

SUMMARY
Long-term synaptic plasticity requires a mechanism that converts short Ca2+ pulses into persistent biochemical signaling to maintain the changes in the synaptic structure and function. Here, we present a novel mechanism of a positive feedback loop, formed by a “reciprocally activating kinase-effector complex” (RAKEC) in dendritic spines, enabling the persistence and confinement of a molecular memory. We found that stimulation of a single spine causes the rapid formation of a RAKEC consisting of CaMKII and Tiam1, a Rac-GEF. This interaction is mediated by a pseudo-autoinhibitory domain on Tiam1, which is homologous to the CaMKII autoinhibitory domain itself. Therefore, Tiam1 binding results in constitutive CaMKII activation, which in turn, persistently phosphorylates Tiam1. Phosphorylated Tiam1 promotes stable actin-polymerization through Rac1, thereby maintaining the structure of the spine during LTP. The RAKEC can store biochemical information in small subcellular compartments, thus potentially serving as a general mechanism for prolonged and compartmentalized signaling.
Keywords: reciprocally activating kinase-effector complex (RAKEC), pseudo-autoinhibitory domain, Rho family small GTPase, guanine-nucleotide exchange factor, Ca2+/calmodulin-dependent protein kinase II, actin cytoskeleton, long-term potentiation, synaptic plasticity, dendritic spine
eTOC Blurb
Saneyoshi et al. find that stimulation of single spine causes rapid formation of a reciprocally activating signaling complex between CaMKII and a Rac-GEF Tiam1, which stably activates Rac1 and maintains enlarged spine structure during LTP.
Introduction
Ever since long-term potentiation (LTP) of synaptic transmission was described in hippocampus (Nicoll, 2017, for review), the central question still remains, which is: how is a persistent molecular memory generated from a transient modulation of neuronal activity? Ca2+/calmodulin-dependent protein kinase II (CaMKII) has been proposed as a potential memory molecule because of its built-in self-activation mechanism. CaMKII is autophosphorylated at Thr 286 as a result of Ca2+ influx through the N-methyl-D-aspartic acid receptor (NMDAR), and as long as Thr 286 remains phosphorylated, CaMKII will remain in the “on” state, even in the absence of Ca2+ (Lisman et al., 2002; Hell, 2014). An in vitro reconstitution study indeed shows bistability of CaMKII (Urakubo et al., 2014), as predicted by the models (Zhabotinsky, 2000; Miller et al., 2005). However, recent imaging studies using a FRET sensor for CaMKII activity revealed that the bulk activity of CaMKII subsides within 1 min after the LTP-inducing stimulation (Takao et al., 2005; Lee et al., 2009). A photoactivatable CaMKII inhibitor paAIP2 is effective only when it is photoactivated during LTP induction but not during the maintenance, which is also consistent with the imaging study (Murakoshi et al., 2017). On the other hand, the increase in AMPA type glutamate receptor (AMPAR) transmission and structural enlargement of dendritic spine (structural LTP or sLTP) can be maintained more than one hour (Bosch and Hayashi, 2012; Nicoll, 2017). Thus, the mechanism that converts a transient Ca2+ signaling into persistent synaptic signaling during LTP has been a long-standing question.
One of the members of the Rho-family small G-protein, Rac1, is a major regulator of F-actin of particular interest in this context because modulation of its activity can alter the structure and function of excitatory synapses (Saneyoshi and Hayashi, 2012). Expression of a dominant negative form of Rac1 or shRNA against Rac1 causes spine shrinkage and elimination, whereas a constitutively active form of Rac1 leads to an increase in spine density and clustering of AMPAR (Tashiro et al., 2000; Wiens et al., 2005). Knocking out Rac1 impairs LTP and spatial learning (Haditsch et al., 2009). One of the final effectors of Rac1 is cofilin, which rapidly and persistently (õne hour) accumulates in spines after LTP induction and regulates the dynamics of actin (Bosch et al., 2014; Noguchi et al., 2016). Indeed, a study monitoring local F-actin/G-actin equilibrium using FRET revealed that the equilibrium moves towards F-actin upon LTP induction for more than 30 min (Okamoto et al., 2004). This shift in F-actin/G-actin equilibrium increases the local amount of F-actin and contributes to an increase in dendritic spine size and enhances the binding capacity to other postsynaptic proteins. Therefore, we deemed Rac1 to be a good candidate to mediate activity-dependent regulation of both the structure and function of excitatory synapses during LTP.
Rac1 is regulated by two major mechanisms (Saneyoshi and Hayashi, 2012). It is converted from an inactive GDP-bound form into an active GTP-bound form by guanine nucleotide exchange factors (GEFs) that stimulate the release of GDP to allow binding of GTP. GTP in the active form is hydrolysed into GDP by their intrinsic GTPase activity, which is enhanced by GTPase-activating proteins (GAPs), resulting in inactivation of the G-proteins. In particular, RacGEFs such as Tiam1 and Kalirin-7/Trio are substrates of CaMKII and act as the site of action where neuronal activity regulates actin cytoskeleton during spine formation and development (Tolias et al., 2005; Xie et al., 2007; Penzes et al., 2008; Herring and Nicoll, 2016). Therefore, RacGEFs are ideally situated as a mediator of Ca2+ signaling to regulate synaptic structure and function.
In the present study, we probed the mechanism underlying the conversion from transient Ca2+ influx to persistent modulation of F-actin via the Rac1 signaling pathway. We propose a concept of a “reciprocally activating kinase-effector complex” (RAKEC). The influx of Ca2+ triggers the formation of a RAKEC between a “pseudo-autoinhibitory” domain on Tiam1 and a binding pocket “T-site” on CaMKII, normally occupied by the autoinhibitory domain of the kinase. Tiam1 binding interrupts autoinhibition of CaMKII resulting in its persistent activity, which, in turn, causes a persistent phosphorylation of Tiam1 and therefore, an activation of Rac1 and downstream actin regulators. The RAKEC thus consists of a positive feedback loop, which enables the conversion of a transient Ca2+ signal triggered by the induction of LTP into a persistent kinase signal. RAKEC can store biochemical information in small subcellular compartments, thus potentially serving as a general mechanism for the production of prolonged and compartmentalized signaling.
RESULTS
Conversion point of temporary into persistent cellular signal is located between CaMKII and Rac1
We monitored Rac1 activity in dendritic spines of CA1 pyramidal neurons in hippocampal organotypic slice cultures using a FRET-fluorescence lifetime imaging microscopy (FLIM) sensor composed of GFP-fused Rac1 and mCherry-fused Pak2-Rac1/Cdc42 binding domain (with R71C, S78A mutations) (Hedrick et al., 2016). Consistent with the previous study, upon induction of structural LTP (sLTP) using uncaging of glutamate on single spines, we observed an activation of Rac1 that lasted more than 30 min (Fig. 1A-C). The activation was significantly less in adjacent spines (average distance 2.7 ± 0.3 µm) and not observed in the dendritic shaft. The change in lifetime was not observed in neurons expressing GFP-Rac1 alone (not shown).
Figure 1. Persistent Rac1 activation is required for sLTP in hippocampal organotypic slice culture.

A. Activation of Rac1 as visualized by FRET-FLIM-based Rac1 activity sensor during sLTP induced by uncaging caged glutamate at 0.5 Hz for 1 min. Rac1 activity and distribution of GFP-Rac1, as a proxy of structure, are shown. Warmer colour hues in FLIM images indicate higher Rac1 activity. Arrowhead indicates the position of photo-uncaging.
B. Averaged time course of Rac1 activation measured as a change in the lifetime of GFP-Rac1 from baseline in stimulated spines (stimulated, red), the dendritic shaft below the stimulated spines (dendrite, green), and adjacent spines (adjacent, yellow). Data from spines stimulated in the presence of CaMKII inhibitor KN-93 are also shown (blue). *, p < 0.05, compared to stimulated spines; one-way ANOVA with the Dunnett’s post-hoc test comparisons.
C. Averaged time course of GFP-Rac1 fluorescence intensity, measured as a proxy of volume change. *, p < 0.05, compared to stimulated spines; one-way ANOVA with the Dunnett’s post-hoc test comparisons.
D-E. Effect of Rac1 inhibitor EHT1864, applied 30 min before (blue) or 5 min after (green) (D), or 15 min after (purple) (E) induction of sLTP. Spines were visualized by expressing untagged GFP. *, p < 0.05, compared to control; ns, not significant; one-way ANOVA with the Dunnett’s post-hoc test comparisons.
Data are represented as mean ± SEM (B-E).
See also Figure S1.
This prompted us to test the requirement of persistent activity of Rac1 in sLTP using a Rac inhibitor (EHT1864), which inhibits GTP loading of Rac (Shutes et al., 2007) (Fig. 1D, E). Addition of this drug before sLTP induction effectively inhibited sLTP. When we added the drug either 5 or 15 min after the induction of sLTP, we found that it also effectively blocked sLTP, once established. These results indicate that the persistent activation of Rac1 by RacGEFs is indeed required for the maintenance of sLTP.
In order to examine the upstream signaling of the Rac1 activation, we used KN-93, an inhibitor of CaMKII that prevents the interaction of CaMKII with Ca2+/calmodulin (Fig. 1B, C). KN-93 also effectively blocked the activation of Rac1, indicating that Rac1 is downstream to CaMKII. Given that the activation of CaMKII returns to baseline levels within 1 min (Fig. S1A-C), as revealed by CaMKII FRET sensor Camui (Takao et al., 2005; Lee et al., 2009), the conversion of transient signal into persistent signal must occur between Rac1 and CaMKII.
Activation of CaMKII induces association with RacGEF Tiam1
GEFs are major players in the process of small G-protein activation. Certain RacGEFs such as Tiam1, Kalirin-7/Trio, and βPIX have been shown to be downstream of the NMDAR and CaMK signaling (Fleming et al., 1999; Tolias et al., 2005; Xie et al., 2007; Saneyoshi et al., 2008; Herring and Nicoll, 2016). We therefore tested involvement of these RacGEFs in sLTP by using specific shRNAs. All of these shRNAs reduced sLTP, though none of them completely abolished it (Fig. 2A, B. p < 0.05 for all shRNAs compared with luciferase shRNA). The efficacy of these three shRNAs was comparable as assessed by immunostaining (Fig. S1D, E).
