

Abstract
Although NPY has potent anxiolytic actions within the BLA, selective activation of BLA NPY Y2 receptors (Y2Rs) acutely increases anxiety by an unknown mechanism. Using ex vivo male rat brain slice electrophysiology, we show that the selective Y2R agonist, [ahx5–24]NPY, reduced the frequency of GABAA-mediated mIPSCs in BLA principal neurons (PNs). [ahx5–24]NPY also reduced tonic activation of GABAB receptors (GABABR), which increased PN excitability through inhibition of a tonic, inwardly rectifying potassium current (KIR). Surprisingly, Y2R-sensitive GABABR currents were action potential-independent, persisting after treatment with TTX. Additionally, the Ca2+-dependent, slow afterhyperpolarizing K+ current (IsAHP) was enhanced in approximately half of the Y2R-sensitive PNs, possibly from enhanced Ca2+ influx, permitted by reduced GABABR tone. In male and female mice expressing tdTomato in Y2R-mRNA cells (tdT-Y2R mice), immunohistochemistry revealed that BLA somatostatin interneurons express Y2Rs, as do a significant subset of BLA PNs. In tdT-Y2R mice, [ahx5–24]NPY increased excitability and suppressed the KIR in nearly all BLA PNs independent of tdT-Y2R fluorescence, consistent with presynaptic Y2Rs on somatostatin interneurons mediating the above effects. However, only tdT-Y2R-expressing PNs responded to [ahx5–24]NPY with an enhancement of the IsAHP. Ultimately, increased PN excitability via acute Y2R activation likely correlates with enhanced BLA output, consistent with reported Y2R-mediated anxiogenesis. Furthermore, we demonstrate the following: (1) a novel mechanism whereby activity-independent GABA release can powerfully dampen BLA neuronal excitability via postsynaptic GABABRs; and (2) that this tonic inhibition can be interrupted by neuromodulation, here by NPY via Y2Rs.
SIGNIFICANCE STATEMENT Within the BLA, NPY is potently anxiolytic. However, selective activation of NPY2 receptors (Y2Rs) increases anxiety by an unknown mechanism. We show that activation of BLA Y2Rs decreases tonic GABA release onto BLA principal neurons, probably from Y2R-expressing somatostatin interneurons, some of which coexpress NPY. This increases principal neuron excitability by reducing GABAB receptor (GABABR)-mediated activation of G-protein-coupled, inwardly rectifying K+ currents. Tonic, Y2R-sensitive GABABR currents unexpectedly persisted in the absence of action potential firing, revealing, to our knowledge, the first report of substantial, activity-independent GABABR activation. Ultimately, we provide a plausible explanation for Y2R-mediated anxiogenesis in vivo and describe a novel and modulatable means of damping neuronal excitability.
Keywords: anxiety, electrophysiology, neuropeptide Y, tdTomato, voltage clamp
Introduction
Fear and anxiety are adaptive emotions experienced by humans and many animals. Although these negative emotions are unpleasant, they orchestrate behavioral and physiological responses to perceived threats and generally facilitate survival. The BLA is critical for fear learning (Rogan et al., 1997; Blair et al., 2001; Herry et al., 2008) and expression (Maren, 1999; Lalumiere, 2014). The BLA receives dense, sensory-derived inputs that are subject to plasticity, allowing it to mount responses to threats via excitatory projections to other brain areas (Walker et al., 2009; Amano et al., 2010). However, excessive or inappropriate anxiety and aberrant fear learning are maladaptive and hallmark debilitating anxiety disorders, such as post-traumatic stress disorder (Shekhar et al., 1999; Lissek et al., 2005; Blechert et al., 2007; Prater et al., 2013).
Glutamatergic pyramidal- and stellate type principal neurons (PNs) mediate BLA output (Fuller and Price, 1988; McDonald et al., 1989) and comprise 85% of its neurons (McDonald, 1996; Rostkowski et al., 2009). Subpopulations of BLA PNs code for negative valence emotions; however, these are intermingled with PNs mediating appetitive and fear/anxiety reducing emotional states (Herry et al., 2008; Senn et al., 2014). The fear/anxiety circuit within the BLA dominates, as nonspecifically exciting or silencing BLA PNs, respectively, increases or reduces anxiety behaviors (Sajdyk and Shekhar, 1997; Bueno et al., 2005). Despite extensive excitatory input from local and extrinsic connections, PN activity is constrained by BLA GABA interneurons (INs) (Fritsch et al., 2009; Capogna, 2014; Wolff et al., 2014); consequently, PNs show low firing rates in vivo and in vitro (Washburn and Moises, 1992; Rosenkranz and Grace, 1999; Likhtik et al., 2006).
In addition to classical neurotransmitters and monoamines (Rainnie, 1999; Kröner et al., 2005), neuropeptides modulate BLA circuits. NPY is potently anxiolytic when infused into the rodent BLA (Sajdyk et al., 1999) and reduces fear and anxiety via a calcineurin-dependent mechanism (Lin et al., 2003; Sajdyk et al., 2008). Moreover, increased NPY expression is linked to human emotional resilience (Yehuda et al., 2006; Sah et al., 2009). The rodent BLA expresses the NPY Y1, Y2, and Y5 receptor subtypes (Y1R, Y2R, Y5R) (Wolak et al., 2003; Stanić et al., 2006; Rostkowski et al., 2009); however, Y1Rs primarily mediate the acute anxiolytic actions of NPY (Sajdyk et al., 1999). In acute brain slices, Y1R activation hyperpolarizes BLA PNs by inhibiting the depolarizing, tonically active H-current (Ih) (Giesbrecht et al., 2010). Y1Rs inhibit lateral amygdala PNs by activating inwardly rectifying potassium currents (KIR) (Sosulina et al., 2008). Paradoxically, targeted activation of BLA Y2Rs increases anxiety behaviors (Nakajima et al., 1998; Sajdyk et al., 2002), by an unknown mechanism. In other brain regions, Y2Rs are typically expressed on presynaptic terminals and inhibit neurotransmitter release (Colmers and Bleakman, 1994; Greber et al., 1994; Chen et al., 1997). Based on this, we hypothesized that BLA INs express presynaptic Y2Rs that disinhibit BLA PNs by reducing tonic GABA release.
We report here that application of the Y2R-selective agonist, [ahx5–24]NPY, reduced miniature GABAA synaptic event frequencies in rat BLA PNs and decreased tonic, postsynaptic currents mediated by GABABRs, which enhanced PN excitability. Moreover, in some PNs, reduced GABABR tone also potentiated the Ca2+-dependent, slow afterhyperpolarization current (IsAHP), suggesting the disinhibition of dendritic voltage-gated Ca2+ influx. Repetitive firing rates were markedly reduced by [ahx5–24]NPY in PNs where it enhanced the IsAHP. Conversely, firing rates increased in PNs where the IsAHP was unaffected by [ahx5–24]NPY. Using a transgenic mouse model in which Y2R-containing neurons expressed the tdTomato reporter, we show that Y2R immunoreactivity was present on somatostatin (SST) INs as well as subsets of BLA PNs.
It is likely that the ongoing, action potential (AP)-independent GABA release from these somatostain INs broadly inhibits BLA PNs through activation of postsynaptic GABABRs. NPY relieves this tonic inhibition via Y2Rs. However, loss of GABABR tone is counterbalanced in some PNs by enhancement of the Ca2+-dependent IsAHP. Y2R effects on BLA PNs are thus widespread, complex, and functionally nonuniform.
Materials and Methods
Animals
Male Sprague Dawley rats (Charles River) 6–12 weeks of age were used for most electrophysiology experiments. All immunohistochemistry experiments and a subset of electrophysiology experiments used transgenic mice that expressed the tdTomato fluorescent protein conditionally under control of the Y2R gene promoter (Y2R-tdTomato). We generated Y2R-tdTomato mice by crossing two commercially available mouse strains: a homozygous tdTomato reporter strain (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, JAX 007914) and heterozygous mice that expressed Cre-recombinase under control of the Y2R gene promoter [Tg(NPY2r-cre) SM20Gsat/Mmucd-Mutant Mouse Resource and Research Center, University of California at Davis, ID #36505]. Y2R-tdTomato mice were identified by PCR, performed on ear notch tissue, using the KAPA HotStart Mouse Genotyping Kit (KAPABiosystems: PCR primers for Npy2r (36505) forward-5′-TTCCTGAGTTCATCCATCCCTAGTTG-3′, CreGS reverse-1–5′-CGGCAAACGGACAGAAGCATT-3′). Male and female Y2R-tdTomato mice between 4 and 16 weeks of age were used for electrophysiology or immunohistochemistry experiments. The care and use of animals were in accordance with standards set by the University of Alberta Care and Use Committee: Health Sciences. Animals were group housed (2 or 3 rats or 2–5 mice per cage) with food and water supplied ad libitum.
Brain slice preparation
Rats or mice were decapitated without prior anesthesia, their brains rapidly removed and submerged in an icy slurry of ACSF optimized for slice preparation, containing the following (in mm): 118 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 5 MgCl2·6H2O, 10 glucose, 26 NaHCO3, and 1.5 CaCl2. This slicing solution was continuously bubbled with carbogen (95% O2, 5% CO2) and was supplemented with kynurenic acid (1 mm) to mitigate glutamate-mediated excitatory toxicity. Coronal brain slices (rats 300 μm; mice 250 μm) containing BLA were prepared with a vibrating slicer (Slicer HR2, Sigmann Elektronik). Slices were then transferred to a carbogenated ACSF (“Bath”) solution, which contained the following (in mm): 124 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 10 glucose, 26 NaHCO3, and 2.5 CaCl2 (300–305 mOsm/L). Slices were maintained in room temperature (22°C) carbogen-bubbled bath solution for at least 30 min following slicing, and were kept in bath solution for the remainder of all experiments. For electrophysiology, slices were placed into a recording chamber attached to a fixed stage of a movable upright microscope (Axioskop FS2, Carl Zeiss) and held submerged by a platinum and polyester fiber “harp.” Slices were continuously perfused with warmed (34.0 ± 0.5°C), carbogenated bath solution at a rate of 2–3 ml/min for at least 20 min before recording.
Electrophysiology
Patch pipettes were pulled from thin-walled borosilicate glass (TW150F; WPI) with a two-stage puller (PP-83; Narishige) to a tip resistance of 5–7 mΩ when back-filled with an internal solution containing the following (in mm): 126 K-gluconate, 10 HEPES, 4 KCl, 5 MgATP, 0.3 NaGTP, 1 EGTA, and 0.3 CaCl2. Neurobiotin (0.05%–0.1%) was added and the pH adjusted to 7.27–7.30 with KOH (275–285 mOsm/L). A modified internal solution containing 126 mm cesium methanesulfonate in place of K-gluconate was used for several series of experiments. In this case, pH was adjusted with CsOH; otherwise, constituents, concentrations, and other properties were identical to K+ internal solution. Recordings were made using a Multiclamp 700B amplifier, and data were acquired using pClamp (version 10.3–10.4) via a Digidata 1322 interface (all Molecular Devices). Liquid junction potentials were calculated for K+ and Cs+ internal solutions as 14.4 and 13.8 mV, respectively; all voltage- and current-clamp data reported here are adjusted to reflect this.
The BLA was identified in slices using criteria based on the Paxinos and Watson (2007) atlas. Here, we define the BLA as including both the basal and basal accessory nuclei, sometimes collectively referred to as the basal amygdala. Recordings were restricted to the BLA and specifically avoided the LA.
BLA neurons were visually identified with infrared-differential interference contrast optics and PNs selected for recordings based on a large, pyramidal- or stellate-shaped soma and often a prominent apical dendrite. Local circuit INs were differentiated from PNs based on their generally smaller, more spherical somata. Once whole-cell patch-clamp configuration was established, electrophysiological properties were used in conjunction with morphology to characterize neurons (Washburn and Moises, 1992; Rainnie et al., 1993). Neurons judged to be PNs exhibited the following: low input resistance (30–100 mΩ), characteristic APs with half-widths >1 ms (approximately twice as long as with most INs) (Mahanty and Sah, 1998), and typically prominent AP spike frequency adaptation when brought above threshold with 800 ms depolarizing current steps.
Neurons were usually held in voltage clamp at −75 mV for 5–10 min before the start of experiments and between experimental manipulations. In recordings using a Cs+ intracellular solution (Cs+i), neurons were held at −15 mV before and between measurements, but otherwise treated similarly. A series of current- and voltage-clamp protocols were performed at 5 min intervals to establish the stability of baseline neuronal properties. Only neurons demonstrating stable resting membrane potential (RMP) and holding current in these control measurements and which showed stable access resistance (±20%) throughout an experiment were selected for analysis. Recordings were analyzed off-line with pClamp version 10.3 or later (Molecular Devices).
Current-clamp experiments
Under our in vitro conditions using a K+-gluconate pipette solution (K+i), PNs rarely, if ever, fired spontaneous APs. To assess neuronal excitability under different experimental conditions, AP firing was elicited with depolarizing current-clamp ramps. Throughout this manuscript, we define rheobase as the depolarizing current required to reach AP threshold, as measured from RMP with a family of 800 ms depolarizing current ramps, initially from 0 to 100 pA, increasing in 100 pA increments (Giesbrecht et al., 2010; Silveira Villarroel et al., 2018). Typically, drug-mediated changes in RMP were small; however, when large changes (>±5 mV) occurred, rheobase was measured at the control RMP.
A family of 800 ms current-clamp steps were used both to measure neuronal input resistance and examine AP properties, including individual waveforms and spike train firing patterns. Input resistance was measured from RMP with a hyperpolarizing step (25–100 pA, chosen to elicit only 2–5 mV hyperpolarizations so as to minimally affect voltage-gated conductances) and calculated at steady state from Ohm's law (r = V/I). When drug applications changed RMP (±5 mV), current was injected to return PNs to their control RMP as for rheobase measurements. AP trains were evoked with successive depolarizing current steps (25–100 pA increments). Depolarizing steps in which PNs fired trains of between 5 and 10 APs were selected for analysis. For within-PN comparisons (in the presence or absence of a test compound), steps eliciting the same number of APs (±1) were compared. Recordings that did not meet the above criteria were excluded from analysis. AP interspike intervals were measured and plotted as a function of their order in the train as were AP AHPs. RMP was also periodically measured by averaging the potential during 30-s-long, passive current-clamp recordings (0 pA, 30 s duration).
Voltage-clamp experiments
For some recordings, we used Cs+i to block multiple types of K+ channels; this increased the neuronal space constant and allowed cells to be held more depolarized (−15 mV in most cases). In these recordings, PNs were ramped from −15 mV to −125 mV (50 mV/s), held at −125 mV for 1 s, ramped to 35 mV (300 mV/s), and returned to −15 mV. The hyperpolarizing ramp from −15 mV allowed us to study conductances, such as those mediated by GABAA receptors and inwardly rectifying K+ (KIR) channels in the absence of voltage-gated Na+ and Ca2+ currents, which were inactivated by holding the neuron at −15 mV before initiating ramps.
For K+i recordings of PNs, drug-mediated changes in current-voltage (I–V) relationships were examined with families of 8 hyperpolarizing voltage steps applied from a −55 mV holding potential (I–V protocol). The I–V protocol began with a −10 mV step; each successive step was incremented by −10 mV to −135 mV. Voltage steps were successively shortened by 100 ms to minimize membrane damage due to larger voltage excursions (Giesbrecht et al., 2010; Silveira Villarroel et al., 2018). Voltage-clamp experiments were performed without capacity or series resistance compensation unless otherwise indicated.
In a subset of voltage-clamp experiments, capacitance and series resistance were compensated using the automated protocols in the Multiclamp Commander software. These resulted in a Cp Fast setting of 17.7 ± 1.1 pF (n = 11), a Cp Slow setting of 3.0 pF (n = 11), and whole-cell capacitance of 150.6 ± 26.3 pF (n = 11). Series resistance compensation was set to 70% (with a lag value of 160 μs); further compensation led to instability and loss of the recording. Results from these experiments did not differ materially from those without the compensation.
Most BLA PNs have a prominent Ih, seen as a slowly activating inward current that increased in magnitude and activation rate with membrane hyperpolarization. Ih does not inactivate and is tonically active in most PNs at their RMP, but is inactive at −55 mV. Ih magnitude at a given potential was thus measured as the difference between the peak positive current (following immediately after the capacitative transient of the voltage step) and the steady-state current at the end of the step and plotted as a function of membrane voltage.
The component of current immediately following the capacitative transient of a voltage-clamp step, before Ih activation, was also measured and plotted as a function of membrane voltage. We termed this conductance the instantaneous inward rectifying K+ current (KIR) because it responded virtually instantaneously to changes in membrane voltage, showed prominent inward rectification, and was revealed to be largely K+-mediated.
