

Abstract
Reliable biomarkers are required to evaluate and manage pancreatic ductal adenocarcinoma. Circulating tumor cells and circulating tumor DNA are shed into blood and can be relatively easily obtained from minimally invasive liquid biopsies for serial assays and characterization, thereby providing a unique potential for early diagnosis, forecasting disease prognosis, and monitoring of therapeutic response. In this review, we provide an overview of current technologies used to detect circulating tumor cells and circulating tumor DNA and describe recent advances regarding the multiple clinical applications of liquid biopsy in pancreatic ductal adenocarcinoma.
Keywords: circulating tumor cells, circulating tumor DNA, liquid biopsy, pancreatic cancer, pancreatic ductal adenocarcinoma, tumor‐derived circulating cell‐free DNA
Abbreviations
- AJCC
American Joint Committee on Cancer
- ALDH
aldehyde dehydrogenase
- ARMS
amplification-refractory mutation system
- ASCO
American Society of Clinical Oncology
- BD‐IPMN
branch duct type intraductal papillary mucinous neoplasm
- BEAMing
beads, emulsion, amplification, and magnetics
- BPER
base‐position error rate
- CAP
College of American Pathologists
- cast‐PCR
competitive allele‐specific TaqMan polymerase chain reaction
- CCGA
Circulating Cell Free Genome Atlas
- cfDNA
circulating cell‐free DNA
- COLD‐PCR
coamplification at lower denaturation temperature polymerase chain reaction
- CRP
cancer resistance pathway
- CSC
cancer stem cell
- CTCs
circulating tumor cells
- ctDNA
circulating tumor DNA
- CTM
circulating tumor microemboli
- DAPI
4′,6‐diamidino‐2‐phenylindole
- ddPCR
droplet digital polymerase chain reaction
- DEP
dielectrophoresis
- DFS
disease‐free survival
- dPCR
digital polymerase chain reaction
- EMT
epithelial–mesenchymal transition
- EpCAM
epithelial cell adhesion molecule
- EPISPOT
Epithelial ImmunoSPOT Assay
- ESA
epithelial‐specific antigen
- EUS‐FNA
endoscopic ultrasound‐guided fine needle aspiration
- EV
extracellular vesicle
- FISH
fluorescence in situ hybridization
- FMSA
flexible micro spring array
- GEDI
geometrically enhanced differential immunocapture
- GEM
geometrically enhanced mixing
- GO
graphene oxide
- GSI
γ‐secretase inhibitor
- HB
herringbone
- HDAC
histone deacetylase
- iDES
integrated digital error suppression
- IF
immunofluorescence
- IHC
immunohistochemical
- IPMN
intraductal papillary mucinous neoplasm
- ISET
isolation by size of epithelial tumor cells
- LNA‐dPNA PCR clamp
locked nucleic acid-dual peptide nucleic acid polymerase chain reaction clamp
- MD‐IPMN
main duct type intraductal papillary mucinous neoplasm
- NGS
next‐generation sequencing
- OS
overall survival
- PARE
personalized analysis of rearranged ends
- PB
peripheral blood
- PDAC
pancreatic ductal adenocarcinoma
- PFS
progression‐free survival
- PNA
peptide nucleic acid
- PV
portal vein
- QMS
quadrupole magnetic sorter
- qPCR
quantitative polymerase chain reaction
- Safe‐SeqS
safe‐sequencing system
- SE‐iFISH
subtraction enrichment and immunostaining-FISH
- SLB
supported lipid bilayer
1. Pancreatic ductal adenocarcinoma
Pancreatic cancer is the fourth leading cause of cancer mortality in the United States (Kamisawa et al., 2016). In 2018, the American Cancer Society estimated that there will be 55 440 newly diagnosed cases and 44 330 deaths from pancreatic cancer (Siegel et al., 2018). Approximately 95% of pancreatic cancers are classified as exocrine cancers, while less than 5% of pancreatic cancers are endocrine cancers, namely, pancreatic neuroendocrine tumors. The exocrine cancers include pancreatic adenocarcinoma, acinar cell carcinoma, cystadenocarcinoma, and pancreatoblastoma: Pancreatic adenocarcinoma, or pancreatic ductal adenocarcinoma (PDAC), is the major histological subtype that comprises about 90% of all pancreatic cancers (Goel and Sun, 2015).
The TNM stages of pancreatic cancer are based on American Joint Committee on Cancer (AJCC) Cancer Staging Manual, which consider primary tumor size (T), regional lymph node involvement (N), and distant metastasis (M) (Allen et al., 2017; Chun et al., 2018; Kamarajah et al., 2017; Kamisawa et al., 2016). Stages I and II are mostly considered as resectable, and stages III and IV are typically classified as locally advanced and metastatic, respectively. PDACs generally carry a very poor prognosis with the 5‐year survival rate for all stages of PDAC as low as 6–8% (Siegel et al., 2018; Ying et al., 2016). While surgical resection remains the only curative therapy, less than 20% of patients are candidates for surgical resection, which increases the 5‐year survival rate to 15–25% (Luketina et al., 2015; Schlitter et al., 2017). Approximately 50–60% of patients are found to have metastasis at diagnosis due to nonspecific or even lack of symptoms that limits earlier diagnosis (Kleeff et al., 2016), with only a 3% 5‐year survival for distant disease (Siegel et al., 2018).
Clinicians are struggling to develop diagnostic strategies for the early detection of the disease. Adequate biopsy is still challenging because of its poor anatomic location. Endoscopic ultrasound‐guided fine needle aspiration (EUS‐FNA) is preferred for obtaining specimens for biopsy, yet its negative predictive value remains at 16–86% (Mohammad Alizadeh et al., 2016). Currently, the serum level of CA 19‐9 is a widely used biomarker for the diagnosis or monitoring of PDAC, but CA19‐9 alone exhibits a wide range of sensitivity (70–95%) and specificity (70–90%) (Ballehaninna and Chamberlain, 2012; Scara et al., 2015). False‐negative results are observed in patients with the Lewis‐negative blood group, Le(a‐b‐) that occurring in about 5–10% of Caucasians, and false‐positive results have been reported in other diseases including obstructive jaundice, acute cholangitis, and chronic pancreatitis (Passerini et al., 2012; Tanaka et al., 2000). Thus, a highly sensitive, reliable, and noninvasive biomarker for evaluating and managing PDAC patients is still required.
2. Circulating tumor cells and circulating tumor DNA
Circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA), as liquid biopsies, are an emerging minimally invasive tool for cancer diagnosis, surveillance, and treatment. CTCs can be traced back to their first description by Thomas Ashworth in 1869 (Ashworth, 1869). CTCs are released from primary tumor and/or metastatic sites into the bloodstream. Since CTCs exist as rare cells in the blood (one CTC among 106–109 blood cells), recent studies focus on the efficient capture of rare CTCs from whole blood (Ferreira et al., 2016). Investigators use CTCs as a guide to (a) determine prognosis, (b) monitor in real‐time therapeutic responses and tumor recurrence, (c) explore therapeutic targets, and (d) potentially develop new drugs by studying metastatic cancer biology and drug resistance mechanisms in CTCs (Ferreira et al., 2016).
ctDNA is a subset of circulating extracellular DNA in plasma (also called cell‐free DNA, cfDNA), specifically released from cancer cells. ctDNA (known as tumor‐derived cfDNA) may originate from apoptotic and necrotic tumor cells, from living tumor cells, or even from CTCs; thus, it has a variable half‐life from 15 minutes up to 2 h (Alix‐Panabieres and Pantel, 2016; Diaz and Bardelli, 2014; Diehl et al., 2008; Kidess and Jeffrey, 2013; Nordgard et al., 2018). While the size of cfDNA released by apoptotic cells represents approximately 166 bp, ctDNA has recently been reported as being more highly fragmented (Mouliere and Rosenfeld, 2015; Underhill et al., 2016). Detecting ctDNA is generally based on the target mutation (e.g., KRAS, BRAF, EGFR, hypermethylation, and multiple gene panels) (Kidess and Jeffrey, 2013). Due to its small fraction (occasionally < 0.01%) among total cfDNA in circulation, approach with sensitive detection methods for ctDNA is highly recommended (Cheng et al., 2016). Recent advances in ctDNA analysis highlight future critical roles in cancer management of this easily and serially accessible assay: (a) monitoring tumor burden, (b) evaluating therapeutic response, and (c) identifying therapeutic targets through minimally invasive molecular profiling (Ignatiadis et al., 2015). Intratumoral heterogeneity exists due to uneven distribution of cancer subclones in the same tumor (spatial heterogeneity) and due to different genetic alterations that may be selected over time (temporal heterogeneity) as a result of microenvironmental selection, genomic instability, and following multiple drug treatments, where such treatments would ablate cancer cells sensitive to the treatment but not block expansion of residual surviving drug‐resistant cancer cell subpopulations (Dagogo‐Jack and Shaw, 2018; Friedman, 2016; Jeffrey and Toner, 2019; McGranahan and Swanton, 2017). Moreover, most patients with metastatic cancer have multiple rather than solitary metastases, some of which may be discordant with the primary tumor and between other metastases. Sequential tissue sampling of every metastatic lesion is impractical and risky. As a liquid biopsy represents cancer cells or cancer cell products/nucleic acids derived from the entire tumor burdens of the patient, liquid biopsy can be a valuable alternative to tissue biopsies. The following discussion summarizes the current technologies of CTCs and ctDNA and application of these tools to manage patients with PDAC.
3. Current technologies in CTCs
Current CTC technologies include two main steps: CTC enrichment and CTC identification. CTC enrichment strategies focus on improving yield of capturing tumor cells, called capture efficiency, and obtaining high‐purity CTCs via depleting the background blood cells (i.e., leukocytes). The most widely used enrichment strategies are based on immunoaffinity, called label dependent, which uses cell surface markers to capture epithelial tumor cells. Immunomagnetic capture is widely used: The specific antibodies are normally conjugated with magnetic nanoparticles, and a magnetic field is then used to capture the CTCs. Tumor‐specific cell surface antigens, such as epithelial cell adhesion molecule (EpCAM), are targeted for the positive enrichment: CellSearch®, which is the only US Food and Drug Administration‐approved platform, MACS®, and MagSweeper are examples that may use EpCAM‐based or other markers, while AdnaTest uses a cocktail of antibodies against multiple antigens (e.g., EpCAM, EGFR, and HER2). In contrast, negative enrichment is the depletion of nonspecific background cells (i.e., leukocytes) using anti‐CD45 antibodies not expressed by tumor cells: MACS®, Quadrupole Magnetic Sorter (QMS), Dynabeads®, and EasySep™ are based on this strategy.
Antibodies can also be attached to microposts and other surfaces for CTC capture. Microfluidic devices have been developed based on the technology controlling the fluid flow, which offers advantages for CTC research such as improved capture efficiency and high purity (Warkiani et al., 2016). The geometrically enhanced differential immunocapture (GEDI) device uses geometrically enhanced microstructures and combines positive enrichment with hydrodynamic chromatography, which additionally enables cell size‐based separation. Surface‐capture microfluidic devices, such as Herringbone (HB) Chip, Geometrically enhanced mixing (GEM) chip, Graphene oxide (GO) Chip, and the modular sinusoidal system (BioFluidica), increase collision events between the cells and the surface‐coated antibodies. The other kind of microfluidic devices, such as CTC‐iChip, IsoFlux™, LiquidBiopsy, Ephesia chip, and Magnetic Sifter, use microfluidic‐ and immunomagnetic‐based strategies, and these devices exhibited higher sensitivity in CTC separation than CellSearch® (Karabacak et al., 2014).
Another major type of CTC enrichment strategies, known as label‐independent enrichments, relies on biophysical properties (e.g., size, including inertial focusing, electrical charge, and density). A substantial number of microfiltration systems are based on the principle that tumor cells (12–25 μm) are basically larger than leukocytes (8–14 μm) (Sollier et al., 2014). Therefore, these systems use 7–8 μm pores [isolation by size of epithelial tumor cells (ISET) filter device, ScreenCell®, and CellSieve™], or less (VyCAP microsieves which have a membrane thickness smaller than the pore size), microfabricated filter membranes [Flexible Micro Spring Array (FMSA) (Harouaka et al., 2014)], or 3‐dimensional microfiltration layers (FaCTChecker, Resettable Cell Trap, and Cluster‐Chip). Inertial focusing microfluidics can be applied for size‐based separations (Vortex and ClearCell® systems). Dielectrophoresis (DEP) uses the polarizabilities of cells in a nonuniform electrical field. In the electrical field, cells are pushed by either negative or positive force and separated based on their cell size and polarizability. Commercialized DEP systems include ApoStream® and DEPArray™. Recently, microfluidic platforms applied both cell size‐ and deformability‐based systems for CTC enrichment: The Parsortix™ (Xu et al., 2015, 2017) and Celsee™ (Gogoi et al., 2016). A density‐based gradient technology has been also commercialized for separating CTCs: Ficoll‐Paque®, RosetteSep™, OncoQuick®, and Lymphoprep™. Viable CTCs can be further characterized through combining functional assay with capturing CTCs (Alix‐Panabieres et al., 2016). The method that targets secreted tumor‐associated analytes [i.e., Epithelial ImmunoSPOT Assay (EPISPOT)] and the assay based on cell adhesion matrix (CAM) (i.e., Vita‐Assay™ and Vita‐Cap™) are commercially available (references for technology platforms described above are cited in (Ferreira et al., 2016).
After enrichment of CTCs, verification of the captured cells is subsequently required. Immunofluorescence (IF) staining, which usually defines 4′,6‐diamidino‐2‐phenylindole (DAPI) + (nuclear stain), CD45− (leukocyte marker), and cytokeratin (CK) + (epithelial marker), which identify epithelial‐like CTCs, is most extensively used, but immunohistochemical (IHC) staining using chromogenic reporters, fluorescence in situ hybridization (FISH), and molecular analyses ranging from reverse transcription polymerase chain reaction (RT–PCR) to aptamer‐based assays to targeted sequencing is also used (Paterlini‐Brechot and Benali, 2007; Smith et al., 2007; Swennenhuis et al., 2009).
4. Clinical application of CTCs in PDAC
Previous CTC studies in pancreatic cancer are summarized in Table 1.
Table 1.
