

Abstract
Krox20/Egr2 is a zinc finger transcription factor that plays essential roles in several developmental processes, including peripheral nervous system myelination by Schwann cells, where it acts as a master gene regulator. Krox20 is known to interact with cofactors of the Nab family and a mutation affecting isoleucine 268, which prevents this interaction, has been shown to result in congenital hypomyelinating neuropathy in humans. To further investigate the role of this interaction, we have introduced such a mutation, Krox20I268F, in the mouse germ line. Clinical, immunohistochemical, and ultrastructural analyses of the homozygous mutants reveal that they develop a severe hypomyelination phenotype that mimics the human syndrome. Furthermore, a time-course analysis of the disease indicates that it follows a biphasic evolution, the hypomyelination phase being followed by a dramatic demyelination. Although for the regulation of most analyzed Krox20 target genes the mutation behaves as a loss of function, this is not the case for a few of them. This differential effect indicates that the molecular function of the Krox20–Nab interaction is target dependent and might explain the degradation of the residual myelin, because of imbalances in its composition. In conclusion, this work provides a novel and useful model for severe human peripheral neuropathies.
Keywords: transcription factor, Charcot–Marie–Tooth disease, animal model, myelin, hindbrain segmentation, cranial ganglia
Introduction
The lineage leading to the generation of myelinating Schwann cells includes several intermediates: Schwann cell precursors, and immature and promyelinating Schwann cells (Mirsky and Jessen, 1996; Zorick and Lemke, 1996; Jessen and Mirsky, 1998). The engagement of immature Schwann cells into the process of myelination has been shown to rely on the expression of a few specific transcription factors. Hence, Oct6/SCIP and then Brn2 are required together with Sox10 for the transition between immature and myelinating Schwann cells (Bermingham et al., 1996; Britsch et al., 2001; Jaegle et al., 2003). A major role of these factors is to synergistically and directly activate the transcription of another transcription factor gene, Krox20/Egr2 (Chavrier et al., 1988; Ghislain et al., 2002; Ghislain and Charnay, 2006). Krox20 is absolutely required for the progression of promyelinating Schwann cells into myelination and for the expression of most myelin-specific genes (Topilko et al., 1994; Le et al., 2005a). Additional evidence suggest that Krox20 constitutes a master regulator of the myelination process, because its forced expression results in an arrest in cell proliferation (Parkinson et al., 2004) and in the induction of genes encoding structural myelin proteins and enzymes involved in lipid synthesis (Nagarajan et al., 2001; Le et al., 2005a), presumably as a result of direct transcriptional activation (Bondurand et al., 2001; Musso et al., 2001; Taveggia et al., 2004; LeBlanc et al., 2005, 2006).
In accordance, various Krox20 mutations have been associated with human peripheral neuropathies, including different forms of Charcot–Marie–Tooth disease, Dejerine–Sottas Syndrome, and congenital hypomyelinating neuropathy (CHN) (Warner et al., 1998; Bellone et al., 1999; Timmerman et al., 1999; Pareyson et al., 2000; Boerkoel et al., 2001; Yoshihara et al., 2001; Szigeti et al., 2007). Whereas most of these mutations affect the DNA binding domain, another type corresponds to the specific replacement of the isoleucine at position 268 (Warner et al., 1998; Szigeti et al., 2007). This amino acid has been shown to be required for the interaction of the closely related factor Egr1/Krox24 with its cofactors Nab1 and Nab2 (Swirnoff et al., 1998; Warner et al., 1999), suggesting that this interaction is also essential for Krox20 function. The Nab proteins were initially described as antagonists of Krox20 activity in several instances (Russo et al., 1995; Swirnoff et al., 1998), including the regulation of the myelin gene Mpz in cultured cells (LeBlanc et al., 2006). However, an agonistic role is supported by the double mutation of Nab1 and Nab2 in mice, which leads to severe hypomyelination of the PNS (Le et al., 2005b). These apparently conflicting data question the precise role of the Krox20–Nab interaction in vivo. To clarify this issue and to develop a bona fide mouse model for the CHN associated with the isoleucine 268 (I268) mutation, we generated and characterized a mouse mutant carrying a point mutation leading to the replacement of I268 by a phenylalanine (F), a change that we have shown abolishes the interaction with Nab proteins.
Materials and Methods
Generation of mutant mice and genotyping.
The Krox20I268Fneo allele was obtained by modification of a construct used previously for the generation of a Krox20floxneo allele (Taillebourg et al., 2002). The KpnI-BamHI fragment containing the loxP site located in the intron was replaced by the wild-type fragment. The I268F mutation (conversion of a TTC codon into ATC) was introduced into an intermediate construct by PCR and the BamHI-SfiI fragment carrying this mutation was used to replace the wild-type fragment in the targeting construct. The Krox20I268Fneo allele was transferred to the mouse germ line in the Institut Clinique de la Souris (Strasbourg, France) by homologous recombination in embryonic stem (ES) cells followed by blastocyst injection. To generate the Krox20I268F allele, Krox20I268FNeo males were mated with PGK-Crem females, which express Cre in the germ line (Lallemand et al., 1998). Presence of the I268F mutation in the founders was verified by PCR amplification of a fragment using primers m1 (5′-TCC ACC ACT TCC ACC TCC TC-3′) and m2 (5′-ACA ACC TTC TGC TGG GCA GG-3′), and subsequent sequencing using the following primer: 5′-CAC GGT GCT GCT GGC CCA - 3′. Genotyping was then performed using primers p1 (5′-GGG CTT GCA TTC TAC AGT GGT GGT C-3′), p2 (5′-AGT TGA CAG CCC GAG TCC AGT GG-3′), and p3 (5′-TTG GCG CCT ACC GGT GGA TGT GG-3′) (see Fig. 1 A). Recombinant DNA and animal manipulations were performed according to French and European Union regulations.
Figure 1.

Generation of the Krox20I268F allele. A, Schematic representation of the different Krox20 alleles, including the wild-type gene, the targeting construct, and the intermediate (Krox20I268Fneo) and final (Krox20I268F) alleles carrying the I268F point mutation. The point mutation is indicated by an asterisk and the loxP sites are symbolized by black arrowheads. Arrows indicate the positions of the different primers used for PCR amplification. The sizes of the amplified fragments used for genotyping are indicated underneath. B, PCR analysis of animals issued from crosses between Krox20I268Fneo/+ or Krox20I268F/+ mice. Wild-type (+/+), heterozygous (+/I268Fneo or +/I268F), and homozygous (I268Fneo/I268Fneo or I268F/I268F) mutant animals are shown. C, Western blot analysis performed on 40 μg of protein extracted from sciatic nerves from P4 wild-type, heterozygous, and homozygous I268F mutant animals. The blot was probed with an antibody directed against Krox20 and reprobed with an anti-actin antibody to verify that equal amounts of proteins had been loaded. The comparison reveals no significant variation in the levels of Krox20 protein between the different genotypes. D, Photograph of wild-type (white arrowhead) and mutant (black arrowhead) littermates at P15. The mutant is smaller and shows tremors and weakness of the hindlimbs. E, At P20, the same mutant has developed a paralysis of both anterior and posterior limbs and is affected by breathing problems. Ba, BamHI; Bg, BglII; RI, EcoRI; S, SpeI; S1, SfiI; K1, KpnI; N1, NotI.
Quantitative RT-PCR.
For quantitative RT-PCR analysis, total RNA was isolated from mouse sciatic nerves using the RNAqueous-micro kit (Ambion) and RNeasy Fibrous Tissue Mini kit (Qiagen) for postnatal day 4 (P4) and (P20) animals, respectively. Total RNA (500 ng) was used to generate cDNA using the pSuperscript III RNase H reverse transcriptase (Invitrogen). Relative levels of gene expression were measured in triplicate by quantitative reverse transcriptase (RT)-PCR on a Lightcycler (Roche) using the Quanti Tect SYBR Green kit (Qiagen) and cyclophilin, pPox (protoporphyrinogenoxidase), and tubulin for normalization, using at least two sets of wild-type and mutant animals for each comparison. Primer sequences are available on request.
