

Abstract
Nanoscale extracellular vesicles (nEVs) have recently demonstrated potential value in cancer diagnostics and treatment monitoring, but translation has been limited by technical challenges in nEV isolation. Thus, we have developed a one-step nEV isolation platform that utilizes nEV size-matched silica nanostructures and surface-conjugated lipid nanoprobe with an integrated microfluidic mixer. The reported platform has 28.8% capture efficiency from pancreatic cancer plasma and can sufficiently enrich nEV for simpler positive identification of point mutations, particularly KRAS, in nEV DNA from plasma of pancreatic cancer patients.
Introduction
Nanoscale extracellular vesicles (nEVs), including exosomes and microvesicles, have been under intense investigation with prospects towards pathophysiological and translational discoveries. More specifically, growing evidence indicates that tumor-derived nEVs can regulate tumor immune responses, initiate the formation of pre-metastatic niches, determine organotropic metastasis, promote epithelial-to-mesenchymal transition, and contribute to therapeutic resistance.1,2 Furthermore, tumor-derived nEVs are increasingly being recognized as the latest addition to the liquid biopsy portfolio for cancer diagnostics and treatment monitoring.3 However, relevant clinical translation has been mainly limited by technical challenges in nEV isolation and enrichment (Table 1). Generally, these techniques have a combination of poor and good qualities, including length protocols, low isolation efficiency, and low purity.4, 5
Table 1:
Comparison of reported lipid nanoprobe nanostructure platform with existing techniques for nEV isolation and enrichment.
| Enrichment Technique | Mechanism | Speed/Rate | Volume | Efficiency | Purity | Cost | Point of Care | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ultracentrifugation5,32–34 | Density | 2–24 h | 0.5–5 ml | 2–25% | Medium/High | $ $ $ | No | |||||
| Immunocapture32,34 | Antigen | 1–5 h | 500 μl | N/A | Medium/High | $ $ | Somewhat | |||||
| Precipitation33 | Solubility | 12–24 h | 1–10 ml | N/A | Low | $ | No | |||||
| Lipid Nanoprobe6 | Affinity | 15 m | up to 1 ml | 50–80% | Medium | $ $ | Somewhat | |||||
| LNP Nanostructure Platform | Affinity & Size | 2 m-3 h 20 m | up to 2 ml | 30% | High | $ | Yes | |||||
| Poor | Acceptable | Good | ||||||||||
Previously, we developed a lipid-nanoprobe system that enables spontaneous labeling of nEVs for subsequent magnetic enrichment in 15 minutes.6 Furthermore, this highly flexible and adoptable method is ideal for various downstream analyses of proteins and nucleic acids due to the extremely pure and high concentrated final nEV sample. Yet, clinical translation has been limited due to small processing capabilities in conjunction with a more technical, two-step nEV enrichment procedure. Thus, we have established this one-step, on-chip lipid-nanoprobe nanostructured platform for a more reliable, feasible, and accurate approach to obtain adequate quantities of pure nEV for EV cargo analysis (Figure 1).
Figure 1:
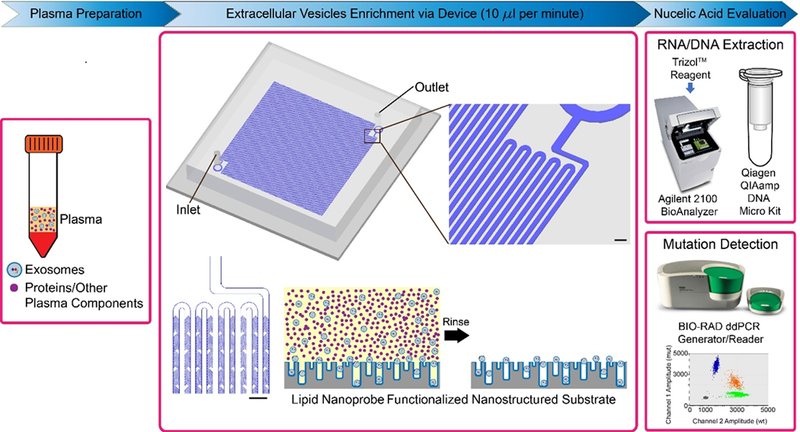
Overall process flow of one-component lipid nanoprobe functionalized nanostructured silica platform. First, plasma sample is prepared, by which it is separated from red and white blood cells via traditional procedures. Next, EVs are enriched, by which 10μl per minute of plasma sample are processed through herringbone micromixer channels. Scale bar are 300 μm. EVs are tethered to the nanostructured silica substrate via the lipid nanoprobe surface functionalization, while the proteins and other plasma components are rinsed away. The tethered EVs are evaluated for their nucleic acid content by performing RNA and DNA extraction followed by mutation detection.
The previously reported system allows the free-floating lipid nanoprobes to spontaneously insert, and thereby tether, to the lipid membrane of the nEV. Following the tethering, magnetic capture probes are used to isolate the nEVs. This newly reported platform allows the lipid nanoprobe to be grafted onto the platform surface, which consisted of precisely fabricated silica nanostructures size-matched to the nEVs. The size-matched silica nanostructures serve dual purposes as to 1) increase the overall surface area available for nEV binding and tethering and 2) the nanostructures create nanoscale pits with size ranges similar to nEVs, which, in some cases, allow nEVs to become fixed in the device structure. Moreover, an integrated, continuous flow micromixer provides two benefits; 1) forces nEV to the platform surface to provide maximal enrichment and (2) forces the unwanted protein contaminants into the device outlet, and thus, waste. By reducing the procedural steps of nEV isolation to one, we have minimized this source of nEV loss. This becomes especially critical when tumor cell-derived nEVs have a low quantity in plasma.
The final sample of isolated and highly enriched nEVs is extremely accommodating to downstream molecular analyses. Tumor-derived nEV DNA mutation allele frequency, or the percentage of DNA fragments that contain a specific point mutation, can be as less than 0.01% in patients.7 Thus detecting this requires (1) a pure and enriched sample and (2) a highly sensitive methodology.7 The first will be provided by the reported platform and the later will be provided by digital droplet polymerase chain reaction (ddPCR).
