

Abstract
Selective cargo transport into axons and dendrites over the microtubule network is essential for neuron polarization. The axon initial segment (AIS) separates the axon from the somatodendritic compartment and controls the microtubule-dependent transport into the axon. Interestingly, the AIS has a characteristic microtubule organization; it contains bundles of closely spaced microtubules with electron dense cross-bridges, referred to as microtubule fascicles. The microtubule binding protein TRIM46 localizes to the AIS and when overexpressed in non-neuronal cells forms microtubule arrays that closely resemble AIS fascicles in neurons. However, the precise role of TRIM46 in microtubule fasciculation in neurons has not been studied. Here we developed a novel correlative light and electron microscopy approach to study AIS microtubule organization. We show that in cultured rat hippocampal neurons of both sexes, TRIM46 levels steadily increase at the AIS during early neuronal differentiation and at the same time closely spaced microtubules form, whereas the fasciculated microtubules appear at later developmental stages. Moreover, we localized TRIM46 to the electron dense cross-bridges and show that depletion of TRIM46 causes loss of cross-bridges and increased microtubule spacing. These data indicate that TRIM46 has an essential role in organizing microtubule fascicles in the AIS.
SIGNIFICANCE STATEMENT The axon initial segment (AIS) is a specialized region at the proximal axon where the action potential is initiated. In addition the AIS separates the axon from the somatodendritic compartment, where it controls protein transport to establish and maintain neuron polarity. Cargo vesicles destined for the axon recognize specialized microtubule tracks that enter the AIS. Interestingly the microtubules entering the AIS form crosslinked bundles, called microtubule fascicules. Recently we found that the microtubule-binding protein TRIM46 localizes to the AIS, where it may organize the AIS microtubules. In the present study we developed a novel correlative light and electron microscopy approach to study the AIS microtubules during neuron development and identified an essential role for TRIM46 in microtubule fasciculation.
Keywords: axon initial segment (AIS), correlative light and electron microscopy (CLEM), microtubule, TRIM46
Introduction
Neurons are highly polarized cells with two main distinct compartments, the somatodendritic compartment and the axonal compartment, which are separated by the axon initial segment (AIS). The AIS is best known for its integration of synaptic inputs and the generation of action potentials (Kole and Stuart, 2012). In addition, it forms a physical barrier at the beginning of the axon to maintain neuronal polarity (Jones and Svitkina, 2016; Leterrier, 2016; Huang and Rasband, 2018). The AIS recruits cell adhesion molecules to create a dense membrane protein region to prevent plasma membrane mixing. And the AIS controls entry of axonal cargoes and prevents erroneous entry of somatodendritic vesicles and redirects them back to the cell body (Lewis et al., 2009; Watanabe et al., 2012; Franker et al., 2016; Kuijpers et al., 2016).
The microtubule cytoskeleton is instrumental to transport cargo into axons and dendrites to establish neuronal polarity (Kapitein and Hoogenraad, 2015). The sorting of cargo into the axon starts in the region just proximal to the AIS and may depend on kinesin motors that recognizes a subset of microtubules present in the proximal axon (Farías et al., 2015). Interestingly the microtubules in the AIS have a very peculiar organization; they are grouped together with very regular spacing connected by electron-dense cross-bridges, generally referred to as fasciculated microtubules (Palay et al., 1968; Peters et al., 1968). A prime candidate for the AIS microtubule fasciculation is the microtubule binding protein TRIM46. We previously found that TRIM46 specifically localizes to the proximal axon (van Beuningen et al., 2015). When overexpressed in cultured cells TRIM46 bundles microtubules in a parallel fashion, which at the electron microscopy (EM) level closely resembles AIS fascicles. Therefore, we suggested that TRIM46 is the organizer of microtubule fascicles in the AIS (Kapitein and Hoogenraad, 2015; van Beuningen et al., 2015). To address this claim more directly in neurons we developed a correlative light and electron microscopy (CLEM) approach to perform high-resolution analysis of fasciculated microtubules in the AIS of cultured neurons and address the role of TRIM46 in AIS microtubule organization.
Materials and Methods
Primary neuronal cultures and transfection.
All experiments with animals were performed in compliance with the guidelines for the welfare of experimental animals issued by the Government of the Netherlands and were approved by the Animal Ethical Review Committee (DEC) of Utrecht University. Primary hippocampal neurons cultures were prepared from embryonic day 18 rat brains (both genders). Cells were plated on Aclar (or coverslips for antibody labeling with TRIM46) coated with poly-l-lysine (37.5 μg/ml) and laminin (1.25 μg/ml) at a density of 50,000/well. Neurons were cultured in Neurobasal medium (NB) supplemented with 2% B27 (Invitrogen), 0.5 mm glutamine (Invitrogen), 15.6 μm glutamate (Sigma-Aldrich), and 1% penicillin/streptomycin (Invitrogen) at 37°C in 5% CO2. For the analysis of the AIS in 2 DIV neurons, hippocampal neurons (400,000 cells) were nucleofected with 2 μg of 480AnkG-GFP using the Amaxa Rat Neuron Nucleofector kit (Lonza) according to the manufacturer's instructions. For the TRIM46 knockdown studies, hippocampal neurons were transfected at 17 DIV with TRIM46-shRNAs (van Beuningen et al., 2015) or scrambled-shRNA together with mCherry as a fill using Lipofectamine 2000 (Invitrogen). For the rescue experiment, GFP or GFP-TRIM46 was cotransfected together with the shRNAs. Briefly, for 1 well of a 12-well plate, 1.8 μg DNA (1.5 μg/well shRNA + 0.3 μg/well mCherry) was mixed with 3.3 μl of Lipofectamine 2000 in 200 μl NB, incubated for 30 min, and then added to the neurons in NB at 37°C in 5% CO2 for 45 min. Next, neurons were washed with NB and transferred to their original medium at 37°C in 5% CO2.
CLEM approach.
To correlatively image Neurons or HeLa cells with fluorescence and electron microscopy we grew cells on Aclar pieces (Electron Microscopy Sciences; 50426-10); 1 cm square pieces were cut, cleaned with acetone, glued in a 12-well plate using Matrigel (Biosciences) and were sterilized by UV light (Jiménez et al., 2010).