Electron microscopy.
Sciatic nerves were fixed for 3 h in 2.5% glutaraldehyde in Soerensen's buffer and osmificated for 1 h in 2% OsO4 (Polysciences). Nerves were rinsed in Soerensen's buffer, dehydrated in graded acetone, and embedded in Epon 812-Araldite. For light microscopy analysis, semithin sections were stained with toluidine blue. Ultrathin sections were stained with uranyl acetate and lead citrate and observed with a Philips CM10 electron microscope. The g-ratio was calculated by division of the axonal diameter by total fiber diameter. Measurements of g-ratio were performed on ∼150 axons with a diameter of at least 1 μm per mouse, using three mice for each genotype. Statistical significance was evaluated using the Student's test.
Cell proliferation and apoptosis assays.
To estimate the rate of cell proliferation, an intraperitoneal injection of bromodeoxyuridin (BrdU; 30 mg/kg body weight; Sigma) was performed and the animals were killed 2 h after the injection. Sciatic nerves were collected and processed for BrdU immunostaining, and nuclei were counterstained with bisbenzimide (Hoechst 33342; Sigma). The rate of apoptosis was estimated using a terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining kit (Apoptag Red In Situ Apoptosis Detection kit; Millipore). The number of BrdU-positive or TUNEL-positive nuclei and the total number of nuclei were determined (∼1500 nuclei for each condition).
Western blotting, immunohistochemistry, and in situ hybridization.
Western blotting with anti-Krox20 (1:2000; Covance) and anti-actin (1:5000; Sigma) polyclonal antibodies have been performed on sciatic nerves as described previously (Decker et al., 2006). For immunochemistry on sections, nerves were collected, fixed in 4% paraformaldehyde for 2 h, incubated overnight at 4°C in 20% sucrose, embedded in Tissue-Tek OCT compound (Gassalem), and frozen in isopentane (−60°C). Longitudinal or transversal sections (16 μm thick) were collected using a Leica cryotome. Sections were incubated for 30 min in 0.1 m PBS containing 5% goat serum and 0.25% Triton X-100 and then overnight at 4°C with the following primary antibodies: a rat polyclonal anti-maltose-binding protein (MBP) antibody (1:100; Millipore), a mouse monoclonal anti-Mpz antibody (1:5, mouse IgG hybridoma) (Yoshimura et al., 1996), and a rabbit anti-neurofilament NF200 antibody (1:100; Sigma). Sections were then incubated for 1 h with appropriate fluorophore-conjugated secondary antibody (Jackson ImmunoResearch). For BrdU detection, sections were treated for 30 min at room temperature with 2N HCl in 0.1 m PBS containing 0.25% Triton X-100, washed twice in 0.1 m borate buffer, pH 8.5, and incubated at 4°C with a rat anti-BrdU antibody (Oxford Biotech), which was subsequently revealed with a Cy3-labeled anti-rat antibody (Jackson ImmunoResearch). Nuclei were counterstained with Hoechst 33342. Sections were mounted in Aqua poly/Mount (Polysciences) and analyzed on Leica DMRD and TCS 4D confocal microscopes. Neurofilament immunochemistry on whole-mount 10.5 d postcoitum embryos was performed as described previously (Helmbacher et al., 2000). Embryos were then photographed using a Nikon SMZ1500 system. For in situ hybridization, postimplantation mouse embryos were recovered at the appropriate stage, fixed overnight at 4°C in PBS containing 4% paraformaldehyde, and progressively dehydrated in methanol and processed for whole-mount in situ hybridization as described previously (Seitanidou et al., 1997). The origins of the digoxigenin probes were as follows: Nab1 and Nab2 (Mechta-Grigoriou et al., 2000), Krox20 (Wilkinson et al., 1989), EphA4 (Gilardi-Hebenstreit et al., 1992). Flat-mounted neural tubes were photographed using a Leica DMRD microscope.
Results
Evolutive and fatal paralysis in Krox20I268F/I268F mutants
To evaluate the effect of the I268F mutation on the interaction between Krox20 and Nab proteins, we performed cotransfections of expression vectors into COS-7 cells, followed by immunoprecipitation of the cell extracts with an antibody directed against Krox20. Western blotting analysis indicated that Nab2 was coimmunoprecipitated with wild-type Krox20, but not with the protein carrying the I268F mutation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This indicates that, as expected, the mutation prevents the interaction with Nab proteins.
To generate the Krox20I268F allele, we first constructed a targeting vector carrying the I268F mutation and the neomycine (neo) resistance gene (Fig. 1 A), which was introduced into ES cells by homologous recombination and then transferred into the mouse germ line to obtain the Krox20I268Fneo allele. In this construct the neo gene was flanked by two loxP sites and the Krox20I268F allele was subsequently obtained by crossing a heterozygous Krox20I268Fneo male with PGK-Crem females, in which Cre is expressed in the germ line (Lallemand et al., 1998). The Krox20I268F allele differs from the wild-type allele only by the presence of the I268F mutation and of a residual loxP site located in the 3′ flanking region (Fig. 1 A). After verification of the presence of the I268F mutation in the founders of the line, the genotyping of the animals was performed on the basis of the presence or absence of the loxP site (Fig. 1 B). To evaluate the effect of the mutation on Krox20 expression, we performed a Western blot analysis on protein extracts from P4 wild-type, heterozygous, and homozygous mutant sciatic nerves. It showed that the presence of the I268F mutation and of the loxP site did not affect the level of Krox20 protein (Fig. 1 C). Therefore these mutations are not likely to directly affect Krox20 expression, in particular its stability, and any phenotype attached to the Krox20I268F allele should be the result of changes in the activity of the protein.
No obvious phenotype was observed in Krox20I268F/+ heterozygous animals, which were normally fertile (data not shown). Homozygous mutant mice were obtained at the expected Mendelian ratios. However, they were usually slightly smaller than wild-type or heterozygous littermates at birth, and the difference increased during the second postnatal week so that at P15 their weight was reduced to ∼80% of wild-type littermates, and deteriorated further in the following days (Fig. 1 D) (data not shown). The same period marked the appearance of obvious locomotion defects, including tremors and abnormal hindlimb coordination. During the third week, the condition of the mutants dramatically worsened. At P17, all homozygous mutant animals showed severe difficulties in muscular coordination and in positioning their hindlimbs and forelimbs (supplemental Video 1, available at www.jneurosci.org as supplemental material). These defects progressed to a paralysis of posterior and, subsequently, anterior limbs, leading to a complete loss of locomotion at 3 weeks (Fig. 1 E, supplemental Video 2, available at www.jneurosci.org as supplemental material). The animals usually died at around that time. High-frequency breathing was also observed, suggesting that the death might be linked to breathing defects. The evolution of Krox20I268F/I268F animals was very reproducible: the progression of the phenotype varied only by a few days from animal to animal and all died between P19 and P22. These data indicate that the I268F substitution leads to evolutive and fatal paralysis at the homozygous state.
Krox20I268F/I268F mice show peripheral hypomyelination
The phenotype of Krox20I268F/I268F animals led us to investigate possible alterations in peripheral nerves. We first performed an analysis of brachial and sciatic nerves at P17, when locomotion is already severely affected. Macroscopic examination of these nerves revealed that they were abnormal in the homozygous mutant: they appeared translucent and lacked the typical striated organization of myelinated fibers from wild-type or heterozygous animals (Fig. 2 A,B,E,F,I,J). Analysis of transverse semithin sections from sciatic nerves stained with toluidine blue (Fig. 2 C,G,K), followed by examination of ultrathin sections by electron microscopy (Fig. 2 D,H,L), established the existence of major myelin alterations in homozygous but not heterozygous mutants. In Krox20I268F/I268F animals, the number of myelinated axons was diminished and more strikingly the thickness of the myelin sheath appeared to be systematically reduced. Hence, amyelinated large-caliber axons were frequently found in homozygous mutant nerves (Fig. 2 L,M), a feature that was never observed in wild-type or heterozygous animals at this age (Fig. 2 D,H). Nevertheless, no sign of axonopathy was detected and neither the diameter nor the number of axons was significantly affected (data not shown). The nonmyelinated axons were associated with Schwann cells, suggesting that these cells had established a one-to-one relationship with axons, a characteristic of the promyelinating stage. In addition, we frequently observed myelin unfoldings in homozygous mutant nerves (Fig. 2 L,N, arrows), but no onion bulb structures. Finally, macrophages were also occasionally found (Fig. 2 O), suggesting the existence of an inflammatory reaction. Although the heterozygous mutants presented no clinical phenotype nor any myelin defect at P17 (Fig. 2 E–H), we extended their investigation by performing ultrastructural analyses of sciatic nerves at P14 and 6 months of age. No abnormalities were observed in these animals compared with wild-type littermates (data not shown).
