

Abstract
Objective
To assess potential mechanisms of cortical superficial siderosis (cSS), a central MRI biomarker in cerebral amyloid angiopathy (CAA), we performed a collaborative meta-analysis of APOE associations with cSS presence and severity.
Methods
We pooled data from published studies reporting APOE genotype and MRI assessment of cSS in 3 distinct settings: (1) stroke clinic patients with symptomatic CAA (i.e., lobar intracerebral hemorrhage, transient focal neurologic episodes) according to the Boston criteria; (2) memory clinic patients; and (3) population-based studies. We compared cSS presence and severity (focal or disseminated vs no cSS) in participants with ε2+ or ε4+ genotype vs the ε3/ε3 genotype, by calculating study-specific and random effects pooled, unadjusted odds ratios (ORs).
Results
Thirteen studies fulfilled inclusion criteria: 7 memory clinic cohorts (n = 2,587), 5 symptomatic CAA cohorts (n = 402), and 1 population-based study (n = 1,379). There was no significant overall association between APOE ε4+ and cSS presence or severity. When stratified by clinical setting, APOE ε4+ was associated with cSS in memory clinic (OR 2.10; 95% confidence interval [CI] 1.11–3.99) but not symptomatic CAA patients. The pooled OR showed significantly increased odds of having cSS for APOE ε2+ genotypes (OR 2.42, 95% CI 1.48–3.95) in both patient populations. This association was stronger for disseminated cSS in symptomatic CAA cohorts. In detailed subgroup analyses, APOE ε2/ε2 and APOE ε2/ε4 genotypes were most consistently and strongly associated with cSS presence and severity.
Conclusion
CAA-related vasculopathic changes and fragility associated with APOE ε2+ allele might have a biologically meaningful role in the pathophysiology and severity of cSS.
Cortical superficial siderosis (cSS) is detected as curvilinear hypointensities following the cortical surface on blood-sensitive T2*-weighted gradient-recalled echo (T2*-GRE) and susceptibility-weighted imaging (SWI) MRI sequences.1 It is generally thought that cSS reflects deposits of blood breakdown products in the outermost cortical layers from, often occult, convexal subarachnoid hemorrhage.1,2 cSS is particularly common in advanced cerebral amyloid angiopathy (CAA) (prevalence 40%–60%),1,3,4 a small vessel disease that results from β-amyloid (Aβ) deposition in cortical and leptomeningeal arterioles. In patients with CAA, cSS seems to be consistently associated with increased risk of incident5 and recurrent6 lobar intracerebral hemorrhage (ICH), including early recurrence,7 as well as future dementia.8 cSS is hence now considered a third cardinal hemorrhagic signature of CAA,1,9 alongside multiple strictly lobar cerebral microbleeds and lobar ICH. It is also included in the modified Boston criteria, as a specific MRI biomarker of the disease.2
The emerging clinical relevance of cSS, either as direct contributor to CAA-related impairment or a biomarker of the disease's presence, severity, and course, raises questions about the mechanisms of this imaging lesion. However, data from neuropathologic studies remain extremely limited. Understanding the underlying mechanisms and vascular pathology contributing to cSS could be facilitated by identifying associations with APOE alleles. Associations between APOE ε2 or ε4 alleles with both lobar ICH risk and CAA presence and severity on neuropathology have been previously described. In fact, APOE genotype seems to be the single most important genetic determinant of CAA pathophysiology identified to date.10,11 The current hypothesis, albeit supported by limited data, is that APOE ε4 enhances vascular Aβ deposition in a dose-dependent fashion,12 while APOE ε2 promotes so-called CAA-related vasculopathic changes (vessel cracking, detachment and delamination of the outermost layer of the tunica media, and fibrinoid necrosis) that can lead to vessel rupture.13 It is hypothesized that cSS results from bleeding-prone leptomeningeal or superficial cortical arterioles that harbor advanced CAA and associated vasculopathic changes.1
To gain further insights into potential mechanisms of cSS in CAA and small vessel disease, we performed a collaborative meta-analysis of all available published studies that provided APOE data according to cSS presence and severity. Since cSS appears to convey high risk of recurrent ICH, it might segregate with APOE genotypes that are associated with CAA-related vasculopathic changes. Hence, we specifically tested the hypothesis that cSS presence and severity is associated with APOE ε2, a marker of CAA-related small vessel fragility.
Methods
Standard protocol approvals, registrations, and patient consents
The study was performed according to a predefined protocol (i.e., before collecting and analyzing data) designed in-house and finalized in January 2016. This report was prepared with reference to Preferred Reporting Items for Systematic Reviews and Meta-Analyses,14 Meta-analysis of Observational Studies in Epidemiology15 guidelines, and the Cochrane Handbook for Systematic Reviews of Interventions.
