

Abstract
Preterm birth is widespread and causes 35% of all neonatal deaths. Infants who survive face potential long-term complications. A major contributing factor of preterm birth is infection. We investigated the role of interleukin 22 (IL22) as a potential clinically relevant cytokine during gestational infection. IL22 is an effector molecule secreted by immune cells. While the expression of IL22 was reported in normal nonpregnant endometrium and early pregnancy decidua, little is known about uterine IL22 expression during mid or late gestational stages of pregnancy. Since IL22 has been shown to be an essential mediator in epithelial regeneration and wound repair, we investigated the potential role of IL22 during defense against an inflammatory response at the maternal–fetal interface. We used a well-established model to study infection and infection-associated inflammation during preterm birth in the mouse. We have shown that IL22 is upregulated to respond to an intrauterine lipopolysaccharide administration and plays an important role in controlling the risk of inflammation-induced preterm birth. This paper proposes IL22 as a treatment method to combat infection and prevent preterm birth in susceptible patients.
Keywords: endometrium, interleukin 22, IL22, pregnancy, preterm birth, uterus
Summary Sentence
IL22 is upregulated in uterine tissue in response to bacterial endotoxin and contributes to defense against inflammatory reaction at the maternal-fetal interface
Introduction
It is estimated that 11% of all live births worldwide are preterm (<37 weeks of gestation) [1]. The preterm birth burden is extensive. It is a single direct cause in 35% of neonatal death cases, while surviving infants often suffer from long-term complications including various physical effects (visual, hearing impairment, chronic lung disease, cardiovascular disease), neurodevelopmental and behavioral deficiencies [2, 3]. The underlying causes of preterm birth vary, but it is estimated that up to 40% of all preterm births are associated with infections [4]. Bacterial vaginosis and urinary tract infections are common sources of pathogens contributing to intrauterine (IU) infection via an ascending route [5]. Hematogenous spread of pathogens to the maternal–fetal interface and to the placenta is implicated in preterm birth linked to periodontal disease and listeria infection [6]. Young or advanced maternal age, compromised socioeconomic status, high blood pressure during pregnancy, stress, tobacco, alcohol or substance abuse are among risk factors associated with preterm birth [3, 7]. The mechanisms of how these risk factors are linked to preterm birth are not fully understood, although inflammation is thought to be implicated in preterm birth associated with obesity and stress [8].
Animal studies support the role of infection and infection-associated inflammation in the pathogenesis of preterm birth [9–11]. It has been demonstrated that both IU and systemic administration of lipopolysaccharide (LPS) causes preterm birth or IU fetal death in mice [12, 13]. Recognition of LPS via pattern-recognition receptor of the toll-like receptor family, TLR4, and CD14, which are expressed by myeloid cells residing in the uterus (macrophages, monocytes, neutrophils, or dendritic cells), induces increased production of proinflammatory cytokines (e.g., tumor necrosis factor [TNF], interleukin 1 beta [IL1B], IL6) [14]. These cytokines can activate signal pathways leading to upregulation of cyclooxygenase-2 (COX-2) and the increasing release of prostaglandin E2 by myometrial cells leading to uterine contractions [15]. The use of anti-TNF or anti-IL6 decreased fetal loss in an LPS-induced murine model of preterm birth [16, 17]. The abundant expression of interleukin 10 (IL10) in uterine tissues implies a strong regulation of the inflammation [18]. Studies with IL10 null mice proved that uncontrolled production of proinflammatory cytokines results in pregnancy loss [13]. Uterine NK (uNK) cells were shown to play a significant role in mediating these adverse effects in IL10-null mice, while TNF neutralization, NK cell depletion or IL10 supplementation could rescue the pregnancy loss in IL10-/- mice [19, 20].
Intrauterine LPS challenge in mice was reported to be associated with neutrophil infiltration at the maternal–fetal interface; however, at the same time it was shown that the absence of neutrophils did not delay preterm birth [21]. Conversely, others have reported relatively little myometrial and decidual leukocyte infiltration upon IU LPS treatment [22]. A decrease in inflammation-induced preterm birth was shown in mice with depleted invariant NKT cells [23]. A resistance to LPS-induced abortion was observed in interleukin 15 (IL15)-deficient mice [24]. IL15 is a cytokine that is essential for development and activation of NK cells, suggesting that NK cells populating the uterus are involved in the mechanism of preterm birth.
uNK cells are a type of innate lymphoid cell (ILC) residing within myometrial and endometrial stroma and play an important role during early and mid-pregnancy. Studies with transgenic mice, including alymphoid mice and Killer cell lectin-like receptor, subfamily A (Klra; previously known as Ly49) knockdown mice, demonstrated that uNK cells are involved in the processes of endometrium decidualization and uterine lumen closure [25, 26]. Interferon gamma (IFNG) production by uNK cells is essential in triggering spiral artery remodeling, a fundamental process that ensures blood supply for the developing embryo [27]. The population of uNK cells is both dynamic and heterogeneous. They are present in the virgin and the preimplantation uterus and their numbers increase substantially at midgestation, when they are abundantly located in decidua interfacing fetal-originated trophoblast cells [28]. The majority of mouse uNK cells can be recognized based on reactivity to lectin Dolichos Biflorus agglutinin (DBA) [29]. DBA-reactive uNK cells appear to be unique to the uterus and are functionally biased towards angiogenesis. They have low levels of Ifng transcripts, but higher expression of angiogenic factors and Il22 [30].
IL22 is a unique cytokine secreted by immune cells including Th17 cells, which are implicated in broad array of inflammatory responses, and by ILC, particularly ILC3, that play important roles in host defense at mucosal surfaces and tissue repair [31]. ILC3 are detectable at low frequency in the uterus of virgin and pregnant mice and were shown to constitutively produce IL22 and IL17 [32]. Similarly, ILC3 were identified in human endometrium and decidua (endometrium during pregnancy) [32, 33]. The role of uterine ILC3 as well as other ILCs in reproduction is not elucidated yet, although it is thought that they play a role in innate defense and tissue homeostasis. Interestingly, uNK cells in human endometrium were also reported to express IL22 [34, 35]. IL22 acts on epithelial and parenchymal cells of nonhematopoietic origin at various barrier surfaces and organs such as skin, intestine, lung, liver, and thymus. It promotes proliferation and survival of epithelial cells, as well as induces secretion of antimicrobial peptides [36]. Trophoblast cells are epithelial cells of fetal origin. They were shown to respond to IL22 by increased proliferation and survival [35]. More data are accumulating on the importance of IL22 in epithelial regeneration and wound repair [37, 38]. Upregulation of IL22 in uterine tissues was reported to be in response to intragastric infection of pregnant mice with Listeria monocytogenes [39]. However, it was not clear if IL22 upregulation could be related to the pathogenesis of pregnancy loss or it plays a different role.
