

Abstract
Reference materials are vital to benchmarking the reproducibility of clinical tests and essential for monitoring laboratory performance for clinical proteomics. The reference material utilized for mass spectrometric analysis of the human proteome would ideally contain enough proteins to be suitably representative of the human proteome, as well as exhibit a stable protein composition in different batches of sample regeneration. Previously, The Clinical Proteomic Tumor Analysis Consortium (CPTAC) utilized a PDX-derived comparative reference (CompRef) materials for the longitudinal assessment of proteomic performance; however, inherent drawbacks of PDX-derived material, including extended time needed to grow tumors and high level of expertise needed, have resulted in efforts to identify a new source of CompRef material. In this study, we examined the utility of using a panel of seven cancer cell lines, NCI-7 Cell Line Panel, as a reference material for mass spectrometric analysis of human proteome. Our results showed that not only is the NCI-7 material suitable for benchmarking laboratory sample preparation methods, but also NCI-7 sample generation is highly reproducible at both the global and phosphoprotein levels. In addition, the predicted genomic and experimental coverage of the NCI-7 proteome suggests the NCI-7 material may also have applications as a universal standard proteomic reference.
Keywords: proteomics reference material, clinical proteomics, mass spectrometry
Graphical Abstract
INTRODUCTION
The clinical proteomic analysis of tumor specimens seeks to improve our understanding of cancer via the integration of genomic information with tumor proteome characterization. Building upon The Cancer Genome Atlas (TCGA) initiative, which sought to identify genomic alterations in multiple tumor types, the Clinical Proteomic Tumor Analysis Consortium (CPTAC) leverages highly innovative proteomic approaches and advanced mass spectrometry instrumentation to comprehensively profile differences in protein expression and protein post-translational modifications, among individual tumors and nontumor tissue samples. Utilizing a proteogenomic approach, linking protein and genomic information, the analysis of colorectal, ovarian, and breast cancers has yielded novel observations in the biology of these respective tumors.1–3
To evaluate the performance of mass spectrometry analysis at Proteome Characterization Centers (PCCs), as well as longitudinally monitor workflow performance, a comparative reference material (“CompRef’) was generated using patient-derived breast cancer tumor xenografts (PDXs). In a previous study, this CompRef material was utilized to examine intralab and cross-center reproducibility of data generation across six different platforms employing distinct mass spectrometry instrumentation and/or protein quantitation strategies.4,5 In addition, in the CPTAC studies examining colorectal, ovarian, and breast cancer, respectively, the xenograft CompRef material was interspersed during clinical tumor analysis as reference standard.6 Although PDXs models are highly representative of the tumor’s heterogeneity and microenvironment,7 implantation requires a high level of technical skill, and tumor growth can be slow.8 In addition, the model animal tumor burden can limit the total tumor size, which can impact overall tumor tissue yield. This latter aspect is especially concerning in the context of generating a sufficient amount of material as a standard reference for multicenter analysis pipelines, and may require the sacrifice of a large number of animals.
The NCI-60 Cell Line Panel was initially established as an in vitro drug-discovery screening platform,9 and more recently the molecular characteristics of the individual cell lines in the panel, including, gene mutation analysis,10 whole exome sequencing,11 methylation patterns,12 mRNA transcription profiles,13 and protein expression,14,15 have been examined. To facilitate access to these large data sets for the wider scientific community, the web-based database annotation tool CellMiner was developed,16 and recently expanded to include web-based tools that allow researchers to integrate these genomic, transcriptomic, protein, and pharmacological profiles.17 Over the past few decades, the NCI-60 Cell Line Panel has been an invaluable resource of the oncology research community, identifying novel anticancer compounds, in addition to its contributions to our understanding of cancer biology.18 Ironically, the longevity of the NCI-60 Cell Line Panel has impacted its future success in drug screening, as the cell lines have adapted to cell culturing and led some researchers to question the panel’s future role in drug discovery, with PDXs models suggested to be more reflective of in vivo tumor response.9 Still, researchers have continued to develop novel strategies to examine the NCI-60 Cell Line Panel, in addition to other cell lines, illustrating additional benefits of using these cell line models in cancer-related studies.19,20
Reference material derived from cell culture models, such as the NCI-60 Cell Line Panel, has the potential to address some of the previous cited drawbacks of PDX models, specifically growth time and tumor tissue yield, in addition to being more economical in terms of expense.21–23 A single cancer cell line may not be sufficient to reflect the protein profile of a tumor in it is entirely, thus utilizing multiple cell lines from a variety of tissue types would allow for extensive coverage of protein expression. At the other extreme, the use of the entire NCI-60 panel as a reference material would be complex and impractical. To determine whether a subset of cell lines selected from the NCI-60 Tumor Cell Line can be used as a new reference material to evaluate proteomic performance, we employed quantitative proteomics to evaluate sample preparation and mass spectrometry analysis reproducibility, as well as examining the coverage of the human cancer proteome using the NCI-7 Cell Line Panel.
METHODS
Chemicals and Materials
Chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless specified otherwise. C18 analytical LC column (Nano-Viper, 75 μm, 150 mm, 2 μm particle size) were from Fisher Scientific (Waltham, MA). C18 SepPak columns were purchased from Waters Corporation (Milford, MA), Trypsin and Trypsin/Lys-C Mix (mass spectrometry (MS) grade) was ordered from Promega Corporation (Madison, WI), and Lys-C was from Wako Laboratory Chemicals, USA (Richmond, VA). All cell lines (A549, COLO205, NCI H226, NCI H23, T-47D, CCRF-CEM and RPMI 8226) were purchased from American Type Culture Collection (ATCC, Manassas, VA). RPMI 1640 media and heat inactivated FBS were from Life Technologies (Grand Island, NY); L-Glutamine was from Lonza (Walkers-ville, MD).
Cell Culturing
All cell lines were grown in RPMI 1640 medium supplemented with 10% HI FBS, and 1% L-Glutamine in a humidified air incubator with 5% CO2 at 37 °C. All cells lines were seeded on T75 flask and grown to 90% confluence before being transferred to a larger T150 flask and maintained.
Generation of NCI-7 Reference Material
All cell lines grown to 90% confluence, washed with a volume of PBS twice, scrapped, collected and centrifuged at 3000g. Individual cell pellets were resuspended in lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris, pH 8.0, 1 mM EDTA, 2 μg/mL aprotinin, 10 μg mL leupeptin, 1 mM PMSF, 10 mM NaF, Phosphatase Inhibitor Cocktail 2 and Phosphatase Inhibitor Cocktail 3 (1:100 dilution), and 20 μM PUGNAc), and homogenized using a Branson Sonifier 250 probe sonicator (Danbury, CT) with a Duty Cycle of 10%, Output Control 1 with a 10 s/50 s ON/OFF cycle (n = 5). Samples were centrifuged at 20 000g and the resulting supernatant retained, and protein concentration was measured via BCA Protein Assay. One mg of protein from each cell line was mixed to generate the NCI-7 reference material.
Generation of PDX (P32/P33) CompRef Material
Growth and preparation of PDX material has been described previously.1,2 Cryopulverized tissue was resuspended in lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 2μg/mL aprotinin, 10 μg mL leupeptin, 1 mM PMSF, 10 mM NaF, Phosphatase Inhibitor Cocktail 2 and Phosphatase Inhibitor Cocktail 3 (1:100 dilution), and 20 μM PUGNAc), and homogenized using a Branson Sonifier 250 probe sonicator (Danbury, CT) with a Duty Cycle of 10%, Output Control 1 with a 10 s/50 s ON/OFF cycle (n = 5). Samples were centrifuged at 20 000g and the resulting supernatant retained, and protein concentration was measured via BCA Protein Assay.