Figure 2.
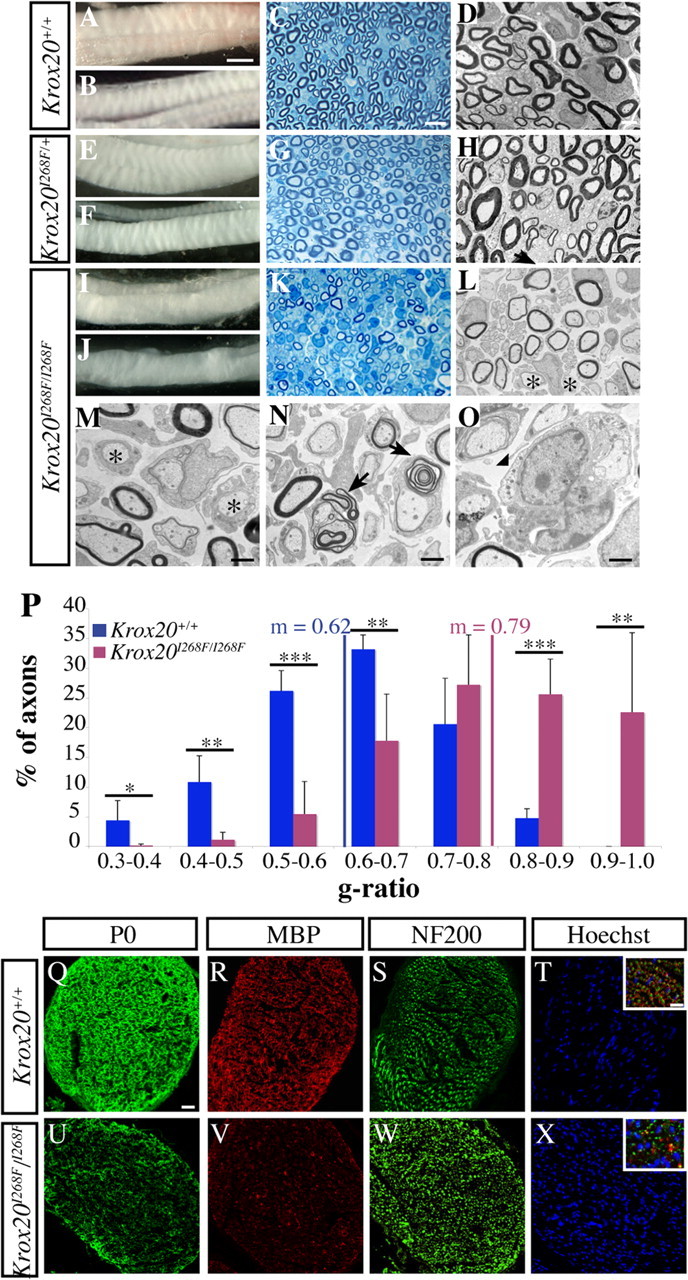
Krox20I268F/I268F mice show a peripheral hypomyelination at P17. Macroscopic examination of brachial (A, E, I) and sciatic nerves (B, F, J) indicates that the homozygous mutant nerves are translucent and show an abnormal organization. Observations of toluidine blue-stained semithin sections (C, G, K) and electron microscopy analysis of ultrathin sections (D, H, L) of sciatic nerves show that the myelin from heterozygous mice is similar to the wild type, whereas it is altered in homozygous mutant animals. M–O, Higher magnifications confirm the presence of totally amyelinated fibers (M, asterisks) and of myelinated fibers with an abnormally thin sheath and of unfoldings (N, arrows) in the mutant mice. Macrophages (O, arrowhead) are also observed. P, Distribution of P17 axons with a caliber superior to 1 μm according to their g-ratio (axon diameter/total myelinated fiber diameter). The statistical significance was analyzed using the Student's t test: *p < 0.05; **p < 0.025; ***p < 0.005. The mean g-ratio is 0.79 in the mutant compared with 0.62 in the wild type (p < 0.005). Error bars indicate SEM. Q–X, Analysis of transverse sciatic nerve sections by immunostaining. P0 and MBP expression is strongly decreased in the mutant (U, V) compared with the wild type (Q, R). Staining for the axonal marker NF200 indicates that axonal density is not significantly affected in the mutant (S, W), whereas nuclear staining (Hoechst 33342) reveals an increase in Schwann cell number (T, X). In the insets (T, X), coimmunostaining with MBP, NF200, and DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride) confirms that MBP is localized around the axons. Scale bars: (in A) A, B, E, F, I, J, 1 mm; (in C) C, G, K, 25 μm; (in D) D, H, L, 5 μm; M, N, 1 μm; O, 0.5 μm; (in Q) Q–X, 50 μm; (in T) insets, 20 μm.
To quantify the variations in myelin sheath thickness of homozygous mutant animals, we performed a morphometric analysis of the ultrastructural data, measuring the g-ratio (axon diameter over myelinated fiber total diameter). A mean value of 0.79 was found in homozygous mutants versus 0.62 in wild-type animals, establishing the existence of a very significant difference (p < 0.005). Consistently, analysis of axonal distribution according to the g-ratio indicated a dramatic shift to higher values in mutant compared with wild-type animals, with in particular >20% of the axons having a g-ratio ranging from 0.9 to 1 in the mutants, corresponding to a very severe to total loss of myelin (Fig. 2 P). In conclusion, this analysis revealed that the I268F mutation results at P17 in a clear hypomyelination of the sciatic nerve, with both a decrease in the number of myelinated fibers and a reduction of myelin sheath thickness. This phenotype is likely to affect other peripheral nerves as suggested by preliminary observations of the brachial nerves (Fig. 2 I) (data not shown) and to be responsible for the observed locomotion defects.
To investigate whether the hypomyelination observed in the homozygous mutants was correlated with a deficiency in myelin protein accumulation, we performed an immunostaining analysis of the distribution of MBP and P0, two major myelin proteins, involved in its compaction and stabilization. Both MBP and P0 levels were strongly reduced in mutant sciatic nerves compared with the wild-type situation (Fig. 2 Q,U,R,V). As expected from the ultrastructural analysis, immunostaining for neurofilaments, which labels axons, did not reveal any major difference in axonal density between homozygous mutant and wild-type nerves (Fig. 2 S,W). In contrast, nuclei staining revealed an increase in cell density in Krox20I268F/I268F nerves (Fig. 2 T,X). Most of these cells are Schwann cells, as indicated by the ultrastructural analysis and immunolabeling for S100 (a marker of immature and mature Schwann cells) at P14 (supplemental Fig. 2B,E, available at www.jneurosci.org as supplemental material) (data not shown). Therefore, the hypomyelination of Krox20I268F/I268F nerves is accompanied by a dramatic reduction in the level of major myelin proteins, despite a likely increase in the number of Schwann cells.
To further investigate the characteristics of the Schwann cells in the mutant nerve, we performed double labeling at P17 for Krox20 and SCIP, which is normally not expressed anymore at this stage in myelinating Schwann cells. We observed an increase in Krox20-positive cells compared with the wild-type situation, presumably reflecting the general increase in the number of Schwann cells (supplemental Fig. 2G,J, available at www.jneurosci.org as supplemental material). In contrast, the increase in SCIP-positive cells was much more dramatic, indicating an elevated proportion of these cells (supplemental Fig. 2H,K, available at www.jneurosci.org as supplemental material). Finally, analysis of the expression of Sox2, an immature Schwann cells marker, also revealed a significant increase in the proportion of this cell type (data not shown). Altogether, these data indicate that the I268F mutation results in an accumulation of immature and promyelinating Schwann cells in the nerves.
