Tg(Nes-cre)1Kln/J | The Jackson Laboratory | Stock No. 003771 |
| Sprague Dawley® Rats | Charles River Laboratories | N/A |
| Rosa26tdTomato, Ai14 | The Jackson Laboratory | Stock No. 007914 |
| Oligonucleotides | ||
| Ppp1r14b siRNA-1 CAAACCCACUGAGGCCUUC[dT][dT] |
Sigma | SASI_Rn01_00039926 |
| Ppp1r14b siRNA-2 GGAAGGUCACCGUCAAGUA[dT][dT] |
Sigma | SASI_Rn01_00039931 |
| Zfp36l1 siRNA-1 GACCUCUUGGGCUCACCUA[dT][dT] |
Sigma | SASI_ Mm01_00063508 |
| Zfp36l1 siRNA-2 GCUUUCGAGACCGCUCUUU[dT][dT] |
Sigma | SASI_Mm01_00063509 |
| Zfp36l1 siRNA-3 GCCUCUUUGCUCCUAGCAU[dT][dT] |
Sigma | SASI_Mm01_00063512 |
| MISSION siRNA Universal Negative Control #1 | Sigma | Cat#: SIC001 |
| See Table S4 for the primers for Genotyping, q-PCR | N/A | N/A |
| Recombinant DNA | ||
| pSicoR-shZfp36l1 | (Nasir et al., 2012) | N/A |
| PB-CAG-DNp53-PDGFB | This paper | N/A |
| PB-CAG-DNp53-PDGFB-Cre | This paper | N/A |
| Software and Algorithms | ||
| Cell Ranger | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Seurat | (Macosko et al., 2015) | http://satijalab.org/seurat/ |
| Altanalyze | Cincinnati Children’s Hospital Medical Center | http://www.altanalyze.org/ |
| Slingshot | Deconstructing Olfactory Stem Cell Trajectories at Single-Cell Resolution | https://github.com/kstreet13/slingshot |
| GraphPad Prism 6.00 | GraphPad | www.graphpad.com |
| PAGODA (SCDE R-package) | (Fan et al., 2016) | http://hms-dbmi.github.io/scde/ |
| BackSPIN algorithm | (Marques et al., 2016) | https://github.com/linnarsson-lab/BackSPIN |
| R language | R Core Team (2016) The R Project for Statistical Computing | http://www.r-project.org |
| Toppfun | Cincinnati Children’s Hospital Medical Center | https://toppgene.cchmc.org/enrichment.jsp |
| Toppcluster | Cincinnati Children’s Hospital Medical Center | https://toppcluster.cchmc.org/ |
| Gene Set Enrichment Analysis (GSEA) | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| MACS | Liu Lab, Harvard University | http://liulab.dfci.harvard.edu/MACS |
| HOMER | Integrative Genomics and Bioinformatics core at the Salk Institute | http://homer.ucsd.edu/homer/ |
| UCSC Genome Browser | The human genome browser at UCSC | http://genome.ucsc.edu/ |
| DRIVE (for finding lineage driving TRs) | This paper | Methods |
Highlights:
scRNA-seq reveals distinct transitional states of glial progenitors in the cortex
Intersectional analysis reveals malignant analogs of glial progenitor intermediates
Reprogramming OPC intermediates into a stem-like state for malignant gliomagenesis
Zfp36l1 is a critical regulator of glial fate specification and gliomagenesis
ACKNOWLEDGMENTS
The authors would like to thank Drs. D. Xin, X. Zhou, A. Lopez-Juarez, E. Rencsok, N. Salomonis, and M. Adam for cell sorting, initial analysis, and drawing. Drs. B. Weiss, E. Simonds, and E. Hurlock for comments, Dr. T. Phenix for the PDGFB vector. This study was funded in part by grants from the National Institutes of Health (R37 NS096359 and R01 NS075243) to Q.R.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and four tables and can be found with this article online.