Peptide Digestion, Tandem-Mass-Tag Labeling, and Off-Line Fractionation
Both NCI-7 cells and PDX CompRef protein lysates were subjected to the same digestion, Tandem-Mass-Tag (TMT) labeling, and off-line fractionation conditions unless otherwise noted. Protein lysates were subjected to reduction with 5 mM 1,4-Dithiothreitol for 30 min at RT, followed by alkylation with 10 mM iodoacetamide for 45 min at RT in the dark. Urea concentration was reduced <2 M using 50 mM Tris-HCl, pH 8.0. For NCI-7 samples, LysC/Trypsin was added in an enzyme to substrate ratio of 1:40 and samples incubated overnight at 37 °C, while PDX CompRef samples were subjected to tandem digestion of Lys-C (1:50) for 2 h at RT followed by trypsin (1:50) overnight at RT. The generated peptides were acidified to a final concentration of 1% formic acid, subjected to cleanup using C-18 SepPak columns (Waters), and then dried. Desalted peptides were labeled with 10-plex TMT reagents following manufacturer’s instructions. Peptides were resuspended in 50 mM HEPES, pH 8.5 and labeled with TMT reagent resuspended in anhydrous acetonitrile for 1 h at RT. The reaction was quenched using 5% hydroxylamine at RT for 15 min, and differentially labeled samples combined. The pooled peptides were then subjected to cleanup using C-18 SepPak columns and dried down. The desalted, TMT-labeled samples were reconstituted in a volume of 20 mM ammonium formate (pH 10) and 2% acetonitrile (ACN) and loaded onto a 4.6 mm × 250 mm RP Zorbax 300 A Extend-C18 column with 3.5 μm size beads (Agilent). Peptides were separated using an Agilent 1200 Series HPLC instrument using basic reversed-phase chromatography with Solvent A (2% ACN, 5 mM ammonium formate, pH 10) and a nonlinear gradient of Solvent B (90% ACN, 5 mM ammonium formate, pH 10) at 1 mL/min as follows: 0% Solvent B (9 min), 6% Solvent B (4 min), 6% to 28.5% Solvent B, (50 min), 28% to 34% Solvent B (5.5 min), 34% to 60% Solvent B (13 min), and then held at 60% Solvent B for 8.5 min. Collected fractions were concatenated as reported previously;24 5% of each the 24 fractions was aliquoted for global proteomic analysis, dried down, and resuspended in 3% ACN, 0.1% formic acid prior to ESI-LC–MS/MS analysis. The remaining sample was utilized for phosphopeptide enrichment.
Phosphopeptide Enrichment
The remaining 95% of the sample were further concatenated prior to phosphopeptide enrichment using immobilized metal affinity chromatography (IMAC) as previously described.24 In brief, N-NTA agarose beads were utilized to prepare Fe3+-NTA agarose beads, and then 300 μg of peptides reconstituted in 80% ACN/0.1% trifluoroacetic acid were incubated with 10 μL of the Fe3+-IMAC beads for 30 min. Sample were then spun down and the supernatant containing unbound peptides was removed. The beads were washed twice and then loaded onto equilibrated C-18 Stage Tips with 80% ACN, 0.1% trifluoroacetic acid. Tips were rinsed twice with 1% formic acid, followed by sample elution off the Fe3+-IMAC beads and onto the C-18 State Tips with 70 μL of 500 mM dibasic potassium phosphate, pH 7.0 three times. C-18 Stage Tips were washed twice with 1% formic acid, followed by elution of the phosphopeptides from the C-18 Stage Tips with 50% ACN, 0.1% formic acid twice. Samples were dried down and resuspended in 3% ACN, 0.1% formic acid prior to ESI-LC–MS/MS analysis.
Nano-ESI-LC-MS/MS Analysis
As noted in the manuscript, two ESI-LC–MS/MS systems were used. For samples run on the Q Exactive HF system, 1 μg of peptide was separated utilizing a nanoACQUITY UPLC system (Waters) on an in-house packed 20 cm × 75 μm diameter C18 column (5 μm ProntoSil C-18-AQbeads (Bishoff Chromatography); Picofrit 10 μm opening (New Objective)). The column was heated to 50 °C using a column heater (Phoenix-ST). The flow rate was 0.300 μL/min with 0.1% formic acid in water (A) and 0.1% formic acid, 90% acetonitrile (B). The peptides were separated with an 8–45% B gradient in 70 min and analyzed using the Thermo Q Exactive HF mass spectrometer (Thermo Scientific). Parameters were as followed MS1: resolution, 120 000; mass range, 350–1800 m/z; AGC Target 3.0 × 106; Max IT, 50 ms; charge state include 2–6; dynamic exclusion, 15 s; top 15 ions selected for MS2. MS2: resolution, 60 000; high-energy collision dissociation activation energy (HCD), 32; isolation width (m/z), 1.0; AGC Target, 1.0 × 105; Max IT, 150 ms. For samples run on the Orbitrap Lumos Fusion system, 1 μg of peptide was separated using Easy nLC 1200 UHPLC system (Thermo Scientific) on an in-house packed 20 cm × 75 μm diameter C18 column (1.9 μm Reprosil-Pur C18-AQbeads (Dr. Maisch GmbH); Picofrit 10 Um opening (New Objective)). The column was heated to 50 °C using a column heater (Phoenix-ST). The flow rate was 0.300 μL/min with 0.1% formic acid and 2% acetonitrile in water (A) and 0.1% formic acid, 90% acetonitrile (B). The peptides were separated with a 6–30% B gradient in 84 min and analyzed using the Thermo Fusion Lumos mass spectrometer (Thermo Scientific). Parameters were as followed MS1: resolution, 60 000; mass range, 350–1800 m/z; RF Lens, 30%; AGC Target 4.0 × 105; Max IT, 50 ms; charge state include 2–6; dynamic exclusion, 45 s; top 20 ions selected for MS2. MS2: resolution, 50 000; high-energy collision dissociation activation energy (HCD), 37; isolation width (m/z), 0.7; AGC Target, 2.0 × 105; Max IT, 105 ms.
Determination of Gene Expression of NCI-60 and NCI-7 Cell Lines
The gene expression of NCI-60 Cell Lines was determined by the transcript data sets download from the publicly available CellMiner Web site (https://discover.nci.nih.gov/cellminer/home.do). The RNA_5_Platform_Gene_Transcript_Average_intensities sheet was selected to be applied in the following analysis. For each cell line, the 25 percentile value of intensities of all the genes across all cell lines was used as a threshold to filter and reserving only the relatively strong signals of mRNA expression (Table S1 and S2). A minimal set of cell lines having most gene IDs represented from the whole genome was generated by a greedy algorithm of calculating the best gene coverage iteratively. After selection of the first ranked cell line, the algorithm added additional cell lines into the set based on the number of uncovered genes compared to all other remaining cells lines until gene coverage was 100% or beyond a predefined threshold (Table S3). Coverage for the selected cell lines for the NCI-7 Cell Line Panel was determined by identifying the gene expression of the individual cell lines using the same filtering criteria (Table S4), divided by the total number of reported genes in the human genome and expressed genes in the NCI-60 Cell Lines (Table S5). To facilitate identifying the coverage of the NCI-7 proteome, the Entrez Gene ID for the human, NCI-60, and NCI-7 genomes were converted to RefSeq IDs using the bioinformatic software tool DAVID (Database for Annotation and Integrated Discovery) (v 6.8).25,26
Data Analysis for Protein Identification and Quantification
All LC–MS/MS files were analyzed by a cloud-based proteomic pipeline developed in Johns Hopkins University to perform database search for spectrum assignments using MS-GF+ in this study against a combined human and mouse RefSeq database (version 20160914).27,28 A decoy database was used to assess the false discovery rate (FDR) at PSM, peptide and protein levels.29 Peptides were searched with two tryptic ends, allowing up to two missed cleavages. Search parameters included 20 ppm precursor tolerance and 0.06 Da fragment ion tolerance, static modification of carbamidomethylation at cysteine (+57.02146), TMT-label modification of N-terminus and lysine (+229.16293) and variable modifications of oxidation at methionine (+15.99491) and phosphorylation at serine, threonine, and tyrosine (+79.96633). Filters used for global data analysis included one PSM per peptide and two peptides per protein, with a 1% FDR threshold at the protein level. Filters used for phosphoproteome data included one PSM per peptide and one peptide per protein, with a 1% FDR threshold at the peptide level. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository30 with the data set identifier PXD008952 and DOI: 10.6019/PXD008952.