Figure 2. Ca2+-dependent formation of a stable Tiam1/CaMKII complex.

A. Sample images of sLTP in neurons in hippocampal organotypic slice culture coexpressing GFP and shRNAs against luciferase (control), Tiam1, Kalirin-7 (Kal7), or βPIX.
B. Summary of the effect of shRNAs. Spine volume was measured by fluorescent intensity of untagged GFP. *, p < 0.05, compared to control; one-way ANOVA with the Dunnett’s post-hoc test comparisons. Data are represented as mean ± SEM.
C. Persistent interaction between Tiam1 and CaMKII but not with Kalirin-7 and βPIX. The Flagtagged RacGEF proteins were individually expressed in HEK293T cells. After lysing the cells, the RacGEFs were immunoprecipitated with Flag antibody and washed in the presence of Ca2+ (+) or absence (-, with EGTA). Endogenous CaMKII co-precipitated with RacGEF proteins were blotted against an anti-CaMKII antibody.
D. A similar experiment with CaMK family kinases. CaMKIα, CaMKIIα, CaMKIV, and CaMKKα tagged with Myc epitope were coexpressed in HEK293T cells with Tiam1-Flag. CaMKs were co-immunoprecipitated with Flag-antibody in the presence of Ca2+ and detected with Myc antibody.
Representative blots were shown from at least three independent experiments (C, and D).
See also Figure S1.
Interestingly, there was one difference among the three RacGEFs tested. When we compared the interaction between CaMKII and RacGEFs by co-immunoprecipitation, we found only Tiam1 formed a stable complex with CaMKII (Fig. 2C). Formation of this complex depends on the presence of Ca2+. Furthermore, once formed, the complex remained intact even when EGTA was used to chelate the Ca2+. In contrast, Kalirin-7 and βPIX did not form stable complexes that could be detected by co-immunoprecipitation. Other members of the CaMK family did not show such complex formation (Fig. 2D), indicating the specificity of this interaction. We also confirmed the interaction between endogenous Tiam1 and CaMKII in brain by coimmunoprecipitation (Fig. S2A).
We then investigated whether Tiam1 and CaMKII interact with each other in a single spine by sLTP induction by measuring FRET between a donor Tiam1-GFP and an acceptor mCherry-CaMKII-mCherry fusion protein using FLIM (Fig. 3A). Following the induction, there was a rapid increase in FRET in the stimulated spine, indicating that these two proteins interact with each other after LTP induction (Fig. 3B-D). The increase in interaction partially subsided after 10 min but a fraction persisted for more than 30 min. This is a stark contrast with the activation time course of CaMKII FRET sensor, Camui, which returns to the baseline within 1 min (Takao et al., 2005; Lee et al., 2009) (Fig. S1A-C). Importantly, the interaction between Tiam1 and CaMKII was blocked by bath application of NMDAR antagonist (AP5), KN-93, and cotransfection of endogenous CaMKII inhibitor protein (CaMKIIN) (Fig. 3C, D and S2B-D). These results indicate that the complex between Tiam1 and CaMKII is dependent on the influx of Ca2+ through the NMDAR and subsequent activation of CaMKII. By plotting the time course of activation, it is clear that the bulk of CaMKII is activated first, followed by the interaction between CaMKII and Tiam1, and finally Rac1 activation (Fig. 3E). This order of activation corroborates the biochemical cascade, indicating causality (Fig. 3F). It should be noted, however, we do not rule out the possibility that two other RacGEFs, Kalirin-7 and βPIX, are also involved in sLTP. They are indeed both situated downstream of CaMK signaling pathway and involved in activity-dependent spine morphogenesis and plasticity (Xie et al., 2007; Saneyoshi et al., 2008; Herring and Nicoll, 2016). Whether these molecules are involved in the maintenance phase of sLTP and if they do, how their signaling is maintained will be of particular interest.
Figure 3. The Tiam1/CaMKII interaction is persistent after sLTP induction in hippocampal organotypic slice culture.

A. FRET-FLIM probe used to detect the interaction between Tiam1 and CaMKII.
B. Interaction between Tiam1 and CaMKII as visualized by FRET-FLIM before and during sLTP (top). Warmer hues indicate more interaction. The distribution of Tiam1-GFP, as a proxy of structure, is shown (bottom).
C, D. Summary of Tiam1/CaMKII interaction (C) and volume of the spine as measured by Tiam1-GFP fluorescent intensity (D). *, p < 0.05, compared to the stimulated spines; one-way ANOVA with the Dunnett’s post-hoc test comparisons. Data are represented as mean ± SEM.
E. Comparison of the time course of CaMKII activation (Fig. S1), Tiam1/CaMKII interaction, and Rac1 activation (Fig. 1). The graphs are normalized by the largest value of lifetime change.
F. Schematic of the signaling cascade leading to LTP. T-shaped bar indicates inhibition. Red arrows indicate positive feedback loop.
See also Figure S2.
Tiam1 competes with the autoinhibitory domain of CaMKII for T-site
We then used Tiam1 deletion mutants to map the interaction region (Fig. 4A, B). We identified the region containing residues 1540–1591 at the carboxyl tail, a region without a known function, to be important for this interaction. Alanine scanning of three consecutive residues at a time further narrowed down the binding region to residues 1543–1557 (Fig. 4C). The sequence around this region is well conserved across vertebrate species (Fig. S3A). In order to obtain quantitative information on the interaction, we performed a fluorescence polarization assay with a fluorescently labelled Tiam1 peptide (residues 1540–1560) and the purified kinase domain of CaMKIIα (residues 6–274 with a kinase null D135N mutation). We found that the peptide and CaMKII directly interact with a KD of 8.1 ± 1.7 µM, which could be competed off with an excess amount of unlabelled peptide (Fig. 4E). Free fluorescein did not show specific binding (Fig. 4D).
Figure 4. Tiam1 interacts with CaMKII T-site.

A. Domain structure of Tiam1. The identified CaMKII binding domain (CBD) is located at the carboxyl tail of Tiam1. PH, pleckstrin-homology domain; CC, coiled-coil region; Ex, extra domain; RBD, Ras-binding domain; PDZ, PSD-95/Dlg/ZO1 domain; DH, Dbl-homology domain.
B. Mapping of the binding domain of Tiam1 by GST-pull-down (PD) assay. Myc-tagged CaMKII expressed in HEK293T cells were incubated with immobilized Tiam1 fragments in the presence or absence of Ca2+. Bound CaMKII was detected by western blotting with Myc antibody. Bars at the left side of bottom image show expected positions of the each Tiam1 fragment fused with GST except 1540–1591, which runs at the dye front.
C. Identification of CaMKII binding region in Tiam1 by alanine scanning. Full-length Tiam1 (FL), 1540–1591 fragment of either wild-type sequence (WT) or alanine mutants introduced in three consecutive residues were co-expressed with Myc-CaMKII in HEK293T cells. Cell lysates were immunoprecipitated with a Flag antibody in the presence of Ca2+. Immunoprecipitated proteins were detected by western blot using Myc and Flag antibodies.
D. Estimate of the binding affinity by fluorescence polarization. Fluorescein-labeled Tiam1 peptide corresponding to residues 1540–1560 (red) and bacterially expressed kinase domain of CaMKII 6–274 with kinase null D135N mutation were used. Fluorescein (green) was used as negative control. N=6 for peptide and 3 for fluorescein.
E. Fluorescence polarization assay with competing unlabeled peptide. The assay was the same as D except for the addition of unlabeled Tiam1 peptide (50 µM). Kinase was at 10 µM. N=12 for CaMKII/Fluorescein-labeled Tiam1 peptide; n=7 for CaMKII/Fluorescein-labeled Tiam1 peptide/unlabeled peptide; n=16 for fluorescein-labeled Tiam1 peptide. *, α = 0.001; ns, not significant; one-way ANOVA followed by the Tukey’s HSD post-hoc test.
F. Similarity between autoinhibitory domain of CaMKII (CaMK2A, residue number, 271–293; NM_171825) and the CaMKII binding domain of Tiam1 (1544–1565; NM_003253.2). Identical amino acids are shown in red and conservative residues were in orange. CBD is underlined.
G. Competition of Tiam1/CaMKII interaction by peptides known to bind either the S- or T-site of CaMKII. HEK293T cells were transfected with Flag-tagged Tiam1. Cell lysates were immunoprecipitated with Flag antibody in the presence of Ca2+ (+) or EGTA (−) with or without the peptide inhibitors (5 µM). AC3: autocamtide 3; AIP, autocamtide-2-related inhibitory peptide; KIINtide, CaMKIINtide; Scrambled, scrambled CBD. After washing, samples were eluted with SDS-PAGE sample buffer and immunoblotted with CaMKII and Flag antibodies.
H. Effects of T-site (F98K, E139R, I205K, or W237K), kinase-null (K42R), autophosphorylation deficient and mimic (T286A and T286D, respectively), and Ca2+/calmodulin binding deficient (T305D/T306D) mutations in CaMKII on interaction with Tiam1. The amount of immunoprecipitated CaMKII mutants was normalized by the total amount of the respective mutant. *, p < 0.05, **, < 0.01, compared to WT CaMKII; one-way ANOVA with the Dunnett’s post-hoc test comparisons.
Representative blots were shown from at least three independent experiments (B, C, and F). Data are represented as mean ± SEM (D, E, H).
See also Figure S3.