The I268F neuropathy evolves in two distinct phases
To better characterize the hypomyelinating phenotype observed in Krox20I268F/I268F mutants, we performed an electron microscopy analysis of the sciatic nerve between P8 and P20. This revealed that the myelination defect involves two phases. In the early one, extending until ∼P17, myelin appears to form, but at a reduced rate compared with wild-type animals, resulting in a delay in the formation of the sheath. Hence, at P8 in Krox20I268F/I268F animals, large-caliber axons are either amyelinated or covered with a very thin myelin sheath, compared with the wild-type situation, where myelination is widely engaged and some sheaths have already reached mature size (Fig. 3 A,E). At P14, the situation has only marginally improved in homozygous mutant animals, with a slight increase in the thickness of myelin sheaths, but still a large number of amyelinated fibers, whereas myelination is almost complete in wild-type animals (Fig. 3 B,F). Furthermore, uncompacted fibers (Fig. 3 I) and unfoldings (data not shown) are observed in the mutant. At P17, although the mutant nerves are still hypomyelinated compared with the wild type, the condition has improved, with a higher proportion of myelinated axons and an increased, although still abnormal, thickness of the myelin sheath (Fig. 3 C,G). However, at ∼P20, when myelination is completed in wild-type animals, the situation appears to have suddenly degraded in mutant mice, with a dramatic reduction of the proportion of myelinated axons and presence of macrophages in the process of eliminating myelin debris (Fig. 3 D,H,J). This marks a second phase in the myelination phenotype, which begins after P17 and continues until death at ∼3 weeks and involves a rapid degradation of previously formed myelin. The destruction of the myelin may be related to the presence of unfoldings and uncompacted myelin observed at P17 and correlated with the presence of infiltrated macrophages that were observed from P17.
Figure 3.

Time course of myelination reveals a biphasic defect in the mutant. A–J, Electron micrographs of sciatic nerve transverse sections between P8 and P20. At P8 and P14, the mutant nerves are hypomyelinated compared with the wild type, with amyelinated fibers and thinner myelin sheaths (A, B, E, F). At P17, myelination in mutant mice has improved, but the nerves are still hypomyelinated (C, G). At P20, when myelination is completed in wild-type animals, mutant mice show a degraded situation compared with P17 (D, H). I, J, Higher magnifications corresponding to homozygous mutant nerves reveal presence of uncompacted myelin at P14 (I) and of macrophages at P20 (J). Scale bar: (in A) A–H, 2.5 μm; I, J, 0.5 μm.
The myelination delay correlates with maintenance of Schwann cell proliferation
As indicated above, at P17, the myelination defect in mutant sciatic nerves is accompanied by a probable increase in the number of Schwann cells. To investigate the origin of this accumulation, we looked for possible differences in the rates of cell proliferation and apoptosis between Krox20I268F/I268F and wild-type sciatic nerves. The proportion of cells in S phase was evaluated by BrdU incorporation after a 2 h pulse, providing an estimate of the rate of cell proliferation. At both P8 and P14, a twofold to threefold increase in the proportion of BrdU-positive cells was observed in mutant compared with wild-type animals (Fig. 4 A–D,I). In contrast, no significant difference was found at P17 and P20 (Fig. 4 I). To characterize these proliferating cells, we performed double labeling for BrdU and S100 at P14. Most of the BrdU-positive cells were found to express S100, indicating that the proliferating cells belong to the Schwann cell lineage (supplemental Fig. 2A–F, available at www.jneurosci.org as supplemental material).
Figure 4.

Persistence of Schwann cell proliferation in Krox20I268F/I268F mutant mice. A–D, The rates of cell proliferation were estimated by BrdU incorporation as revealed by immunostaining 2 h after intraperitoneal injection. The micrographs show BrdU staining alone (A, C) or combined with nuclei counterstaining using Hoechst 33342 (B, D) on sciatic nerve sections from wild-type or mutant mice at P14. E–H, Apoptosis was estimated using TUNEL staining. The micrographs show TUNEL staining alone (E, G) or combined with nuclei counterstaining using Hoechst 33342 (F, H) on sciatic nerve sections from wild-type or mutant mice at P20. Arrows in B, D, F, and H point to double-labeled cells. I, J, Representation of the percentage of BrdU- (I) or TUNEL-positive (J) nuclei at different stages. There is an increase in the rate of proliferation in mutant mice compared with wild-type animals at P8 and P14, whereas no significant difference in apoptosis was observed at any stage. Histogram data are given as mean ± SEM for three animals. Statistical significance was determined using the Student's t test: *p < 0.05; **p < 0.01. Error bars indicate SEM. Scale bar: 25 μm.
The level of apoptosis was estimated by TUNEL. No significant difference was observed between Krox20I268F/I268F and wild-type mice at any stage (Fig. 4 E–H,J). Altogether, these data suggest that the accumulation of Schwann cells observed at P17 in the mutants is essentially caused by an increased cell proliferation rate at earlier stages, possibly resulting from a defect in arrest of proliferation. Hence, there is a striking correlation between the delay in myelination and this increase in cell proliferation at P8 and P14. In contrast, when most Schwann cells have engaged into myelination at P17, they have also stopped proliferating. Apoptosis does not appear to play a role in the variation in Schwann cells number, even during the period of active myelin degradation. In addition, this degradation process does not lead to reactivation of cell proliferation.
Molecular phenotype of the Krox20I268F/I268F mutant
Because Krox20 is a transcription factor, the I268F mutation is expected to result in modifications in gene expression, some of which being responsible for the observed Schwann cell phenotype. To identify such molecular alterations, we used quantitative RT-PCR to compare the levels of mRNA of a subset of genes between Krox20I268F/I268F and wild-type animals. These genes were selected on the basis of their known implication in myelination or Schwann cell development, or of their reported regulation during these processes (Jessen and Mirsky, 2005; Le et al., 2005a). In a first series of experiments, we performed the measurements at P4, an early stage in the myelination process, and at P20, when the myelin sheath is being degraded in the mutant. The outcome of this analysis allowed us to classify the tested genes into three groups (Fig. 5). The first one corresponds to genes showing an at least twofold higher expression in the mutant at P4 and P20 or only at P20 (Fig. 5 A), the second one to genes showing an at least twofold reduced expression at P4 or P20 (Fig. 5 B), and the third to genes with more limited variations (Fig. 5 C). It is striking to observe that the behavior of the vast majority of the tested genes is consistent with the I268F mutation reducing the activity of the Krox20 protein. Hence, most of the genes belonging to the first group (overexpressed in the mutant) are associated with Schwann cell proliferation, as c-jun and cyclin D1 (CcnD1) (Parkinson et al., 2004), or correspond to markers of immature (FoxD3, Id2, Id4, Sox2) (Le et al., 2005a), promyelinating (SCIP/Oct6) (Zorick et al., 1996), or nonmyelinating (Krox24) (Topilko et al., 1997) Schwann cells. These genes are normally downregulated during myelination driven by Krox20. The genes of the second group, whose expression is reduced in the mutant, are known or expected to be upregulated by Krox20: they either encode myelin proteins (Mpz, Prx) (Wood and Engel, 1976; Gillespie et al., 1994), are associated with the inhibition of c-Jun activity and of cell proliferation (Jip1) (Parkinson et al., 2004), or are known to be downregulated in a Krox20 hypomorphic mutant (Igf2) (Le et al., 2005a). To follow the evolution of the expression of some of the misregulated genes at a stage of hypomyelination we also performed RT-PCR analyses at P17 on a subset of genes. The results were fully consistent with the data obtained at P20 (Fig. 5 A,B) showing that these altered genes are not specifically linked in the mutant to the second phase of the phenotype (myelin degradation). Finally, we also tested genes associated with cholesterol and lipid synthesis (Hmgcr and Cgt) at P17 and found that they were downregulated fourfold to eightfold in the mutant (data not shown). These data indicate that most of the disregulated genes in the mutant behave in a manner consistent with an inability of Schwann cells to efficiently differentiate and engage into the myelination process. They are consistent with the immunolabeling experiments that revealed a higher proportion of SCIP- and Sox2-positive cells in the mutant nerves (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (data not shown).