RESULTS AND DISCUSSION
Determining Coverage of the Human Genome by the NCI-7 Cell Line Panel
The NCI-7 Cell Line Panel is comprised of the cell lines NCI-H23, RPMI-8226, T47D, A549, COLO205, NCI-H226, and CCRF CEM, which are representative of several tissues and various genetic mutations (Table 1). Extensive genomic profiling of the NCI-60 cell lines has been performed previously utilizing a variety of analysis platforms and pipelines.10,11,13 our analysis, we examined the “5 Platform Gene Transcript” data set from CellMiner to determine the genes expressed by the individual cell lines in the NCI-60 Cell Line Panel. Utilizing our filtering criteria as described in the Methods section to identify gene transcripts, we plotted the individual cell lines against the number of their respective unique genes expressed; ranking the NCI-60 Cell Line Panel in order of their coverage of the human genome (Figure 1), and selected seven cell lines that were readily available from ATCC. We found that 7 cell lines (NCI-7 Cell Line Panel) from the NCI-60 Cell Line Panel represented 92% of the NCI-60 Cell Line expressed genome, as well as representing 88% coverage of the entire human genome (Table S5).
Table 1.
NCI-7 CompRef Cell Line Composition with Respective Histology and Gene Mutation Profile
| cell line | tissue (histology) | gene mutation |
|---|---|---|
| NCI-H23 | Lung (adenocarcinoma) | KRAS, TP53 |
| RPMI-8226 | Peripheral blood (plasmacytoma, myeloma) | KRAS, EGFR, TP53 |
| T47D | Breast (carcinoma, ductal) | PI3KA, TP53 |
| A549 | Lung (adenocarcinoma) | KRAS, CDKN2A |
| COLO205 | Colon (adenocarcinoma) | BRAF, SMAD4, APC, TP53 |
| NCI-H226 | Lung (mesothelioma, squamous cell) | CDKN2A |
| CCRF-CEM | Peripheral blood (leukemia, acute lymphoblastic) | KRAS, CDKN2A, PTEN, TP53 |
Figure 1.
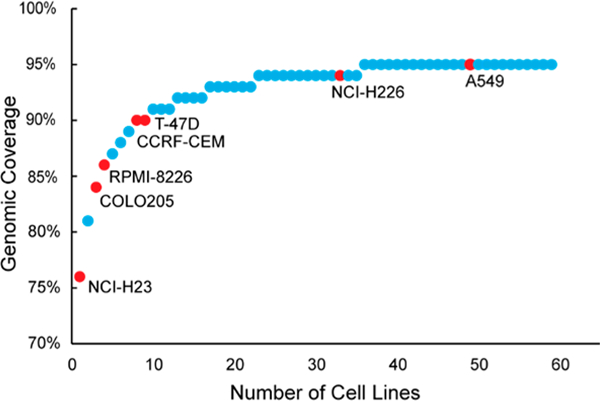
NCI-60 Cell Line Panel ranked by coverage of the human genome. NCI-7 cell lines are labeled and identified with red dots.
Assessing Regeneration Reproducibility of NCI-7 Cell References
One of the critical assessment for reference materials is whether the materials can be stably regenerated even after an extended period of time. Extended passaging of a single cell line has been shown to result in phenotypic and genotypic alterations that can influence protein expression.31 To circumvent this drawback of cell lines, recommendations include limiting cell culturing to time periods less than three months and obtaining new cell aliquots from a cell bank.32 Moreover, we hypothesized that by generating a reference material from several cell lines we might further reduce differences related to protein expression, as well as varied protein expression specific to the individual cell lines.33 We obtained three distinct vials of each individual cell line from ATCC to generate three preparations of the NCI-7 Cell Line Panel (Table S6), then employed quantitative proteomics to determine reproducibility of the NCI-7 cell reference generation from different seed cells as illustrated in Figure 2A. Following tryptic digestion, three channels from each NCI-7 Cell Line Panel preparation (NCI-7 Replicate #1, #2, and #3) were TMT labeled, and subjected to basic reverse-phase fractionation, phosphopeptide enrichment, and nano-LC–MS/MS analysis on an Orbitrap Lumos Fusion mass spectrometer. In total, 179 298 peptides from 10 926 proteins (Table S7) and 51 505 phosphopeptides (Table S8) were identified. We plotted the median protein ratio value for all of the individual channels, observing close correlation between our expected values and observed values (Figure S1). Using reported TMT ratios from the ten TMT-channels, we constructed correlation matrices for both global protein expression and phosphopeptide expression. As shown in Figure 3, Pearson’s correlation coefficients were high between the technical replicates for each of the NCI-7 preparations (>0.95) When evaluating the protein expression profile across the three NCI-7 replicates, a high correlation coefficient (>0.93) was also observed, demonstrating high reproducibility of the individual NCI-7 preparations. Examination of phosphopeptide expression revealed a similar pattern of high correlation between the technical (0.94–0.99) and biological replicates (0.88–0.95) of the NCI-7 material (Figure 3). The stable global and phosphoprotein profiles between the three distinct sample preparations of NCI-7 is extremely promising in the context of using this it as a universal protein reference material. Not only would using cell lines allow for large scale production of NCI-7, but as long as primary cell stocks are used, cell culturing methods remain consistent, and passage number monitored, the protein profile would remain relatively unchanged. This characteristic is not only important for studies that may span months, or even years, and require a longitudinal standard reference, but also identifying a potential universal reference standard for benchmarking various sample preparation pipelines, mass spectrometry instrumentation, and data analysis methodology.34,35 Overall, these results show that sample generation of the NCI-7 Cell Line Panel material is highly reproducible at both global protein and phosphopeptide levels.
Figure 2.
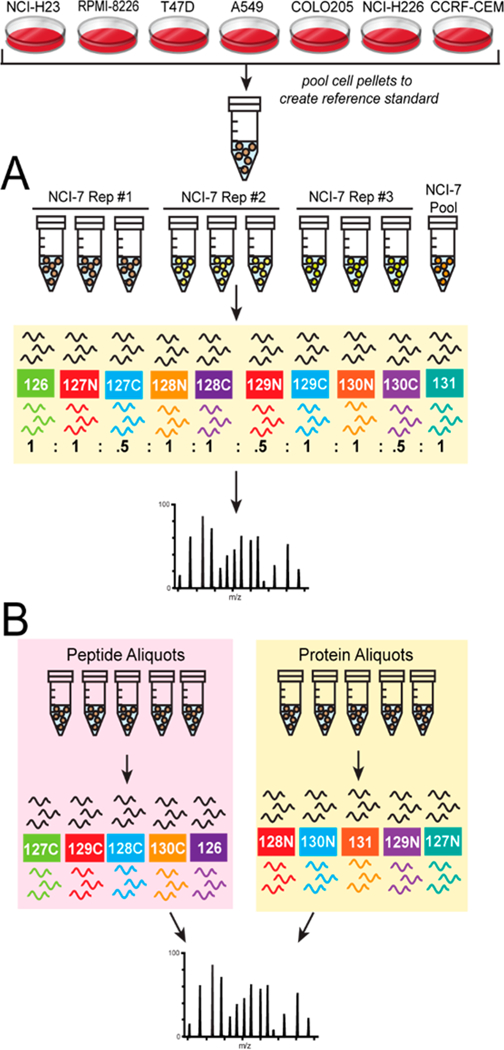
Schematic of NCI-7 CompRef workflows. Equal amounts of protein from each individual cell line was pooled to generate protein reference standard. The pooled reference standard was then utilized to evaluate NCI-7 sample generation reproducibility (A) and sample digestion and mass tag labeling(B).
Figure 3.
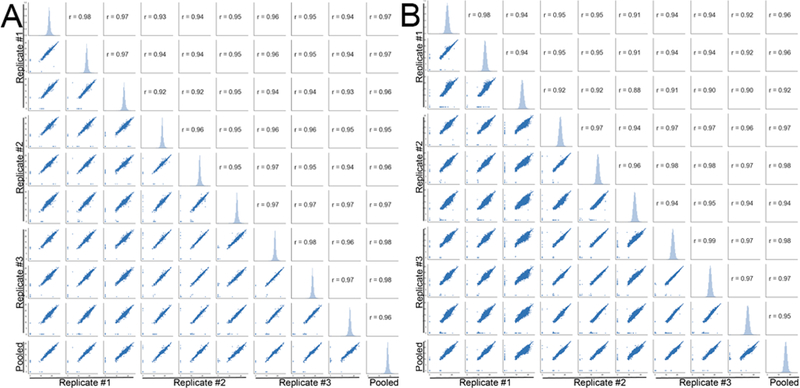
Reproducibility of NCI-7 CompRef sample generation. Correlation matrix of three distinct NCI-7 CompRef pooled reference replicates using reported protein ratios for global proteomics (A) and peptide ratios for phosphoproteomics (B).