Previous CTC studies focusing on pancreatic cancer
| Enrichment strategy | Refs | N | Stage | Detection strategy | Detection rate | Enumeration | ||
|---|---|---|---|---|---|---|---|---|
|
IM CellSearch® |
EpCAM | Dotan et al. (2016) | 48 | IV | IF | DAPI+/CD45−/panCK+, MUC‐1+ | 48% (23/48) (≥ 1 CTC): 13% (6/48) (≥ 2 CTCs); 8% (3/37) (≥ 2 CTCs) at first evaluation after Tx | NA |
| Piegeler et al. (2016) | 8 |
IB (n = 1) IIB (n = 1) III (n = 2) IV (n = 4) |
IF | DAPI+/CD45−/CK+ | 87.5% (7/8): 100% (1/1) in Stage IB; 100% (1/1) in Stage IIB; 100% (2/2) in Stage III; 75% (3/4) in Stage IV | Median 4.5 CTCs/7.5 mL, range 0–83 CTCs/7.5 mL | ||
| Bissolati et al. (2015) | 20 | R | IF | DAPI+/CD45−/panCK+ |
20% (4/20) in PB 40% (8/20) in PV |
NA | ||
| Catenacci et al. (2015) | 14 | IIB‐IV | IF | DAPI+/CD45−/EpCAM+ |
21.4% (3/14) in PB 100% (14/14) in PV |
(1) In PB, mean 0.7 CTCs/7.5 mL, median 0 CTCs/7.5 mL, range 0–7 CTCs/7.5 mL. (2) In PV, mean 125.64 CTCs/7.5 mL, median 68.5 CTCs/7.5 mL, range 1–516 CTCs/7.5 mL | ||
| Earl et al. (2015) | 35 |
R (n = 10) LA (n = 11) M (n = 14) |
IF | DAPI+/CD45−/CK+ |
20% (7/35) in total 10% (1/10) in R 42.8% (6/14) in M |
Mean 0.77 CTCs/7.5 mL in total, mean 0.1 CTCs/7.5 mL in R, mean 1.9 CTCs/7.5 mL in M | ||
| Bidard et al. (2013) | 79 | III | IF | CD45−/CK+, EGFR |
5% (4/75) at baseline 9% (5/56) at first evaluation 11% (9/79) in total 50% (2/4) in Stage IV (control) |
1–15 CTCs/7.5 mL | ||
| Kurihara et al. (2008) | 26a |
II (n = 1) III (n = 1) IVA (n = 10) IVB (n = 14) |
IF | DAPI+/CD45−/panCK+ |
42% (11/26) in total 45.8% (11/24) in Stage IV |
Mean 16.9 CTCs/7.5 mL, range 1–105 CTCs/7.5 mL | ||
| Allard et al. (2004) | 16a | IV | IF | DAPI+/CD45−/panCK+ | 19% (4/21) (≥ 2 CTCs) of the samples | Mean 2 ± 6 CTCs/7.5 mL, median 3.5 CTCs/7.5 mL | ||
|
IM CellCollector® |
EpCAM | El‐Heliebi et al. (2018) | 15 |
I (n = 7) II‐III (n = 6) NA (n = 2) |
RCA, IF | KRAS, DAPI+/CD45−/CK18 or PanCK± | 47% (7/15) KRAS mut 40% (6/15) |
Range 1–3 CTCs/patient, KRAS mut 1–8 RCPs/CTC, KRAS wt 1–2 RCPs/CTC |
|
IM MACS |
EpCAM | Effenberger et al. (2018) | 69 | I (n = 2) II (n = 30) III (n = 10) IV (n = 27) |
IF | DAPI+/CD45−/CK+ | 33.3% (23/69) | Range 1–19 CTCs/7.5 mL |
| Zhou et al. (2011) | 25a |
I‐II (n = 5) III (n = 8) IV (n = 12) |
RT–PCR | h‐TERT, CK20, CEA, C‐MET | 100% (25/25) | NA | ||
|
IM Dynabeads® |
MUC1 EpCAM |
de Albuquerque et al. (2012) | 34 | II‐IV | RT–PCR | KRT19, MUC1, EPCAM, CEACAM5, BIRC5 | 47.1% (16/34): 20.6% for KRT19 and MUC1; 23.5% for EPCAM; 2.9% for CEACAM5; 17.6% for BIRC5 | NA |
| IM | anti‐cMET | Zhang et al. (2016b) | 7 | NA | IF, FISH | DAPI+/CD45−/c‐MET+, MET FISH |
0% with c‐MET CTC assay 14% (1/7) with CellSearch® |
Range 0–1 CTCs/7.5 mL (CellSearch®) |
|
IM SE |
CD45(−) | Zhang et al. (2015b) | 22a |
I (n = 2) II (n = 10) III (n = 4) IV (n = 6) |
IF, FISH | DAPI+/CD45−/CK+ and/or CEP8 signal number > 2, DAPI+/CD45−/CK− and CEP8 signal number > 2 | 68.2% (15/22) (≥ 2 CTCs) in total: 9.1% (2/22) with CK+; 59.1% (13/22) with CK−; 9.1% (2/22) (> 10 CTCs); 78.6% (11/14) (≥ 2 CTCs) in PDAC | Median 3 CTCs/3.5 mL, range 0–60 CTCs/3.5 mL, 60 CTCs/3.5 mL in a Pt with stage II, 14 CTCs/3.5 mL in a Pt with stage IV |
|
IM SE |
CD45 (−) | Wu et al. (2018) | 19 |
IIA (n = 3) IIB (n = 11) III (n = 4) IV (n = 1) |
IF, FISH | DAPI+/CD45−/CK+ and/or CEP8 signal number > 2 | 26.3% (5/19) CTMs in total: 21.1% (4/19) at baseline; 27.3% (3/11) in stage IIB; 25% (1/4) in stage III; 100% (1/1) in stage IV | Median 5 CTCs/7.5 mL (at baseline), range 1–30 CTCs/7.5 mL (at baseline) |
|
IM SE |
CD45 (−) | Gao et al. (2016) | 25 |
I (n = 5) II (n = 8) III (n = 6) IV (n = 6) |
IF, FISH | DAPI+/CD45−/CK18 + or CEP8 signal number > 2 | 88% (22/25) | Median 3 CTCs/7.5 mL, range 0–13 CTCs/7.5 mL |
|
IM MACS |
CD45 (−) | Zhang et al. (2015a) | 13 | NA | IF, Aptamer, FISH | DAPI+/CD45−/panCK+, DAPI+/CD45−/BC‐15+ | 84.6% (11/13) | Mean 34.4 CTCs/7.5 mL (panCK+), mean 24 CTCs/7.5 mL (BC‐15 + ) |
| Ren et al. (2011) | 41 | III‐IV | IF | DAPI+/CA19‐9 + /CK+ | 80.5% (33/41) (≥ 2 CTCs) | Mean 16.8 ± 16.0 CTCs/7.5 mL, range 0–59 CTCs/7.5 mL | ||
|
SLB, μF CMx chip |
EpCAM | Chang et al. (2016) | 63 |
I (n = 1) II (n = 32) III (n = 10) IV (n = 20) |
IF | DAPI+/CD45−/panCK+ |
81% (51/63) CTCs 81% (51/63) CTMs (multiple cells ≥ 2 CTCs) |
Mean 70.2 CTCs/2 mL, mean 29.5 CTMs/2 mL |
| Tien et al. (2016) | 41 | IA‐III | IF | DAPI+/CD45−/panCK+ |
39% (16/41) in PB 58.5% (24/41) in PV |
(1) In PB, mean CTCs 92.0/2 mL, median CTCs 52.0/2 mL. (2) In PV, mean CTCs 313.4/2 mL, median CTCs 116.5/2 mL | ||
| IM, μF Parallel flow micro aperture chip |
EpCAM anti‐CEA Size‐based filtration |
Chang et al. (2015) | 12a | IV | IF | DAPI+/CD45−/CK+ | 91.7% (11/12) | Mean 26 ± 11 CTCs/8 mL, range 0–42 CTCs/8 mL, mean 31 CTCs/8 mL (untreated Pts), mean 22 CTCs/8 mL (treated Pts) |
|
μF Nanostructured capture NanoVelcro chip |
EpCAM | Court et al. (2018) | 100 |
I (n = 9) II (n = 31) III (n = 31) IV (n = 29) |
IF | DAPI+/CD45−/CK+ | 78% (78/100): 44.4% (4/9) in stage I; 74.2% (23/31) in stage II; 77.4% (24/31) in stage III; 93.1% (27/29) in stage IV | Median 2 (IQR 1–6) CTCs/4 mL in total, median 7 (IQR 3–13) CTCs/4 mL in occult metastatic Pts |
|
μF Micropost GEDI |
Size‐based filtration EpCAM |
Rhim et al. (2014) | 11 |
I (n = 1) IIA (n = 1) IIB (n = 1) III (n = 1) IV (n = 7) |
IF | DAPI+/CD45−, DAPI+/CD45−/CK+ |
73% (8/11) in PDAC 40% (8/21) in Cystic lesion |
Mean 14.1 ± 18.1 CTCs/mL (PDAC), mean 4.5 ± 7.3 CTCs/mL (Cystic lesion) |
|
μF Cell surface capture GEM |
EpCAM | Sheng et al. (2014) | 18a | IV | IF | DAPI+/CD45−/CK+ | 94.4% (17/18) | Range 0–23 CTCs/7.5 mL |
|
μF Cell surface capture BioFluidica |
EpCAM | Kamande et al. (2013) | 12 |
R (n = 5) M (n = 7) |
IF | DAPI+/CD45−/EpCAM+ | 100% (7/7) in M | Mean 53CTCs/mL in M, median 51 CTCs/mL in M, range 9–95 CTCs/mL in M, mean 11 CTCs/mL in R |
| μF Cell surface capture Slit filtrationeDAR |
EpCAM Size‐based filtration |
Zhao et al. (2013) | 10a | IV | IF | Hoechst+/CD45 + /EpCAM+/CK+ | 80% (8/10) | Range 2–872 CTCs/mL |
| Size‐based filtration ISET | Pore size 8.0 μm | Poruk et al. (2016) | 50 | I (n = 8) II (n = 38)IV (n = 4) |
IF | DAPI+/CD45−/panCK+, DAPI+/CD45−/vimentin+ |
78% (39/50) with eCTCs 52% (26/50) with mCTCs |
Median 30 eCTCs/mL, range 1–251 eCTCs/mL, median 3 mCTCs/mL, range 1–16 mCTCs/mL |
| Khoja et al. (2012) | 53 |
M or Inoperable |
Light microscope, IHC | CD45−, Morphology |
88.9% (24/27) (ISET) 39.6% (21/53) (CellSearch®) |
Mean 26 CTCs/7.5 mL (ISET), median 9 CTCs/7.5 mL (ISET), range 0–240 CTCs/7.5 mL (ISET), mean 2 CTCs/7.5 mL (CellSearch®), median 0 CTCs/7.5 mL (CellSearch®) range 0–15 CTCs/7.5 mL (CellSearch®) | ||
| Size‐based filtration ScreenCell | Pore size 7.5 μm | Sefrioui et al. (2017) | 58a |
L (n = 16) LA (n = 18) M (n = 24) |
Light microscope | Morphology | 56% (33/49) in available samples: 57% (16/28) in L‐LA; 81% (17/21) in M | Median 1 CTC/mL, range 0–151 CTCs/mL |
| Kulemann et al. (2016) | 21 |
IIA (n = 2) IIB (n = 8) III (n = 4) IV (n = 7) |
IF, Light microscope, IHC, PCR | Hoechst+/CK+, Hoechst+/ZEB‐1 + , Morphology, KRAS |
86% (18/21) including KRAS
mut: 100% (2/2) in Stage IIA; 75% (6/8) in Stage IIB; 75% (3/4) in Stage III; 100% (7/7) in Stage IV 66.7% (14/21) with cytology only 23.8% (5/21) CTC clusters 57.1% (4/7) ZEB1 + in Stage IV |
Mean 0.5 CTCs/3 mL, range 0–37 CTC/3 mL | ||
| Cauley et al. (2015) | 105 | IA‐IV | Light microscope | Morphology | 49% (51/105) | NA | ||
| Kulemann et al. (2015) | 11 |
IIB (n = 4) III (n = 3) IV (n = 4) |
Light microscope, RT–PCR | Morphology, KRAS |
18% (2/11) with cytology 73% (8/11) with KRAS mut: 75% (3/4) in Stage IIB; 100% (3/3) in Stage III; 50% (2/4) in Stage IV |
NA | ||
| Iwanicki‐Caron et al. (2013) | 27 |
R (n = 9) LA (n = 9) M (n = 9) |
Light microscope | Morphology | 55.6% (15/27) in total: 44.4% (4/9) in R; 66.7% (6/9) in LA; 55.6% (5/9) in M | NA | ||
| Size‐based filtration FMSA (vs. CellSearch®) | Microfiltration | Ma et al. (2015) | 2a |
IIB (n = 1) IV (n = 1) |
Ad5GTSe infection/GFP, IF | GFP+, CK+/CD45− | 100% (2/2) (FMSA), 50% (1/2) (CellSearch®) | 13–30 CTCs/7.5 mL (FMSA), 0–1 CTCs/7.5 mL (CellSearch®) |
|
Size‐based filtration MetaCell |
Pore size 8.0 μm | Bobek et al. (2014) | 17 |
I (n = 1) IIA (n = 4) IIB (n = 4) III (n = 5) IV (n = 3) |
IF, Light microscope, IHC | DAPI+/CK18 + , Morphology, MGS, CK, CEA, Vimentin | 76.5% (13/17) in total: 78.6% (11/14) in Stage I‐III; 66.7% (2/3) in Stage IV | NA |
| Density Gradient Ficoll‐Paqueplus |
Density Gradient | Gorner et al. (2015) | 6 |
II (n = 2) III (n = 1) IV (n = 3) |
FACS RT–PCR |
Hoechst+/CD45−/EpCAM+, Integrin+, or MUC+, c‐MET, AGR2, EpCAM, Krt‐19, CD45 | 66.6% (4/6): 66.6% (2/3) in Stage II‐III; 66.6% (2/3) in Stage IV | NA |
| CAM assay | Premasekharan et al. (2016) | 2 | IV | FACS | DAPI+/CD45−/CAMhigh/CD14low | 100% (2/2) | NA | |
| oHSV1‐hTERT‐GFP |
Telomerase RT positive cancer cells GFP+ (viable cells) |
Zhang et al. (2016a) | 17 |
IIB (n = 1) III (n = 4) IV (n = 12) |
IF FACS |
CD45−/GFP+ | 88.2% (15/17) | Mean 43.1 CTCs/4 mL |
| No enrichment | Marrinucci et al. (2012) | 18a | IV | IF | DAPI+/CD45−/CK+ | 61% (11/18) (≥ 2 CTCs), 50% (9/18) (≥ 5 CTCs) | Mean 15.8 CTCs/mL |
CAM, cell adhesion matrix; CTC, circulating tumor cell; CTM, Circulating tumor microemboli; eCTC, epithelial‐like CTC; FISH, fluorescent in situ hybridization; IF, immunofluorescence; IHC, immunohistochemistry; IM, immunomagnetic; IQR, interquartile range; LA, locally advanced; M, metastatic; mCTC, mesenchymal‐like CTC; N, number of patients; NA, not available; PB, peripheral blood; PDAC, pancreatic ductal adenocarcinoma; PV, portal vein; R, resectable; RCA, rolling‐circle amplification using padlock probe; RCP, rolling‐circle product; Refs, references; SE, subtraction enrichment; SLB, supported lipid bilayer; Tx, treatment; Pt, patient; μF, microfluidic.
Various tumor types of pancreatic cancers are included.
4.1. Detection
The detection of CTCs in patients of pancreatic cancer has been compared with that in patients with other cancers in previous studies. Using the CellSearch® system, Allard et al. enumerated CTCs in 2183 blood samples from 946 metastatic patients with 12 different cancer types, which included 21 blood samples from 16 patients with pancreatic cancer. Lower number of CTCs was detected in pancreatic cancer (mean, 2 CTCs/7.5 mL) than any other carcinomas, such as prostate cancer, ovarian cancer, breast cancer, gastric cancer, colorectal cancer, bladder cancer, rental cancer, and lung cancer. CTCs above the cutoff level (≥2 CTCs) were detected in only 4 out of 21 samples (19%) (Allard et al., 2004).
In contrast, recent works using state‐of‐the‐art techniques demonstrated comparable detection rates of CTCs in pancreatic cancer when compared with those in different types of carcinomas. Zhang et al. (2016a) used hTERT promoter‐regulated oncolytic herpes simplex virus‐1 that targets telomerase reverse transcriptase‐positive tumor cells, and identified CTCs in 88.2% (15/17) of patients with various stages of pancreatic cancer. Chang et al. developed a parallel flow microfluidic chip that is combined with different strategies such as immunomagnetics and size‐based filtration. This device performed well for isolating of CTCs in patients with metastatic pancreatic cancer (91.7%, 11/12 in pancreatic cancer; 100%, 38/38 in non‐small‐cell lung cancer) (Chang et al., 2015). Another study by Ting et al. applied the microfluidic CTC‐iChip, which depletes normal blood cells by inertial focusing size‐based sorting and separates CTCs immunomagnetically, for single‐cell RNA sequencing. In this study, median 118 CTCs/mL (range, 0–1694) were detected in pancreatic tumor‐bearing mice (KPC mice) (Ting et al., 2014). Varillas et al. (2017) have introduced a detailed procedure for using a microfluidic chip with a herringbone structure and reported that this device could consistently detect a low number of CTCs in pancreatic cancer. Interestingly, El‐Heliebi et al. applied KRAS as a marker for CTC enumeration and molecular characterization. They used an in vivo isolation of CTCs (GILUPI CellCollector®) directly from the vein of patients and applied signal amplification of in situ padlock probes via rolling‐circle amplification: 47% (7/15) of patients were CTC‐positive (range, 1–3 CTCs/patient), and 40% (6/15) of patients had KRAS mutant CTCs (El‐Heliebi et al., 2018).
With regard to the enrichment strategies, size‐based filtering strategies exhibited higher sensitivity in isolating CTCs compared with EpCAM‐based approaches in patients with metastatic or inoperable pancreatic cancer: ISET and CellSearch® detected CTCs in 88.9% (38/50) and in 39.6% (21/53) of patients, respectively (Khoja et al., 2012). A recent study by Brychta et al. compared the performance of these two strategies by cell spiking experiments [EpCAM‐based CTC isolation (IsoFlux) vs. automated size‐based filtration (Siemens Healthineers)]: Especially for low EpCAM expressing cells, the filtration‐based strategy exhibited higher recovery rate (52%) than the IsoFlux device (1%). Additional experiments using the filtration‐based strategy were able to capture CTCs in 42% of frozen diagnostic leukapheresis (DLA) samples from 19 patients with pancreatic cancer. Although there was no difference in prevalence of CTCs in samples from patients with and without metastases (44% vs 40%, respectively), CTC numbers were somewhat higher when distant metastases were present (0–7 for Stage IV disease versus 0–2 for stages 2b‐III) (Brychta et al., 2017).
4.2. Early diagnosis
The potential role of CTCs as an early diagnostic marker has recently been revealed by Rhim et al. (Table 2). Using GEDI chip, CTCs were captured in three different subject groups [PDAC patients at all stages, patients with precancerous cystic lesion, that is, intraductal papillary mucinous neoplasm (IPMN) or mucinous cystic neoplasm, and cancer‐free controls]. Interestingly, CTCs were detected in 40% (8/21) of the patients with precancerous lesions: Circulating pancreas epithelial cells may precede the detectable tumors. The detection rates of CTCs were 73% (8/11) and 0% (0/19) in PDAC patients and cancer‐free group, respectively (Rhim et al., 2014).
Table 2.
Studies investigating the role of CTC/ctDNA detection in early cancer diagnosis
| References | Patients | Analyte | Methods | Results | Comments |
|---|---|---|---|---|---|
| Rhim et al. (2014) |
PDAC (n = 11), Precancerous cystic lesions (n = 21): Side‐branch IPMN (n = 18); MCN (n = 3) Cancer‐free controls (n = 19) |
CTC | microfluidic platform GEDI |
CTCs were captured in: 8 of 11 (73%) patients with PDAC 8 of 21 (40%) patients with cystic lesions; 0 of 19 (0%) cancer‐free controls |
Pancreas epithelial cells can be detected in patients with cystic lesions of pancreas before the clinical diagnosis of cancer. |
| Berger et al. (2016) | PDAC (stage IV) (n = 24), IPMN (n = 21), Borderline IPMN (n = 16), SCA (n = 26), Cancer‐free controls (n = 38) | ctDNA | ddPCR (Bio‐Rad) |
mean cfDNA value of:
GNAS mut ctDNA:
KRAS mut ctDNA:
|
cfDNA discriminates IPMN patients from controls Detection of GNAS and KRAS mutations discriminates IPMN patients from those with harmless pancreatic tumors |
cfDNA, cell‐free DNA; CTC, circulating tumor cell; ctDNA, circulating tumor DNA; ddPCR, droplet digital PCR; IPMN, intraductal papillary mucinous neoplasm; MCN, mucinous cystic neoplasm; PDAC, pancreatic ductal adenocarcinoma; SCA, serous cystadenoma.