Figure 5.
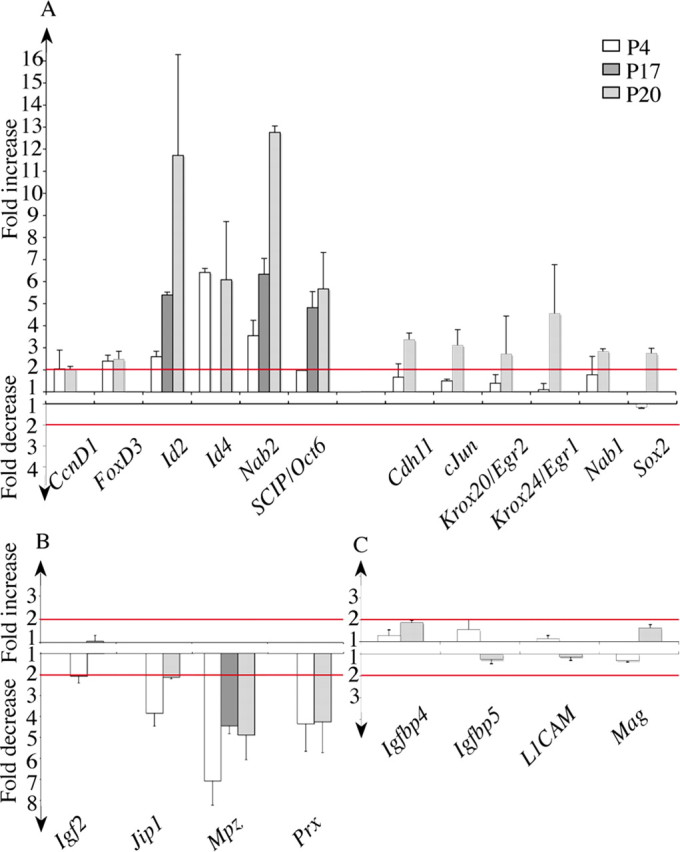
The myelination defect correlates with abnormal levels of expression of several genes involved in Schwann cell differentiation or proliferation. The mRNA levels were determined by RT-PCR analysis on samples of sciatic nerves at P4, P17, and P20. The graphs show the ratios of mRNA levels in homozygous mutant versus wild-type animals, presented as fold increase or decrease. A, The expression of a first subset of genes is increased in mutant compared with wild-type animals. The ratios superior to the threshold of twofold (indicated by the top red line) were considered significant. B, For a second subset of genes, the expression is decreased in mutant compared with wild-type animals. The ratios superior to the threshold of twofold (indicated by the lower red line) were considered significant. C, A last subset of genes showed only limited variations in expression. Error bars indicate SEM.
There are, however, a few genes that do not comply with this interpretation, implying that the I268F mutation acts as a simple loss of function. Nab1 and Nab2 are known to be positively regulated by Krox20 both in the developing hindbrain (Mechta-Grigoriou et al., 2000) (A. Desmazières, P. Charnay, and P. Gilardi-Hebenstreit, unpublished data) and in Schwann cells (Le et al., 2005a). Nevertheless, they are overexpressed in Krox20I268F/I268F animals (Fig. 5 A,B), rather dramatically in the case of Nab2 (12-fold increase), suggesting that the Krox20I268F mutation could reinforce the activation of these genes. Another exception is provided by L1CAM, an immature and nonmyelinating Schwann cell marker (Nieke and Schachner, 1985), which is expected to be repressed in presence of Krox20 (Le et al., 2005a). In this case, the expression is not significantly affected by the mutation, suggesting that I268F is neutral for the regulation of this gene.
In conclusion, most of the modifications in gene expression observed in Krox20I268F/I268F animals are consistent with a reduction of the activity of the Krox20 protein. However, the behavior of a few genes suggest that the situation is more complex and that in these cases the mutation has either no or an opposite effect.
The I268F mutation also affects cranial nerve organization
Because Krox20 plays an essential role in hindbrain patterning (Schneider-Maunoury et al., 1993, 1997, 1998; Seitanidou et al., 1997; Giudicelli et al., 2001; Voiculescu et al., 2001), we investigated the possibility that the I268F mutation might also affect this process. More specifically, because Krox20 is necessary to the development of rhombomere 3 (r3) and r5, we examined the expression of different hindbrain segmental markers to analyze the morphology and molecular identity of these rhombomeres. In situ hybridizations were performed between the 3 and 26 somite stages (ss) with probes for Krox20, Nab1, Nab2, and EphA4, which are all expressed in r3 and r5, and for Hoxb1, which is expressed in r4 and in the posterior hindbrain and spinal cord. Throughout this period, no difference was found between Krox20I268F/I268F and wild-type embryos in the position, size, and morphology of r3 and r5 (Fig. 6 A–L). Furthermore, no changes were observed in the expression of EphA4 and Hoxb1 (Fig. 6 K,L) (data not shown). However, minor modifications were detected in the levels of expression of Krox20 and Nab genes, with a persistence of Krox20 ventral expression (Fig. 6 E,F) and slight elevations in the levels of Nab1 mRNA in r3 and of Nab2 mRNA in r5 (Fig. 6 G–J). Intermediate levels were observed in heterozygous animals (data not shown). Altogether, these data suggest that in the hindbrain the situation is radically different from the PNS, because the Krox20I268F mutation does not have dramatic consequences on gene expression and development in the hindbrain, in contrast to the Schwann cells and to the null mutation, which affects both cell types. Moreover, as Krox20 is known to positively regulate its own expression as well as that of the Nabs in the developing hindbrain (Seitanidou et al., 1997; Mechta-Grigoriou et al., 2000; Giudicelli et al., 2001) (A. Desmazières, P. Charnay, and P. Gilardi-Hebenstreit, unpublished data), the slight upregulation of these genes can be interpreted as the I268F mutation resulting in a Krox20 gain of function in this tissue.
Figure 6.

The I268F mutation affects cranial nerve organization. A–F, Time-course analysis of Krox20 expression by in situ hybridization between E8 and E9.5. The only difference observed between Krox20I268F/I268F (B, D, F) and wild-type (A, C, E) embryos is a slight delay in the extinction of Krox20 expression in ventral r3 and r5 at 26 ss (E9.5) (F, arrows). G–J, Analysis of Nab1 and Nab2 expression by in situ hybridization at E8.5. Normalization of the signal by colabeling of the midhindbrain junction with an Fgf8 (fibroblast growth factor 8) probe suggests that Nab1 and Nab2 expression levels in r3 and r5 are increased in homozygous mutant (H, J) compared with wild-type (G, I) embryos. K, L, Analysis of EphA4 expression by in situ hybridization shows no modification in the mutant at E9.5. M–O, Cranial nerve organization was revealed by neurofilament immunolabeling performed on whole-mount embryos at E10.5. Homozygous mutant embryos show cranial nerve abnormalities: close apposition and partial intermingling of nerve roots IX (glossopharyngial) and X (vague) (N, white arrow) and fusion of ganglia VII/VIII (vestibuloacoustic) and V (trigeminal) (O, white arrow). pr3, Prerhombomere 3; mhb, midhindbrain junction.
Another phenotype associated with the Krox20 null mutation is a modification of the organization of cranial nerves and ganglia, with a fusion of trigeminal (V) and facial/acoustic (VII/VIII) ganglia and an abnormal proximity of glossopharyngeal (IX) and vagal (X) nerves (Schneider-Maunoury et al., 1993; Le et al., 2005a). We therefore also investigated whether the I268F mutation might affect the cranial nerve and ganglia organization. For this purpose, we revealed this organization by performing an immunostaining analysis of embryonic day 10.5 (E10.5) embryos with an antibody directed against neurofilaments (2H3). Whereas cranial nerve organization is not affected in heterozygous embryos (data not shown), it is altered in Krox20I268F/I268F embryos (Fig. 6 M–O), with a variable penetrance. The most common defect is a close apposition and partial intermingling of nerves IX and X (n = 5/6) (Fig. 6 N), whereas a fusion of ganglia V and VII/VIII is only occasionally observed (n = 1/6) (Fig. 6 O). Therefore, in this case, in contrast to the hindbrain, the I268F mutation appears to partially mimic the null mutation, consistent with a partial loss of function.