Determining the Coverage of the NCI-7 Proteome
Since previous CPTAC-related studies utilized a PDX CompRef material to assess instrument performance, we wanted to compare the NCI-7 to a PDX CompRef material comprised of the xenografts WHIM2-P32 (basal) and WHIM16-P33 (luminal-B), which have been well characterized previously.36,37 The material from the two xenografts followed a similar protocol of sample preparation and digestion, basic reverse-phase fractionation, phosphopeptide enrichment, and nano-LC–MS/MS analysis, with a distinct TMT-labeling schema (Figure S2). In this analysis, we identified a total of 170 120 peptides from 13 182 protein groups when searched against a combined mouse + human database (Table S9), and 147 981 peptides from 9596 protein groups when searched against a human-only database (Table S10). When examining the phosphoproteome, we identified 44 168 and 39 353 phosphopeptides when searching against a mouse + human (Table S11) and human-only (Table S12), respectively. The total number of identified peptides/proteins and phosphosites are summarized in Table 2. For our comparative purposes, we only evaluated proteins identified using the NCBI human ref-Seq database, observing 7707 protein groups and 17 964 phosphopeptides shared between the NCI-7 and P32/P33 CompRef data sets (Figure 4). As the results indicate, there is a large percentage (~26%) of mouse proteins present in the PDX CompRef material, which is to be expected due to human stromal cell component of the primary tumor being replaced by murine cells as the tumor is passaged.38 The occupancy of mouse proteins in the MS spectrum did not appear to significantly impact the overlap of global protein coverage between the NCI-7 and P32/P33 material, and commonly identified proteins between the two data sets potentially provide further evidence of a core human proteome.39 However, the low overlap of phosphopeptides between the two data sets suggests either a distinct pattern of phosphosite occupancy between the two reference materials, or that stromal cells are a significant contributor to the phosphoproteome in heterocellular models.40 In contrast, we found few bovine-derived proteins in the NCI-7 material (sourced from the fetal bovine serum utilized in the cell culturing), identifying less than 0.2% peptide spectral matches (PSMs) in our data set (data not shown).
Table 2.
Number of Peptide, Protein, and Phosphopeptide Identifications for the PDX and NCI-7 CompRef Materials
| sample | databasea | protein # | peptide # | phosphopeptide # |
|---|---|---|---|---|
| P32/P33 | M + H | 13 182 | 170 120 | 44168 |
| P32/P33 | H | 9596 | 147 981 | 39 353 |
| NCI-7 | H | 10 926 | 179 298 | 51 505 |
M, mouse database; H, human database.
Figure 4.
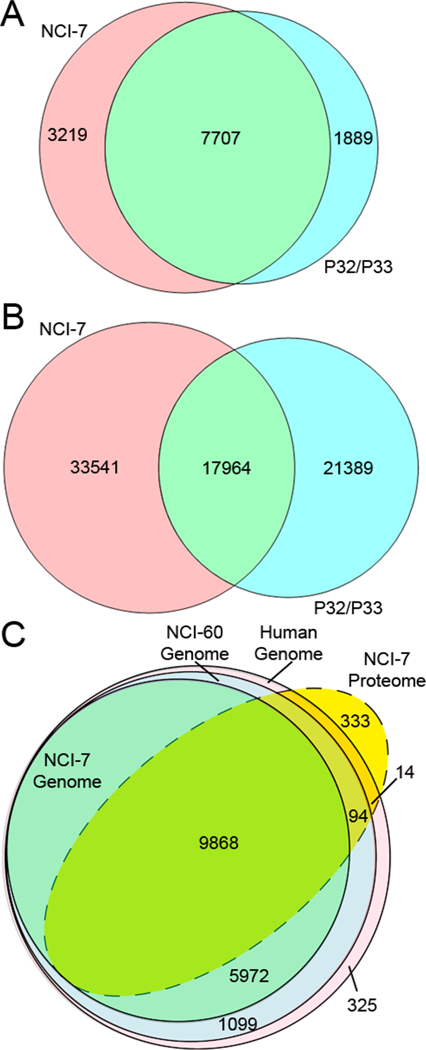
Coverage of the NCI-7 Cell Line proteome. Overlap of human proteome between NCI-7 CompRef and PDX P32/P33 CompRef for global proteins (A) and phosphopeptides (B). Total coverage of the human, NCI-60, and NCI-7 genome by the NCI-7 Cell Line material (C).
Next, we wanted to define the coverage of the NCI-7 material to determine whether the cell line-derived material might be comparable to clinical tumor specimens. Previously, yeast S. cerevisiae has been examined as a proteomics reference standard, citing benefits including low cost, high protein yield, and protein complexity and dynamic range.41 However, with improvements in mass spectrometry instrumentation and proteome data set exceeding 8000 protein identifications,42 the total proteome of yeast (comprising ~6600 proteins),43 may be insufficient as a reference material for comprehensive assessment of laboratory performance. The NCI-7 material shares many of the positive attributes of the yeast reference standard, while exceeding the number of identifiable gene products, as indicated by our mass spectrometry analysis of the NCI-7 material identifying over 10 000 protein groups, which is similar to the number of total number of proteins identified in the proteomic analysis of the entire NCI-60 Cell Line Panel.15 Utilizing the corresponding RefSeq IDs, further evaluation of our proteome data set revealed coverage of the for the Human, NCI-60, and NCI-7 genomes to be 57.4%, 58.5% and 62.2%, respectively (Figure 4C). Interestingly, we identified 333 proteins that were not observed in the “Human Genome” (Table S13); however, additional investigation revealed that the CellMiner data sets we utilized were from studies performed prior to 2011,13,44,45 and subsequent NCBI Annotated Releases (including the RefSeq database used for protein identification) have been updated with newly annotated proteins.
With CPTAC already having examined colorectal, ovarian, and breast cancer via mass spectrometry,1–3 and future plans to examine additional cancers, there will be a desire to link these various data sets to facilitate “pan-cancer” analyses. The Cancer Genome Atlas (TCGA) Research Network has already proposed and begun integrating the various levels of genomics information obtained in the multiple TCGA analyses,46,47 and small scaled protein “pan-cancer” study has already been carried out.48 With the ability to identify and quantify thousands of proteins and integrate the genetic information from hundreds of clinical samples, the results of the CPTAC studies have the potential to significantly expand our understanding of underlying molecular basis of both individual and multiple tumor types. Similar to the previous CPTAC proteogenomic studies which included one isobaric channel in each quantitative set as an experimental reference, the inclusion of several NCI-7 channels interspersed in the quantitative sets of future CPTAC analyses would allow for linking the data from distinct cancer types and enable pan-cancer analyses. Overall, our results show the NCI-7 material has a complex dynamic range of protein expression, and sufficient coverage of the human proteome to serve as a standard proteomic reference material.
