

Abstract
Solid pseudopapillary neoplasms (SPNs) are rare and relatively indolent tumors of the pancreas. While primary SPNs can be surgically resected, there are currently no therapies available for patients with advanced stage disease. Given that these tumors frequently carry CTNNB1 hotspot (recurrently mutated loci in a gene) mutations resulting in β‐catenin nuclear accumulation, it has been speculated that the Wnt pathway may be a driver in this disease. Here, we present a comprehensive “multi‐omics” study where the genome, transcriptome, and methylome of SPNs were analyzed. We found that SPNs are characterized by a low‐complexity genome where somatic mutations in CTNNB1, present in 100% of the cases, are the only actionable genomic lesions. Compared to more common subtypes of pancreatic tumors (adenocarcinomas and pancreatic neuroendocrine tumors), SPNs show high expression levels of genes belonging to the Wnt pathway. Their methylome was consistent with an epithelial cell origin and a general upregulation of Wnt pathway genes. Clinical studies to evaluate the exquisite sensitivity of SPNs to inhibitors of the Wnt pathway are warranted.
Keywords: beta‐catenin, gene expression, methylation, pancreas, SPN, Wnt
Abbreviations
- ADC
adenocarcinoma
- ENCODE
encyclopedia of DNA elements
- indel
small insertion and deletion
- PNET
pancreatic neuroendocrine tumors
- SNV
single nucleotide variant
- SPN
solid pseudopapillary neoplasms
1. Introduction
Solid pseudopapillary neoplasms (SPNs) are rare pancreatic tumors of unknown etiology, accounting for ~ 1% of all pancreatic exocrine neoplasms. SPNs typically present as large, solitary, well‐circumscribed lesions with indolent clinical course and a low propensity to metastasize (Klimstra et al., 2000; Ren et al., 2014). Although they are commonly managed surgically, some SPNs can exhibit a more aggressive behavior and metastasize. Currently, there is no consensus on effective systemic treatments for malignant SPNs, and given the rarity of advanced disease, prospective clinical trials testing novel therapies have not been performed as yet.
Solid pseudopapillary neoplasms are characterized by the presence of somatic CTNNB1 exon 3 hotspot mutations (Abraham et al., 2002; Guo et al., 2017; Tanaka et al., 2001; Wu et al., 2011), leading to stabilization and nuclear localization of β‐catenin, which can be detected by immunohistochemistry (Tanaka et al., 2001). Given these premises, the role of the Wnt pathway in SPN development has been suggested. The Wnt pathway is a signal transduction cascade that under normal physiological conditions regulates development and stemness but has also been tightly associated with multiple growth‐related pathologies and cancer (Nusse and Clevers, 2017). A key component of the Wnt pathway is the protein β‐catenin encoded by the gene CTNNB1, which regulates transcription of downstream target genes (e.g., LEF1, AXIN2). Hotspot mutations in CTNNB1 can cause abnormal accumulation of nuclear β‐catenin through stabilization of the protein and eventually constitutive activation of the pathway.
Here, we assessed whether SPNs are dependent on the Wnt signaling pathway and performed a comprehensive “multi‐omics” profiling of SPNs collected at our institution during the last two decades to define their repertoire of somatic mutations and copy number alterations, the transcriptional activation of the Wnt pathway and their methylation profiles.
2. Methods
2.1. Cases
Patients with written informed consent for research that had a diagnosis of SPN treated at Memorial Sloan Kettering Cancer Center (MSKCC) were identified from our institutional database. Clinicopathological characteristics of the SPN patients are shown in Table 1. Electronic medical records were retrospectively reviewed for data on patient demographics, pathology, presentation (benign versus malignant SPN), treatment (surgical and systemic therapies), and outcomes. In addition, pancreatic adenocarcinomas (ADCs) and pancreatic neuroendocrine tumors (PNETs) were identified as controls. All pathology was reviewed by gastrointestinal pathologists at MSKCC experienced in the diagnosis of SPN. Approval for data collection and analysis was obtained from the MSKCC Institutional Review Board. The study conforms to the guidelines set by the Declaration of Helsinki.
Table 1.