Study identification and selection criteria
We sought all studies of adult humans published in any language that reported APOE genotype data and had cSS assessment on MRI, regardless of whether any association between the 2 was reported. We searched PubMed and Embase (from inception to January 2016 and updated in November 2017) using a combination of keyword search and MeSH terms, i.e. (“cortical superficial siderosis” or “convexity siderosis” or “convexal siderosis” or “cortical hemosiderosis” or siderosis or hemosiderosis) and (APOE or “apolipoprotein E”). We also screened the references lists of all relevant studies and reviews identified, and searched Google Scholar for other studies citing potentially eligible relevant studies. We included relevant studies with >20 participants, including studies that recruited individuals from 3 distinct settings: (1) patients presenting to stroke clinics with symptomatic sporadic CAA (i.e., lobar ICH, transient focal neurological episodes) according to the validated classic Boston criteria (i.e., cSS was not part of CAA diagnosis); (2) memory clinic patients; and (3) participants from population-based studies. The rationale for including individuals from these different clinical settings was twofold. First, they represent the most likely clinical scenarios in which cSS in small vessel disease is detected on MRI (1 and 2) and are of potential clinical significance. Second, they capture the spectrum of cardinal CAA phenotypes: relatively “pure” stroke presentations (including lobar ICH), cognitive impairment/dementia, or incidental findings in elderly healthy populations.
We excluded case reports, small case series, and studies including hereditary/familial forms of CAA. Two authors independently selected eligible studies, resolving disagreements by discussion. When 2 or more studies with overlapping cohorts existed, we included only the study providing the most data about the association and the largest number of participants.
Data extraction
Eligible studies were classified according to the primary clinical setting as (1) symptomatic CAA; (2) memory clinics; or (3) population-based. For each included study, we extracted information using standard pro formas on publication year, country in which the study was conducted, study design, participant source, and baseline demographic/clinical characteristics.
For our planned meta-analyses, we extracted, or required from authors, summary-level data on numbers of participants with each APOE genotype (i.e., ε3ε3, ε2ε3, ε3ε4, ε4ε4, ε2ε2, ε2ε4) according to cSS presence and severity (focal or disseminated) where available. A structured data extraction form was created and completed as far as possible by entering data from the relevant publications or circulated to authors and a collaborative group was established. We also extracted information on the MRI sequence characteristics used for cSS detection and the rating methods used for cSS classification.
Quality and risk of bias assessment
We assessed each study against a list of quality criteria we devised based on study size, cohort recruitment method (prospective vs other), blinding of cSS ratings and APOE genotype data, quality of genotyping, blood-sensitive MRI sequence type used, criteria of cSS assessment, and inter-rater agreement. These criteria were created using elements with reference to the Strengthening the Reporting of Genetic Association Studies (STREGA),16 Meta-analysis of Observational Studies in Epidemiology (MOOSE)15 recommendations, and consensus standards for cSS assessment and rating.1
Statistical analysis and synthesis
We performed meta-analyses using Stata 13.0 (StataCorp LP, College Station, TX) and considered a p value of <0.05 to imply statistical significance. In our primary analyses, we calculated study-specific and random effects pooled, unadjusted odds ratios (ORs) for cSS presence vs absence among ε4 allele carriers (ε4+) vs the reference genotype ε3ε3 and among ε2 carriers (ε2+) vs the wild type ε3ε3. This comparison was selected to avoid potential confounding by mixed effects of ε2 and ε4 in the comparison group. In secondary analyses, we compared cSS severity (focal vs no cSS and disseminated vs no cSS) in participants with an ε2+ or ε4+ genotype vs the ε3/ε3 genotype. In analyses looking at cSS severity (i.e., focal or disseminated cSS), the comparison groups included only patients without cSS. Meta-analyses were performed both separately by study setting/population and overall. In all analyses, we used a random effects model with DerSimonian-Laird weights,17 using ORs and their corresponding 95% confidence intervals (CIs), with the inverse variance method for weighting. We assessed statistical heterogeneity using I2 statistics and visually through inspection of the forest plot. Values of ≤25%, 25%–50%, and ≥50% were defined as low, moderate, and high degrees of heterogeneity, respectively. We explored publication bias with funnel plots. As a subanalysis, and to reveal potential dose–effect relationships, we have also explored the association between all different APOE genotypes (i.e., ε2ε3, ε3ε4, ε4ε4, ε2ε2, ε2ε4 vs ε3ε3) and cSS presence and severity in the whole population and across different clinical settings. To assess robustness of the methods, we repeated all the above analyses using the fixed effects method.
Data availability statement
All relevant data and methods are reported in the article. No data are available in public repositories.
