

Abstract
Mechanical anisotropy is an essential property for biomolecules to assume structural and functional roles in mechanobiology. However, there is insufficient information on the mechanical anisotropy of ligand-biomokcule complexes. Herein, we investigated the mechanical property of individual human telomeric G-quadruplexes bound with a telomestatin ligand by optical tweezers. Stacking of the ligand to the G-tetradplanes changes the conformation of the G-quadruplex, which resembles a balloon squeezed at certain directions. Such a squeezed balloon effect strengthens the G-tetrad planes whereas dislocates and weakens the loops in the G-quadruplex upon ligand binding. These dynamic interactions indicate that the binding between the ligand and G-quadruplex follows the induced-fit model. We anticipate that the altered mechanical anisotropy of the ligand-G-quadruplex complex can add additional level of regulations on the motor enzymes that process DNA or RNA molecules.
Graphical Abstract
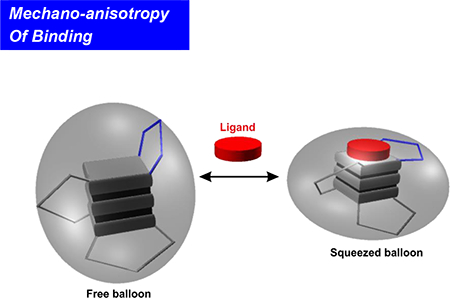
Binding of a telom estatin analogue strengthens the m echanical stability of G - tetrads while w eakens that of loops in human telom eric G-quadruplex. The m echanical anisotropy effect is sustained by the uneven structural variation of the G-quadruplex after ligand binding, sim ilar to a balloon squeezed at certain directions (the squeezed balloon effect).
For many materials, mechanical anisotropy is an inherent property in which mechanical behavior of a material varies with the direction of applied force. Mechanical anisotropy in biomacromolecules such as proteins[1] and nucleic acids[2] is of high biological significance. In structural proteins, for example, strengthened mechanical stability along a specific direction offers tolerance to the force perturbation at that direction, a special feature important to direct the movement of cells, cellular compartments, or molecular assemblies. For nucleic acid structures such as G-quadruplexes,[3] mechanical anisotropy differentiates their capability to stall motor proteins, such as DNA and RNA polymerases,[4] along the polymerization direction (i.e. 5’−3’ of nucleic acid strands) with respect to other directions.
Ligand binding to biomolecules is the first step in many biological processes. During the binding, a ligand is associated with a receptor by the formation of non-covalent bonds including H-bonding and ionic attractions, as well as polar and hydrophobic interactions. From energetic perspective, these newly formed networks of intermolecular forces (IMFs) strengthen the change in free energy of the binding (ΔGbinding), which leads to an overall increased mechanical stability of the receptor molecule. On the other hand, ligand binding may change the conformation of the receptor, especially in the induced-fit binding mode.[5] Such a conformational change is expected to vary the mechanical anisotropy of the receptor. As a result, the increased mechanical stability of the ligand-bound receptor may not persist in all directions. In extreme cases, it is possible that local conformational changes may even weaken the mechanical stability of specific regions.
However, such a weakening effect upon ligand binding has not been observed previously. In this work, we evaluated the mechanical anisotropy of a G-quadruplex with or without a telomestatin analogue L2H2–6OTD.[6] We designed mechanical unfolding experiments in which a G-quadruplex in human telomere sequence 5’-TTA(GGGTTA)4 was subjected to force at different directions (Figure 1). In each unfolding geometry, we modified two anchoring residues with click chemistry functional groups. These two anchoring residues were then linked to two double-stranded DNA handles, which were further attached to two optically trapped polystyrene beads. Each mechanical unfolding was carried out along the direction defined by the two anchoring residues. Two sets of unfolding geometries were designed. In the first set, residues in the loops or the 5’/3’ flanking sequence served as two anchors (5’-L2, 5’-L3, L1-L3, L3–3’, and L2–3’). In the second set, two guanine residues in the same G-tetrad unit (designated as the top, middle, and bottom G-tetrad) served as anchors. We named such a strategy as molecular mosaicking, which allows us to piece together properties of the two main structural elements in the G-quadruplex: G-tetrad stacking and loop-loop interaction.
Figure 1.