4.3. A marker of advanced disease
The correlation of CTC levels with more aggressive pathologic features and with advanced disease is still debated. A multicenter randomized clinical trial suggested that CTC detection with CellSearch® significantly correlated with aggressive tumor differentiation (Bidard et al., 2013). In another study, which used a modular microfluidic system, CTC levels isolated from metastatic PDAC patients (mean 53 CTCs/mL, n = 7 patients) was significantly higher than those from resectable PDAC patients (mean 11 CTCs/mL, n = 5 patients), although further testing will be required because of the small numbers of patients tested in this first proof‐of‐principle assay (Kamande et al., 2013). The expression of C‐MET, CK20, and CEA mRNA detected by RT–PCR after MACS purification correlated with TNM stage (Zhou et al., 2011). More recently, Court et al. (2018) preoperatively enumerated CTC using the microfluidic NanoVelcro chip and reported that PDAC patients with occult metastatic disease had significantly more CTCs than PDAC patients with localized disease (median 7 CTCs vs. 1 CTC, P < 0.0001).
In contrast, Cauley et al. (2015) described that CTC positivity was not associated with tumor characteristics, lymph node metastasis, respectability, and advanced TNM stage. Similarly, the percentage of CTC detection using size‐based filtration was not associated with the TNM stage or distant metastasis (Bobek et al., 2014; Kulemann et al., 2015).
4.4. Prognosis
Studies investigating the role of CTC detection as a prognostic marker are summarized in Table 3. Research efforts on CTC enumeration for better prognostic classification are well underway. Several studies discussed below performed multivariable analysis using the Cox regression model, which exhibits CTCs as an independent prognostic factor. Bidard et al. (2013) conducted multicenter randomized clinical trial evaluating 79 patients with locally advanced nonmetastatic PDAC. Patients were randomly assigned to receive gemcitabine alone, or gemcitabine plus erlotinib. The CTC positivity was measured by CellSearch® at two different time points (at baseline and at two months): The overall detection rate of CTCs (either at baseline or at two months) was 11%. CTC positivity in locally advanced pancreatic adenocarcinoma at any time point was an independent prognostic factor for overall survival (OS) in multivariable analysis but not for progression‐free survival (PFS). A more recent study by Effenberger et al. enrolled 69 patients with PDAC and identified CTCs using MACS enrichment: Here, CTC positivity was an independent risk factor of reduced PFS (HR = 4.543, P = 0.006) and OS (HR = 2.093, P = 0.028) (Effenberger et al., 2018). Studies using different platforms in PDAC patients exhibited association of CTCs with survival rates. Chang et al. used a supported lipid bilayer (SLB) surface‐coated microfluidic chip (CMx platform): Patients with unfavorable circulating tumor microemboli (CTM) levels exhibited shorter PFS and OS when compared with patients with favorable CTM levels (PFS, 2.7 months vs. 12.1 months, P < 0.0001; OS, 6.4 months vs. 19.8 months, P < 0.0001). These associations were still observed in each subgroup (early stage and advanced stage) (Chang et al., 2016). Gao et al. (2016) applied EpCAM independent subtraction enrichment and immunostaining‐FISH (SE‐iFISH) to enumerate CTCs and demonstrated that the presence of ≥ 3 CTCs/7.5 mL was the strong predictive factor for worse OS (HR = 4.547, P = 0.016). Poruk et al. compared epithelial CTCs and mesenchymal‐like CTCs using IF staining for panCK and vimentin markers, respectively, after the size‐based CTC separation. The epithelial CTCs (CK‐positive) were strongly associated with poorer survival but not mesenchymal‐like CTCs (P < 0.01 vs. P = 0.39). With regard to median time to recurrence, detection of CTCs expressing both CK and vimentin was the significant predictive factor for earlier recurrence (P = 0.01) (Poruk et al., 2016). A recent useful meta‐analysis described that detectable baseline CTCs including disseminated tumor cells in the bone marrow was associated with worse disease‐free survival (DFS)/PFS (HR = 1.93, P = 0.007) and OS in pancreatic cancer (HR = 1.84, P ≤ 0.0001) (Stephenson et al., 2017).
Table 3.
Studies investigating the role of CTC/ctDNA detection as a prognostic marker
| References | N | Analyte | Methods | Sampling points at | Results |
|---|---|---|---|---|---|
| Wu et al. (2018) | 19 | CTC | SET‐iFISH | Before the start of Tx, 10 days after Op, 1 month after Op, 3 months after Op, 7 months after Op |
The median OS of the CTM (+) and CTM (−) patients (at baseline) were 7.3 and 25.4 months (P = 0.001). The median DFS of the CTM (+) and CTM (−) patients (at baseline) were 1.8 and 18.97 months (P = 0.037 ) |
| Court et al. (2018) | 100 | CTC | NanoVelcro chip | Before the start of Tx |
CTC positivity was a multivariate predictor of OS (HR, 1.38, P = 0.040). CTC count was a univariate predictor of recurrence‐free survival (HR, 2.36, P = 0.017). |
| Effenberger et al. (2018) | 69 | CTC | MACS | Before the start of Tx |
CTC positivity was independent risk factor of reduced PFS (HR, 4.543, P = 0.006). CTC positivity was independent risk factor of shortened OS (HR, 2.093, P = 0.028). |
| Gao et al. (2016) | 25 | CTC | SE‐iFISH | Before the start of Tx | The median OS of the CTC ≥ 3 and CTC < 3 patients were 10.2 and 15.2 months (P = 0.023) |
| Chang et al. (2016) | 63 | CTC | SLB μF CMx | Before the start of Tx | Survival difference between favorable (CTM < 30) patients and unfavorable (CTM ≥ 30) patients (PFS, 12.1 vs. 2.7 months; OS, 19.8 vs. 6.4 months) |
| Poruk et al. (2016) | 50 | CTC | ISET | Before the start of Tx | Epithelial CTC positivity was associated with worse survival rate (median survival, 13.7 months vs. not reached, P = 0.008) |
| Zhang et al. (2015b) | 22 | CTC | SE‐iFISH | Before the start of Tx | CTC positivity (≥2/3.75 mL) correlated with worse survival rate (P = 0.0458) |
| Bidard et al. (2013) | 79 | CTC | CellSearch® | Before the start of Tx. After 2 months of Tx | CTC positivity (at baseline and/or at 2 months) correlated with poor OS (RR = 2.5, P = 0.01) |
| de Albuquerque et al. (2012) | 34 | CTC | Dynabeads® | Before the start of Tx | The median PFS of the CTC (+) and CTC (−) patients were 66.0 and 138.0 days (P < 0.01) |
| Kurihara et al. (2008) | 26 | CTC | CellSearch® | Before the start of Tx | The MSTs of the CTC (+) and CTC (−) patients were 110.5 and 375.8 days (P < 0.001) |
| Bernard et al. (2019) | 194 | ctDNA | ddPCR (Bio‐Rad) | Before the start of Tx (n = 175): Serially monitored during Tx (n = 68) |
Baseline ctDNA (+) was associated with shorter PFS (HR = 1.8, P = 0.019) in metastatic PDAC. Baseline ctDNA (+) was associated with shorter OS (HR = 2.8, P = 0.0045) in metastatic PDAC. Baseline ctDNA and exoDNA MAF ≥ 5% was a significant predictor of OS (HR = 7.73, P = 0.00002) in metastatic PDAC |
| Perets et al. (2018) | 17 | ctDNA | Targeted sequencing (Ion PGM™) | Before the start of Tx |
The OS of KRAS
mut ctDNA(+) and ctDNA(−) patients were 8 and 37.5 months (P < 0.004). The OS negatively correlated with the change in ctDNA levels (between each pair of consecutive samples) (r = ‐0.76, P = 0.03). |
| Kim et al. (2018) | 106 | ctDNA | ddPCR (Bio‐Rad) | Before the start of Tx: Every 3 months after Tx |
Baseline KRAS mutation concentration (HR = 2.08, P = 0.009) and KRAS fraction (HR = 1.73, P = 0.042) were significant prognostic factors for PFS. Baseline KRAS mutation concentration (HR = 1.97, P = 0.034) was a significant prognostic factor for OS. Increase of cfDNA concentration, KRAS mut ctDNA concentration and KRAS fraction (in the sample collected at 6 months after Tx) were correlated with OS (P < 0.001, P = 0.013, and P = 0.036, respectively). |
| Cheng et al. (2017) | 188 | ctDNA | Targeted sequencing (Hi‐Seq 2500), ddPCR (Bio‐Rad) | Before the start of Tx: For a subset of cases, multiple time points after Tx | ERBB2 exon 17 mutation (HR = 1.61, P = 0.035) and KRAS G12V mutation (HR = 1.45, P = 0.019) were independent prognostic factors for OS. |
| Adamo et al. (2017) | 26 | ctDNA | Targeted sequencing (Ion PGMTM), ddPCR (Bio‐Rad) | Before the start of Tx | The KRAS mut ctDNA correlated with poorer disease‐specific survival (P = 0.018). |
| Del Re et al. (2017) | 27 | ctDNA | ddPCR (Bio‐Rad) | Before the start of Tx: Subsequently after 15 days of Tx and at first radiologic evaluation |
Increase of ctDNA (in the sample collected at day 15) is correlated with PFS and OS (PFS, 2.5 vs 7.5 months, P = 0.03; OS 6.5 vs 11.5 months, P = 0.009). Baseline KRAS mut was not associated with PFS and OS (P = 0.24 and P = 0.16). |
| Pietrasz et al. (2017) | 135 | ctDNA | Targeted sequencing (Ion Proton™) digital PCR (RainDrop™) | Before the start of adjuvant CTx, (n = 31). Before the start of Tx (n = 104) |
The DFS of ctDNA (+) and ctDNA (−) patients were 4.6 and 17.6 months (P = 0.03) in resectable PDAC (n = 31). The OS of ctDNA(+) and ctDNA(−) patients were 19.3 and 32.2 months (P = 0.027) in resectable PDAC (n = 31). The OS of ctDNA(+) and ctDNA(−) patients were 6.5 and 19.0 months (P < 0.001) in advanced PDAC (n = 104 ) |
| Pishvaian et al. (2017) | 34 | ctDNA | Targeted sequencing (Hi‐Seq 2500) | Not mentioned | Detectable ctDNA correlated with poorer OS (P = 0.045). |
| Sefrioui et al. (2017) | 68 | ctDNA | ddPCR (Bio‐Rad) | Before the start of Tx | The median OS of KRAS mut ctDNA(+) and ctDNA(−) patients were 5.2 and 11 months (P = 0.01) |
| Hadano et al. (2016) | 105 | ctDNA | ddPCR (Bio‐Rad) | Before the start of Tx |
The DFS of ctDNA (+) and ctDNA (−) patients were 6.1 and 16.1 months (P < 0.001). The OS of ctDNA (+) and ctDNA (−) patients were 13.6 and 27.6 months (P < 0.0001) |
| Earl et al. (2015) | 31 | ctDNA | ddPCR (Bio‐Rad) | Before the start of Tx (n = 24). After the start of Tx (n = 7) | The OS of KRAS mut ctDNA(+) and ctDNA(−) patients were 60 and 772 days (P = 0.001). |
| Kinugasa et al. (2015) | 75a | ctDNA | ddPCR (Bio‐Rad) | Before the start of Tx | The MST of KRAS mut ctDNA(+) and ctDNA(−) patients were 276 and 413 days (P = 0.02) – KRAS G12V mutation was most well correlated (219 days vs. 410 days) |
| Sausen et al. (2015) | 51 | ctDNA | ddPCR (Bio‐Rad) | Before the start of Tx: For a subset of cases, multiple time points after surgery |
The PFS of ctDNA (+) and ctDNA (−) patients (at baseline) were 7.9 and 15.2 months (P = 0.0151). The PFS of ctDNA (+) and ctDNA (−) patients (after surgery) were 9.9 months and not reached (P = 0.0199). |
| Tjensvoll et al. (2016) | 14a | ctDNA | PNA‐mediated real‐time PCR clamping | Before the start of Tx: Subsequently every month during Tx |
ctDNA shows trends toward reduced PFS and OS (P = 0.064 and 0.066). ctDNA levels before initiation of Tx is independent prognostic factor for PFS and OS (HR 1.31, P = 0.047). |
| Takai et al. (2015) | 259 | ctDNA | digital PCR (RainDrop™) | Before the start of Tx | The KRAS mut ctDNA correlated with poorer OS (P < 0.0001). |
| Singh et al. (2015) | 127a, b | ctDNA | Nested PCR | Not mentioned | The median OS of high cfDNA and low cfDNA patients were 3 and 11 months (P = 0.002). KRAS mut was not associated with survival pattern of patients (P = 0.398). |
| Chen et al. (2010) | 91 | ctDNA | Direct sequencing | Before the start of Tx | The MST of KRAS mut ctDNA(+) and ctDNA(−) patients were 3.9 and 10.2 months (P < 0.001) |
CTC, circulating tumor cell; ctDNA, circulating tumor DNA; CTM, circulating tumor microemboli; CTx, chemotherapy; ddPCR, droplet digital PCR; DFS, disease‐free survival; exoDNA, exosome DNA; MAF, mutant allele fraction; MST, median survival time; N, number of patients; Op, operation; OS, overall survival; PFS, progression‐free survival; Tx, treatment.
Various tumor types of pancreatic cancers are included
KRAS mutation test was available for 110 samples.
4.5. Different sampling sites
Research comparing CTCs in portal vein (PV) and those in peripheral blood (PB) is in progress (Table 4). Bissolati et al. evaluated PV samplings in 20 patients with nonmetastatic PDAC undergoing surgical resection. Five out of nine CTC‐positive patients had CTCs in PV but not in systemic circulation, detected by CellSearch®. At 3‐year follow‐up, patients with detectable CTCs in PV exhibited higher rate of liver metastasis than patients without detectable CTCs in PV (53% vs. 8%, P = 0.038) (Bissolati et al., 2015). Catenacci et al. evaluated CTCs in EUS‐guided PV sampling. Using CellSearch®, they detected CTCs in PV blood samples from 100% (18/18) of patients, while only four patients (22.2%) had CTCs in the PB. Even in patients with nonmetastatic and localized or borderline‐resectable pancreatic cancer, high levels of CTCs were detected (mean 83.2 CTCs/7.5 mL) in PV (Catenacci et al., 2015). Further recently, Tien et al. (2016) collected intraoperative PB and PV samples from 41 PDAC patients. CTC count (CMx platform) in PV was a strong predictor for liver metastasis in a 6‐month follow‐up after surgery (P = 0.002). The PV is the main entrance for distant metastasis of PDAC, and tumor cells spread into blood circulation before radiologically detected. CTCs in PV seem to more closely reflect the metastatic potential, although prospective studies with large cohorts are still required.
Table 4.
Studies investigating the role of CTCs detected in portal vein samples
| References | N | Methods | Sampling points at | Results |
|---|---|---|---|---|
| Bissolati et al. (2015) | 20 | CellSearch® | At surgery, before any manipulation of cancer | Liver metastases occurred more frequently 2–3 years after surgery in portal vein CTC (+) patients (57.1% vs. 8.3%, P = 0.038). |
| Tien et al. (2016) | 41 | SLB μF CMx | At surgery, before any manipulation of cancer | CTCs count in portal venous blood is the significant predictor for liver metastases within 6 months after surgery (P = 0.0042). |
CTC, circulating tumor cell; N, number of patients; SLB, supported lipid bilayer; μF, microfluidic.
4.6. Additional markers for CTCs in PDAC
Epithelial–mesenchymal transition (EMT) may explain how the epithelial tumor cells disseminate from primary site and penetrate the endothelium of blood vessel (Chaffer and Weinberg, 2011). Even though the extent of tumor cells undergoing EMT still remains unclear, the epithelial markers (e.g., EpCAM and CK) of epithelial cells are downregulated by EMT‐inducing signals; thus, CTC capture strategies targeting expression of epithelial markers may fail to isolate a subset of CTCs (Krebs et al., 2014). The expression of epithelial markers such as EpCAM, CK, and E‐cadherin has been reported to be reduced lower than 40% in CTCs of PDAC (Rhim et al., 2012). Similarly, CellSearch® detected CTCs in 39.6% (21/53) of patients with metastatic PDAC, while ISET exhibited better enrichment of CTCs (CTC positivity in 88.9% of patients with metastatic PDAC) (Khoja et al., 2012). Combining additional markers for capturing mesenchymal‐like CTCs remain to be identified. Potential mesenchymal markers include the following: ZEB1, SNAI1, vimentin, N‐cadherin, FGFR2, PLS3, Twist1, and PI3K/AKT (Barriere et al., 2014). A few recent studies have reported the application of mesenchymal markers to detect CTCs in PDAC. CTCs enriched by ScreenCell® filtration devices were stained with ZEB1 and CK. ZEB1‐positive CTCs were found in almost exclusively in patients with metastatic PDAC (P = 0.01) (Kulemann et al., 2016). Dotan et al. evaluated 23 patients with metastasis who had at least one CTC detected at baseline by using CellSearch®. They assessed for the expression of MUC‐1, which play a role of inducing EMT: MUC‐1 expression was observed in 43% (10/23) of the patients, and patients with CTCs positive for MUC‐1 had shorter median OS than those with CTCs negative for MUC‐1 (2.7 months vs. 9.6 months, P = 0.044) (Dotan et al., 2016). Another study, which compared epithelial CTCs and mesenchymal‐like CTCs using a vimentin marker, was discussed above (Poruk et al., 2016). However, blood cells including monocytes and granulocytes retain vimentin expression during the maturation, which warrant additional confirmation of tumor‐specific markers (Dellagi et al., 1983).