Results
Characteristics and quality of included studies
From 33 publications identified in our literature search, we identified 13 relevant studies fulfilling our inclusion criteria and pooled in meta-analyses (figure 1). Study populations comprised 7 memory clinic cohorts (n = 2,587), 5 symptomatic nonoverlapping CAA cohorts (n = 402), and 1 population-based study including healthy people (n = 1,379) (table 1). The memory clinic studies had different inclusion criteria and dementia prevalence (table 1). The symptomatic CAA studies included 2 cohorts presenting with stroke syndromes other than ICH, 1 with pure CAA-ICH, and 2 with both CAA-ICH and CAA non-ICH presentations. Four out of these 5 CAA cohorts were derived from different studies completed at the same center,4,18–20 but included largely nonoverlapping patient cohorts (i.e., different clinical settings/recruitment, clinical presentation, inclusion criteria, inception). In detail (see footnote in table 1), 1 of these 4 single-center cohorts,4 an advanced research MRI study of CAA-related ICH, included around 10% of overlapping patients with a separate consecutive clinical ICH cohort,19 from which CAA-related lobar ICH were included in our analysis, based on our best estimates. This latter clinical CAA cohort19 might have slightly overlapped (∼10%) with a pathology-based cohort of CAA patients.20 Mean age was between 63 and 75 years and about half of all participants were male. Most studies were conducted in predominantly white populations in centers from Europe, the United States, or Australia, while 2 studies were conducted in Asia. The distribution of the ethnicity of participants in each study was not available.
Figure 1. Flow chart of study identification and selection.
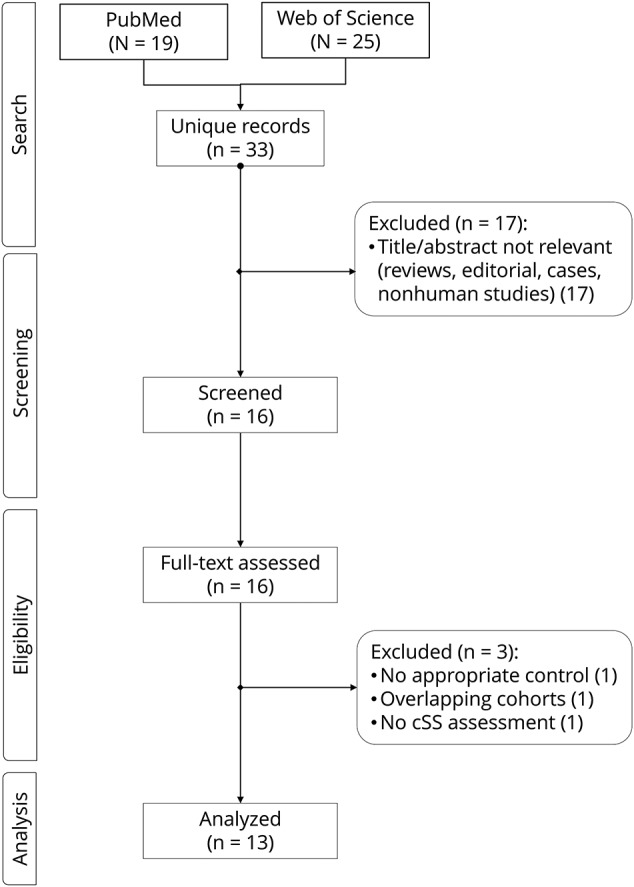
cSS = cortical superficial siderosis.
Table 1.
Basic characteristics and methodologic aspects of included studies


There was variation in overall study quality (table 2), including sample size and retrospective vs prospective designs. The genotyping quality was generally good when assessed against current reporting standards. Studies varied in the type of blood-sensitive sequences used for cSS detection (e.g., T2*-GRE vs SWI), as well as MRI field strength, with all cohorts on symptomatic CAA patient populations using 1.5T MRI, while memory clinic studies were performed at 3T MRI. The methods of cSS assessment were reliable and largely in line with current consensus recommendations in the field.1 For details on quality assessment and individual scores of included studies in the meta-analysis, see table 2.
Table 2.
Key quality indicators of pooled studies

Pooled prevalence and severity of cSS in included studies
The pooled prevalence of cSS presence was 2% (95% CI 2%–3%, I2 59%, p = 0.02) in memory clinic patients, 47% (95% CI 38%–56%, I2 66%, p = 0.02) in symptomatic CAA patients, and 1% (95% CI 1%–2%) in the single population-based study included in our analysis. The overall pooled prevalence of focal cSS in memory clinic vs symptomatic CAA patients was 1% (95% CI 1%–2%, I2 48%, p = 0.07) and 17% (95% CI 13%–22%, I2 24%, p = 0.26), respectively. The overall pooled prevalence of disseminated cSS was 1% (95% CI 0%–1%, I2 9%, p = 0.36) vs 28% (95% CI 20%–36%, I2 60%, p = 0.04) in memory clinic vs symptomatic CAA cohorts, respectively. In all comparisons, the prevalence of cSS (presence and severity) was higher in symptomatic CAA vs memory clinic patients (p < 0.0001). Among patients with any cSS, the prevalence of focal cSS was 67% (95% CI 55%–79%, I2 17%, p = 0.30) in memory clinic patients and 37% (95% CI 26%–49%, I2 60%, p = 0.04) in symptomatic CAA cases (p < 0.001 between the 2 groups).