Investigation of mechanical anisotropy of DNA G-quadruplex by molecular mosaicking strategies. A human telomeric G-quadruplex was tethered between two optically trapped particles via two duplex DNA handles that are conjugated to two residues in the G-quadruplex using click chemistry (see Figures 2A and S1 as an example). Top and bottom insets in bubbles depic G-quadruplexes without and with the telomestatin derivative (circularplate), respectively. Big and small 3D arrows inside the two bubbles respectively depict strong and weak mechanical stabilities of G-terads or loops along certain directions. Loop 3 is depicted as L3 while loops 1 and 2 are marked as L1 and L2, respectively. Chemical structures of telomestatin ligand L2H2–6OTD and G-tetrad are shown to the bottom.
Next, we performed force ramping experiments along specific directions in each geometry set. We gradually increased the force with a loading rate of 5.5 pN/s by moving one of the trapped particles away from another using a steerable mirror.[7] Unfolding of a G-quadruplex was manifested by a sudden rupture event in force-extension curves recorded during the experiments (Figure S2). The change-in-extension (Δx) of each rupture event was measured to retrieve the change-in-contour-length (ΔL, see Materials and Methods), which reflected the size of a folded structure. On the other hand, the rupture forces of these rupture events depicted the mechanical stability of the structure along specific unfolding directions. Analyses of ΔL (Figures S3&S4) for all unfolding geometries confirmed it was the telomeric G-quadruplex that was unfolded between two anchoring residues.
To reveal the effect of a ligand on the property of G-quadruplex, we mechanically unfolded the telomeric G-quadruplex with and without 100 nM telomestatin analogue L2H2–6OTD.[6] Compared to the free G-quadruplex, mechanical unfolding by holding two G-residues in a particular G-tetrad showed much increased mechanical stabilities in presence of the L2H2–6OTD, which are consistent with previous reports (Figure 2).[8] When we changed the anchoring residues to the loop residues or those in the 5’ or 3’ flanking sequence, we found that mechanical stability of the ligand-bound G-quadruplex was reduced significantly (Figure 3 and Table 1). Such a finding is rather surprising as ligand binding is expected to strengthen the overall structure as demonstrated previously.[8a] To confirm this finding, we analyzed unfolding kinetics by fitting each unfolding force histogram with the equation proposed by Dudko.[9] As shown in Table 1, when the telomestatin derivative was bound to the G-quadruplex, we found that the unfolding rate constant (Kunfold) along the trajectory confined by the two G-residues in a particular G-tetrad became slower than that without ligand. If pulled from the loop residues, however, the Kunfold in the presence of the ligand became faster than the free G-quadruplex. Consistent with this finding, when pulled from the G-tetrad residues, the unfolding activation energy (ΔGunfold†) in presence of the ligand was increased compared to the free G-quadruplex; whereas it was decreased when pulled from the loop residues. Given that ΔGunfoid† and Kunfold are correlated with the mechanical stability of a structure, these data suggested mechanical anisotropy for a telomestatin-bound telomeric G-quadruplex: while the ligand strengthens the mechanical stability of the G-tetrads, it weakens the loop domains.
Figure 2.

(A) Schematic of the click chemistry coupling between two modified guanines in the top G-tetrad and two duplex DNA handles for mechanical unfolding experiments. Dotted arrows depict unfolding directions. (B) Rupture force histograms of the constructs unfolded through the Top, Middle, and Bottom G-tetrads. The histograms are fitted with the Dudko equation (solid, see SI) without (top) and with (bottom)100 nM L2H2–6OTD in a 10 m M Tris buffer (pH 7.4, 100 mM KCl). Residues of each fitting are shown to the top. The black dotted lines depict average values
Figure 3.

Rupture force histograms of the constructs unfolded through loop regions designated as L2–3’ 5’ -L2, L1–3’,5’ -L3’, and L1-L3 (see Figure S1 fo r detailed click chemistry coupling) in presence of 100 nM L2H2–6OTD in a 10 mM Tris buffer (pH 7.4 with 100 mM KCl). Dotted arrows show unfolding directions. The histogram s are fitted with the Dudko equation (solid curves). Fitting residues are shown to the top in which the black dotted lines indicate average values.
Table 1.