Here, we have reported on the development, fabrication, and optimization of both the silica nanostructured substrate, as well as, the re-design and optimization of the lipid nanoprobe system from the previous report. The feasibility and reliability of the platform was validated with nEV spiked plasma samples mimicking plasma from cancer patients. Lastly, up to 2-ml plasma from three pancreatic cancer patients, with known KRAS mutation within the tissue sample, and two healthy controls were respectively processed with this platform. The analysis of KRAS mutations in isolated nEV-derived ddPCR demonstrated nEVs DNA mutations were concordant with that of corresponding tissue specimen, while no KRAS mutation was identified from the two healthy controls. Overall, the potential translation of this promising nano-enabled nEV isolation platform could facilitate improved cancer diagnosis and treatment monitoring techniques.
Materials & Methods
Plasma Sample Collection
Clinical samples were obtained, with consent, from advanced pancreatic cancer patients at the Penn State Hershey Medical Center according to an institutional-review-board-approved protocol (IRB 40267EP). Samples were drawn into 10 ml Vacutainer K2-EDTA tube (Becton Dickinson) from peripheral venipuncture. After centrifugation at 300 g for 5 min and then 16,500 g for 20 min at 4°C, plasma was collected. Samples were filtered using a 0.22 μm pore filter to remove larger cell debris and large vesicles. Filtered plasma samples were stored at −80°C until processing. Heterogeneous focusing on a sub population.
Cell Culture and Cell Culture-derived nEV Isolation
Pancreatic carcinoma cell line, Panc-1 cells, containing KRAS G12D mutation and breast carcinoma cell line, MDA-MB-231 cells, without KRAS mutation were purchased from the American Type Culture Collection. All cells passed testing for mycoplasma contamination and were maintained in phenol-red-free-DMEM medium (Corning) supplemented with 10% (v/v) FBS, 100 units ml−1 penicillin and 100 μg ml−1 streptomycin. Cells were cultured in a humidified atmosphere of 5% CO2 at 37°C. Panc-1 and MDA-MB-231 cells were grown in three T225 flasks (Falcon) for two to three days until they reached a confluency of 80%. Next, cells were cultured in medium without FBS for 48 h. The medium was collected and centrifuged at 300 g for 5 min followed by 16,500 g for 20 min at 4°C. Plasma was then collected and filtered using a 0.22 μm pore filter. A total of 108 ml of medium was collected and continuously ultracentrifuged at 100,000 g at 4°C for 2 h. The EV pellets were suspended in 200 μl of PBS. Samples were incubated with 10 μl of DNase I (1 unit μl−1; Life Technologies) or 5 μg ml−1 RNase at 37°C for 2 h. The supernatant was collected and stored at −80°C.
Fabrication of nEV Size-Matched Silica Nanostructures
nEV size-matched silica nanostructures require a size-tunable iron nanoparticle (FeNP) dry-etching mask to precisely control nanostructure size for maximize nEV isolation. Silica nanostructures were fabricated by physical vapor deposition (PVD), high-temperature annealing, and reactive ion etching (RIE) techniques (Figure S2). Fused silica substrate, chosen for both transparency during sample processing and resistance towards FeNP diffusion, which could negatively influence FeNP during the high-temperature annealing process required for FeNP formation,8 were cleaned in Nano-strip® (Cyantek®) to ensure a particle- and organic-free surface for iron deposition. Iron, 1.5 to 9 nm thick, was deposited onto the surface by electron-beam PVD at 0.5 Å/sec and 10−6 Torr vacuum. Film thickness was verified with atomic force microscopy (AFM).
The homogenously dispersed FeNP mask was formed by annealing the iron film at 850°C for 30 minutes under 2.5 L/min of reductive gas (4% H2 bal. N2). Substrates were then gradually cooled to room temperature. The silica was etched to nanostructures using an RIE tool (SLR 720 RIE, Plasma-Therm). The process consisted of a 2 to 8 minute etch at 400 W RF power, 100 mTorr pressure, and a mixture of CF4, Ar, O2, and SF6 (5:3:2:5).
The silica wafer was diced into individual platform substrates by spin-coating a thin layer of protective photoresist, P4620 (AZ® MicroChemicals) and baked at 90°C. The wafer was then diced into 4 equivalent pieces via a saw (Advanced Dicing Technologies). The protective photoresist was removed by soaking in Nano™ REMOVER PG (microChem) at 60°C. Residual iron nanoparticles and organics were removed via piranha solution (1 H2O2: 3 H2SO4) at 90°C for 30 minutes, followed by a water rinse and N2 dry.
Lipid Nanoprobe Optimization with Cell Assay
In the previously reported study, we identified that polyethylene glycol (PEG) tagged 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) can effectively eliminate, on average, 68.5% of the total proteins during nEV enrichment.6 However, PEG-DSPE requires a specific iso-osmotic solution containing no physiological salts or buffers. Moreover, we determined that DSPE can bind to albumin and other lipoproteins, which is problematic when processing clinical plasma samples. Although the binding constant of PEG-DPSE is ~1×103 M−1 at RT,9 the abundant plasma proteins significantly decrease isolation efficiency of nEVs.
Hence, three types of lipids (monoacyl lipid, diacyl lipid, and cholesterol) bearing various ethyleneoxide (EO) units, from 9 to 228, were comprehensively screened using a cell assay. Optimization of both the lipid tail and the EO units, or length, or the nanoprobe spacers is critical as the nanoprobe must demonstrate strong non-covalent interactions with nEV lipid bilayer membrane and stable retention as well as optimal reagent solubility, respectively.