In a subset of recordings, using the above I–V protocol, a prominent outward current was clearly visible upon returning the membrane potential to −55 mV following steps negative to −85 mV. This current was quantified by subtracting the mean steady-state holding current at −55 mV from the peak outward current (evoked by hyperpolarizing neurons to −135 mV and then stepping back to −55 mV). The I–V relationship of this outward current was also estimated by performing a series of fast (10 ms) voltage steps during the peak of this outward current evoked following an 850 ms, −135 mV voltage step.
Synaptic currents were measured during passive voltage-clamp recordings (2 min) with neurons held at −15 mV using Cs+i, so that bicuculline-sensitive GABAA events appeared as large-amplitude, outward, IPSCs. At this potential, glutamate-mediated inward synaptic currents were negligible. Events were recorded under baseline conditions, in the presence of drugs and periodically during washout. Unitary synaptic interevent intervals and amplitudes were quantified with Mini Analysis (Synaptosoft). Because of the high event frequencies observed under these conditions, only the first 800 events of each trace were analyzed (typically 30–60 s).
Immunohistochemistry
Animals.
tdTomato-Y2R mice were anesthetized with pentobarbital (50 mg/kg, i.p.) and perfused with PBS, pH 7.4, and 4% PFA. Brains were removed and placed in fixative overnight before transfer to PBS/sodium azide (0.01%). Sections through the mouse brain were sliced (40 μm) on a vibratome, and free-floating sections were processed for immunohistochemical analysis of NPY, SST, parvalbumin (PV), and CaMKII as described previously (Rostkowski et al., 2009).
CaMKII/PV-CaMKII (mouse monoclonal, clone 6G9, #MAB8699, RRID:AB_2067919, Millipore) was used as a marker for BLA glutamatergic projection neurons (Rostkowski et al., 2009), and PV (polyclonal goat, Swant; RRID:AB_10000344) for the major class of BLA INs. Free-floating sections were rinsed through 3 changes of PBS over 10 min, followed by a 15 min wash in 1% H2O2 in PBS to diminish endogenous peroxidase activity. Next, tissues were blocked for 3 h in immunocytochemistry (ICC) buffer (0.1 m PBS containing 0.2% gelatin, 0.01% thimerosal, and 0.002% neomycin, pH 7.5) with 5% normal donkey serum (NDS; Equitech-Bio) to block nonspecific binding. Sections were incubated at 4°C for 72 h in primary antibody directed against CaMKII (1:13,000) in ICC buffer with 2% NDS and subsequently washed through 5 changes of ICC buffer over 50 min followed by a 3 h incubation in ICC buffer containing Alexa-488 donkey anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories; 1:250). After a series of washes, sections were subsequently incubated with primary antibody for PV (1:10,000; diluted in ICC buffer with 2% NDS and 0.1% Triton X-100). PV signal was visualized using Cy5-conjugated donkey anti-goat secondary antibody. Following washes in TBS, pH 7.5, sections were mounted onto Superfrost Plus slides (Thermo Fisher Scientific) and coverslips were rapidly applied with polyvinyl-alcohol; 1,4 diazobicyclo[2.2.2]octane antifade mounting medium.
SST and NPY
To further determine colocalization of tdTomato signal within specific interneuronal populations (SST and NPY) in the BLA, sections were incubated with primary antibodies for SST (1:500; Millipore, monoclonal rat, clone YC7 AB345; RRID:AB_2315346) and NPY (1: 2000; Peninsula T-4069.0400: RRID:AB_518503) diluted in ICC buffer with 2% NDS and 0.1% Triton X-100. Interneuronal markers were visualized using the following fluorophore-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories; 1:250): Cy5 donkey anti-mouse (SST) and Alexa488 anti-rabbit (NPY). Following washes in 4 changes of TBS over 20 min, sections were mounted onto Superfrost Plus slides (Thermo Fisher Scientific), and coverslips were rapidly applied with polyvinyl-alcohol; 1,4 diazobicyclo[2.2.2]octane mounting medium.
Antibody specificity
The mouse monoclonal CaMKII antibody was raised against the α subunit of CaMKII, and recognizes both phosphorylated and nonphosphorylated forms of CaMKII and has been well characterized in prior studies (Erondu and Kennedy, 1985). Additionally, the NPY, PV, and CaMKII antibodies used here are markers for specific neuronal subpopulations in the BLA, and each produces a characteristic pattern of immunostaining seen in numerous previous studies of the rat BLA (Kemppainen and Pitkänen, 2000; McDonald and Betette, 2001; McDonald and Mascagni, 2001, 2002; Mascagni and McDonald, 2003).
Accordingly, sections were analyzed using scanning laser confocal microscopy (Rosalind Franklin University Multiuser Microscopy and Imaging Facility). The following excitation wavelengths were used: 488 nm for the secondary fluorophore FITC, 568 nm for Cy3, and 647 nm for Cy5. Colocalization was determined by overlapping signals observed at several focal planes throughout each cell. Brightness and contrast of the photomicrographs presented here were adjusted using Photoshop version 12 (Adobe) to ensure the highest quality images for publication.
Determination of NPY Y2R gene expression in tdTomato-expressing cells
Animals.
Control mice were anesthetized with isoflurane (4%) and rapidly decapitated. Brains were quickly removed, frozen in isopentane, and stored at −80°C. Fresh frozen mouse brains were sectioned on a cryostat (20 μm) and placed on gelatin-coated slides. After sectioning, they were stored at −80°C. On the day of labeling, the slides were removed from the freezer and the sections thawed and air-dried for 20 min. Afterward, sections were postfixed in 4% PFA for 1 h. They were subsequently dehydrated through a graded series of ethanols (50%, 70%, 100%) and air-dried overnight. The next day, sections were processed according to the manufacturer's directions (RNAscope, ACD Bio-Techne); the slides were incubated for 10 min in RNAscope hydrogen peroxide, followed by 2 rinses in 1× PBS. Tissues were incubated in Protease IV for 30 min at room temperature, followed by 2 rinses in 1× PBS. Probes (RNAscope Probe-tdTomato, catalog #317041-C3; NPY Y2R, RNAscope Probe-Mm-Npy2r; catalog #315951-C2) were applied to the sections, which incubated for 2 h at 40°C. Sections were washed in wash buffer (2 × 2 min, room temperature), then incubated in Hybridize AMP1 for 30 min at 40°C, followed by washes, and final incubations with Hybridize AMP2 (30 min) and Hybridize AMP3 (15 min). Tissues were incubated with RNAscope MultiplexFL version 2HRP-C1 at 40°C (15 min); this was followed by incubation with TSA Plus fluorescein (30 min at 40°C) and RNAscope MultiplexFL version 2 blocker. This was followed by 15 min in RNAscope MultiplexFL version 2HRP-C2 and then 30 min in TSA Plus Cyanine 3, both at 40°C. After washes, the sections were incubated for 30 s with DAPI to stain nuclei. After final rinsing, coverslips were applied with mounting medium, and the slides were air dried overnight.
Analysis of signal
Brain sections were selected from each animal based on anatomical features of the BLA and surrounding areas to match predetermined bregma levels corresponding to anterior, middle, and posterior aspects of the BLA as indicated in Figure 12 (n = 3–5 animals) (Paxinos and Franklin, 2012). Images were acquired with confocal microscopy and imported into NIS Elements (Nikon Instruments) for determination of cell counts. For each image, the BLA was identified and the total number of cells in the BA was counted unilaterally throughout the rostral-caudal sections. Specificity of the signal was assessed by eliminating either the primary or secondary antibodies; further characterization of antibody specificity was previously described (Rostkowski et al., 2009). For presentation purposes, images were imported into PhotoShop, and brightness and contrast were optimized.
Figure 12.
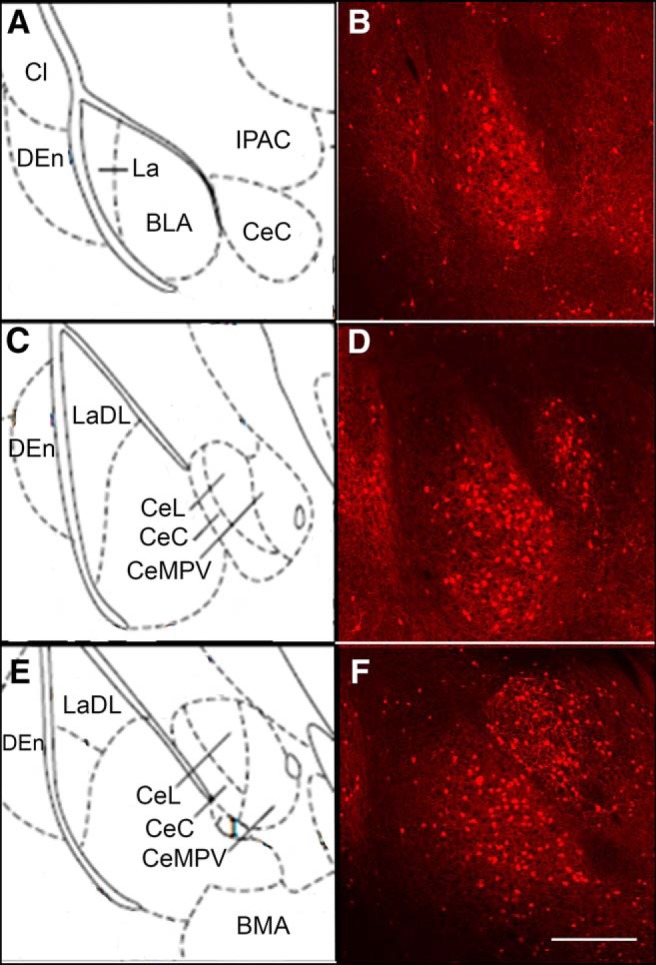
Distribution of tdTomato fluorescent signal in the BLA of Y2R-tdTomato mice. tdTomato-fluorescent cells were present throughout the rostral-caudal extent of the BLA (A–F). Cells were generally located more medially in the BLA with very few cells present in the La (lateral amygdala) or LaDL (dorsolateral amygdala). Significant numbers of tdTomato-expressing cells were also present in the central nucleus of amygdala (CeC) and the dorsal endopiriform cortex (DEn). Cl, Claustrum; IPAC, interstitial nucleus of posterior limb of anterior commissure; CeL, central amygdala lateral division; CeMPV, central amygdala medial posterior ventral division; BMA, basomedial amygdala. Scale bar, 500 μm.
Materials
The selective Y2-R agonist [6-aminohexanoic5–24]NPY ([ahx5–24]NPY), was a generous gift from Dr. Annette G. Beck-Sickinger (Leipzig, Germany). Baclofen was a gift from Ciba-Geigy. Kyneurenic acid was purchased from Abcam Biochemicals. GTP was purchased from Roche Diagnostics. CsCl, Cs+-methanesulfonate, K+-gluconate, EGTA, MgATP, CsOH, ivabradine, UCL 2077, CdCl2, apamin, and formalin fixative were all obtained from Sigma-Aldrich. TTX was purchased from Alomone Labs. Bicuculline, SCH 23390, and CGP 52432 were purchased from Tocris Bioscience. KOH was purchased from BDH Chemicals. All other chemicals were obtained from Thermo Fisher Scientific. All drugs were either stored in aliquots of concentrated stock solutions at −20°C and diluted in ASCF bath solution, or made fresh from powder in bath solution immediately before application.
Experimental design and statistical analysis
Prism software (versions 5–8, GraphPad) and SPSS (version 20, IBM) were used for statistical analysis and creation of figures. All data are expressed as mean ± SEM. Data distributions were tested for normality with the D'Agostino-Pearson Omibus Normality Test. Although power analyses were not performed, sample sizes were chosen in keeping with publications using similar experimental designs.
A one-way repeated-measures ANOVA with the Bonferroni post-test was used for analyzing effects of pharmacological agents on a single electrophysiological parameter at multiple time points (control, peak drug effects, and after prolonged washout [10–20 min]) or for comparing multiple sequential drug applications. This was used to assess drug effects on mean synaptic event parameters (frequencies or amplitudes). An unbiased hierarchical cluster analysis was performed to evaluate the presence of multiple sIPSC frequency response profiles following drug application. A between-groups linkage clustering method was used, with Squared Euclidean distance for the measurements. A Student's paired t test was used for within-neuron comparisons of a single parameter under two conditions (typically control and drug). This was used to assess drug effects on parameters, such as rheobase, input resistance, and RMP. With RMP responses to [ahx5–24]NPY, we also performed a cluster analysis as above to determine whether there were two populations of PN responses. A Student's unpaired t test was used when comparing a single parameter between neurons: for example, comparing electrophysiological parameters between fluorescent and nonfluorescent PNs. In instances where data distributions substantially deviated from normality, statistical significance was assessed with the Mann–Whitney U test. A two-way, repeated-measures ANOVA with the Bonferroni post-test was used for within-neuron comparison of I–V plots (IIR and Ih) under control and drug conditions. A two-way ANOVA without repeated measures was used when comparing I–V plots of currents blocked by drugs in separate groups of PNs (between neuron comparisons) and when comparing I–V plots between fluorescent and nonfluorescent PNs.
Differences in mean values were considered significant at p < 0.05. Comprehensive descriptions of statistical analysis are included in the figure legends.
Results
Rat BLA PNs are hyperpolarized in vitro
Using a K+-based intracellular solution (K+i), a representative sample of BLA PNs displayed a strongly hyperpolarized mean RMP [−81 ± 0.5 mV; n = 64 cells (42 rats)]. This value is more negative than the mean PN RMP reported in a previous study from our group (−76 ± 0.6 mV) (Giesbrecht et al., 2010), perhaps due to the older animals used here. PNs also showed characteristically low input resistance under control conditions [59.5 ± 2.5 mΩ; n = 57 cells (37 rats)] and only very rarely fired spontaneous APs in passive current-clamp recordings.
Y2Rs affect unitary IPSC frequencies in rat BLA PNs
As reported previously, GABAA-mediated sIPSC frequencies in BLA PNs are underestimated when measured using K+ gluconate-based intracellular solutions (Silveira Villarroel et al., 2018). Therefore, we used a Cs+ methanesulfonate intracellular solution (Cs+i), which allowed PNs to be held depolarized at −15 mV, where IPSCs were unambiguously large, outward currents. Consistent with previous reports (Smith and Dudek, 1996; Silveira Villarroel et al., 2018), virtually every PN tested showed substantial GABAA synaptic input. (Fig. 1). The mean sIPSC frequency was 18.7 ± 2.5 Hz [n = 20 cell (16 rats)]. Although application of the Y2R agonist, [ahx5–24]NPY (1 μm), reduced the PN sIPSC frequency to 14.7 ± 1.8 Hz, this did not reach statistical significance [F(1.468,27.89) = 3.606; p = 0.0531; n = 20 cells (16 rats); repeated-measures one-way ANOVA]. Surprisingly, [ahx5–24]NPY (1 μm) significantly and reversibly increased the mean sIPSC amplitude from 21 ± 1.5 to 23 ± 2.0 pA [p = 0.0438; n = 20 cells (16 rats); repeated-measures one-way ANOVA].
Figure 1.