Discussion
In the present work, we investigated the consequences of the disruption of the interaction between Krox20 and its Nab cofactors in the mouse. We found that it leads to a peripheral neuropathy with a biphasic evolution that could constitute a novel and interesting model for human pathologies. In addition, this mutation induces a disorganization of cranial nerves, but no significant phenotype in the hindbrain. Finally, the molecular analysis of these phenotypes and their interpretation provides some clues about the functions of the Nab proteins in the process of Krox20-driven transcriptional control. These different issues are discussed in detail below.
Physiological consequences of the disruption of Krox20–Nab interaction
Krox20 is known to play an essential role in PNS myelination because total or partial loss-of-function mutations lead to complete absence of myelin (Topilko et al., 1994; Le et al., 2005a). In the present work, we established that the I268F mutation, which we have shown to prevent Krox20–Nab interaction, results in a severe hypomyelination. The pathology shows original characteristics, in particular a biphasic evolution. During the first two postnatal weeks, homozygous mutant animals suffer from peripheral hypomyelination, with progressive impairment and paralysis of the limbs. The hypomyelination is accompanied by an elevated proliferation rate of the Schwann cells, although it is not clear whether this abnormal proliferation is in part a consequence or a cause of the differentiation block. The situation deteriorates seriously during the third week with the development of a severe demyelination leading to animal death. This demyelination phase is accompanied by infiltration of the nerves by macrophages not observed at earlier stages. However it is not yet clear whether the inflammatory response is the direct cause of myelin degradation or the macrophages are simply recruited to clear the myelin debris resulting from another mechanism.
The initial hypomyelination observed in Krox20I268F homozygous mutants is similar to the phenotype reported for the double knock-out of Nab1 and Nab2 (Le et al., 2005b), suggesting that the main role of Nab factors may involve their interaction with Krox20. However, no time-course analysis was performed in the latter study and the possible occurrence of a demyelination phase has not been reported. Additional studies of the double Nab knock-out will therefore be required to determine whether the phenotypes are identical or whether the demyelination is specific to the Krox20I268F/I268F mutant. This latter possibility would suggest that the replacement of I268 not only affects Krox20–Nab interactions, but another Krox20 property as well.
We also shown that, in contrast to the loss-of-function mutations, the Krox20I268F mutation only results in minor modifications of gene expression in the developing hindbrain, affecting Krox20 itself and the Nabs. Whether these alterations have any morphological or physiological consequences remains to be determined. In any case, this indicates that, despite the specific presence of the Nab proteins in r3 and r5, the Krox20–Nab interaction is not likely to play a significant role in modulating the transcriptional activity of Krox20 in the hindbrain, in contrast to Schwann cells. Finally, the Krox20I268F mutation results in an abnormal organization of cranial nerves and ganglia, a phenotype that is reminiscent of the one associated with the complete loss of function, although less penetrant. Initially, this latter phenotype was proposed to constitute a consequence of the elimination of r3 and r5 in the Krox20 null mutant (Schneider-Maunoury et al., 1993, 1997). However, because these rhombomeres are maintained in the Krox20I268F mutant, another mechanism has to be found.
Molecular functions of the Nab factors
According to previous studies, the molecular function of the Nab proteins in the transcriptional regulation by Krox20 appears confusing: several studies (mostly involving cell culture systems) suggest that Nabs act as antagonists of Krox20 and, more generally, of Krox20 family members (Russo et al., 1995; Swirnoff et al., 1998), whereas the analysis of the Nab double knock-out mutant suggests that in Schwann cells they may constitute agonists of Krox20 (Le et al., 2005b). Our analysis establishes that the situation is indeed complex. In Schwann cells, prevention of Krox20–Nab interaction mostly appears as a partial loss of function of Krox20, suggesting that the Nabs are indeed acting as agonists of Krox20: the hypomyelination of the Krox20I268F mutant is fully consistent with this interpretation and, in addition, at the transcriptional level, this mutation affects the expression of most of the genes regulated during the myelination process in the same direction as the Krox20 null mutation. Krox20 has been proposed to directly control the transcription of several genes expressed in Schwann cells (Zorick et al., 1999; Bondurand et al., 2001; Musso et al., 2001; Taveggia et al., 2004; LeBlanc et al., 2006). The fact that we observe modifications in gene expression in Krox20I268F/I268F mutants both for upregulated and downregulated genes suggests that Nab proteins may cooperate with Krox20 for both activation and repression of its targets. However, we noticed that in a few cases Krox20 appears to act independently of Nabs or to be even antagonized by these factors: the regulation of the L1-CAM gene, which is derepressed in the absence of Krox20 (Le et al., 2005a), is not affected by the Krox20I268F mutation; Nab1 and Nab2, which are themselves under positive regulation by Krox20, are overexpressed in the Krox20I268F mutant. These latter data suggest a versatile molecular function for Nabs, an idea that is reinforced by the analysis of their role in hindbrain development. They also suggest that in some respects the I268F mutation may act as a gain of function. However, no phenotype could be associated with the heterozygous state in the PNS. We also shown in this study that prevention of Krox20–Nab interaction has only minor consequences on hindbrain patterning, such as a slight overexpression of Nab genes and persistence of Krox20 expression in the ventral hindbrain. Therefore, the function of Nabs in transcriptional activation or repression by Krox20 depends on the target gene and on the cell type considered. In Schwann cells, Nabs operate as Krox20 agonists for most target genes, whereas in the hindbrain most of the transcription controlled by Krox20 is independent from Nabs. In both systems, Nabs appear to antagonize their own expression.
A novel model for peripheral neuropathy
As expected, the pathology of Krox20I268F/I268F mice presents characteristics similar to those of its human counterpart, a rare disease that affects patients carrying a replacement of I268. This is an early and very severe peripheral neuropathy that has been classified as congenital hypomyelinating neuropathy (Warner et al., 1998). It leads to a near complete loss of myelin sheath with severe reduction of MBP immunoreactivity (Szigeti et al., 2007), like in the Krox20I268F/I268F mice. In both cases, the mutation appears recessive. The Krox20I268F mutant mice will therefore constitute a useful animal model for this form of CHN. However, as discussed above, the myelinopathy in Krox20I268F/I268F mutants is biphasic, a hypomyelination phase preceding the demyelination. It is not known whether the human disease also involves such an evolution. This possibility should now be investigated. Myelin degradation has been observed previously in others hypomyelinating mouse models, but it usually occurs much more slowly and at a later stage (Giese et al., 1992). In humans, acute or subacute myelin degradation has been described in several pathologies (Rudnik-Schoneborn et al., 1993; Ginsberg et al., 2004), and our mutant should therefore provide an interesting model to study this process as well.
The fact that Nabs act as agonists of Krox20 for most but not all of its target genes in Schwann cells may explain the biphasic evolution of the Krox20I268F/I268F phenotype. The involvement of Nabs in the activation of myelin genes or in the repression of genes required for Schwann cell proliferation accounts for the hypomyelination phase. However, our data raise the possibility that the expression of all myelin components is not affected in the same manner by the mutation and, therefore, that the myelin would not only be synthesized at low levels but also be of abnormal composition, resulting in its instability or in the induction of an immune response. In both cases, this would lead to myelin destruction and explain the demyelination that occurs during the third postnatal week. An alternative explanation would be that the relatively low level of Krox20 activity associated with the Krox20I268F mutation is sufficient for the initiation of myelination, but not for the maintenance of the myelin sheath. As a result, myelin would be degraded, in agreement with our previous observation that lack of Krox20 activity in the adult results in rapid demyelination (Decker et al., 2006).