To localize the AIS we detached the Aclar from the well plate and incubated the neurons upside-down with anti-NF186 (1:200; NeuroMab, A12/18) in 60 μl of its own growth medium for 30 min, which targets the neurofascin membrane protein extracellularly. Cells were washed 3× for 2 min with 100 μl growth medium and placed back in 400 μl growth medium for 30 min in a 12-well plate. For this procedure the neurons were kept in the incubator on a 37°C stretching table (OTS40) when changing the media.
The neurons were fixed by gently adding 400 μl of prewarmed 2× concentrated fixing solution (4% paraformaldehyde + 0.4% glutaraldehyde in 0.2 m PHEM buffer, pH 6.9) for 10 min at 37°C, after which the growth medium + fixative was removed and exchanged for 1 ml 1× fixing solution (2% paraformaldehyde + 0.2% glutaraldehyde in 0.1 m PHEM buffer, pH 6.9) for 10 min at room temperature. All following steps were performed at room temperature. The neurons were washed 6× for 5 min with PBS, incubated with 100 mm NH4Cl in PBS for 10 min to quench free aldehyde groups, washed 6× for 5 min with PBS, and incubated for 1 h with a bridging secondary antibody (1:300; DakoCytomation, Z0412) in PBS + 1% bovine serum albumin (BSA). Afterward the samples were washed 3× for 10 min with PBS + 0.1% BSA, incubated with a mix of protein-A gold 15 nm (1:60; CMC Utrecht) with 1:100 rabbit AlexaFluor 488 secondary antibody (Life Technologies, A11034) in PBS + 1% BSA for 45 min, and finally washed 3× for10 min in PBS.
Candidate neurons were fluorescently imaged using an EVOS inverted microscope (AMG) and the positions of selected neurons were roughly marked by engraving the Aclar using a 26G needle (21 G needles were used to pin down the Aclar).
Cells were further fixed with 3.5% glutaraldehyde + 1% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.4, postfixed with a 1% Osmium + 1.5% KFeCN solution in 0.1 m cacodylate buffer, pH 7.4, washed with water, dehydrated with ethanol, and infiltrated with increasing amounts of Epon resin. Small pieces of Aclar with the regions-of-interest (marked by the needle engravings) were cut using a razor blade and Epon-embedded for perpendicular sectioning. The neurons were orientated in such a way that the soma of the neuron is sectioned first before reaching the AIS.
After Epon polymerization, Aclars were peeled off using a razor blade leaving the neurons in the Epon. The needle engraving can be seen on the Epon surface, which can be used to find the target neuron, but are lost once a drop of Epon is added on top. To further mark the position of the target neuron, landmarks were placed around the target cell using a microdissection setup (Zeiss, PALM microbeam; Kolotuev et al., 2009). To visualize these marks under the electron microscope a 3 nm gold layer was applied using a sputter coater. Excess gold was wiped off using a cotton-stab and a drop of Epon was added on top of the cells and polymerized for 48 h at 60°C. The Epon samples were trimmed toward the target cell using the landmarks and sectioned with a Leica Utracut E microtome. Serial sections were placed on grids (Cu 50M-H coated with Formvar film and carbon), stained with uranyl acetate and lead citrate and examined in a Tecnai10 or Tecnai12 electron microscope (FEI) operating at 100 kV and equipped with a SIS CCD Megaview II camera (Tecnai10) or a Tietz TVIPS TemCam F-214 (Tecnai12). Landmarks were used to identify the target cell.
Immunostaining and imaging.
For immunocytochemistry for TRIM46, neurons were fixed for 10 min with 4% paraformaldehyde + 4% sucrose. Primary rabbit anti-TRIM46 antibody (van Beuningen et al., 2015) was incubated 1:200 overnight at 4°C in GDB buffer (0.2% BSA, 0.8 m NaCl, 0.5% Triton X-100, 30 mm phosphate buffer, pH 7.4). After three washes in PBS, anti-rabbit AlexaFluor 488 secondary antibody (Life Technologies, A11034) was incubated in the same buffer for 1 h at RT. Coverslips were washed 3× and mounted using VECTASHIELD (Vector Laboratories). Neurons were imaged using a LSM700 confocal laser-scanning microscope (Zeiss) with a EC Plan-Neofluar 40× NA 1.30 oil objective. Z series were acquired with a 0.5 μm step size and sum projections were made using ImageJ for quantifications and maximum projections were made for the images in the paper. The average TRIM46 intensity was measured by making a 3 × 12 μm selection in ImageJ along the AIS.
For the TRIM46 knockdown validation, the mouse anti-Neurofascin antibody (1:500; NeuroMab, 75-172) was incubated 10 min in the neuron culture medium in the incubator, followed by 10 min incubation with anti-mouse AlexaFluor 647 secondary antibody (Life Technologies, A21236). Neurons were then fixed and stained with anti-TRIM46 and guinea pig anti-AnkG (1:500; SYSY, 386004) and imaged on a LSM880 confocal laser scanning microscope (Zeiss) with a α Plan-Apochromat 100×/1.46 oil objective.
Western blot.
Hippocampal neurons were plated in precoated 6-well plates (500,000/well) and directly lysed in 100 μl 2× SDS/DTT sample buffer and boiled for 5 min at 2, 4, 10, or 21 DIV. Equal amounts of cell lysate were loaded on a 10% SDS-PAGE gel, transferred to PVDF membranes and blocked with 2% BSA in PBS/0.05% Tween 20. Primary rabbit anti-TRIM46 antibody (1:2000) was diluted in blocking buffer and incubated overnight at 4°C, washed three times with PBS/0.05% Tween 20 and incubated with secondary IRDye 800LT antibodies (LI-COR Biosciences) for 45 min at room temperature. Membranes were then washed three times with PBS/0.05% Tween 20 and scanned on Odyssey Infrared Imaging system (LI-COR Biosciences).
GFP-nanobody immunogold labeling of TRIM46.