Meta-analyses: APOE ε4 and ε2 and cSS presence and severity
The results of the main analyses of the association between APOE ε4 and ε2 with cSS presence are summarized in figure 2. Compared to participants with an APOE ε3/ε3 genotype, pooled overall results showed no increased odds of cSS presence in participants with APOE ε4+ genotype (figure 2A). When stratified by clinical subgroups, APOE ε4+ genotype was associated with cSS presence in memory clinic patients, but not symptomatic CAA patients (figure 2A). The pooled OR showed increased odds of having cSS for APOE ε2+ genotypes (OR 2.42, 95% CI 1.48–3.95) with no statistical heterogeneity between study results (figure 2B). This association was strong in both memory clinic and symptomatic CAA cohorts (figure 2B).
Figure 2. Forest plots: cSS presence and APOE e4+ or APOE e2+ vs APOE e3/e3.

Forest plots of the association between cortical superficial siderosis (cSS) presence and (A) APOE ε4+ vs APOE ε3/ε3 genotype and (B) APOE ε2+ vs APOE ε3/ε3 genotype.Meta-analyses were performed using a random effects model. Studies are displayed in order of publication date. The squares represent study-specific odds ratios (ORs), with their size proportional to their statistical weight. Diamonds represent pooled ORs and their 95% confidence intervals (CI), stratified by clinical setting and overall. CAA = cerebral amyloid angiopathy.
All clinical studies provided relevant data for meta-analyses of APOE genotypes and cSS severity (focal or disseminated). These data were not available to pool in the single population-based study that reported on APOE and cSS. Overall, pooled results showed that, compared to patients with an ε3/ε3 genotype, those with an ε4+ genotype did not have increased odds of having either focal or disseminated cSS (figure 3A). Memory clinic patients showed only a nonsignificant trend for an association with APOE ε4+ genotype and cSS burden (figure 3A). Overall, patients with APOE ε2+ genotype had increased odds for an association with disseminated, but not focal, cSS (figure 3B). When stratified by clinical setting, focal cSS was associated with APOE ε2+ genotype in memory clinic patients, and showed a strong trend with disseminated cSS in this population (figure 3B). In symptomatic CAA cohorts, only disseminated cSS was associated with APOE ε2+ genotype. There was no evidence of publication bias and statistical heterogeneity was low to moderate across analyses (data provided in each forest plot).
Figure 3. Subanalysis summary results.

(A) Subanalysis summary results of the effect of APOE ε4+ vs APOE ε3/ε3 genotype on the presence of focal and disseminated cortical superficial siderosis (cSS), according to clinical setting and overall.(B) Subanalysis summary results of the effect of APOE ε2+ vs APOE ε3/ε3 genotype on the presence of focal and disseminated cSS, according to clinical setting and overall. The comparison groups included only patients without cSS. The columns on the right of the forest plots denote the number of participants included in each analysis. Diamonds represent the overall pooled odds ratios (ORs) and their 95% confidence interval (CI) for each comparison. Study-specific ORs are not shown. CAA = cerebral amyloid angiopathy.
Subgroup meta-analyses: Different APOE genotypes and cSS presence and severity
The detailed results of subanalyses exploring the association between all different APOE genotypes (vs ε3/ε3) and cSS presence and burden are summarized in table 3. In the overall analysis of all the cohorts together, the most consistent associations with higher effect sizes were seen with APOE ε2/ε2 and APOE ε2/ε4 genotypes (table 3). The associations were different when stratified by the clinical setting. In memory clinic cohorts, cSS presence and severity was also associated with APOE ε4/ε4, while the stronger link was with the APOE ε2/ε4 genotype (table 3). Among symptomatic CAA cohorts, only APOE ε2/ε3 and APOE ε2/ε4 were associated with disseminated cSS (table 3). Of note, the APOE ε4/ε4 genotype was associated with marginally lower odds of having disseminated cSS in symptomatic CAA patients (OR 0.24, 95% CI 0.06–0.92, p = 0.038, see table 3). The statistical heterogeneity for these subanalyses ranged from low to moderate and high (table 3).
Table 3.
Detailed meta-analysis results for the association between different APOE genotypes (vs ε3/ε3) and cSS presence and burden


Discussion
The current meta-analysis provides a comprehensive assessment on cSS,1 CAA,29 and their association with APOE genotype. Drawing data from >4,000 participants of relevant studies and different clinical setting, our main results indicated that cSS, especially disseminated cSS, is most strongly associated with APOE ε2+ genotype. This overall association was consistent and did not vary significantly according to clinical setting. We found that APOE ε4+ genotype was overall not associated with cSS presence or burden. However, APOE ε4+ genotype results varied depending on the clinical setting, with memory clinic patients (but not symptomatic ICH patients) showing an association with cSS, albeit weaker compared to APOE ε2+ genotype based on the unadjusted pooled ORs (2.10 vs 3.28, respectively). In the detailed subanalyses looking at individual APOE genotypes and cSS burden, the most consistent associations with higher effect sizes were seen with the ε2/ε2 and ε2/ε4 genotypes.