[ahx5–24]NPY reduces the frequency of BLA principal neuron miniature inhibitory synaptic events. A, Voltage-clamp recordings (Vh = −15 mV, Cs+-methanesulfonate internal solution) from a representative PN, which responded to bath-applied [ahx5–24]NPY (1 μm; blue) with a substantial (>10%) reduction in the sIPSC frequency (black). sIPSCs, seen as upward current deflections (asterisks), occurred frequently (∼20 Hz) under control conditions (blue). Following bath application of the Y2R agonist to the same PN, the sIPSC frequency was substantially reduced (peak drug effect ∼5 min wash). B, Mean sIPSC frequencies from the subset of PNs in which the Y2R agonist decreased spontaneous event frequencies (>10%; 14 of 20 tested). sIPSC frequencies in control, during peak effect of [ahx5–24]NPY (1 μm), and after prolonged washout (p = 0.0027; n = 14 cells, 10 rats; one-way repeated-measures ANOVA; F(1.096,14.25) = 13.62, p = 0.0020). In these cells, the Y2R agonist significantly and reversibly reduced the mean sIPSC frequency from 21.6 ± 3.1 to 13.4 ± 2.2 Hz (Bonferroni's post-test). C, Mean amplitudes of sIPSC events reported in B. The Y2R agonist did not significantly affect sIPSC amplitude in these PNs (control, 19.7 ± 1.6 pA; [ahx5–24]NPY, 20.2 ± 1.8 pA; one-way repeated-measures ANOVA; F(1.437,18.68) = 3.566, p = 0.0616; n = 14 cells, 10 rats). D, Voltage-clamp recordings as in A from a representative PN, which responded to [ahx5–24]NPY (1 μm) with a substantial (>10%) increase in mean sIPSC frequency (blue). The Y2R agonist increased the frequency of large-amplitude events that typically clustered into clear bursts. E, Mean sIPSC frequencies from the subset of PNs where the Y2R agonist increased spontaneous event frequencies (>10% of control values; 6 of 20 tested) in control, during peak effects of [ahx5–24]NPY (1 μm), and after prolonged washout (one-way repeated-measures ANOVA; F(1.401,7.006) = 12.06, p = 0.0076; n = 6 cells, 6 rats). In these PNs, the Y2R agonist significantly and reversibly increased mean sIPSC frequency from 12.1 ± 2.3 to 17.8 ± 3.3 Hz (Bonferroni's post-test). F, Mean sIPSC amplitudes from the events reported in E (one-way repeated-measures ANOVA; F(1.540,7.699) = 10.72, p = 0.0078; n = 6 cells, 6 rats). In these PNs, the Y2R agonist significantly and reversibly increased sIPSC amplitude from 23.1 ± 3.3 to 31.0 ± 4.2 pA (Bonferroni's post-test). G, Dendrogram of a hierarchical cluster analysis performed on Δ sIPSC frequency of PNs following application of [ahx5–24]NPY (1 μm). H, Proximity matrix of a hierarchical cluster analysis using squared Euclidean distance on Δ sIPSC frequency of PNs following application of [ahx5–24]NPY (1 μm). Neurons were reclassified based on similarity for graphical representation. I, Scatter plot depicting the heterogeneous response profile of sIPSC frequency of PNs following application of [ahx5–24]NPY (1 μm). Responses fall robustly into two distinct clusters (t = 9.116, df = 18, p < 0.0001). J, Voltage-clamp recordings as in A from a representative PN in the presence of TTX (500 nm). mIPSC frequencies (black) remained comparable to event frequencies observed without TTX (∼20 Hz) under control conditions (blue). Application of [ahx5–24]NPY (1 μm) substantially reduced mIPSC frequency in this cell. K, Mean mIPSC frequency in control, with [ahx5–24]NPY (1 μm) and after washout (one-way repeated-measures ANOVA; treatment: F(1.559,26.50) = 28.18, p < 0.0001; n = 18 cells, 14 rats). In all PNs tested (18 of 18), the Y2R agonist significantly reduced the mIPSC frequency from 20.2 ± 2.1 to 15.6 ± 1.7 Hz (Bonferroni's post-test). L, Mean mIPSC amplitudes from the events in H. The Y2R agonist did not significantly affect mIPSC amplitudes (control, 19.4 ± 1.1 pA; [ahx5–24]NPY, 19.1 ± 1.1 pA; one-way repeated-measures ANOVA; treatment: F(1.338,22.75) = 2.818, p = 0.0975 n = 18 cells, 14 rats). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant.
Despite decreasing event frequencies in most cases, effects of the Y2R agonist on PN sIPSCs appeared heterogeneous, with some PNs showing clear Y2R-mediated decreases (Fig. 1A–C) and some showing increases in sIPSC frequency (Fig. 1D–F). To determine whether [ahx5–24]NPY elicited multiple sIPSC frequency response profiles, we calculated the percentage sIPSC frequency change following [ahx5–24]NPY application and performed an unbiased hierarchical cluster analysis. Squared Euclidean distance was used to determine dissimilarity between clusters, and between-groups linkage was used for the clustering method. This analysis revealed that that responses were indeed heterogeneous and distributed into two separate clusters. In the larger cluster [14 of 20 cells (10 rats)], [ahx5–24]NPY (1 μm) elicited a substantial (>10%) and reversible reduction in sIPSC frequency, but did not affect sIPSC amplitude (Fig. 1A-C,G–I). By contrast, in the second cluster [6 of 20 cells (6 rats)], [ahx5–24]NPY (1 μm) elicited a substantial (>10%) and reversible increase in both mean sIPSC frequency and amplitude (Fig. 1D–F,G–I); this effect typically manifested as more frequent, large-amplitude sIPSC bursts (Fig. 1D,F).
To determine whether the above effects of [ahx5–24]NPY on PN GABA events depended on AP activity in INs, we isolated mIPSCs with the application of 500 nm TTX. Interestingly, under these conditions, mIPSC frequencies remained high [20.2 ± 2.1 Hz (n = 18)], suggesting substantial, AP-independent GABA release in the BLA. With TTX in the bath solution, application of [ahx5–24]NPY (1 μm) significantly and reversibly decreased the mean PN mIPSC frequency from 20.2 ± 2.1 to 15.6 ± 1.7 Hz [p < 0.0001; n = 18 cells (14 rats)] (Fig. 1J–L), but did not affect mIPSC amplitudes (Fig. 1J,L). In contrast to [ahx5–24]NPY's effects on sIPSCs, its actions on mIPSCs were strikingly uniform. Virtually every PN tested responded with a >10% reduction in mIPSC frequency, and none of these cells responded with increased mIPSC amplitude. Thus, although Y2R activation increases large-amplitude, AP-dependent GABAA-mediated synaptic events in a minority of PNs, the Y2R agonist predominantly reduces an otherwise high basal rate of AP-independent GABA release onto PNs, most likely via a presynaptic mechanism.
Activation of Y2Rs increases excitability of BLA PNs
Using K+i pipettes, we then tested in current clamp whether activation of the Y2R affected PN excitability. Treatment with [ahx5–24]NPY (1 μm) decreased the depolarizing current required to initiate AP firing from rest (rheobase current), from 387 ± 24 to 295 ± 19 pA, [p < 0.0001; n = 25 cells (21 rats); 25 of 30 cells tested] (Fig. 2A,B). Effects on rheobase were relatively rapid, peaking by 5–10 min wash but, interestingly, failed to reverse in most cells despite prolonged drug washout, in contrast to the rapid washout of effects on sIPSCs or mIPSCs above. Changes in excitability were accompanied by a pronounced, and likewise irreversible, increase in input resistance [control, 59.0 ± 4.2 mΩ to 81 ± 5.4 mΩ; p < 0.0001; n = 34 cells (21 rats) in 34 of 41 PNs tested] (Fig. 2C).
Figure 2.

[ahx5–24]NPY increases BLA principal neuron excitability. A, Responses of a representative PN to a depolarizing current ramp (bottom) under control conditions (black) and following bath application of [ahx5–24]NPY (1 μm) (blue) (K+-gluconate internal). Following Y2R agonist treatment, AP firing from the RMP was evoked with substantially less current. B, Depolarizing current magnitude (rheobase) required to elicit PN AP firing from RMP under control conditions and following bath application of [ahx5–24]NPY (1 μm). Twenty-five of 30 PNs responded to the Y2R agonist with a significant reduction in rheobase (control, 387.2 ± 24.2 pA; [ahx5–24]NPY, 294.7 ± 18.8 pA; t(24) = 7.365; p < 0.0001; n = 25 cells, 21 rats; Student's paired t test). C, PN input resistance under control conditions (black) and following bath application of [ahx5–24]NPY (1 μm). Thirty-four of 41 PNs tested responded to the Y2R agonist with significantly increased input resistance from 59.03 ± 4.17 to 80.63 ± 5.40 mΩ (t(33) = 9.467; p < 0.001; n = 34 cells, 21 rats; Student's paired t test). In these PNs, the Y2R agonist also elicited a small but significant overall depolarization from −81.3 ± 0.8 to −79.6 ± 1.1 mV (t(33) = 2.757; p = 0.0094; n = 34 cells, 21 rats; Student's paired t test). D, Although effects of the Y2R agonist on PN RMP were inconsistent, overall [ahx5–24]NPY (1 μm) caused a modest but significant RMP depolarization from −80.9 ± 0.7 to −78.2 ± 0.8 mV (t(44) = 3.482; p < 0.0011; n = 45 cells, 25 rats; Student's paired t test). E, Dendrogram of a hierarchical cluster analysis performed on ΔRMP of PNs following application of [ahx5–24]NPY (1 μm). F, Proximity matrix of a hierarchical cluster analysis using squared Euclidean distance on Δ RMP of PNs following application of [ahx5–24]NPY (1 μm). Neurons were reclassified based on similarity for graphical representation. G, Scatter plot depicting the heterogeneous RPM response profile in BLA PNs following application of [ahx5–24]NPY (1 μm). Responses fall robustly into two distinct clusters (t = 10.01, df = 41, p < 0.0001). **p < 0.01, ***p < 0.001, ****p < 0.0001.
While application of [ahx5–24]NPY (1 μm) depolarized some PNs, this effect was typically modest and not as consistent as the effects on excitability and input resistance. Application of [ahx5–24]NPY (1 μm) to 45 PNs resulted in a modest but significant depolarization of 2.7 ± 0.8 mV [p = 0.0011; n = 45 cells (25 rats)] from the control value of −81 ± 0.7 mV (Fig. 2D). However, only 20 of 45 PNs (16 rats) responded to the Y2R agonist with a >2 mV depolarization [6.3 ± 1.1 mV (p < 0.0001; n = 20)]; 2 PNs even measurably hyperpolarized (>2 mV). Such a dichotomy in membrane potential responses to [ahx5–24]NPY is reminiscent of the differential effects of the Y2R agonist on sIPSCs reported above. We therefore applied a cluster analysis similar to that used above for sIPSCs to the RMP response data (Fig. 2E–G). As with the sIPSC data, two very clear clusters of responses emerged, with a majority (34 of 43) of neurons demonstrating a modest but significant depolarization (1.2 ± 0.28 mV, t = 4.257, df = 33, p < 0.0002), whereas a minority (9 of 43) exhibited a robust depolarization (7.9 ± 0.8 mV, t = 10.02, df = 8, p < 0.0001). Thus, as with sIPSC responses to the Y2R agonist, so too were the RMP responses of BLA PNs segregated into two populations.
Y2Rs decrease a KIR in BLA PNs
We next performed K+i recordings in voltage clamp to determine how [ahx5–24]NPY altered BLA PN membrane properties. [ahx5–24]NPY (1 μm) decreased the steady-state outward current in PNs held at −55 mV, consistent with the loss of a tonic hyperpolarizing current (Fig. 3A). I–V plots, generated from families of hyperpolarizing voltage-clamp steps, revealed two distinct current components affected by [ahx5–24]NPY. First, we observed an “instantaneous” current with pronounced inward rectification, which reversed at −82.0 ± 1.0 mV [n = 30 cells (20 rats)] (Fig. 3B,C). Second, we observed the much slower, hyperpolarization-dependent inward current we previously identified as the Ih (Womble and Moises, 1993; Giesbrecht et al., 2010) (Fig. 3B,C), which was inhibited by 30 μm ivabradine, an Ih blocker (Bucchi et al., 2006) [n = 5 cells (5 rats)] (not illustrated).
Figure 3.

[ahx5–24]NPY reduces an inwardly rectifying current in BLA PNs. A, Voltage-clamp (Vh = −55 mV) trace from a representative PN during bath application of [ahx5–24]NPY (1 μm). Y2R activation reduced the steady-state holding current at −55 mV for this cell. B, Representative current responses to a family of hyperpolarizing voltage-clamp steps (inset) from −55 mV. [ahx5–24]NPY (1 μm; blue) reduced both the instantaneous activating KIR (arrows, left) and the Ih (arrows, right). Traces from control and drug measurements were aligned at the −55 mV holding current for this and all similar figures. C, Current responses to a voltage step from −55 to −135 mV in control (black) and in [ahx5–24]NPY (blue) illustrating how the KIR and Ih were differentiated. The KIR was measured as the current immediately following decay of the step capacitative transient, but before activation of Ih. Ih was measured as the difference between the KIR and the steady-state current at the end of the step. D, I–V plots of the PN KIR current (C) under control conditions (in the absence of any drugs, K+-gluconate internal) (black) and following treatment with [ahx5–24]NPY (1 μm) (blue; two-way repeated-measures ANOVA; treatment: F(1,234) = 46.15, p < 0.0001; voltage: F(8,234) = 221.2, p < 0.0001; interaction: F(8,234) = 35.97, p < 0.0001; n = 27 cells, 20 rats). The Y2R agonist significantly reduced the KIR as indicated (Bonferroni's post-test). E, An I–V current plot illustrating the net PN KIR blocked following [ahx5–24]NPY (1 μm) treatment. This plot was generated by subtracting within-cell KIR plots following [ahx5–24]NPY treatment from control (see D; blue-black). This Y2R-sensitive current showed inward rectification and reversed at −87.4 ± 3.0 mV. This subtraction method was used when illustrating the KIR current blocked by [ahx5–24]NPY (or other drugs) in subsequent figures. F, I–V plot of the KIR current blocked by1 μm [ahx5–24]NPY in the presence of TTX (500 nm; two-way repeated-measures ANOVA; treatment: F(1,45) = 37.77, p < 0.0001; voltage: F(8,45) = 82.08, p < 0.0001; interaction: F(8,45) = 14.75, p < 0.0001; n = 6 cells, 5 rats). Under these conditions, the Y2R agonist significantly reduced the KIR as indicated (Bonferroni's post-test). G, Representative PN current responses to a family of hyperpolarizing voltage-clamp steps from −55 mV (inset) in the presence of the Y2R antagonist BIIE0246 (100 nm). Under these conditions, effects of [ahx5–24]NPY (1 μm) on PN conductance were entirely blocked. H, I–V plots of the KIR blocked by [ahx5–24]NPY (1 μm) in the absence of other drugs (cells from D, E; n = 27 cells, 20 rats, blue) and in the presence of BIIE0246 (100 nm; n = 4 cells, 2 rats, gray; two-way ANOVA; treatment: F(1,261), p < 0.0001; voltage: F(8,261), p < 0.0001; interaction: F(8,261), p = 0.0003). The Y2R agonist reduced significantly less KIR current in the presence of the Y2R antagonist as indicated (Bonferroni's post-test). *p < 0.05, **p < 0.01, ***p < 0.001.
In the great majority of PNs tested [27 of 30 cells (20 rats)], application of [ahx5–24]NPY (1 μm) substantially decreased the amplitude of the instantaneous inward rectifier (Fig. 3B). The current inhibited by [ahx5–24]NPY reversed at −87.0 ± 3.0 mV (n = 22) and showed clear inward rectification (Fig. 3D). An inwardly rectifying current that reversed at −83.9 ± 8.3 mV (n = 6) was similarly inhibited by [ahx5–24]NPY in PNs where series resistance was compensated (70%) (not illustrated). Because [ahx5–24]NPY demonstrated differential effects on PN sIPSCs, we isolated its presynaptic inhibitory actions with the application of TTX (500 nm); in 6 of 8 PNs tested, again we observed clear and substantial reductions in this inwardly rectifying current (Fig. 3E). This Y2R-mediated action did not readily reverse upon peptide washout, in the absence or presence of TTX, but was blocked by the Y2R antagonist BIIE0246 (100 nm) (Fig. 3F,G).
Because the Y2R-sensitive inwardly rectifying current resembled a tonically active, inwardly rectifying K+ current (KIR), we tested the effects of SCH23390, a dopamine D1 receptor antagonist and direct blocker of G-protein coupled, inwardly rectifying K+ channels (GIRK) (Kuzhikandathil and Oxford, 2002; Sosulina et al., 2008). Bath application of SCH23390 (15 μm) substantially reduced the KIR in the majority of PNs tested (14 of 15) (Fig. 4A); this blocked current reversed at −94.2 ± 3.2 mV [n = 14 cells (9 rats)] (Fig. 4B). Moreover, the effects of [ahx5–24]NPY (1 μm) on PN conductance were largely occluded by pretreatment with SCH 23390 (Fig. 4B,C). Because SCH23390 also inhibits dopamine D1 receptors, we used Cs+ as an alternative approach to block PN KIR currents. Cs+i only blocks the outward current component of a KIR, while K+ influx through this conductance persists at membrane potentials hyperpolarized to EK (Greif et al., 2000). Consistent with a KIR, in Cs+i recordings the effects of the Y2R agonist were occluded at depolarized potentials, although it continued to significantly reduce current at potentials negative to −90 mV (Fig. 4D); similar results were seen when this was repeated in the presence of TTX (500 nm) (Fig. 4E).
Figure 4.