Baseline patient characteristics
| SPNs | All n = 14 N (%) |
|---|---|
| Age, mean (range), years | 37 (22–71) |
| Sex | |
| Female | 11 (79) |
| Male | 3 (21) |
| Smoker/prior smoker | 1 (7) |
| Symptomatic at presentation | 14 (100) |
| Pain | 11 (79) |
| Diarrhea | 2 (14) |
| Other | 1 (7) |
| Primary tumor location | |
| Head | 3 (21) |
| Body | 5 (36) |
| Tail | 6 (43) |
| Primary tumor size, mean (range), centimeters | 5.9 (1.2–11) |
| Primary tumor resected | 14 (100) |
| Surgery | |
| Pancreaticoduodenectomy | 3 (21) |
| Distal pancreatectomy | 11 (79) |
| Hepatic debulking | 1 (7) |
| Metastatic disease | 3 (21) |
| Location of metastases | |
| Liver | 2 (14) |
| Omentum/peritoneum | 2 (14) |
| Lung | 1 (7) |
| Lymph nodes | 1 (7) |
| Systemic therapy | 1 (7) |
| Hepatic arterial embolization | 1 (7) |
2.2. Immunohistochemistry
Immunohistochemical analysis for β‐catenin (Clone 14, 1 : 200; BD Transduction, San Jose, CA) was performed on 4‐μm‐thick sections from representative formalin‐fixed, paraffin‐embedded SPN tissue blocks, as previously described (Basturk et al., 2016). Positive and negative controls were included in each slide run. The expression of β‐catenin was assessed in the membrane and cytoplasmic and nuclear subcellular compartments, and considered abnormal if cytoplasmic and nuclear accumulation were present.
2.3. Whole‐exome sequencing analysis
DNA samples extracted from 14 flash‐frozen SPNs and matched normal tissues were subjected to whole‐exome sequencing at the MSKCC Integrated Genomics Operation as previously described (Ng et al., 2017). Briefly, raw sequence reads were aligned to the reference human genome GRCh37 using the burrows‐wheeler aligner (BWA 0.7.15) (Li and Durbin, 2009). Local realignment, duplicate read removal, and base quality score recalibration were performed using the genome analysis toolkit (GATK 3.7) (McKenna et al., 2010). Somatic single nucleotide variants (SNVs) were called using mutect (1.1.7) (Cibulskis et al., 2013), and small insertions and deletions (indels) were identified using strelka (1.0.15) (Saunders et al., 2012), varscan2 (2.3.7) (Koboldt et al., 2012), lancet (1.0.0) (Narzisi et al., 2018), and scalpel (0.5.3) (Narzisi et al., 2014) and further curated by manual inspection. SNVs and indels outside of target regions were filtered out, as were SNVs and indels for which the variant allele fraction (VAF) in the tumor sample was < 5 times that of the paired normal VAF as previously described (Ng et al., 2017; Weigelt et al., 2018). Finally, SNVs and indels found at > 5% global minor allele frequency in dbSNP (build 137) and > 5% global allele frequency in exac (0.3.1) were discarded. Somatic copy number alterations and loss of heterozygosity were obtained using facets (Shen and Seshan, 2016). The cancer cell fractions (CCF) of all mutations were computed using absolute (1.0.6) (Carter et al., 2012). A mutation was classified as clonal if its probability of being clonal was > 50% (Landau et al., 2013) or if the lower bound of the 95% confidence interval of its CCF was > 90% (Ng et al., 2017; Weigelt et al., 2018). Mutations that did not meet the above criteria were considered subclonal. A combination of in silico functional predictors was used to define the potential functional impact of each missense SNV as previously described (Martelotto et al., 2014; Ng et al., 2017). Mutation hotspots were assigned according to Chang et al. (2016).
CTNNB1 mutation frequencies were assessed in 10 100 common cancers from The Cancer Genome Atlas (TCGA) (Bailey et al., 2018; Gao et al., 2013), including 175 pancreatic ADCs. In addition, mutational data from 8 SPNs from Wu et al. (2011) and 344 non‐SPN pancreatic tumors, including 23 acinar cell carcinomas from Jiao et al. (2014), 98 PNETs from Scarpa et al. (2017), and 24 cystic tumors of the pancreas Wu et al. (2011). The mutational data were retrieved from cBioPortal (Gao et al., 2013). CTNNB1 mutation and hotspot mutation frequencies were assessed following exclusion of hypermutated cases, defined as cancers harboring more than 1000 nonsynonymous mutations, microsatellite‐unstable, or harboring POLE or POLD1 exonuclease domain mutations, as previously described (Pareja et al., 2018). Mutation diagrams (‘lollipop’ plots) were generated using MutationMapper on cbioportal (Gao et al., 2013) and manually curated.