The GFP nanobody (vhhGFP) including a His-tag was cloned in a pET28a vector using the vhhGFP4 sequence (Caussinus et al., 2011) and transformed into BL21DE3 bacteria. Cells were induced with 1 mm IPTG at OD0.6 and grown overnight at 20°C. The bacterial pellet was resuspended in PBS supplemented with lysozyme and cOmplete Protease Inhibitor cocktail (EDTA free, Roche). After five rounds of sonication the soluble fraction, containing the nanobodies, was separated by centrifugation at 20,000 × g for 40 min at 4°C. Meanwhile, 1 ml of cOmplete His-tag Purification Resin (Sigma-Aldrich) was washed with resuspension buffer. After centrifugation, the soluble fraction was supplemented with DTT (final concentration, 1 mm) and was incubated with the washed beads for 1 h at 4°C. The beads were washed 3× in washing buffer (PBS supplemented with 1 mm DTT) and 1× with washing buffer + 40 mm Imidazole. Finally the recombinant protein was eluted by 15 min incubation with washing buffer supplemented with 300 mm imidazole. The supernatant was collected and immediately dialyzed overnight against PBS at 4°C and diluted to 0.5 mg/ml in PBS. The gold conjugation was done following the procedure developed by the van Bergen en Henegouwen lab (unpublished; P. Jain, R. Damman, K. Vocking, F. van Rijsingen, J. Motshagen, J. van Amstel, S. Howes, F. Förster, J.A. Post, and P.M.P. van Bergen en Henegouwen.
Testing of the gold-conjugated antibody was performed using Li Silver enhancement Kit (Nanoprobes) on HeLa cells expressing GFP-TRIM46 grown on glass coverslips (van Beuningen et al., 2015). HeLa cells were cultured in DMEM/Ham's F10 (45/45%) supplemented with 10% fetal calf serum and 1% penicillin/streptomycin at 37°C and 5% CO2. Cell lines were not authenticated by authors after purchase. The cell lines were routinely checked for mycoplasma contamination using LT07-518 Mycoalert assay (Lonza). For EM, cells were plated on pre-engraved Aclar pieces (Jiménez et al., 2010) and transfected. HeLa cells transfected with Fugene6 (Promega) according to the manufacturer's protocol.
For the membrane extraction, cells were incubated for 2 min in extraction buffer [1 m sucrose + 0.15% Triton-X in PEM80 (80 mm PIPIES pH 6.8, 4 mm MgCl2, 1 mm EGTA)] at 37°C, fixed for 10 min in 4% PFA in PEM80 at 37°C. Cells were washed 4× 1 min with PEM80 + 100 nm taxol, blocked for 10 min with 1% BSA in PEM80 + 100 nm taxol, and incubated with the nanobody for 1 h (1:50 in blocking solution). Cells were washed 3× for 2 min and further fixed in 3% PFA + 0.5% glutaraldehyde in PEM80.
Image processing and quantifications.
Image processing was performed using ImageJ (NIH) and Photoshop (Adobe). To quantify inter-microtubule distances and the distance to the membrane, microtubule coordinates were acquired using the ImageJ multipoint tool and the membrane coordinates were acquired using the polygon selection tool with spline fit. These coordinates were used for the nearest neighbor analysis and the membrane distance analysis using a custom written MATLAB script (MATLAB R2011b; MathWorks). Scoring of bridged microtubules was performed by visual inspection.
The model (Movie 1) was made using IMOD v4.10.16 (Kremer et al., 1996). The original tiff images were aligned using the IMOD cross-correlation option and subsequently segmented. The plug-in iMorph in ImageJ (developed by Hajime Hirase) was used to “smoothen” the sequential images and QuickTime was used to assemble individual movies.
Reconstruction of six consecutive ∼100-nm-thick slices of the AIS of a 21 DIV neuron. Microtubules are in turquoise, the cross-bridges in yellow, and the mitochondria in transparent light red.
Statistical analysis.
Statistical analyses were performed using GraphPad Prism software v7.0. All bar diagrams represent mean with SEM, and in the scatterplots the median is indicated with a black line. The statistical analysis of microtubule distribution during neuronal development was performed using a Kruskal–Wallis test with Dunn's correction, except for the analysis of the fraction of bridged microtubules analysis for which a one-way ANOVA with Holm-Sidak's multiple-comparison test was used. The statistical analysis for the TRIM46 levels was performed with a one-way ANOVA with Dunn's correction and for the TRIM46 knockdown effect on bridged microtubules and the nearest neighbor analysis a Mann–Whitney test was used.
Results
CLEM approach to study the AIS
To localize the AIS and to efficiently find the position back by electron microscopy we developed a novel CLEM approach (Fig. 1). To identify the AIS we used an antibody against the extracellular part of neurofascin to avoid the need for permeabilization to preserve the ultrastructure. Using a fluorescent secondary antibody we selected target neurons and localized the AIS before embedding the samples in Epon resin for further EM processing. To find back the target neuron by EM we marked the region-of-interest with a laser dissection microscope (Kolotuev et al., 2009) and filled the laser engravings with evaporated gold to make these visible during sectioning and in the EM microscope (Fig. 1A–C). In addition the neurofascin antibody was labeled with protein-A gold 15 nm, which decorates the outside membrane of the AIS in EM images (Fig. 1D).
Figure 1.

Correlative light and electron microscopy approach to image the AIS. A, Top, Schematic representation of the CLEM approach depicting a side-view of the sample preparation; bottom, example images (top view). Rat hippocampal neurons were grown on pieces of Aclar film (Jiménez et al., 2006). Cells were labeled with anti-neurofascin, fixed, and labeled with fluorescent antibodies as well as 15 nm gold. Target neurons were imaged and the positions on the Aclar were marked using a needle. After further fixing and postfixing, Aclar pieces with the regions-of-interest (ROIs) were cut out and embedded into Epon resin. Then the Aclar pieces were removed, leaving the neurons and the needle markings upside down into the Epon block (Jiménez et al., 2010). The positions of the target neurons were further marked using a microdissection setup (Kolotuev et al., 2009). We drew 1 and 2 lines along the AIS to easily distinguish the left-right orientation and drew a 45° angled line pointing to the target neuron, so that while approaching the ROI, the distance between tow laser marks is the same as the distance remaining toward the ROI (see also B and C). To make these laser engravings visible during the trimming and sectioning of the Epon block as well as during the imaging by EM we applied a thin layer of gold using a sputter coater and wiped the excess away of the flat surface before covering the sample with a drop of Epon, leaving only the engravings filled with gold (visible in C). B, High-magnification of the neurons in Epon after marking the position using a laser dissection microscope. Red dashed lines represent the positions of the sections in C with the arrowheads pointing to the laser engravings seen in C. The small red line is the position of the EM image in D. C, 300 nanometer sections made while approaching the AIS to assess the position. Arrowheads point to the laser marks, and the neuronal cell bodies are indicated. D, EM image of a dendrite and the AIS from neuron II, where the AIS is decorated with 15 nm gold particles targeted to the extracellular part of the neurofascin membrane protein. Scale bars: A–C, 50 μm; D, 200 nm. CB, cell body.