Upon inspection of the sequence of Tiam1 involved in CaMKII binding, we noticed that it has homology with the autoinhibitory domain of CaMKII, which normally prevents substrate binding by docking on a region called the “T-site” that is adjacent to the substrate binding “S-site” (Fig. 4F) (Lisman et al., 2002). Given the homology, we wondered if Tiam1 interacts with CaMKII at the T-site. To test this, we first used several peptides known to bind either S- or T-sites in order to compete with the Tiam1/CaMKII interaction (Fig. 4G). Syntide 2, an S-site binding peptide, did not interfere with the Tiam1/CaMKII interaction. In contrast, autocamtide-3 (AC3) and autocamtide 2-related inhibitory peptide (AIP), which mimic the autoinhibitory domain of CaMKII (Ishida and Fujisawa, 1995), and CaMKIIN, which is known to interact with the T-site (Vest et al., 2007), effectively blocked the Tiam1/CaMKII interaction. This indicates that Tiam1 interacts with CaMKII at the T-site in a manner competing with the autoinhibitory domain.
To gain more molecular insight, we modeled the Tiam1/CaMKII interaction using the crystal structure of CaMKIIN bound to CaMKII as a starting point (Fig. S3B-F) (Chao et al., 2010) and performed an energy minimization of the Tiam1 peptide. The resultant model shows that the basic residues (H1545, R1548 and K1554) interact with acidic residues on CaMKII. Also, hydrophobic residues (L1552 and L1558) fit into hydrophobic grooves in the kinase. These residues, which appear to be critical for binding, are largely consistent with the results from alanine scanning of CaMKIIN (Coultrap and Bayer, 2011). Consistent with this model, mutations in the T-site of CaMKII (F98K, E139R, I205K and W237K) (Fig. 4H) abolished the interaction with Tiam1.
We also carried out an additional mutagenesis to test how the interaction with Tiam1 is regulated (Fig. 4H). A CaMKII mutant deficient in Ca2+/calmodulin binding (T305D/T306D) and one deficient in kinase activity (K42R) both showed diminished binding to Tiam1. A constitutively active CaMKII mutant (T286D) showed increased binding whereas an autophosphorylation deficient mutant (T286A) and a double mutant (K42R/T286D) showed binding comparable to wild-type. The reduced binding in the T305D/T306D mutant confirmed that the T-site must first be exposed by Ca2+/calmodulin binding before Tiam1 can interact with CaMKII at the T-site. The enhanced binding in the T286D mutant indicates that an autophosphorylation at T286 facilitates Tiam1 binding. However, the observation that the T286A mutant showed comparable binding indicates T286 phosphorylation is not an absolute requirement. The observation that the catalytically inactive mutant (K42R) showed diminished binding suggests that there may be CaMKII phosphorylation sites on Tiam1 and/or nucleotide binding to CaMKII that facilitate binding (Coultrap and Bayer, 2011). In the K42R/T286D mutant, the effect of T286D was canceled out by the K42R mutation, and thus showed intermediate binding.
Reciprocal activation of CaMKII and Tiam1 in the complex
If Tiam1 competes with the autoinhibitory domain of CaMKII for the same T-site, the binding may modulate the activity of the kinase. To test the activity of CaMKII complexed with Tiam1, purified wild-type or T286A mutant CaMKII was mixed with a GST-Tiam1 1540–1591 fragment in the presence of Ca2+/calmodulin. ATP was omitted at this step to avoid inducing CaMKII autophosphorylation. Subsequently, Ca2+ was chelated using EGTA, ATP was added, and then the constitutive kinase activity was measured using Homer3 as a substrate (Fig. 5A, left) (Bayer et al., 2006; Kim et al., 2015). CaMKII without Tiam1 (GST only), did not show any activity. However, when complexed to Tiam1, CaMKII exhibited pronounced kinase activity leading to Homer3 phosphorylation. Importantly, this activity was independent of T286 autophosphorylation because the T286 phosphorylation was not detected under this condition as revealed by blotting with pT286 antibody. Also, the activity of Tiam1-bound T286A CaMKII mutant was comparable to wild-type in the presence of EGTA. In the presence of Ca2+/calmodulin, Homer3 was phosphorylated at a similar level under these conditions, indicating that each reaction has a comparable level of the enzyme (Fig. 5A, right). The interaction with purified full-length Tiam1 also induced Ca2+/calmodulin-independent activity of CaMKII (Fig. S4A). These results confirm that once the complex between Tiam1 and the T-site of CaMKII has formed, CaMKII maintains activity, even in the absence of Ca2+/calmodulin in a T286 phosphorylation independent manner. The T-site binding region of Tiam1 works as a pseudo-autoinhibitory domain to dislodge the autoinhibitory domain of CaMKII and, instead of inhibiting, maintains CaMKII in its active conformation.
Figure 5. RAKEC formation between CaMKII and Tiam1 is required for Tiam1 phosphorylation during LTP.

A. In vitro kinase assay of Tiam1-bound CaMKII. CaMKII was affinity-purified with a Flag antibody from HEK293T cells expressing Flag-tagged CaMKII and incubated with GST-Tiam1 fragment (residue 1540–1591) or GST in the presence of Ca2+/calmodulin to form a complex. Then EGTA was added to chelate Ca2+. ATP was omitted in these steps to avoid autophosphorylation. In vitro phosphorylation reaction was carried out in the absence (EGTA) or presence of Ca2+/calmodulin (Ca2+/CaM) using purified GST-Homer3 as a substrate after adding ATP. The reaction products were subjected to western blotting with antibodies against phosphorylated-Homer3 (pHomer3) (Mizutani et al., 2008), GST, T286 phosphorylated CaMKII (pT286 CaMKII), and CaMKII.
B. Activation of Tiam1 by CaMKII. Tiam1 was coexpressed with a constitutively active form of CaMKII (1–290) in HEK293T cells. Activated Rac1 was pulled down with Pak1-p21-bindingdomain, which specifically binds to the GTP-bound form of Rac1. *, p < 0.05, compared to nontransfected cells; t-test.
C. Persistent activation of Tiam1 RacGEF after chemical LTP induction in neurons in dissociated culture demonstrated by Rac1-G15A pull-down assay. Activated Tiam1 in the lysate of neurons underwent glycine stimulation and were subject to a pull-down assay using a nucleotide-free Rac1 G15A mutant, which selectively binds to activated RacGEFs. Bound Tiam1 was detected by western blotting with anti-Tiam1 antibody. *, p < 0.05, compared to non-stimulated neurons; Wilcoxon signed-rank test.
D. Phos-tag SDS-PAGE analysis of Tiam1 protein. Tiam1-Flag proteins were purified from transfected HEK293T cells with Flag-agarose and incubated with Mg2+/ATP, and 20 nM of CaMKII. The samples were treated with 10 U of λ phosphatase for 60 min at 30 °C. Samples were resolved either 7.5 µM Phos-tag SDS-PAGE (top) or conventional SDS-PAGE (bottom). Tiam1 and CaMKII proteins were detected by western blotting with anti-Tiam1 or CaMKII antibodies, respectively. Slowly migrating species represent phosphorylated proteins. The reason why shifted band does not form a discrete ladder is likely because of the existence of multiple phosphorylation sites on Tiam1.
E. Persistent phosphorylation of Tiam1 after chemical LTP induction in neurons in dissociated culture. Lysate from neurons which underwent chemical LTP induction with glycine for 10 min in the absence or presence of CaMKII inhibitor KN-93 (10 µM) were subject to Phos-tag SDS-PAGE to separate phosphorylated protein from unphosphorylated protein on a gel and then blotted with anti-Tiam1 antibody. Lower blot is a conventional SDS-PAGE. *, p < 0.05, **, p < 0.01, compared to control; one-way ANOVA followed by the Tukey’s HSD post-hoc test.
F. The persistent phosphorylation requires Tiam1/CaMKII complex formation. Flag-tagged Tiam1 (wild-type or AAA mutant) was introduced into neurons in dissociated culture using a lentiviral vector. Phosphorylated Tiam1 was detected similarly to E. except that anti-Flag antibody was used. *, p < 0.05, **, p < 0.01, compared to non-stimulated WT; one-way ANOVA with the Dunnett’s post-hoc test comparisons.
Representative blots are shown from at least three independent experiments in A and D. Data are represented as mean ± SEM (B, C, E, F).
See also Figure S4.
Finally, we confirmed that phosphorylation of Tiam1 by CaMKII activates its RacGEF activity by a pull-down assay with the Pak1-p21-binding-domain, which preferentially binds to active Rac1/Cdc42 (Fig. 5B). Indeed, Tiam1 phosphorylation by CaMKII significantly increased RacGEF activity, consistent with a previous study (Fleming et al., 1999). In sum, these two proteins form a reciprocally activating kinase-effector complex (RAKEC); Ca2+-dependent Tiam1 binding to T-site of CaMKII via its pseudo-autoinhibitory domain induces Ca2+-independent kinase activity, which, in turn, maintains Tiam1 phosphorylation and its RacGEF activity.
RAKEC formed between Tiam1 and CaMKII maintains RacGEF activity after chemical LTP induction by persistent phosphorylation
We next tested whether binding of Tiam1 with CaMKII contributes to the regulation of Tiam1 RacGEF activity in neurons. We induced chemical LTP in hippocampal dissociated culture preparations, which has been shown to increase AMPAR transmission as well as the volume of dendritic spines (Bosch et al., 2014). We found Tiam1 RacGEF activity was persistently increased after chemical LTP induction with 200 µM glycine for 10 min by using a Rac1 G15A pull-down assay, which preferentially isolates activated RacGEFs (Fig. 5C) (Garcia-Mata et al., 2006; Um et al., 2014). We then monitored Tiam1 phosphorylation under these conditions using Phos-tag SDS-PAGE, which allows separation of phosphorylated protein (slower migration) from unphosphorylated proteins (faster migration) on the gel (Fig. 5D). When phosphorylated Tiam1 samples were treated with λ phosphatase, the slowly migrating population was diminished, confirming that this population was indeed phosphorylated. Chemical LTP induction in the culture resulted in a persistent increase in the slowly migrating component, which was observed as a smeary pattern on the Phos-tag SDS-PAGE. This was blocked by CaMKII inhibitor KN-93 (Fig. 5E), which indicates that multiple sites on Tiam1 are phosphorylated by CaMKII. To test the contribution of the RAKEC formed between Tiam1 and CaMKII on Tiam1 phosphorylation in neurons, we created a Tiam1 mutant where three residues 1552–1554 (LKK) were replaced by alanine (Tiam1-AAA) to render it unable to bind CaMKII (Fig. 4C). The mutant showed a significant reduction in phosphorylation even though all phosphorylation sites were intact, indicating that the RAKEC formed between Tiam1 and CaMKII is required to maintain the phosphorylated state of Tiam1 (Fig. 5F). These results indicate that chemical induction of LTP induces the persistent phosphorylation of Tiam1 through the CaMKII/Tiam1 RAKEC even after the Ca2+ signal dissipates.