[ahx5–24]NPY inhibits a KIR in BLA PNs. A, Representative PN current responses to a family of hyperpolarizing voltage steps (as in Fig. 3) in the continual presence of SCH23390 (15 μm), before (gray) and following bath application of [ahx5–24]NPY (1 μm; blue). The Y2R agonist had little to no effect on this PN conductance in the presence of SCH23390. B, I–V plot of the PN KIR current blocked (Net IKir) by 15 μm SCH23390 (gray), which showed inward rectification and reversed at −94.2 ± 3.2 mV, consistent with a GIRK (two-way repeated-measures ANOVA; treatment: F(1,117) = 3.738, p = 0.0556; voltage: F(8,117) = 30.36, p < 0.0001; interaction: F(8,117), p < 0.0001; n = 14 cells, 9 rats). SCH23390 significantly reduced the KIR as indicated (Bonferroni's post-test). C, I–V plots of the PN KIR current blocked by1 μm [ahx5–24]NPY in the absence of other drugs (cells from D, E) (blue; n = 27 cells, 20 rats) and in the presence of SCH23390 (15 μm) (gray + blue; n = 8 cells, 6 rats; two-way ANOVA; treatment: F(1,297) = 23.07, p < 0.0001; voltage: F(8,297) = 19.35, p < 0.0001; interaction: F(8,297) = 2.064, p = 0.0329). [ahx5–24]NPY reduced significantly less PN current with SCH23390 present as indicated (Bonferroni's post-test). D, I–V plot of the PN KIR blocked by [ahx5–24]NPY (1 μm; blue), as recorded with a Cs+i pipette and using voltage-clamp ramps (not steps as in B, C) depolarizing from −115 to −15 mV (two-way repeated-measures ANOVA; treatment: F(1,55) = 30.91, p < 0.0001; voltage: F(10,55) = 4.599, p < 0.0001; interaction: F(10,55) = 5.336, p < 0.0001; n = 6 cells, 5 rats). Under these conditions, [ahx5–24]NPY significantly reduced conductance as indicated (Bonferroni's post-test) but had no significant effect on membrane conductance at potentials depolarized to −95 mV. E, I–V plots of the KIR blocked by [ahx5–24]NPY (1 μm) as in D, with TTX (500 nm) added to the perfusate (two-way repeated-measures ANOVA; treatment: F(1,44) = 75.28, p < 0.0001; voltage: F(10,44) = 14.44, p < 0.0001; interaction: F(10,44) = 10.07, p < 0.0001; n = 5 cells, 4 rats). [ahx5–24]NPY continued to significantly reduce conductance as indicated (Bonferroni's post-test) and had no significant effect on membrane conductance at potentials depolarized to −85 mV. *p < 0.05, **p < 0.01, ***p < 0.001.
Y2R activation reduces tonic GABAB-mediated inhibition
While the inhibition of postsynaptic PN GIRK currents by [ahx5–24]NPY with TTX present would be consistent with a postsynaptic Y2R effect, the Y2R typically acts presynaptically (El Bahh et al., 2005). PNs in the BLA express both GABAA and GABABRs, the latter of which commonly activate neuronal GIRK channels (Bettler et al., 2004). As Y2R activation reduced synaptic GABA release onto most PNs studied, we hypothesized that this could result in a reduction of ongoing activation of GABABRs.
In K+i recordings, we tested the effects of the GABABR antagonist CGP 52432 at 1 μm [a concentration considered selective for postsynaptic receptors (Olpe et al., 1993; Pozza et al., 1999)]. Bath application of CGP 52432 itself significantly reduced the KIR [15 of 18 cells tested (12 rats)] (Fig. 5A,B). Furthermore, in the presence of 1 μm CGP 52432, the effects of [ahx5–24]NPY (1 μm) on the KIR were largely occluded (Fig. 5C). Results were similar with TTX present (Fig. 5D); indeed, the Y2R agonist was significantly less effective in the presence of both CGP 52432 and TTX, compared with TTX alone (Fig. 5E).
Figure 5.
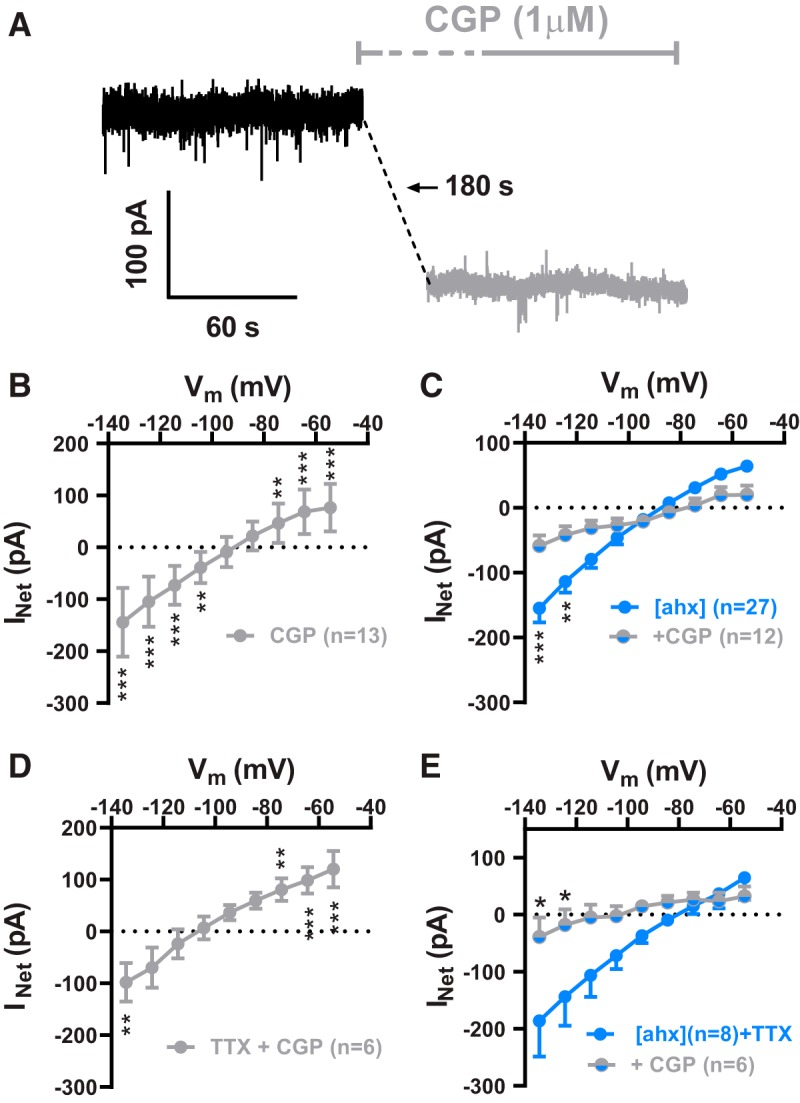
Y2Rs decrease tonic activation of PN GABABRs. A, Voltage-clamp (Vh = −55 mV) traces from a representative PN in control (black) and during application of the GABABR antagonist, CGP 52432 (1 μm; gray). The steady-state holding current was reduced by CGP 52432 in this cell, similar to actions of the Y2R agonist (compare Fig. 3). B, I–V plot of the KIR current blocked by 1 μm CGP 52432 (gray), which showed inward rectification and reversed at −92.2 ± 3.2 mV (two-way repeated-measures ANOVA; treatment: F(1,108) = 24.65, p < 0.0001; voltage: F(8,108) = 186.5, p < 0.0001; interaction: F(8,108) = 44.89, p < 0.0001; n = 13 cells, 11 rats). CGP 52432 significantly decreased the KIR as indicated (Bonferroni's post-test). C, I–V plots of the KIR blocked by [ahx5–24]NPY (1 μm) in the absence of other drugs (cells from D, E) (blue; n = 27, 20 rats) and in the presence (gray + blue) of CGP 52432 (1 μm; n = 12 cells, 9 rats; two-way ANOVA; treatment: F(1,333) = 3.280, p = 0.0710; voltage: F(8,333), p < 0.0001; interaction: F(8,333), p < 0.0001). In the presence of CGP 52432, [ahx5–24]NPY reduced significantly less current as indicated (Bonferroni's post-test). D, I–V plot of the KIR blocked by CGP 52432 (1 μm; gray) in the presence of TTX (500 nm), which reversed at −104.5 ± 3.5 mV (two-way repeated-measures ANOVA; treatment: F(1,36) = 7.204, p = 0.0109; voltage: F(8,36) = 70.95, p < 0.0001; interaction: F(8,36) = 6.440, p < 0.0001; n = 5 cells, 4 rats). CGP 52432 continued to significantly decrease the KIR in the presence of TTX as indicated (Bonferroni's post-test). E, I–V plots of the KIR blocked by [ahx5–24]NPY (1 μm) in TTX (500 nm), in the absence (blue; n = 8 cells, 6 rats) or presence of CGP 52432 (1 μm) (blue + gray; n = 6 cells, 4 rats; two-way ANOVA; treatment: F(1,108) = 13.61, p = 0.0004; voltage: F(8,104) = 5.956, p < 0.0001; interaction: F(8,108) = 1.944, p = 0.0606). With CGP and TTX both present, [ahx5–24]NPY was significantly less effective than in TTX alone as indicated (Bonferroni's post-test). *p < 0.05, **p < 0.01, ***p < 0.001.
Blockade of the GABABR itself significantly increased PN excitability in current-clamp experiments (K+i). Thus, CGP 52432 (1 μm) alone not only reduced the rheobase from 417 ± 25 to 354 ± 28 pA [p = 0.0042; n = 10 cells (8 rats)] but also occluded further changes to rheobase by subsequent application of [ahx5–24]NPY (Fig. 6A,B). Furthermore, CGP 52432 increased PN input resistance, from 58.0 ± 3.3 to 76.0 ± 1.7 mΩ [p < 0.0001; n = 12 cells (10 rats)] (Fig. 6C). However, even with CGP 52432 present, [ahx5–24]NPY (1 μm) still elicited a modest but significant increase in input resistance, from 76.0 ± 1.7 to 85.0 ± 1.6 mΩ [p = 0.0099; n = 12 cells (10 rats)] (Fig. 6C). Because the Y2R agonist reduced unitary IPSCs, we repeated the above experiments in the presence of both CGP 52432 and the GABAA receptor antagonist bicuculline (10 μm). When GABAA and GABABRs were both blocked, the Y2R agonist had no effect either on PN rheobase or input resistance [n = 7cells (4 rats)] (Fig. 6D,E).
Figure 6.

Loss of tonic GABABR activation underlies Y2R effects on PN excitability. A, Representative response to depolarizing current ramps (compare Fig. 2A) under control conditions (black), following bath application of CGP 52432 (1 μm) (gray), then with addition of [ahx5–24]NPY (1 μm) treatment in CGP (blue). Blockade of the GABABR not only reduced the rheobase but also largely occluded further effects of the Y2R agonist. B, Rheobase measured in control, in CGP 52432 (1 μm), and following subsequent addition of [ahx5–24]NPY in CGP 52432 (one-way repeated-measures ANOVA; F(1.692,15.23) = 14.02, p < 0.0001; n = 10 cells, 8 rats). CGP 52432 (1 μm) significantly reduced rheobase from 417.1 ± 25.3 to 354.4 ± 28.5 pA, whereas further addition of [ahx5–24]NPY (1 μm) in CGP did not significantly reduce rheobase (Bonferroni's post-test). C, PN input resistance in control, in CGP 52432 (1 μm; gray), and following subsequent addition of [ahx5–24]NPY (1 μm) in CGP 52432 (blue; one-way repeated-measures ANOVA; F(1.659,18.25), p < 0.0001; n = 12 cells, 10 rats). CGP 52432 (1 μm) significantly increased PN RIN from 58.1 ± 4.2 to 76.5 ± 5.0 mΩ, whereas addition of [ahx5–24]NPY (1 μm) in CGP elicited a further significant increase in RIN from 76.5 ± 5.0 to 85.2 ± 4.6 mΩ (Bonferroni's post-test). D, Rheobase current measured as in B in control, in the presence of both CGP 52432 (1 μm; gray) and bicuculline (10 μm; green) and after further addition of [ahx5–24]NPY (1 μm; blue; one-way repeated-measures ANOVA; F(1.144,6.863) = 15.07, p = 0.0055; n = 7 cells, 4 rats). Coapplication of the GABAB and GABAA receptor antagonists significantly reduced PN rheobase from 375.1 ± 46.6 to 320.3 ± 49.7 pA, whereas further treatment with the Y2R agonist had no significant further effects (Bonferroni's post-test). E, PN input resistance as in C, in control, in the presence of both CGP 52432 (1 μm) and bicuculline (10 μm) and after further addition of [ahx5–24]NPY (1 μm; one-way repeated-measures ANOVA; F(1.451,8.706) = 9.435, p = 0.0094; n = 7 cells, 4 rats). Coapplication of the GABAB and GABAA receptor antagonists significantly increased input resistance from 63.0 ± 9.5 to 79.4 ± 10.5 mΩ, whereas further addition of the Y2R agonist had no additional effect (Bonferroni's post-test). *p < 0.05, **p < 0.01, ***p < 0.001, NS, not significant.
We next reasoned that, if Y2Rs affect PNs by reducing ongoing GABABR activation, then maximally activating GABABRs with an exogenous agonist should bypass this Y2R-mediated effect. We therefore applied a high (30 μm) concentration of the prototypic GABABR agonist, baclofen (Sodickson and Bean, 1998), to PNs (K+i). Baclofen treatment caused a substantial (>100%) increase in steady-state holding current at −55 mV, from 311 ± 32 to 650 ± 84 pA, in all PNs tested [t(5) = 3.623; p = 0.0152; n = 6 cells (3 rats); Student's paired t test] (not illustrated). Importantly, baclofen-mediated KIR currents exhibited little desensitization in the continued presence of baclofen (Fig. 7A). Addition of the Y2R agonist in the presence of baclofen (30 μm) caused no additional effect on the PN KIR current [n = 5 cells (2 rats)] (Fig. 7B,C).
Figure 7.
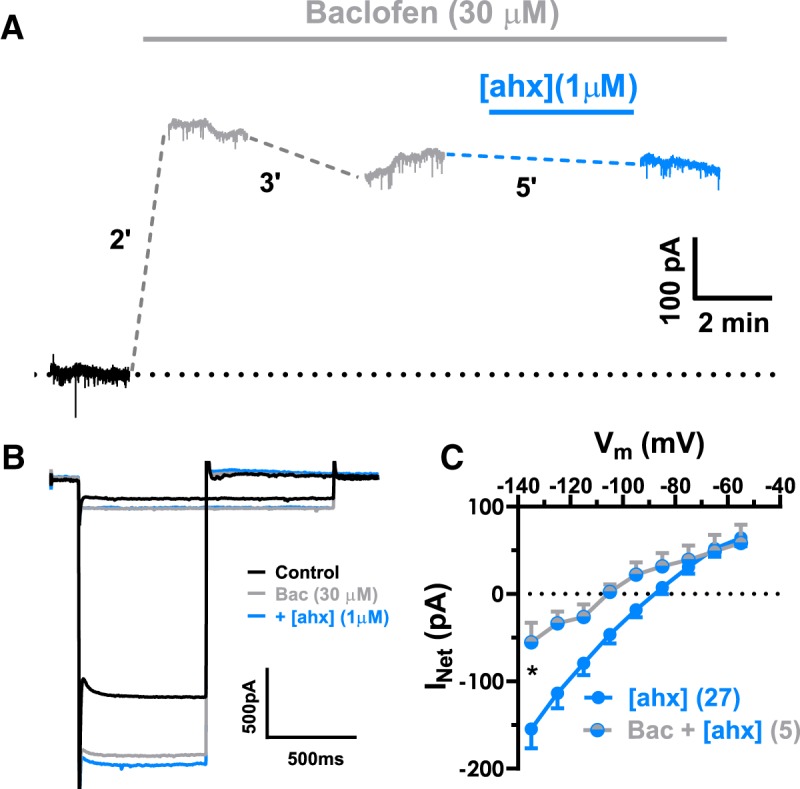
GABAB receptor activation prevents Y2R effects on PN conductance. A, Voltage-clamp (Vh = −55 mV) traces from a representative neuron during application of the GABABR agonist, baclofen (30 μm; gray). The steady-state holding current was substantially increased by baclofen. The baclofen-mediated outward current desensitized only minimally after prolonged (>5 min) drug treatment. In the continued presence of baclofen, addition of the Y2R agonist (blue) had little effect on the holding current. B, Representative PN current responses to a family of hyperpolarizing voltage-clamp steps from −55 mV (for clarity, only the steps from −55 mV to −65 mV and from −55 mV −135 mV are illustrated). Bath application of baclofen (30 μm) (gray) substantially enhanced this PN′s conductance relative to control (black), in the continued presence of baclofen, subsequent [ahx5–24]NPY (1 μm) application (blue) had little effect. C, I–V plots of the KIR current blocked by the Y2R agonist in the absence of other drugs [n = 27 (20 rats); blue] or in the presence of baclofen [n = 5 cells, 2 rats (blue + gray)] (two-way ANOVA; treatment: F(1,207) = 27.12, p < 0.0001; voltage: F(8,207) = 18.01, p < 0.0001; interaction: F(8,207) = 1.810, p = 0.0767). In the presence of baclofen, [ahx5–24]NPY reduced significantly less KIR current as indicated (Bonferroni's post-test). *p < 0.05.