Applying NCI-7 Cell Line Panel To Assess the Reproducibility of Sample Preparation and Protein Digestion
Sample preparation is critical aspect in proteomic analyses, and can be a major source of experimental variation that can impact the accuracy of downstream quantitation.49–51 One of the benefits of quantitative proteomics using tandem-mass-tag (TMT) labeling is the ability to multiplex biological samples and analyze them in a single mass spectrometry analysis, which reduces the analytical variation of LC–MS/MS; however, this analytical strategy is especially sensitive to variations in sample preparation, due to its incorporation protein digestion and purification.52 To evaluate lab performance of protein sample preparation using NCI-7 Cell Line Panel, we devised the experimental procedure described in Figure 2B. First, the cell lines comprising the NCI-7 Cell Line Panel were cultured and cell pellets generated. Following lysis, equal protein amounts from each cell line were mixed to generate the NCI-7 cell lysate mixture. To assess the efficiency of sample preparation and digestion, we generated several protein aliquots of various concentrations to be digested individually. In parallel, we generated a single digest, wherein the sample postdigest would be aliquoted at the peptide level. After the complete digest of all protein samples, the derived peptides were labeled with TMT reagents to facilitate protein quantitation, followed by basic reverse-phase fractionation, and nano-LC–MS/MS analysis. Global peptide fractions were analyzed on an Orbitrap QE HF mass spectrometer, identifying a total of 68 578 peptides derived from 6428 protein groups (Table S14). To assess sample preparation reproducibility, we evaluated the TMT reporter channels corresponding to sample aliquoted at the protein level, where as to assess the accuracy of TMT-labeling, we utilized the TMT reporter channels corresponding to sample aliquoted at the peptide level. We plotted the median protein ratio value for all of the individual channels, observing close correlation between our expected values and observed values (Figure 5). Moreover, we utilized two distinct TMT channels to normalize our data, TMT-127C which corresponded to the 1-fold aliquoted peptide sample (Figure 5A) and TMT-128N, which corresponded to the 1-fold aliquoted protein sample (Figure 5B). Interestingly, we did not observe additional variation in the overall distribution of protein ratios comparing to peptide ratios when we normalized to our 1-fold protein aliquoted sample. With the accuracy of TMT reporter ratio being influenced by TMT reporter ion intensity in MS2, we evaluated the threshold in which accurate TMT reporter intensity was reduced or loss, finding an ion intensity below 10 000 could impact the accuracy of TMT ratios (Figure 5C, Figure S3A). When we included additional variables of cumulative PSM count and Coefficient of Variation (CV), we found a similar threshold cutoff of 10 000 ion intensity, recording a CV of approximately 14.1% that continued to drop with increasing ion intensity (Figure S3B). Finally, we wanted to determine the impact of sample dilution on the total number of peptides and proteins identified in each respective TMT channel. Our results indicated similar peptide and protein identification rates between our 1-fold (400 μg), 2-fold (200 μg), and 5-fold (80 μg) peptide and protein sample aliquots (Figure 6). In our 10-fold (40 μg) peptide sample aliquot we observed a reduction in the total number of peptides and protein identified, which was mirrored in our 10-fold protein sample aliquot. In total, these quantitative proteomic results indicate a high level of reproducibility for sample preparation and protein digestion, in addition to accurate peptide labeling using TMT isobaric reagents. Moreover, our experimental design evaluating sample preparation and TMT-labeling simultaneously is suitable for laboratory benchmarking to assess a lab’s technical and mass spectrometry performance when utilizing the NCI-7 Cell Line Panel as a reference standard.
Figure 5.
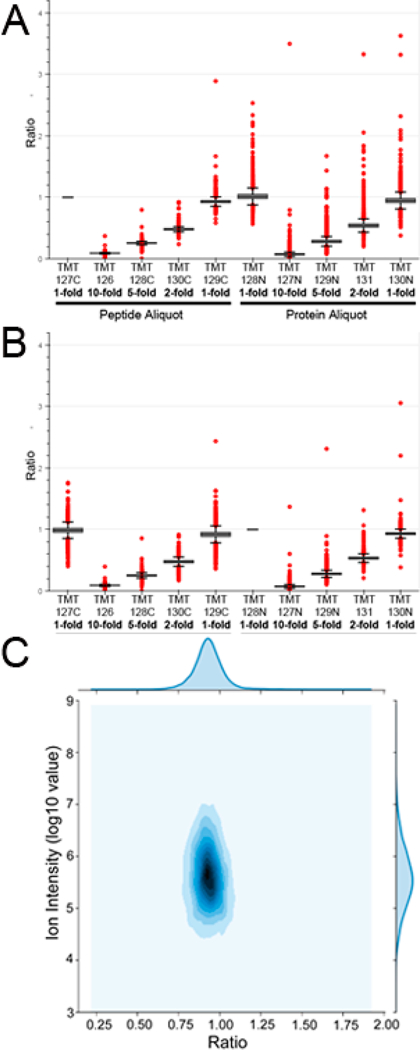
Accuracy of tandem-mass-tag (TMT) quantitation and reproducibility of sample digestion based by peptide aliquots and protein aliquots protein ratios, respectively. Box plots were generated for reported protein ratios using either a peptide aliquot channel (A) and protein aliquot channel (B) for data normalization. (C) Influence of ion intensity on reporter tag accuracy. Data plotted is from Replicate #1, Fraction #1.
Figure 6.
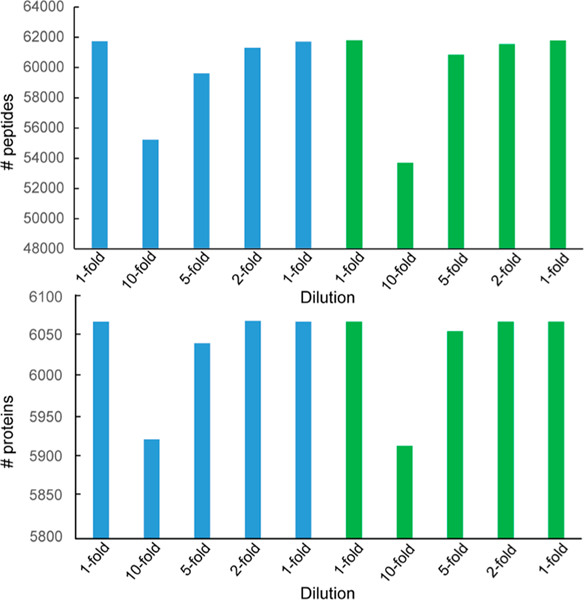
Starting sample amount influences identification rate. The number of identified peptides (top) and proteins (bottom) are reported for each TMT channel. Analysis revealed an ion threshold intensity of 10 000 produced accurate quantitation and was utilized as a filter for the number of identifications per channel.
CONCLUSION
In this study, we evaluated the use of a new comparative reference material, NCI-7 Cell Line Panel, comprised of seven cancer cell lines from the NCI-60 cell line panel to be utilized as a longitudinal reference standard for CPTAC-related studies, as well as potentially be utilized for interlaboratory instrumentation QC and intralaboratory benchmarking and comparison. In our analysis, we first determined the reproducibility of generating the NCI-7 reference material, showing consistent global proteome and phosphoproteome expression profiles across three distinct sample preparations. Next, we estimated the NCI-7 cell line panel was representative of 88% of the human genome, and proteomic analysis was able to identify 63% of the predicted NCI-7 genome in a single data set. Finally, we devised a quantitative strategy that could assess sample preparation metrics, including sample digestion as well as labeling accuracy, that could be applied in future benchmarking studies as a measurement of laboratory performance. Together, this study has shown the NCI-7 cell line material not only is suitable as a reference standard for assessing the performance of CPTAC PCCs, but may also have future applications as a universal proteomic reference material.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health, National Cancer Institute, the Clinical Proteomic Tumor Analysis Consortium (CPTAC, U24CA210985).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteo-me.8b00165.