FITC-tagged C18-PEG, DSPE-PEG, and cholesterol-PEG powders were purchased (NanoCS) and purified without further purification. The FITC-tagged PEGylated lipids were dissolved in pure anhydrous ethanol at a final, 1mM concentration and stored at −80°C. Approximately 107 MDA-MB-231 breast cancer cells were collected and resuspended in 250 μl of 10% human albumin in PBS. In the first-round screening, 10 nanomoles of C18-PEG2000, DSPE-PEG2000, and cholesterol-PEG2000 were added to 250 μl of PBS before being added to the cell suspension. The samples were mixed gently at 4°C for 5 min followed by centrifugation at 500 g for 5 min to remove redundant lipids and then fixed with 4% paraformaldehyde at 4°C for 10 min. The cells were resuspended in 1.5 ml of PBS, stained with DAPI solution (1 μg ml−1) at room temperature (RT) for 10 min, thoroughly rinsed thrice with PBS, and finally resuspended in 500 μl of PBS. 20 μl of cell suspension was added onto glass cover slips for fluorescent imaging under a 40× objective lens using an Olympus IX71 microscope. The fluorescence intensities were analyzed using the ImageJ software package (National Institutes of Health). In the second-round screening, 10 nanomoles of cholesterol-PEG400, cholesterol-PEG1000, cholesterol-PEG2000, cholesterol-PEG3400, cholesterol-PEG5000, and cholesterol-PEG10000 were investigated with cell assays following the same protocol.
Lipid Nanoprobe Nanostructure Surface Functionalization
Nanostructure silica substrates were silanized via immersion in 2% 3-aminopropyltirethoyxylsilane (3-APTES) in ethanol for 1 hour at RT. Approximate thickness of silane layer after silanization after 1 hour is 4Å.10 This was followed by an ethanol and water rinse and cure at 120°C for 30 minutes. Next, the substrates were immersed in dimethylformamide (DMF) solution containing 10% pyridine and 1 mM phenyldiisothiocyanate (PDITC) for 2 h followed by washing with DMF and 1,2-dichloroethane. 30 μM of amine group tagged cholesterol-PEG1000 in DMF was placed on the substrate and allowed to incubate at 45°C for 2 hours. After rinsing with ethanol, the functionalized surface was deactivated by capping unreacted PDITC moieties by immersion in 50 mM 6-amino-1-hexanol in DMF for 5 hours. The negative control devices were grafted with 6-amino-1-hexanol only. Finally, each substrate was thoroughly rinsed with DMF, ethanol, and DI water. All substrates were kept at −20°C until further use.11, 12
Characterization of Nanostructures
Nanostructure substrates were characterized via scanning electron microscopy (SEM) (Zeiss SIGMA VP-FESEM). Substrates were coated with several nanometers of iridium to prevent surface charging, and the morphology of the substrate at various fabrication points was examined. For AFM, surface topography was evaluated quantitatively via a Dimension 5000 AFM (Bruker Nano Inc.). The changes in surface area and root mean square surface roughness were measured. Height images of samples were captured at a tapping frequency of approximately 300 kHz. The analyzed field measured 1×1 μm at a scan rate of 1 Hz with 256 scanning lines. After surface functionalization with lipids, contact angles were measured. A droplet of DI water was placed on the surface of the substrate at RT, and after 30 seconds the contact angle was measured using a contact angle goniometer (NRL-100, Rame-Hart). Five measurements were performed and statistically presented for each run.
Micromixer Device Assembly
A PDMS herringbone micromixer with a total microchannel surface area of ~360 mm2 (length = 3.6 m, height = 100 μm, width 25 μm), which can accommodate ~1011 nEVs, was fabricated via casting and curing on an SU8 master mold. More specifically, a 2-layer herringbone structure was fabricated; first with SU-8 50 spin coated onto a clean silicon wafer to obtain a 50-μm thick layer followed by soft bake at 60°C for 6 minutes and hard bake at 90°C for 20 minutes. After exposure to UV through a mask at 400 mJ cm−2, the second layer of 50-μm-thick SU-8 50 was spin-coated onto the first layer followed by soft bake, UV exposure, and post-exposure bake. Following development and cleaning, the master mold was produced. Polydimethylsiloxane (PDMS) (10:1 w/w) was poured, degassed, casted onto the mold, and allowed to cure in an 80°C oven for 2 h. The PDMS micromixer chambers were bonded onto the nanostructured silica via a PDMS pre-polymer and toluene mixture as a glue.13 Validation of bonding parameters was performed as followed, toluene and liquid PDMS was thoroughly mixed at 2:1, 3:1, 4:1, 5:1, and 6:1 (w/w), respectively, followed by silicon wafer coating at 1500 rpm for 1 min. The PDMS replica was then placed on the wafer surface in full contact with the thin layer of glue. Glue thickness was characterized by AFM. An optimal glue thickness was determined, and the PDMS replicas carrying approximately 300 nm-thick glue were placed onto the nanostructured substrates. This was followed by bonding at 60°C overnight to cure the glue, and thereby forming the final devices. Device leakage tests were performed with higher flow rates of 100 μl/min.
Enrichment of nEVs via Device Processing
Isolation efficiency of nEVs via the platform was quantified with nEV model samples, and then test with clinical samples. nEV model samples were prepared by collecting nEVs from two different cancer cell lines, breast adenocarcinoma MDA-MB-231 cells and pancreatic carcinoma Panc-1 cells, which carry wild-type KRAS and KRAS G12D mutation, respectively. The model samples were characterized for both size and surface markers. Either ~109 MDA-MB-231 cell-derived nEVs spiked in 1-ml exosome-depleted plasma or up to 2 ml of clinical plasma were processed with the platform using various flow rate ranging from 5 μl/min to 40 μl/min.14 After nEV sample processing, the device was rinsed with PBS containing 1% BSA and 1mM EDTA at 10 μl/min. All nEVs, both trapped and grafted, were either fixed for SEM analysis or nucleic acid extraction.