Together, these results indicate that BLA PNs are tonically inhibited via the activation of postsynaptic GABABRs. This inhibition persists when APs are blocked but was constrained by Y2R activation.
Y2Rs indirectly modulate Ih in BLA PNs
Although virtually all PNs responded to [ahx5–24]NPY with a reduction in mIPSC frequency and loss of tonic GABABR-mediated currents, additional effects of the Y2R agonist were clearly seen in subsets of PNs. Specifically, voltage step protocols using K+i pipettes revealed that [ahx5–24]NPY (1 μm) also significantly decreased the Ih in 15 of 32 PNs tested (12 rats) (Fig. 8A,B), although it somewhat increased Ih magnitude in a minority [7 of 32 PNs (5 rats)] (not illustrated). Application of CGP 52432 (1 μm) largely replicated the effects of [ahx5–24]NPY on Ih, significantly decreasing its magnitude in 7 of 16 PNs (5 rats) (Fig. 8C,D). Therefore, like the effects of the Y2R agonist on PN KIR currents, modulation of PN Ih appears to be an indirect response, mediated by the loss of tonic GABABR signaling. Interestingly, bath application of the GABABR agonist baclofen (30 μm) also substantially reduced Ih magnitude in all PNs tested (not illustrated).
Figure 8.

Y2R modulates Ih by suppressing tonic GABAB receptor activation. A, Representative current responses (top left), to a family of hyperpolarizing voltage-clamp steps (Vh = −55 mV, bottom left). Right, Superimposed are current responses to voltage-clamp steps from Vh to −135 mV in control conditions (black) and in [ahx5–24]NPY (1 μm) (blue), indicating a substantial, Y2R-mediated reduction in Ih. B, [ahx5–24]NPY (1 μm) significantly decreased Ih in 15 of 32 PNs tested as indicated (Bonferroni's post-test; two-way repeated-measures ANOVA; treatment: F(1,112) = 112.5, p < 0.0001; voltage: F(7,112) = 32.42, p < 0.0001; interaction: F(7,112) = 15.17, p < 0.0001; n = 15 cells, 12 rats). C, Superimposed current responses to voltage-clamp steps from −55 mV to −135 mV in control (black) and in CGP 52432 (1 μm) treatment (gray), illustrating a substantial GABABR antagonist effect on Ih similar to that of the Y2R agonist. D, CGP 52432 (1 μm) significantly decreased Ih (7 of 15 PNs tested) as indicated (Bonferroni's post-test; two-way repeated-measures ANOVA; treatment: F(1,48) = 4.869, p < 0.0001; voltage: F(7,48) = 25.61, p < 0.0001; interaction: F(7,48) = 4.869, p = 0.0003; n = 7 cells, 5 rats). **p < 0.01, ***p < 0.001.
Y2Rs reduce firing rates in a subset of PNs by potentiating the Ca2+-dependent IsAHP
In current clamp, AP AHPs were also clearly enhanced by [ahx5–24]NPY in approximately half of the PNs tested. To study the effect of Y2R activation on the AHP and on AP firing rates, we elicited trains of APs using 800 ms depolarizing current steps, individually adjusting their amplitudes to elicit similar (±1) numbers of APs in the absence and the presence of [ahx5–24]NPY (1 μm) in each neuron studied. Treatment with the Y2R agonist increased the amplitude of the first AHP by >30% in 10 of 21 PNs tested; from 6.0 ± 1.0 to 11.0 ± 1.1 mV [p < 0.0001; n = 10 cells (8 rats)] (Fig. 9A,B). Typically, [ahx5–24]NPY potentiated the AHPs after each of the first 5 AHPs in a train (Fig. 9A,C). Blockade of GABABRs with CGP 52432 (1 μm) likewise enhanced the magnitude of the first AHP from 8.0 ± 1.2 to 11.5 ± 2.0 mV [t(6) = 3.500; 0.0128; n = 7 cells (5 rats); Student's paired t test] in 7 of 13 PNs tested (not illustrated).
Figure 9.

[ahx5–24]NPY increases the AHP in a subset of PNs. A, Left, Superimposed AP trains (current step-evoked) from a cell where [ahx5–24]NPY (1 μm; blue) enhanced the AP AHP seen in control (black). Similar numbers of step-evoked APs were compared in control and in [ahx5–24]NPY: here 8 and 7 Aps, respectively. Right, The first step-evoked AP from the above traces expanded and aligned, illustrating the substantial AHP enhancement. B, AHP amplitudes of initial APs in control and in [ahx5–24]NPY (1 μm). In 10 of 21 PNs tested, the Y2R agonist significantly increased the first AHP from 5.97 ± 1.00 to 11.19 ± 1.13 mV (t(9) = 8.709; p < 0.0001; n = 10 cells, 8 rats; Student's paired t test). C, Amplitudes of the first 5 AHPs in a train evoked in control and in [ahx5–24]NPY (1 μm) (two-way repeated-measures ANOVA; treatment: F(1,25) = 109.2, p < 0.0001; voltage: F(4,25) = 0.05778, p = 0.9934; interaction: F(4,24) = 0.5242, p < 0.7189; n = 6 cells, 5 rats). AP amplitudes were significantly increased by the Y2R agonist as indicated (Bonferroni's post-test). Four further PNs, which responded to [ahx5–24]NPY with a >30% enhancement of the first AHP, were excluded because they failed to fire 5 successive APs following Y2R agonist treatment (regardless of increasing step amplitude). D, Current responses to a hyperpolarizing voltage-clamp step (−55 to −135 mV) from a PN in which [ahx5–24]NPY (1 μm; blue) enhanced AHP amplitude seen in control (black), as in A. Following Y2R agonist treatment, a slowly decaying “rebound” outward current (visible at the end of the hyperpolarizing step) was substantially enhanced. Inset, Right, The expanded view (blue) illustrates this outward current's characteristic slow decay and preceding inward current. Inset, Bottom right, Only a modest effect on Ih was seen. E, Rebound outward current peak amplitudes from PNs whose first AHP was increased (>30%) in response to the Y2R agonist. In such neurons, [ahx5–24]NPY (1 μm) significantly increased this outward current from 77.5 ± 26.4 to 223.8 ± 47.8 pA (t(9) = 4.094; p = 0.0027; n = 10 cells, 8 rats; Student's paired t test). In PNs where [ahx5–24]NPY (1 μm) decreased the KIR, but did enhance the AHP, this outward current was not significantly changed [28.8 ± 13.8 pA (control) to 37.9 ± 19.5 pA (Y2R); t(10) = 1.246; p = 0.2413; n = 11 cells, 7 rats; Student's paired t test] (not illustrated). F, Current responses to a hyperpolarizing voltage-clamp step (−55 to −135 mV) from a PN in which the GABABR antagonist CGP 52432 (1 μm; gray) enhanced AHP amplitude relative to control (black), similar to effects of the Y2R agonist. Following GABABR blockade, a slowly decaying outward rebound current (visible upon returning to −55 from −135 mV) is substantially enhanced. G, Rebound outward current peak amplitudes from PNs where the GABABR antagonist increased the first AHP (>30%). In AHP-responsive PNs, CGP 52432 (1 μm) significantly increased this outward current from 39.5 ± 28.1 to 190.6 ± 58.1 pA (t(6) = 2.735; p = 0.0340; n = 7 cells, 5 rats; Student's paired t test). *p < 0.05, **p < 0.01, ***p < 0.001.
In PNs in which [ahx5–24]NPY potentiated the AHP, voltage-clamp steps (K+i) demonstrated the characteristic Y2R-mediated inhibition of the KIR. In such PNs, the Y2R agonist additionally elicited or enhanced an outward current seen after the membrane potential was returned to −55 mV at the end of larger hyperpolarizing steps (Fig. 9D). As measured at −55 mV following a hyperpolarizing step to −135 mV, this “rebound” current increased significantly, from 78 ± 26 to 224 ± 48 pA [p = 0.0027; n = 10 cells (8 rats)] in neurons in which [ahx5–24]NPY (1 μm) also markedly (>30%) enhanced the first AHP (Fig. 9E). Conversely, in PNs whose AHPs were unaffected by the Y2R agonist, the above rebound current was not enhanced (not illustrated).
As with the Y2R agonist, the GABABR antagonist inhibited the KIR in most PNs [15 of 18 cells (12 rats)], but only enhanced this rebound current in 7 of the 18 cells. Here too, enhancement of the rebound current by CGP 52432 was linked to its potentiation of the AHP. In these cells, the rebound current increased from 40 ± 11 to 191 ± 58 pA [p = 0.034; n = 7 cells (5 rats)] (Fig. 9F,G). Thus, Y2R-mediated potentiation of the AHP also appears to result from reduced GABABR tone.
In the presence of [ahx5–24]NPY, PNs almost always required less depolarizing current to reach threshold than in control. However, in cells in which Y2R activation enhanced the AHP (and the rebound current amplitude) there was, as expected, an accompanying reduction in repetitive AP firing rates. Specifically, when [ahx5–24]NPY (1 μm) increased the rebound current by >10%, the interval between the two first APs caused by a current step more than doubled, from 33.8 ± 9.5 to 80.1 ± 9.5 ms [p = 0.0002; n = 9 cells (9 rats)] (Fig. 10A,B). By contrast, in PNs where [ahx5–24]NPY inhibited the KIR, but did not affect or induce a rebound current, we saw a significant decrease in interval between the first two APs induced by the current step, from 50.5 ± 7.1 to 29.3 ± 7.0 ms [p = 0.0047; n = 10 cells (7 rats)] (Fig. 10C,D).
Figure 10.

[ahx5–24]NPY elicits opposing effects on AP firing patterns. A, AP trains evoked by depolarizing current steps in a PN in which [ahx5–24]NPY (1 μm; inset, top left; blue) enhanced the rebound outward current relative to control (inset; black). Similar numbers of step-evoked APs (here, 9 APs each; full trace not shown) were compared in control (black) and in [ahx5–24]NPY (blue). Treatment with the Y2R agonist nearly doubled the latency between the first two APs (black and blue bars above traces). B, Interspike interval plotted against AP number in a train, compared in control and in [ahx5–24]NPY (1 μm), from neurons responding to the Y2R agonist as in A (rebound current increased >10%; two-way repeated-measures ANOVA; treatment: F(1,40) = 4.124, p = 0.0490; AP number: F(4,40) = 7.021, p = 0.0002; interaction: F(4,40) = 4.617, p = 0.0037; n = 9 cells, 9 rats). In these PNs, [ahx5–24]NPY (1 μm) significantly increased the interval between the first two current step-evoked APs, from 33.8 ± 9.5 to 80.1 ± 9.5 ms (Bonferroni's post-test). C, Representative AP trains evoked by depolarizing current steps in a PN as in A in which [ahx5–24]NPY (1 μm; blue) inhibited the KIR and Ih but did not affect the outward rebound current (inset, top left). In this example, 7 and 8 APs were evoked by current steps in control and drug, respectively (full trace not shown). Inset, Bottom left, Change in latency of initial APs. D, Interspike interval plotted against AP number in trains, compared in control and in [ahx5–24]NPY (1 μm), from neurons where [ahx5–24]NPY did not enhance the outward rebound current (two-way repeated-measures ANOVA; treatment: F(1,45) = 29.95, p < 0.0001; AP number: F(4,45) = 20.03, p < 0.0001; interaction: F(4,45) = 0.5365, p = 0.7096; n = 10 cells, 7 rats). In these PNs, Y2R activation significantly decreased the interval between the first and second APs and between the second and third APs (Bonferroni's post-test). *p < 0.05, **p < 0.01, ***p < 0.001.
We next performed voltage step experiments to examine the rebound current's I–V relationship. Repetitively evoking a rebound current with a single step from −55 to −135 mV under control conditions then following [ahx5–24]NPY treatment, we introduced additional 10 ms hyperpolarizing steps during the peak rebound current (ranging from −65 to −135 mV, in −10 mV increments; Fig. 11A, inset). Net I–V plots of the current enhanced by [ahx5–24]NPY were then generated by subtracting the current measured in control from that measured after drug treatment. The rebound current induced or enhanced by [ahx5–24]NPY showed clear outward rectification and reversed near −80 mV, suggesting Y2R-mediated enhancement of an outwardly rectifying K+ conductance (Fig. 11A).
Figure 11.
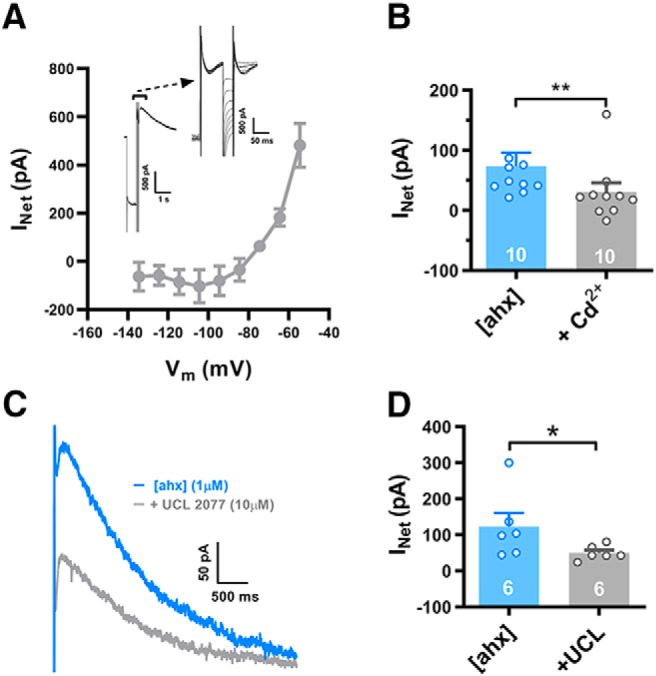
Loss of GABAB tone unmasks a Ca2+-dependent IsAHP. A, I–V relationship of the rebound outward current enhanced by Y2R activation. We isolated this with a series of brief (30 ms) hyperpolarizing voltage steps applied during its peak (insets), both in control and in [ahx5–24]NPY (1 μm; not illustrated; n = 3 cells, 3 rats). Net rebound current was measured as the difference between resting current before initiation of the voltage protocol and at the arrow placed immediately before the end of the hyperpolarizing steps, below the expanded trace in the inset. B, In [ahx5–24]NPY (1 μm), Cd2+ (100–200 μm) significantly decreased the rebound outward current (Irebound) from 73.3 ± 23.2 pA to 30.3 ± 15.5 pA (t(9) = 4.698; p = 0.0011; n = 10 cells, 8 rats; Student's paired t test). Seven PNs received 100 μm Cd2+; 3 received 200 μm Cd2+. As both doses had similar effects, we pooled these data. C, A representative outward rebound current following [ahx5–24]NPY (1 μm; blue), as in A. Treatment with UCL 2077 (10 μm; gray) substantially decreased this outward current. D, UCL 2077 (10 μm) treatment significantly decreased Irebound from 122.2 ± 38.3 pA to 49.6 ± 8.2 pA (p = 0.0260; n = 6 cells, 4 rats; Mann–Whitney U test). *p < 0.05, **p < 0.01.
Dendritic GABABRs commonly activate both GIRK channels (Leung and Peloquin, 2006) and inhibit VGCCs (Pérez-Garci et al., 2013), which can synergistically decrease dendritic Ca2+ influx during AP firing. We thus hypothesized that Y2R-mediated enhancement of the AHP resulted from the recruitment of a Ca2+-dependent K+ conductance (GK,Ca), following removal of basal GABAB tone and consequent enhancement of AP-mediated Ca2+ influx into PN dendrites. Indeed, a transient inward current was often seen preceding the rebound current, suggesting that Ca2+ influx was mediated by anodal break excitation (Fig. 9D). Consistent with this, the nonspecific VGCC blocker Cd2+ (100–200 μm) sharply reduced rebound outward current amplitude from 73 ± 23 to 30 ± 16 pA [p = 0.0011; n = 10 cells (8 rats)] when applied in the presence of the Y2R agonist (Fig. 11B).
The decay kinetics of the rebound current were notably slow, fitting a single exponential with time constants >1 s in the presence of either [ahx5–24]NPY or CGP 52432. This property was consistent with that of the slow afterhyperpolarizing current (IsAHP), a GK,Ca previously described in the BLA and elsewhere (Womble and Moises, 1993; Power et al., 2011). We therefore tested the selective IsAHP blocker UCL 2077 at 10 μm (its maximal water-soluble concentration) (Shah et al., 2006) on PNs expressing the rebound current upon [ahx5–24]NPY treatment. Application of UCL 2077 to such rebound currents markedly reduced their amplitudes from 122 ± 38 to 50 ± 8 pA [p < 0.05; n = 6 cells (4 rats); Mann–Whitney U test] (Fig. 11C,D). Because 10 μm is UCL 2077's approximate IC50 in brain slices (Shah et al., 2006), a higher concentration (if soluble) might have more fully blocked this current. Conversely, the SK channel blocker apamin (1 μm) did not inhibit the rebound current (n = 3; not illustrated).