2.4. Gene expression analysis
Total RNA extracted from nine SPNs, seven pancreatic ADCs, and 8 PNETs was subjected to expression analysis of 240 selected genes using the nCounter platform from Nanostring Technologies, Seattle, WA (Table S3). The raw counts were normalized using the nSolver analysis software. We performed background subtraction using the negative control and normalized the background corrected counts using the housekeeping genes ACTB, MRPL19, PSMC4, RPLP0, and SF3A1. Comparison between SPN samples and ADCs or PNETs was done using the r/bioconductor limma package, and genes with P values < 0.05 were considered as significantly differentially expressed.
2.5. Genome‐wide methylation analysis
DNA extracted from 13 SPNs was subjected to Illumina Infinium Methylation 850K array profiling as previously described (Chiang et al., 2016). The r/bioconductor minfi package was used to process the raw files and obtain the normalized beta values. The methylation data in the form of beta values of cells of epithelial (n = 22), fibroblast (n = 10), and muscle cell (n = 6) origin were downloaded from encyclopedia of DNA elements (ENCODE) project (ENCODE Project Consortium et al., 2007) and processed as described above. To avoid the confounding effects of gender, probes mapping to sex chromosomes were discarded while batch effect was removed using the combat method (Johnson et al., 2007) implemented in the r/bioconductor package sva. Probes which were differentially methylated between ENCODE samples of epithelial, fibroblast, and muscle cell origin were identified using the Kruskal–Wallis test and an FDR‐adjusted P‐value < 0.1 (Table S4). The SPN methylation data were projected onto the first two principal components estimated using the ENCODE samples. Probes of the Wnt pathway found to be differentially methylated between SPNs and ENCODE samples were subjected to hierarchical cluster analysis using Pearson correlation and Ward's distance.
2.6. Statistical analysis
Using the transcriptomic data, genes differentially expressed between SPNs and ADCs or PNETs were identified using a linear model fitted to the expression data. We considered genes with a P‐value < 0.05 and an absolute fold change value > 2 as statistically significant. In the case of methylation data, the data were filtered by identifying probes with multimodal distributions of beta values in the ENCODE dataset. A mixture of Gaussian distributions was fitted to the pooled data using the r/bioconductor package mclust. Probes which were differentially methylated between ENCODE samples of epithelial, fibroblast, and muscle cell origin were identified using the Kruskal–Wallis test and an FDR‐adjusted P‐value < 0.1. The SPN methylation data were projected onto the first two principal components estimated using the ENCODE samples. All hypothesis tests were unpaired, and P‐values were two‐sided. Unless otherwise stated, all computations were performed in r/bioconductor.
3. Results
3.1. Landscape of somatic mutations in SPNs
Whole‐exome sequencing analysis revealed clonal somatic hotspot CTNNB1 mutations in all 14 SPNs analyzed (Fig. 1A). Only two additional recurrently mutated genes were identified in more than one patient sample (THADA and TPRXL; Fig. 1A, Tables [Link], [Link]). All SPNs displayed low levels of copy number alterations, consistent with a lack of genomic instability characteristic of SPNs (Fig. S1). A Pan‐Cancer analysis of 10 100 nonhypermutated cancers across 31 common cancer types from TCGA (Bailey et al., 2018; Gao et al., 2013) revealed that while CTNNB1 exon 3 mutations were present at 15–20% of liver, uterine, and adrenocortical carcinomas, mutations affecting CTNNB1 exon 3 are rare in other cancer types (< 3%; Fig. 1B). Furthermore, of the 328 pancreatic tumors included in the cBioPortal database (Bailey et al., 2018; Gao et al., 2013; Jiao et al., 2014; Scarpa et al., 2017; Wu et al., 2011) only 12 (3.7%) harbored CTNNB1 exon 3 mutations (Fig. 1C). Eight of these 12 cases were SPNs, whereas the remaining tumors bearing CTNNB1 hotspot mutations were ADCs (n = 2) or pancreatoblastomas (n = 2) (Wu et al., 2011). Thus, we conclude that in the context of pancreatic neoplasms in adults, mutations in β‐catenin (amino acids 32 through 45) strongly prompt a diagnosis of SPN. SPNs displayed low mutational burdens (median: 19, range: 7–50) compared to β‐catenin wild‐type pancreatic tumors (median: 50, range: 1–185; P < 0.0001, Mann–Whitney U) (Bailey et al., 2018; Gao et al., 2013; Jiao et al., 2014; Scarpa et al., 2017; Wu et al., 2011), which is consistent with the finding that a subset of endometrioid endometrial carcinomas driven by CTNNB1 mutations and Wnt pathway activation are characterized by a relatively low tumor mutational burden (Liu et al., 2014).