AIS microtubules are increasingly fasciculated during neuron development
Previously, it was reported that the AIS-specific microtubule fasciculation was not observed in cultured neurons (Bartlett and Banker, 1984). However, when sectioning through 21 DIV (21 d in vitro) hippocampal neurons we clearly observed fasciculated microtubules (Fig. 2). This example neuron has its AIS (marked by neurofascin) running parallel to a dendrite, which we sectioned and imaged at multiple positions along the AIS. Only in the AIS, marked by gold particles on the membrane, and not in the parallel running dendrite do the microtubules acquire very regular spacing and are often connected by electron dense cross-bridges (Fig. 2B,D, arrowheads). Even before the axon splits from the dendrite, lateral segregation of the AIS membrane marker, neurofascin, and fasciculated microtubules can be seen, as was also seen by Peters et al. (1968) in tissue. These data suggest that there may be a close connection between the AIS (sub-)membrane organization and microtubule fasciculation.
Figure 2.
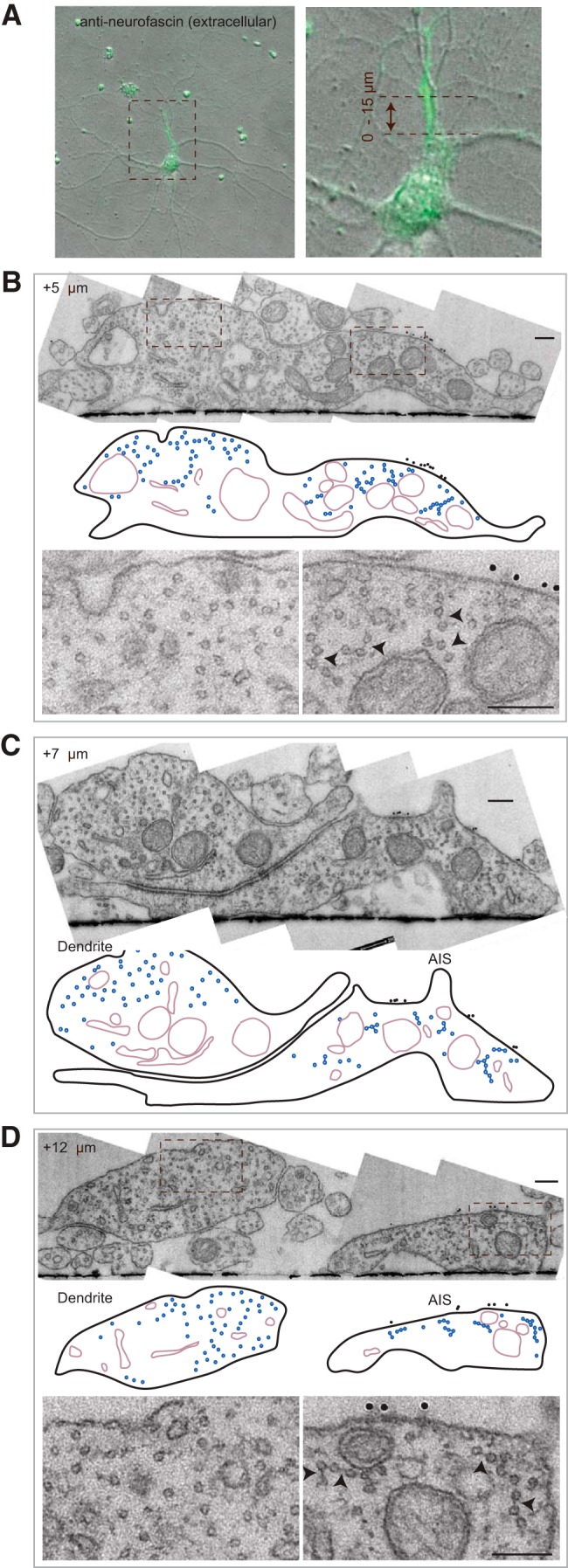
21 DIV neurons have fasciculated microtubules in the AIS. A–D, Typical example of a 21 DIV neuron processed by our correlative approach to analyze the AIS microtubules. Extracellular neurofascin staining to identify the AIS by fluorescence (A) and by EM (B–D). Note that there is a dendrite running parallel to the AIS, which at +5 μm did not split yet from the axon although the neurofascin marker and fasciculated microtubules already group at the right side of the neurite (B). In the schematic representations the microtubules and cross-bridges are represented in blue and organelles in violet. In the AIS magnifications arrowheads point to electron dense cross-bridges. Scale bar, 200 nm.
To address how the microtubules acquire this typical organization in the AIS, we analyzed and quantified the microtubule distribution at different developmental stages using the above described CLEM approach. For 2 DIV neurons cells were transfected with the AIS marker Ankyrin-G480-GFP at 0 DIV (Fréal et al., 2016) as neurofascin is not well enriched at the AIS at this stage (Albrecht et al., 2016). Whereas at 2 DIV the AIS microtubules are quite randomly distributed, already at 5 DIV the microtubules acquire very regular spacing, which is maintained during further development (Fig. 3A–D). This is in agreement with previous work detecting alignment of AIS microtubules already at 4 DIV using platinum replica electron microscopy (Jones et al., 2014). Moreover, this correlates in time with neuron polarization and the assembly of the AIS (Zhong et al., 2014; Albrecht et al., 2016; Banker, 2018). We performed a nearest neighbor analysis and found that at 5 DIV nearly all microtubules are closely spaced together with a median distance of 37 nm measured from the middle of each microtubule (the microtubule diameter is 25 nm; Figs. 3E, 4A). This microtubule spacing hardly changes during later developmental stages where we measured a median spacing of 39 nm. This spacing is very close to what was reported before for AIS microtubules in rat brains (Peters et al., 1968). In dendrites, microtubules are not obviously grouped together and a more scattered distribution is observed with a median nearest-neighbor distance of 67 nm.