Although it can be challenging to infer specific pathophysiologic mechanisms from genetic associations, the most straightforward and parsimonious explanation for our results is that cSS is indeed a strong and specific MRI biomarker for more advanced or active CAA. These results confirm our prespecified hypothesis, and are consistent with prior observations in the field, with what is presumed to be the effect of APOE ε2 vs ε4 on underlying CAA-related vasculopathic changes, and with the emerging clinical relevance of cSS as an independent risk factor for future symptomatic ICH. APOE genotype is the single most important genetic determinant in CAA pathophysiology.10,11 APOE ε4 appears to enhance vascular Aβ deposition in a dose-dependent fashion,12 while APOE ε2 promotes vasculopathic changes (vessel cracking, vessel-within-vessel appearance, and fibrinoid necrosis), which can lead to vessel rupture.13 A previous meta-analysis investigating APOE associations with cerebral microbleeds found that strictly lobar microbleeds (a putative marker of CAA presence) was related to APOE ε4+ allele (OR 1.35, 95% CI 1.10–1.66, p = 0.005), but not APOE ε2.30 This result was consistent for any cerebral microbleeds presence in a more recent comprehensive meta-analysis.31 The dissociation of the relationship between APOE genotype and cerebral microbleeds vs cSS has implications for discerning potential mechanisms. APOE ε4 might predispose to the particular kind of CAA-related vessel thickening postulated to lead to microscopic intraparenchymal hemorrhage (e.g., strictly lobar cerebral microbleeds).32 In contrast, APOE ε2 might promote the most severe stages or aggressive phenotype of CAA pathology that precede rupture, especially in leptomeningeal vessels, hence contributing to MRI-visible superficial CAA-related bleeds (aka cSS) in multiple spatially separated foci.4,20 A recent neuropathologic study used a detailed grading system for assessing CAA in parenchymal and leptomeningeal vessels separately.33 In a sample of Alzheimer disease (AD) and elderly control brains without dementia, APOE ε2 was a much stronger risk factor for CAA development, especially in leptomeningeal vessels, compared to APOE ε4 (OR 10.93, 95% CI 4.33–27.57 vs 1.69, 0.92–3.10, respectively).33 Fittingly, an inverse relationship has been observed between greater lobar cerebral microbleed counts and cSS in advanced CAA, suggesting the possibility of differing CAA phenotypes that are driven in part by APOE genotype, and marked by the predominance of either cSS or cerebral microbleed MRI patterns.4,30
These inferences would also be in line with several clinical observations in the field. APOE ε2 is already known to be associated with CAA-related ICH, perhaps causally,34 also predisposing to larger volumes of CAA-related bleeding.35 cSS is independently associated with future ICH risk in CAA, both recurrent6,36 and first lobar ICH.5 In fact, clinical cohorts that incorporated and investigated both cSS and strictly lobar microbleeds in relation to future ICH demonstrated that cSS, but not microbleeds, is the strongest independent risk factor for CAA–ICH.5,6,36 Also, there is preliminary evidence that APOE ε4 may be associated with CAA type 1 (where CAA is found in cortical capillaries) and APOE ε2 with CAA type 2 (where cerebrovascular amyloid is primarily deposited in leptomeningeal and cortical vessels sparing cortical capillaries).37 A recent neuropathologic study that validated a detailed CAA grading system showed that all individuals with the APOE ε2/ε2 genotype had CAA type 2, while the APOE ε4/ε4 genotype was associated with CAA type 1 (OR 8.0, 95% CI 2.8–23.3).33 Finally, though the exact pathophysiologic mechanisms underlying cSS remain debatable,1 MRI-detected cSS seems to reflect repeated episodes of superficial bleeding from CAA-laden bleeding-prone leptomeningeal vessels.1 Thus cSS may be a strong marker of not only more severe CAA, but CAA with more fragile, rupture-prone vessels, thereby heralding a risk of subsequent ICH.6,38 APOE ε2–driven vascular injury in CAA, in combination with other risk factors, could influence pathways providing initiation sites for cSS and hence future lobar ICH risk.
Our study benefited from thorough ascertainment of the totality of evidence to date on the topic, within the 2 clinical settings in which CAA is most commonly considered. The first setting consists of patients diagnosed in stroke clinics with relatively advanced symptomatic CAA, while memory clinic patients and healthy elderly are heterogeneous participants who mostly do not have advanced CAA (instead, mild to moderate CAA commonly accompanies Alzheimer neurodegenerative pathology in memory clinic patients). This fact explains the much higher proportion of cSS in the symptomatic CAA group and might partly account for the different relationship with APOE ε4. We note the higher frequency of APOE ε4 in patients from memory clinics, at least in part, due to the relationship between this genotype and its well-known predisposition to AD (instead, APOE ε2 variant confers reduced risk for AD and thus has lower frequency in memory clinics).39 Similar to AD, as noted above, histopathologic studies in CAA indicate that APOE ε4 has an equivalent role in CAA by promoting vascular Aβ deposition; APOE ε2, however, has a different effect—it increases vessel wall damage caused by cerebrovascular amyloid deposition. In other words, the hypothesis is that APOE ε4 increases the likelihood of having CAA among memory clinic patients (identified clinically by the presence of cSS). Among symptomatic CAA patients with already advanced (or enriched in) underlying cerebrovascular amyloid deposition, it does not increase the likelihood of cSS—in this group, APOE ε2 comes into play (especially in a dose-depended and synergistic fashion with APOE ε4 gene).