A subset of tumor cells, so‐called cancer stem cells (CSCs), have properties of stem cells and display self‐renewing and multipotency capabilities, which are considered to be responsible for metastasis, chemoresistance, and recurrence of tumors (Krebs et al., 2014; Satoh et al., 2015). It has been reported that CSC and EMT share common molecular pathways (e.g., Wnt/ß‐catenin and Notch signaling), and epithelial cells undergoing EMT acquire CSC features (Igawa et al., 2014). Key markers for identifying pancreatic CSCs include CD133 and aldehyde dehydrogenase (ALDH) (Fitzgerald and McCubrey, 2014). Marker combinations of CD44, CD24, and epithelial‐specific antigen (ESA) were also identified as indicators of pancreatic CSCs (Li et al., 2007). Other putative markers for pancreatic CSCs include c‐Met, doublecortin‐like kinase 1, and CD44v6 (Polireddy and Chen, 2016). A recent study by Poruk et al. evaluated 60 consecutive PDAC patients undergoing surgery. CTCs were detected by IF staining using CK, CD133, CD44, and ALDH, after isolated by ISET. CK+/ALDH+ CTCs and CK+/CD133 + /CD44 + CTCs were detected in 77% (46/60) and in 57% (46/60) of patients, respectively. For the 59 nonmetastatic patients, ALDH‐positive CTCs and CK+/CD133 + /CD44 + CTCs were significantly associated with decreased DFS and higher risk of tumor recurrence (Poruk et al., 2017).
5. Current technologies in ctDNA
Since ctDNA is present in minute quantity in the bloodstream, extraction of cfDNA without contamination of plasma with genomic DNA is a major challenge in ctDNA analysis. Preanalytical variables that include specimen types (plasma or serum), specimen collection procedures (time to processing of whole blood), blood collection tubes, specimen handling (including centrifugation protocols and temperature), and methods of cfDNA isolation and purification are the most important factors to control this success (Diefenbach et al., 2018; Markus et al., 2018; Sato et al., 2018). Plasma has been preferred as a source for extracting circulating DNA. Even though serum contains 2–24 times higher amount of cfDNA than plasma, serum is not recommended due to the possible contamination from white blood cells during the clotting process (Heitzer et al., 2015; Parpart‐Li et al., 2017; Trigg et al., 2018; Zhao et al., 2019). If specimen processing can be performed within 6 h from collection, standard K2EDTA collection tubes are suitable for blood sampling. However, when the processing is delayed by up to 48 h, specialized cell‐stabilizing blood collection tubes should be used to reduce contamination by genomic DNA released from leukocyte lysis (Alidousty et al., 2017; Medina Diaz et al., 2016; Merker et al., 2018; Risberg et al., 2018; Ward Gahlawat et al., 2019; Warton et al., 2017). Current evidence recommends that isolated plasma, not whole blood, can be stored frozen up to 9 months or up to a few years, depending on analytical goals (van Dessel et al., 2017; Meddeb et al., 2019). The isolated plasma is preferably aliquoted into a single use fraction: A single freeze–thaw cycle had no significant effect on cfDNA stability (Bronkhorst et al., 2015; Merker et al., 2018). Several issues regarding DNA isolation and nonmalignant conditions that induce the release of cfDNA should be considered, but the following discussion focuses more on the techniques in progress for sensitive detection of the small fraction of ctDNA (Heitzer et al., 2015; Qin et al., 2016).
Based on PCR technology, new technologies including real‐time quantitative PCR (qPCR) (Brown, 2016), amplification‐refractory mutation system (ARMS)‐based qPCR (Zhang et al., 2015c), competitive allele‐specific TaqMan PCR (cast‐PCR) (Ashida et al., 2016; Reid et al., 2015), coamplification at lower denaturation temperature PCR (COLD‐PCR) (Milbury et al., 2011) have been introduced. More recently, digital PCR (dPCR), which uses droplets to compartmentalize individual DNA strands, reached the high sensitivity ranging from 0.1% to 0.001% and is therefore beneficial to detect low allele frequency variants (Gorgannezhad et al., 2018; Vogelstein and Kinzler, 1999). dPCR includes droplet PCR, Bio‐Rad droplet dPCR (ddPCR) platform (Hindson et al., 2011), and BEAMing (beads, emulsion, amplification and magnetics) (Chen et al., 2013): This method is currently among the most promising of targeted approaches, which focuses on the detection of rare mutations in DNA samples with prior knowledge of genetic changes at specific loci of the tumor (e.g., KRAS, BRCA2, ERBB2, and EGFR) (Alix‐Panabieres and Pantel, 2016; Cheng et al., 2017) and exhibits high analytical sensitivity. BEAMing combines emulsion PCR amplification and flow cytometry and therefore can be assessed in the standard laboratory setting (Dressman et al., 2003). BEAMing quantifies independently the fluorescently labeled particles, which is able to detect the rare variants with allele frequency < 0.01%. This method enables the counting of error rate of DNA polymerases (Gorgannezhad et al., 2018). The ddPCR platform performs PCR amplification within water‐in‐oil emulsion droplets where individual DNA molecules are dispersed in. Using fluorescently labeled probes, droplets can be identified as a binary (mutant‐positive or mutant‐negative) system. The Bio‐Rad QX‐200 platform produces 20 000 droplets and is one of the most commonly used dPCR systems for ctDNA detection (Gorgannezhad et al., 2018).
Next‐generation sequencing (NGS), or a massively parallel sequencing, detects a wider range of mutation with higher coverage, but with lower sensitivity (approximately 1%) than dPCR. The targeted NGS approach sequences multiple cancer‐associated genes (Zill et al., 2015). Platforms such as safe‐sequencing system (Safe‐SeqS) (Kinde et al., 2011), TAm‐Seq (Forshew et al., 2012), Ion‐AmpliSeq (Rothe et al., 2014), CAPP‐Seq (Newman et al., 2014), and sensitive mutation detection using sequencing (SiMSen‐seq) (Stahlberg et al., 2017) have been developed. Zill et al. used Guardant360 assay to sequence cfDNA in 21 867 advanced cancer patients including 867 PDAC samples and reported the genomic findings and the response outcomes (Zill et al., 2018). Recent progress enabled whole‐genome sequencing to be applied to a liquid biopsy (Dawson et al., 2013). These NGS approaches largely extended noninvasive profiling of tumors not only focus on single nucleotide variants but also identify structural variants and copy number variations [e.g., personalized analysis of rearranged ends (PARE)] (Leary et al., 2012). Recent advances in NGS technology enable similar sensitivity to detection of ctDNA as by digital PCR. A recent study showed a statistical method based on each base‐position error rate (BPER), which detects variants with low allele frequency as low as 0.003 (single nucleotide variation) and 0.001 (insertions/deletions) (Pécuchet et al., 2016). Newman et al. recently developed an integrated digital error suppression (iDES)‐enhanced CAPP‐Seq, which incorporates in silico removal of artifacts detected in cfDNA sequencing data. This strategy enabled very sensitive detection of tumor‐derived DNA down to 0.002% for generalized iDES‐enhanced CAPP‐Seq and 0.00025% using a customized panel (Newman et al., 2016). Other newer methods include the use of bar‐coded amplicon‐based NGS rather than hybrid capture‐based plasma NGS (Guibert et al., 2018) or an improved method using dual peptide nucleic acid (PNA) clamping‐mediated locked nucleic acid‐dual peptide nucleic acid PCR clamp (LNA‐dPNA PCR clamp) with sensitivities in the 0.01%‐0.1% range (Zhang et al., 2019). Figure 1 summarizes the various technologies and the rages of their limit of detection.
Figure 1.
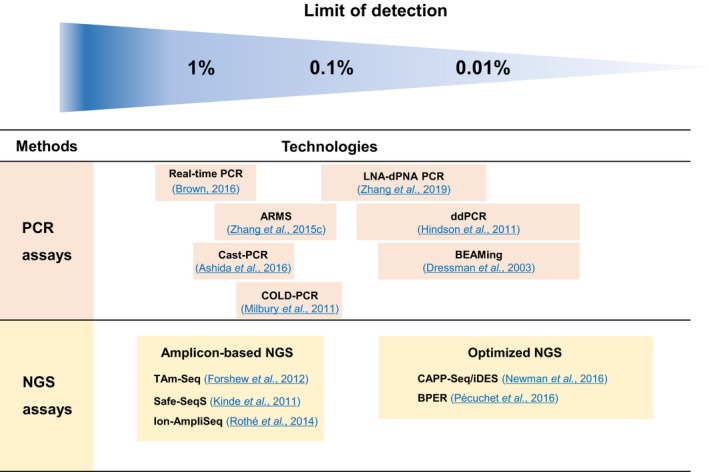
Examples of technology platforms for detecting circulating tumor DNA and limit of detection ranges. These depend on number of mutations measured and quantity of DNA present in a blood sample. Optimized NGS techniques provide sequencing error correction. Other ctDNA assays being applied to pancreatic cancer include personalized panels and commercially available tests. PCR: polymerase chain reaction; NGS: next‐generation sequencing; ARMS: amplification‐refractory mutation system; COLD‐PCR: coamplification at lower denaturation temperature PCR; Cast‐PCR: competitive allele‐specific TaqMan PCR; LNA‐dPNA PCR: locked nucleic acid‐dual peptide nucleic acid PCR clamp; ddPCR: droplet digital PCR; BEAMing: beads, emulsion, amplification, and magnetics digital PCR; TAm‐Seq: tagged‐amplicon deep sequencing; CAPP‐Seq/iDES: cancer personalized profiling by deep sequencing with integrated digital error suppression; BPER: base‐position error rate.
There has been encouraging improvement in the quest for early detection of pancreatic cancer. CancerSEEK, a multi‐analyte blood test, combines multiplex PCR (16 genes) and immunoassay (8 protein biomarkers) (Cohen et al., 2018). This method has shown over 69% sensitivity and over 99% specificity for five cancers including pancreatic cancer aiming to screen different cancers in the general population (Kalinich and Haber, 2018).
6. Clinical application of ctDNA in PDAC
Previous ctDNA studies in pancreatic cancer are summarized in Table 5. Several studies demonstrated that the amounts of plasma DNA in patients with cancer is higher than those in healthy individuals (Anker et al., 1999; Sozzi et al., 2003). With regard to PDAC, multiple studies have reported that cfDNA concentration was higher in pancreatic cancer patients compared with normal controls and in advanced stages compared with early stages (Berger et al., 2016; Singh et al., 2015; Takai et al., 2015).
Table 5.
Previous ctDNA studies focusing on pancreatic cancer
| Method | Refs | N | Stage | Paired tissue | Sample | Target | Detection rate |
|---|---|---|---|---|---|---|---|
| ddPCR Bio‐Rad | Bernard et al. (2019) | 194 | R (n = 71), M (n = 123) | 14 | Plasma | KRAS: ‐ G12D; G12V; G12R; G12C; G12S; G13D | (1) Tissue mutation: 85.7% (12/14), (2) Concordance rate (Tissue vs. ctDNA): 68.2% (15/22), (3) ctDNA: 52.0% (53/102) in R (therapy naïve patients), 31.8% (21/66) in M (therapy naïve patients) |
| Kim et al. (2018) | 106 | R (n = 41), LA (n = 25), M (n = 40) | 77 | Plasma | KRAS: G12D; G12V; G12R; G12S; G13D | (1) Tissue mutation: 96.1% (74/77), (2) Concordance rate (Tissue vs. ctDNA): 76.6% (59/77), (3) ctDNA: 77.9% (60/77) in available samples: 68.6% (24/35) in R; 83.3% (5/6) in LA; 86.1% (31/36) in M | |
| Del Re et al. (2017) | 27 | III (n = 4), IV (n = 23) | NA | Plasma | KRAS: G12D; G12V; G12R; G13D | ctDNA, 70.4% (19/27): 25% (1/4) in stage III; 78% (18/23) in stage IV; G12D 74% (14/19); G12V 11% (2/19); G12R 11% (2/19); G13D 5% (1/19) | |
| Sefrioui et al. (2017) | 58b | L (n = 16), LA (n = 18), M (n = 24) | 27 | Plasma | KRAS | (1) Tissue mutation: 63% (17/27), (2) Concordance rate (Tissue vs. ctDNA): 70.4% (19/27), (3) cfDNA concentration, median 59.5 ng/mL (range 12.9–925.3 ng/mL): 73.8 ± 45.6 in L; 77.2 ± 41.1 in LA; 122.4 ± 44 in M, (4) ctDNA: 56% (31/55) in available samples, (5) Diagnosis of PDAC using ctDNA: Sensitivity: 65%; Specificity: 75% | |
| Hadano et al. (2016) | 105 | R (n = 105): I (n = 2); II (n = 82); III (n = 3); IV (n = 18) | 105 | Plasma | KRAS: G12D; G12V; G12R | (1) Tissue mutation – 82% (86/105): G12D 42% (44/86); G12V 29% (30/86); G12R 11% (12/86). (2) ctDNA – 31% (33/105): G12D 73% (24/33); G12V 21% (7/33); G12R 6% (2/33). (3) ctDNA concentration: mean 10.1 copies of ctDNA/mL | |
| Berger et al. (2016) | 87b , c | IV | 16 | Plasmaor Serum | GNAS: codon 201; KRAS; G12D; G12V | (1) cfDNA concentration: 4.220 ± 2.501 ng·mL−1 in PDAC; 0.2887 ± 0.0319 ng·mL−1 in IPMN; 0.1360 ± 0.0203 ng·mL−1 in NC. (2) GNAS mut ctDNA: 71.4% (15/21) in IPMN; 25.0% (6/24) in PDAC. (3) KRAS mut ctDNA: 41.7% (10/24) in PDAC; 0% in SCA, IPMN and NC. (4) Concordance rate (Tissue vs. ctDNA): 56.3% | |
| Earl et al. (2015) | 31 | R (n = 10), LA (n = 8), M (n = 13) | 12 | Plasma | KRAS: G12D; G12V; G12R | (1) cfDNA concentration: median 93 RNaseP/20 μL, range 6–1663 RNaseP/20 μL. (2) ctDNA – 26% (8/31): 30% (3/10) in R; 12.5% (1/8) in LA; 30.8% (4/13) in M; G12D (6/8); G12R (1/8); G12V (1/8). (3) Tissue mutation: 58.3% (7/12). (4) ctDNA/Tissue mutation: 60% (3/5) | |
| Kinugasa et al. (2015) | 75b | II (n = 2), III (n = 5), IV (n = 68) | 75 | Serum | KRAS: G12D; G12V; G12R | (1) Tissue mutation – 74.7% (56/75): G12D 29.3% (22/75); G12V 37.3% (28/75); G12R 8.0% (6/75); (2) ctDNA – 62.6% (47/75): G12D 38.6% (29/75); G12V 34.6% (26/75); G12R 5.3% (4/75). (3) Concordance rate (Tissue vs. ctDNA) – 77.3% (58/75). (4) Specificity – 5% (1/20) in NCs: G12V | |
| Sausen et al. (2015) | 51 | I (n = 2), II (n = 45), III (n = 4) | 44 | Plasma | KRAS | (1) ctDNA – 43% (22/51). (2) Specificity: > 99.9% | |
| Chip‐based digital PCR Quanta Studio® | Brychta et al. (2016) | 50b | I (n = 4), II (n = 37), III (n = 6), IV (n = 3) | 50 | Plasma | KRAS: G12D; G12V; G12C | (1) Tissue mutation – 72% (36/50) for KRAS status: G12D 44% (22/50); G12V 20% (10/50); G12C 10% (5/50). (2) ctDNA/Tissue mutation – 35% (13/37): G12D 36% (8/22); G12V 50% (5/10); G12C 0% (0/5). (3) Specificity: 100% |
| Whole‐exome sequencing Hi‐Seq 2500 ddPCR Bio‐Rad | Cheng et al. (2017) | 188 | M | NA | Plasma | Focused on 60 genes, KRAS, BRCA2, EGFR, KDR,ERBB2 | ctDNA – 83% (156/188): KRAS 72.3% (136/188); BRCA2 11.7% (22/188); KDR 13.8% (26/188); EGFR 13.3% (25/188); ERBB2 exon 17 13.3% (25/188); ERBB2 exon 27 6.4% (12/188) |
| Targeted Sequencing Ion PGM™ ddPCR Bio‐Rad | Adamo et al. (2017) | 26 | R (n = 6), LA (n = 5), M (n = 15) | 11 | Plasma | 50 gene panel. KRAS: codons 12 and 13 | (1) Tissue mutation – 73% (8/11): G12D 50% (4/8); G12V 38% (3/8). (2) cfNDA concentration: 585 ng·mL−1 (PDAC); 300 ng·mL−1 (CP); 175 ng·mL−1 (NC). (3) KRAS mut ctDNA (targeted sequencing, validated by ddPCR) – 26.9% (7/26): 16.7% (1/6) in R; 40% (6/15) in M |
| Targeted Sequencing Ion Proton™ digital PCR RainDrop™ | Pietrasz et al. (2017) | 135 | R (n = 31), LA (n = 36), M (n = 68 | NA | Plasma | 22 gene panel. KRAS: G12D, G12V, G12R | (1) cfDNA concentration: 52.5 ± 79.5 ng·mL−1 in R; 105.8 ± 227.25 ng·mL−1 in LA‐M. (2) ctDNA – 48% (50/104) in LA‐M: KRAS (n = 43); TP53 (n = 23); SMAD4 (n = 8); NRAS (n = 2); PIK3CA (n = 1); STK11 (n = 1) |