Figure 1.
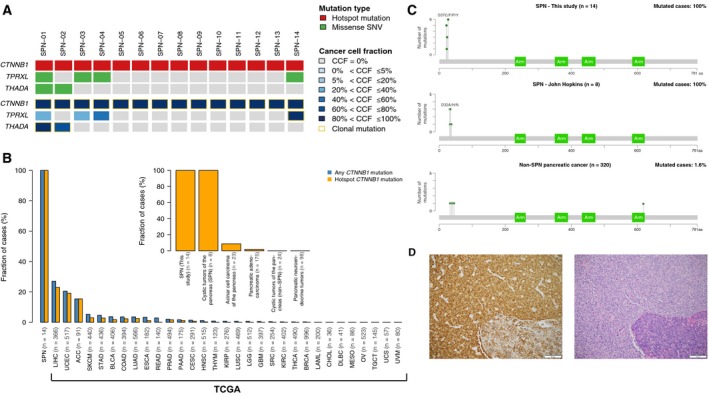
Prevalence of CTNNB1 mutations in SPNs and other common cancer types. (A) Mutation type (top) and CCF (bottom) of all recurrently mutated genes in SPNs subjected to whole‐exome sequencing in this study. (B) Bar plots depicting the frequency of CTNNB1 and CTNNB1 hotspot mutations in 14 SPNs from this study and in 10 100 samples from 31 common cancer types from TCGA. The insert shows the prevalence of CTNNB1 hotspot mutations in SPNs analyzed in this study compared to SPNs and other pancreatic tumors from this study and from TCGA. (C) Lollipop plot representing the β‐catenin protein domains and the spectra of the CTNNB1 mutations in 14 SPNs profiled in this study (top), in eight SPNs from Wu et al. (2011) (middle), and in 320 non‐SPN pancreatic tumors available from cBioPortal (Gao et al., 2013). Mutations are shown on the x‐axis, and the frequency of a particular mutation is represented by the height of each “lollipop” (y‐axis). (D) Representative micrographs of an SPN with adjacent normal tissue (bottom right in both images). Hematoxylin‐ and eosin‐stained section (left) and β‐catenin immunohistochemical analysis (right).
3.2. Activation of the Wnt pathway in SPNs
From a genomics standpoint, SPNs appear to be driven solely by CTNNB1 activating somatic mutations, which result in nuclear accumulation of the protein (Polakis, 1999). Accordingly, we detected strong β‐catenin protein expression in both cytosol and nuclei of all SPNs analyzed (Fig. 1D). This, in turn, initiates a transcriptional program primed to activate the Wnt pathway (Mosimann et al., 2009). To investigate the components of the Wnt signaling pathway activated in SPNs, we performed a differential gene expression analysis of nine SPNs, four of which were metastatic, seven pancreatic ADCs, and eight PNETs using the Nanostring platform (Table S3). Compared to ADCs and PNETs, SPNs displayed significantly higher expression levels of genes encoding components of the Wnt pathway, including DKK4, which has recently been detected by mass spectrometry in SPNs (Park et al., 2015), and direct TCF/β‐catenin target genes such as LEF1 (Aoki et al., 1999) or AXIN2 (Jho et al., 2002) (Fig. 2A–D, Fig. S2). Other genes whose expression has been described to be induced by Wnt/β‐catenin signaling activation such as LGR5, ASCL2, or CCND1 were also expressed at significantly higher levels in SPNs (Fig. S2).
Figure 2.