Our findings in the subanalyses of detailed APOE genotypes and associations with cSS presence and burden probably account for the prevalence and differential effects of ε4 and ε2 in AD and CAA. However, these subanalyses need to be interpreted with caution, under the prism of the following additional considerations. The relatively small sample size contributing to each subanalysis (e.g., according to clinical setting, focal or disseminated cSS) and rarity of certain alleles and genotypes resulted in wide 95% CIs. Due to the increased number of subanalyses, we run the risk of multiple comparisons. Hence, they should be considered hypothesis-generating, highlighting possible trends, effect sizes, and potential mechanistic pathways to be explored in further studies on cSS. It is important to again point out that the associations between cSS presence/burden and specific APOE genotypes require, to some extent, different interpretation in memory clinic vs symptomatic CAA cohorts. In memory clinic patients, APOE genotype–cSS associations are driven by the presence of substantial underlying CAA pathology that is denoted by cSS. In other words, cSS in memory clinic cohorts identifies patients with advanced CAA (i.e., beyond mild CAA, often a common “innocent bystander” in this setting)—the known risk factor of originally developing cerebrovascular Aβ accumulation is APOE ε4 and ε2. Symptomatic CAA cohorts are by definition enriched with cerebrovascular Aβ pathology and hence APOE–cSS associations are driven/indicating more specific (or predominant) pathophysiologic mechanisms at play, especially within the most severe cases of cSS (i.e., disseminated cSS). It is possible that the APOE ε2/ε4 genotype may represent double hit for the superficial vessels, promoting not only amyloid deposition but also vessel wall cracking in the most vulnerable arterioles in CAA. In the setting of advanced cerebrovascular Aβ pathology, it is not surprising that only this double hit genotype of APOE ε2/APOE ε4 shows a strong link with disseminated cSS. These hypotheses require external validation and direct support from experimental studies. Future studies should also investigate the effect of APOE genotypes on cSS progression and interactions with future CAA-related ICH risk.
Some limitations of our study need to be acknowledged. The design of included studies, case selection, and MRI measures for cSS detection were variable. Patients from symptomatic CAA cohorts underwent brain MRI at I.5T, other cohorts at 3T. While the consisted MRI field strength within clinical setting subgroups is reassuring, in the overall analyses, the difference could influence the sensitivity for cSS detection and rating. In the same vein, blood-sensitive sequences used for cSS detection were also variable across studies, including both T2*-GRE and SWI. Though the variable MRI measures is a potentially important limitation for this sort of meta-analysis, no data exist on how sensitivity for cSS classification is affected by different MRI sequences. Considering that cSS represents a much higher volume of blood-breakdown products (i.e., hemosiderin) compared for example to cerebral microbleeds, the differences in MRI sensitivity might not be very pronounced. None of the included studies fulfilled all our methodologic quality indicators. There are likely a number of studies that could not be included in the current meta-analysis simply because they did not report on either APOE or cSS, reflecting the fact that cSS is a relatively new addition in the spectrum of CAA MRI markers. This raises the issue of potential confounding and selection bias, which is difficult to address. It should be emphasized that, despite including all available data from relevant publications, the overall sample size for certain subgroup meta-analyses was relatively small. This limits the precision of the pooled results, especially in combination with the low prevalence of cSS in the memory clinic studies. In a memory clinic setting, one should recognize the known relative rarity of APOE ε2 genotypes and the confounding effect of the presence of dementia and Alzheimer type pathology. While CAA and AD are linked in the context of cognitive impairment populations, their precise relationship remains poorly understood.40 This and other considerations could partly explain the weak association observed between cSS presence and APOE ε4 in the memory clinic cohorts. For example, both CAA and neurodegenerative pathology contribute to cognitive impairment in the elderly41 and APOE ε4 is a well-known risk factor for both cerebrovascular and parenchymal amyloid accumulation. Nevertheless, reassuringly, the association with APOE ε2 and cSS was consistently detected in memory clinic patients and with stronger effect size than the APOE ε4 association. Finally, our meta-analysis was performed at a group level, which means that generated pooled estimates are not adjusted for any confounders that might influence the association between APOE status and cSS, including age, sex, and other MRI markers of small vessel disease. It is thus possible that reported associations are overstated. It could be argued that our unadjusted group-level meta-analysis is exploratory, providing a rough indication of likely effect sizes across populations for the APOE–cSS link, setting the scene for a more detailed individual patient-level meta-analysis. We are hoping to pursue this in the future in a large collaborative study.