In the current study, we hypothesized that IL22 could contribute to defense against inflammatory responses at the maternal–fetal interface in response to IU infection. We used a well-established LPS-induced preterm labor model [9, 12, 40] in the mouse to test this hypothesis. We found that IL22 is upregulated in uterine tissue in response to bacterial endotoxin and prevents apoptosis of placental cells. Importantly, supplementation with recombinant IL22 (rIL22) significantly improved the pregnancy outcome in mice that are challenged with IU LPS treatment.
Materials and methods
Mice
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of Rosalind Franklin University and by the NorthShore University HealthSystem Animal Care and Use Committee and conform to the Guide for Care and Use of Laboratory Animals (1996, National Academy of Sciences). Mice used in the present studies were C57BL/6J (B6, The Jackson Laboratory Cat# 000664); 129S5-Il22tm1.1Lex/Mmucd (Il22KO, MMUCD Cat# 036745-UCD), and the CD1 strain (Harlan laboratories, Madison, WI). Female mice in estrus were selected by the gross appearance of the vaginal epithelium [41] and were impregnated naturally. Mating was confirmed by the presence of a vaginal plug and the day of plug formation was counted as day 0.5 of pregnancy. Males were left with females for a maximum of 2 days to ensure accuracy of gestation date.
Animal treatment and tissue harvest
C57BL/6J and Il22KO mice
Intrauterine injection of LPS (extracted from Salmonella enterica, L2262, Sigma, St. Louis, MO, with doses of 25, 10, 1, or 0.1 μg/animal) was performed into the right uterine horn on day 14.5 of a 19–20-day gestation period, as previously described [9, 40, 42]. In the B6 mouse, the 25 μg intervention reliably induced preterm delivery within 24–48 h, while 10 or 1 μg doses were generally tolerated. The Il22KO mouse was unable to tolerate any dose of LPS. Recombinant mouse IL22 (rIL22, BioLegend, San Diego, CA, 10 μg/animal) was injected intravenously (IV) before IU LPS treatment and 24 h after. During IU LPS treatment procedure, the number of implantation sites was recorded. The timing of preterm delivery and number of live pups were assessed during harvest, 48 h post IU LPS treatment. For tissue harvests, a separate group of animals were euthanized 6 h after surgery. The injected/right uterine horn was incised longitudinally along the antimesenteric border. Gestational tissues, mesometrial decidua, and junction zones or placenta were harvested and flash-frozen in liquid nitrogen and stored at –80°C for mRNA and protein extraction. Whole implantation sites with pups removed were fixed in 4% paraformaldehyde for immunohistochemistry.
CD-1 mice
Intrauterine injection of LPS (extracted from Salmonella enterica, L2262, Sigma, St. Louis, MO, 100 μg/animal) was performed into the right uterine horn on day 14.5 of a 19–20-day gestation period, as previously described [9, 40, 12]. This intervention reliably induced preterm delivery within 18–24 h. Recombinant mouse IL22 (rIL22, BioLegend, San Diego, CA, 10 μg/animal) was injected IV 3 h after IU LPS. The timing of preterm delivery and number of live pups delivered at term were observed.
Immunofluorescence and immunohistochemistry
For the detection of uterine NK cells DBA-lectin staining was done using standard procedures [29, 30, 42]. Fixed-frozen implantation site tissue sections (5 μm) were blocked with an Avidin/Biotin Blocking Kit (Vector Labs Cat# SP-2001). Following blocking sections were incubated with biotin-conjugated DBA-lectin (Vector Labs Cat# B-1035) (1 μg/ml in 3% phosphate buffered saline [PBS] supplemented with 3% bovine serum albumin [BSA]) overnight at 4°C, followed by incubation for 1 h at RT with Avidin-conjugated Texas Red (Vector Labs Cat# A-2006). All antibodies used in the study are listed in Supplementary Table 1. Negative control sections were similar treatments that did not contain biotin-conjugated DBA-lectin. For other immunofluorescence staining, sections were incubated with mouse FITC-anti-Pan Cytokeratin (Sigma-Aldrich Cat# F0397), rabbit anti-interleukin 22 receptor, alpha 1 (IL22RA1) (Millipore Cat# 06-1077), rat anti-IL22 (R&D Cat# MAB582) or isotype-matched controls, overnight at 4°C. Primary antibodies were followed by secondary antibodies, FITC-rabbit anti-rat (Vector Labs Cat# FI-4000), FITC-goat anti-rabbit (Vector Labs Cat# FI-1000), or Alexa-647-goat anti-rabbit (Invitrogen Cat# A21246). Cells were fixed in Prolong gold antifade reagent with DAPI (Invitrogen Cat# P36934) to visualize nuclei. Antigen distribution was examined using a Nikon Eclipse TE2000-S fluorescence microscope (Nikon Instrument INC).