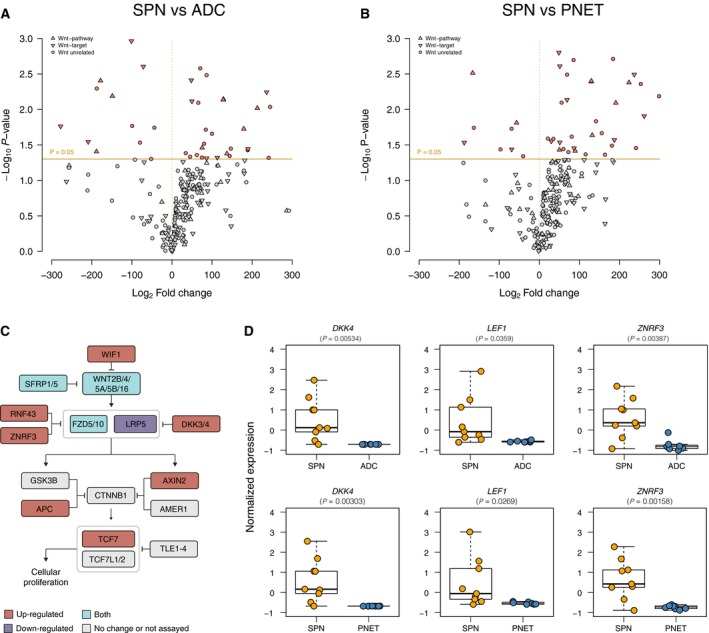
Patterns of gene expression in SPNs, pancreatic ADCs, PNETs, and normal tissue samples. (A) Volcano plot showing the genes differentially expressed between SPNs and ADCs. The Log2 fold change refers to the ratio of mean expression between SPNs and ADCs. Orange line depicts P‐value cut‐off of 0.05. (B) Volcano plot showing the genes differentially expressed genes between SPNs and PNETs. The Log2 fold change refers to the ratio of mean expression between SPNs and PNETs. Orange line depicts P‐value cut‐off of 0.05. (C) Schematic illustration of the main components of the canonical Wnt pathway. The genes found to be significantly differentially expressed (P < 0.05) in (A) and (B) are color‐coded according to the legend. (D) Boxplots depicting the expression levels of the Wnt pathway genes DKK4,LEF1, and ZNRF3 in SPNs, ADCs, and PNETs. Modified t‐test.
3.3. Methylome analysis of SPNs
Given the unknown histogenesis of SPNs, we analyzed the global methylation profiles of 12 of the 14 SPNs analyzed by whole‐exome sequencing and compared their methylation patterns with those of normal cells of various anatomical sites from the ENCODE database (ENCODE Project Consortium et al., 2007). The majority of the SPNs profiled in this study clustered with epithelial cells (Fig. 3A). We further performed a differential analysis between ENCODE samples of epithelial origin and SPNs, which identified 2511 differentially methylated probes (Table S3). Probes that were significantly differentially methylated between samples of epithelial, fibroblast, and muscle cell origin in the ENCODE database were mapped to genes in the Wnt pathway and showed differential methylation in the gene bodies (intronic and exonic areas of a gene), which, based on ontology, would have resulted in upregulation of the respective transcript (e.g. WNT5A). Conversely, hypermethylated probe sets corresponded with genes whose transcripts were downregulated in SPNs (e.g., CD44; Fig. 3B). Two exceptions were probes mapping to ZIC2 and WNT5B, which were found to be hypermethylated in SPN compared to ENCODE samples. Unlike CD44, these genes had a significantly higher expression in SPNs compared to ADCs or PNETs. Also, the methylation pattern is not unexpected, given that the hypermethylated probes mapped to the 3′UTR of the corresponding gene, a phenomenon not uncommonly associated with overexpression of the target gene.
Figure 3.
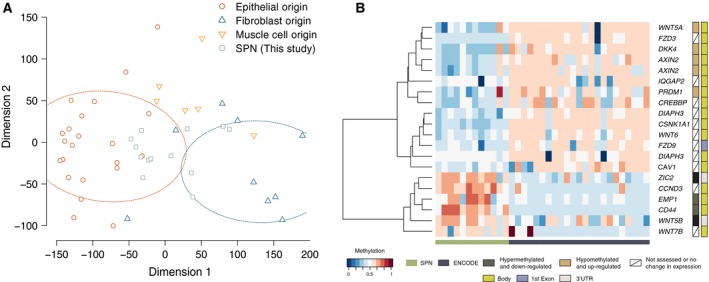
Patterns of methylation in SPNs and normal tissue samples. (A) Principal component analysis of methylation data showing the clustering of samples of different histological origins from the ENCODE project together with SPNs profiled in this study. (B) Heatmap of methylation data from SPN samples profiled in this study and ENCODE samples of epithelial origin. Probes found to be differentially methylated (FDR < 0.1) between SPN and ENCODE samples were mapped to the Wnt pathway. With few exceptions, the probes mapped to gene bodies where hypomethylation corresponded to upregulation of the corresponding transcript. The two exceptions were probes mapping to ZIC2 and WNT5B which were located in 3′UTR regions where hypermethylation also corresponded to upregulation of the upstream gene.