Validation of NPY Y2Rs and tdTomato expression in brains of transgenic Y2-tdTomato mice
The above results were all from rat BLA, but the recent availability of the Y2R-tdTomato mouse (see Materials and Methods) allowed us to develop concordant information on the distribution and cell types expressing NPY2Rs in the BLA, in addition to determining the alignment of Y2R actions in rat and mouse. Overall, expression of the tdTomato signal was robust in the brains of these mice but did vary among individual animals. The distribution of tdTomato expression agreed in general with published reports of Y2R mRNA expression (Fetissov et al., 2004) and, within the BLA, showed a pattern of distribution that was present in the rostral to more caudal regions of the nucleus (Fig. 12). In general, there were more tdTomato-Y2R-expressing cells within the medial basal aspects of the nucleus, with few fluorescent cells present within the lateral aspects of the BLA. We observed numerous fluorescent cell bodies that ranged in size from small (10 μm) to medium (15–20 μm), suggesting that Y2Rs are expressed by multiple cell types.
To validate the correlation of tdTomato signal with Y2R expression, we used double-label ISH to determine the presence of Y2R mRNA within cells expressing tdTomato mRNA. Because much of the NPY Y2R protein is expressed on nerve terminals, away from the cell body, the use of a Y2R antibody to identify neuronal cell bodies expressing the receptor is not optimal. Figure 13 demonstrates the expression of tdTomato mRNA in the BLA (Fig. 13A) and the overlapping expression of Y2R mRNA (Fig. 13B,C). Higher magnification of the BLA (Fig. 13D–F) indicates a strong coexpression of Y2R mRNA in tdTomato mRNA-expressing cells. Specifically, virtually every cell (∼99.3%; 298 of 300 cells) that expressed tdTomato mRNA also expressed signal for Y2R mRNA (Fig. 13A–F, arrows); however, not all Y2R mRNA-expressing cells exhibited a tdTomato signal. Thus, although the expression of tdTomato did not report the Y2R expression in a 1:1 fashion, its presence reliably identified cells that do express Y2R mRNA within the BLA. This relationship was further identified in other brain regions, such as the pyramidal cells of the hippocampus (Fig. 13G–I) where the Y2R potently modulates presynaptic glutamate release (El Bahh et al., 2002; 2005).
Figure 13.
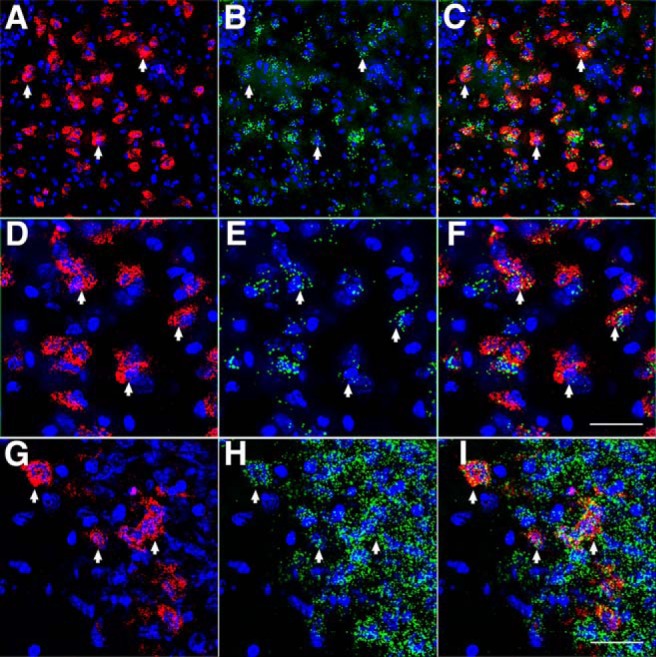
Coexpression of Y2R mRNA and tdTomato mRNA in Y2R-tdTomato mouse brain. tdTomato mRNA (A) and Y2R mRNA (B) expression in the BLA. C, Merged image demonstrates complete Y2R mRNA coexpression in tdTomato cells. Arrows indicate coexpression. DAPI (blue) demarcates cell nuclei. Scale bar: C, 25 μm. D–F, Higher magnification of cells presented in A–C demonstrating the presence of Y2R mRNA (green puncta) in tdTomato mRNA-containing cells. Scale bar: F, 30 μm. tdTomato (G) and Y2R (H) gene expression in the CA3 region of the hippocampus. While many double-labeled cells are present (arrows), diffuse Y2R labeling is also present that does not associate with tdTomato signaling (I). These panels demonstrate that tdTomato-synthesizing neurons coexpress Y2R but that not all Y2R mRNA-expressing neurons coexpress tdTomato. Scale bar: I, 30 μm.
In Y2R-tdTomato mice, NPY/SST INs and a subset of PNs express Y2Rs
The electrophysiological results in rat (above) suggest that Y2Rs are expressed by, and inhibit, GABA release from INs in or near the BLA. Having established the validity of tdTomato as a marker of Y2R expression in the Y2R-tdTomato mouse, we next used immunohistochemistry in these mice to determine whether tdTomato colocalized with markers for specific interneuronal populations within the BLA. The colocalization of tdTomato, NPY, and SST in neurons of the BLA is presented in Figure 14A–D. As previously described in rats, NPY cells coexpressed SST, although single-labeled SST cells were also evident (McDonald, 1989). The tdTomato signal was present within both populations (NPY+ and NPY−) of SST-immunoreactive cells; notably, SST expression defines a GABAergic IN population that preferentially innervates the distal dendrites of PNs (McDonald, 1989; Muller et al., 2007). Importantly, with rare exceptions (4 colabeled cells observed in 3 animals; 339 total PV cells), tdTomato fluorescence did not colocalize with labeling for PV (Fig. 14E,G,H), which identifies a sizable interneuronal population in the BLA (McDonald and Mascagni, 2001).
Figure 14.

Coexpression of tdTomato immunoreactivity in NPY- and SST-immunoreactive INs and CaMKII-immunopositive neurons in the BLA. High magnification of tdTomato- (A), NPY- (B), and SST- (C) immunoreactive neurons in the BLA. D, The merged image indicates that tdTomato is expressed in NPY+/SST+ (double arrowhead) and NPY−/SST+ (arrows) neuronal populations. Subsets of tdTomato-immunoreactive cells (E) in the BLA also contain immunoreactivity for CaMKII (F, H, arrows) but not the interneuronal marker PV (G, H). Single-labeled cells for CaMKII (carat- v) and tdTomato (single arrowhead) are also present; in general, all (>99%) of the PV population is single-labeled with no significant coexpression with tdTomato. Scale bar, 100 μm.
The overall size and number of tdTomato-expressing cells in the BLA suggested that it labeled more than just a subset of INs, because INs represent only ∼15% of the total BLA neuronal population (Paré and Smith, 1998; Rostkowski et al., 2009). Indeed, the majority of tdTomato-expressing cells within the medial aspect of the BLA were immunopositive for CaMKII (Fig. 14E,F,H), a marker for glutamatergic projection neurons (Rostkowski et al., 2009). This is consistent with observations in other brain regions, particularly the hippocampus, where pyramidal neurons represent the source of the vast majority of Y2Rs (El Bahh et al., 2005). Therefore, although our immunohistochemical data do not indicate whether receptor expression is presynaptic or postsynaptic, it is clear that Y2Rs are expressed both by NPY/SST INs and a subset of PNs in the BLA.
Y2R responses in mouse PNs
We next performed patch-clamp experiments in brain slices prepared from Y2R-tdTomato mice. Overall, electrophysiological properties of mouse and rat BLA PNs were similar, although the mouse neurons were considerably smaller in size, based on capacitance measurements (Table 1). Comparisons between fluorescent and nonfluorescent BLA PNs in the Y2R-tdTomato mouse also revealed largely similar electrophysiological properties. More specifically, whole-cell capacitance, RMP, input resistance, rheobase, and I–V plots were not significantly different between fluorescent and nonfluorescent PNs (Table 1).
Table 1.
Comparative electrophysiological properties of fluorescent (Y2R+) and nonfluorescent (Y2R−) BLA pyramidal neurons from rat and from Y2R-tdTomato mice
| Property | Rat | Mouse fluorescent | Mouse nonfluorescent |
|---|---|---|---|
| RMP | −81.0 ± 0.5 mV (n = 77) | −81.1 ± 1.2 mV (n = 28) | −83.8 ± 1.1 mV (n = 23) |
| Input resistance | 59.1 ± 2.6 mΩ (n = 77) | 91.0 ± 4.3 mΩ (n = 32) | 94.3 ± 3.1 mΩ (n = 23) |
| Rheobase | 370.4 ± 15.8 pA (n = 59) | 239.5 ± 21.0 pA (n = 29) | 252.5 ± 22.5 pA (n = 24) |
| First AHP (C-step-evoked) | 6.0 ± 0.7 mV (n = 22) | 4.4 ± 0.7 mV (n = 23) | 3.5 ± 0.6 mV (n = 21) |
| Capacitance | 434.9 ± 21.4 pF (n = 24) | 144.4 ± 7.5 pF (n = 28) | 146.7 ± 6.4 pF (n = 24) |
| Rebound current | 52.0 ± 15.1 pA (n = 21) | 89.6 ± 7.9 pA (n = 33) | 80.3 ± 8.3 pA (n = 25) |
| Ih (−135 mV) | 252.3 ± 15.5 pA (n = 70) | 117.6 ± 8.0 pA (n = 33) | 109.4 ± 8.5 pA (n = 25) |
Indeed, the only difference consistently observed between electrical properties of fluorescent and nonfluorescent cells was the interval between the first two APs of trains evoked by current steps. Specifically, this interspike interval was 54.9 ± 7.7 ms [n = 25 cells (12 mice)] in nonfluorescent PNs and 88.9 ± 9.0 ms [n = 25 cells (18 mice)] in fluorescent PNs (p = 0.035; two-way ANOVA; Bonferroni's post-test) (Fig. 15A–C). Both fluorescent and nonfluorescent cells responded to [ahx5–24]NPY (1 μm) with the loss of KIR currents (Fig. 15D,E) and increased rheobase, similar to the responses in rat PNs (Fig. 15F,G). In fluorescent PNs, [ahx5–24]NPY significantly reduced the rheobase current from 229 ± 28 to 191 ± 21 pA [p = 0.01;n = 19 cells (14 mice); Student's paired t test]; and in nonfluorescent PNs, the Y2R agonist significantly reduced the rheobase current from 287 ± 27 to 213 ± 32 pA [p < 0.0001; n = 13 cells (10 mice); Student's paired t test]. The rheobase response to the Y2R agonist was not significantly different in fluorescent and nonfluorescent PNs (p = 0.082, two-way ANOVA). As was the case in rat PNs, a subset of mouse PNs also responded to the Y2R agonist with an induction or enhancement of the IsAHP-mediated rebound current. This response occurred preferentially in fluorescent PNs; in these PNs, [ahx5–24]NPY significantly increased the rebound current from 76 ± 10 to 119 ± 18 pA (p = 0.0115; n = 16) (Fig. 15I). The overall response rate (defined as a >10% increase in rebound current amplitude) in fluorescent PNs was 10 of 16; in these responsive neurons, the Y2R agonist increased rebound amplitude from 76 ± 14 to 150 ± 21 pA [n = 10 cell (10 mice)]. By contrast, [ahx5–24]NPY did not significantly alter rebound current amplitude in nonfluorescent PNs (only 2 of 14 showed >10% increase in the IsAHP).
Figure 15.

Y2R-mediated responses in Y2R+ and Y2R− BLA PNs in Y2R-tdTomato mice. A1, An AP train evoked in a nonfluorescent PN in BLA of a Y2R-tdTomato mouse, with an initial AP doublet burst. A2, Shown on a faster timescale, initial doublet APs (interspike interval < 10 ms) were relatively common (7 of 25 tested) in nonfluorescent PNs. B1, An AP train evoked in a fluorescent (Y2R-expressing; orange) PN in the BLA of a Y2R-tdTomato mouse. B2, Shown at a faster timescale, the first AP of this train. Initial AP doublets (as in A) were less common (3 of 25 tested) in fluorescent PNs. C, AP interspike intervals, plotted against position in a train, from fluorescent (n = 25 cells, 18 mice; orange) and nonfluorescent (n = 25 cells, 12 mice; black) BLA PNs from Y2R-tdTomato mice (two-way ANOVA; cell type: F(1,304) = 0.6109, p = 0.4350; AP number: F(8,304) = 10.44, p < 0.0001; interaction: F(8,304) = 1.718, p = 0.0936). The first interspike interval was significantly shorter in nonfluorescent than in fluorescent PNs (54.9 ± 7.7 ms vs 88.9 ± 9.0 ms; p = 0.0023, Bonferroni's post-test). D1, Current responses to hyperpolarizing voltage steps from a representative nonfluorescent Y2R-tdTomato mouse BLA PN, in control (black) and in [ahx5–24]NPY (1 μm) (blue). D2, Current responses from a fluorescent BLA PN in a Y2R-tdTomato mouse, in control (orange) and in [ahx5–24]NPY (1 μm) (blue). E, KIR current blocked by [ahx5–24]NPY (1 μm) in nonfluorescent PNs (blue; n = 13) and fluorescent PNs (orange; n = 19) in BLA from Y2R-tdTomato mice. The Y2R agonist significantly reduced the KIR current in both fluorescent and nonfluorescent PNs. Nonfluorescent PNs: two-way repeated-measures ANOVA; treatment: F(1,108) = 14.22, p = 0.0003; voltage: F(8,108) = 88.76, p < 0.0001; interaction: F(8,108) = 28.75, p < 0.0001; n = 13 cells, 10 mice. The Y2R agonist significantly reduced the KIR as indicated (Bonferroni's post-test). Fluorescent PNs: two-way repeated-measures ANOVA; treatment: F(1,162) = 14.45, p = 0.0002; voltage: F(8,165) = 111.7, p < 0.0001; interaction: F(8,165) = 7.762, p < 0.0001, n = 19 cells, 14 mice. The Y2R agonist significantly reduced the KIR as indicated (Bonferroni's post-test). F1, Responses of a nonfluorescent PN to a depolarizing current ramp from RMP in control (black) and in [ahx5–24]NPY (1 μm) (blue). F2, The Y2R agonist significantly reduced rheobase current in nonfluorescent PNs, from 287.1 ± 26.7 pA to 213 ± 31.8 pA (t(12) = 4.902; p < 0.0004; n = 13 cells, 10 mice; Student's paired t test). G1, Responses of a fluorescent PN to a depolarizing current ramp from RMP in control (orange) and in [ahx5–24]NPY (1 μm) (blue). G2, The Y2R agonist significantly reduced rheobase current in fluorescent PNs from 228.7 ± 27.7 pA to 190.9 ± 20.5 pA (t(18) = 2.878; p < 0.0100; n = 19 cells, 14 mice; Student's paired t test). H1, Response of a nonfluorescent PN to a hyperpolarizing voltage step (as in Fig. 9) in control (black) and in [ahx5–24]NPY (1 μm) (blue). Although the Y2R agonist substantially decreased the KIR in this PN, it did not enhance the IsAHP-mediated rebound outward current. H2, In nonfluorescent PNs, [ahx5–24]NPY (1 μm) did not enhance the rebound current (control: 82.8 ± 12.2 pA, [ahx]: 85.9 ± 16.2 (t(13) = 0.3176; p = 0.7558; n = 14 cells, 11 mice; Student's paired t test). I1, Response of a fluorescent PN to a hyperpolarizing voltage step in control (orange) and in [ahx5–24]NPY (1 μm) (blue). The Y2R agonist substantially decreased the KIR and enhanced the IsAHP-mediated rebound current. I2, In fluorescent PNs, [ahx5–24]NPY (1 μm) significantly increased the rebound current from 76.2 ± 10.5 pA to 119.1 ± 18.4 pA (t(15) = 2.878; p = 0.0115; n = 16 cells, 13 mice; Student's paired t test). *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Previous work has identified neuronal mechanisms underlying the acutely anxiolytic, largely Y1R-mediated effects of NPY in the BLA (Sosulina et al., 2008; Giesbrecht et al., 2010; Molosh et al., 2013). BLA also expresses Y2Rs (Stanić et al., 2006), and selective Y2R activation there unexpectedly increases anxiety (Sajdyk et al., 2002), via a previously unknown mechanism(s). Here we demonstrate that Y2R activation reduces GABA release from a select group of INs, most likely via presynaptic Y2Rs. This highly specific action reduced tonic GABABR signaling in most PNs, both in rat and mouse, thereby eliciting unexpectedly broad functional consequences. Loss of the GABABR-induced KIR currents greatly increased PN excitability, providing a plausible mechanism for Y2R-induced anxiety. Furthermore, reduced GABABR tone facilitated Ca2+ entry, which potentiated the IsAHP in approximately half the PNs. Although this provides a mechanism whereby Y2R activation increases anxiety, it appears to contradict the overall anxiolytic outcome associated with either acute or repeated administration of NPY.