Real-time reverse transcription polymerase chain reaction (RT-PCR)
Total RNA from either the mesometrial decidua and junction zone or the placental tissues were extracted. Homogenization was performed in either Ambion TRIzol Reagent (Life Technologies Cat# 15-596-018) or using an RNasy Micro Kit (Qiagen Cat# 74004) according to the manufacturer's protocol. For ex vivo experiments, cell suspensions were collected, centrifuged; the medium was separated and the pellet was lysed in Trizol. Complementary DNA was prepared using TaqMan Fast Advanced Master Mix (Applied Biosystems Cat# 4444557). Duplex RT-PCR was performed with one primer pair amplifying the gene of interest and the other an internal reference (Actb or Gapdh) in the same tube using the Applied Biosystems Step One Real-time PCR system. Semiquantitative analysis of gene expression was done using the comparative CT (ΔΔCT) method, normalizing expression of the gene of interest to actin, beta (Actb) or glyceraldehyde-3-phosphate dehydrogenase (Gapdh). The prevalidated Taqman gene expression assays for Il22 (Mm01226722_g1), Il22ra1 (Mm01192943_m1), Ifng (Mm01168134_m1), Tnf (Mm00443258_m1), chemokine (C-C motif) ligand 3 (Ccl3) (Mm00438260_s1), BCL2-associated X protein (Bax) (Mm00432051_m1), B cell leukemia/lymphoma 2 (Bcl2) (Mm0047763_m1) (ThermoFisher Scientific), and internal control Actb (Mm02619580_g1) (ThermoFisher Scientific) or Gapdh (4352339E) (Applied Biosystems) were used. Real-time PCR was performed using the universal PCR master mix reagent (Applied Biosystems).
Placental cells preparation and ex vivo treatment
Uteri were dissected on day 14.5 of pregnancy, and placentas were dissected from decidual caps; a single-cell suspension was prepared as described previously [43]. Briefly, tissues were minced in Hanks balanced salt solution (Life Technologies), mechanically dispersed through a 100-μm nylon filter, and centrifuged at 1500 rpm. The remaining pellet was dispersed in Roswell Park Memorial Institute (RPMI) medium at 107cells/ml in 48-well plates. Prior to plating, placental suspensions underwent red cell lysis by incubation with red blood cell lysis buffer (BioLegend). The above specimens were incubated at 37°C in 5% CO2/95% air for 1 h. Viability of ex vivo cultured cells was >95% as assessed using the trypan blue dye exclusion test. Placental cells underwent treatment for 2 h with PBS or LPS (5 ng/ml) followed by supplementation with rIL22 (5 μg/ml) for an additional 12 h.
Protein extraction
For protein extraction, cells were homogenized in ice-cold 1X radio-immunoprecipitation assay (RIPA) buffer (Santa Cruz Biotechnology) containing protease and phosphatase inhibitors (Roche Applied Science). Lysates were incubated on ice for 30 min and centrifuged at 10 000× g for 10 min at 4°C. Supernatant fluid was collected and used as a total cell lysate for protein assays. Protein concentration was measured by bicinchoninic acid (BCA) protein assay.
Caspase activity and fas ligand (FASL) measurement
Activity of Caspase9, -8, -3, and -1 (CASP9, -8, -3, -1) was measured using SensoLyte AFC Caspase Profiling Kit (AnaSpec, Fremont, CA) [44] in total protein extracted from decidual cells and placental cells treated ex vivo with LPS, with or without rIL22. Caspase activity was assayed on a fluorometer (Bio-Tek Instruments) as per the instructions provided by manufacture. Equal amounts of protein (50 μg) were used. Caspase activity was measured in relative fluorescence units/μg of protein. FASL was analyzed by Milliplex map kit (Millipore, St. Charles, MO) according to the manufacturer's instruction.
Statistical analysis
The data are expressed as mean ± SEM. The statistical analyses were performed using MedCalc software (version 13.0.4.0). Data were assessed with one-way or two-way analysis of variance (ANOVA). When the data were not normally distributed, two groups were compared with the Kruskal–Wallis test. P < 0.05 was considered significant.
Results
Intrauterine lipopolysaccharide administration induces uterine interleukin 22 expression
To reveal if the expression of IL22 is affected during preterm birth, we used a well-defined mouse model that is based on IU LPS administration to a prepartum mouse [12]. C57BL6 mice (referred further as WT) were treated with different doses of LPS on gestation day (gd) 14.5 to determine the dose that reliably induces abortion in this strain within 48 h after the injection. The administration of LPS 25 μg/mouse caused complete loss of fetuses by the time of tissue collection either due to preterm delivery or IU death (Figure 1). In contrast, the LPS dose of 1 μg/mouse was tolerated well, suggesting that protective mechanisms were effective in prevention of preterm delivery.
Figure 1.

Intrauterine LPS administration in C57BL6 mice reliably induces preterm birth with the 25 μg/mouse dose, while the 1 μg/mouse dose is well tolerated. LPS was injected into the uterus on gd 14.5 (LPS dose of 1ug (n = 5), 10ug (n = 5), 25ug (n = 3), PBS (n = 5)), and the uterus was collected 48 h post-treatment. The number of live or dead fetuses and aborted implantation sites was documented. Data analyzed by ANOVA followed by Tukey–Kramer post hoc analysis, **P < 0.01.
The expression of IL22 was analyzed in uterine samples collected 6 h after the administration of either 25 or 1 μg of LPS and compared to control samples from animals that received IU infusions of PBS. None of the control samples revealed the presence of Il22 transcripts, but we found the expression of Il22 in all LPS-treated animals (Figure 2A). Interestingly, the animals administered with low-dose LPS (1 ug/mouse) revealed the highest Il22 levels.
Figure 2.

Effect of IU LPS administration to C57BL6 mice on gd 14.5 on cytokine expression in uterine tissue and spleen. (A) Expression of Il22, Ifng, Tnf, and Ccl3 mRNA in the uterine samples collected 6 h post-LPS injection. (B) mRNA expression of these cytokines in spleen collected 6 h post-LPS injection. Animals administered an LPS dose of 1 μg (n = 5) or 25 μg (n = 5) per mouse were compared with mice receiving PBS (n = 5). Data presented as Mean ± SEM. Data analyzed by Kruskal–Wallis test or ANOVA followed by Tukey–Kramer post hoc analysis, *P < 0.05, **P < 0.01.
The LPS administration also caused increased uterine expression of Ifng (Figure 2A). Other proinflammatory cytokines that were analyzed in uterine tissues demonstrated either no change (Tnf) or significant decrease (Ccl3) (Figure 2A). To check the systemic response to IU LPS administration, spleen samples were analyzed similarly (Figure 2B). No significant difference was found in mRNA levels of Ifng, Tnf, or Ccl3 in the spleen of LPS-treated animals compared to PBS-treated controls. Interestingly, a substantial expression of Il22 was observed in the spleen of animals that received a high dose of LPS (25 ug/mouse). Il22 was also detected in the spleen of animals treated with 1 μg LPS, but not in the spleen of control animals.