Most of the symptomatic CAA cohorts suitable for meta-analysis came from a single center. We acknowledge the potential for some overlap among these cohorts—while we do not have all the detailed data on overlapping patients, based on the different clinical setting/recruitment, clinical presentation, inclusion criteria, and inception points, this overlap is minimal and ranges between 5% and 10%, as summarized in the table 1 footnote. Any potential overlap between these cohorts is random and unlikely to have affected our main results. However, due to this limitation, our findings will benefit from further validation and updated meta-analyses. The plausible suggestion of different APOE influences on the severity and type of amyloid deposition in the vessel wall and advanced vasculopathic changes suggested in the interpretation of our current findings are based on limited data.42 In particular, further studies on the proposed differential effects of ε4 and ε2 alleles will be valuable. However, it is reasonable to provide an informed discussion based on these biological hypotheses and assumptions, in an effort to start building a pathophysiology model of cSS that could explain both clinical and research findings and help develop hypothesis-driven studies in the field. Finally, the vascular damage pathways leading to cSS must be only partly influenced by APOE ε2 as evidenced by the fact that a number of cSS cases are also found within the APOE ε3/ε3 genotype.
Notwithstanding these caveats, our results provide useful data to partly settle the question in favor of a link between APOE ε2+ genotype and cSS in CAA. The pathophysiologic implication is that APOE ε2 influences the risk of cSS through promoting the most severe stages of CAA pathology that are associated with rupture. These results are also in line with cSS being a strong hemorrhagic MRI signature in CAA and the likely smoking gun of bleeding risk. Future research efforts on the topic require methodologically robust, large studies adhering to current reporting standards,1,9 and collaborative data pooling efforts. Based on the totality of current evidence, our study suggests a biologically meaningful association between the APOE ε2+ genotype and severe cSS in patients with CAA, probably as a result of the role of ε2 in the severity of vasculopathy in CAA-affected leptomeningeal and very superficial cortical vessels. The exact pathophysiologic mechanisms that underlie these associations should be investigated, as they might define targets for therapeutic interventions in CAA.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- CAA
cerebral amyloid angiopathy
- CI
confidence interval
- cSS
cortical superficial siderosis
- ICH
intracerebral hemorrhage
- OR
odds ratio
- SWI
susceptibility-weighted imaging
- T2*-GRE
T2*-weighted gradient-recalled echo
Appendix. Authors

Study funding
This work was supported by The Genetics of Cerebral Hemorrhage with Anticoagulation study, funded by NIH/NINDS grant R01NS059727. This study is not industry sponsored. Andreas Charidimou is supported by a Bodossaki Foundation postdoctoral scholarship.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Charidimou A, Linn J, Vernooij MW, et al. Cortical superficial siderosis: detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain 2015;138:2126–2139. [DOI] [PubMed] [Google Scholar]
- 2.Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010;74:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charidimou A, Jäger RH, Fox Z, et al. Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology 2013;81:626–632. [DOI] [PubMed] [Google Scholar]
- 4.Shoamanesh A, Martinez-Ramirez S, Oliveira-Filho J, et al. Interrelationship of superficial siderosis and microbleeds in cerebral amyloid angiopathy. Neurology 2014;83:1838–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charidimou A, Boulouis G, Xiong L, et al. Cortical superficial siderosis and first-ever cerebral hemorrhage in cerebral amyloid angiopathy. Neurology 2017;88:1607–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charidimou A, Peeters AP, Jäger R, et al. Cortical superficial siderosis and intracerebral hemorrhage risk in cerebral amyloid angiopathy. Neurology 2013;81:1666–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roongpiboonsopit D, Charidimou A, William CM, et al. Cortical superficial siderosis predicts early recurrent lobar hemorrhage. Neurology 2016;87:1863–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moulin S, Labreuche J, Bombois S, et al. Dementia risk after spontaneous intracerebral haemorrhage: a prospective cohort study. Lancet Neurol 2016, 15:820–829. [DOI] [PubMed] [Google Scholar]
- 9.Greenberg SM, Salman RA, Biessels GJ, et al. Outcome markers for clinical trials in cerebral amyloid angiopathy. Lancet Neurol 2014;13:419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol 1995;38:254–259. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg SM, Briggs ME, Hyman BT, et al. Apolipoprotein E epsilon 4 is associated with the presence and earlier onset of hemorrhage in cerebral amyloid angiopathy. Stroke 1996;27:1333–1337. [DOI] [PubMed] [Google Scholar]
- 12.Rannikmäe K, Samarasekera N, Martînez-Gonzâlez NA, Al-Shahi Salman R, Sudlow CL. Genetics of cerebral amyloid angiopathy: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2013;84:901–908. [DOI] [PubMed] [Google Scholar]