SEM Analysis of Isolated nEVs
After enrichment and device rinsing, the PDMS chamber was removed from the device, via cutting, leaving only trapped and grafted nEV on the silica nanostructured surface. The nEVs were fixed on the substrate with 2.5% glutaraldehyde in PBS at 4°C for overnight. Following a triplicate wash with PBS, the fixed nEVs were dehydrated on the nanostructured surface. Dehydration consisted of submersion in a series of ethanol solutions ranging from 25% to 100%. Substrates were then dried via critical point drying (Leica EM CPD300). Substrates were stored in a desiccator until microscopy. Immediately prior to imaging, substrates were sputter coated with Iridium. Substrates were imaged at 10 keV.
Nucleic Acid Extraction & Isolation Efficiency Quantification
After enrichment and device rinsing, trapped nEVs were directly lysed by flowing lysis buffers (Qiagen), Trizol (Thermo Fisher Scientific) and proteinase K and Buffer ATL (Qiagen), through the device microchannels at flow rates consistent with the device processing. RNA extraction was performed via a phase separation methodology with Trizol and then chloroform according to the manufacturer’s instructions. The final RNA pellet was dissolved in 20 μl of RNase-free water. The RNA concentration was measured using an Agilent 2100 Bioanalyzer. The DNA was extracted using the QIAamp DNA micro kit (Qiagen) according to the manufacturer’s instructions. The final DNA was eluted in 20 μl of AE buffer and stored at −80°C until PCR amplification. DNA quantification was performed via Nanodrop (Thermo Fisher Scientific). To determine the isolation efficiency of nEVs, an RNA mass compassion approach was used. RNA was extracted from the original nEV sample and the flow-through nEVs. The mass of the extracted RNA was measured and quantified and compared from before and after sample processing with the platform. Of note, there is currently no agreed upon standard method to unanimously quantify isolation efficiency of nEV enrichment methods.15,16 A combination of particle characterization and western blotting for EV markers before and after capture could be indicative. However, this method has not steadily demonstrated the accuracy and feasibility in processing clinical samples. Hence, we have adopted an RNA mass comparison approach, which although it has its flaws has been adopted in studies.17,18
Detection of EV-derived DNA Mutations
Extracted and purified DNA was first digested with 40 U of HaeIII (New England Biosciences) in 100 μl of 1 x NEB buffer 4 and BSA at 37°C for 1 hour, followed by deactivation at 80°C for 20 minutes. EV cargo DNA mutation detection was performed with the PrimerPCR™ ddPCR™ mutation detection assay kit (Bio-Rad). The EV DNA concentrations were normalized to the precise concentration of a genomic DNA sample and amplified with the primer set. Panc-1-derived EV DNA was used as a positive control and MDA-MB-231-derived EV DNA was used as a negative control. The background was analyzed with PBS. For statistical purposes, all samples were amplified and analyzed in triplicate. Amplifications were performed in 20 μl reaction volumes on a QX100 ddPCR (Bio-Rad). The PCR reaction volume was comprised of 10 μl Bio-Rad Supermix TaqMan, 1 μl if each (target and reference) amplification primer/probe mix, and 8 μl extracted EV DNA. Thermal cycling was comprised of an initial denaturing and polymerase hot-start activating step at 95°C for 10 minutes, followed by 40 repeated cycles of 95°C for 30 seconds and 55°C for 60 seconds. Results were analyzed with QuantaSoft v.1.3.2 software (Bio-Rad) and reported as copies per milliliter of plasma.
Statistical analysis
Results were presented at mean ± SD. Statistical comparisons were performed by paired Student’s t-test with a two-tailed p value to compare selected data pairs when two groups were compared.
Results and Discussions
Size-Tunable FeNP Mask Morphology and EV Size-Matching
In general, there was a direct relationship observed between iron film thickness, inter-particle distance of FeNPs, and FeNP diameter (Figure 2a–b). Consistently, there was an inverse relationship between iron film thickness and FeNP area density. The average inter-particle distance formed in each group was 71.6 ± 30.5 nm, 89.0 ± 45.7 nm, 238.3 ± 99.7 nm, and 361.1 ± 145.5 nm, respectively (Figure 2c). As the unmasked silica was etched to create the nanostructured cavities for nEV trapping, it was critical that the inter-particle distance matches the matches the average size of the nEV population. Thus, we chose 3-nm thick films for further technology optimization and isolation of nEVs ~120 nm.19 Accordingly, average FeNP area density of 3-nm thick film is 116.4 ± 14.5 per μm2 (Figure 2c).
Figure 2:
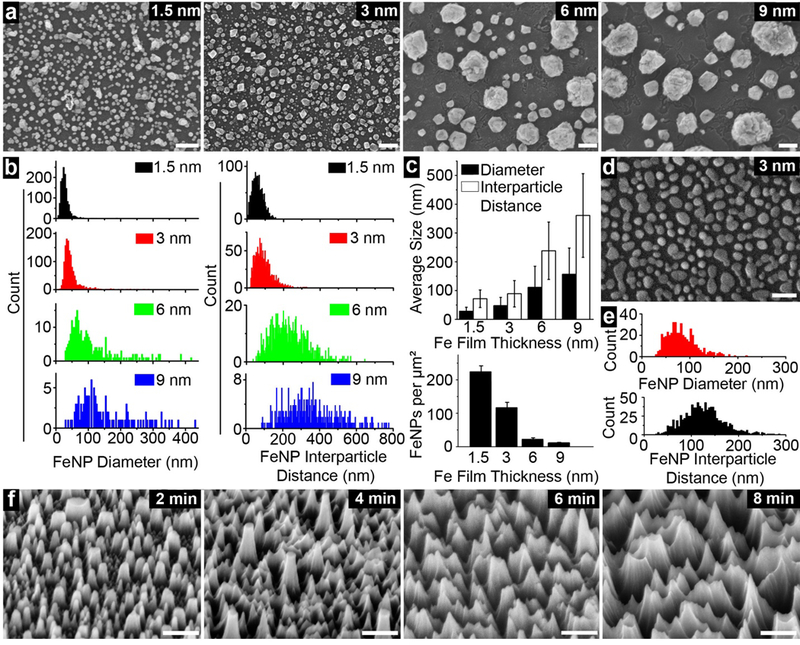
Preparation of FeNP coated surface for nanostructure fabrication. a) FeNPs of various sizes and inter-particle distances formed from iron film with various thicknesses. Scale bars are 100 nm. b) FeNP size and inter-particle distance distribution in each film thickness. c) quantitative analyses of average FeNP size and inter-particle distance (top), and density of FeNPs for each film thickness (bottom). (Top) Error bars are standard deviation of average nanoparticle size across respective figures of 2a. Large error bars are a result of size distribution. (Bottom) Error bars are standard deviation of 5 square area density across respective figures of 2a. d) SEM image (top view) of FeNPs formed under 4% hydrogen. Scale bar is 200 nm e) quantitative analyses of the average size of FeNPs and inter-particle distance formed under 4% hydrogen. f) generated nanostructures after etching for 2, 4, 6, and 8 min. Scale bars are 200 nm.