RAKEC formed between Tiam1 and CaMKII is required for sLTP
We were curious whether the RAKEC formed between the Tiam1 carboxyl tail and CaMKII T-site is necessary for sLTP. For this purpose, we first overexpressed a GFP-tagged Tiam1 WT or AAA mutant in neurons in slice culture and monitored sLTP as above. At the same time, we monitored Tiam1/CaMKII interaction by FRET-FLIM with mCherry-CaMKII-mCherry while measuring the volume change by using Tiam1-GFP fluorescence intensity as a proxy. We confirmed that CaMKII and Tiam1-AAA did not interact in this assay (Fig. 6A-D). Concomitantly, sLTP was severely diminished compared to the neurons expressing wild-type Tiam1. This indicates that the interaction between the carboxyl tail of Tiam1 and CaMKII is important for sLTP.
Figure 6. RAKEC formation between Tiam1 and CaMKII is required for persistence of sLTP and Rac1 activity in hippocampal organotypic slice culture.

A, B. Interaction of Tiam1 with CaMKII T-site is required for sLTP. Interaction between Tiam1 wild-type (A) or AAA mutant (B, residues 1552–1554 were replaced by alanine) and CaMKII were measured using a FRET-FLIM sensor for Tiam1/CaMKII interaction similarly to Figure 1, but by using slice kept in culture for two weeks. Only the early phase of sLTP was tested.
C, D. Summary of FRET change and volume (using amount of Tiam1-GFP as a proxy of structure, (as opposed to Figure 1D and E, where free GFP was used) from multiple spines. **, p < 0.01, compared to WT and AAA; t-test.
E. Activation of Rac1 as visualized by FRET-FLIM-based Rac1 activity sensor during sLTP in neurons coexpressing shRNA against Tiam1, without or with Tiam1 rescue constructs. of either wild-type or AAA mutant.
F, G. Averaged time course of Rac1 activation (F) and GFP-Rac1 fluorescent intensity change (G). Other conventions are similar to Figure. 1. *, p < 0.05, compared to parenthesized pair; ns, not significant; one-way ANOVA followed by the Tukey’s HSD post-hoc test.
Data are represented as mean ± SEM (C, D, F, G).
We then tested whether the persistent activation of Rac1 observed during sLTP actually requires the RAKEC formation between Tiam1 and CaMKII. We monitored Rac1 activity by FRET-FLIM in the presence of the Tiam1-AAA mutant to abolish complex formation while down-regulating endogenous Tiam1 with shRNA. The rescue constructs of both Tiam1 WT and AAA were expressed at a comparable level in neurons in slice culture (Fig. S4B, C). We found that Rac1 activation and sLTP were both significantly impaired (Fig. 6E). These results confirm that the RAKEC formed between Tiam1 and CaMKII is indeed required for persistent Rac1 activation and underlies sLTP.
A study using photoactivatable CaMKII inhibitor AIP (paAIP2) reported that photoactivation of paAIP2 failed to reverse established sLTP although it is effective in blocking sLTP if activated during the induction (Murakoshi et al., 2017). It was therefore concluded that there is no contribution of CaMKII activity in sLTP maintenance, which seemingly is contradictory to current results. CaMKII inhibitor peptide AIP and Tiam1 both interact with the T-site of CaMKII (Ishida and Fujisawa, 1995). Therefore, we hypothesized that Ca2+/calmodulin-activated versus Tiam1-activated CaMKII may have different sensitivities to AIP. To test this possibility, we monitored Tiam1/CaMKII interaction by FRET while we activated paAIP2. As a result, we found that Tiam1/CaMKII interaction was insensitive to paAIP2 (Fig. S5). In separate experiments, we confirmed that the activation of paAIP2 during glutamate uncaging did block sLTP (Fig. S5A, B). This indicates that the experiment using paAIP2 cannot rule out the possibility of persistent activation of CaMKII through a mechanism mediated by T-site. When the CaMKII T-site is already occupied by Tiam1, AIP needs to compete it off in order to bind. This likely reduces the potency of AIP and explains the inability of paAIP2 to block LTP during maintenance phase.
DISCUSSION
The mechanism of forming a persistent molecular memory from a transient Ca2+ signal has been a long-standing mystery. Here, we aimed to elucidate this mechanism and as a result we investigated the signaling pathways which lead to actin remodeling in the dendritic spine.
A synaptic Ca2+ signal is converted into RAKEC formation
We identified a novel cellular mechanism, RAKEC, which controls the spatiotemporal pattern of a specific kinase signaling mechanism (Fig. 7). We propose that the binding of a kinase to a class of effectors that harbour a specific sequence homologous to the autoinhibitory domain of the kinase (here termed pseudo-autoinhibitory domain) displaces the autoinhibitory domain to relieve the kinase from autoinhibition. Consequently, the activated kinase maintains the phosphorylated state of the effector. This creates a reciprocally activating positive feedback loop between the effector and the kinase, which significantly prolongs the duration of downstream cellular signaling. We demonstrated that the RacGEF Tiam1 maintains activity of CaMKII as well as its own phosphorylation status through this mechanism even after intracellular Ca2+ has diminished. This maintains Rac1 activity, F-actin organization, and eventually, the enlarged spine structure. This is the first experimentally identified positive feedback loop with an inherent bistability defined by bound/unbound status underlying LTP. Indeed, an early modeling study proposed that a positive feedback loop can sustain cellular signaling (Bhalla and Iyengar, 1999; Urakubo et al., 2014).
Figure 7. Schematic model of RAKEC between Tiam1 and CaMKII for persistence of sLTP.
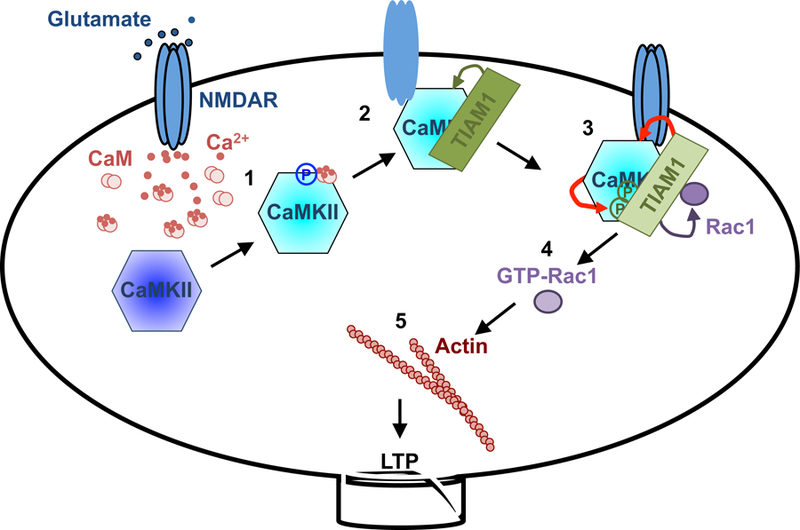
1. Ca2+ influx through NMDAR increases Ca2+/CaM binding to CaMKII, which activates the kinase and results in T286 autophosphorylation. Phosphorylated T286 disinhibits the kinase from autoinhibition.
2. Ca2+/CaM-activated CaMKII binds to Tiam1 and NR2B subunit of NMDAR. Pseudoautoinhibitory domain on Tiam1 dislodges the CaMKII autoinhibitory domain and prevents it from inhibiting the kinase.
3. In the protein complex with Tiam1, CaMKII phosphorylates and activates Tiam1 (RAKEC). This mechanism prolongs the kinase activity, which in turn maintains the phosphorylated status of Tiam1 and, therefore, its RacGEF activity.
4. Phosphorylated Tiam1 activates Rac1.
5. Rac1 promotes actin remodeling to maintain spine morphology.
Blue: inactive CaMKII, cyan: activated CaMKII, Red arrows: reciprocal activation.
The proposed RAKEC mechanism maintains the activity of CaMKII/Tiam1 complex during LTP maintenance. This is contradictory to the previous work using a CaMKII FRET biosensor, Camui, which demonstrated that CaMKII activity increases only transiently during LTP induction and returns to basal level during maintenance (Lee et al., 2009). We believe this is because of the sensitivity the Camui probe. There are ~2600 CaMKII dodecamers at single synapse, making CaMKII one of the most abundant postsynaptic proteins (Sheng and Hoogenraad, 2007; Feng et al., 2011). In contrast, most other signal transduction molecules such as Tiam1 are considerably less abundant (Sheng and Hoogenraad, 2007; Feng et al., 2011). Therefore, the proportion of CaMKII forming RAKEC with Tiam1 during LTP maintenance is expected to be small compared to the total amount of CaMKII. Additionally, biochemical studies detected basal CaMKII activity in unstimulated neuronal tissue (Barria et al., 1997; Yamagata et al., 2009). This background activity may mask the CaMKII that is activated by RAKEC mechanism in the Camui FRET measurement. Even though the FRET study shows CaMKII activity returns to the baseline level (Lee et al., 2009), the synaptic translocation of CaMKII (Shen and Meyer, 1999; Shen et al., 2000; Otmakhov et al., 2004; Bosch et al., 2014) would lead to a net increase in the number of active CaMKII molecules per synapse. This would account for the formation of the CaMKII RAKEC during LTP maintenance.