To determine the IL22 source at the maternal–fetal interface, implantation sites were stained with DBA-lectin, which reacts with uterine NK cells, and with anti-IL22. DBA-lectin-positive cells were localized in decidua (i.e., maternal tissues, which will be referred to as uterine tissues), but not in placenta (a fetal tissue). Demarcation of different zones within an implantation site is shown in Figure 3A, where the junctional zone can be distinguished by bright cytokeratin staining specific for trophoblasts and the decidual area by DBA-lectin staining characteristic of uterine NK cells. Samples from LPS-treated animals but not control PBS-treated animals revealed positive IL22 staining (Figure 3B and D). The IL22 staining was observed in decidua in areas adjacent to large blood vessels and to the junctional zone of placenta and was either colocalized with DBA-lectin staining or not (Figure 3D). Also, some weak but persistent diffuse staining of IL22 was observed within the junctional zone.
Figure 3.
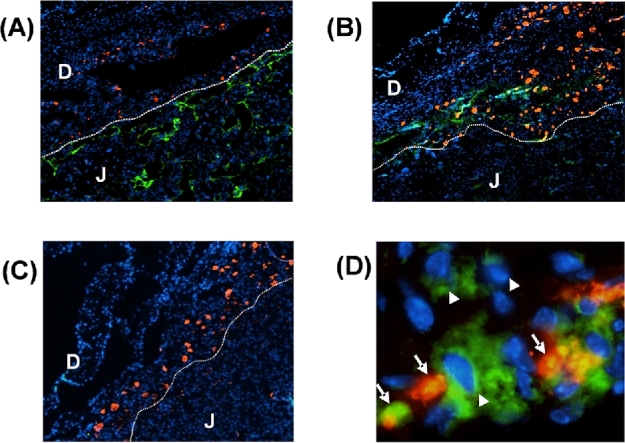
Intrauterine LPS administration in mice on gd 14.5 induces the expression of IL22 at the maternal–fetal interface. (A) Red—DBA lectin, green—pan-cytokeratin, blue—DAPI staining for nuclei. Line is drawn to indicate demarcation of different zones within the maternal–fetal interface. Decidua can be identified by presence of DBA-positive uNK cells. Junctional zone of the placenta is identified by pan-cytokeratin-positive trophoblasts. Original magnification 4×. (B, C, D) Red—DBA lectin, green—IL22, blue—DAPI. (B) IL22 staining is observed in samples from mice treated with 25 μg/mouse LPS. IL22 expression is localized in decidua along with DBA-lectin (red) positive uNK cells in areas adjacent to the junctional zone. Original magnification 4×. (C) No IL22 staining was detected in samples from control PBS-treated mice. Original magnification 4×. (D) A representative image of IL22 expression in decidua from LPS-treated mouse. Both DBA-lectin positive uNK cells (arrow) and DBA-lectin negative cells (arrow head) are found to express IL22. Original magnification 40×. D—decidua, J- junctional zone of placenta.
Interleukin 22-deficient mice are highly susceptible to lipopolysaccharide-induced preterm birth
To investigate whether the observed uterine IL22 increase after LPS challenge is a part of the pathogenic mechanism that leads to pregnancy loss or if it is an important factor in preterm birth prevention, mice deficient for IL22 were tested with IU LPS administration. Il22-/- mice were found to be very sensitive to LPS treatment as indicated by the dose response data (Figure 4A). The large dose of LPS (25 μg/mouse) caused complete abortion of fetuses within the first 24 h, and the uterus was significantly reduced in size by the time of tissue collection (Figure 4B). However, in contrast to WT mice, the LPS dose of 1 μg/mouse was not tolerated by Il22-/- mice and caused complete pregnancy loss. Moreover, the LPS dose of 0.1 μg/mouse was also efficient in induction of preterm birth in Il22-/- mice. A comparison of cytokine expression in uterine tissues from PBS- or LPS-treated animals did not reveal a significant difference for Ifng or Ccl3, although significantly higher Tnf expression was detected in mice administered with LPS (Figure 4C).
Figure 4.

IL22-deficient mice are highly susceptible to LPS-induced preterm birth. The risk of LPS-triggered abortion and IU fetal death is reduced by rIL22 supplementation. (A) Pregnant Il22-/- mice received IU injection of different doses of LPS (25 μg (n = 1), 10 μg (n = 5), 1 μg (n = 4), or 0.1 μg/mouse (n = 4)) or PBS (n = 4) on gd 14.5. The uterus was collected 48 h post-treatment and the number of live or dead fetuses and aborted implantation sites was documented. Percent of live fetuses per pregnancy in LPS-treated animals was compared to PBS-treated mice. (B) Images of uteri from Il22-/- mice collected 48 h post-LPS or -PBS injection. Left image: uterus from PBS-treated mouse. Right upper: uterus from mouse treated with 1 μg LPS. Right lower: uterus from mouse treated with 1 μg LPS along with rIL22 supplementation. (C) Expression of Ifng, Tnf, and Ccl3 mRNA in the uterine samples from Il22-/- mice. Uteri were collected 6 h post-LPS (1 μg, n = 5) or PBS injection (n = 4). (D) Pregnant (gd14.5) Il22-/-mice received IU injection of LPS (1 or 0.1 μg) with or without rIL22 supplementation. Percent of live fetuses per pregnancy was calculated 48 h post-treatment. N = 8 in LPS group, n = 7 in LPS + rIL22 group. Data analyzed by Kruskal–Wallis test or ANOVA followed by Tukey–Kramer post-hoc analysis, *P < 0.05, **P < 0.01.