Rupture force (Funfold, pN), unfolding rate constant (kunfod, s−1), unfolding energy barrier (ΔG†unfold, kcal/mol)’ and distance from the folded to the transition state (x†) of telomeric DNA G-quadruplex via different unfolding geometries without and with 100 nM L2H2–6OTD monomer
| Variable | Ligand | Top | Middle | Bottom | L2–3' | 5’-L2 | L1–3' | 5’-L3 | L1-L3 |
|---|---|---|---|---|---|---|---|---|---|
|
Funfold (pN) |
No | 33±2.4 | 21±0.5 | 32±1.9 | 44±1.6 | 42±0.9 | 34±0.9 | 34±1.3 | 46±0.5 |
| Yes | 39.3±1.1 | 32.2±1.8 | 36.5±1.9 | 40.4±1.5 | 32.7±0.5 | 34.3±1.8 | 34.7±1.0 | 38±1.2 | |
|
kunfold (s−1) |
No | 0.019±0.001 | 0.053±0.005 | 0.018±0.001 | 0.0048* | 0.0055* | 0.0111* | 0.015* | 0.0009* |
| Yes | 0.013±0.003 | 0.029±0.004 | 0.013±0.002 | 0.007±0.003 | 0.016±0.005 | 0.015±0.004 | 0.019±0.001 | 0.006±0.001 | |
|
ΔG†unfold,
(kcal/mol) |
No | 14.8±0.8 | 15.3±0.2 | 12.9±0.7 | 19±3.0* | 17.5±0.4* | 17.1±2.0* | 21* | 21±1.0* |
| Yes | 19.8±0.3 | 20.6±1.3 | 19.7±0.9 | 14.8±1.0 | 14.3±0.7 | 15.1±1.2 | 15.6±0.6 | 13.3±1.2 | |
|
x† (nm) |
No | 0.10±0.01 | 0.12±0.01 | 0.11±0.01 | 0.10* | 0.10* | 0.11* | 0.08* | 0.14* |
| Yes | 0.09±0.01 | 0.08±0.01 | 0.09±0.01 | 0.12±0.02 | 0.11±0.02 | 0.11±0.01 | 0.09±0.01 | 0.14±0.04 | |
data taken from reference[2].
This observation represents the first example of mechanical anisotropy in a biomolecule bound with a specific ligand.[2c] To understand the molecular mechanism of this anisotropy, we compared NMR structures of telomeric G-quadruplex with[6] and without[10] the L2H2–6OTD ligand. Previous investigations by multiple unfolding geometries[2a] as well as CD spectra (Figure S6) have indicated that the G-quadruplex formed in the same telomeric sequence adopts a hybrid-1 conformation.[10] Here, after incorporation of 3 more unfolding geometries, we revealed that the single-molecule hybrid-1 structure is the best matching conformation for both ligand-bound and free G-quadruplexes by comparing with known structures (Figure S5). The structural difference for our method and others can be ascribed to the difference in buffer conditions as well as the sequence and concentration of the DNA (see SI). In addition, to evaluate the effect of the click chemistry modification on the conformation and function of G-quadruplexes, we compared binding constants of the L2H2–6OTD with the wild-type and the azide modified telomere sequences (Figure S10). We found identical binding affinities for these two sequences, suggesting the maintenance of the same conformations. Therefore, we overlaid the ligand-bound hybrid-1 G-quadruplex (PDB: 2MB3)[6] with the free hybrid-1 G-quadruplex (PDB: 2HY9).[10] As shown in Figures 4 & S7, the conformational difference between the two structures is obvious. When the L2H2–6OTD binds to the G-quadruplex, the loops (loop 3 in particular) are displaced to accommodate the ligand. This accommodation is characteristic of the induced-fit model for ligand binding.[5] To quantify different conformations of G-quadruplexes with and without L2H2–6OTD, we measured all phosphorus-to-phosphorus distances in the two PDB structures. To reveal the effect of the ligand binding on the telomeric G-quadruplex, we calculated the difference in the distance of each phosphorus pair in the ligand-bound versus free G-quadruplex (Figure 4A, see Materials and Methods for details). The effects of the ligand on different parts of the G-quadruplex were then mosaicked together by interrogating the change-in-the-distances in three domains, the G-tetrads, the loops, and the crossover tetrad-loop regions.
Figure 4.