A report by Chang et al. (2017) showed that in T286A knock-in mice, LTP was not induced using a standard LTP induction protocol. But with a stronger stimulation protocol, both structural and functional LTP could be induced, proposing that the phosphorylation of CaMKII on T286 is not required for LTP for maintenance. We found that the CaMKII phospho-block T286A can still form RAKEC between CaMKII and Tiam1 while phospho-mimic T286D mutant enhances formation (Fig. 4H, 5A), indicating that the T286 phosphorylation is facilitative to the formation of RAKEC but not an absolute requirement. Therefore, it is possible that a certain proportion of CaMKII remains active through RAKEC mechanism even in T286A context and maintains LTP in phosphor-T286-independent manner.
In addition to temporal coding, we found that the Tiam1/CaMKII complex remained within the dendritic spine after sLTP inducing stimulation and did not diffuse to adjacent spines (Fig. 3B, C). One explanation for this is that the interaction of CaMKII with other postsynaptic molecules such as NR2B maintains the Tiam1/CaMKII complex at the stimulated spine (Bayer et al., 2001; Barria and Malinow, 2005). CaMKII is an oligomeric enzyme that has been shown as dodecameric or tetradecameric (Chao et al., 2011; Bhattacharyya et al., 2016). Therefore, a single CaMKII holoenzyme can simultaneously interact with NR2B and Tiam1. Since CaMKII is turned on downstream of NMDAR activation, this would ensure assembly of binding partners only at the activated synapse. Given that the T-site interactions of CaMKII are required to enhance synaptic transmission by constitutively active CaMKII (Incontro et al., 2018), the importance of the T-site interaction would not only be for a kinase activity but also for localization or forming RAKEC. Therefore, CaMKII may function as a hub for both structural and cellular signaling.
At this point, it is not clear how much we can generalize the mechanism. The expression of Tiam1 is widespread and especially high in dentate granule cells, pyriform cortex, olfactory tubercle, and cerebellar granule cells (Allen Brain Atlas). Therefore, it is intriguing to test the involvement of Tiam1 in these regions.
An atomic model of the interaction between CaMKII and Tiam1
At an atomic level, the interaction between CaMKII and Tiam1 was modeled using the X-ray crystal structure of a CaMKII kinase domain bound to CaMKIIN (Chao et al., 2010). Many of the residues in CaMKIIN important for the interaction are preserved in Tiam1 as well (Fig. S3B). Mutation of those residues and their counterpart on CaMKII abolishes the binding (Fig. 4C and 6C). However, it is curious that Tiam1 binding leads to activation while CaMKIIN binding causes inhibition. One possible explanation may be related to whether binding involves the S-site (substrate binding site, located next to the T-site) or not. Most of the residues conserved between Tiam1 and CaMKIIN are located at the core consensus and N-terminal side, which interacts with the T-site (Fig. S3B). There is less conservation at the C-terminal side of the binding region, which interacts with the S-site (Fig. S3B). Because of this, Tiam1 binding may not interfere with the ability of substrates to bind the S-site whereas CaMKIIN binding masks the S-site. Indeed, alanine substitutions on the C-terminal side of the binding region of Tiam1 did not affect binding to CaMKII (Fig. 4C), whereas a similar manipulation of CaMKIIN did diminish its inhibitory activity. Also, an S-site binding peptide, Syntide 2, did not affect Tiam1 binding (Fig. 4G). This difference in S-site interaction may explain the differential effects on kinase activity. Further studies to investigate this point will require more detailed atomic information of the interaction between Tiam1 and CaMKII.
Formation of RAKEC as a general mechanism to regulate the spatial-temporal pattern of the kinase activity
There are several known CaMKII T-site interacting proteins including NR2B, ether à-go-go (eag) potassium channel (in Drosophila), GJD2/connexin 36, LRRC7/densin-180 and CaMKIIN, in addition to Tiam1 (Bayer et al., 2001; Walikonis et al., 2001; Sun et al., 2004; Vest et al., 2007; Alev et al., 2008). Indeed, the carboxyl tail of NR2B locks CaMKII in an active conformation independently of T286 autophosphorylation, which is necessary for LTP (Bayer et al., 2001; Barria and Malinow, 2005; Halt et al., 2012). Furthermore, a mass spectrometric study found that CaMKII interacts with multiple proteins, some of which might be mediated by the T-site as well. We propose that these interactors may also form RAKECs when CaMKII activity is temporally and spatially modulated, which in turn would maintain their phosphorylation state and, therefore, the cellular signaling they mediate. This study thus demonstrates that RAKECs can store biochemical information in small subcellular compartments, and thus potentially serve as a mechanism to produce prolonged and compartmentalized signaling. Indeed, this revives an earlier view that CaMKII functions as a memory molecule (Lisman et al., 2002; Hell, 2014; Lisman, 2017).
Although Tiam1 specifically interacts with CaMKII but not with other members of the CaMK family (Fig. 2D), autoinhibition is a common regulatory mechanism characteristic of many kinase families (Soderling, 1990; Huse and Kuriyan, 2002; Pufall and Graves, 2002). It is therefore possible that RAKECs represent a general mechanism for the regulation of specific cellular signaling pathways. Only effectors that can relieve autoinhibition can maintain the activated state of the kinase, thereby attaining spatiotemporal specificity of signaling and substrate specificity at the same time. In this way, the mechanism presented here provides a novel way to regulate specific cellular signaling cascades with precise spatiotemporal patterns.
STAR Methods:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Takeo Saneyoshi (saneyoshi.takeo.3v@kyoto-u.ac.jp).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experimental protocols were approved by the RIKEN and Kyoto University Committee for Animal Care guidelines.
Animals
Rats from Sprague-Dawley strain (purchased Japan SLC, both males and females) were used for hippocampal slice culture and dissociated neuronal cultures.
Cell culture
HEK293T cells were cultured at 37 °C in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Transfections were performed using the PEI max (Polysciences, PA, USA). Hippocampal slices and dissociated neuronal cultured were prepared as described (Bosch et al., 2014). P6–8 and P0–1 Sprague-Dawley rats (both males and females) were used for slice culture and dissociated culture, respectively. Slices were transfected by a biolistic method (Gene-Gun, Bio-Rad, CA, USA). Dissociated neurons were infected using lentivirus vector (Bosch et al., 2014).
METHOD DETAILS
Reagents
KN-93, bicuculline, strychnine, D-(−)-2-Amino-5-phosphonopentanoic acid (AP5), and MNI-caged-L-glutamate were from Tocris Bioscience (Bristol, UK); TTX from Latoxan (Valence, France) or Wako (Osaka, Japan); Glutathione agarose (GST-Accept) from Nacalai Tesque (Kyoto, Japan); Phos-tag acrylamide from Wako; Syntide 2, AC3, AIP, and CaMKIINtide were from Merck-Millipore (Darmstadt, Germany). CaMKII-binding-peptide of Tiam1, RTLDSHASRMTQLKKQAALSG, and its randomized sequence, HLRMRLGSAKSQSTKLATADQ, fluorescein-RTLDSHASRMTQLKKQAA-amide were synthesized in the Research Resource Center of RIKEN Brain Science Institute. CaMKII (clone G1), phosphorylated T-286 CaMKII, Tiam1 (clones N15, C16), and Myc (clone 9E10) were purchased from Santa Cruz (TX, USA). CaMKII (clone 45) and Rac1 (clone 102) from BD Bioscience (NJ, USA). Anti-Tiam1 from sheep from R&D systems (MN, USA). Flag-M2 monoclonal antibody and Flag-M2-agarose from Sigma (MO, USA). Anti-MAP2 (HM2) and anti-Kalirin-7 antibodies from Novus Biologicals (CO, USA). GST antibody from Nacalai. Rabbit anti-phospho-S120 Homer3 antibody (Mizutani et al., 2008) was a kind gift from Dr. Katsuhiko Mikoshiba.
Plasmids
Tiam1 cDNA was isolated from mouse cDNA library. Rac1 biosensor was reported previously (Hedrick et al., 2016). For Tiam1 molecular replacement experiment (Fig. 6E-G), shRNA for Tiam1 was subcloned into FHUGW and GFP was replaced with mCherry-Pak2 Rac1/Cdc42 binding domain (RBD) (with R71C, S78A mutations)-mCherry. The shRNA sequences are follows: For Tiam1, GAGGGAGAAGGAAGTGGTC (Tolias et al., 2005), luciferase, GTACGCGGAATACTTCGA (Bosch et al., 2014), Kalirin-7, TACTTGAGTTGCAGACTTT (Ma et al., 2008), βPIX, GGTAGTACGAGCCAAGTTT (Saneyoshi et al., 2008). shRNAs were subcloned in pSuper (Oligoengine), pmU6pro, or FHUGW (Lois et al., 2002). For Tiam1 lentivirus production, shRNA for Tiam1 was subcloned into FHUGW, and GFP was replaced with shRNA-resistant Tiam1 wild-type or AAA mutant. Flag-tagged Kalirin-7 was generated from pEAK10-His-Myc-Kal7 (Addgene plasmid # 25454). Flag-tagged βPIX, CaMKIα, CaMKIIα, CaMKIV, and CaMKKα were described previously (Saneyoshi et al., 2008). CaMKII mutants, Tiam1 fragments and mutants were made by standard method.
Lenti-virus production
HEK293T cells were transfected with FHUGW plasmids along with pCAG_HIV_gp and pCMV_VSVG_RSV-Rev vectors using PEI method (Kim et al., 2015). Virus-containing culture supernatants were concentrated by ultracentrifugation. Virus solutions were aliquoted and kept at −80°C until use.
2-Photon laser-scanning microscopy and structural LTP induction
Imaging was carried out with a two-photon microscope (FV1000-MPE, Olympus, Tokyo, Japan) with Ti-sapphire lasers (Spectra-Physics, CA, USA). Fluorescent lifetime was measured as previously described (Bosch et al., 2014) using time-correlated photon-counting technology (SPC-830, Becker and Hickl, Berlin, Germany; H7422P-40, Hamamatsu Photonics, Hamamatsu, Japan) at 910 nm excitation. Detection was synchronized with excitation light pulses using an external detector. Emission light was filtered with a 680 nm short-pass and 510/70 nm band-pass filters. Bleed-through of the acceptor fluorescence into the emission channel was negligible. The lifetime images were analyzed using a custom written macro in Igor-Pro (Wavemetrics, OR, USA). Averaged fluorescence lifetime in the spine head was calculated and presented as the difference from baseline (Bosch et al., 2014). For experiments visualizing Rac1 activity, neurons were cultured in extra MgCl2 (final concentration 10.8 mM) on the day before imaging as previously described (Harvey et al., 2008).