Ongoing, AP-independent GABA inhibition of BLA PN dendrites
We observed strikingly high mIPSC frequencies (∼20 Hz) in BLA PNs at −15 mV (Cs+i), consistent with previous observations (Silveira Villarroel et al., 2018). [ahx5–24]NPY substantially decreased mIPSC frequencies, but not amplitudes in nearly all PNs, suggesting a presynaptic mechanism. As most PNs responded to [ahx5–24]NPY in this manner, it moreover suggests that most receive Y2R-sensitive GABA input. This mechanism is consistent with our immunohistochemical data demonstrating Y2R expression on SST-INs (some of which express NPY), whereas Y2R expression was absent in PV INs, the largest IN population in the BLA. SST-INs preferentially innervate PN distal dendrites, a site of signal integration and synaptic plasticity (McDonald and Mascagni, 2002; Wolff et al., 2014), Thus, Y2R activation selectively suppresses dendritic GABA input. Consistent with this, postsynaptic GABABRs are exclusively expressed at PN distal dendritic spines and shafts (McDonald et al., 2004).
GABABRs are largely extrasynaptic on BLA PN dendrites (Kulik et al., 2003; McDonald et al., 2004), and their activation, by synaptic spillover, usually requires substantial GABA release (Scanziani, 2000; Kohl and Paulsen, 2010). Rat neocortical pyramidal neurons display tonic, GABABR-mediated currents only in elevated extracellular K+ and reduced Mg2+ (Wang et al., 2010). Mature dentate granule cells are inhibited via tonic GIRK currents, which arise mainly from constitutive channel activity (Gonzalez et al., 2018). Here we report that BLA PNs show robust, tonic GABABR-dependent currents under typical acute slice conditions that persisted in TTX. This is, to our knowledge, the first report of tonic, activity-independent GABAB responses, and so, represents a novel form of tonic, neuromodulator-sensitive inhibition.
Disinhibition of PN dendrites: implications of space clamp
In complex neuronal dendrites, voltage-control declines with distance from the somatic pipette. Layer 5 PN apical dendrites exhibit complete loss of voltage-control at distances >180 μm from the soma (Williams and Mitchell, 2008). Because Y2R-sensitive GABA events likely originate near GABABRs on PN dendrites, voltage-clamp fidelity is probably poor. However, this KIR current is tonically active, so essentially time-independent, and somatic recordings reasonably capture slower dendritic currents, such as the KIR (Williams and Mitchell, 2008). Shunting associated with K+ currents also reduces dendritic space clamp (Bar-Yehuda and Korngreen, 2008), implying that, by reducing conductance, Y2R activation may indeed enhance space clamp.
Potential control in voltage clamp (space clamp) depends strongly on a neuron's geometry (e.g., Williams and Mitchell, 2008). Although direct interactions between GABABR and Ih have not been reported, inhibition of local K+ currents must reduce hyperpolarization-dependent activation of Ih, as seen previously (Schweitzer et al., 2004; Day et al., 2005). Given the poor distal space clamp predicted in a BLA PN, suppression of a dendritic, GABABR-mediated KIR could readily result in deactivation of neighboring Ih. The uniform reductions in Ih seen with maximal GABABR activation are likely due to the massive increase in KIR conductance in these experiments, shunting all dendritic currents and essentially rendering Ih invisible at the soma, consistent with results in CA1 PNs (Schweitzer et al., 2004).
These results imply that reduced tonic GABABR-mediated activation of the KIR (subsequent to Y2R activation) in PNs should depolarize dendrites and deactivate Ih. Such Ih deactivation would mitigate dendritic depolarization resulting from KIR inhibition, possibly accounting for the relatively modest [ahx5–24]NPY-mediated depolarizations seen in current clamp (although an electrotonically distant site likely also plays a role). Given that GABABRs and dendritic Ih shape integration of incoming excitatory inputs (Nolan et al., 2004; Otmakhova and Lisman, 2004), this has potential implications for signal processing and synaptic plasticity.
BLA PNs are not all disinhibited equally
Y2R activation led to potentiation of the Ca2+-dependent IsAHP K+ current in a subset of PNs, although other IK,Cas may also contribute. This Y2R-mediated postsynaptic effect was also frequently mimicked by GABABR antagonism, suggesting an ongoing inhibition of the IsAHP tonic by GABAB activity. These channels are expressed mainly on PN dendrites (Power et al., 2011). Dendritic GABABRs suppress local Ca2+ signaling, both by membrane hyperpolarization (Leung and Peloquin, 2006) and direct VGCC inhibition (Chalifoux and Carter, 2011; Pérez-Garci et al., 2013). The observed IsAHP enhancement is thus consistent with disinhibition of dendritic Ca2+ influx.
Using the Y2R-TdTomato reporter mouse, IsAHP-responsive PNs were identified to disproportionately express Y2R. Direct Y2R-mediated enhancement of IsAHP, although possible, is inconsistent with the typically presynaptic expression and inhibitory nature of Y2Rs, and with the occlusion of Y2R effects by GABABR antagonism. Likewise, our findings in rat suggest that reduced PN GABABR tone underlies IsAHP enhancement. Y2R expression might thus signify a functionally distinct subset of BLA PNs differentially responsive to reduced GABAB tone.
Depending on whether or not [ahx5–24]NPY potentiated the IsAHP, Y2R activation differentially affected firing patterns in a given PN. Specifically, where IsAHP was enhanced by Y2R activation, the interval between the first two APs of a train also increased substantially, and any initial spike doublets were suppressed. However, in PNs where GABABR-mediated K+ currents were reduced without affecting the IsAHP, the interval between the first two APs decreased significantly, resulting often in an initial AP doublet. In any case, throttling GABA release onto PN dendrites apparently reveals two populations of AP output responses.
Increasing evidence suggests BLA PNs are functionally heterogeneous. Nonoverlapping groups, differentiated by connectivity, mediate opposing regulation of fear-based behaviors (Tye et al., 2008; Namburi et al., 2015). Senn et al. (2014) showed that behavioral fear conditioning increases doublet firing in fear-coding BLA PNs, while reducing similar bursts in the extinction-coding PN population, the opposite effect followed extinction training (Senn et al., 2014). In PNs where [ahx5–24]NPY enhanced the IsAHP, increased excitability was offset, consistent with fear-encoding cells. If so, concurrent Y1R activation by NPY would help suppress output of these PNs. Conversely, numerous anxiogenic neuromodulators, including corticotrophin releasing factor inhibit the IsAHP (Rainnie et al., 1992; Womble and Moises, 1993; Power and Sah, 2008), potentially limiting the recruitment of this inhibitory current by Y2R-mediated enhancement of dendritic Ca2+ influx.
Implications for NPY-mediated plasticity
Repeated NPY infusions into the BLA induce long-term, anxiety-reducing effects mediated by the Ca2+-dependent phosphatase, calcineurin (Sajdyk et al., 2008). Likewise, NPY enhances conditioned fear extinction (Gutman et al., 2008), which also requires calcineurin (Lin et al., 2003). Calcineurin is expressed by BLA PNs (Leitermann et al., 2012) and localizes to dendritic spines in other PNs, including those of neocortex and hippocampus (Kuno et al., 1992; Colledge et al., 2000), where it mediates LTD (Mulkey et al., 1994; Torii et al., 1995). This suggests that NPY facilitates LTD-like plasticity in the BLA. By moderately enhancing dendritic Ca2+ influx, Y2R-inhibition of GABAB tone could facilitate calcineurin-mediated plasticity. Conversely, fear acquisition, and the long-term, elevated anxiety state induced by repeated BLA corticotrophin releasing factor infusions, require activation of Ca2+-calmodulin-dependent kinase type II (CaMKII) (Rainnie et al., 2004; Rodrigues et al., 2004). CaMKII also localizes to dendritic spines in pyramidal type neurons (Matsuzaki et al., 2004) but mediates LTP (Lisman et al., 2012). Calcineurin's higher Ca2+ affinity favors LTD induction by small rises in dendritic Ca2+; larger dendritic Ca2+ elevations preferentially recruit CaMKII and promote LTP (Mansuy, 2003). The shared dependence on dendritic Ca2+ both of anxiety-reducing and anxiety-provoking plasticity implies that GABABR-mediated dendritic inhibition must be constrained in both cases. Selective activation of the Y2R in isolation from the moderating actions of Y1Rs thus may explain its anxiety-provoking actions.
In conclusion, dendrites of BLA PNs receive strong, tonic, largely AP-independent, GABABR-mediated inhibition in vitro. Y2R-mediated suppression of dendritic GABA input relieves this GABABR tone, decreasing KIR currents and increasing PN excitability. In some cells, this was counterbalanced by a Ca2+-dependent increase in the IsAHP. Thus, Y2R activation elicits a widespread but nuanced influence on BLA circuitry. The increased dendritic Ca2+ entry permitted via this mechanism could mediate Ca2+-dependent plasticity, a likely component of NPY's long term anxiety-reducing actions (Lin et al., 2003; Sajdyk et al., 2008). Consequences of Y2R activation for PN dendrites likely contributes to NPY's acute and long-term anxiolytic actions and highlights PN dendrites as a site of NPY's actions within the BLA.
Footnotes
This work was supported by National Institutes of Health Grants MH081152 and MH090297 to J.H.U. and W.F.C. J.P.M. was supported by Alberta Innovates Health Solutions Doctoral studentship and Canadian Institute of Health Research Doctoral and Masters studentships. S.D.M. was supported by Alberta Innovates Health Solutions Doctoral studentship. W.F.C. was a Medical Scientist of the Alberta Heritage Foundation for Medical Research.
The authors declare no competing financial interests.
References
- Amano T, Unal CT, Paré D (2010) Synaptic correlates of fear extinction in the amygdala. Nat Neurosci 13:489–494. 10.1038/nn.2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Yehuda D, Korngreen A (2008) Space-clamp problems when voltage clamping neurons expressing voltage-gated conductances. J Neurophysiol 99:1127–1136. 10.1152/jn.01232.2007 [DOI] [PubMed] [Google Scholar]
- Bettler B, Kaupmann K, Mosbacher J, Gassmann M (2004) Molecular structure and physiological functions of GABA(B) receptors. Physiol Rev 84:835–867. 10.1152/physrev.00036.2003 [DOI] [PubMed] [Google Scholar]
- Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE (2001) Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem 8:229–242. 10.1101/lm.30901 [DOI] [PubMed] [Google Scholar]
- Blechert J, Michael T, Vriends N, Margraf J, Wilhelm FH (2007) Fear conditioning in posttraumatic stress disorder: evidence for delayed extinction of autonomic, experiential, and behavioural responses. Behav Res Ther 45:2019–2033. 10.1016/j.brat.2007.02.012 [DOI] [PubMed] [Google Scholar]
- Bucchi A, Tognati A, Milanesi R, Baruscotti M, DiFrancesco D (2006) Properties of ivabradine-induced block of HCN1 and HCN4 pacemaker channels. J Physiol 572:335–346. 10.1113/jphysiol.2005.100776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno CH, Zangrossi H Jr, Viana MB (2005) The inactivation of the basolateral nucleus of the rat amygdala has an anxiolytic effect in the elevated T-maze and light/dark transition tests. Braz J Med Biol Res 38:1697–1701. 10.1590/S0100-879X2005001100019 [DOI] [PubMed] [Google Scholar]
- Capogna M. (2014) GABAergic cell type diversity in the basolateral amygdala. Curr Opin Neurobiol 26:110–116. 10.1016/j.conb.2014.01.006 [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG (2011) GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. J Neurosci 31:4221–4232. 10.1523/JNEUROSCI.4561-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, DiMaggio DA, Han SP, Westfall TC (1997) Autoreceptor-induced inhibition of neuropeptide Y release from PC-12 cells is mediated by Y2 receptors. Am J Physiol 273:H1737–H1744. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD (2000) Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron 27:107–119. 10.1016/S0896-6273(00)00013-1 [DOI] [PubMed] [Google Scholar]
- Colmers WF, Bleakman D (1994) Effects of neuropeptide Y on the electrical properties of neurons. Trends Neurosci 17:373–379. 10.1016/0166-2236(94)90046-9 [DOI] [PubMed] [Google Scholar]
- Day M, Carr DB, Ulrich S, Ilijic E, Tkatch T, Surmeier DJ (2005) Dendritic excitability of mouse frontal cortex pyramidal neurons is shaped by the interaction among HCN, Kir2, and Kleak channels. J Neurosci 25:8776–8787. 10.1523/JNEUROSCI.2650-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bahh B, Cao JQ, Beck-Sickinger AG, Colmers WF (2002) Blockade of neuropeptide Y(2) receptors and suppression of NPY's anti-epileptic actions in the rat hippocampal slice by BIIE0246. Br J Pharmacol 136:502–509. 10.1038/sj.bjp.0704751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bahh B, Balosso S, Hamilton T, Herzog H, Beck-Sickinger AG, Sperk G, Gehlert DR, Vezzani A, Colmers WF (2005) The anti-epileptic actions of neuropeptide Y in the hippocampus are mediated by Y and not Y receptors. Eur J Neurosci 22:1417–1430. 10.1111/j.1460-9568.2005.04338.x [DOI] [PubMed] [Google Scholar]
- Erondu NE, Kennedy MB (1985) Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J Neurosci 5:3270–3277. 10.1523/JNEUROSCI.05-12-03270.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetissov SO, Byrne LC, Hassani H, Ernfors P, Hökfelt T (2004) Characterization of neuropeptide Y Y2 and Y5 receptor expression in the mouse hypothalamus. J Comp Neurol 470:256–265. 10.1002/cne.11047 [DOI] [PubMed] [Google Scholar]
- Fritsch B, Qashu F, Figueiredo TH, Aroniadou-Anderjaska V, Rogawski MA, Braga MF (2009) Pathological alterations in GABAergic interneurons and reduced tonic inhibition in the basolateral amygdala during epileptogenesis. Neuroscience 163:415–429. 10.1016/j.neuroscience.2009.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller TA, Price JL (1988) Putative glutamatergic and/or aspartatergic cells in the main and accessory olfactory bulbs of the rat. J Comp Neurol 276:209–218. 10.1002/cne.902760205 [DOI] [PubMed] [Google Scholar]
- Giesbrecht CJ, Mackay JP, Silveira HB, Urban JH, Colmers WF (2010) Countervailing modulation of Ih by neuropeptide Y and corticotrophin-releasing factor in basolateral amygdala as a possible mechanism for their effects on stress-related behaviors. J Neurosci 30:16970–16982. 10.1523/JNEUROSCI.2306-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JC, Epps SA, Markwardt SJ, Wadiche JI, Overstreet-Wadiche L (2018) Constitutive and synaptic activation of GIRK channels differentiates mature and newborn dentate granule cells. J Neurosci 38:6513–6526. 10.1523/JNEUROSCI.0674-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber S, Schwarzer C, Sperk G (1994) Neuropeptide Y inhibits potassium-stimulated glutamate release through Y2 receptors in rat hippocampal slices in vitro. Br J Pharmacol 113:737–740. 10.1111/j.1476-5381.1994.tb17055.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greif GJ, Sodickson DL, Bean BP, Neer EJ, Mende U (2000) Altered regulation of potassium and calcium channels by GABA(B) and adenosine receptors in hippocampal neurons from mice lacking Galpha(o). J Neurophysiol 83:1010–1018. 10.1152/jn.2000.83.2.1010 [DOI] [PubMed] [Google Scholar]
- Gutman AR, Yang Y, Ressler KJ, Davis M (2008) The role of neuropeptide Y in the expression and extinction of fear-potentiated startle. J Neurosci 28:12682–12690. 10.1523/JNEUROSCI.2305-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A (2008) Switching on and off fear by distinct neuronal circuits. Nature 454:600–606. 10.1038/nature07166 [DOI] [PubMed] [Google Scholar]
- Kemppainen S, Pitkänen A (2000) Distribution of parvalbumin, calretinin, and calbindin-D28k immunoreactivity in the rat amygdaloid complex and colocalization with gamma-aminobutyric acid. J Comp Neurol 426:441–467. [DOI] [PubMed] [Google Scholar]
- Kohl MM, Paulsen O (2010) The roles of GABAB receptors in cortical network activity. Adv Pharmacol 58:205–229. 10.1016/S1054-3589(10)58009-8 [DOI] [PubMed] [Google Scholar]
- Kröner S, Rosenkranz JA, Grace AA, Barrionuevo G (2005) Dopamine modulates excitability of basolateral amygdala neurons in vitro. J Neurophysiol 93:1598–1610. 10.1152/jn.00843.2004 [DOI] [PubMed] [Google Scholar]