In conclusion, in the present work we have shown that the disruption of Krox20–Nab interaction in the mouse should constitute a novel and interesting model for different forms of severe peripheral neuropathies in humans.
Footnotes
This work was supported by grants from Institut National de la Santé et de la Recherche Médicale, Ministère de l'Education Nationale de la Recherche et de la Technologie (MENRT), Agence Nationale pour la Recherche, Association pour la Recherche sur le Cancer, Association Française contre les Myopathies (AFM), and GIS-Institut des Maladies Rares. A.D. was supported by predoctoral fellowships from MENRT and AFM. We are grateful to Laurence Richard and Martine Piaser for technical assistance in electron microscopy, and to Piotr Topilko and Julien Ghislain for critical reading of this manuscript.
References
- Bellone E, Di Maria E, Soriani S, Varese A, Doria LL, Ajmar F, Mandich P. A novel mutation (D305V) in the early growth response 2 gene is associated with severe Charcot-Marie-Tooth type 1 disease. Hum Mutat. 1999;14:353–354. doi: 10.1002/(SICI)1098-1004(199910)14:4<353::AID-HUMU17>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Bermingham JR, Jr, Scherer SS, O'Connell S, Arroyo E, Kalla KA, Powell FL, Rosenfeld MG. Tst-1/Oct-6/SCIP regulates a unique step in peripheral myelination and is required for normal respiration. Genes Dev. 1996;10:1751–1762. doi: 10.1101/gad.10.14.1751. [DOI] [PubMed] [Google Scholar]
- Boerkoel CF, Takashima H, Bacino CA, Daentl D, Lupski JR. EGR2 mutation R359W causes a spectrum of Dejerine-Sottas neuropathy. Neurogenetics. 2001;3:153–157. doi: 10.1007/s100480100107. [DOI] [PubMed] [Google Scholar]
- Bondurand N, Girard M, Pingault V, Lemort N, Dubourg O, Goossens M. Human Connexin 32, a gap junction protein altered in the X-linked form of Charcot–Marie–Tooth disease, is directly regulated by the transcription factor SOX10. Hum Mol Genet. 2001;10:2783–2795. doi: 10.1093/hmg/10.24.2783. [DOI] [PubMed] [Google Scholar]
- Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, Wegner M. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15:66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavrier P, Zerial M, Lemaire P, Almendral J, Bravo R, Charnay P. A gene encoding a protein with zinc fingers is activated during G0/G1 transition in cultured cells. EMBO J. 1988;7:29–35. doi: 10.1002/j.1460-2075.1988.tb02780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker L, Desmarquet-Trin-Dinh C, Taillebourg E, Ghislain J, Vallat JM, Charnay P. Peripheral myelin maintenance is a dynamic process requiring constant Krox20 expression. J Neurosci. 2006;26:9771–9779. doi: 10.1523/JNEUROSCI.0716-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain J, Charnay P. Control of myelination in Schwann cells: a Krox20 cis-regulatory element integrates Oct6, Brn2 and Sox10 activities. EMBO Rep. 2006;7:52–58. doi: 10.1038/sj.embor.7400573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain J, Desmarquet-Trin-Dinh C, Jaegle M, Meijer D, Charnay P, Frain M. Characterisation of cis-acting sequences reveals a biphasic, axon-dependent regulation of Krox20 during Schwann cell development. Development. 2002;129:155–166. doi: 10.1242/dev.129.1.155. [DOI] [PubMed] [Google Scholar]
- Giese KP, Martini R, Lemke G, Soriano P, Schachner M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. 1992;71:565–576. doi: 10.1016/0092-8674(92)90591-y. [DOI] [PubMed] [Google Scholar]
- Gilardi-Hebenstreit P, Nieto MA, Frain M, Mattei MG, Chestier A, Wilkinson DG, Charnay P. An Eph-related receptor protein tyrosine kinase gene segmentally expressed in the developing mouse hindbrain. Oncogene. 1992;7:2499–2506. [PubMed] [Google Scholar]
- Gillespie CS, Sherman DL, Blair GE, Brophy PJ. Periaxin, a novel protein of myelinating Schwann cells with a possible role in axonal ensheathment. Neuron. 1994;12:497–508. doi: 10.1016/0896-6273(94)90208-9. [DOI] [PubMed] [Google Scholar]
- Ginsberg L, Malik O, Kenton AR, Sharp D, Muddle JR, Davis MB, Winer JB, Orrell RW, King RH. Coexistent hereditary and inflammatory neuropathy. Brain. 2004;127:193–202. doi: 10.1093/brain/awh017. [DOI] [PubMed] [Google Scholar]
- Giudicelli F, Taillebourg E, Charnay P, Gilardi-Hebenstreit P. Krox-20 patterns the hindbrain through both cell-autonomous and non cell-autonomous mechanisms. Genes Dev. 2001;15:567–580. doi: 10.1101/gad.189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmbacher F, Schneider-Maunoury S, Topilko P, Tiret L, Charnay P. Targeting of the EphA4 tyrosine kinase receptor affects dorsal/ventral pathfinding of limb motor axons. Development. 2000;127:3313–3324. doi: 10.1242/dev.127.15.3313. [DOI] [PubMed] [Google Scholar]
- Jaegle M, Ghazvini M, Mandemakers W, Piirsoo M, Driegen S, Levavasseur F, Raghoenath S, Grosveld F, Meijer D. The POU proteins Brn-2 and Oct-6 share important functions in Schwann cell development. Genes Dev. 2003;17:1380–1391. doi: 10.1101/gad.258203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Origin and early development of Schwann cells. Microsc Res Tech. 1998;41:393–402. doi: 10.1002/(SICI)1097-0029(19980601)41:5<393::AID-JEMT6>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Lallemand Y, Luria V, Haffner-Krausz R, Lonai P. Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res. 1998;7:105–112. doi: 10.1023/a:1008868325009. [DOI] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Araki T, Schmidt RE, Milbrandt J. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci USA. 2005a;102:2596–2601. doi: 10.1073/pnas.0407836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Svaren J, LaPash C, Araki T, Schmidt RE, Milbrandt J. Nab proteins are essential for peripheral nervous system myelination. Nat Neurosci. 2005b;8:932–940. doi: 10.1038/nn1490. [DOI] [PubMed] [Google Scholar]
- LeBlanc SE, Srinivasan R, Ferri C, Mager GM, Gillian-Daniel AL, Wrabetz L, Svaren J. Regulation of cholesterol/lipid biosynthetic genes by Egr2/Krox20 during peripheral nerve myelination. J Neurochem. 2005;93:737–748. doi: 10.1111/j.1471-4159.2005.03056.x. [DOI] [PubMed] [Google Scholar]
- LeBlanc SE, Jang SW, Ward RM, Wrabetz L, Svaren J. Direct regulation of myelin protein zero expression by the Egr2 transactivator. J Biol Chem. 2006;281:5453–5460. doi: 10.1074/jbc.M512159200. [DOI] [PubMed] [Google Scholar]
- Mechta-Grigoriou F, Garel S, Charnay P. Nab proteins mediate a negative feedback loop controlling Krox-20 activity in the developing hindbrain. Development. 2000;127:119–128. doi: 10.1242/dev.127.1.119. [DOI] [PubMed] [Google Scholar]
- Mirsky R, Jessen KR. Schwann cell development, differentiation and myelination. Curr Opin Neurobiol. 1996;6:89–96. doi: 10.1016/s0959-4388(96)80013-4. [DOI] [PubMed] [Google Scholar]
- Musso M, Balestra P, Bellone E, Cassandrini D, Di Maria E, Doria LL, Grandis M, Mancardi GL, Schenone A, Levi G, Ajmar F, Mandich P. The D355V mutation decreases EGR2 binding to an element within the Cx32 promoter. Neurobiol Dis. 2001;8:700–706. doi: 10.1006/nbdi.2001.0397. [DOI] [PubMed] [Google Scholar]