Furthermore, as hydrogen embrittlement is a popular method to produce very fine powder materials of iron,20 we further decreased the volume percent of hydrogen used in the reductive gas environment from 15% to 4% to more finely adjust the FeNP size and inter-particle distance. The average FeNP diameter and inter-particle distance correspondingly increased to 83.6 ± 32.4 nm and 125.8 ± 42.7 nm, respectively (Figure 2d and 2e), which was even more agreeable with average size of nEVs. The density of FeNPs also slightly decreased to 77.5 ± 25.8 per μm2. Moreover, the requirement of a reductive gas environment was validated by annealing the 3 nm iron film in an oxygenated ambient environment. In this instance, FeNPs layer did not form due to severe oxidization (Figure S2b).
Of note, the FeNP etch mask can be produced on both non-patterned and patterned 100 mm and 150 mm wafers with pattern, line width resolution upwards of 2 μm (Figure S2a). In comparison, neither colloidal silver nor polystyrene nanoparticles coated by spin-casting or dip-coating can reach 2-μm resolution with uniform dispersion.21,22 Also, the production of FeNP-covered substrates can be easily scaled up to batch fabrication. Typically, dozens of silica wafers can be processed in one batch with minimal variation, and thus, can significantly reduce the fabrication cost of the microdevice.
Silica nanostructures were produced via RIE, and etching time was optimized to create nanostructures with depths sufficient for at least individual nEV trapping. With an etch rate determined as ~42 nm/min, etch times between 2 and 8 minutes were evaluated (Figure S3). After etching, size specific nanostructures are obvious on the respective silica substrates (Figure 2f). In contrast, on control planar surface without the FeNP layer (Figure S2c), such structures were not formed (Figure S2d). As evident in Figures S2e–f, the residual FeNP layer was still evident from the top and side view of nanostructures immediately after etching suggesting that FeNPs can be a reliable metal mask.
Multiple angle images were obtained to demonstrate the resultant silica nanostructures morphology. Figure S2g illustrates a heavy contrasted imaged, by which the bottom of the generated nanostructures corresponds to the dark areas of the cavities. The average size of these cavities was consistent with the inter-particle distance of the FeNPs. Furthermore, elemental analysis performed after soaking in piranha solution indicated that the iron mask was completely removed (Figure S2h), and the nanostructured surface was now ready for chemical modification. Atomic force microscopy (AFM) was used to quantify roughness and surface profile. Images were processed with 2nd order flattening and the roughness tool on NanoScope Analysis (Bruker). Results indicated that the root-mean-squared value (Rq), the arithmetical mean deviation (Ra), and the increase in surface area increased with increasing etch time (Figure S3). A 6-nm etch time was ultimately selected as it ensured a nanostructure depth of ~240 nm. This was optimal as it allowed for at least one nEV to be trapped in each nanostructure while still allowing for easy access in downstream analysis and collection processes.
Fluorescent Behaviors of Lipid Nanoprobes in Cell Assay
It was found that cells labeled with the equivalent molar amount of cholesterol displayed the highest fluorescent intensity (Figure 3). It was speculated that C18 and DSPE may readily self-assemble, forming micelles or liposomes in ionic solution and absorb to plasma protein surfaces. Thus, the lipid nanoprobe cannot sufficiently isolate the nEV, and hence, the capture efficiency will significantly decrease. In contrast, cholesterol, a short and rigid molecule, is less likely to self-assemble and is ideal for nEV membrane insertion.23 In the second round of screening, the optimal lipid head, cholesterol, was investigated with PEG spacers ranging from 9 to 228 EO units. Furthermore, the average fluorescent intensity gradually decreased with the increasing EO units (Figure 3), and no significant difference was found between cholesterol-PEG400 and cholesterol-PEG1000. For further optimization, PEG length was considered to reduce steric hindrance in surface immobilization of lipid nanoprobes. An optimal length of a ~7 nm minimizes steric hinderance, thus we decided to use cholesterol-PEG1000 of ~6.4 nm in length.24
Figure 3.
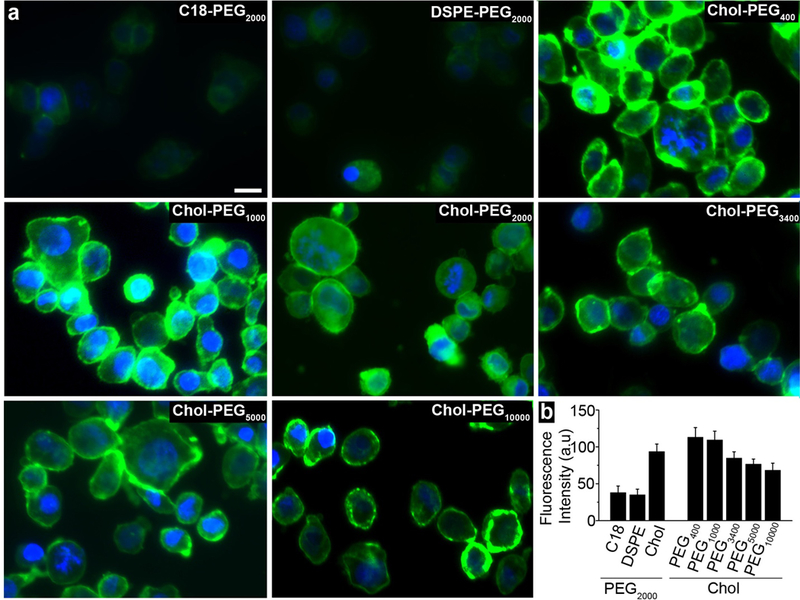
Optimization of lipid nanoprobes for nEV isolation. a) various fluorophore labelled lipid nanoprobes inserted into cell membrane. Scale bar is 10 μm. b) quantitative data of average fluorescent intensity in each group. Error bars correspond to standard deviation (n=3).