Recombinant interleukin 22 reduces the risk of lipopolysaccharide-triggered abortion and intrauterine fetal death
To explore IL22 as a therapeutic agent for the prevention of LPS-induced preterm labor, the Il22-/- mice were treated with two doses of rIL22, 10 μg each, injected IV before and 24 h after LPS administration. As shown in Figure 4B and D, IL22 supplementation in animals that received 1 μg or 0.1 μg/mouse of LPS significantly increased the number of live fetuses or prevented preterm labor compared to animals that received the same LPS treatment alone. To our knowledge, this is the first report to demonstrate that IL22 is a potential mediator to prevent LPS-induced preterm delivery. Subsequent experiments with rIL22 supplementation were performed using CD1 mice. Compared to inbred C57BL/6J strain, the CD-1 is an outbred strain with much higher fecundity. Therefore, use of this strain expedites reaching the required sample size. Intrauterine 100 μg/mouse LPS administration is known to reliably induce preterm birth in this strain [12, 40]. As shown in Table 1, IL22 supplementation prevented LPS-induced preterm labor by ∼78% and significantly increased the number of live pups delivered at term compared to animals without supplementation.
Table 1.
IL22 supplementation inhibits LPS-induced preterm delivery.
| Treatment groups dose/mouse | Preterm delivery (%) | Number of pups delivered at term |
|---|---|---|
| LPS IU (100 μg) + PBS IV (100 μl) | 8/8 (100) | 0 |
| LPS IU (100 μg) + rIL-22 IV (10 μg) | 2/9 (22)** | 9.57 ± 1.13**,¶ |
| PBS IU (100 μl) + rIL-22 IV (10 μg) | 0/3 (0) | 12.02 ± 1.23 |
| PBS IU + PBS IV (100 μl) | 0/3 (0) | 12.14 ± 1.14 |
**P ≤ 0.01, LPS + PBS vs LPS + rIL22.
¶Includes only pups from pregnancies delivered at term.
IV injections performed 3 h after IU inoculation.
Interleukin 22 target cells are located within junctional and labyrinth zones of placenta
IL22 acts via the IL22 receptor, which is a heterodimer consisting of unique alpha subunit, IL22RA1, and a common IL10 beta subunit, IL10RB. We evaluated Il22ra1 mRNA expression in uterine samples from WT and Il22-/- animals treated with LPS or PBS (Figure 5A). Il22ra1 levels in WT as well as Il22-/- mice were not affected by LPS treatment. However, it was found that Il22-deficient mice, despite the absence of Il22, demonstrated significantly higher Il22ra1 expression in comparison to WT mice. Immunofluorescent staining was performed to investigate IL22RA1 expression within the implantation site. Cells stained positive for IL22RA1 were mainly distributed in the junctional zone of placenta consisting of various cytokeratin positive trophoblast populations (Figure 5B). Moreover, IL22RA1 expression was observed throughout the labyrinth zone of the placenta (Figure 5C).
Figure 5.

IL22RA1 expression in murine implantation site. (A) Il22ra1 mRNA expression in uterine samples from pregnant WT and Il22-/- mice. Mice were treated with PBS (n = 5 for WT, n = 4 for Il22-/-) or LPS dose that reliably induces preterm birth (25 μg for WT (n = 5) and 1 μg (n = 5) for Il22-/- mice) and tissues were collected 6 h post LPS treatment. Data presented as Mean ± SEM. Data analyzed by two-way ANOVA with Bonferroni correction, *P < 0.05. (B) and (C) Immunofluorescent staining with antibodies against IL22RA1 (red) and pan-cytokeratin (green) in gd14.5 implantation site sections from WT mice. Small panels: upper—DAPI (blue) for nuclei, middle—IL22RA1, bottom—pan-cytokeratin; large panels: merge. Decidua and junctional zone of placenta are shown in (B), placental labyrinth is shown in (C). Original magnification 20×.
rIL22 supplementation prevents activation of the extrinsic pathway of apoptosis in placental cells
To further assess the mechanism of the observed protective effect of IL22, we characterized the intrinsic and extrinsic pathways of apoptosis [44, 45] in ex vivo cultured placental cells incubated with LPS or PBS for 2 h, followed by treatment with or without rIL22 for 12 h. The factors involved in the intrinsic pathway of apoptosis (BAX, BCL2, and CASP9) remained unchanged in placental cells (Figure 6A). However, supplementation with rIL22 significantly reduced the LPS-induced level of proteins involved in the extrinsic pathway of apoptosis (CASP8 and CASP3) (Figure 6B). Therefore, the data suggest that IL22 prevents LPS-induced activation of the extrinsic pathway of apoptosis at the feto–maternal interface.
Figure 6.

rIL22 supplementation prevents activation of extrinsic pathway of apoptosis in placental cells. mRNA expression of Bax and Bcl2, and activity of CASP9, molecules involved in the intrinsic pathway of apoptosis (A) and, protein concentration of FASL and activity of CASP8 and CASP3, molecules involved in extrinsic pathway of apoptosis (B) in placental cells cultured ex vivo with PBS (control) or LPS for 2 h, followed by treatment with rIL22 or control for 12 h. Depicted are representative figures from three repeat experiments. Data presented as Mean ± SEM. Data analyzed by ANOVA followed by Tukey–Kramer post hoc analysis. *P < 0.05 compared to LPS with and without IL22.
Discussion
We have identified the important role of IL22 in controlling the uterine reaction to bacterial endotoxin exposure during late pregnancy in mice. IL22 is produced by decidual cells in response to IU LPS administration but primarily targets placental cells. Our data suggest that IL22 prevents preterm birth and IU fetal death and promotes survival of placental cells. IL22 is a cytokine expressed by various immune cells but targets primarily epithelial and stromal cells [36]. Cells with potential to secrete IL22 were described both in mouse and human endometrium. In human early pregnancy decidua, the ILC3 were shown to release IL22 [33]. The authors demonstrated that these ILC3 could partially differentiate into uNK cells. In mouse, the DBA-lectin-positive uNK cells, which constitute a major lymphocyte population during midgestation, were reported as potential IL22 producers [30]. The ILC3 are also present in the murine uterus where they are sparsely distributed in the myometrial layer of uterus [32, 46].