| Targeted Sequencing MiSeq ddPCR Bio‐Rad | Berger et al. (2018) | 20 | IV | 11 | Plasma | 7 gene panel. KRAS, TP53 | (1) Tissue mutation: 63.6% (7/11) for KRAS status. (2) ctDNA: 100% (11/11) in therapy naïve patients; 55.6% (5/9) in pretreated patients |
| digital PCR RainDrop™ Targeted Sequencing Ion Proton™ | Pecuchet et al. (2016) | 100b | R (n = 23), LA‐M (n = 77) | NA | Plasma | KRAS, EGFR, 22 gene panel | (1) Amplicon Sequencing (digital PCR as a reference method)a: Sensitivity 97.6%; Specificity 94.0%; Accuracy 97.4%. (2) Method comparison (Amplicon Sequencing vs. digital PCR): Highly correlated mutation AF (R 2 = 0.95) |
| digital PCR RainDrop™ Targeted Sequencing HiSeq 2500 Ion PGM™ | Takai et al. (2016); Takai et al. (2015) | 259 | IA (n = 3), IB (n = 2), IIA (n = 29), IIB (n = 44), III (n = 17), IV (n = 163), NA (n = 1) | NA | Plasma | KRAS: G12D; G12V; G12R; G13D; 60 gene panel | (1) ctDNA (digital PCR based screening): 32% (83/259). (2) ctDNA (confirmed by targeted sequencing): 93.7% (45/48); 93.3% (42/45) was detected by digital PCR as well |
| Targeted Sequencing HiSeq 2500 | Pishvaian et al. (2017) | 34 | NA | 23 | Plasma | 68 gene panel | (1) Tissue mutation: 87% (20/23) for KRAS status. (2) ctDNA – 56% (19/34): mutations in median 2 genes/patient; 29% (10/34) for KRAS status. (3) Concordance rate (Tissue vs. ctDNA): 39% (9/23) for KRAS status |
| Zill et al. (2015) | 26b | III (n = 3), IV (n = 23) | 26 | Plasma | 54 gene panel | ctDNA/Tissue mutation: sensitivity 92.3%; specificity 100%; accuracy 97.7% | |
| Targeted Sequencing MiSeq HiSeq 4000 | Cohen et al. (2017) | 221 | R | 152 (TP53)50 (KRAS) | Plasma | KRAS, TP53 (n = 152) | (1) Tissue mutation: 100% (50/50) for KRAS status; 42% (64/152) for TP53 status. (2) ctDNA – KRAS 30% (66/221) in plasma: 94% (62/66) in codon 12; 6% (4/66) in codon 61; (3) ctDNA/Tissue mutation: TP53 20% (13/64) in paired plasma |
| Targeted Sequencing Ion PGM™ | Perets et al. (2018) | 17 | M | NA | Plasma | KRAS: exon 2 | ctDNA: 29.4% (5/17) |
| Calvez‐Kelm et al. (2016) | 437 | L (n = 39), Reg (n = 143), Sys (n = 135), NA (n = 120) | NA | Plasma | KRAS: codons 4–16, 51–69 | ctDNA – 21.1% (92/437) in total: 10.3% (4/39) in L; 17.5% (25/143) in Reg; 33.3% (45/135) in Sys | |
| CancerSEEK | Cohen et al. (2018) | 1005a | I‐III | NA | Plasma | 16 gene panel, 8 proteins | ctDNA: Sensitivities ranged from 69 to 98% for detection of five cancer types including pancreatic cancer; Specificity > 99% |
|
BEAMing (n = 2) PCR ligation (n = 50) Safe‐SeqS (n = 103) |
Bettegowda et al. (2014) | 155 | I (n = 22), II (n = 94), III (n = 5), IV (n = 34) | 155 | Plasma | KRAS: codons 12,13, 59, 60 and 61 | ctDNA – 57.4% (89/155) in total: 48.8% (59/121) in Stage I‐III; 88.2% (30/34) in Stage IV |
|
PNA‐mediated real‐time PCR clamping |
Tjensvoll et al. (2016) | 14b | LA (n = 2), M (n = 12) | NA | Plasma | KRAS | ctDNA: 71% (10/14) |
| Dabritz et al. (2009) | 56b | Inop (n = 23), Op (n = 25), NA (n = 8) | NA | Plasma | KRAS | ctDNA: 36% (20/56) | |
| Microarray‐mediated methylation assay MethDet56 | Liggett et al. (2010) | 30b | NA | NA | Plasma | Methylation | Differentiate PC from CP: Sensitivity 91.2%; Specificity 90.8% |
| Melnikov et al. (2009) | 34 | R (n = 25), NR (n = 9) | NA | Plasma | Methylation | Differentiate PC from NC: Sensitivity 76%; Specificity 59% | |
| MSP Nested PCR Direct sequencing | Jiao et al. (2007) | 83 | L (n = 16), LA (n = 37), M (n = 30) | 9 | Plasma | Methylation: p16; ‐ ppENK; KRAS; codon 12 | ctDNA – 62.6% (52/83) with ≥ 1 alteration: KRAS 32.5% (25/77); ppENK 29.3% (22/75); p16 24.6% (14/57) |
| Nested PCR | Singh et al. (2015) | 127b , d | No M (n = 74), M (n = 53) | NA | Plasma | KRAS: codon 12 | (1) cfDNA concentration: mean 85.2 ± 49.1 ng·mL−1 in patients; mean 35.4 ± 7.4 ng·mL−1 in NC. (2) ctDNA – 30.9% (34/110) in available samples: GAT 55.9% (19/34); TGT 17.6% (6/34); CGT 26.5% (9/34) |
| COLD‐PCR combined with unlabeled‐probe HRM approach | Wu et al. (2014) | 36 | NA | 36 | Plasma | KRAS: codon 12, 13 | ctDNA – 72.2% (26/36): All of 26 tissue DNA were KRAS mut. |
| Colorimetric‐based assay STA™ | Ollar et al. (2010) | 14 | NA | 14 | Peripheral blood | KRAS: codon 12 (GGT>TGT) | ctDNA/Tissue mutation: 21.4% (3/14): Tissue (+), PB (−); 7.1% (1/14): Tissue (−), PB (+); 71.4% (10/14): Tissue (−), PB (−) |
| MLA | Uemura et al. (2004) | 28b | I (n = 2), II (n = 8), III (n = 7), IVA (n = 7), IVB (n = 4) | 28 | Plasma | KRAS: exon 1 | (1) ctDNA/Tissue mutation: 93% (26/28) in tissue; 35% (9/26) in paired plasma. (2) Specificity – No mutation in normal DNA |
| Direct sequencing | Chen et al. (2010) | 91 | III (n = 29), IV (n = 62) | NA | Plasma | KRAS: codon 12 | ctDNA – 33% (30/91): G12D 56.7% (17/30); G12V 36.7% (11/30); G12R 6.7% (2/30); 17.2% (5/29) in Stage III; 40.3% (25/62) in Stage IV |
| PCR‐RFLP | Dianxu et al. (2002) | 41 | I (n = 2), II (n = 6), III (n = 5), IV (n = 26), NA (n = 2) | 36 | Plasma | KRAS: codon 12 | (1) Tissue mutation: 91.7% (33/36). (2) ctDNA – 70.7% (29/41); (3) ctDNA/Tissue mutation – 75.8% (25/33) in paired plasma. (4) Specificity – 100% (3/3): Tissue (−) and ctDNA (−) |
|
PCR‐RFLP Direct sequencing |
Mulcahy et al. (1998) | 21 | NR | 10 | Plasma | KRAS: codon 12 | ctDNA – 81% (17/21): before clinical diagnosis in 4 patients |
AF, allele frequency; cfDNA, cell‐free DNA; CP, chronic pancreatitis; ctDNA, circulating tumor DNA; ddPCR, droplet digital PCR; Inop, inoperable; IPMN, intraductal papillary mucinous neoplasm; L, local; LA, locally advanced; M, metastatic; MLA, mismatch ligation assay; MSP, methylation‐specific PCR; N, number of patients; NA, not available; NC, normal control; NR, nonresectable; Op, operable; PC, pancreatic cancer; PDAC, pancreatic ductal adenocarcinoma; PNA, peptide nucleic acid; R, resectable; Refs, references; Reg, regional; Safe‐SeqS, Safe‐Sequencing System; SCA, serous cystadenoma; Sys, systematic.
Results from other types of cancer patients are included.
Various tumor types of pancreatic cancers are included.
Five study cohorts, PDAC (n = 24); IPMN (n = 21); borderline IPMN (n = 16); SCA (n = 26)
KRAS mutation test was available for 110 samples.
6.1. Method comparison
Pécuchet et al. evaluated 77 patients with pancreatic cancer and compared a microfluidic dPCR (RainDrop®) and NGS analysis (Ion Proton™) in detecting KRAS and EGFR mutations. 97.4% (75/77) of results were concordant. KRAS mutation was only detected by dPCR in two samples (allele frequency 0.003 and 0.006, respectively) (Pécuchet et al., 2016). Similarly, Pietrasz et al. assessed 135 patients with PDAC and compared the two methods in detecting KRAS mutant ctDNA. They reported high concordance (R 2 = 0.94) between the targeted NGS analysis (Ion Proton™) and dPCR (RainDrop®) in detecting KRAS mutant ctDNA: One sample considered as KRAS mutation‐negative in NGS analysis was positive in dPCR (allele frequency 0.0061) (Pietrasz et al., 2017). Takai et al. applied a two‐stage strategy to analyze KRAS mutant ctDNA in PDAC patients. They used ddPCR (Bio‐Rad) as a prescreening method and then performed NGS analysis (Illumina HiSeq 2000) for 60 genes including KRAS (Takai et al., 2015). The use of NGS analysis as a prescreening method, combined with ddPCR (Bio‐Rad) for further validation, has also been successfully applied for ctDNA analysis (Adamo et al., 2017; Cheng et al., 2017). The combined strategy was suggested as cost‐effective and efficient method for analyzing ctDNA in PDAC patients (Takai et al., 2015). Further approaches to establish efficient strategies for analyzing tumor genomes in plasma DNA are highly warranted.
6.2. Early diagnosis
According to the recent joint review by the American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP), further studies are still required to prove the clinical utility of ctDNA in early diagnosis (Merker et al., 2018). IPMNs are the most frequent potentially malignant pancreatic cysts and classified into main duct type (MD‐IPMN) and branch duct type (BD‐IPMN). Since only 15–20% of BD‐IPMN will develop malignancy and nonsurgical management is recommended for low‐risk BD‐IPMNs, we need to correctly identify malignant IPMNs. Recently, an imaging tool that is combined with the identification of genomic patterns, coined ‘radiomics’, has been proposed by several studies (Hanania et al., 2016; Permuth et al., 2016). Similarly, Berger et al. detected GNAS mutant plasma DNA in 71.4% (15/21) of IPMN patients, but neither in serous cyst adenoma patients nor in healthy controls. ctDNA assay can be a useful tool for the discrimination of IPMN with malignant potential from other harmless pancreatic tumors, even though additional approaches to differentiate low from high‐grade IPMN is still required (Berger et al., 2016).
For realizing early cancer detection using ctDNA‐based screening tests, an interesting clinical trial (https://clinicaltrials.gov/ct2/show/NCT02889978) by a company (GRAIL, Inc) is currently ongoing and recruiting 15 000 participants including cancer subjects with multiple types and healthy subjects. This project, called the Circulating Cell Free Genome Atlas (CCGA), aims to identify potential cancer mutations and to complete a reference database of the mutations in circulating DNA in plasma (Aravanis et al., 2017).
6.3. Prognostic marker
Previous ctDNA studies mostly focused on KRAS hotspot (codon 12) mutations and its association with clinical outcomes of patients with PDAC. Sausen et al. (2015) demonstrated that patients with KRAS mutant ctDNA after surgery were more likely to relapse than those without KRAS mutant ctDNA (9.9 months vs. not reached, P = 0.02). Another study evaluated PDAC patients undergoing surgery and reported that the detection of ctDNA by ddPCR at baseline correlated with shorter DFS and OS (DFS, 6.1 months vs. 16.1 months; OS, 13.6 months vs. 27.6 months; P < 0.001 and P < 0.0001, respectively) (Hadano et al., 2016). This was also confirmed by Earl et al. (2015) in which patients with ctDNA detected by ddPCR had significantly shorter OS than patients with no detectable ctDNA. In metastatic PDAC, undetectable KRAS mutant ctDNA was significantly associated with survival benefit (8 months vs. 37.5 months, P < 0.004) (Perets et al., 2018). For patients with resectable disease, MST of patients in whom ctDNA was detected were significantly shorter than those of patients in whom ctDNA was not detected (3.9 months vs. 10.2 months, P < 0.001) (Chen et al., 2010). Furthermore, it has been reported that high amount of cfDNA is a relevant prognostic marker for pancreatic cancer patients (Singh et al., 2015; Tjensvoll et al., 2016). A recent meta‐analysis by Creemers et al. (2017) showed that the ctDNA in pancreatic cancer is significantly associated with a poor prognosis. In contrast, Bernard et al. (2019) analyzed longitudinal KRAS mutant allele fraction from ctDNA and exosome DNA and determined that longitudinal monitoring through exosome DNA rather than ctDNA provides prognostic information.
6.4. Predictive marker
So far, the role of ctDNA as a relevant predictive marker in PDAC remains to be identified. Recently, reported predictive markers for gemcitabine response are limited to the germline variants (Innocenti et al., 2012; Li et al., 2016). In a phase III trial, comparing gemcitabine alone with erlotinib plus gemcitabine, the OS was significantly prolonged on the combined therapy, yet EGFR status did not predict the response to the therapy (Moore et al., 2007). As the frequency of KRAS mutation in PDAC ranges from 88 to 100%, current efforts are underway to target KRAS pathway to make therapeutic progress in PDAC (Collisson et al., 2012; Krantz and O'Reilly, 2018; Rao et al., 2004; Van Cutsem et al., 2004; Ying et al., 2016). Additionally, targeting pancreatic CSCs, γ‐secretase inhibitors (GSI) to inhibit Notch signaling pathway have been developed (Abel et al., 2014; Whitehead et al., 2012). With regard to the epigenetic regulation, deregulation of histone deacetylases (HDACs) has been reported to play a role in pancreatic cancer development (Polireddy and Chen, 2016). HDAC inhibitors are currently tested for pancreatic cancer treatment, but there seems to be no benefit in clinical outcomes (Millward et al., 2012; Richards et al., 2006; Tinari et al., 2012). In this context, ctDNA assay will have clinical utility in noninvasive molecular profiling for the novel druggable mutations. An NGS approach targeting 60 cancer‐associated genes identified potentially targetable mutations in plasma DNA of PDAC patients (Takai et al., 2015).
7. Future perspectives
Early detection, real‐time disease monitoring, molecular profiling for targeted therapy are applications that promise to improve pancreatic cancer management. Liquid biopsy is a potentially valuable tool for in this regard. Multiple studies revealed the clinical use of liquid biopsy in monitoring patients (Table 6). ctDNA analysis may be more sensitive, easily accessible, and suitable not only for monitoring tumor dynamics during treatment, but for noninvasive molecular profiling of tumors due to the high incidence of nongermline (as well as some germline) genetic variations (Cicenas et al., 2017). Several recent studies have performed ctDNA analysis targeting noncoding repetitive DNA sequence such as ALU and described the possible use of noncoding DNA as additional prognostic marker in cancer monitoring (Chang et al., 2017; Lehner et al., 2013). CTC analysis, however, has its own strengths in that CTCs enable functional analyses such as drug testing, particularly as they represent cells still remaining after previous treatment during the course of disease. Thus, we suggest that both CTCs and ctDNA can be used in future parallel or complementary analyses (Kidess‐Sigal et al., 2016) and it is hoped that both these technologies will influence future diagnosis and treatment of this currently devastating disease. In addition to CTCs and ctDNA, there is increasing attention for emerging role of extracellular vesicles (EVs). Exosomes are a well‐studied EV population and can be a source for tumor‐specific proteins and RNAs (i.e., mRNA, noncoding RNA, and miRNA). Exosomes that carry cargo consisting of disease‐specific nucleic acids and proteins can provide a promising tool for characterizing cancer specific features as well as targeted treatment in pancreatic cancer (Kamerkar et al., 2017; Massoumi et al., 2019; Qian et al., 2019; Qiu et al., 2018; Siravegna et al., 2017).