(A) Change in the distance of the phosphorus-phosphorus atoms between the free and the L2H 2–60T D bound G-quadruplexes. GQ represents the G-tetrad planes. LP depicts the loop region, which was marked by L1, L2, and L3 to represent the first, second, and the third loops (counted from the 5’ end), respectively. L3 (slashed histogram) is the most distorted. (B) Squeezed balloon model. Binding of a ligand to the top of a G-quadruplex (sphere) leads to the compression of the bound region a t the expense of squeezed-out loop regions (Lp). This effect increases the mechanical strength of the bound region while compromising the mechanical stability of loop regions, which resembles a squeezed balloon.
Among these three regions, the change in the conformation of the three G-tetrads is minimal when the ligand is bound to the G-quadruplex. Not only is the distribution of the change-in-distance narrow, it is symmetrically distributed around zero as well. These suggest that the ligand binding has a minimal effect on the G-tetrad stacking, a core structural element in the G-quadruplex. On the other hand, significant conformational deviation has been observed for the loops (Figure 4A, bottom). To identify the loop with the largest conformational change, we compared the change in the phosphorus-phosphorus distance between the G-tetrad core and each of the three loops. As G-tetrads do not significantly vary conformation with the binding of the ligand, they serve as reference points against which each loop was evaluated for its conformation variation. This analysis revealed that loop 3 has the most pronounced structural change (Figure 4). Given that phosphorus-phosphorus distances are longer for the loop 3 residues when the ligand is bound, it indicates that loop 3 is pushed away from the G-tetrad core (see Figure S7 for direct visualization). NMR structures at the atomic level confirmed that loop 3 is disturbed most by the binding of the L2H2–6OTD ligand. In the free G-quadruplex, Watson-Crick base pairing exists between the thymine in the 5’ flanking sequence and the adenine in the loop 3, which locks loop 3 in place, rendering a rigid G-quadruplex structure. Binding of the ligand destroys this base pairing, making the loop 3 more flexible in the ligand-bound G-quadruplex.
The dislocated loops have reduced mechanical stability after binding of the telomestatin derivative to the G-quadruplex. During mechanical unfolding of the G-quadruplex from loop residues, we propose a two-barrier energy diagram (Figure S8A) in which a proximal energy barrier responsible for the loop-loop disruption is followed by a distal barrier to disassemble the G-tetrad stacking. In the absence of a ligand, the stronger loop-loop interaction with respect to the G-tetrad stacking makes the first barrier (loops) predominant over the second barrier (G-tetrad). This allows the former to serve as a rate determining step. In the presence of a ligand, the first barrier of the loop-loop interactions is compromised since loops are disturbed by the ligand binding. On the other hand, the second barrier of the G-tetrad stacking becomes stronger due to the π-π stacking between the terminal G-tetrad and the polyoxazole ring of the L2H2–6OTD ligand (see below). Application of mechanical force (F), however, reduces the energy diagram with a value of Fx.x[11], here x is the reaction coordinate along the applied force. Since the distance between the folded state to the second barrier (x†) is longer than that of the first barrier, it leads to a more reduced energy for the second barrier. As a result, overall unfolding force from the loop residues becomes smaller as both energy barriers are much reduced.
Mechanical unfolding of the G-quadruplex via the G-tetrad residues, however, can be governed by the energy barrier for the G-tetrad stacking, which is immediately followed by the disruption of the loop-loop interactions (Figure S8B). Binding of the L2H2–6OTD to the terminal G-tetrad reinforces the G-tetrad stacking. In the free state, the polyoxazole ring in the ligand is arranged in a slightly bended plane.[6] Upon the stack-binding of the ligand to the G-tetrad, however, the polyoxazole ring becomes flatter, facilitating a stronger π-π interaction between the ligand and the G-tetrad. As a result, the mechanical stability of the overall G-tetrad stacking increases.[2b] Such a conformation adjustment in ligand presents another evidence for the induced-fit binding between the L2H2–6OTD and the telomeric G-quadruplex. It is possible that the selected-fit[12] may also exist, in which a ligand binds to one of pre-existing structures. Given the fact that we did not observe different G-quadruplex conformations in the unfolding force (Figures 2&3) or ΔL (Figures S3&S4) histograms without ligand, such a mechanism is less likely for the binding between the telomeric G-quadruplex and the L2H2–6OTD ligand.