While there was no detectable IL22 in gd 14.5 uterine tissues from control mice, we found IL22 in all uterine samples from LPS challenged mice. IL22 staining within the utero-placental unit demonstrated that IL22 is secreted by cells distributed along the junctional zone of the placenta. In addition, some weak diffuse IL22 staining was observed within the spongiotrophoblast/junctional zone, which could be the secreted form and not cell-associated IL22. Simultaneous staining with anti-IL22 Abs and DBA-lectin revealed that besides DBA-lectin-positive uNK cells, IL22 is also produced by DBA-lectin negative cells as well. Those might include NK cells, ILC, T cells, or myeloid cells [30, 32, 47, 48]. The source of the IL22 in the decidua should be characterized. For instance, a secretion of IL22 upon LPS stimulation of bone marrow-derived DC was described [48]. Because myeloid cells present in high numbers in murine uterus during pregnancy, they indeed can be explored as potential IL22 producers.
In our studies, the LPS-induced upregulation of Il22 in uterine tissues collected 6 h postsurgery was accompanied by increased expression of Ifng and Tnf, although for Tnf the difference was not statistically significant. Interestingly, a similar relatively low response was seen with a leukocyte influx to the uterus assessed shortly after IU LPS challenge in pregnant gd 16 mice [22]. The analysis at 7 h post-LPS administration revealed a marked leukocyte infiltration into the lungs and liver but little into the uterus; the increase in a number of mobilized leukocytes was observed only at the onset of parturition. Further, Edey et al. [22] demonstrated significantly higher numbers of infiltrating inflammatory monocytes and neutrophils at 7 h postsurgery in the uterus of control PBS-treated animals when compared to LPS-treated mice. These data are consistent with our data for higher Ccl3 levels in uterus of PBS-treated animals than in animals treated with LPS, as CCL3 is the main chemokine for neutrophil trafficking. Upregulation of uterine Il22 expression was also reported in response to systemic LPS challenge (intraperitoneal administration) in pregnant gd 8.5 mice [49]. Within the first 8 h post-LPS treatment, a ten-fold increase of Il22 transcript expression was found, while 16 h post-LPS treatment there was no upregulation of Il22. Thus, it seems that Il22 upregulation in the uterus of pregnant mice is an element of the body's early response to an inflammatory agent, like bacterial LPS. In addition to uterine upregulation of Il22, a significant increase in expression of Il22 was observed in the spleen. The systemic IL22 response was also reported after intraperitoneal LPS challenge [50]. Th17 cells or a discrete innate lymphoid population with CD25 + CCR6+ phenotype could be considered among cellular sources of IL22 in the spleen [51]. IL22 is known as a cytokine with the properties of a double-edged sword cytokine, capable of mediating both inflammatory and anti-inflammatory responses (reviewed in [36, 52]). Il22-/- mice were tested by IU LPS administration to investigate whether the observed uterine Il22 upregulation in response to LPS challenge is a part of the pathogenic mechanism that leads to pregnancy loss or if it is an important factor in preterm birth prevention. Indeed, pregnant Il22-/- mice were highly susceptible to LPS treatment and responded with increased uterine Tnf levels. Importantly, the risk of LPS-triggered abortion and IU fetal death in Il22-/- mice was significantly reduced with rIL22 supplementation. Moreover, the use of rIL22 successfully inhibited LPS-induced preterm birth in normal CD1 mice. Interestingly, LPS challenge of Il22-/- mice did not upregulate uterine Ifng expression nor modified Ccl3 transcripts level. However, when the cytokine response was compared between LPS-treated Il22-/- and WT mice, no significant differences between two groups were found. In contrast, LPS treatment in Il10-/- mice was reported to cause a dramatic increase in proinflammatory cytokines when compared to WT mice [13]. These results suggest different mechanisms of preterm birth in Il10-/- and Il22-/- mice.
Duffin et al. 2016 [50] demonstrated that rIL22 treatment reduces the risk of systemic inflammation in mice injected with LPS intraperitoneally. The authors determined that IL22 production by intestinal ILC3 is essential in protecting against gut barrier dysfunction to prevent systemic inflammation. LPS-induced PGE2 was shown to directly activate intestinal ILC3 and induce IL22 secretion. IL22 targets intestinal mucosal epithelium and promotes its survival and regeneration [37, 50].
With regard to the IL22 receptor, previously it was reported that Il22ra1 mRNA in mice was detected at very low levels in restricted organs (such as the kidney, liver, lung), but was upregulated upon LPS stimulation [53]. Our data demonstrate that expression of IL22RA1 in murine late gestation uterus does not require LPS stimulation. Immunofluorescent staining revealed similar staining in control and LPS-treated mice that was mainly localized to placental cells, namely cells in the junctional zone and the labyrinth. Higher expression of uterine IL22RA1 was detected in Il22-/- mice when compared to WT mice, possibly as a mechanism to account for a lack of IL22. Human trophoblast cells were also reported to express IL22RA1 [35]. It was shown that IL22 promotes survival of trophoblasts, while reduced secretion of IL22 by decidual NK cells affects trophoblast growth and is involved in the occurrence of spontaneous miscarriage [35]. Our results suggest that IL22 supplementation is important to prevent LPS-induced apoptosis in trophoblasts.
Some limitations of our findings include relatively small sample size of the experimental groups and not a completely defined cellular source of IL22 at the implantation site. Further experiments with LPS challenge at different gestation time-points, e.g., gd 7.5–10.5, when DBA-lectin-positive uNK cells were the most abundant, will be informative to determine IL22-producing cells in murine uterus.
In conclusion, our study provides evidence that IL22 plays an important role in controlling the risk of inflammation-induced preterm birth. The upregulation of IL22 in response to LPS challenge contributes to defense against inflammatory responses at the maternal–fetal interface and prevents apoptosis of placental cells. This study features IL22 as a new approach to treat and prevent preterm birth in patients at risk.
Supplementary data
Supplementary data are available at BIOLRE online.
Supplementary Table S1. Antibodies used in the study.
Acknowledgments
Author contributions: SD, SS, and MKJ designed and performed research, analyzed data, and wrote the paper. VA and GKK performed research, AGS helped in writing the paper. EH and KDB designed research, analyzed data, and helped in writing the paper.
Conflict of Interest: The authors have declared that no conflict of interest exists.
Footnotes
Grant Support: This study was funded in part by the Clinical Immunology, Rosalind Franklin University of Medicine and Science, North Chicago, Illinois, USA, NIH (#1R01 HD056118 and 3R01HD056118-03S1), the March of Dimes Birth Defects Foundation (#21-FY10-202), and the Satter Foundation for Perinatal Research.