4. Discussion
Here, we report a multi‐omics analysis of SPNs compared to the other two more common subtypes of pancreatic cancer, ADC and PNET. In agreement with the existing literature, we found that SPNs have low genomic complexity and are characterized by the invariable presence of CTNNB1 exon 3 mutations. Accordingly, we found consistent increased expression of genes encoding components of the Wnt pathway in these tumors. Nuclear β‐catenin accumulation promotes the expression of a panel of Wnt target genes codifying for proteins involved in a myriad of cell processes. Among them, some codify components of the Wnt/β‐catenin pathway itself such as LEF1, AXIN2, or RNF43, which induce a positive feedback loop that enhances signaling activation (Zhan et al., 2017). Here, we observed that SPNs with mutations in CTNBB1 that provoke constitutive accumulation of nuclear β‐catenin express higher levels of some of these Wnt target genes that are also Wnt pathway components such as LEF1, AXIN2, and RNF43 (Fig. 3).
We also observed that other genes codifying components of the Wnt pathway are highly expressed in SPN than ADC, such as ligands (WNT5A, WNT2B, or RSPO4), receptor components (LRG6 or FZD10), or other key proteins for signal transduction such as APC. Although these are not formally demonstrated to be Wnt/β‐catenin pathway target genes, their higher expression could suggest and enhanced activation of the pathway additional to that promoted by CTNNB1/β‐catenin activating mutations.
The lack of somatic genetic alterations other than CTNNB1 mutations and low levels of genomic complexity may provide an explanation for the indolent behavior of SPNs, akin to colorectal polyps associated with familial adenomatous polyposis in patients with germline APC mutations developing multiple KRAS wild‐type, benign tumors where a dysfunctional APC can be the only driver (Leoz et al., 2015; Takane et al., 2016). In mice, activation of β‐catenin is indeed sufficient to produce large pancreatic tumors that resemble human SPNs both morphologically and by immunohistochemical analysis (Heiser et al., 2008).
Of note, a recent report identified also inactivating mutations of the epigenetic regulators KDM6A, TET1, and BAP1 to be associated with metastatic spread (Amato et al., 2019). Whether these alterations are also playing a role in regulating the expression of Wnt‐dependent genes remains to be elucidated.
While inhibition of the Wnt pathway may be an intuitive therapeutic option for this disease, its rarity and the lack of available preclinical models are clear limitations in the advancement toward an effective treatment for metastatic SPNs. Furthermore, the more clinically advanced Wnt pathway inhibitors target components that are upstream of β‐catenin transcriptional activity and would therefore be futile for the treatment of SPN patients.
5. Conclusions
Taken together, our data suggest that SPNs have simple genomes and appear to be uniformly driven by CTNNB1 exon 3 mutations with consequent activation of the Wnt signaling. Future drugs designed to target directly β‐catenin may have therapeutic potential in this disease.
Conflict of interest
MS received honoraria from ADC Pharma and Menarini Ricerche; research funds received from Puma Biotechnology, Daiichi‐Sankyo, Immunomedics, and Menarini Ricerche; is a co‐founder of Medendi Medical Travel; and is in the scientific board of the Bioscience Institute. JSR‐F reports personal/ consultancy fees from VolitionRx, Page.AI, Invicro, Roche, Genentech, Ventana and Goldman Sachs, outside the submitted work.
Author contributions
JSR‐F and MS conceived the study. DK performed the pathology review. RK, PS, and DNB performed bioinformatics analyses. NR provided the tumor samples with their clinical annotation; HP and OA performed and interpreted the nanostring analysis. MSn and JS performed the methylation analysis. PS, DNB, BW, NR, JSR‐F, and MS interpreted the data. PS, DNB, BW, JSR‐F, and MS wrote the first draft of the manuscript, which was edited and approved by all authors.
Supporting information
Fig. S1. Copy number profiles of SPNs analyzed in this study.