Figure 3.

Microtubule fasciculation during AIS maturation. A–D, Typical examples of AIS microtubule organization in 2 DIV (A), 5 DIV (B), 11 DIV (C), and 21 DIV (D) neurons. In the schematic representation below, microtubules and cross-bridges are in blue and the organelles in violet. For 2 DIV we transfected neurons at 0 DIV with Ankyrin-G-GFP and identified the AIS based on the GFP accumulation in one of the neurites because neurofascin is largely absent at that stage, hence there is no gold labeling. Scale bar, 200 nm. E, Nearest-neighbor analysis of inter-microtubule distances during neuron development, measured from mid-microtubule. The graph represents all microtubules within 150 nm range, leaving out a few distant single microtubules, however all microtubules were included for the statistics. The black lines represent the median values. Statistics, Kruskal–Wallis test with Dunn's correction. F, Quantification of bridged microtubules during neuron development. Each neuron was imaged at multiple positions along the AIS or dendrite, and the average of bridged microtubules was quantified per image. Error bar represent SEM and for the statistics a one-way ANOVA with Dunn's correction was performed and compared with 2 DIV. G, Serial sections of fasciculated microtubules showing that the cross-bridges do not form a continuous structure (distance in between images is indicated). Scale bar, 200 nm. H, Nearest-neighbor analysis for cross-bridged microtubules at the AIS during development. The black lines represent the median values. Statistics, Kruskal–Wallis test with Dunn's correction. I, Zoom of the plasma membrane of a 21 DIV neuron AIS showing the close apposition of fasciculated microtubules with the dense undercoating of the membrane and gold labeled neurofascin. Scale bar, 200 nm. J, Quantification of the microtubule distance to the plasma membrane in the AIS during development. Only microtubules in close proximity to the plasma membrane (100 nm) are represented here and are measured from the middle of the microtubule. The black lines represent the median values. Statistics, Kruskal–Wallis test with Dunn's correction. Number of neurons (N) and total number of quantified sections (n) analyzed for E–G: 2 DIV: N = 3, n = 7; 5 DIV: N = 2, n = 8; 11 DIV: N = 3 (of which one 10 DIV), n = 11; 21 DIV: N = 5, n = 17. ns, Nonsignificant; *p < 0.05, ***p < 0.005, ****p < 0,0001.
Figure 4.

Quantification of AIS microtubules for individual neurons. A, Nearest-neighbor analysis of inter-microtubule distances for individual neurons, measured from mid-microtubule. The graph represents all microtubules within 150 nm range, leaving out a few distant single microtubules; however, all microtubules were included for the statistics. The black lines represent the median values. Statistics, Kruskal–Wallis test with Dunn's correction. B, Quantification of bridged microtubules for individual neurons at multiple positions during neuron development. C, Quantification of the microtubule distance to the plasma membrane in the AIS for individual neurons. Only microtubules in close proximity to the plasma membrane (100 nm) are represented here and are measured from the middle of the microtubule. The black lines represent the median values. Statistics, Kruskal–Wallis test with Dunn's correction. D, Nearest-neighbor analysis for all microtubules in individual neuron at 21 DIV upon control of TRIM46 knockdown. The graph represents the microtubules within 150 nm range, cutting off several values, which were included for the statistics. Black line represents the median value. Statistics, Mann–Whitney test. E, Quantification of bridged microtubules in the AIS for individual neurons at multiple positions upon control or TRIM46 knockdown. ns, Nonsignificant; *p < 0.05, **p < 0.01, ****p < 0,0001.
Although already at 5 DIV microtubules have consistent spacing, we could hardly detect electron dense cross-bridges at that stage. However, at later stages these cross-bridges become more prominent and abundant. The cross-bridges are best observed at 21 DIV in the AIS, but never in dendrites (Figs. 3A–D,F, 4B). This may explain the lack of fasciculated AIS microtubules reported before in cultured neurons, which analyzed neurons at 7 DIV (Bartlett and Banker, 1984). The microtubules cross-bridges are not continuous structures as can be seen in sequential images (Fig. 3G; Movie 1). Moreover, only analyzing the microtubule spacing of cross-bridged microtubules did not significantly change the median spacing (5 DIV: 36 and 37 nm for 11 and 21 DIV; Fig. 3H). Altogether the regular microtubule spacing of AIS microtubules correlates in time with neuron polarization and AIS assembly, and the typical electron dense cross-bridges become more prominent and abundant during later development.
Because the master-organizer of the AIS, Ankyrin-G, can associate with microtubules (Leterrier et al., 2011; Fréal et al., 2016) and with membrane proteins such as neurofascin (Michaely and Bennett, 1995), we analyzed the microtubule distribution with respect to the membrane during AIS maturation. Indeed we frequently observed microtubule fascicles in close proximity with electron dense submembrane patches, often also enriched for neurofascin (gold particles; Fig. 3G,I). The microtubules distance to the membrane slightly increases during AIS maturation (5–21 DIV; Figs. 3J, 4C).
TRIM46 is part of the electron dense microtubule cross-bridges
A prime candidate protein responsible for AIS-specific microtubule fasciculation is TRIM46. We previously found that TRIM46 localizes to the AIS and that it is important for axon formation (van Beuningen et al., 2015). Moreover, when expressed in non-neuronal cells, it induces microtubule bundles with parallel polarity and at the same time electron dense cross-bridges can be observed in between the microtubules mimicking the AIS ultrastructure (van Beuningen et al., 2015). To address whether TRIM46 is indeed important for microtubule fasciculation in the AIS, we first determined the relative levels of TRIM46 during neuron maturation (Fig. 5A–C). Although TRIM46 can be detected in some of the 2 DIV neurons, the levels are weak. At 5 DIV, a time point where the microtubules acquired their typical spacing, all neurons have clear TRIM46 localization at the AIS. At later time points there is a steady increase in the TRIM46 levels at the AIS.