The strengthened G-tetrad core and weakened loops in G-quadruplex demonstrate the plasticity of the G-quadruplex-ligand complex. Ligand binding increases the overall free energy cost to unfold the G-quadruplex, which is a thermodynamic variable contributed by the unfolding’s from all possible directions with different energy profiles. While the stability of a bound structure increases along certain directions, it may decrease among others. Such an anisotropic response is sustained by the uneven variations of the G-quadruplex structure after ligand binding, which resembles the behavior of a balloon squeezed at certain directions (the squeezed balloon effect, see Figure 4B).
It has been proposed that G-quadruplex may serve as a mechanical block to replication or transcription processes[13] The blocking can be considered as a tug-of-war between the load force of a polymerase and mechanical stability of the G-quadruplex along the polymerizing directions. Polymerase is expected to stall if the G-quadruplex in front of the enzyme can withstand the load force of the polymerase. As the polymerase moves along the nucleic acid template in the 3’−5’ direction, the enzyme exerts mechanical force directly on the stacked G-tetrad core in the G-quadruplex (Figure S9). Since the L2H2–6OTD ligand strengthens the G-tetrad core, it ensures increased potency to stall polymerases. However, for other G-quadruplex ligands that do not share similar stacking mechanism to the terminal G-tetrads, the mechanical anisotropy should be separately evaluated to elucidate their regulatory functions from mechanical perspective.
In summary, by mechanical unfolding a DNA telomeric G-quadruplex bound with a telomestatin analogue from different directions, we observed mechanical anisotropy of a ligand-bound biomolecule for the first time. The mechanical anisotropy was explained by the squeezed balloon effect in which the G-quadruplex structure varies unevenly upon ligand binding. The dynamic interaction between the ligand and the G-quadruplex suggests their binding follows the induced-fit model. These unprecedented findings provide new insights for the design of small-molecule ligands to DNA secondary structures, which, from mechanical perspective, can modulate replications or transcriptions along the DNA template.
Supplementary Material
Acknowledgments
This research work was supported by NSF CHE-1415883&CHE-1609504 to H. M.
K. N express sincere thanks for a Grants-in-Aid for Scientific Research (B) from JSPS (23310158 and 26282214) and a Grant-in Aid for Challenging Exploratory Research from JSPS (21655060). H. S thanks Grant-in-Aid Priority Research from Japan Society for the Promotion of Science (JSPS) and the grant from the WPI program (iCeMS, Kyoto University).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
Contributor Information
Sagun Jonchhe, Department of Chemistry & Biochemistry and School of Biomedical Sciences, Kent State University, Kent, OH 44240 (USA).
Chiran Ghimire, Department of Chemistry & Biochemistry and School of Biomedical Sciences, Kent State University, Kent, OH 44240 (USA).
Yunxi Cui, Department of Chemistry & Biochemistry and School of Biomedical Sciences, Kent State University, Kent, OH 44240 (USA).
Shogo Sasaki, Department of Biotechnology and Life Science Faculty of Technology, Tokyo University of Agriculture and Technology (TUAT), Koganei, Tokyo 184-8588, Japan..
Mason McCool, Department of Chemistry & Biochemistry and School of Biomedical Sciences, Kent State University, Kent, OH 44240 (USA).
Soyoung Park, Department of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-oiwakecho, Sakyo-ku, Kyoto 606-8502, Japan.; Institute for Integrated Cell Material Sciences (iCeMS), Kyoto University, Yoshida-ushinomiyacho, Sakyo-ku, Kyoto 606-8501, Japan.
Keisuke lida, Department of Biotechnology and Life Science Faculty of Technology, Tokyo University of Agriculture and Technology (TUAT), Koganei, Tokyo 184-8588, Japan..
Kazuo Nagasawa, Department of Biotechnology and Life Science Faculty of Technology, Tokyo University of Agriculture and Technology (TUAT), Koganei, Tokyo 184-8588, Japan..
Hiroshi Sugiyama, Department of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-oiwakecho, Sakyo-ku, Kyoto 606-8502, Japan.; Institute for Integrated Cell Material Sciences (iCeMS), Kyoto University, Yoshida-ushinomiyacho, Sakyo-ku, Kyoto 606-8501, Japan.
Hanbin Mao, Department of Chemistry & Biochemistry and School of Biomedical Sciences, Kent State University, Kent, OH 44240 (USA).