Fig. S2. Differential gene expression analysis of SPNs, ADCs and PNETs.
Table S1. Whole‐exome sequencing statistics.
Table S2. Nonsynonymous somatic mutations identified in SPNs using whole‐exome sequencing.
Table S3. Genes in the Wnt pathway assessed by Nanostring nCounter.
Table S4. Probes differentially methylation between SPN samples (this study) and ENCODE samples of epithelial origin.
Acknowledgements
MS is funded by Cycle for Survival. MS and JSR‐F are funded in part by the Breast Cancer Research Foundation. Research reported in this paper was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (grant No P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The study was in part supported by the Friedberg Charitable Foundation grant to MSn.
Pier Selenica, Nitya Raj and Rahul Kumar contributed equally to this article.
Contributor Information
Jorge S. Reis‐Filho, Email: reisfilj@mskcc.org
Maurizio Scaltriti, Email: scaltrim@mskcc.org.
References
- Abraham SC, Klimstra DS, Wilentz RE, Yeo CJ, Conlon K, Brennan M, Cameron JL, Wu TT and Hruban RH (2002) Solid‐pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor beta‐catenin mutations. Am J Pathol 160, 1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato E, Mafficini A, Hirabayashi K, Lawlor RT, Fassan M, Vicentini C, Barbi S, Delfino P, Sikora K, Rusev B et al (2019) Molecular alterations associated with metastases of solid pseudopapillary neoplasms of the pancreas. J Pathol 247, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki M, Hecht A, Kruse U, Kemler R and Vogt PK (1999) Nuclear endpoint of Wnt signaling: neoplastic transformation induced by transactivating lymphoid‐enhancing factor 1. Proc Natl Acad Sci USA 96, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta‐Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B et al (2018) Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385.e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basturk O, Chung SM, Hruban RH, Adsay NV, Askan G, Iacobuzio‐Donahue C, Balci S, Zee SY, Memis B, Shia J et al (2016) Distinct pathways of pathogenesis of intraductal oncocytic papillary neoplasms and intraductal papillary mucinous neoplasms of the pancreas. Virchows Arch 469, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA et al (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, Gao J, Socci ND, Solit DB, Olshen AB et al (2016) Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 34, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang S, Weigelt B, Wen HC, Pareja F, Raghavendra A, Martelotto LG, Burke KA, Basili T, Li A, Geyer FC et al (2016) IDH2 mutations define a unique subtype of breast cancer with altered nuclear polarity. Cancer Res 76, 7118–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES and Getz G (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET et al (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Luo G, Jin K, Long J, Cheng H, Lu Y, Wang Z, Yang C, Xu J, Ni Q et al (2017) Somatic genetic variation in solid pseudopapillary tumor of the pancreas by whole exome sequencing. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiser PW, Cano DA, Landsman L, Kim GE, Kench JG, Klimstra DS, Taketo MM, Biankin AV and Hebrok M (2008) Stabilization of beta‐catenin induces pancreas tumor formation. Gastroenterology 135, 1288–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN and Costantini F (2002) Wnt/beta‐catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22, 1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Yonescu R, Offerhaus GJ, Klimstra DS, Maitra A, Eshleman JR, Herman JG, Poh W, Pelosof L, Wolfgang CL et al (2014) Whole‐exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol 232, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C and Rabinovic A (2007) Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Klimstra DS, Wenig BM and Heffess CS (2000) Solid‐pseudopapillary tumor of the pancreas: a typically cystic carcinoma of low malignant potential. Semin Diagn Pathol 17, 66–80. [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L et al (2013) Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoz ML, Carballal S, Moreira L, Ocana T and Balaguer F (2015) The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl Clin Genet 8, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H and Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Patel L, Mills GB, Lu KH, Sood AK, Ding L, Kucherlapati R, Mardis ER, Levine DA, Shmulevich I et al (2014) Clinical significance of CTNNB1 mutation and Wnt pathway activation in endometrioid endometrial carcinoma. J Natl Cancer Inst 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelotto LG, Ng CK, De Filippo MR, Zhang Y, Piscuoglio S, Lim RS, Shen R, Norton L, Reis‐Filho JS and Weigelt B (2014) Benchmarking mutation effect prediction algorithms using functionally validated cancer‐related missense mutations. Genome Biol 15, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M et al (2010) The genome analysis toolkit: a mapreduce framework for analyzing next‐generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann C, Hausmann G and Basler K (2009) Beta‐catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol 10, 276–286. [DOI] [PubMed] [Google Scholar]