Hippocampal organotypic slice cultures 5–12 days in culture (DIV) were transfected with plasmids at the following ratio; for Rac1 activity imaging, mEGFP-Rac1:mCherry-Pak2-RBD-mCherry at 1:2 ratio and for detection of Tiam1-CaMKII interaction, Tiam1-mEGFP:mCherry-CaMKIIα-mCherry at 1:4 ratio. All imaging experiments were carried out at 30 °C in Mg2+-free artificial cerebral spine fluid (ACFS) containing 4 mM CaCl2, 1 µM tetrodotoxin, 50 µM picrotoxin, and 2.5 mM MNI-caged-L-glutamate aerated with 95% O2 and 5% CO2. Imaging was performed at 8–16 DIV in primary or secondary dendrites from the distal part of the main apical dendrite of CA1 pyramidal neurons. We induced structural LTP on spines with a clear head and neck by uncaging MNI-glutamate with 2 to 6 ms laser pulses (720 nm at 5 mW under objective lens) repeated at 0.5 or 1 Hz for 30 pulses. The sLTP stimulation with 0.5 Hz for 30 pulses were used for Camui and Tiam1/CaMKII imaging in Fig. 3E, and Rac1 activity imaging. For other experiments, 1 Hz for 30 pulses was used to induce sLTP.
For photoactivation of paAIP2 experiment, we monitored structure with GFP fluorescence or Tiam1-GFP as a proxy. Images were taken at 1010 nm to avoid photoactivation of paAIP2. Photoactivation was applied using 473 nm laser (100 mW/cm2, Shanghai Laser, Shanghai, China) with 200 ms pulses at 1 Hz for 1 min.
Chemical LTP
Chemical LTP was performed as describe (Fortin et al., 2010; Bosch et al., 2014). Dissociated hippocampal neurons at 14 to 21 DIV were preincubated with HEPES-based ACSF (130 mM NaCl, 2.5 mM KCl, 0 mM Mg2+, 2 mM CaCl2, 25 mM HEPES [pH 7.3], 30 mM glucose, 310–320 mOsm/L), containing 1 µM tetrodotoxin for 30 min. Neurons were stimulated with ASCF supplemented with 200 µM glycine, 20 µM bicuculline, 3 µM strychnine for 10 min, then returned to ACSF without glycine.
Immunoprecipitation from HEK293T cells
HEK293T cell lysates were prepared in lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% TritonX-100, 10% glycerol, 1 mM Na3VO4, 10 mM NaF, 1 mM β-glycerophosphate, 1 x phosphatase inhibitor cocktail [Nacalai], 1 x complete tablet [Roche, Basel, Switzerland]) and centrifugation at 10,000 x g for 10 min at 4 °C. The supernatant was subjected to immunoprecipitation using 10 µL of the anti-Flag antibody-beads (Sigma) for 2–4 hours at 4 °C. Beads were washed with 1 mL of lysis buffer for three times. Bound proteins were eluted with SDS-PAGE sample buffer or Flag-peptide and subjected to western blotting.
CaMKII activity induced by T-site interaction
Flag-tagged human CaMKIIα wild-type and T286A were purified from HEK293T cell using FlagM2-agarose. After elution with Flag peptide, concentration of CaMKII was quantified by western blotting using purified CaMKIIβ protein as a standard (Kim et al., 2015). Kinase assay was performed according to Bayer et al. (2001). GST or GST-Tiam1–1540-1591 were preincubated for 12 min at 30 °C with CaMKII (100 nM CaMKII, 10 µM GST or GST-Tiam1–1540-1591 protein, 4.5 µM calmodulin, 10 mM PIPES pH 7.0, 0.1 mg/ml BSA, 0.25 mM CaCl2). EGTA was added to a final concentration of 2.5 mM, and samples were used for kinase reaction. Kinase reaction was performed for 30 min at 30 °C using 20 nM CaMKII from above reaction and 5 µg of GST-Homer3 as substrate (Mizutani et al., 2008), in a buffer containing 50 mM HEPES (pH 7.5), 10 mM MgCl2, 2.5 mM DTT, 0.005% Tween 20, 0.1 mg/ml BSA, 50 µM ATP, and either 1 mM CaCl2 and 1.5 µM calmodulin or 5 mM EGTA. The reaction was stopped by adding SDS-PAGE sample buffer.
GST-pull down assay
Ten µg of GST or GST-fusion proteins were immobilized on 25 µl of glutathione-agarose. HEK293T cell lysates were added to the beads, and incubated for 2 hours at 4 °C. After washing, bound proteins were extracted with SDS-PAGE sample buffer and detected by western blotting.
Pak-PBD pull-down assay
GST-Pak1-p21 binding domain (PBD) was prepared from bacteria as previously described (Saneyoshi et al., 2008). For Pak-PBD pull-down assay, HEK293T cells were detached by trypsin/EDTA (Nacalai) before cell lysis. Soluble cell lysate was prepared in 1 ml of BOS lysis buffer (1% NP40, 50 mM Tris-HCl [pH 7.5], 200 mM NaCl, 10 mM MgCl2, 10% glycerol, 1 x Complete EDTA-free tablet [Roche], 1 x phosphatase inhibitor cocktail [Nacalai]), supplemented with 40 µg/ml of GST-Pak1-PBD per 10-cm diameter culture dish (Corning, NY, USA). Soluble cell extracts were incubated with 25 µl of glutathione-agarose for 30 min at 4 °C, and washed three times with BOS lysis buffer. Bound activated endogenous Rac1 was detected by western blotting using Rac1 antibody.
Rac1G15A pull-down assay
GST-Rac1G15A protein was prepared from bacterial strain BL21(DE3)pLysS as previously described (Saneyoshi et al., 2008). After chemical LTP was induced, dissociated hippocampal lysates were prepared in deoxycholate lysis buffer (50 mM Tris-HCl [pH 8.5], 150 mM NaCl, 1% deoxycholate, 5 mM MgCl2, 10% Glycerol, 1 mM Na3VO4, 10 mM NaF, 1 mM β-glycerophosphate, 1 x phosphatase inhibitor cocktail [Nacalai], 1 x Complete tablet [Roche]), and centrifuged at 20,000 x g for 10 min at 4 °C. Soluble fractions were incubated with 10 µg of GST-Rac1G15A immobilized on glutathione-agarose for 2–4 hours at 4 °C and washed three times with deoxycholate lysis buffer. Bound activated Tiam1 was detected by western blotting using Tiam1 antibody.
Phos-tag SDS-PAGE
Five % acrylamide gel, 15 µM of MnCl2 and 7.5 µM of Phos-tag acrylamide (AAL-107, Wako) (Fig. 5D, E), or 5% acrylamide gel with 10 µM of MnCl2 and 7.5 µM Phos-tag acrylamide (Fig. 5F) were used (Hosokawa et al., 2015). The samples were separated by electrophoresis using the gels above. The gels were washed with 20 mM EDTA to remove Mn2+ before western blotting.
Purification of CaMKII kinase domain for fluorescence polarization assay
Human CaMKIIα kinase domain was expressed using E. coli, and prepared as described previously (Chao et al., 2010). Briefly, the cDNA fragment (amino acids 6–274 with D135N mutation) was subcloned in a pSMT-3 vector containing an N-terminal sumo expression tag (LifeSensors, Pennsylvania, PA) and protein expression was done in BL21(DE3)pLysS cells. Protein expression was induced by addition of 1 mM isopropyl β-D-1-thiogalactopyranoside and grown overnight at 18 °C. All subsequent purification steps were carried out at 4 °C. Cell pellets were resuspended in Buffer A (25 mM Tris, pH 8.5, 150 mM potassium chloride (KCl), 1 mM DTT, 50 mM imidazole, and 10% glycerol) and lysed using a cell disrupter. Cleared lysate was loaded on a 5 mL Ni-NTA column, eluted with 0.5 M imidazole, desalted using a HiPrep 26/10 desalting column into Buffer A with 0 mM imidazole and 2 mM tris(2-carboxyethyl)phosphine) (TCEP), and cleaved with Ulp1 protease overnight. The cleaved samples were loaded onto the Ni-NTA column and the flow through was loaded onto a Q-FF 5 mL column, and then eluted with a KCl gradient. Eluted proteins were concentrated and then further purified using a Superdex 200 gel-filtration column equilibrated in 25 mM Tris, pH 8.0, 150 mM KCl, 2 mM TCEP and 10% glycerol. Fractions with purified protein (>95 % pure) were frozen at −80 °C. All columns were purchased from GE Healthcare (Piscataway, NJ).
Fluorescence polarization
The fluorescence polarization experiments were carried out by adding 10 µl of 120 nM fluorescein-labeled Tiam1 peptides (fluorescein-RTLDSHASRMTQLKKQAA-amide dissolved in 25 mM Tris pH 7.5, 150 mM KCl, 0.02 % Tween, 0.02 % Triton) to 10 µl D135N CaMKII kinase domain at varying concentrations (25 mM Tris pH 7.5, 150 mM KCl, 10 % glycerol, 1 mM TCEP). A Synergy H1 hybrid plate reader (Biotek) was then used to measure the fluorescence polarization with a filter of 485/20 nm excitation and 528/20 nm emission. Error bars were calculated from the standard error of mean between replicates. Data were fit using a single-site binding equation (Bhattacharyya et al., 2016):
where,
[CaMKII]: concentration of monomeric kinase domain of CaMKII, varied from 0 to 7.5 × 10−5 M.
and
[peptide]: concentration of Tiam1 peptide, fixed at 6 × 10−8 M.