Table 6.
Studies that revealed the clinical use of CTCs/ctDNA in monitoring patients
| Reference | Analyte | Time point measuring CTCs/ctDNA | Results |
|---|---|---|---|
| Dotan et al. (2016) | CTCs | First disease evaluation (6–10 weeks after treatment initiation) | For patients with ≥ 1 CTCs at diagnosis, 47% (7/15 patients) had no CTCs detected at first disease evaluation. |
| Sheng et al. (2014) | CTCs | First day of each subsequent treatment cycle. | The CTC number correlated proportionally with CT scan measured tumor size in each of the three patients. |
| Bernard et al. (2019) | ctDNA |
Baseline Immediately after neoadjuvant therapy completion (n = 34) in resectable PDAC At least two consecutive samples within the same treatment regimen (n = 34) in metastatic PDAC |
Reduction in ctDNA after completion of neoadjuvant therapy did not correlate with progression (resectable PDAC). Reduction in exoDNA MAF after completion of neoadjuvant therapy correlated with progression (OR = 38.4; P = 0.0002) (resectable PDAC). Serial ctDNA MAF did not correlate with progression in metastatic PDAC. Any on‐treatment serial exoDNA sample was significantly associated with eventual progression (P < 0.0001) in metastatic PDAC. |
| Berger et al. (2018) | ctDNA |
Baseline 4 weeks after treatment at disease progression |
The median CMAF level significantly decreased during treatment (P = 0.0027) and increased during progression (P = 0.0104). CMAF levels during treatment significantly correlated with PFS (P = 0.0013 ) |
| Del Re et al. (2017) | ctDNA | Subsequently after 15 days of Tx and at first radiologic evaluation |
KRAS
mut ctDNA change (at the 15‐day sample) correlated with PFS (increase, 2.5 months vs. stability/reduction, 7.5 months; P = 0.03). KRAS mut ctDNA change (at the time of first radiologic evaluation) correlated with PFS (increase, 2.8 months vs. reduction, 7.5 months; P = 0.028). |
| Tjensvoll et al. (2016) | ctDNA | Subsequently every month during treatment | ctDNA measurements could reveal disease progression at an earlier stage for some patients compared to conventional monitoring methods. |
| Sausen et al. (2015) | ctDNA | Multiple time points after surgery | Patients with detectable ctDNA after surgical resection were more likely to relapse than those with undetectable alterations (P = 0.02) |
CMAF, combined mutational allele frequency; CT, computed tomography; CTC, circulating tumor cell; ctDNA, circulating tumor DNA; exoDNA, exosome DNA; MAF, mutant allele fraction; PFS, progression‐free survival.
At this time, however, based on an extensive joint review on ctDNA by the American Society of Clinical Oncology and the College of American Pathologist, there are still many questions regarding the clinical validity and clinical utility of ctDNA assays in cancer screening, early‐stage disease, and treatment monitoring (Merker et al., 2018). Further research, development of tools utilizing ctDNA, and clinical practice guidance are warranted.
The liquid biopsy field will require further investigation with particular emphasis on clinical utility, not only clinical validation. This means demonstrating that liquid biopsy assay results will affect patient care in specific ways (e.g., changes in surgical approach and/or use of neoadjuvant therapies, changes in drug treatments) and that such changes will improve the morbidity and mortality of pancreatic cancer that we hope will occur in the near future. In particular, prospective clinical trials will be required that show that ctDNA may predict which patients are more likely to respond to chemotherapy and/or immune therapy and/or radiation therapy, or whether specific genetic aberrations identified in blood products (CTCs, ctDNA, EVs) predict response to particular therapies. For example, an exosomes test from Exosome Diagnostics is being tested as a companion diagnostic for Intezyne's phase 1/2 clinical trials of IT‐139, a novel cancer resistance pathway (CRP) inhibitor for the treatment of pancreatic, gastric, and other cancers in combination with existing anticancer therapies. Equally exciting it that the potential use of targeted EVs as a systemic treatment. A phase I trial that studies the best dose and side effects of mesenchymal stromal cells‐derived exosomes with KRAS G12D siRNA (iExosomes) has been approved by the U.S. National Cancer Institute (https://clinicaltrials.gov/ct2/show/NCT03608631) but not yet started. It will be used for the treatment of participants with metastatic pancreatic cancer with KRAS G12D mutation, hoping that iExosomes may prove a better treatment for this dismal disease.
In summary, while liquid biopsy is promising, it still remains a burgeoning field. However, there are many positive signs that it will have a strong impact on the diagnosis, monitoring, and treatment of pancreatic cancer.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
J‐SL and SSJ wrote the manuscript and created the figure and graphical abstract. SSP, YKL, and JAN edited the manuscript. All authors reviewed and approved the final version.
Acknowledgements
The authors would like to thank Prof. Ash Alizadeh for his review and suggestions.
References
- Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E, Wang L, Dziubinski ML and Simeone DM (2014) The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS ONE 9, e91983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamo P, Cowley CM, Neal CP, Mistry V, Page K, Dennison AR, Isherwood J, Hastings R, Luo J, Moore DA et al (2017) Profiling tumour heterogeneity through circulating tumour DNA in patients with pancreatic cancer. Oncotarget 8, 87221–87233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Albuquerque A, Kubisch I, Breier G, Stamminger G, Fersis N, Eichler A, Kaul S and Stölzel U (2012) Multimarker gene analysis of circulating tumor cells in pancreatic cancer patients: a feasibility study. Oncology 82, 3–10. [DOI] [PubMed] [Google Scholar]
- Alidousty C, Brandes D, Heydt C, Wagener S, Wittersheim M, Schäfer SC, Holz B, Merkelbach‐Bruse S, Büttner R, Fassunke J et al (2017) Comparison of blood collection tubes from three different manufacturers for the collection of cell‐free DNA for liquid biopsy mutation testing. J Mol Diagnost 19, 801–804. [DOI] [PubMed] [Google Scholar]
- Alix‐Panabieres C, Bartkowiak K and Pantel K (2016) Functional studies on circulating and disseminated tumor cells in carcinoma patients. Mol Oncol 10, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alix‐Panabieres C and Pantel K (2016) Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov 6, 479–491. [DOI] [PubMed] [Google Scholar]
- Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW and Terstappen LW (2004) Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res 10, 6897–6904. [DOI] [PubMed] [Google Scholar]
- Allen PJ, Kuk D, Castillo CF, Basturk O, Wolfgang CL, Cameron JL, Lillemoe KD, Ferrone CR, Morales‐Oyarvide V, He J et al (2017) Multi‐institutional validation study of the American Joint Commission on Cancer (8th Edition) Changes for T and N Staging in Patients With Pancreatic Adenocarcinoma. Ann Surg 265, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anker P, Mulcahy H, Chen XQ and Stroun M (1999) Detection of circulating tumour DNA in the blood (plasma/serum) of cancer patients. Cancer Metastasis Rev 18, 65–73. [DOI] [PubMed] [Google Scholar]
- Aravanis AM, Lee M and Klausner RD (2017) Next‐generation sequencing of circulating tumor DNA for early cancer detection. Cell 168, 571–574. [DOI] [PubMed] [Google Scholar]
- Ashida A, Sakaizawa K, Mikoshiba A, Uhara H and Okuyama R (2016) Quantitative analysis of the BRAF (V600E) mutation in circulating tumor‐derived DNA in melanoma patients using competitive allele‐specific TaqMan PCR. Int J Clin Oncol 21, 981–988. [DOI] [PubMed] [Google Scholar]
- Ashworth T (1869) A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust Med J 14, 146–149. [Google Scholar]
- Ballehaninna UK and Chamberlain RS (2012) The clinical utility of serum CA 19‐9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: an evidence based appraisal. J Gastroint Oncol 3, 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriere G, Fici P, Gallerani G, Fabbri F, Zoli W and Rigaud M (2014) Circulating tumor cells and epithelial, mesenchymal and stemness markers: characterization of cell subpopulations. Annals Trans Med 2, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AW, Schwerdel D, Costa IG, Hackert T, Strobel O, Lam S, Barth TF, Schröppel B, Meining A, Büchler MW et al (2016) Detection of hot‐spot mutations in circulating cell‐free DNA from patients with intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology 151, 267–270. [DOI] [PubMed] [Google Scholar]
- Berger AW, Schwerdel D, Ettrich TJ, Hann A, Schmidt SA, Kleger A, Marienfeld R and Seufferlein T (2018) Targeted deep sequencing of circulating tumor DNA in metastatic pancreatic cancer. Oncotarget 9, 2076–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard V, Kim DU, San Lucas FA, Castillo J, Allenson K, Mulu FC, Stephens BM, Huang J, Semaan A, Guerrero PA et al (2019) Circulating nucleic acids associate with outcomes of patients with pancreatic cancer. Gastroenterology 156, 108–118.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM et al (2014) Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Trans Med 6, 224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidard FC, Huguet F, Louvet C, Mineur L, Bouché O, Chibaudel B, Artru P, Desseigne F, Bachet JB, Mathiot C et al (2013) Circulating tumor cells in locally advanced pancreatic adenocarcinoma: the ancillary CirCe 07 study to the LAP 07 trial. Annals Oncol 24, 2057–2061. [DOI] [PubMed] [Google Scholar]
- Bissolati M, Sandri MT, Burtulo G, Zorzino L, Balzano G and Braga M (2015) Portal vein‐circulating tumor cells predict liver metastases in patients with resectable pancreatic cancer. Tumour Biol 36, 991–996. [DOI] [PubMed] [Google Scholar]
- Bobek V, Gurlich R, Eliasova P and Kolostova K (2014) Circulating tumor cells in pancreatic cancer patients: enrichment and cultivation. World J Gastroenterol 20, 17163–17170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronkhorst AJ, Aucamp J and Pretorius PJ (2015) Cell‐free DNA: preanalytical variables. Clinica Chim Acta 450, 243–253. [DOI] [PubMed] [Google Scholar]
- Brown P (2016) The Cobas(R) EGFR mutation test v2 assay. Fut Oncol (London, England) 12, 451–452. [DOI] [PubMed] [Google Scholar]
- Brychta N, Drosch M, Driemel C, Fischer JC, Neves RP, Esposito I, Knoefel W, Möhlendick B, Hille C, Stresemann A et al (2017) Isolation of circulating tumor cells from pancreatic cancer by automated filtration. Oncotarget 8, 86143–86156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brychta N, Krahn T and von Ahsen O (2016) Detection of KRAS mutations in circulating tumor DNA by digital PCR in early stages of pancreatic cancer. Clin Chem 62, 1482–1491. [DOI] [PubMed] [Google Scholar]
- Calvez‐Kelm FL, Foll M, Wozniak MB, Delhomme TM, Durand G, Chopard P, Pertesi M, Fabianova E, Adamcakova Z, Holcatova I et al (2016) KRAS mutations in blood circulating cell‐free DNA: a pancreatic cancer case‐control. Oncotarget 7, 78827–78840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catenacci DV, Chapman CG, Xu P, Koons A, Konda VJ, Siddiqui UD and Waxman I (2015) Acquisition of portal venous circulating tumor cells from patients with pancreaticobiliary cancers by endoscopic ultrasound. Gastroenterology 149, 1794–1803.e1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauley CE, Pitman MB, Zhou J, Perkins J, Kuleman B, Liss AS, Fernandez‐Del Castillo C, Warshaw AL, Lillemoe KD and Thayer SP (2015) Circulating epithelial cells in patients with pancreatic lesions: clinical and pathologic findings. J Am Coll Surg 221, 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL and Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Chang MC, Chang YT, Chen JY, Jeng YM, Yang CY, Tien YW, Yang SH, Chen HL, Liang TY, Wang CF et al (2016) Clinical significance of circulating tumor microemboli as a prognostic marker in patients with pancreatic ductal adenocarcinoma. Clin Chem 62, 505–513. [DOI] [PubMed] [Google Scholar]
- Chang CL, Huang W, Jalal SI, Chan BD, Mahmood A, Shahda S, O'Neil BH, Matei DE and Savran CA (2015) Circulating tumor cell detection using a parallel flow micro‐aperture chip system. Lab Chip 15, 1677–1688. [DOI] [PubMed] [Google Scholar]
- Chang Y, Tolani B, Nie X, Zhi X, Hu M and He B (2017) Review of the clinical applications and technological advances of circulating tumor DNA in cancer monitoring. Clin Risk Manag 13, 1363–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Balaj L, Liau LM, Samuels ML, Kotsopoulos SK, Maguire CA, Loguidice L, Soto H, Garrett M, Zhu LD et al (2013) BEAMing and droplet digital PCR analysis of mutant IDH1 mRNA in glioma patient serum and cerebrospinal fluid extracellular vesicles. Mol Ther Nucleic Acids 2, e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Tu H, Meng ZQ, Chen Z, Wang P and Liu LM (2010) K‐ras mutational status predicts poor prognosis in unresectable pancreatic cancer. Europ J Surg Oncol 36, 657–662. [DOI] [PubMed] [Google Scholar]
- Cheng H, Liu C, Jiang J, Luo G, Lu Y, Jin K, Guo M, Zhang Z, Xu J, Liu L et al (2017) Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int J Cancer 140, 2344–2350. [DOI] [PubMed] [Google Scholar]
- Cheng F, Su L and Qian C (2016) Circulating tumor DNA: a promising biomarker in the liquid biopsy of cancer. Oncotarget 7, 48832–48841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun YS, Pawlik TM and Vauthey JN (2018) 8th Edition of the AJCC Cancer Staging Manual: pancreas and hepatobiliary cancers. Ann Surg Oncol 25, 845–847. [DOI] [PubMed] [Google Scholar]
- Cicenas J, Kvederaviciute K, Meskinyte I, Meskinyte‐Kausiliene E, Skeberdyte A and Cicenas J (2017) KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers 9, E42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JD, Javed AA, Thoburn C, Wong F, Tie J, Gibbs P, Schmidt CM, Yip‐Schneider MT, Allen PJ, Schattner M et al (2017) Combined circulating tumor DNA and protein biomarker‐based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci USA 114, 10202–10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox A et al (2018) Detection and localization of surgically resectable cancers with a multi‐analyte blood test. Science 359, 926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort D et al (2012) A central role for RAF–>MEK–>ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov 2, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court CM, Ankeny JS, Sho S, Winograd P, Hou S, Song M, Wainberg ZA, Girgis MD, Graeber TG, Agopian VG et al (2018) Circulating tumor cells predict occult metastatic disease and prognosis in pancreatic cancer. Ann Surg Oncol 25, 1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creemers A, Krausz S, Strijker M, van der Wel MJ, Soer EC, Reinten RJ, Besselink MG, Wilmink JW, van de Vijver MJ, van Noesel CJM et al (2017) Clinical value of ctDNA in upper‐GI cancers: a systematic review and meta‐analysis. Biochem Biophys Acta 1868, 394–403. [DOI] [PubMed] [Google Scholar]
- Dabritz J, Preston R, Hanfler J and Oettle H (2009) Follow‐up study of K‐ras mutations in the plasma of patients with pancreatic cancer: correlation with clinical features and carbohydrate antigen 19–9. Pancreas 38, 534–541. [DOI] [PubMed] [Google Scholar]
- Dagogo‐Jack I and Shaw AT (2018) Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 15, 81–94. [DOI] [PubMed] [Google Scholar]
- Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler‐Araujo B et al (2013) Analysis of circulating tumor DNA to monitor metastatic breast cancer. New Engl J Med 368, 1199–1209. [DOI] [PubMed] [Google Scholar]
- Del Re M, Vivaldi C, Rofi E, Vasile E, Miccoli M, Caparello C, d'Arienzo PD, Fornaro L, Falcone A and Danesi R (2017) Early changes in plasma DNA levels of mutant KRAS as a sensitive marker of response to chemotherapy in pancreatic cancer. Sci Rep 7, 7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellagi K, Vainchenker W, Vinci G, Paulin D and Brouet JC (1983) Alteration of vimentin intermediate filament expression during differentiation of human hemopoietic cells. EMBO J 2, 1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dessel LF, Beije N, Helmijr JC, Vitale SR, Kraan J, Look MP, de Wit R, Sleijfer S, Jansen MP, Martens JW et al (2017) Application of circulating tumor DNA in prospective clinical oncology trials – standardization of preanalytical conditions. Mol Oncol 11, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianxu F, Shengdao Z, Tianquan H, Yu J, Ruoqing L, Zurong Y, Xuezhi W (2002) A prospective study of detection of pancreatic carcinoma by combined plasma K‐ras mutations and serum CA19‐9 analysis. Pancreas 25, 336–341. [DOI] [PubMed] [Google Scholar]
- Diaz LA Jr and Bardelli A (2014) Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 32, 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diefenbach RJ, Lee JH, Kefford RF and Rizos H (2018) Evaluation of commercial kits for purification of circulating free DNA. Cancer Genet 228–229, 21–27. [DOI] [PubMed] [Google Scholar]
- Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA et al (2008) Circulating mutant DNA to assess tumor dynamics. Nat Med 14, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotan E, Alpaugh RK, Ruth K, Negin BP, Denlinger CS, Hall MJ, Astsaturov I, McAleer C, Fittipaldi P, Thrash‐Bingham C et al (2016) Prognostic significance of MUC‐1 in circulating tumor cells in patients with metastatic pancreatic adenocarcinoma. Pancreas 45, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressman D, Yan H, Traverso G, Kinzler KW and Vogelstein B (2003) Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci USA 100, 8817–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl J, Garcia‐Nieto S, Martinez‐Avila JC, Montans J, Sanjuanbenito A, Rodriguez‐Garrote M, Lisa E, Mendía E, Lobo E, Malats N et al (2015) Circulating tumor cells (Ctc) and kras mutant circulating free Dna (cfdna) detection in peripheral blood as biomarkers in patients diagnosed with exocrine pancreatic cancer. BMC Cancer 15, 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KE, Schroeder C, Hanssen A, Wolter S, Eulenburg C, Tachezy M, Gebauer F, Izbicki JR, Pantel K and Bockhorn M (2018) Improved risk stratification by circulating tumor cell counts in pancreatic cancer. Clin Cancer Res 24, 2844–2850. [DOI] [PubMed] [Google Scholar]
- El‐Heliebi A, Hille C, Laxman N, Svedlund J, Haudum C, Ercan E, Kroneis T, Chen S, Smolle M, Rossmann C et al (2018) In situ detection and quantification of AR‐V7, AR‐FL, PSA, and KRAS point mutations in circulating tumor cells. Clin Chem 64, 536–546. [DOI] [PubMed] [Google Scholar]
- Ferreira MM, Ramani VC and Jeffrey SS (2016) Circulating tumor cell technologies. Mol Oncol 10, 374–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald TL and McCubrey JA (2014) Pancreatic cancer stem cells: association with cell surface markers, prognosis, resistance, metastasis and treatment. Advances Biol Reg 56, 45–50. [DOI] [PubMed] [Google Scholar]
- Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez‐Linan M and Bentley D (2012) Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Trans Med 4, 136ra68. [DOI] [PubMed] [Google Scholar]
- Friedman R (2016) Drug resistance in cancer: molecular evolution and compensatory proliferation. Oncotarget 7, 11746–11755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhu Y, Zhang Z, Zhang C, Huang X and Yuan Z (2016) Clinical significance of pancreatic circulating tumor cells using combined negative enrichment and immunostaining‐fluorescence in situ hybridization. J Exp Clin Cancer Res 35, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel G and Sun W (2015) Novel approaches in the management of pancreatic ductal adenocarcinoma: potential promises for the future. J Hematol Oncol 8, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogoi P, Sepehri S, Zhou Y, Gorin MA, Paolillo C, Capoluongo E, Gleason K, Payne A, Boniface B, Cristofanilli M et al (2016) Development of an automated and sensitive microfluidic device for capturing and characterizing circulating tumor cells (CTCs) from clinical blood samples. PLoS ONE 11, e0147400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgannezhad L, Umer M, Islam MN, Nguyen NT and Shiddiky MJA (2018) Circulating tumor DNA and liquid biopsy: opportunities, challenges, and recent advances in detection technologies. Lab Chip 18, 1174–1196. [DOI] [PubMed] [Google Scholar]
- Görner K, Bachmann J, Holzhauer C, Kirchner R, Raba K, Fischer JC, Martignoni ME, Schiemann M and Alunni‐Fabbroni M (2015) Genetic analysis of circulating tumor cells in pancreatic cancer patients: a pilot study. Genomics 106, 7–14. [DOI] [PubMed] [Google Scholar]
- Guibert N, Hu Y, Feeney N, Kuang Y, Plagnol V, Jones G, Howarth K, Beeler JF, Paweletz CP and Oxnard GR (2018) Amplicon‐based next‐generation sequencing of plasma cell‐free DNA for detection of driver and resistance mutations in advanced non‐small cell lung cancer. Annals Oncol 29, 1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadano N, Murakami Y, Uemura K, Hashimoto Y, Kondo N, Nakagawa N, Sueda T and Hiyama E (2016) Prognostic value of circulating tumour DNA in patients undergoing curative resection for pancreatic cancer. Br J Cancer 115, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanania AN, Bantis LE, Feng Z, Wang H, Tamm EP, Katz MH, Maitra A and Koay EJ (2016) Quantitative imaging to evaluate malignant potential of IPMNs. Oncotarget 7, 85776–85784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harouaka RA, Zhou MD, Yeh YT, Khan WJ, Das A, Liu X, Christ CC, Dicker DT, Baney TS, Kaifi JT et al (2014) Flexible micro spring array device for high‐throughput enrichment of viable circulating tumor cells. Clin Chem 60, 323–333. [DOI] [PubMed] [Google Scholar]
- Heitzer E, Ulz P and Geigl JB (2015) Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem 61, 112–123. [DOI] [PubMed] [Google Scholar]
- Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC et al (2011) High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 83, 8604–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igawa S, Gohda K, Fukui T, Ryuge S, Otani S, Masago A, Sato J, Murakami K, Maki S, Katono K et al (2014) Circulating tumor cells as a prognostic factor in patients with small cell lung cancer. Oncol Lett 7, 1469–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatiadis M, Lee M and Jeffrey SS (2015) Circulating tumor cells and circulating tumor DNA: challenges and opportunities on the path to clinical utility. Clin Cancer Res 21, 4786–4800. [DOI] [PubMed] [Google Scholar]
- Innocenti F, Owzar K, Cox NL, Evans P, Kubo M, Zembutsu H, Jiang C, Hollis D, Mushiroda T, Li L et al (2012) A genome‐wide association study of overall survival in pancreatic cancer patients treated with gemcitabine in CALGB 80303. Clin Cancer Res 18, 577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanicki‐Caron I, Basile P, Toure E, Antonietti M, Lecleire S, Di Fiore A, Oden‐Gangloff A, Blanchard F, Lemoine F, Di Fiore F et al (2013) Usefulness of circulating tumor cell detection in pancreatic adenocarcinoma diagnosis. Am J Gastroenterol 108, 152–155. [DOI] [PubMed] [Google Scholar]
- Jeffrey SS and Toner M (2019) Liquid biopsy: a perspective for probing blood for cancer. Lab Chip 19, 548–549. [DOI] [PubMed] [Google Scholar]
- Jiao L, Zhu J, Hassan MM, Evans DB, Abbruzzese JL and Li D (2007) K‐ras mutation and p16 and preproenkephalin promoter hypermethylation in plasma DNA of pancreatic cancer patients: in relation to cigarette smoking. Pancreas 34, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinich M and Haber DA (2018) Cancer detection: seeking signals in blood. Science 359, 866–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamande JW, Hupert ML, Witek MA, Wang H, Torphy RJ, Dharmasiri U, Njoroge SK, Jackson JM, Aufforth RD, Snavely A et al (2013) Modular microsystem for the isolation, enumeration, and phenotyping of circulating tumor cells in patients with pancreatic cancer. Anal Chem 85, 9092–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarajah SK, Burns WR, Frankel TL, Cho CS and Nathan H (2017) Validation of the American Joint Commission on Cancer (AJCC) 8th Edition Staging system for patients with pancreatic adenocarcinoma: a surveillance, epidemiology and end results (SEER) Analysis. Ann Surg Oncol 24, 2023–2030. [DOI] [PubMed] [Google Scholar]
- Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ and Kalluri R (2017) Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisawa T, Wood LD, Itoi T and Takaori K (2016) Pancreatic cancer. Lancet 388, 73–85. [DOI] [PubMed] [Google Scholar]
- Karabacak NM, Spuhler PS, Fachin F, Lim EJ, Pai V, Ozkumur E, Martel JM, Kojic N, Smith K, Chen PI et al (2014) Microfluidic, marker‐free isolation of circulating tumor cells from blood samples. Nat Protoc 9, 694–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoja L, Backen A, Sloane R, Menasce L, Ryder D, Krebs M, Board R, Clack G, Hughes A, Blackhall F et al (2012) A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker. Br J Cancer 106, 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidess E and Jeffrey SS (2013) Circulating tumor cells versus tumor‐derived cell‐free DNA: rivals or partners in cancer care in the era of single‐cell analysis? Genome Med 5, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidess‐Sigal E, Liu HE, Triboulet MM, Che J, Ramani VC, Visser BC, Poultsides GA, Longacre TA, Marziali A, Vysotskaia V et al (2016) Enumeration and targeted analysis of KRAS, BRAF and PIK3CA mutations in CTCs captured by a label‐free platform: comparison to ctDNA and tissue in metastatic colorectal cancer. Oncotarget 7, 85349–85364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MK, Woo SM, Park B, Yoon KA, Kim YH, Joo J, Lee WJ, Han SS, Park SJ and Kong SY (2018) Prognostic implications of multiplex detection of KRAS mutations in cell‐free DNA from patients with pancreatic ductal adenocarcinoma. Clin Chem 64, 726–734. [DOI] [PubMed] [Google Scholar]
- Kinde I, Wu J, Papadopoulos N, Kinzler KW and Vogelstein B (2011) Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA 108, 9530–9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinugasa H, Nouso K, Miyahara K, Morimoto Y, Dohi C, Tsutsumi K, Kato H, Matsubara T, Okada H and Yamamoto K (2015) Detection of K‐ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer 121, 2271–2280. [DOI] [PubMed] [Google Scholar]
- Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, Neale RE, Tempero M, Tuveson DA, Hruban RH et al (2016) Pancreatic cancer. Nat Rev Dis Primers 2, 16022. [DOI] [PubMed] [Google Scholar]
- Krantz BA and O'Reilly EM (2018) Biomarker‐based therapy in pancreatic ductal adenocarcinoma: an emerging reality? Clin Cancer Res 24, 2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH and Dive C (2014) Molecular analysis of circulating tumour cells‐biology and biomarkers. Nat Rev Clin Oncol 11, 129–144. [DOI] [PubMed] [Google Scholar]
- Kulemann B, Liss AS, Warshaw AL, Seifert S, Bronsert P, Glatz T, Pitman MB and Hoeppner J (2016) KRAS mutations in pancreatic circulating tumor cells: a pilot study. Tumour Biol 37, 7547–7554. [DOI] [PubMed] [Google Scholar]
- Kulemann B, Pitman MB, Liss AS, Valsangkar N, Fernandez‐Del Castillo C, Lillemoe KD, Hoeppner J, Mino‐Kenudson M, Warshaw AL and Thayer SP (2015) Circulating tumor cells found in patients with localized and advanced pancreatic cancer. Pancreas 44, 547–550. [DOI] [PubMed] [Google Scholar]
- Kurihara T, Itoi T, Sofuni A, Itokawa F, Tsuchiya T, Tsuji S, Ishii K, Ikeuchi N, Tsuchida A, Kasuya K et al (2008) Detection of circulating tumor cells in patients with pancreatic cancer: a preliminary result. J Hepato‐Biliary‐Pancreatic Surg 15, 189–195. [DOI] [PubMed] [Google Scholar]
- Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, O'Shaughnessy J, Kinzler KW, Parmigiani G, Vogelstein B et al (2012) Detection of chromosomal alterations in the circulation of cancer patients with whole‐genome sequencing. Sci Trans Med 4, 162ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner J, Stotzer OJ, Fersching D, Nagel D and Holdenrieder S (2013) Circulating plasma DNA and DNA integrity in breast cancer patients undergoing neoadjuvant chemotherapy. Clin Chim Acta 425, 206–211. [DOI] [PubMed] [Google Scholar]
- Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF and Simeone DM (2007) Identification of pancreatic cancer stem cells. Cancer Res 67, 1030–1037. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang JW, Jenkins G, Xie F, Carlson EE, Fridley BL, Bamlet WR, Petersen GM, McWilliams RR and Wang L (2016) Genetic variations associated with gemcitabine treatment outcome in pancreatic cancer. Pharmacogenet Gen 26, 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett T, Melnikov A, Yi QL, Replogle C, Brand R, Kaul K, Talamonti M, Abrams RA and Levenson V (2010) Differential methylation of cell‐free circulating DNA among patients with pancreatic cancer versus chronic pancreatitis. Cancer 116, 1674–1680. [DOI] [PubMed] [Google Scholar]
- Luketina RR, Hackert T and Buchler MW (2015) Vascular resection in pancreatic cancer. Indian J Surg 77, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hao S, Wang S, Zhao Y, Lim B, Lei M, Spector DJ, El‐Deiry WS, Zheng SY and Zhu J (2015) A combinatory strategy for detection of live CTCs using microfiltration and a new telomerase‐selective adenovirus. Mol Cancer Ther 14, 835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus H, Contente‐Cuomo T, Farooq M, Liang WS, Borad MJ, Sivakumar S, Gollins S, Tran NL, Dhruv HD, Berens ME et al (2018) Evaluation of pre‐analytical factors affecting plasma DNA analysis. Sci Rep 8, 7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrinucci D, Bethel K, Kolatkar A, Luttgen MS, Malchiodi M, Baehring F, Voigt K, Lazar D, Nieva J, Bazhenova L et al (2012) Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys Biol 9, 016003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoumi RL, Hines OJ, Eibl G and King JC (2019) Emerging evidence for the clinical relevance of pancreatic cancer exosomes. Pancreas 48, 1–8. [DOI] [PubMed] [Google Scholar]
- McGranahan N and Swanton C (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613–628. [DOI] [PubMed] [Google Scholar]
- Meddeb R, Pisareva E and Thierry AR (2019) Guidelines for the preanalytical conditions for analyzing circulating cell‐free DNA. Clin Chem 65, 623–633. [DOI] [PubMed] [Google Scholar]
- Medina Diaz I, Nocon A, Mehnert DH, Fredebohm J, Diehl F and Holtrup F (2016) Performance of streck cfDNA blood collection tubes for liquid biopsy testing. PLoS ONE 11, e0166354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov AA, Scholtens D, Talamonti MS, Bentrem DJ and Levenson VV (2009) Methylation profile of circulating plasma DNA in patients with pancreatic cancer. J Surg Oncol 99, 119–122. [DOI] [PubMed] [Google Scholar]
- Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, Lindeman N, Lockwood CM, Rai AJ, Schilsky RL et al (2018) Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol 36, 1631–1641. [DOI] [PubMed] [Google Scholar]
- Milbury CA, Chen CC, Mamon H, Liu P, Santagata S and Makrigiorgos GM (2011) Multiplex amplification coupled with COLD‐PCR and high resolution melting enables identification of low‐abundance mutations in cancer samples with low DNA content. J Mol Diagnost 13, 220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millward M, Price T, Townsend A, Sweeney C, Spencer A, Sukumaran S, Longenecker A, Lee L, Lay A, Sharma G et al (2012) Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest New Drugs 30, 2303–2317. [DOI] [PubMed] [Google Scholar]
- Mohammad Alizadeh AH, Shahrokh S, Hadizadeh M, Padashi M and Zali MR (2016) Diagnostic potency of EUS‐guided FNA for the evaluation of pancreatic mass lesions. Endoscop Ultrasound 5, 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA et al (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25, 1960–1966. [DOI] [PubMed] [Google Scholar]
- Mouliere F and Rosenfeld N (2015) Circulating tumor‐derived DNA is shorter than somatic DNA in plasma. Proc Natl Acad Sci USA 112, 3178–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy HE, Lyautey J, Lederrey C, qi Chen X, Anker P, Alstead EM, Ballinger A, Farthing MJ, Stroun M (1998) A prospective study of K‐ras mutations in the plasma of pancreatic cancer patients. Clin Cancer Res 4, 271–275. [PubMed] [Google Scholar]
- Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE et al (2014) An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 20, 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, Stehr H, Liu CL, Bratman SV, Say C et al (2016) Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 34, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgård O, Tjensvoll K, Gilje B and Søreide K (2018) Circulating tumour cells and DNA as liquid biopsies in gastrointestinal cancer. Br J Surg 105, e110–e120. [DOI] [PubMed] [Google Scholar]
- Ollar RA, Cooperman AM, Wayne ME, Barrecchia JF, Sonpal N, Duddempudi S and Kasmin FE (2010) A colorimetric method for detection of K‐ras codon 12 point mutations in DNA extracted from tissue and peripheral blood in pancreatic disorders. Biochem Genet 48, 577–589. [DOI] [PubMed] [Google Scholar]
- Parpart‐Li S, Bartlett B, Popoli M, Adleff V, Tucker L, Steinberg R, Georgiadis A, Phallen J, Brahmer J, Azad N et al (2017) The effect of preservative and temperature on the analysis of circulating tumor DNA. Clin Cancer Res 23, 2471–2477. [DOI] [PubMed] [Google Scholar]
- Passerini R, Cassatella MC, Boveri S, Salvatici M, Radice D, Zorzino L, Galli C and Sandri MT (2012) The pitfalls of CA19‐9: routine testing and comparison of two automated immunoassays in a reference oncology center. Am J Clin Pathol 138, 281–287. [DOI] [PubMed] [Google Scholar]
- Paterlini‐Brechot P and Benali NL (2007) Circulating tumor cells (CTC) detection: clinical impact and future directions. Cancer Lett 253, 180–204. [DOI] [PubMed] [Google Scholar]
- Pécuchet N, Rozenholc Y, Zonta E, Pietrasz D, Didelot A, Combe P, Gibault L, Bachet JB, Taly V, Fabre E et al (2016) Analysis of base‐position error rate of next‐generation sequencing to detect tumor mutations in circulating DNA. Clin Chem 62, 1492–1503. [DOI] [PubMed] [Google Scholar]