References
- 1. Blencowe H, Cousens S, Chou D, Oestergaard M, Say L, Moller AB, Kinney M, Lawn J, Born Too Soon Preterm Birth Action Group. Born too soon: the global epidemiology of 15 million preterm births. Reprod Health 2013; 10Suppl 1:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. MacDorman MF, Matthews TJ, Mohangoo AD, Zeitlin J. International comparisons of infant mortality and related factors: United States and Europe, 2010. Natl Vital Stat Rep 2014; 63:1–6. [PubMed] [Google Scholar]
- 3. Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet 2008; 371:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agrawal V, Hirsch E. Intrauterine infection and preterm labor. Semin Fetal Neonatal Med 2012; 17:12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mendz GL, Kaakoush NO, Quinlivan JA. Bacterial aetiological agents of intra-amniotic infections and preterm birth in pregnant women. Front Cell Infect Microbiol 2013; 3:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baud D, Greub G. Intracellular bacteria and adverse pregnancy outcomes. Clin Microbiol Infect 2011; 17:1312–1322. [DOI] [PubMed] [Google Scholar]
- 7. Ferre C, Callaghan W, Olson C, Sharma A, Barfield W. Effects of maternal age and age-specific preterm birth rates on overall preterm birth rates - United States, 2007 and 2014. MMWR Morb Mortal Wkly Rep 2016; 65:1181–1184. [DOI] [PubMed] [Google Scholar]
- 8. Rogers LK, Velten M. Maternal inflammation, growth retardation, and preterm birth: insights into adult cardiovascular disease. Life Sci 2011; 89:417–421. [DOI] [PubMed] [Google Scholar]
- 9. Hirsch E, Saotome I, Hirsh D. A model of intrauterine infection and preterm delivery in mice. Am J Obstet Gynecol 1995; 172:1598–1603. [DOI] [PubMed] [Google Scholar]
- 10. Gravett MG, Witkin SS, Haluska GJ, Edwards JL, Cook MJ, Novy MJ. An experimental model for intraamniotic infection and preterm labor in rhesus monkeys. Am J Obstet Gynecol 1994; 171:1660–1667. [DOI] [PubMed] [Google Scholar]
- 11. Surve MV, Anil A, Kamath KG, Bhutda S, Sthanam LK, Pradhan A, Srivastava R, Basu B, Dutta S, Sen S, Modi D, Banerjee A. Membrane vesicles of group B Streptococcus disrupt feto-maternal barrier leading to preterm birth. PLoS Pathog 2016; 12:e1005816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elovitz MA, Wang Z, Chien EK, Rychlik DF, Phillippe M. A new model for inflammation-induced preterm birth. Am J Pathol 2003; 163:2103–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robertson SA, Skinner RJ, Care AS. Essential role for IL-10 in resistance to lipopolysaccharide-induced preterm labor in mice. J Immunol 2006; 177:4888–4896. [DOI] [PubMed] [Google Scholar]
- 14. Cappelletti M, Della Bella S, Ferrazzi E, Mavilio D, Divanovic S. Inflammation and preterm birth. J Leukoc Biol 2016; 99:67–78. [DOI] [PubMed] [Google Scholar]
- 15. Pollard JK, Mitchell MD. Intrauterine infection and the effects of inflammatory mediators on prostaglandin production by myometrial cells from pregnant women. Am J Obstet Gynecol 1996; 174:682–686. [DOI] [PubMed] [Google Scholar]
- 16. Holmgren C, Esplin MS, Hamblin S, Molenda M, Simonsen S, Silver R. Evaluation of the use of anti-TNF-alpha in an LPS-induced murine model. J Reprod Immunol 2008; 78:134–139. [DOI] [PubMed] [Google Scholar]
- 17. Wakabayashi A, Sawada K, Nakayama M, Toda A, Kimoto A, Mabuchi S, Kinose Y, Nakamura K, Takahashi K, Kurachi H, Kimura T. Targeting interleukin-6 receptor inhibits preterm delivery induced by inflammation. Mol Hum Reprod 2013; 19:718–726. [DOI] [PubMed] [Google Scholar]
- 18. Lin H, Mosmann TR, Guilbert L, Tuntipopipat S, Wegmann TG. Synthesis of T helper 2-type cytokines at the maternal-fetal interface. J Immunol 1993; 151:4562–4573. [PubMed] [Google Scholar]
- 19. Murphy SP, Fast LD, Hanna NN, Sharma S. Uterine NK cells mediate inflammation-induced fetal demise in IL-10-null mice. J Immunol 2005; 175:4084–4090. [DOI] [PubMed] [Google Scholar]
- 20. Murphy SP, Hanna NN, Fast LD, Shaw SK, Berg G, Padbury JF, Romero R, Sharma S. Evidence for participation of uterine natural killer cells in the mechanisms responsible for spontaneous preterm labor and delivery. Am J Obstet Gynecol 2009; 200:308.e1–308.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rinaldi SF, Catalano RD, Wade J, Rossi AG, Norman JE. Decidual neutrophil infiltration is not required for preterm birth in a mouse model of infection-induced preterm labor. J Immunol 2014; 192:2315–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edey LF, O’Dea KP, Herbert BR, Hua R, Waddington SN, MacIntyre DA, Bennett PR, Takata M, Johnson MR. The local and systemic immune response to intrauterine LPS in the prepartum mouse. Biol Reprod 2016; 95:125–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li LP, Fang YC, Dong GF, Lin Y, Saito S. Depletion of invariant NKT cells reduces inflammation-induced preterm delivery in mice. J Immunol 2012; 188:4681–4689. [DOI] [PubMed] [Google Scholar]
- 24. Lee AJ, Kandiah N, Karimi K, Clark DA, Ashkar AA. Interleukin-15 is required for maximal lipopolysaccharide-induced abortion. J Leukoc Biol 2013; 93:905–912. [DOI] [PubMed] [Google Scholar]
- 25. Lima PD, Tu MM, Rahim MM, Peng AR, Croy BA, Makrigiannis AP. Ly49 receptors activate angiogenic mouse DBA+ uterine natural killer cells. Cell Mol Immunol 2014; 11:467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hofmann AP, Gerber SA, Croy BA. Uterine natural killer cells pace early development of mouse decidua basalis. Mol Hum Reprod 2014; 20:66–76. [DOI] [PubMed] [Google Scholar]