Figure 5.

TRIM46 localizes to the electron dense cross-bridge. A, Immunolabeling against TRIM46 at the AIS at different developmental stages. Scale bar, 20 μm. B, Quantification of TRIM46 labeling during neuron development, normalized to 5 DIV. N > 3 experiments of min 10 neurons each. Error bars represent SEM. Statistics, one-way ANOVA with Dunn's correction (compared with 5 DIV). ns, Nonsignificant; *p < 0.05, ****p < 0.0001. C, Western blot analysis of hippocampal neurons at different developmental stages. Neurons were grown at 500,000/well, lysed in 100 μl lysis buffer and equal amounts of cell lysate were loaded and stained for TRIM46. D, GFP-TRIM46 expression in HeLa cells. E, EM image of the transfected HeLa cell in D (as marked by the red dotted line). F, G, High-magnification of the boxed area in E, with further zooms showing the fasciculated microtubules upon GFP-TRIM46 overexpression in HeLa cells with immunolabeling with the gold-conjugated anti-gfp nanobody (F). G, The quantification of the gold to mid-microtubule distance and the black lines represent the median value. Scale bar, 200 nm.
To test more directly whether TRIM46 localizes to the microtubule fascicles and electron dense cross-bridges we performed immune-gold electron microscopy. Because of the densely packed structure of the microtubule fascicles we decided to use nanobodies for detection. Nanobodies are much smaller in size than conventional antibodies and thus also get much closer to the epitope. We expressed GFP-TRIM46 in cultured HeLa cells and detected the GFP using gold coupled gfp-nanobodies (Fig. 5D; Kijanka et al., 2017).
To analyze the AIS microtubules the addition of a small amount of glutaraldehyde greatly improved the microtubule ultrastructure preservation, however this also blocked the binding of the gfp nanobody (data not shown). Therefore we used a membrane extraction protocol (Tas et al., 2017) to label GFP-TRIM46 with the gfp-nanobodies (Fig. 5E–G). We performed the CLEM approach to section the microtubules under a 90° angle and found that the gold particles nicely labeled the bundled microtubules at the base of the electron dense cross-bridges 23 nm from the mid-microtubule (Fig. 5F,G). This shows that TRIM46 is part of the electron dense cross-bridge.
TRIM46 is critical for AIS microtubule fasciculation
To test whether TRIM46 is required for microtubule fasciculation in neurons, we performed TRIM46 knockdown experiments. We transfected neurons with control or previously validated shRNA constructs against TRIM46 (van Beuningen et al., 2015) at 17 DIV and fixed at 21 DIV. The knockdown of TRIM46 led to a fragmented Ankyrin-G and neurofascin labeling at the AIS as previously observed (Fig. 6A–D; van Beuningen et al., 2015). Moreover, this phenotype could be rescued by re-expression of GFP-TRIM46, validating the specificity of the knockdown. At the electron microscopy level the knockdown of TRIM46 led to a reduction in the cross-bridged microtubule fraction and to a marked increase in microtubule spacing in the AIS compared with control knockdown neurons (Fig. 6D–F). Therefore TRIM46 is required for the maintenance of AIS microtubule fascicles.
Figure 6.

TRIM46 depletion results in the loss of microtubule fasciculation. Several examples for control knockdown + gfp (A), TRIM46 shRNAs + GFP (B), or TRIM46 shRNAs + GFP-TRIM46 (C) transfected neurons stained for TRIM46, AnkG, and neurofascin. Scale bar, 5 μm. D, AIS microtubule organization at 21 DIV upon control or TRIM46 knockdown with on top examples of the analyzed neurons stained with extracellular neurofascin (green, marked by arrowheads) to identify the AIS and filled with mCherry (magenta) to identify transfected neurons. In the schematic representation, microtubules and cross-bridges are in blue and organelles in violet. Scale bar, 100 nm. E, Quantification of bridged microtubules upon TRIM46 knockdown. Each neuron was imaged at multiple positions along the AIS, and the average of bridged microtubules was quantified per image. Error bars represent SEM. Statistics, Mann–Whitney test. F, Nearest-neighbor analysis for all microtubules during neuron development. The graph represents the microtubules within 150 nm range, cutting off several values, which were included for the statistics. Black lines represent the median values. Statistics, Mann–Whitney test. Number of neurons (N) and total number of quantified sections (n) analyzed for (D–F): Control knockdown: N = 2, n = 7; TRIM46 knockdown: N = 4, n = 15. ns, Nonsignificant; ****p < 0.0001. For variation between neurons, see Figure 4, D and E.
Discussion
The AIS is essential for action potential generation and to separate the axon from the somatodendritic compartment for polarity maintenance. At the ultra-structural level the AIS is characterized by an electron dense granular undercoating at the plasma membrane as well as fasciculated microtubules, which are closely spaced microtubules interconnected by electron dense cross-bridges (Palay et al., 1968; Peters et al., 1968). Whereas the granular membrane undercoating is considered to represent Ankyrin-G with all its membrane and cytoskeleton associated proteins (Jones and Svitkina, 2016), the nature of the microtubules fascicles is largely unknown.