Validation of Lipid Nanoprobe Grafting via Wetting Properties
Bare, non-functionalized silica nanostructure surface was measured as 0° due to its super-hydrophilic nature. The cholesterol-PEG1000 modified surface had a contact angle of 26.5° ± 1.4°, as it is an amphiphilic molecule (Figure S4a). Whereas, cholesterol-PEG2000, a molecule containing more hydrophilic EO units than cholesterol-PEG1000, modified the surface to a smaller contact angle of 16.2° ± 0.7°. Furthermore, the contact angles of surface grafted with C18-PEG2000 and DSPE-PEG2000 containing long hydrophobic tail(s) were 42.1° ± 1.8° and 85.7° ± 2.6°, respectively (Figure S4a). The changes in contact angle among the various groups reflects the surface property after nanoprobe modification and indicates that the lipid nanoprobes were successfully grafted onto the nanostructured surface.
Device Performance with nEV Model Samples
Size distribution was measured with nanoparticle tracking analysis (NTA, Nanosight) and indicated average nEV diameters of 115 nm and 107 nm for MDA-MB-231 cell and Panc-1 cells, respectively. In addition, we routinely identified typical EV protein markers, including CD63 and CD9, and a well-known housekeeping protein β-actin in the two types of nEVs (Figure S5b).
General micromixer flow was observed with die and leak tested. (Figure 4a). With the nanostructured substrate, the isolation efficiency reached 28.8 ± 5.3% at a flow rate of 10 μl/min, and ~74.1 ng RNA was extracted from isolated nEVs (Figure 4b). Furthermore, increasing the flow rate decreased the isolation efficiency. This was likely due to insufficient time for cholesterol-lipid membrane binding and firm tethering to occur, the mixing effect increases with flow rate though. In contrast, on a planar surface, the highest isolation efficiency of 11.4 ± 3.5% was obtained at a flow rate of 5 μl/min. Similarly, increasing the flow rate significantly decreased the isolation efficiency. Hence, these results, in conjunction with SEM images (Figure 4c and 4d), are indicative that the silica nanostructures do indeed provide an additional nEV enrichment mechanism via trapping. In fact, enrichment is increased by nearly three times due to the nanostructures.
Figure 4:
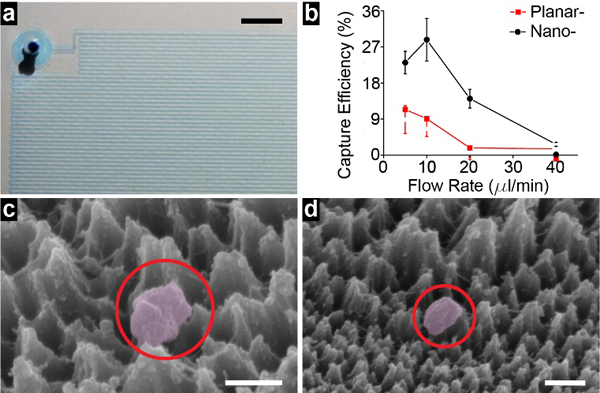
Isolation of nEVs and device performance a) assembled micromixer device demonstrated with a blue dye. Scale bar is 250 μm. b) isolation efficiency of nEVs from model samples under various flow rate ranging from 5 μl/min to 40 μl/min. Error bars correspond to standard deviation (n=3). c) an isolated nEV on lipid nanoprobe functionalized surface. Scale bar is 100 nm. d) an isolated nEV trapped within the size-matched silica nanostructures instead (upper) of the non-size-matched silica nanostructures (lower). Scale bar is 200 nm.
Of note, in our previous study of the 2-component lipid nanoprobe, 100 nmol DSPE was used to label and isolate nEVs from plasma and achieved 48.3% isolation efficiency for 100 μl plasma.6 So, although the isolation efficiency of nEVs using the micromixer platform is relatively lower, it can process up to 2-ml plasma samples. Therefore, in comparison with nEVs amount isolated from 100-μl plasma, the isolated nEVs via the microdevice can still increase the isolation quantity by an order of magnitude.
Moreover, after isolation, the total plasma protein amount decreased from an average of 125.8 mg to 4.4 mg, indicating tat 96.5% of plasma protein, were removed. According to the guideline of EV methodology, the ratio of 3×1010 EVs per μg of protein or greater has been proposed as high purity, indicating the nEVs we harvested still have protein contamination.25 However, in our previous study the nEVs isolated with lipid nanoprobe system still satisfy protein/peptide measurement with ultrasensitive mass spectrometer.6 Altogether, this isolation approach provides purer nEV contents than that isolated with 2-h ultracentrifugation at 100,000 g, which can only remove 71.2% of total protein. Thus, we have indicated a nearly 25% improvement from the EV enrichment standard technique.
Of note, although the reduction of plasma protein does not directly correlate to increase in isolation efficiency. This is still a requirement in nEV sample preparation. Many downstream applications, such as qPCR and sequencing, are very sensitive to unwanted proteins. However, a reduction of nearly 96.5% plasma protein will improve the general quality of most downstream analysis results. More specifically, this downstream application, ddPCR, is less sensitive to factors (i.e. proteins) that inhibit the target gene amplification. Thus, 96.5% plasma protein removal is satisfactory.