Reference
- [1].a) Carrion-Vazquez M, Li H, Lu H, Marszalek PE, Oberhauser AF, Fernandez JM, Nat. Struct. Biol 2003, 10, 738–743; [DOI] [PubMed] [Google Scholar]; b) Yongnan D. Li, Lamour G, Gsponer J, Zheng P, Li H, Biophys. J 2012, W3, 2361–2368; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brockwell DJ,Paci E, Zinober RC, Beddard GS, Olmsted PD, Smith DA, Perham RN, Radford SE, Nat. Struct. Biol 2003, 10, 731; [DOI] [PubMed] [Google Scholar]; d) Dietz H, Berkemeier F, Bertz M, Rief M, Proc. Natl. Acad. Sci. U. S. A 2006, 103,12724–12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Yu Z, Koirala D, Cui Y, Easterling LF, Zhao Y, Mao H, J. Am. Chem. Soc 2012, 134, 12338–12341; [DOI] [PubMed] [Google Scholar]; b) Ghimire C, Park S, Iida K, Yangyuoru P, Otomo H, Yu Z, Nagasawa K, Sugiyama H, Mao H, J. Am. Chem. Soc 2014, 136, 15537–15544; [DOI] [PubMed] [Google Scholar]; c) Sun Y, Di W, Li Y, Huang W, Wang X, Qin M, Wang W, Cao Y, Angew. Chem., Int. Ed 2017, 56, 9376–9380. [DOI] [PubMed] [Google Scholar]
- [3].Gellert M, Lipsett MN, Davies DR, Proc. Natl. Acad. Sci. U. S. A 1962, 48, 2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Galburt EA, Grill SW, Wiedmann A, Lubkowska L, Choy J, Nogales E, Kashlev M, Bustamante C, Nature 2007, 446, 820–823; [DOI] [PubMed] [Google Scholar]; b) Mejia YX, Mao H, Forde NR, Bustamante C, J. Mol. Biol 2008, 382, 628–637; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yin H, Wang MD, Svoboda K, Landick R, Block SM, Gelles J, Science 1995, 270, 1653–1657. [DOI] [PubMed] [Google Scholar]
- [5].Koshland DE, Angew. Chem., Int. Ed 1995, 33, 2375–2378. [Google Scholar]
- [6].Chung WJ, Heddi B, Tera M, Iida K, Nagasawa K, Phan AT, J. Am. Chem. Soc 2013, 135,13495–13501. [DOI] [PubMed] [Google Scholar]
- [7].a) Mao H, Luchette P, Ses. Actuators, B 2008, 129, 764–771; [Google Scholar]; b) Luchette P, Abiy N, Mao H, Ses. Actuators, B 2007, 128, 154–160. [Google Scholar]
- [8].a) Koirala D, Dhakal S, Ashbridge B, Sannohe Y, Rodriguez R, Sugiyama H, Balasubramanian S, Mao H, Nat. Chem 2011,3, 782–787; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Punnoose JA, Ma Y, Li Y, Sakuma M, Mandai S, Nagasawa K, Mao H, J. Am. Chem. Soc 2017, 139, 7476–7484. [DOI] [PubMed] [Google Scholar]
- [9].Dudko OK, Hummer G, Szabo A, Proc. Natl. Acad. Sci. U. S. A 2008, 105, 15755–15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dai J, Punchihewa C, Ambrus A, Chen D, Jones RA, Yang D, Nucleic Acids Res. 2007, 35, 2440–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Koirala D, Yangyuoru PM, Mao H, Rev. Anal. Chem 2013, 32, 197–208. [Google Scholar]
- [12].a) Chung-Jung T, Buyong M, Yin SY, Sandeep K, Ruth N, Proteins: Struct., Funct., Bioinf 2001, 44, 418–427; [Google Scholar]; b) R. WT, Carola v. D., Proteim: Struct., Funct., Bioinf 2009, 75,104–110. [Google Scholar]
- [13].a) Catasti P, Chen X, Mariappan SSV, Bradbury EM, Gupta G, Genetica 1999, 106, 15–36; [DOI] [PubMed] [Google Scholar]; b) Catasti P, Chen X, Moyzis RK, Bradbury EM, Gupta G, J. Mol. Biol 1996, 264, 534–545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.