- Perets R, Greenberg O, Shentzer T, Semenisty V, Epelbaum R, Bick T, Sarji S, Ben‐Izhak O, Sabo E and Hershkovitz D (2018) Mutant KRAS circulating tumor DNA is an accurate tool for pancreatic cancer monitoring. Oncologist 23, 566–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Permuth JB, Choi J, Balarunathan Y, Kim J, Chen DT, Chen L, Orcutt S, Doepker MP, Gage K, Zhang G et al (2016) Combining radiomic features with a miRNA classifier may improve prediction of malignant pathology for pancreatic intraductal papillary mucinous neoplasms. Oncotarget 7, 85785–85797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piegeler T, Winder T, Kern S, Pestalozzi B, Schneider PM and Beck‐Schimmer B (2016) Detection of circulating tumor cells in patients with esophagogastric or pancreatic adenocarcinoma using the Cell Search(R) system: an observational feasibility study. Oncol Lett 12, 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrasz D, Pécuchet N, Garlan F, Didelot A, Dubreuil O, Doat S, Imbert‐Bismut F, Karoui M, Vaillant JC, Taly V et al (2017) Plasma circulating tumor DNA in pancreatic cancer patients is a prognostic marker. Clin Cancer Res 23, 116–123. [DOI] [PubMed] [Google Scholar]
- Pishvaian MJ, Joseph Bender R, Matrisian LM, Rahib L, Hendifar A, Hoos WA, Mikhail S, Chung V, Picozzi V, Heartwell C et al (2017) A pilot study evaluating concordance between blood‐based and patient‐matched tumor molecular testing within pancreatic cancer patients participating in the Know Your Tumor (KYT) initiative. Oncotarget 8, 83446–83456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polireddy K and Chen Q (2016) Cancer of the pancreas: molecular pathways and current advancement in treatment. J Cancer 7, 1497–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poruk KE, Blackford AL, Weiss MJ, Cameron JL, He J, Goggins MG, Rasheed ZA, Wolfgang CL, Wood LD (2017) Circulating tumor cells expressing markers of tumor initiating cells predict poor survival and cancer recurrence in patients with pancreatic ductal adenocarcinoma. Clin Cancer Res 23, 2681–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poruk KE, Valero V 3rd, Saunders T, Blackford AL, Griffin JF, Poling J, Hruban RH, Anders RA, Herman J, Zheng L et al (2016) Circulating tumor cell phenotype predicts recurrence and survival in pancreatic adenocarcinoma. Ann Surg 264, 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premasekharan G, Gilbert E, Okimoto RA, Hamirani A, Lindquist KJ, Ngo VT, Roy R, Hough J, Edwards M, Paz R et al (2016) An improved CTC isolation scheme for pairing with downstream genomics: demonstrating clinical utility in metastatic prostate, lung and pancreatic cancer. Cancer Lett 380, 144–152. [DOI] [PubMed] [Google Scholar]
- Qian L, Yu S, Chen Z, Meng Z, Huang S and Wang P (2019) Functions and clinical implications of exosomes in pancreatic cancer. Biochim Biophys Acta Rev Cancer 1871, 75–84. [DOI] [PubMed] [Google Scholar]
- Qin Z, Ljubimov VA, Zhou C, Tong Y and Liang J (2016) Cell‐free circulating tumor DNA in cancer. Chin J Cancer 35, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Yang G, Feng M, Zheng S, Cao Z, You L, Zheng L, Zhang T and Zhao Y (2018) Extracellular vesicles as mediators of the progression and chemoresistance of pancreatic cancer and their potential clinical applications. Mol Cancer 17, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Cunningham D, de Gramont A, Scheithauer W, Smakal M, Humblet Y, Kourteva G, Iveson T, Andre T, Dostalova J et al (2004) Phase III double‐blind placebo‐controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol 22, 3950–3957. [DOI] [PubMed] [Google Scholar]
- Reid AL, Freeman JB, Millward M, Ziman M and Gray ES (2015) Detection of BRAF‐V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin Biochem 48, 999–1002. [DOI] [PubMed] [Google Scholar]
- Ren C, Han C, Zhang J, He P, Wang D, Wang B, Zhao P and Zhao X (2011) Detection of apoptotic circulating tumor cells in advanced pancreatic cancer following 5‐fluorouracil chemotherapy. Cancer Biol Ther 12, 700–706. [DOI] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH et al (2012) EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Thege FI, Santana SM, Lannin TB, Saha TN, Tsai S, Maggs LR, Kochman ML, Ginsberg GG, Lieb JG et al (2014) Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology 146, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DA, Boehm KA, Waterhouse DM, Wagener DJ, Krishnamurthi SS, Rosemurgy A, Grove W, Macdonald K, Gulyas S, Clark M et al (2006) Gemcitabine plus CI‐994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double‐blind, placebo‐controlled, multicenter study. Ann Oncol 17, 1096–1102. [DOI] [PubMed] [Google Scholar]
- Risberg B, Tsui DWY, Biggs H, Ruiz‐Valdepenas Martin de Almagro A, Dawson SJ, Hodgkin C, Jones L, Parkinson C, Piskorz A et al (2018) Effects of collection and processing procedures on plasma circulating cell‐free DNA from cancer patients. J Mol Diagnost 20, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothé F, Laes JF, Lambrechts D, Smeets D, Vincent D, Maetens M, Fumagalli D, Michiels S, Drisis S, Moerman C et al (2014) Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol 25, 1959–1965. [DOI] [PubMed] [Google Scholar]
- Sato A, Nakashima C, Abe T, Kato J, Hirai M, Nakamura T, Komiya K, Kimura S, Sueoka E and Sueoka‐Aragane N (2018) Investigation of appropriate pre‐analytical procedure for circulating free DNA from liquid biopsy. Oncotarget 9, 31904–31914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh K, Hamada S and Shimosegawa T (2015) Involvement of epithelial to mesenchymal transition in the development of pancreatic ductal adenocarcinoma. J Gastroenterol 50, 140–146. [DOI] [PubMed] [Google Scholar]
- Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart‐Li S, Murphy D, Kay Li Q et al (2015) Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun 6, 7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarà S, Bottoni P and Scatena R (2015) CA 19‐9: Biochemical and clinical aspects. Adv Exp Med Biol 867, 247–260. [DOI] [PubMed] [Google Scholar]
- Schlitter AM, Segler A, Steiger K, Michalski CW, Jager C, Konukiewitz B, Pfarr N, Endris V, Bettstetter M, Kong B et al (2017) Molecular, morphological and survival analysis of 177 resected pancreatic ductal adenocarcinomas (PDACs): Identification of prognostic subtypes. Sci Rep 7, 41064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefrioui D, Blanchard F, Toure E, Basile P, Beaussire L, Dolfus C, Perdrix A, Paresy M, Antonietti M, Iwanicki‐Caron I et al (2017) Diagnostic value of CA19.9, circulating tumour DNA and circulating tumour cells in patients with solid pancreatic tumours. Br J Cancer 117, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, Ogunwobi OO, Chen T, Zhang J, George TJ, Liu C and Fan ZH (2014) Capture, release and culture of circulating tumor cells from pancreatic cancer patients using an enhanced mixing chip. Lab Chip 14, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD and Jemal A (2018) Cancer statistics, 2018. CA Cancer J Clin 68, 7–30. [DOI] [PubMed] [Google Scholar]
- Singh N, Gupta S, Pandey RM, Chauhan SS and Saraya A (2015) High levels of cell‐free circulating nucleic acids in pancreatic cancer are associated with vascular encasement, metastasis and poor survival. Cancer Invest 33, 78–85. [DOI] [PubMed] [Google Scholar]
- Siravegna G, Marsoni S, Siena S and Bardelli A (2017) Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 14, 531–548. [DOI] [PubMed] [Google Scholar]
- Smith JE, Medley CD, Tang Z, Shangguan D, Lofton C and Tan W (2007) Aptamer‐conjugated nanoparticles for the collection and detection of multiple cancer cells. Anal Chem 79, 3075–3082. [DOI] [PubMed] [Google Scholar]
- Sollier E, Go DE, Che J, Gossett DR, O'Byrne S, Weaver WM, Kummer N, Rettig M, Goldman J, Nickols N et al (2014) Size‐selective collection of circulating tumor cells using Vortex technology. Lab Chip 14, 63–77. [DOI] [PubMed] [Google Scholar]
- Sozzi G, Conte D, Leon M, Ciricione R, Roz L, Ratcliffe C, Roz E, Cirenei N, Bellomi M, Pelosi G et al (2003) Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol 21, 3902–3908. [DOI] [PubMed] [Google Scholar]
- Ståhlberg A, Krzyzanowski PM, Egyud M, Filges S, Stein L and Godfrey TE (2017) Simple multiplexed PCR‐based barcoding of DNA for ultrasensitive mutation detection by next‐generation sequencing. Nat Protoc 12, 664–682. [DOI] [PubMed] [Google Scholar]
- Stephenson D, Nahm C, Chua T, Gill A, Mittal A, de Reuver P and Samra J (2017) Circulating and disseminated tumor cells in pancreatic cancer and their role in patient prognosis: a systematic review and meta‐analysis. Oncotarget 8, 107223–107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swennenhuis JF, Tibbe AG, Levink R, Sipkema RC and Terstappen LW (2009) Characterization of circulating tumor cells by fluorescence in situ hybridization. Cytometry A 75, 520–527. [DOI] [PubMed] [Google Scholar]
- Takai E, Totoki Y, Nakamura H, Kato M, Shibata T and Yachida S (2016) Clinical utility of circulating tumor DNA for molecular assessment and precision medicine in pancreatic cancer. Adv Exp Med Biol 924, 13–17. [DOI] [PubMed] [Google Scholar]
- Takai E, Totoki Y, Nakamura H, Morizane C, Nara S, Hama N, Suzuki M, Furukawa E, Kato M, Hayashi H et al (2015) Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci Rep 5, 18425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Okada S, Ueno H, Okusaka T and Ikeda M (2000) The usefulness of serial changes in serum CA19‐9 levels in the diagnosis of pancreatic cancer. Pancreas 20, 378–381. [DOI] [PubMed] [Google Scholar]
- Tien YW, Kuo HC, Ho BI, Chang MC, Chang YT, Cheng MF, Chen HL, Liang TY, Wang CF, Huang CY et al (2016) A high circulating tumor cell count in portal vein predicts liver metastasis from periampullary or pancreatic cancer: a high portal venous CTC count predicts liver metastases. Medicine 95, e3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinari N, De Tursi M, Grassadonia A, Zilli M, Stuppia L, Iacobelli S and Natoli C (2012) An epigenetic approach to pancreatic cancer treatment: the prospective role of histone deacetylase inhibitors. Curr Cancer Drug Targets 12, 439–452. [DOI] [PubMed] [Google Scholar]
- Ting DT, Wittner BS, Ligorio M, Vincent Jordan N, Shah AM, Miyamoto DT, Aceto N, Bersani F, Brannigan BW, Xega K et al (2014) Single‐cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep 8, 1905–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen‐Ottosen Berry K, Gilje B, Søreide JA, Javle M, Nordgård O and Smaaland R (2016) Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol 10, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigg RM, Martinson LJ, Parpart‐Li S and Shaw JA (2018) Factors that influence quality and yield of circulating‐free DNA: a systematic review of the methodology literature. Heliyon 4, e00699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Hibi K, Kaneko T, Takeda S, Inoue S, Okochi O, Nagasaka T and Nakao A (2004) Detection of K‐ras mutations in the plasma DNA of pancreatic cancer patients. J Gastroenterol 39, 56–60. [DOI] [PubMed] [Google Scholar]
- Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, Gligorich KM, Rostomily RC, Bronner MP and Shendure J (2016) Fragment length of circulating tumor DNA. PLoS Genet 12, e1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype C, Neumann H et al (2004) Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol 22, 1430–1438. [DOI] [PubMed] [Google Scholar]
- Varillas JI, Chen K, Zhang J, George TJ Jr and Hugh Fan Z (2017) A novel microfluidic device for isolation of circulating tumor cells from pancreatic cancer blood samples. Methods Mol Biol 1634, 33–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B and Kinzler KW (1999) Digital PCR. Proc Natl Acad Sci USA 96, 9236–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Gahlawat A, Lenhardt J, Witte T, Keitel D, Kaufhold A, Maass KK, Pajtler KW, Sohn C and Schott S (2019) Evaluation of storage tubes for combined analysis of circulating nucleic acids in liquid biopsies. Int J Mol Sci 20, E704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warkiani ME, Khoo BL, Wu L, Tay AK, Bhagat AA, Han J and Lim CT (2016) Ultra‐fast, label‐free isolation of circulating tumor cells from blood using spiral microfluidics. Nat Protoc 11, 134–148. [DOI] [PubMed] [Google Scholar]
- Warton K, Yuwono NL, Cowley MJ, McCabe MJ, So A and Ford CE (2017) Evaluation of Streck BCT and PAXgene stabilised blood collection tubes for cell‐free circulating DNA studies in plasma. Mol Diag Therapy 21, 563–570. [DOI] [PubMed] [Google Scholar]
- Whitehead J, Thygesen H, Jaki T, Davies S, Halford S, Turner H, Cook N and Jodrell D (2012) A novel Phase I/IIa design for early phase oncology studies and its application in the evaluation of MK‐0752 in pancreatic cancer. Stat Med 31, 1931–1943. [DOI] [PubMed] [Google Scholar]
- Wu J, Zhou Y, Zhang CY, Song BB, Wang BL, Pan BS, Lou WH and Guo W (2014) Co‐amplification at lower denaturation‐temperature PCR combined with unlabled‐probe high‐resolution melting to detect KRAS codon 12 and 13 mutations in plasma‐circulating DNA of pancreatic adenocarcinoma cases. Asian Pacific J Cancer Prevent 15, 10647–10652. [DOI] [PubMed] [Google Scholar]
- Wu G, Zhu R, Li Y, Zhao Y and Dai M (2018) Prognostic significance of circulating tumor microemboli in patients with pancreatic ductal adenocarcinoma. Oncol Lett 15, 7376–7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Mao X, Guo T, Chan PY, Shaw G, Hines J, Stankiewicz E, Wang Y, Oliver RTD, Ahmad AS et al (2017) The novel association of circulating tumor cells and circulating megakaryocytes with prostate cancer prognosis. Clin Cancer Res 23, 5112–5122. [DOI] [PubMed] [Google Scholar]
- Xu L, Mao X, Imrali A, Syed F, Mutsvangwa K, Berney D, Cathcart P, Hines J, Shamash J and Lu YJ (2015) Optimization and evaluation of a novel size based circulating tumor cell isolation system. PLoS ONE 10, e0138032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A and DePinho RA (2016) Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 30, 355–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bao L, Yang S, Qian Z, Dong M, Yin L, Zhao Q, Ge K, Deng Z, Zhang J et al (2016a) Tumor‐selective replication herpes simplex virus‐based technology significantly improves clinical detection and prognostication of viable circulating tumor cells. Oncotarget 7, 39768–39783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Boominathan R, Foulk B, Rao C, Kemeny G, Strickler JH, Abbruzzese JL, Harrison MR, Hsu DS, Healy P et al (2016b) Development of a novel c‐MET‐based CTC detection platform. Mol Cancer Res 14, 539–547. [DOI] [PubMed] [Google Scholar]
- Zhang S, Chen Z, Huang C, Ding C, Li C, Chen J, Zhao J and Miao L (2019) Ultrasensitive and quantitative detection of EGFR mutations in plasma samples from patients with non‐small‐cell lung cancer using a dual PNA clamping‐mediated LNA‐PNA PCR clamp. Analyst 144, 1718–1724. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li S, Liu F, Zhou L, Shao N and Zhao X (2015a) SELEX aptamer used as a probe to detect circulating tumor cells in peripheral blood of pancreatic cancer patients. PLoS ONE 10, e0121920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang F, Ning N, Chen Q, Yang Z, Guo Y, Xu D, Zhang D, Zhan T and Cui W (2015b) Patterns of circulating tumor cells identified by CEP8, CK and CD45 in pancreatic cancer. Int J Cancer 136, 1228–1233. [DOI] [PubMed] [Google Scholar]
- Zhang BO, Xu CW, Shao Y, Wang HT, Wu YF, Song YY, Li XB, Zhang Z, Wang WJ, Li LQ et al (2015c) Comparison of droplet digital PCR and conventional quantitative PCR for measuring EGFR gene mutation. Exp Therap Med 9, 1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Li Y, Chen P, Li S, Luo J and Xia H (2019) Performance comparison of blood collection tubes as liquid biopsy storage system for minimizing cfDNA contamination from genomic DNA. J Clin Lab Anal 33, e22670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Nelson WC, Wei B, Schiro PG, Hakimi BM, Johnson ES, Anand RK, Gyurkey GS, White LM, Whiting SH et al (2013) New generation of ensemble‐decision aliquot ranking based on simplified microfluidic components for large‐capacity trapping of circulating tumor cells. Anal Chem 85, 9671–9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Hu L, Yu Z, Zheng J, Yang D, Bouvet M and Hoffman RM (2011) Marker expression in circulating cancer cells of pancreatic cancer patients. J Surg Res 171, 631–636. [DOI] [PubMed] [Google Scholar]
- Zill OA, Banks KC, Fairclough SR, Mortimer SA, Vowles JV, Mokhtari R, Gandara DR, Mack PC, Odegaard JI, Nagy RJ et al (2018) The landscape of actionable genomic alterations in cell‐free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res 24, 3528–3538. [DOI] [PubMed] [Google Scholar]
- Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M, Hendifar AE, Wang Z, Atreya CE, Kelley RK et al (2015) Cell‐free DNA next‐generation sequencing in pancreatobiliary carcinomas. Cancer Discov 5, 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]