REFERENCES:
- Beckervordersandforth R, Tripathi P, Ninkovic J, Bayam E, Lepier A, Stempfhuber B, Kirchhoff F, Hirrlinger J, Haslinger A, Lie DC, et al. (2010). In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell 7, 744–758. [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Chen Y, Cai WH, Hurlock EC, Wu H, Kernie SG, Parada LF, and Lu QR (2007). A crucial role for Olig2 in white matter astrocyte development. Development 134, 1887–1899. [DOI] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, and Jegga AG (2009). ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, and Lu QR (2007). Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protoc 2, 1044–1051. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, and Barres BA (2006). Functional genomic analysis of oligodendrocyte differentiation. J Neurosci 26, 10967–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulken BW, Leeman DS, Boutet SC, Hebestreit K, and Brunet A (2017). Single-Cell Transcriptomic Analysis Defines Heterogeneity and Transcriptional Dynamics in the Adult Neural Stem Cell Lineage. Cell Rep 18, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan JB, Zhang K, Chun J, et al. (2016). Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat Methods 13, 241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, Neftel C, Frank N, Pelton K, Hebert CM, et al. (2018). Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher RB, Das D, Gadye L, Street KN, Baudhuin A, Wagner A, Cole MB, Flores Q, Choi YG, Yosef N, et al. (2017). Deconstructing Olfactory Stem Cell Trajectories at Single-Cell Resolution. Cell Stem Cell 20, 817–830 e818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, Ellisman MH, and Verma IM (2012). Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, and Deneen B (2014). Glial development: the crossroads of regeneration and repair in the CNS. Neuron 83, 283–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge WP, Miyawaki A, Gage FH, Jan YN, and Jan LY (2012). Local generation of glia is a major astrocyte source in postnatal cortex. Nature 484, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochgerner H, Zeisel A, Lonnerberg P, and Linnarsson S (2018). Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat Neurosci 21, 290–299. [DOI] [PubMed] [Google Scholar]
- Hodson DJ, Janas ML, Galloway A, Bell SE, Andrews S, Li CM, Pannell R, Siebel CW, MacDonald HR, De Keersmaecker K, et al. (2010). Deletion of the RNA-binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat Immunol 11, 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, and Bergles DE (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinghoffer RA, Hamilton TG, Hoch R, and Soriano P (2002). An allelic series at the PDGFalphaR locus indicates unequal contributions of distinct signaling pathways during development. Dev Cell 2, 103–113. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, and Alvarez-Buylla A (2009). The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32, 149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, Bruce JN, and Canoll P (2011). Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS One 6, e20041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J,C, Yu K, Hatcher A, Huang TW, Lee HK, Carlson J, Weston MC, Chen F, Zhang Y, Zhu W, et al. (2017). Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci 20, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, et al. (2011). Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Chen Y, Zhao C, Wang H, He D, Xu L, Wang J, He X, Deng Y, Lu EE, et al. (2016). Olig2-Dependent Reciprocal Shift in PDGF and EGF Receptor Signaling Regulates Tumor Phenotype and Mitotic Growth in Malignant Glioma. Cancer Cell 29, 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, and Rowitch DH (2002). Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109, 75–86. [DOI] [PubMed] [Google Scholar]
- Lu QR, Yuk D, Alberta JA, Zhu Z, Pawlitzky I, Chan J, McMahon AP, Stiles CD, and Rowitch DH (2000). Sonic hedgehog--regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 25, 317–329. [DOI] [PubMed] [Google Scholar]
- MacDonald TJ, Aguilera D, and Kramm CM (2011). Treatment of high-grade glioma in children and adolescents. Neuro Oncol 13, 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques S, van Bruggen D, Vanichkina DP, Floriddia EM, Munguba H, Varemo L, Giacomello S, Falcao AM, Meijer M, Bjorklund AK, et al. (2018). Transcriptional Convergence of Oligodendrocyte Lineage Progenitors during Development. Dev Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcao A, Xiao L, Li H, Haring M, Hochgerner H, Romanov RA, et al. (2016). Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, and Rowitch DH (2012). Astrocytes and disease: a neurodevelopmental perspective. Genes Dev 26, 891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, Attema JL, Li G, Haddix T, Edwards MS, et al. (2011). Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A 108, 4453–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir A, Norton JD, Baou M, Zekavati A, Bijlmakers MJ, Thompson S, and Murphy JJ (2012). ZFP36L1 negatively regulates plasmacytoid differentiation of BCL1 cells by targeting BLIMP1 mRNA. PLoS One 7, e52187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, et al. (2017). Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624 e1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, and Richardson WD (2008). PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci 11, 1392–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageot CM, and Stiles CD (2002). Molecular mechanisms controlling cortical gliogenesis. Curr Opin Neurobiol 12, 244–249. [DOI] [PubMed] [Google Scholar]
- Stumpo DJ, Byrd NA, Phillips RS, Ghosh S, Maronpot RR, Castranio T, Meyers EN, Mishina Y, and Blackshear PJ (2004). Chorioallantoic fusion defects and embryonic lethality resulting from disruption of Zfp36L1, a gene encoding a CCCH tandem zinc finger protein of the Tristetraprolin family. Mol Cell Biol 24, 6445–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. (2016). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, et al. (2017). Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff RH, Tekki-Kessaris N, Stiles CD, Rowitch DH, and Richardson WD (2001). Oligodendrocyte development in the spinal cord and telencephalon: common themes and new perspectives. Int J Dev Neurosci 19, 379–385. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Wang S, and Anderson DJ (2000). Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 25, 331–343. [DOI] [PubMed] [Google Scholar]
- Zhu X, Zuo H, Maher BJ, Serwanski DR, LoTurco JJ, Lu QR, and Nishiyama A (2012). Olig2-dependent developmental fate switch of NG2 cells. Development 139, 2299–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Sun B, Zhang CL, Fine A, Chiu SY, and Messing A (1997). Live astrocytes visualized by green fluorescent protein in transgenic mice. Dev Biol 187, 36–42. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, and Messing A (2001). hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31, 85–94. [DOI] [PubMed] [Google Scholar]
- Zong H, Parada LF, and Baker SJ (2015). Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb Perspect Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Lists of driving transcriptional regulators. Related to Figure 4.
Table S2. Lists of differentially expressed genes for identified clusters. Related to Figures 1, 3 and 5.
Table S4. Primer sequences for q-PCR and genotyping. Related to STAR Methods.
Data Availability Statement
All the scRNA-seq and ATAC-seq data have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE122871.