Box plots of reported TMT ratios; schematic of TMT-labeling for PDX model; plot illustrating influence of ion intensity on reporter tag accuracy (PDF)
NCI-60 Cell Lines Gene Transcript values; ranking of NCI-60 Cell Lines by gene expression; ranking of NCI-60 Cell Lines by genomic coverage; NCI-7 Gene Expression profiles; coverage of the human and NCI-60 genomes by NCI-7 Cell Line Panel; passage number and harvest dates of three NCI-7 Cell Line preparations; identified proteins from NCI-7 Cell Line sample regeneration reproducibility global proteome analysis; identified proteins from NCI-7 Cell Line sample regeneration reproducibility phosphoproteome analysis; identified proteins from PDX-model P32/P33 CompRef sample global proteome analysis (human + mouse database); identified proteins from PDX-model P32/P33 CompRef sample phosphoproteome analysis (human + mouse database); identified proteins from PDX-model P32/P33 CompRef sample global proteome analysis (human database); identified proteins from PDX-model P32/P33 CompRef sample phosphoproteome analysis (human database); NCI-7 proteins not identified in CellMiner data set; identified proteins from NCI-7 Cell Line sample preparation and digestion reproducibility global proteome analysis (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Zhang B; Wang J; XiaojingWang; Zhu J; Liu Q; Shi Z; Chambers MC; Zimmerman LJ; Shaddox KF; Kim S; Davies SR; Wang S; PeiWang; Kinsinger CR; Rivers RC; Rodriguez H; Townsend RR; Ellis MJC; Carr S. a.; Tabb DL; Coffey RJ; Slebos RJC; Liebler DC Proteogenomic Characterization of Human Colon and Rectal Cancer. Nature 2014, 513, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mertins P; Mani DR; Ruggles KV; Gillette MA; Clauser KR; Wang P; Wang X; Qiao JW; Cao S; Petralia F; Kawaler E; Mundt F; Krug K; Tu Z; Lei JT; Gatza ML; Wilkerson M; Perou CM; Yellapantula V; Huang K; Lin C; McLellan MD; Yan P; Davies SR; Townsend RR; Skates SJ; Wang J; Zhang B; Kinsinger CR; Mesri M; Rodriguez H; Ding L; Paulovich AG; Fenyo D; Ellis MJ; Carr SA Proteogenomics Connects Somatic Mutations to Signalling in Breast Cancer. Nature 2016, 534 (7605), 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang H; Liu T; Zhang Z; Payne SH; Zhang B; McDermott JE; Zhou JY; Petyuk VA; Chen L; Ray D; Sun S; Yang F; Chen L; Wang J; Shah P; Cha SW; Aiyetan P; Woo S; Tian Y; Gritsenko MA; Clauss TR; Choi C; Monroe ME; Thomas S; Nie S; Wu C; Moore RJ; Yu KH; Tabb DL; Feny D; Bafna V; Wang Y; Rodriguez H; Boja ES; Hiltke T; Rivers RC; Sokoll L; Zhu H; Shih IM; Cope L; Pandey A; Zhang B; Snyder MP; Levine DA; Smith RD; Chan DW; Rodland KD; Carr SA; Gillette MA; Klauser KR; Kuhn E; Mani DR; Mertins P; Ketchum KA; Thangudu R; Cai S; Oberti M; Paulovich AG; Whiteaker JR; Edwards NJ; McGarvey PB; Madhavan S; Wang P; Chan DW; Pandey A; Shih IM; Zhang H; Zhang Z; Zhu H; Cope L; Whiteley GA; Skates SJ; White FM; Levine DA; Boja ES; Kinsinger CR; Hiltke T; Mesri M; Rivers RC; Rodriguez H; Shaw KM; Stein SE; Fenyo D; Liu T; McDermott JE; Payne SH; Rodland KD; Smith RD; Rudnick P; Snyder M; Zhao Y; Chen X; Ransohoff DF; Hoofnagle AN; Liebler DC; Sanders ME; Shi Z; Slebos RJC; Tabb DL; Zhang B; Zimmerman LJ; Wang Y; Davies SR; Ding L; Ellis MJC; Townsend RR Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166 (3), 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tabb DL; Wang X; Carr SA; Clauser KR; Mertins P; Chambers MC; Holman JD; Wang J; Zhang B; Zimmerman LJ; Chen X; Gunawardena HP; Davies SR; Ellis MJC; Li S; Townsend RR; Boja ES; Ketchum KA; Kinsinger CR; Mesri M; Rodriguez H; Liu T; Kim S; McDermott JE; Payne SH; Petyuk VA; Rodland KD; Smith RD; Yang F; Chan DW; Zhang B; Zhang H; Zhang Z; Zhou JY; Liebler DC Reproducibility of Differential Proteomic Technologies in CPTAC Fractionated Xenografts. J. Proteome Res. 2016, 15 (3), 691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhou J; Chen L; Zhang B; Tian Y; Liu T; Thomas SN; Chen L; Schnaubelt M; Boja E; Hiltke T; Kinsinger CR; Rodriguez H; Davies SR; Li S; Snider JE; Erdmann-gilmore P; Tabb DL; Townsend RR; Ellis MJ; Rodland KD; Smith RD; Carr SA; Zhang Z; Chan DW; Zhang H Quality Assessments of Long-Term Quantitative Proteomic Analysis of Breast Cancer Xenograft Tissues. J. Proteome Res. 2017, 16, 4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rudnick PA; Markey SP; Roth J; Mirokhin Y; Yan X; Tchekhovskoi DV; Edwards NJ; Thangudu RR; Ketchum KA; Kinsinger CR; Mesri M; Rodriguez H; Stein SE A Description of the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Common Data Analysis Pipeline. J. Proteome Res. 2016, 15 (3), 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Whittle JR; Lewis MT; Lindeman GJ; Visvader JE Patient-Derived Xenograft Models of Breast Cancer and Their Predictive Power. Breast Cancer Res. 2015, 17 (1), 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Workman P; Aboagye EO; Balkwill F; Balmain A; Bruder G; Chaplin DJ; Double JA; Everitt J; Farningham DAH; Glennie MJ; Kelland LR; Robinson V; Stratford IJ; Tozer GM; Watson S; Wedge SR; Eccles SA Guidelines for the Welfare and Use of Animals in Cancer Research. Br. J. Cancer 2010, 102 (11), 1555–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shoemaker RH The NCI60 Human Tumour Cell Line Anticancer Drug Screen. Nat. Rev. Cancer 2006, 6 (10), 813–823. [DOI] [PubMed] [Google Scholar]
- (10).Ikediobi ON; Davies H; Bignell G; Edkins S; Stevens C; O’Meara S; Santarius T; Avis T; Barthorpe S; Brackenbury L; Buck G; Butler A; Clements J; Cole J; Dicks E; Forbes S; Gray K; Halliday K; Harrison R; Hills K; Hinton J; Hunter C; Jenkinson A; Jones D; Kosmidou V; Lugg R; Menzies A; Mironenko T; Parker A; Perry J; Raine K; Richardson D; Shepherd R; Small A; Smith R; Solomon H; Stephens P; Teague J; Tofts C; Varian J; Webb T; West S; Widaa S; Yates A; Reinhold W; Weinstein JN; Stratton MR; Futreal PA; Wooster R Mutation Analysis of 24 Known Cancer Genes in the NCI-60 Cell Line Set. Mol. Cancer Ther. 2006, 5 (11), 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Abaan OD; Polley EC; Davis SR; Zhu YJ; Bilke S; Walker RL; Pineda M; Gindin Y; Jiang Y; Reinhold WC; Holbeck SL; Simon RM; Doroshow JH; Pommier Y; Meltzer PS The Exomes of the NCI-60 Panel: A Genomic Resource for Cancer Biology and Systems Pharmacology. Cancer Res. 2013, 73 (14), 4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Reinhold WC; Varma S; Sunshine M; Rajapakse V; Luna A; Kohn KW; Stevenson H; Wang Y; Nogales V; Moran S; et al. The NCI-60 Methylome and Its Integration into CellMiner. Cancer Res. 2017, 77 (3), 601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shankavaram UT; Reinhold WC; Nishizuka S; Major S; Morita D; Chary KK; Reimers M. a.; Scherf U; Kahn A; Dolginow D; Cossman J; Kaldjian EP; Scudiero D. a.; Petricoin E; Liotta L; Lee JK; Weinstein JN Transcript and Protein Expression Profiles of the NCI-60 Cancer Cell Panel: An Integromic Microarray Study. Mol. Cancer Ther. 2007, 6 (3), 820–832. [DOI] [PubMed] [Google Scholar]