Modeling of Tiam1 peptide and CaMKII kinase domain
We used Coot (Emsley et al., 2010) to create the Tiam1 peptide using CaMKIIN as a model and performed a global energy minimization of the peptide itself. We then modeled the Tiam1 peptide onto the CaMKII kinase domain using the crystal structure of CaMKII bound to CaMKIIN as a starting point (Chao et al., 2010), The modeled peptide was docked onto the CaMKII kinase domain using PyMol, and side chain orientations were adjusted to mimic the CaMKIIN interaction mode.
QUANTIFICATION AND STASTICAL ANALYSIS
Quantification results were expressed as the mean ± S.E.M. The statistical significance of the results was assessed by analysis of variance (ANOVA) with post-hoc comparisons using the Tukey’s HSD test or Dunnett’s test, Wilcoxon signed-rank test, or two-tailed student’s t-test using Statcel2 for Excel (Microsoft) or KaleidaGraph (Synergy Software). For all tests of statistical significance, P < 0.05 was considered significant.
For spine imaging quantification, numbers of observation represent the spine number which was imaged from single neurons. In western blot analysis, number represents numbers of independent experiments.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Flag-M12 antibody | Sigma-Aldrich | Cat#. F1804 |
| Flag-M2 agarose | Sigma-Aldrich | Cat#. A2220 |
| Rabbit polyclonal anti-Tiam1 C16 | Santa-cruz biotechnology | Cat#. Sc-872 |
| Rabbit polyclonal anti-Tiam1 N15 | Santa-cruz biotechnology | Cat#. Sc-871 |
| Sheep polyclonal anti-Tiam1 | R&D systems | Cat#. AF-5038 |
| Mouse monoclonal anti-CaMKII (G-1) | Santa-cruz biotechnology | Cat#. Sc-5306 |
| Rabbit anti-phosphor-CaMKII T286 | Santa-cruz biotechnology | Cat#. Sc-12886 |
| Mouse monoclonal anti-Myc-9E10 antibody | Santa-cruz biotechnology | Cat#. Sc-40 |
| Mouse monoclonal anti-GST antibody (GS019) | Nacalai tesque | Cat#. 04435–26 |
| Mouse monoclonal anti-Rac1 antibody (clone 102) | BD biosciences | Cat#. 610651 |
| Mouse monoclonal anti-MAP2 antibody (HM2) | Novus Biologicals | Cat#. #NB120–11267 |
| Goat polyclonal anti-Kalirin-7 antibody | Novus Biologicals | Cat#. #NB100–41371 |
| Rabbit polyclonal anti-βPIX antibody | Cell Signaling technologies | Cat#. #4515S |
| Rabbit polyclonal anti-Phosphorylate Homer3 S120 | Mizutani et al. 2008 | N/A |
| Rabbit polyclonal anti- Phospho-(Ser/Thr) Akt Substrate Antibody | Cell Signaling technologies | Cat#. #9611S |
| Alexa647-goat anti-mouse IgG | Invitrogen | Cat#. A21236 |
| Alexa594-goat anti-rabbit IgG | Invitrogen | Cat#. A11037 |
| Alexa546-donkey anti-goat IgG | Invitrogen | Cat#. A11056 |
| Anti-mouse IgG HRP-linked whole Ab Sheep | GE healthcare | Cat#. NA931–1ML |
| Anti-rabbit IgG HRP-linked whole Ab donkey | GE healthcare | Cat#. NA934–1ML |
| Anti-sheep IgG HRP-linked | Invitrogen | Cat#. A16041 |
| Bacterial and Virus Strains | ||
| Lenti-FUGHW-shTiam1-Tiam1-Flag WT | This paper | N/A |
| Lenti-FUGHW-shTiam1-Tiam1-Flag AAA | This paper | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tetrodotoxin | Latoxan laboratory | Cat#. L8503 |
| Picrotoxin | Nacalai tesque | Cat#. 28004–71 |
| MNI-caged glutamate | Tocris | Cat#. 1490 |
| KN-93 | Tocris | Cat#. 1278 |
| EHT1864 | Tocris | Cat#. 3872 |
| AP-5 | Tocris | Cat#. 0106 |
| Phostag acrylamide | FUJIFILM Wako Pure Chemical Corporation | Cat#. AAL-107 |
| PEI max | Polyscience, Inc | Cat#. 24765–1 |
| Tiam1 CBD: RTLDSHASRMTQLKKQAALSG |
This paper | N/A |
| Tiam1 CBD scrambled: HLRMRLGSAKSQSTKLATADQ |
This paper | N/A |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human: HEK293T cell | ATCC | CRL3216 |
| Experimental Models: Organisms/Strains | ||
| Rat; SD | SLC | |
| Oligonucleotides | ||
| Recombinant DNA | ||
| pmGFP-Rac1 | Hedrick et al., 2016 | N/A |
| pmCherry-PBD-mCherry | Hedrick et al., 2016 | N/A |
| pSuper-shTiam1 | Tolias et al., 2005 | N/A |
| pSuper-shKalirin-7 | Ma et al., 2008 | N/A |
| pmU6pro-sh-betaPIX | Saneyoshi et al., 2008 | N/A |
| pCAGGS-mGFP | This paper | N/A |
| pCAGGS-Tiam1-Flag | This paper | N/A |
| pCAGGS-Kalirin-7-Flag | This paper | Addgene plasmid#25454 |
| pCAGGS-betaPIX-Flag | Saneyoshi et al., 2008 | N/A |
| pCAGGS-Myc-CaMKI-alpha | Saneyoshi et al., 2008 | N/A |
| pCAGGS-Myc-CaMKII-alpha | Saneyoshi et al., 2008 | N/A |
| pCAGGS-Myc-CaMKIV | Saneyoshi et al., 2008 | N/A |
| pCAGGS-Myc-CaMKK-alpha | Saneyoshi et al., 2008 | N/A |
| pCAGGS-mCherry-CaMKII-alpha-mCherry | This paper | N/A |
| pCAGGS-Tiam1-EGFP | This paper | N/A |
| pCS2-Flag-Tiam1 1540–1591 | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc wild-type | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc F98K | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc E139R | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc I205K | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc W237K | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc K42R | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc T286A | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc T286D | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc K42R,T286D | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc T305DT306D | This paper | N/A |
| pCAGGS-CaMKII-alpha-myc 1–290 | This paper | N/A |
| pFHUGW-shLuc-LacZ-Flag | This paper | N/A |
| pFHUGW-shTiam1-LacZ-Flag | This paper | N/A |
| pFHUGW-shTiam1-Tiam1-Myc WT | This paper | N/A |
| pFHUGW-shTiam1-Tiam1-Myc AAA | This paper | N/A |
| p-mRFP1CaMKII-alpha-mGFP (camui) | Kwok et al., 2008 | N/A |
| pJPA-CaMKIIN | Chang et al., 1998 | N/A |
| pmCherry-PA-AIP2 | Murakoshi et al., 2016 | N/A |
| GST | pGEX4T1, GE healthcare | Cat#. 28954549 |
| GST-Tiam1 421–1591 | This paper | N/A |
| GST-Tiam1 421–852 | This paper | N/A |
| GST-Tiam1 840–1025 | This paper | N/A |
| GST-Tiam1 1025–1591 | This paper | N/A |
| GST-Tiam1 1540–1591 | This paper | N/A |
| Flag peptide | Sigma-Aldrich | Cat#. F3290 |
| Syntide2 | Calbiochem | Cat#. 05–23-4910 |
| AC3 | Anaspec Inc. | Cat#. 64927 |
| AIP | Anaspec Inc. | Cat#. 64926 |
| CaMKIINtide | Calbiochem | Cat#. 208920 |
| CBD | This paper | N/A |
| CBD scrambled | This paper | N/A |
| GST-Pak1RBD | Saneyoshi et al. 2008 | N/A |
| GST-Rac1 G15A | This paper | N/A |
| Lambda protein phosphatese | NEB | P0753S |
| GST-AC2 | This paper | N/A |
| GST-Homer3 | Mizutani et al. 2008 | N/A |
| Software and Algorithms | ||
| Igor Pro | Bosch et al., 2014 | https://www.wavemetrics.com/ |
| Statcel2 | OMS publishing Inc. | |
| KaleidaGraph | This paper | http://www.synergy.com/wordpress_650164087/kaleidagraph/ |
| ImageJ | Bosch et al., 2014 | https://imagej.nih.gov/nih-image/ |
| Other | ||
Highlights.
Persistent Rac1 activity is required for structural LTP of dendritic spines
CaMKII stably binds on pseudo-autoinhibitory domain of Tiam1, a Rac-GEF
This binding disinhibits autoinhibition of CaMKII and maintains its activity
Persistently activated CaMKII phosphorylates Tiam1 and maintains Rac1 activity
ACKNOWLEDGMENTS
We would like to dedicate this work to the memory of John Lisman (1944–2017), who has always inspired us. We thank Drs. Karl Ulrich Bayer, Morgane Rosendale, and Lily Yu for comments on the manuscript. We also thank Drs. Mikoshiba and Eipper for providing anti-phospho-Homer3 antibody and Kalirin-7 construct, respectively. This work was supported by RIKEN, RIKEN Presidents Fund, NIH grant R01DA17310, Grant-in-Aid for Scientific Research (A, 20240032, 16H02455) and for Scientific Research on Innovative Area “Foundation of Synapse and Neurocircuit Pathology” (22110006) and “Constructive understanding of multi-scale dynamism of neuropsychiatric disorders” (18H04733) from the MEXT, Japan, Human Frontier Science Programme, The Uehara Memorial Foundation, The Naito Foundation, The Takeda Science Foundation, Japan Foundation for Applied Enzymology, Novartis Foundation (Japan) for the Promotion of Science, Research Foundation for Opto-Science and Technology, Brain Science Foundation, and Guangdong Key International Visiting Program, China (Y.H.); Grant-in-Aid for Young Scientist (A, 854508), for Scientific Research (B, 1031691), and for Scientific Research on Innovative Area “Multi-dimensional fluorescence live imaging of cellular functions and molecular activities (374539, 817144)” from the MEXT, Japan, The Kyoto University Foundation, The Shimadzu Science Foundation, The Takeda Science Foundation, and The Pharmacological Research Foundation, Tokyo (T.S.); NIH grant DP1-NS096787, R01-MH080047 (R.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Y.H. is partly supported by Takeda Pharmaceutical Co. Ltd., Fujitsu Laboratories, and Dwango.