- Nagarajan R, Svaren J, Le N, Araki T, Watson M, Milbrandt J. EGR2 mutations in inherited neuropathies dominant-negatively inhibit myelin gene expression. Neuron. 2001;30:355–368. doi: 10.1016/s0896-6273(01)00282-3. [DOI] [PubMed] [Google Scholar]
- Nieke J, Schachner M. Expression of the neural cell adhesion molecules L1 and N-CAM and their common carbohydrate epitope L2/HNK-1 during development and after transection of the mouse sciatic nerve. Differentiation. 1985;30:141–151. doi: 10.1111/j.1432-0436.1985.tb00525.x. [DOI] [PubMed] [Google Scholar]
- Pareyson D, Taroni F, Botti S, Morbin M, Baratta S, Lauria G, Ciano C, Sghirlanzoni A. Cranial nerve involvement in CMT disease type 1 due to early growth response 2 gene mutation. Neurology. 2000;54:1696–1698. doi: 10.1212/wnl.54.8.1696. [DOI] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Droggiti A, Dickinson S, D'Antonio M, Mirsky R, Jessen KR. Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J Cell Biol. 2004;164:385–394. doi: 10.1083/jcb.200307132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnik-Schoneborn S, Rohrig D, Nicholson G, Zerres K. Pregnancy and delivery in Charcot-Marie-Tooth disease type 1. Neurology. 1993;43:2011–2016. doi: 10.1212/wnl.43.10.2011. [DOI] [PubMed] [Google Scholar]
- Russo MW, Sevetson BR, Milbrandt J. Identification of NAB1, a repressor of NGFI-A- and Krox20-mediated transcription. Proc Natl Acad Sci USA. 1995;92:6873–6877. doi: 10.1073/pnas.92.15.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Maunoury S, Topilko P, Seitandou T, Levi G, Cohen-Tannoudji M, Pournin S, Babinet C, Charnay P. Disruption of Krox-20 results in alteration of rhombomeres 3 and 5 in the developing hindbrain. Cell. 1993;75:1199–1214. doi: 10.1016/0092-8674(93)90329-o. [DOI] [PubMed] [Google Scholar]
- Schneider-Maunoury S, Seitanidou T, Charnay P, Lumsden A. Segmental and neuronal architecture of the hindbrain of Krox-20 mouse mutants. Development. 1997;124:1215–1226. doi: 10.1242/dev.124.6.1215. [DOI] [PubMed] [Google Scholar]
- Schneider-Maunoury S, Gilardi-Hebenstreit P, Charnay P. How to build a vertebrate hindbrain. Lessons from genetics. C R Acad Sci III. 1998;321:819–834. doi: 10.1016/s0764-4469(99)80022-5. [DOI] [PubMed] [Google Scholar]
- Seitanidou T, Schneider-Maunoury S, Desmarquet C, Wilkinson DG, Charnay P. Krox-20 is a key regulator of rhombomere-specific gene expression in the developing hindbrain. Mech Dev. 1997;65:31–42. doi: 10.1016/s0925-4773(97)00051-8. [DOI] [PubMed] [Google Scholar]
- Swirnoff AH, Apel ED, Svaren J, Sevetson BR, Zimonjic DB, Popescu NC, Milbrandt J. Nab1, a corepressor of NGFI-A (Egr-1), contains an active transcriptional repression domain. Mol Cell Biol. 1998;18:512–524. doi: 10.1128/mcb.18.1.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szigeti K, Wiszniewski W, Saifi GM, Sherman DL, Sule N, Adesina AM, Mancias P, Papasozomenos SC, Miller G, Keppen L, Daentl D, Brophy PJ, Lupski JR. Functional, histopathologic and natural history study of neuropathy associated with EGR2 mutations. Neurogenetics. 2007;8:257–262. doi: 10.1007/s10048-007-0094-0. [DOI] [PubMed] [Google Scholar]
- Taillebourg E, Buart S, Charnay P. Conditional, floxed allele of the Krox20 gene. Genesis. 2002;32:112–113. doi: 10.1002/gene.10062. [DOI] [PubMed] [Google Scholar]
- Taveggia C, Pizzagalli A, Fagiani E, Messing A, Feltri ML, Wrabetz L. Characterization of a Schwann cell enhancer in the myelin basic protein gene. J Neurochem. 2004;91:813–824. doi: 10.1111/j.1471-4159.2004.02745.x. [DOI] [PubMed] [Google Scholar]
- Timmerman V, De Jonghe P, Ceuterick C, De Vriendt E, Lofgren A, Nelis E, Warner LE, Lupski JR, Martin JJ, Van Broeckhoven C. Novel missense mutation in the early growth response 2 gene associated with Dejerine-Sottas syndrome phenotype. Neurology. 1999;52:1827–1832. doi: 10.1212/wnl.52.9.1827. [DOI] [PubMed] [Google Scholar]
- Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, Babinet C, Charnay P. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–799. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- Topilko P, Levi G, Merlo G, Mantero S, Desmarquet C, Mancardi G, Charnay P. Differential regulation of the zinc finger genes Krox-20 and Krox-24 (Egr-1) suggests antagonistic roles in Schwann cells. J Neurosci Res. 1997;50:702–712. doi: 10.1002/(SICI)1097-4547(19971201)50:5<702::AID-JNR7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Voiculescu O, Taillebourg E, Pujades C, Kress C, Buart S, Charnay P, Schneider-Maunoury S. Hindbrain patterning: Krox20 couples segmentation and specification of regional identity. Development. 2001;128:4967–4978. doi: 10.1242/dev.128.24.4967. [DOI] [PubMed] [Google Scholar]
- Warner LE, Mancias P, Butler IJ, McDonald CM, Keppen L, Koob KG, Lupski JR. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet. 1998;18:382–384. doi: 10.1038/ng0498-382. [DOI] [PubMed] [Google Scholar]
- Warner LE, Svaren J, Milbrandt J, Lupski JR. Functional consequences of mutations in the early growth response 2 gene (EGR2) correlate with severity of human myelinopathies. Hum Mol Genet. 1999;8:1245–1251. doi: 10.1093/hmg/8.7.1245. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG, Bhatt S, Chavrier P, Bravo R, Charnay P. Segment-specific expression of a zinc-finger gene in the developing nervous system of the mouse. Nature. 1989;337:461–464. doi: 10.1038/337461a0. [DOI] [PubMed] [Google Scholar]
- Wood JG, Engel EL. Peripheral nerve glycoproteins and myelin fine structure during development of rat sciatic nerve. J Neurocytol. 1976;5:605–615. doi: 10.1007/BF01175573. [DOI] [PubMed] [Google Scholar]
- Yoshihara T, Kanda F, Yamamoto M, Ishihara H, Misu K, Hattori N, Chihara K, Sobue G. A novel missense mutation in the early growth response 2 gene associated with late-onset Charcot–Marie–Tooth disease type 1. J Neurol Sci. 2001;184:149–153. doi: 10.1016/s0022-510x(00)00504-9. [DOI] [PubMed] [Google Scholar]
- Yoshimura K, Negishi T, Kaneko A, Sakamoto Y, Kitamura K, Hosokawa T, Hamaguchi K, Nomura M. Monoclonal antibodies specific to the integral membrane protein P0 of bovine peripheral nerve myelin. Neurosci Res. 1996;25:41–49. doi: 10.1016/0168-0102(96)01026-7. [DOI] [PubMed] [Google Scholar]
- Zorick TS, Lemke G. Schwann cell differentiation. Curr Opin Cell Biol. 1996;8:870–876. doi: 10.1016/s0955-0674(96)80090-1. [DOI] [PubMed] [Google Scholar]
- Zorick TS, Syroid DE, Arroyo E, Scherer SS, Lemke G. The transcription factors SCIP and Krox-20 mark distinct stages and cell fates in Schwann cell differentiation. Mol Cell Neurosci. 1996;8:129–145. doi: 10.1006/mcne.1996.0052. [DOI] [PubMed] [Google Scholar]
- Zorick TS, Syroid DE, Brown A, Gridley T, Lemke G. Krox-20 controls SCIP expression, cell cycle exit and susceptibility to apoptosis in developing myelinating Schwann cells. Development. 1999;126:1397–1406. doi: 10.1242/dev.126.7.1397. [DOI] [PubMed] [Google Scholar]