- (14).Nishizuka S; Charboneau L; Young L; Major S; Reinhold WC; Waltham M; Kouros-Mehr H; Bussey KJ; Lee JK; Espina V; Munson PJ; Petricoin E; Liotta LA; Weinstein JN Proteomic Profiling of the NCI-60 Cancer Cell Lines Using New High-Density Reverse-Phase Lysate Microarrays. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (24), 14229–14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gholami AM; Hahne H; Wu Z; Auer FJ; Meng C; Wilhelm M; Kuster B Global Proteome Analysis of the NCI-60 Cell Line Panel. Cell Rep. 2013, 4 (3), 609–620. [DOI] [PubMed] [Google Scholar]
- (16).Shankavaram UT; Varma S; Kane D; Sunshine M; Chary KK; Reinhold WC; Pommier Y; Weinstein JN CellMiner: A Relational Database and Query Tool for the NCI-60 Cancer Cell Lines. BMC Genomics 2009, 10, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Reinhold WC; Sunshine M; Liu H; Varma S; Kohn KW; Morris J; Doroshow J; Pommier Y CellMiner: A Web-Based Suite of Genomic and Pharmacologic Tools to Explore Transcript and Drug Patterns in the NCI-60 Cell Line Set. Cancer Res. 2012, 72 (14), 3499–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chabner BA NCI-60 Cell Line Screening: A Radical Departure in Its Time. J. Natl. Cancer Inst. 2016, 108 (5), 1–7. [DOI] [PubMed] [Google Scholar]
- (19).Beroukhim R; Mermel CH; Porter D; Wei G; Raychaudhuri S; Donovan J; Barretina J; Boehm JS; Dobson J; Urashima M; Mc Henry KT; Pinchback RM; Ligon AH; Cho Y-J; Haery L; Greulich H; Reich M; Winckler W; Lawrence MS; Weir BA; Tanaka KE; Chiang DY; Bass AJ; Loo A; Hoffman C; Prensner J; Liefeld T; Gao Q; Yecies D; Signoretti S; Maher E; Kaye FJ; Sasaki H; Tepper JE; Fletcher JA; Tabernero J; Baselga J; Tsao M-S; Demichelis F; Rubin MA; Janne PA; Daly MJ; Nucera C; Levine RL; Ebert BL; Gabriel S; Rustgi AK; Antonescu CR; Ladanyi M; Letai A; Garraway LA; Loda M; Beer DG; True LD; Okamoto A; Pomeroy SL; Singer S; Golub TR; Lander ES; Getz G; Sellers WR; Meyerson M The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463 (7283), 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bignell GR; Greenman CD; Davies H; Butler AP; Edkins S; Andrews JM; Buck G; Chen L; Beare D; Latimer C; Widaa S; Hinton J; Fahey C; Fu B; Swamy S; Dalgliesh GL; Teh BT; Deloukas P; Yang F; Campbell PJ; Futreal PA; Stratton MR Signatures of Mutation and Selection in the Cancer Genome. Nature 2010, 463 (7283), 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Adeegbe DO; Liu Y Ex Vivo Engineering of the Tumor Microenvironment; Aref AR, Barbie D; Humana Press: New York City, NY, 2017. [Google Scholar]
- (22).Aparicio S; Hidalgo M; Kung AL Examining the Utility of Patient-Derived Xenograft Mouse Models. Nat. Rev. Cancer 2015, 15 (5), 311–316. [DOI] [PubMed] [Google Scholar]
- (23).Lee JY; Kim SY; Park C; Kim NKD; Jang J; Park K; Yi JH; Hong M; Ahn T; Rath O; Schueler J; Kim ST; Do I-G; Lee S; Park SH; Ji YI; Kim D; Park JO; Park YS; Kang WK; Kim K-M; Park W-Y; Lim HY; Lee J; Tae Kim S; Ji Yun Lee S; et al. Patient-Derived Cell Models as Preclinical Tools for Genome-Directed Targeted Therapy. Oncotarget 2015, 6 (28), 25619–25630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mertins P; Qiao JW; Patel J; Udeshi ND; Clauser KR; Mani DR; Burgess MW; Gillette MA; Jaffe JD; Carr SA Integrated Proteomic Analysis of Post-Translational Modifications by Serial Enrichment. Nat. Methods 2013, 10 (7), 634–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Huang Sherman BT; Lempicki RADW; Huang DW; Sherman BT; Lempicki R. a. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2008, 4 (1), 44–57. [DOI] [PubMed] [Google Scholar]
- (26).Huang DW; Sherman BT; Lempicki R. a. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kim S; Pevzner PA MS-GF+ Makes Progress towards a Universal Database Search Tool for Proteomics. Nat. Commun. 2014, 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kim S; Gupta N; Pevzner PA Spectral Probabilities and Generating Functions of Tandem Mass Spectra a Strike Against the Decoy Database-Supplement-Supplementary_Info. J. Proteome Res. 2008, 7 (8), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Elias JE; Gygi SP Target-Decoy Search Strategy for Increased Confidence in Large-Scale Protein Identifications by Mass Spectrometry. Nat. Nat. Methods 2007, 4 (3), 207–214. [DOI] [PubMed] [Google Scholar]
- (30).Vizcaíno J; Deutsch E; Wang R ProteomeXchange Provides Globally Coordinated Proteomics Data Submission and Dissemination. Nat. Biotechnol. 2014, 32 (3), 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Jin W; Penington CJ; McCue SW; Simpson MJ A Computational Modelling Framework to Quantify the Effects of Passaging Cell Lines. PLoS One 2017, 12 (7), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Masters JR; Stacey GN Changing Medium and Passaging Cell Lines. Nat. Protoc. 2007, 2 (9), 2276–2284. [DOI] [PubMed] [Google Scholar]
- (33).Geiger T; Wehner A; Schaab C; Cox J; Mann M Comparative Proteomic Analysis of Eleven Common Cell Lines Reveals Ubiquitous but Varying Expression of Most Proteins. Mol. Cell. Proteomics 2012, 11 (3), M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bunk DM Design Considerations for Proteomic Reference Materials. Proteomics 2010, 10 (23), 4220–4225. [DOI] [PubMed] [Google Scholar]
- (35).Klimek J; Eddes JS; Hohmann L; Jackson J; Peterson A; Letarte S; Gafken PR; Katz JE; Mallick P; Lee H; Schmidt A; Ossola R; Eng JK; Aebersold R; Martin DB The Standard Protein Mix Database: A Diverse Data Set to Assist in the Production of Improved Peptide and Protein Identification Software Tools. J. Proteome Res. 2008, 7 (1), 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ding L; Ellis MJ; Li S; Larson DE; Chen K; Wallis JW Genome Remodelling in a Basal-like Breast Cancer Metastasis and Xenograft. Nature 2010, 464, 999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Li S; Shen D; Shao J; Crowder R; Liu W; Prat A; He X; Liu S; Hoog J; Lu C; Ding L; Griffith OL; Miller C; Larson D; Fulton RS; Harrison M; Mooney T; McMichael JF; Luo J; Tao Y; Goncalves R; Schlosberg C; Hiken JF; Saied L; Sanchez C; Giuntoli T; Bumb C; Cooper C; Kitchens RT; Lin A; Phommaly C; Davies SR; Zhang J; Kavuri MS; McEachern D; Dong YY; Ma C; Pluard T; Naughton M; Bose R; Suresh R; McDowell R; Michel L; Aft R; Gillanders W; DeSchryver K; Wilson RK; Wang S; Mills GB; Gonzalez-Angulo A; Edwards JR; Maher C; Perou CM; Mardis ER; Ellis MJ Endocrine-Therapy-Resistant ESR1 Variants Revealed by Genomic Characterization of Breast-Cancer-Derived Xenografts. Cell Rep. 2013, 4 (6), 1116–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).DeRose YS; Wang G; Lin YC; Bernard PS; Buys SS; Ebbert MT Tumor Grafts Derived from Women with Breast Cancer Authentically Reflect Tumor Pathology, Growth, Metastasis and Disease Outcomes. Nat. Med. 2011, 17, 1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Schirle M; Heurtier M-A; Kuster B Profiling Core Proteomes of Human Cell Lines by One-Dimensional PAGE and Liquid Chromatography-Tandem Mass Spectrometry. Mol. Cell. Proteomics 2003, 2 (12), 1297–1305. [DOI] [PubMed] [Google Scholar]