Mutation Detection of Device-enriched nEVs from Clinical Samples
Before applying clinical samples for detection of nEV cargo DNA mutation, the limit of detection (LOD) and reliability of the ddPCR platform was validated in two steps. First, a total of ~1010 nEVs derived from MDA-MB-231 cells and Panc-1 cells in gradient ratios (5%, 1%, 0.2%, 0.04%, 0.01%, and 0.002% of nEVs derived from Panc-1 cells) was spiked into 1-ml exosome-depleted plasma. The nEV-derived DNA was analyzed with Agilent TapeStation (Figure S5e). The nEV DNA samples contained DNA fragments longer than 10 kbp, apart from mitochondrial and circulating cell-free DNA.6,26,27 The known KRAS G12D mutation from Panc-1 cells derived nEVs was analyzed with ddPCR for determining its mutation allele frequency (MAF). The plotted curve shows the experimental data was in line with spike ratio (R2= 0.987) (Figure 5a).
Figure 5:
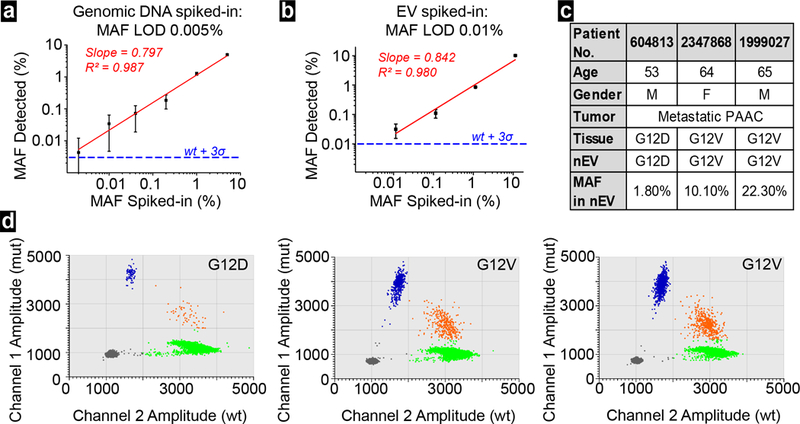
a) LOD of ddPCR measured with spike samples of nucleic acids containing KRAS G12D mutation in various allele frequency. Error bars correspond to standard deviation (n=5). b) LOD of ddPCR measured with spike samples of nEVs derived from Panc-1 cell at various allele frequency. Error bars corresponds to standard deviation (n=5). c) summary of 3 patients with metastatic pancreatic adenocarcinoma (PAAC) d) ddPCR plots for KRAS mutations detection in the 3 respective PAAC patients.
Next, nEVs, isolated from spiked samples of mixed nEVs (10%, 1%, 0.1%, 0.01%, and 0.001% of nEVs derived from Panc-1 cells), were extracted for nEV-derived DNA, and KRAS mutation analysis was performed. Although the R-squared value decreased to 0.980 in regression analysis, the entire curve still showed a good linear relationship (Figure 5b). These two curves also demonstrated that ddPCR is reliable in detecting 0.01% or higher MAF in given samples.
Subsequently, we processed three plasma samples of pancreatic cancer patients with confirmed KRAS G12D or G12V mutations from tissue samples and two healthy controls, using the lipid nanoprobe functionalized nanostructured silica device. The nEVs were isolated from up to 2 ml plasma of each subject. In total, we collected 61.6 ng, 435.4 ng, and 336.6 ng DNA from three cancer patients (#604813, #2347868, and #1999027, Figure 5c), respectively. Meanwhile, only 6.9 ng and 4.9 ng DNA were collected from two healthy controls. Of note, this low mass of DNA from two healthy controls was not unexpected. EV concentration, and thus, genomic DNA concentration can be greater in cancer patients than in healthy donors.28 The ddPCR data confirmed the corresponding mutation detected in patients’ tissue samples (Figure 5d). Moreover, the 2D amplitude plots revealed average allele frequency of respective KRAS mutation were 1.8%, 10.1%, and 22.3%, while no KRAS G12D or G12V mutations were found in healthy controls (Figure S6). This data indicated that the allele frequency of mutation in nEV-derived DNA could have a very broad range. The mutation allele frequency may depend on cancer type, stage, progression, and many other factors.
Conclusions
In conclusion, the presented device platform provided the ability for rapid and straightforward nEV isolation and enrichment. Moreover, due to these capabilities in combination with ddPCR, we were able to quickly determine the mutation allele frequency from clinical samples and make a previously unknown conclusion. Specifically, the mutation allele frequency can have a very broad range and is most definitely patient-specific. By the platform providing us with this newfound observation, we can now implement this technology on more and varying clinical samples to understand trends between mutation allele frequency and pathophysiology.
Furthermore, the utilization of the platform, in combination with ddPCR, is extremely beneficial for the application of early cancer diagnostics. In early stage cancer, the allele frequency may be lower than 0.01%.29,30 Therefore, this detection platform and ddPCR system provide the necessary high sensitivity and high specificity.31 Second, ddPCR makes this detection platform very suitable for treatment monitoring, especially in detection of known mutation loci such as EGFR T790M and L858R that can guide targeted therapy. Third, the high-throughput aspect of this platform is ideal for processing patient plasma and makes clinical translation extremely feasible. All things considered, the nanostructured substrate-integrated micromixer combined with high-sensitive ddPCR could have great potential in cancer treatment monitoring in the future.
Supplementary Material
Acknowledgements
This work was supported by the National Cancer Institute of the National Institutes of Health under grant number 1R01CA230339 to S. Y. Zheng.