REFERENCES
- Alev C, Urschel S, Sonntag S, Zoidl G, Fort AG, Hoher T, Matsubara M, Willecke K, Spray DC, and Dermietzel R (2008). The neuronal connexin36 interacts with and is phosphorylated by CaMKII in a way similar to CaMKII interaction with glutamate receptors. Proc Natl Acad Sci USA 105, 20964–20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, and Malinow R (2005). NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, and Soderling TR (1997). Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276, 2042–2045. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, and Schulman H (2001). Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805. [DOI] [PubMed] [Google Scholar]
- Bayer KU, LeBel E, McDonald GL, O’Leary H, Schulman H, and De Koninck P (2006). Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci 26, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla US, and Iyengar R (1999). Emergent properties of networks of biological signaling pathways. Science 283, 381–387. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya M, Stratton MM, Going CC, McSpadden ED, Huang Y, Susa AC, Elleman A, Cao YM, Pappireddi N, Burkhardt P, et al. (2016). Molecular mechanism of activation-triggered subunit exchange in Ca2+/calmodulin-dependent protein kinase II. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, and Hayashi Y (2014). Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82, 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, and Hayashi Y (2012). Structural plasticity of dendritic spines. Curr Opin Neurobiol 22, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Parra-Bueno P, Laviv T, Szatmari EM, Lee SR, and Yasuda R (2017). CaMKII autophosphorylation is necessary for optimal integration of Ca2+ signals during LTP induction, but not maintenance. Neuron 94, 800–808 e804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LH, Pellicena P, Deindl S, Barclay LA, Schulman H, and Kuriyan J (2010). Intersubunit capture of regulatory segments is a component of cooperative CaMKII activation. Nat Struct Mol Biol 17, 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LH, Stratton MM, Lee IH, Rosenberg OS, Levitz J, Mandell DJ, Kortemme T, Groves JT, Schulman H, and Kuriyan J (2011). A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin- dependent kinase II holoenzyme. Cell 146, 732–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, and Bayer KU (2011). Improving a natural CaMKII inhibitor by random and rational design. PLoS One 6, e25245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Raghavachari S, and Lisman J (2011). Quantitative estimates of the cytoplasmic, PSD, and NMDAR-bound pools of CaMKII in dendritic spines. Brain Res 1419, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming IN, Elliott CM, Buchanan FG, Downes CP, and Exton JH (1999). Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J Biol Chem 274, 12753–12758. [DOI] [PubMed] [Google Scholar]
- Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, and Soderling TR (2010). Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci 30, 11565–11575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, and Burridge K (2006). Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol 406, 425–437. [DOI] [PubMed] [Google Scholar]
- Haditsch U, Leone DP, Farinelli M, Chrostek-Grashoff A, Brakebusch C, Mansuy IM, McConnell SK, and Palmer TD (2009). A central role for the small GTPase Rac1 in hippocampal plasticity and spatial learning and memory. Mol Cell Neurosci 41, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, and Hell JW (2012). CaMKII binding to GluN2B is critical during memory consolidation. EMBO J 31, 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Yasuda R, Zhong H, and Svoboda K (2008). The spread of Ras activity triggered by activation of a single dendritic spine. Science 321, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick NG, Harward SC, Hall CE, Murakoshi H, McNamara JO, and Yasuda R (2016). Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 538, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW (2014). CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81, 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE, and Nicoll RA (2016). Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc Natl Acad Sci USA 113, 2264–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa T, Mitsushima D, Kaneko R, and Hayashi Y (2015). Stoichiometry and phosphoisotypes of hippocampal AMPA type glutamate receptor phosphorylation. Neuron 85, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, and Kuriyan J (2002). The conformational plasticity of protein kinases. Cell 109, 275–282. [DOI] [PubMed] [Google Scholar]
- Incontro S, Diaz-Alonso J, Iafrati J, Vieira M, Asensio CS, Sohal VS, Roche KW, Bender KJ, and Nicoll RA (2018). The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase-dependent and independent mechanisms. Nature communications 9, 2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida A, and Fujisawa H (1995). Stabilization of calmodulin-dependent protein kinase II through the autoinhibitory domain. J Biol Chem 270, 2163–2170. [DOI] [PubMed] [Google Scholar]
- Kim K, Lakhanpal G, Lu HE, Khan M, Suzuki A, Kato-Hayashi M, Narayanan R, Luyben TT, Matsuda T, Nagai T, et al. (2015). A temporary gating of actin remodeling during synaptic plasticity consists of the interplay between the kinase and structural functions of CaMKII. Neuron 87, 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, and Yasuda R (2009). Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J (2017). Criteria for identifying the molecular basis of the engram (CaMKII, PKMzeta). Mol Brain 10, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, and Cline H (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3, 175–190. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, and Baltimore D (2002). Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872. [DOI] [PubMed] [Google Scholar]
- Ma XM, Wang Y, Ferraro F, Mains RE, and Eipper BA (2008). Kalirin-7 is an essential component of both shaft and spine excitatory synapses in hippocampal interneurons. J Neurosci 28, 711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller P, Zhabotinsky AM, Lisman JE, and Wang XJ (2005). The stability of a stochastic CaMKII switch: dependence on the number of enzyme molecules and protein turnover. PLoS Biol 3, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani A, Kuroda Y, Futatsugi A, Furuichi T, and Mikoshiba K (2008). Phosphorylation of Homer3 by calcium/calmodulin-dependent kinase II regulates a coupling state of its target molecules in Purkinje cells. J Neurosci 28, 5369–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Shin ME, Parra-Bueno P, Szatmari EM, Shibata AC, and Yasuda R (2017). Kinetics of endogenous CaMKII required for synaptic plasticity revealed by optogenetic kinase inhibitor. Neuron 94, 37–47 e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA (2017). A Brief History of Long-Term Potentiation. Neuron 93, 281–290. [DOI] [PubMed] [Google Scholar]
- Noguchi J, Hayama T, Watanabe S, Ucar H, Yagishita S, Takahashi N, and Kasai H (2016). State-dependent diffusion of actin-depolymerizing factor/cofilin underlies the enlargement and shrinkage of dendritic spines. Scientific reports 6, 32897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, and Hayashi Y (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7, 1104–1112. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Tao-Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, and Lisman J (2004). Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci 24, 9324–9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, and Srivastava DP (2008). Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol 18, 405–413. [DOI] [PubMed] [Google Scholar]
- Pufall MA, and Graves BJ (2002). Autoinhibitory domains: modular effectors of cellular regulation. Annu Rev Cell Dev Biol 18, 421–462. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, and Hayashi Y (2012). The Ca2+ and Rho GTPase signaling pathways underlying activity-dependent actin remodeling at dendritic spines. Cytoskeleton (Hoboken) 69, 545–554. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, and Soderling TR (2008). Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/βPIX signaling complex. Neuron 57, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, and Meyer T (1999). Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284, 162–166. [DOI] [PubMed] [Google Scholar]
- Shen L, Liang F, Walensky LD, and Huganir RL (2000). Regulation of AMPA receptor GluR1 subunit surface expression by a 4. 1N-linked actin cytoskeletal association. J Neurosci 20, 7932–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, and Hoogenraad CC (2007). The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 76, 823–847. [DOI] [PubMed] [Google Scholar]
- Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, and Der CJ (2007). Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem 282, 35666–35678. [DOI] [PubMed] [Google Scholar]
- Soderling TR (1990). Protein kinases. Regulation by autoinhibitory domains. J Biol Chem 265, 1823–1826. [PubMed] [Google Scholar]
- Sun XX, Hodge JJ, Zhou Y, Nguyen M, and Griffith LC (2004). The eag potassium channel binds and locally activates calcium/calmodulin-dependent protein kinase II. J Biol Chem 279, 10206–10214. [DOI] [PubMed] [Google Scholar]
- Takao K, Okamoto K, Nakagawa T, Neve RL, Nagai T, Miyawaki A, Hashikawa T, Kobayashi S, and Hayashi Y (2005). Visualization of synaptic Ca2+/calmodulindependent protein kinase II activity in living neurons. J Neurosci 25, 3107–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Minden A, and Yuste R (2000). Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex 10, 927–938. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, and Greenberg ME (2005). The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 45, 525–538. [DOI] [PubMed] [Google Scholar]
- Um K, Niu S, Duman JG, Cheng JX, Tu YK, Schwechter B, Liu F, Hiles L, Narayanan AS, Ash RT, et al. (2014). Dynamic control of excitatory synapse development by a Rac1 GEF/GAP regulatory complex. Dev Cell 29, 701–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakubo H, Sato M, Ishii S, and Kuroda S (2014). In vitro reconstitution of a CaMKII memory switch by an NMDA receptor-derived peptide. Biophys J 106, 1414–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, Davies KD, O’Leary H, Port JD, and Bayer KU (2007). Dual mechanism of a natural CaMKII inhibitor. Mol Biol Cell 18, 5024–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walikonis RS, Oguni A, Khorosheva EM, Jeng CJ, Asuncion FJ, and Kennedy MB (2001). Densin-180 forms a ternary complex with the α-subunit of Ca2+/calmodulin-dependent protein kinase II and α-actinin. J Neurosci 21, 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens KM, Lin H, and Liao D (2005). Rac1 induces the clustering of AMPA receptors during spinogenesis. J Neurosci 25, 10627–10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, and Penzes P (2007). Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 56, 640–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, Watanabe M, Hatanaka N, Totsuka M, Yagi T, et al. (2009). Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIα in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci 29, 7607–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhabotinsky AM (2000). Bistability in the Ca2+/calmodulin-dependent protein kinase-phosphatase system. Biophys J 79, 2211–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.