- Kulik A, Vida I, Luján R, Haas CA, López-Bendito G, Shigemoto R, Frotscher M (2003) Subcellular localization of metabotropic GABA(B) receptor subunits GABA(B1a/b) and GABA(B2) in the rat hippocampus. J Neurosci 23:11026–11035. 10.1523/JNEUROSCI.23-35-11026.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno T, Mukai H, Ito A, Chang CD, Kishima K, Saito N, Tanaka C (1992) Distinct cellular expression of calcineurin A alpha and A beta in rat brain. J Neurochem 58:1643–1651. 10.1111/j.1471-4159.1992.tb10036.x [DOI] [PubMed] [Google Scholar]
- Kuzhikandathil EV, Oxford GS (2002) Classic D1 dopamine receptor antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) directly inhibits G protein-coupled inwardly rectifying potassium channels. Mol Pharmacol 62:119–126. 10.1124/mol.62.1.119 [DOI] [PubMed] [Google Scholar]
- Lalumiere RT. (2014) Optogenetic dissection of amygdala functioning. Front Behav Neurosci 8:107. 10.3389/fnbeh.2014.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitermann RJ, Sajdyk TJ, Urban JH (2012) Cell-specific expression of calcineurin immunoreactivity within the rat basolateral amygdala complex and colocalization with the neuropeptide Y Y1 receptor. J Chem Neuroanat 45:50–56. 10.1016/j.jchemneu.2012.07.005 [DOI] [PubMed] [Google Scholar]
- Leung LS, Peloquin P (2006) GABA(B) receptors inhibit backpropagating dendritic spikes in hippocampal CA1 pyramidal cells in vivo. Hippocampus 16:388–407. 10.1002/hipo.20168 [DOI] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Popescu AT, Paré D (2006) Identification of basolateral amygdala projection cells and interneurons using extracellular recordings. J Neurophysiol 96:3257–3265. 10.1152/jn.00577.2006 [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Leu TH, Chang WC, Wang ST, Gean PW (2003) Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci 23:1574–1579. 10.1523/JNEUROSCI.23-05-01574.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S (2012) Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci 13:169–182. 10.1038/nrn3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissek S, Powers AS, McClure EB, Phelps EA, Woldehawariat G, Grillon C, Pine DS (2005) Classical fear conditioning in the anxiety disorders: a meta-analysis. Behav Res Ther 43:1391–1424. 10.1016/j.brat.2004.10.007 [DOI] [PubMed] [Google Scholar]
- Mahanty NK, Sah P (1998) Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 394:683–687. 10.1038/29312 [DOI] [PubMed] [Google Scholar]
- Mansuy IM. (2003) Calcineurin in memory and bidirectional plasticity. Biochem Biophys Res Commun 311:1195–1208. 10.1016/j.bbrc.2003.10.046 [DOI] [PubMed] [Google Scholar]
- Maren S. (1999) Neurotoxic basolateral amygdala lesions impair learning and memory but not the performance of conditional fear in rats. J Neurosci 19:8696–8703. 10.1523/JNEUROSCI.19-19-08696.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascagni F, McDonald AJ (2003) Immunohistochemical characterization of cholecystokinin containing neurons in the rat basolateral amygdala. Brain Res 976:171–184. 10.1016/S0006-8993(03)02625-8 [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. 10.1038/nature02617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ. (1989) Coexistence of somatostatin with neuropeptide Y, but not with cholecystokinin or vasoactive intestinal peptide, in neurons of the rat amygdala. Brain Res 500:37–45. 10.1016/0006-8993(89)90297-7 [DOI] [PubMed] [Google Scholar]
- McDonald AJ. (1996) Glutamate and aspartate immunoreactive neurons of the rat basolateral amygdala: colocalization of excitatory amino acids and projections to the limbic circuit. J Comp Neurol 365:367–379. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Betette RL (2001) Parvalbumin-containing neurons in the rat basolateral amygdala: morphology and co-localization of calbindin-D28k. Neuroscience 102:413–425. 10.1016/S0306-4522(00)00481-4 [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F (2001) Colocalization of calcium-binding proteins and GABA in neurons of the rat basolateral amygdala. Neuroscience 105:681–693. 10.1016/S0306-4522(01)00214-7 [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F (2002) Immunohistochemical characterization of somatostatin containing interneurons in the rat basolateral amygdala. Brain Res 943:237–244. 10.1016/S0006-8993(02)02650-1 [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Beitz AJ, Larson AA, Kuriyama R, Sellitto C, Madl JE (1989) Co-localization of glutamate and tubulin in putative excitatory neurons of the hippocampus and amygdala: an immunohistochemical study using monoclonal antibodies. Neuroscience 30:405–421. 10.1016/0306-4522(89)90261-3 [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F, Muller JF (2004) Immunocytochemical localization of GABABR1 receptor subunits in the basolateral amygdala. Brain Res 1018:147–158. 10.1016/j.brainres.2004.05.053 [DOI] [PubMed] [Google Scholar]
- Molosh AI, Sajdyk TJ, Truitt WA, Zhu W, Oxford GS, Shekhar A (2013) NPY Y1 receptors differentially modulate GABAA and NMDA receptors via divergent signal-transduction pathways to reduce excitability of amygdala neurons. Neuropsychopharmacology 38:1352–1364. 10.1038/npp.2013.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC (1994) Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369:486–488. 10.1038/369486a0 [DOI] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ (2007) Postsynaptic targets of somatostatin-containing interneurons in the rat basolateral amygdala. J Comp Neurol 500:513–529. 10.1002/cne.21185 [DOI] [PubMed] [Google Scholar]
- Nakajima M, Inui A, Asakawa A, Momose K, Ueno N, Teranishi A, Baba S, Kasuga M (1998) Neuropeptide Y produces anxiety via Y2 type receptors. Peptides 19:359–363. 10.1016/S0196-9781(97)00298-2 [DOI] [PubMed] [Google Scholar]
- Namburi P, Beyeler A, Yorozu S, Calhoon GG, Halbert SA, Wichmann R, Holden SS, Mertens KL, Anahtar M, Felix-Ortiz AC, Wickersham IR, Gray JM, Tye KM (2015) A circuit mechanism for differentiating positive and negative associations. Nature 520:675–678. 10.1038/nature14366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A (2004) A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119:719–732. 10.1016/j.cell.2004.11.020 [DOI] [PubMed] [Google Scholar]
- Olpe HR, Steinmann MW, Ferrat T, Pozza MF, Greiner K, Brugger F, Froestl W, Mickel SJ, Bittiger H (1993) The actions of orally active GABAB receptor antagonists on GABAergic transmission in vivo and in vitro. Eur J Pharmacol 233:179–186. 10.1016/0014-2999(93)90048-M [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE (2004) Contribution of Ih and GABAB to synaptically induced afterhyperpolarizations in CA1: a brake on the NMDA response. J Neurophysiol 92:2027–2039. 10.1152/jn.00427.2004 [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y (1998) Intrinsic circuitry of the amygdaloid complex: common principles of organization in rats and cats. Trends Neurosci 21:240–241. 10.1016/S0166-2236(98)01240-5 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K (2012) The mouse brain in stereotaxic coordinates, Ed 4 San Diego: Academic Press. [Google Scholar]
- Paxinos G, Watson C, (2007) The rat brain in stereotaxic coordinates, Ed 6 San Diego:Academic Press. [Google Scholar]
- Pérez-Garci E, Larkum ME, Nevian T (2013) Inhibition of dendritic Ca2+ spikes by GABAB receptors in cortical pyramidal neurons is mediated by a direct Gi/o-β-subunit interaction with Cav1 channels. J Physiol 591:1599–1612. 10.1113/jphysiol.2012.245464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P (2008) Competition between calcium-activated K+ channels determines cholinergic action on firing properties of basolateral amygdala projection neurons. J Neurosci 28:3209–3220. 10.1523/JNEUROSCI.4310-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Bocklisch C, Curby P, Sah P (2011) Location and function of the slow afterhyperpolarization channels in the basolateral amygdala. J Neurosci 31:526–537. 10.1523/JNEUROSCI.1045-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozza MF, Manuel NA, Steinmann M, Froestl W, Davies CH (1999) Comparison of antagonist potencies at pre- and post-synaptic GABA(B) receptors at inhibitory synapses in the CA1 region of the rat hippocampus. Br J Pharmacol 127:211–219. 10.1038/sj.bjp.0702498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prater KE, Hosanagar A, Klumpp H, Angstadt M, Phan KL (2013) Aberrant amygdala-frontal cortex connectivity during perception of fearful faces and at rest in generalized social anxiety disorder. Depress Anxiety 30:234–241. 10.1002/da.22014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG. (1999) Serotonergic modulation of neurotransmission in the rat basolateral amygdala. J Neurophysiol 82:69–85. 10.1152/jn.1999.82.1.69 [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Fernhout BJ, Shinnick-Gallagher P (1992) Differential actions of corticotropin releasing factor on basolateral and central amygdaloid neurones, in vitro. J Pharmacol Exp Ther 263:846–858. [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P (1993) Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol 69:1350–1362. 10.1152/jn.1993.69.4.1350 [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A (2004) Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci 24:3471–3479. 10.1523/JNEUROSCI.5740-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE (2004) Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci 24:3281–3288. 10.1523/JNEUROSCI.5303-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Stäubli UV, LeDoux JE (1997) Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390:604–607. 10.1038/37601 [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA (1999) Modulation of basolateral amygdala neuronal firing and afferent drive by dopamine receptor activation in vivo. J Neurosci 19:11027–11039. 10.1523/JNEUROSCI.19-24-11027.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostkowski AB, Teppen TL, Peterson DA, Urban JH (2009) Cell-specific expression of neuropeptide Y Y1 receptor immunoreactivity in the rat basolateral amygdala. J Comp Neurol 517:166–176. 10.1002/cne.22143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah R, Ekhator NN, Strawn JR, Sallee FR, Baker DG, Horn PS, Geracioti TD Jr (2009) Low cerebrospinal fluid neuropeptide Y concentrations in posttraumatic stress disorder. Biol Psychiatry 66:705–707. 10.1016/j.biopsych.2009.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajdyk TJ, Shekhar A (1997) Excitatory amino acid receptors in the basolateral amygdala regulate anxiety responses in the social interaction test. Brain Res 764:262–264. 10.1016/S0006-8993(97)00594-5 [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Vandergriff MG, Gehlert DR (1999) Amygdalar neuropeptide Y Y1 receptors mediate the anxiolytic-like actions of neuropeptide Y in the social interaction test. Eur J Pharmacol 368:143–147. 10.1016/S0014-2999(99)00018-7 [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Schober DA, Smiley DL, Gehlert DR (2002) Neuropeptide Y-Y2 receptors mediate anxiety in the amygdala. Pharmacol Biochem Behav 71:419–423. 10.1016/S0091-3057(01)00679-7 [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Johnson PL, Leitermann RJ, Fitz SD, Dietrich A, Morin M, Gehlert DR, Urban JH, Shekhar A (2008) Neuropeptide Y in the amygdala induces long-term resilience to stress-induced reductions in social responses but not hypothalamic-adrenal-pituitary axis activity or hyperthermia. J Neurosci 28:893–903. 10.1523/JNEUROSCI.0659-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M. (2000) GABA spillover activates postsynaptic GABA(B) receptors to control rhythmic hippocampal activity. Neuron 25:673–681. 10.1016/S0896-6273(00)81069-7 [DOI] [PubMed] [Google Scholar]
- Schweitzer P, Roberto M, Madamba SG, Siggins GR (2004) Gamma-hydroxybutyrate increases a potassium current and decreases the H-current in hippocampal neurons via GABAB receptors. J Pharmacol Exp Ther 311:172–179. 10.1124/jpet.104.069534 [DOI] [PubMed] [Google Scholar]
- Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Gründemann J, Fadok JP, Müller C, Letzkus JJ, Lüthi A (2014) Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 81:428–437. 10.1016/j.neuron.2013.11.006 [DOI] [PubMed] [Google Scholar]
- Shah MM, Javadzadeh-Tabatabaie M, Benton DC, Ganellin CR, Haylett DG (2006) Enhancement of hippocampal pyramidal cell excitability by the novel selective slow-afterhyperpolarization channel blocker 3-(triphenylmethylaminomethyl)pyridine (UCL2077). Mol Pharmacol 70:1494–1502. 10.1124/mol.106.026625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar A, Sajdyk TS, Keim SR, Yoder KK, Sanders SK (1999) Role of the basolateral amygdala in panic disorder. Ann N Y Acad Sci 877:747–750. 10.1111/j.1749-6632.1999.tb09315.x [DOI] [PubMed] [Google Scholar]
- Silveira Villarroel H, Bompolaki M, Mackay JP, Miranda Tapia AP, Michaelson SD, Leitermann RJ, Marr RA, Urban JH, Colmers WF (2018) NPY induces stress resilience via downregulation of Ih in principal neurons of rat basolateral amygdala. J Neurosci 38:4505–4520. 10.1523/JNEUROSCI.3528-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BN, Dudek FE (1996) Amino acid-mediated regulation of spontaneous synaptic activity patterns in the rat basolateral amygdala. J Neurophysiol 76:1958–1967. 10.1152/jn.1996.76.3.1958 [DOI] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP (1998) Neurotransmitter activation of inwardly rectifying potassium current in dissociated hippocampal CA3 neurons: interactions among multiple receptors. J Neurosci 18:8153–8162. 10.1523/JNEUROSCI.18-20-08153.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosulina L, Schwesig G, Seifert G, Pape HC (2008) Neuropeptide Y activates a G-protein-coupled inwardly rectifying potassium current and dampens excitability in the lateral amygdala. Mol Cell Neurosci 39:491–498. 10.1016/j.mcn.2008.08.002 [DOI] [PubMed] [Google Scholar]
- Stanić D, Brumovsky P, Fetissov S, Shuster S, Herzog H, Hökfelt T (2006) Characterization of neuropeptide Y2 receptor protein expression in the mouse brain: I. Distribution in cell bodies and nerve terminals. J Comp Neurol 499:357–390. 10.1002/cne.21046 [DOI] [PubMed] [Google Scholar]
- Torii N, Kamishita T, Otsu Y, Tsumoto T (1995) An inhibitor for calcineurin, FK506, blocks induction of long-term depression in rat visual cortex. Neurosci Lett 185:1–4. 10.1016/0304-3940(94)11210-A [DOI] [PubMed] [Google Scholar]
- Tye KM, Stuber GD, de Ridder B, Bonci A, Janak PH (2008) Rapid strengthening of thalamo-amygdala synapses mediates cue-reward learning. Nature 453:1253–1257. 10.1038/nature06963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Miles LA, Davis M (2009) Selective participation of the bed nucleus of the stria terminalis and CRF in sustained anxiety-like versus phasic fear-like responses. Prog Neuropsychopharmacol Biol Psychiatry 33:1291–1308. 10.1016/j.pnpbp.2009.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Neubauer FB, Lüscher HR, Thurley K (2010) GABAB receptor-dependent modulation of network activity in the rat prefrontal cortex in vitro. Eur J Neurosci 31:1582–1594. 10.1111/j.1460-9568.2010.07191.x [DOI] [PubMed] [Google Scholar]
- Washburn MS, Moises HC (1992) Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci 12:4066–4079. 10.1523/JNEUROSCI.12-10-04066.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ (2008) Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci 11:790–798. 10.1038/nn.2137 [DOI] [PubMed] [Google Scholar]
- Wolak ML, DeJoseph MR, Cator AD, Mokashi AS, Brownfield MS, Urban JH (2003) Comparative distribution of neuropeptide Y Y1 and Y5 receptors in the rat brain by using immunohistochemistry. J Comp Neurol 464:285–311. 10.1002/cne.10823 [DOI] [PubMed] [Google Scholar]
- Wolff SB, Gründemann J, Tovote P, Krabbe S, Jacobson GA, Müller C, Herry C, Ehrlich I, Friedrich RW, Letzkus JJ, Lüthi A (2014) Amygdala interneuron subtypes control fear learning through disinhibition. Nature 509:453–458. 10.1038/nature13258 [DOI] [PubMed] [Google Scholar]
- Womble MD, Moises HC (1993) Hyperpolarization-activated currents in neurons of the rat basolateral amygdala. J Neurophysiol 70:2056–2065. 10.1152/jn.1993.70.5.2056 [DOI] [PubMed] [Google Scholar]
- Yehuda R, Brand S, Yang RK (2006) Plasma neuropeptide Y concentrations in combat exposed veterans: relationship to trauma exposure, recovery from PTSD, and coping. Biol Psychiatry 59:660–663. 10.1016/j.biopsych.2005.08.027 [DOI] [PubMed] [Google Scholar]