- 27. Ashkar AA, Di Santo JP, Croy BA. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J Exp Med 2000; 192:259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Croy BA, Zhang J, Tayade C, Colucci F, Yadi H, Yamada AT. Analysis of uterine natural killer cells in mice. Methods Mol Biol 2010; 612:465–503. [DOI] [PubMed] [Google Scholar]
- 29. Zhang JH, Yamada AT, Croy BA. DBA-lectin reactivity defines natural killer cells that have homed to mouse decidua. Placenta 2009; 30:968–973. [DOI] [PubMed] [Google Scholar]
- 30. Chen Z, Zhang J, Hatta K, Lima PD, Yadi H, Colucci F, Yamada AT, Croy BA. DBA-lectin reactivity defines mouse uterine natural killer cell subsets with biased gene expression. Biol Reprod 2012; 87:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev 2013; 252:116–132. [DOI] [PubMed] [Google Scholar]
- 32. Doisne JM, Balmas E, Boulenouar S, Gaynor LM, Kieckbusch J, Gardner L, Hawkes DA, Barbara CF, Sharkey AM, Brady HJ, Brosens JJ, Moffett A et al. . Composition, development, and function of uterine innate lymphoid cells. J Immunol 2015; 195:3937–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vacca P, Montaldo E, Croxatto D, Loiacono F, Canegallo F, Venturini PL, Moretta L, Mingari MC. Identification of diverse innate lymphoid cells in human decidua. Mucosal Immunol 2015; 8:254–264. [DOI] [PubMed] [Google Scholar]
- 34. Male V, Hughes T, McClory S, Colucci F, Caligiuri MA, Moffett A. Immature NK cells, capable of producing IL-22, are present in human uterine mucosa. J Immunol 2010; 185:3913–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y, Xu B, Li MQ, Li DJ, Jin LP. IL-22 secreted by decidual stromal cells and NK cells promotes the survival of human trophoblasts. Int J Clin Exp Pathol 2013; 6:1781–1790. [PMC free article] [PubMed] [Google Scholar]
- 36. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol 2015; 33:747–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, Ivanov JA, Fu YY et al. . Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015; 528:560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McGee HM, Schmidt BA, Booth CJ, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA, Horsley V. IL-22 promotes fibroblast-mediated wound repair in the skin. J Invest Dermatol 2013; 133:1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Poulsen KP, Faith NG, Steinberg H, Czuprynski CJ. Bacterial load and inflammation in fetal tissues is not dependent on IL-17a or IL-22 in 10–14 day pregnant mice infected with Listeria monocytogenes. Microb Pathog 2013; 56:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Agrawal V, Smart K, Jilling T, Hirsch E. Surfactant protein (SP)-A suppresses preterm delivery and inflammation via TLR2. PLoS One 2013; 8:e63990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Agrawal V, Jaiswal MK, Chaturvedi MM, Tiwari DC, Jaiswal YK. ORIGINAL ARTICLE: Lipopolysaccharide alters the vaginal electrical resistance in cycling and pregnant mice. Am J Reprod Immunol 2009; 61:158–166. [DOI] [PubMed] [Google Scholar]
- 42. Zhang J, Chen Z, Smith GN, Croy BA. Natural killer cell-triggered vascular transformation: maternal care before birth? Cell Mol Immunol 2011; 8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Agrawal V, Jaiswal MK, Ilievski V, Beaman KD, Jilling T, Hirsch E. Platelet-activating factor: a role in preterm delivery and an essential interaction with toll-like receptor signaling in mice. Biol Reprod 2014; 91:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jaiswal MK, Agrawal V, Mallers T, Gilman-Sachs A, Hirsch E, Beaman KD. Regulation of apoptosis and innate immune stimuli in inflammation-induced preterm labor. J Immunol 2013; 191:5702–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharp AN, Heazell AE, Crocker IP, Mor G. Placental apoptosis in health and disease. Am J Reprod Immunol 2010; 64:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mori M, Bogdan A, Balassa T, Csabai T, Szekeres-Bartho J. The decidua-the maternal bed embracing the embryo-maintains the pregnancy. Semin Immunopathol 2016; 38:635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gibbs A, Leeansyah E, Introini A, Paquin-Proulx D, Hasselrot K, Andersson E, Broliden K, Sandberg JK, Tjernlund A. MAIT cells reside in the female genital mucosa and are biased towards IL-17 and IL-22 production in response to bacterial stimulation. Mucosal Immunol 2017; 10:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fumagalli S, Torri A, Papagna A, Citterio S, Mainoldi F, Foti M. IL-22 is rapidly induced by pathogen recognition receptors stimulation in bone-marrow-derived dendritic cells in the absence of IL-23. Sci Rep 2016; 6:33900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao H, Kalish F, Schulz S, Yang Y, Wong RJ, Stevenson DK. Unique roles of infiltrating myeloid cells in the murine uterus during early to midpregnancy. J Immunol 2015; 194:3713–3722. [DOI] [PubMed] [Google Scholar]
- 50. Duffin R, O’Connor RA, Crittenden S, Forster T, Yu C, Zheng X, Smyth D, Robb CT, Rossi F, Skouras C, Tang S, Richards J et al. . Prostaglandin E2 constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science 2016; 351:1333–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dumoutier L, de Heusch M, Orabona C, Satoh-Takayama N, Eberl G, Sirard JC, Di Santo JP, Renauld JC. IL-22 is produced by gammaC-independent CD25+ CCR6+ innate murine spleen cells upon inflammatory stimuli and contributes to LPS-induced lethality. Eur J Immunol 2011; 41:1075–1085. [DOI] [PubMed] [Google Scholar]
- 52. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol 2011; 12:383–390. [DOI] [PubMed] [Google Scholar]
- 53. Tachiiri A, Imamura R, Wang Y, Fukui M, Umemura M, Suda T. Genomic structure and inducible expression of the IL-22 receptor alpha chain in mice. Genes Immun 2003; 4:153–159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.