- 13.Greenberg SM, Vonsattel JP, Segal AZ, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology 1998;50:961–965. [DOI] [PubMed] [Google Scholar]
- 14.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 2009;339:b2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stroup DF, Berlin JA, Morton SC, et al. Meta-analysis of observational studies in epidemiology: a proposal for reporting: Meta-analysis of Observational Studies in Epidemiology (MOOSE) group. JAMA 2000;283:2008–2012. [DOI] [PubMed] [Google Scholar]
- 16.Little J, Higgins JP, Ioannidis JP, et al. Strengthening the Reporting of Genetic Association Studies (STREGA): an extension of the STROBE statement. Genet Epidemiol 2009;33:581–598. [DOI] [PubMed] [Google Scholar]
- 17.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986;7:177–188. [DOI] [PubMed] [Google Scholar]
- 18.Boulouis G, Charidimou A, Jessel MJ, et al. Small vessel disease burden in cerebral amyloid angiopathy without symptomatic hemorrhage. Neurology 2017;88:878–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charidimou A, Boulouis G, Haley K, et al. White matter hyperintensity patterns in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 2016;86:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charidimou A, Martinez-Ramirez S, Shoamanesh A, et al. Cerebral amyloid angiopathy with and without hemorrhage: evidence for different disease phenotypes. Neurology 2015;84:1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Lizana E, Carmona-Iragui M, Alcolea D, et al. Cerebral amyloid angiopathy-related atraumatic convexal subarachnoid hemorrhage: an ARIA before the tsunami. J Cereb Blood Flow Metab 2015;35:710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shams S, Martola J, Charidimou A, et al. Cortical superficial siderosis: prevalence and biomarker profile in a memory clinic population. Neurology 2016;87:1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charidimou A, Ni J, Martinez-Ramirez S, et al. Cortical superficial siderosis in memory clinic patients: further evidence for underlying cerebral amyloid angiopathy. Cerebrovasc Dis 2016;41:156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Na HK, Park JH, Kim JH, et al. Cortical superficial siderosis: a marker of vascular amyloid in patients with cognitive impairment. Neurology 2015;84:849–855. [DOI] [PubMed] [Google Scholar]
- 25.Zonneveld HI, Goos JD, Wattjes MP, et al. Prevalence of cortical superficial siderosis in a memory clinic population. Neurology 2014;82:698–704. [DOI] [PubMed] [Google Scholar]
- 26.Yates PA, Desmond PM, Phal PM, et al. Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology 2014;82:1266–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kantarci K, Gunter JL, Tosakulwong N, et al. Focal hemosiderin deposits and beta-amyloid load in the ADNI cohort. Alzheimers Dement 2013;9:S116–S123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pichler M, Vemuri P, Rabinstein AA, et al. Prevalence and natural history of superficial siderosis: a population-based study. Stroke 2017;48:3210–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maxwell SS, Jackson CA, Paternoster L, Cordonnier V, Al-Shahi Salman R, Sudlow CL. Genetic associations with brain microbleeds: systematic review and meta-analyses. Neurology 2011;77:158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schilling S, DeStefano AL, Sachdev PS, et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology 2013;81:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenberg SM, Nandigam RN, Delgado P, et al. Microbleeds versus macrobleeds: evidence for distinct entities. Stroke 2009;40:2382–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Love S, Chalmers K, Ince P, et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener Dis 2014;3:19–32. [PMC free article] [PubMed] [Google Scholar]
- 34.Biffi A, Anderson CD, Jagiella JM, et al. APOE genotype and extent of bleeding and outcome in lobar intracerebral haemorrhage: a genetic association study. Lancet Neurol 2011;10:702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brouwers HB, Biffi A, Ayres AM, et al. Apolipoprotein E genotype predicts hematoma expansion in lobar intracerebral hemorrhage. Stroke 2012;43:1490–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charidimou A, Boulouis G, Roongpiboonsopit D, et al. Cortical superficial siderosis multifocality in cerebral amyloid angiopathy: a prospective study. Neurology 2017;89:2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thal DR, Ghebremedhin E, Rüb U, Yamaguchi H, Del Tredici K, Braak H. Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 2002;61:282–293. [DOI] [PubMed] [Google Scholar]
- 38.Linn J, Wollenweber FA, Lummel N, et al. Superficial siderosis is a warning sign for future intracranial hemorrhage. J Neurol 2013;260:176–181. [DOI] [PubMed] [Google Scholar]
- 39.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol 2011;10:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer's disease: innocent observation or key player? Brain 2011;134:335–344. [DOI] [PubMed] [Google Scholar]
- 41.Boyle PA, Yu L, Nag S, Leurgans RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 2015;85:1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rannikmäe K, Kalaria RN, Greenberg SM, et al. APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J Neurol Neurosurg Psychiatry 2014;85:300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data and methods are reported in the article. No data are available in public repositories.