- (40).Tape CJ; Ling S; Dimitriadi M; McMahon KM; Worboys JD; Leong HS; Norrie IC; Miller CJ; Poulogiannis G; Lauffenburger DA; Jørgensen C Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165 (4), 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Paulovich AG; Billheimer D; Ham A-JL; Vega-Montoto L; Rudnick PA; Tabb DL; Wang P; Blackman RK; Bunk DM; Cardasis HL; Clauser KR; Kinsinger CR; Schilling B; Tegeler TJ; Variyath AM; Wang M; Whiteaker JR; Zimmerman LJ; Fenyo D; Carr SA; Fisher SJ; Gibson BW; Mesri M; Neubert TA; Regnier FE; Rodriguez H; Spiegelman C; Stein SE; Tempst P; Liebler DC Interlaboratory Study Characterizing a Yeast Performance Standard for Benchmarking LC-MS Platform Performance. Mol. Cell. Proteomics 2010, 9 (2), 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Davis S; Charles PD; He L; Mowlds P; Kessler BM; Fischer R Expanding Proteome Coverage with CHarge Ordered Parallel Ion aNalysis (CHOPIN) Combined with Broad Specificity Proteolysis. J. Proteome Res. 2017, 16 (3), 1288–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Picotti P; Clement-Ziza M; Lam H; Campbell DS; Schmidt A; Deutsch EW; Rost H; Sun Z; Rinner O; Reiter L; Shen Q; Michaelson JJ; Frei A; Alberti S; Kusebauch U; Wollscheid B; Moritz RL; Beyer A; Aebersold R A Complete Mass-Spectrometric Map of the Yeast Proteome Applied to Quantitative Trait Analysis. Nature 2013, 494 (7436), 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Reinhold WC; Mergny JL; Liu H; Ryan M; Pfister TD; Kinders R; Parchment R; Doroshow J; Weinstein JN; Pommier Y Exon Array Analyses across the NCI-60 Reveal Potential Regulation of TOP1 by Transcription Pausing at Guanosine Quartets in the First Intron. Cancer Res. 2010, 70 (6), 2191–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Liu H; D’Andrade P; Fulmer-Smentek S; Lorenzi P; Kohn KW; Weinstein JN; Pommier Y; Reinhold WC mRNA and microRNA Expression Profiles of the NCI-60 Integrated with Drug Activities. Mol Cancer Ther. 2010, 9 (5), 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Chang K; Creighton CJ; Davis C; Donehower L; Drummond J; Wheeler D; Ally A; Balasundaram M; Birol I; Butterfield YSN; Chu A; Chuah E; Chun H-JE; Dhalla N; Guin R; Hirst M; Hirst C; Holt R; Jones SJM; Lee D; Li HI; Marra M. a.; Mayo M; Moore R. a.; Mungall AJ; Robertson a G.; Schein JE; Sipahimalani P; Tam A; Thiessen N; Varhol RJ; Beroukhim R; Bhatt AS; Brooks AN; Cherniack AD; Freeman SS; Gabriel SB; Helman E; Jung J; Meyerson M; Ojesina AI; Pedamallu CS; Saksena G; Schumacher SE; Tabak B; Zack T; Lander ES; Bristow C. a.; Hadjipanayis A; Haseley P; Kucherlapati R; Lee S; Lee E; Luquette LJ; Mahadeshwar HS; Pantazi A; Parfenov M; Park PJ; Protopopov A; Ren X; Santoso N; Seidman J; Seth S; Song X; Tang J; Xi R; Xu AW; Yang LL; Zeng D; Auman JT; Balu S; Buda E; Fan C; Hoadley K. a.; Jones CD; Meng S; Mieczkowski P. a.; Parker JS; Perou CM; Roach J; Shi Y; Silva GO; Tan D; Veluvolu U; Waring S; Wilkerson MD; Wu J; Zhao W; Bodenheimer T; Hayes DN; Hoyle AP; Jeffreys SR; Mose LE; Simons JV; Soloway MG; Baylin SB; Berman BP; Bootwalla MS; Danilova L; Herman JG; Hinoue T; Laird PW; Rhie SK; Shen H; Triche T; Weisenberger DJ; Carter SL; Cibulskis K; Chin L; Zhang JJ; Getz G; Sougnez C; Wang M; Dinh H; Doddapaneni HV; Gibbs R; Gunaratne P; Han Y; Kalra D; Kovar C; Lewis L; Morgan M; Morton D; Muzny D; Reid J; Xi L; Cho J; Dicara D; Frazer S; Gehlenborg N; Heiman DI; Kim J; Lawrence MS; Lin P; Liu YY; Noble MS; Stojanov P; Voet D; Zhang H; Zou L; Stewart C; Bernard B; Bressler R; Eakin A; Iype L; Knijnenburg T; Kramer R; Kreisberg R; Leinonen K; Lin J; Liu YY; Miller MML; Reynolds SM; Rovira H; Shmulevich I; Thorsson V; Yang D; Zhang W; Amin S; Wu CCC-J; Wu C-CC-J; Akbani R; Aldape K; Baggerly K. a.; Broom B; Casasent TD; Cleland J; Creighton CJ; Dodda D; Edgerton M; Han L; Herbrich SM; Ju Z; Kim H; Lerner S; Li J; Liang H; Liu W; Lorenzi PL; Lu Y; Melott J; Mills GB; Nguyen L; Su X; Verhaak R; Wang W; Weinstein JN; Wong A; Yang Y; Yao J; Yao R; Yoshihara K; Yuan Y; Yung AK; Zhang N; Zheng S; Ryan M; Kane DW; Aksoy BA; Ciriello G; Dresdner G; Gao J; Gross B; Jacobsen A; Kahles A; Ladanyi M; Lee W; Lehmann K-V; Miller MML; Ramirez R; Ratsch G; Reva B; Sander C; Schultz N; Senbabaoglu Y; Shen R; Sinha R; Sumer SO; Sun Y; Taylor BS; Weinhold N; Fei S; Spellman P; Benz C; Carlin D; Cline M; Craft B; Ellrott K; Goldman M; Haussler D; Ma S; Ng S; Paull E; Radenbaugh A; Salama S; Sokolov A; Stuart JM; Swatloski T; Uzunangelov V; Waltman P; Yau C; Zhu J; Hamilton SR; Abbott S; Abbott R; Dees ND; Delehaunty K; Ding L; Dooling DJ; Eldred JM; Fronick CC; Fulton R; Fulton LL; Kalicki-Veizer J; Kanchi K-L; Kandoth C; Koboldt DC; Larson DE; Ley TJ; Lin L; Lu C; Magrini VJ; Mardis ER; McLellan MD; McMichael JF; Miller C. a.; O’Laughlin M; Pohl C; Schmidt H; Smith SM; Walker J; Wallis JW; Wendl MC; Wilson RK; Wylie T; Zhang Q; Burton R; Jensen M. a.; Kahn A; Pihl T; Pot D; Wan Y; Levine D. a.; Black AD; Bowen J; Frick J; Gastier-Foster JM; Harper H. a.; Helsel C; Leraas KM; Lichtenberg TM; McAllister C; Ramirez NC; Sharpe S; Wise L; Zmuda E; Chanock SJ; Davidsen T; Demchok J. a.; Eley G; Felau I; Ozenberger B. a.; Sheth M; Sofia H; Staudt L; Tarnuzzer R; Wang Z; Yang LL; Zhang JJ; Omberg L; Margolin A; Raphael BJ; Vandin F; Wu H-T; Leiserson MDM; Benz SC; Vaske CJ; Noushmehr H; Wolf D; Veer LV; Collisson E. a.; Anastassiou D; Ou Yang T-H; Lopez-Bigas N; Gonzalez-Perez A; Tamborero D; Xia Z; Li W; Cho D-Y; Przytycka T; Hamilton M; McGuire S; Nelander S; Johansson P; Jornsten R; Kling T; Sanchez J; Shaw KRM The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45 (10), 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Leiserson MDM; Vandin F; Wu H-T; Dobson JR; Eldridge JV; Thomas JL; Papoutsaki A; Kim Y; Niu B; McLellan M; Lawrence MS; Gonzalez-Perez A; Tamborero D; Cheng Y; Ryslik GA; Lopez-Bigas N; Getz G; Ding L; Raphael BJ Pan-Cancer Network Analysis Identifies Combinations of Rare Somatic Mutations across Pathways and Protein Complexes. Nat. Genet. 2015, 47 (2), 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Akbani R; Ng PKS; Werner HMJ; Shahmoradgoli M; Zhang F; Ju Z; Liu W; Yang J-Y; Yoshihara K; Li J; Ling S; Seviour EG; Ram PT; Minna JD; Diao L; Tong P; Heymach JV; Hill SM; Dondelinger F; Stadler N; Byers LA; Meric-Bernstam F; Weinstein JN; Broom BM; Verhaak RGW; Liang H; Mukherjee S; Lu Y; Mills GB A Pan-Cancer Proteomic Perspective on The Cancer Genome Atlas. Nat. Commun. 2014, 5 (May), 3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhang G; Fenyö D; Neubert T. a. Evaluation of the Variation in Sample Preparation for Comparative Proteomics Using Stable Isotope Labeling by Amino Acids in Cell Culture. J. Proteome Res. 2009, 8, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Piehowski PD; Petyuk VA; Orton DJ Sources of Technical Variability in Quantitative LC-MS Proteomics: Human Brain Tissue Sample Analysis. J. Proteome Res. 2013, 12 (5), 2128–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Scheerlinck E; Dhaenens M; Van Soom A; Peelman L; De Sutter P; Van Steendam K; Deforce D Minimizing Technical Variation during Sample Preparation prior to Label-Free Quantitative Mass Spectrometry. Anal. Biochem. 2015, 490, 14–19. [DOI] [PubMed] [Google Scholar]
- (52).Rauniyar N; Yates JR Isobaric Labeling-Based Relative Quanti Fi Cation in Shotgun Proteomics. J. Proteome Res. 2014, 13, 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.