Footnotes
Conflicts of interest
S.-Y. Z. is the founder of Captis Diagnostics.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Andaloussi SE, Mäger I, Breakefield XO, Wood MJ, Nature Reviews Drug Discovery. 2013, 12, 5. [DOI] [PubMed] [Google Scholar]
- 2.Théry C, Zitvogel L, Amigorena S, Nature Reviews Immunology. 2002, 2, 8. [DOI] [PubMed] [Google Scholar]
- 3.Siravegna G, Marsoni S, Siena S, Bardelli A, Nature Reviews Clinical Oncology. 2017. [DOI] [PubMed] [Google Scholar]
- 4.Yasui T, Yanagida T, Ito S, Konakade Y, Takeshita D, Naganawa T, Nagashima K, Shimada T, Kaji N, Nakamura Y, Thiodorus IA, He Y, Rahong S, Kanai M, Yukawa H, Ochiya T, Kawai T, Baba Y. Science Advances. 2017, 3, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liga A, Vliegenthart ADB, Oosthuyzen W, Dear JW, Kersaudy-Kerhoas M, Lab on a Chip. 2015, 15, 11. [DOI] [PubMed] [Google Scholar]
- 6.Wan Y, Cheng G, Liu X, Hao SJ, Nisic M, Zhu CD, Xia YQ, Li WQ, Wang ZG, Zhang WL, Rice SJ, Sebastian A, Albert I, Belani CP, Zheng SY. Nature Biomedical Engineering. 2017, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krug A, Enderle D, Karlovich C, Priewasser T, Bentink S, Spiel A, Brinkmann K, Emenegger J, Grimm D, Castellanos E, Rizaldos. Annals of Oncology. 2017. [Google Scholar]
- 8.Yeh YT, Tang Y, Sebastian A, Dasgupta A, Perea-Lopez N, Albert I, Lu H, Terrones M, Zheng SY, Science Advances. 2016, 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charbonneau DM, Tajmir-Riahi HA, The Journal of Physical Chemistry B. 2010, 114, 2. [DOI] [PubMed] [Google Scholar]
- 10.Wan Y, Kim Y, Li N, Cho SK, Bachoo R, Ellington AD, Iqbal SM, Cancer Research. 2010, 70, 22. [DOI] [PubMed] [Google Scholar]
- 11.Smith E, Chan W, Langmuir. 2008, 24, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wan Y, Mahmood MAI, Li N, Allen PB, Kim Y, Bachoo R, Ellington AD, Iqbal SM, Cancer. 2012, 118, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu H, Huang B, Zare RN, Lab on a Chip. 2005, 5, 12. [DOI] [PubMed] [Google Scholar]
- 14.Chen C, Skog J, Hsu CH, Lessard RT, Balaj L, Wurdinger T, Carter BS, Breakefield XO, Toner M, Irimia D, Lab on a Chip. 2010, 10, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konoshenko MY, Lekchnov EA, Vlassov AV, Laktionov PP, Biomedical Research International. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramirez MI, Amorim MG, Gadelha C, Milic I, Welsh JA, Freitas VM, Nawaz M, Akbar N, Couch Y, Makin L, Cooke F, Vettore AL, Batista PX, Freezor R, Pezuk JA, Rosa-Fernandes L, Silva IT, Coakley G, Nunes DN, Carter D, Palmisano G, Dias-Neto E, Nanoscale. 2018, 10. [DOI] [PubMed] [Google Scholar]
- 17.Zeringer E, Li M, Barta T, Schageman J, Pederson KW, Neurater A, Magdaleno S, Setterquist R, Vlassov AV, World Journal of Methodology. 2013, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang YT, Huang YY, Zheng L, Qin SH, Xu XP, An TX, Xu Y, Wu YS, Hu XM, Ping BH, Wang Q, International Journal of Molecular Medicine. 2017, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raposo G, Stoorvogel W, The Journal of Cell Biology. 2013, 200, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song J, Curtin WA, Nature Materials. 2012, 12. [DOI] [PubMed] [Google Scholar]
- 21.Xia Y, Tang Y, Yu X, Wan Y, Chen Y, Lu H, Zheng SY, Small. 2017, 13, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, Wan Y, Liu Y, Nanoscale. 2014, 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan Y, Wang L, Zhu C, Zheng Q, Wang G, Tong J, Fang Y, Xia Y, Cheng G, He X, Cancer Research. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saiki RK, Walsh PS, Levenson CH, Erlich HA, Proceedings of the National Academy of Sciences. 1989, 86, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coumans FA, Brisson AR, Buzas EI, Dignat-George F, Drees EEE, El-Andaloussi S, Emanueli C, Gasecka A, Hendrix A, Hill AF, Lacroix R, Lee Y; van Leeuwen TG, Mackman N, Mäger I, Nolan JP, van der Pol E, Pegtel DM, Sahoo S, Siljander PRM, Strurk G, de Wever O, Nieuwland R, Circulation Research. 2017, 120. [DOI] [PubMed] [Google Scholar]
- 26.Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, Zhang J, Weitz J, Chin L, Futreal A. Journal of Biological Chemistry. 2014, 289, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, Zheng Y, Hoshino A, Brazier H, Xiang J, Cell Research. 2014, 24, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lazaro-Ibanez E, Sanz-Garcia A, Visakorpi T, Escobeedo-Lucea C, Sijuander P, Ayuso-Sacido A, Yliperttula M, Prostate. 2014, 74, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Nature Medicine. 2014, 24, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenex-Linan M, Bentley D, Science Translational Medicine. 2012, 4, 136. [DOI] [PubMed] [Google Scholar]
- 31.Shaw JA, Page K, Blighe K, Hava N, Guttery D, Ward B, Brown J, Ruangpratheep C, Stebbing J, Payne R, Genome Research. 2012, 22, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalra H, Adda CG, Liem M, Ang C-S, Mechler A, Simpson RJ, Hulett MD, Mathivanan S, Proteomics. 2013, 13. [DOI] [PubMed] [Google Scholar]
- 33.Rekker K, Saare M, Roost AM, Kubo A-L, Zarovni N, Chiesi A, Salumets A, Peters M, M, Clinical Biochemistry. 2014, 47. [DOI] [PubMed] [Google Scholar]
- 34.Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, Simpson RJ, Methods, 2012, 56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.