Here we developed a novel correlative light and electron microscopy approach to analyze subcellular structures in cultured cells. We found that in cultured neurons the AIS microtubules align and acquire regular spacing during early development (5 DIV), which is maintained during further development. This is in agreement with previous work from the Svitkina laboratory, which used platinum replica electron microscopy to analyze the AIS microtubules (Jones et al., 2014). Previously we have identified the protein TRIM46 to localize to the AIS, and when overexpressed in cultured HeLa cells TRIM46 leads to bundled parallel microtubules with very similar electron dense cross-bridges as in the neuronal AIS (van Beuningen et al., 2015). Here, we localize TRIM46 to the electron dense cross-bridges and show that it is important for the maintenance of these structures. Moreover, we observed a loss of regular microtubule spacing upon TRIM46 depletion, suggesting that these cross-bridges regulate microtubule spacing. Because TRIM46 is detected at the AIS at early stages of neuronal development this suggests that TRIM46 also regulates microtubule spacing during early neuronal development. However at these early neuronal stages, we could not observe the electron dense cross-bridges by EM, which is consistent with previous EM studies (Bartlett and Banker, 1984; Jones et al., 2014). It is likely that the microtubule bundles in young neurons do not have sufficient amounts of TRIM46 protein to be detected by EM as electron dense cross-bridges. Because we made use of chemical fixation to prepare the samples, this might affect the appearance of the electron-dense cross-bridges. To gain additional insight in the structural organization of the cross-bridges, future experiments using correlative cryo-EM are required.
Several members of the TRIM protein family are known to self-associate by forming anti-parallel dimers and other higher-order complexes (Li et al., 2014; Sanchez et al., 2014). Whether TRIM46 forms antiparallel dimers to regulate microtubule spacing in the AIS would require in vitro reconstitution experiments and structural analysis. Moreover, it is interesting to note that TRIM46 is not the only protein that can organize microtubules in the AIS. In Purkinje cells the microtubule stabilizing protein MTCL1 localizes to the AIS and is important to maintain neuronal polarity (Satake et al., 2017). It is so far unclear whether this MTCL1 function is specific to Purkinje cells and whether it might work together with TRIM46 or has a redundant function. What might be the function of the TRIM46-positive microtubule fascicles in the AIS? The microtubule fascicles are not exclusive to the AIS, but also extend into the proximal axon (Palay et al., 1968; Peters et al., 1968), where the initial sorting of vesicles into the axon starts (Farías et al., 2015). Previously, we have found that motor proteins, such as kinesin-1, specifically target cargo into the axon, whereas other kinesins target both the axon as well as dendrites (Lipka et al., 2016). This difference is thought to in part be because of a difference in microtubule organization (Cai et al., 2009; Konishi and Setou, 2009; Tas et al., 2017). Therefore this suggests that the fasciculation of microtubules, which extend into the proximal axon serve as specialized tracks to sort axonal cargo into the axon. Indeed, cargo transport into the axon was markedly reduced in the absence of TRIM46 (van Beuningen et al., 2015), suggesting that the uniform microtubule arrays in the axon organized by TRIM46 drive efficient cargo delivery and trafficking into axons of differentiated neurons.
Footnotes
This work was supported by the Netherlands Organization for Scientific Research (NWO) (NWO-ALW-TOP to M.H., NWO-ALW-VIDI to L.C.K., NWO-ALW-VICI to C.C.H.) and by the European Research Council (ERC Starting Grant to L.C.K., ERC Consolidator Grant to C.C.H.). We thank Eugene Katrukha for guidance with MATLAB, Elly van Donselaar and Rob Mesman for valuable advice, Purvi Jain, Hans Meeldijk, and Chris Schneijdenberg for technical assistance, and the neuron culture team for the cultures.
The authors declare no competing financial interests.
References
- Albrecht D, Winterflood CM, Sadeghi M, Tschager T, Noé F, Ewers H (2016) Nanoscopic compartmentalization of membrane protein motion at the axon initial segment. J Cell Biol 215:37–46. 10.1083/jcb.201603108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker G. (2018) The development of neuronal polarity: a retrospective view. J Neurosci 38:1867–1873. 10.1523/JNEUROSCI.1372-16.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett WP, Banker GA (1984) An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture: I. Cells which develop without intercellular contacts. J Neurosci 4:1944–1953. 10.1523/JNEUROSCI.04-08-01944.1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, McEwen DP, Martens JR, Meyhofer E, Verhey KJ (2009) Single molecule imaging reveals differences in microtubule track selection between kinesin motors. PLoS Biol 7:e1000216. 10.1371/journal.pbio.1000216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caussinus E, Kanca O, Affolter M (2011) Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat Struct Mol Biol 19:117–121. 10.1038/nsmb.2180 [DOI] [PubMed] [Google Scholar]
- Farías GG, Guardia CM, Britt DJ, Guo X, Bonifacino JS (2015) Sorting of dendritic and axonal vesicles at the pre-axonal exclusion zone. Cell Rep 13:1221–1232. 10.1016/j.celrep.2015.09.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franker MA, Esteves da Silva M, Tas RP, Tortosa E, Cao Y, Frias CP, Janssen AFJ, Wulf PS, Kapitein LC, Hoogenraad CC (2016) Three-step model for polarized sorting of KIF17 into dendrites. Curr Biol 26:1705–1712. 10.1016/j.cub.2016.04.057 [DOI] [PubMed] [Google Scholar]
- Fréal A, Fassier C, Le Bras B, Bullier E, De Gois S, Hazan J, Hoogenraad CC, Couraud F (2016) Cooperative interactions between 480 kDa ankyrin-G and EB proteins assemble the axon initial segment. J Neurosci 36:4421–4433. 10.1523/JNEUROSCI.3219-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Rasband MN (2018) Axon initial segments: structure, function, and disease. Ann N Y Acad Sci 1420:46–61. 10.1111/nyas.13718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez N, Humbel BM, van Donselaar E, Verkleij AJ, Burger KN (2006) Aclar discs: a versatile substrate for routine high-pressure freezing of mammalian cell monolayers. J Microsc 221:216–223. 10.1111/j.1365-2818.2006.01558.x [DOI] [PubMed] [Google Scholar]