- Narzisi G, Corvelo A, Arora K, Bergmann EA, Shah M, Musunuri R, Emde A‐K, Robine N, Vacic V and Zody MC (2018) Genome‐wide somatic variant calling using localized colored de Bruijn graphs. Commun Biol 1, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narzisi G, O'Rawe JA, Iossifov I, Fang H, Lee YH, Wang Z, Wu Y, Lyon GJ, Wigler M and Schatz MC (2014) Accurate de novo and transmitted indel detection in exome‐capture data using microassembly. Nat Methods 11, 1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CKY, Piscuoglio S, Geyer FC, Burke KA, Pareja F, Eberle CA, Lim RS, Natrajan R, Riaz N, Mariani O et al (2017) The landscape of somatic genetic alterations in metaplastic breast carcinomas. Clin Cancer Res 23, 3859–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R and Clevers H (2017) Wnt/beta‐catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999. [DOI] [PubMed] [Google Scholar]
- Pareja F, Brandes AH, Basili T, Selenica P, Geyer FC, Fan D, Da Cruz Paula A, Kumar R, Brown DN, Gularte‐Merida R et al (2018) Loss‐of‐function mutations in ATP6AP1 and ATP6AP2 in granular cell tumors. Nat Commun 9, 3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Lim JS, Lee HJ, Na K, Lee MJ, Kang CM, Paik YK and Kim H (2015) Distinct protein expression profiles of solid‐pseudopapillary neoplasms of the pancreas. J Proteome Res 14, 3007–3014. [DOI] [PubMed] [Google Scholar]
- Polakis P (1999) The oncogenic activation of beta‐catenin. Curr Opin Genet Dev 9, 15–21. [DOI] [PubMed] [Google Scholar]
- Ren Z, Zhang P, Zhang X and Liu B (2014) Solid pseudopapillary neoplasms of the pancreas: clinicopathologic features and surgical treatment of 19 cases. Int J Clin Exp Pathol 7, 6889–6897. [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ and Cheetham RK (2012) Strelka: accurate somatic small‐variant calling from sequenced tumor‐normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, Lawlor RT, Johns AL, Miller DK, Mafficini A et al (2017) Whole‐genome landscape of pancreatic neuroendocrine tumours. Nature 543, 65–71. [DOI] [PubMed] [Google Scholar]
- Shen R and Seshan VE (2016) FACETS: allele‐specific copy number and clonal heterogeneity analysis tool for high‐throughput DNA sequencing. Nucleic Acids Res 44, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takane K, Matsusaka K, Ota S, Fukuyo M, Yue Y, Nishimura M, Sakai E, Matsushita K, Miyauchi H, Aburatani H et al (2016) Two subtypes of colorectal tumor with distinct molecular features in familial adenomatous polyposis. Oncotarget 7, 84003–84016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kato K, Notohara K, Hojo H, Ijiri R, Miyake T, Nagahara N, Sasaki F, Kitagawa N, Nakatani Y et al (2001) Frequent beta‐catenin mutation and cytoplasmic/nuclear accumulation in pancreatic solid‐pseudopapillary neoplasm. Cancer Res 61, 8401–8404. [PubMed] [Google Scholar]
- Weigelt B, Bi R, Kumar R, Blecua P, Mandelker DL, Geyer FC, Pareja F, James PA, kConFab Investigators , Couch FJ et al (2018) The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J Natl Cancer Inst 110, 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, Eshleman JR, Goggins MG, Wolfgang CL, Canto MI et al (2011) Whole‐exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin‐dependent pathways. Proc Natl Acad Sci USA 108, 21188–21193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan T, Rindtorff N and Boutros M (2017) Wnt signaling in cancer. Oncogene 36, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Copy number profiles of SPNs analyzed in this study.
Fig. S2. Differential gene expression analysis of SPNs, ADCs and PNETs.
Table S1. Whole‐exome sequencing statistics.
Table S2. Nonsynonymous somatic mutations identified in SPNs using whole‐exome sequencing.
Table S3. Genes in the Wnt pathway assessed by Nanostring nCounter.
Table S4. Probes differentially methylation between SPN samples (this study) and ENCODE samples of epithelial origin.