- Jiménez N, Van Donselaar EG, De Winter DA, Vocking K, Verkleij AJ, Post JA (2010) Gridded aclar: preparation methods and use for correlative light and electron microscopy of cell monolayers, by TEM and FIB-SEM. J Microsc 237:208–220. 10.1111/j.1365-2818.2009.03329.x [DOI] [PubMed] [Google Scholar]
- Jones SL, Svitkina TM (2016) Axon initial segment cytoskeleton: architecture, development, and role in neuron polarity. Neural Plast 2016:6808293. 10.1155/2016/6808293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SL, Korobova F, Svitkina T (2014) Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments. J Cell Biol 205:67–81. 10.1083/jcb.201401045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, Hoogenraad CC (2015) Building the neuronal microtubule cytoskeleton. Neuron 87:492–506. 10.1016/j.neuron.2015.05.046 [DOI] [PubMed] [Google Scholar]
- Kijanka M, van Donselaar EG, Müller WH, Dorresteijn B, Popov-Čeleketić D, El Khattabi M, Verrips CT, van Bergen En Henegouwen PMP, Post JA (2017) A novel immuno-gold labeling protocol for nanobody-based detection of HER2 in breast cancer cells using immuno-electron microscopy. J Struct Biol 199:1–11. 10.1016/j.jsb.2017.05.008 [DOI] [PubMed] [Google Scholar]
- Kole MH, Stuart GJ (2012) Signal processing in the axon initial segment. Neuron 73:235–247. 10.1016/j.neuron.2012.01.007 [DOI] [PubMed] [Google Scholar]
- Kolotuev I, Schwab Y, Labouesse M (2009) A precise and rapid mapping protocol for correlative light and electron microscopy of small invertebrate organisms. Biol Cell 102:121–132. 10.1042/BC20090096 [DOI] [PubMed] [Google Scholar]
- Konishi Y, Setou M (2009) Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat Neurosci 12:559–567. 10.1038/nn.2314 [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR (1996) Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116:71–76. 10.1006/jsbi.1996.0013 [DOI] [PubMed] [Google Scholar]
- Kuijpers M, van de Willige D, Fréal A, Chazeau A, Franker MA, Hofenk J, Rodrigues RJ, Kapitein LC, Akhmanova A, Jaarsma D, Hoogenraad CC (2016) Dynein regulator NDEL1 controls polarized cargo transport at the axon initial segment. Neuron 89:461–471. 10.1016/j.neuron.2016.01.022 [DOI] [PubMed] [Google Scholar]
- Leterrier C. (2016) The axon initial segment, 50Years later: a nexus for neuronal organization and function. Curr Top Membr 77:185–233. 10.1016/bs.ctm.2015.10.005 [DOI] [PubMed] [Google Scholar]
- Leterrier C, Vacher H, Fache MP, d'Ortoli SA, Castets F, Autillo-Touati A, Dargent B (2011) End-binding proteins EB3 and EB1 link microtubules to ankyrin G in the axon initial segment. Proc Natl Acad Sci U S A 108:8826–8831. 10.1073/pnas.1018671108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL Jr, Mao T, Svoboda K, Arnold DB (2009) Myosin-dependent targeting of transmembrane proteins to neuronal dendrites. Nat Neurosci 12:568–576. 10.1038/nn.2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wu H, Wu W, Zhuo W, Liu W, Zhang Y, Cheng M, Chen YG, Gao N, Yu H, Wang L, Li W, Yang M (2014) Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res 24:762–765. 10.1038/cr.2014.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipka J, Kapitein LC, Jaworski J, Hoogenraad CC (2016) Microtubule-binding protein doublecortin-like kinase 1 (DCLK1) guides kinesin-3-mediated cargo transport to dendrites. EMBO J 35:302–318. 10.15252/embj.201592929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaely P, Bennett V (1995) Mechanism for binding site diversity on ankyrin: comparison of binding sites on ankyrin for neurofascin and the cl-/HCO3- anion exchanger. J Biol Chem 270:31298–31302. 10.1074/jbc.270.52.31298 [DOI] [PubMed] [Google Scholar]
- Palay SL, Sotelo C, Peters A, Orkand PM (1968) The axon hillock and the initial segment. J Cell Biol 38:193–201. 10.1083/jcb.38.1.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Proskauer CC, Kaiserman-Abramof IR (1968) The small pyramidal neuron of the rat cerebral cortex: the axon hillock and initial segment. J Cell Biol 39:604–619. 10.1083/jcb.39.3.604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez JG, Okreglicka K, Chandrasekaran V, Welker JM, Sundquist WI, Pornillos O (2014) The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc Natl Acad Sci U S A 111:2494–2499. 10.1073/pnas.1318962111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake T, Yamashita K, Hayashi K, Miyatake S, Tamura-Nakano M, Doi H, Furuta Y, Shioi G, Miura E, Takeo YH, Yoshida K, Yahikozawa H, Matsumoto N, Yuzaki M, Suzuki A (2017) MTCL1 plays an essential role in maintaining Purkinje neuron axon initial segment. EMBO J 36:1227–1242. 10.15252/embj.201695630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas RP, Chazeau A, Cloin BMC, Lambers MLA, Hoogenraad CC, Kapitein LC (2017) Differentiation between oppositely oriented microtubules controls polarized neuronal transport. Neuron 96:1264–1271.e5. 10.1016/j.neuron.2017.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beuningen SFB, Will L, Harterink M, Chazeau A, van Battum EY, Frias CP, Franker MAM, Katrukha EA, Stucchi R, Vocking K, Antunes AT, Slenders L, Doulkeridou S, Sillevis Smitt P, Altelaar AFM, Post JA, Akhmanova A, Pasterkamp RJ, Kapitein LC, de Graaff E, et al. (2015) TRIM46 controls neuronal polarity and axon specification by driving the formation of parallel microtubule arrays. Neuron 88:1208–1226. 10.1016/j.neuron.2015.11.012 [DOI] [PubMed] [Google Scholar]
- Watanabe K, Al-Bassam S, Miyazaki Y, Wandless TJ, Webster P, Arnold DB (2012) Networks of polarized actin filaments in the axon initial segment provide a mechanism for sorting axonal and dendritic proteins. Cell Rep 2:1546–1553. 10.1016/j.celrep.2012.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, He J, Zhou R, Lorenzo D, Babcock HP, Bennett V, Zhuang X (2014) Developmental mechanism of the periodic membrane skeleton in axons. eLife 3:e04581. 10.7554/eLife.04581 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reconstruction of six consecutive ∼100-nm-thick slices of the AIS of a 21 DIV neuron. Microtubules are in turquoise, the cross-bridges in yellow, and the mitochondria in transparent light red.