

Abstract
Microglial cells maintain the immunological integrity of the healthy brain and can exert protection from traumatic injury. During ischemic tissue damage such as stroke, peripheral immune cells acutely infiltrate the brain and may exacerbate neurodegeneration. Whether and how microglia can protect from this insult is unknown. Polymorphonuclear neutrophils (PMNs) are a prominent immunologic infiltrate of ischemic lesions in vivo. Here, we show in organotypic brain slices that externally applied invading PMNs massively enhance ischemic neurotoxicity. This, however, is counteracted by additional application of microglia. Time-lapse imaging shows that microglia exert protection by rapid engulfment of apoptotic, but, strikingly, also viable, motile PMNs in cell culture and within brain slices. PMN engulfment is mediated by integrin- and lectin-based recognition. Interference with this process using RGDS peptides and N-acteyl-glucosamine blocks engulfment of PMNs and completely abrogates the neuroprotective function of microglia. Thus, engulfment of invading PMNs by microglia may represent an entirely new mechanism of CNS immune privilege.
Keywords: neuroinflammation, stroke, microglia, polymorphonuclear granulocytes, PMN, phagocytosis, time-lapse imaging
Introduction
Abundant evidence exists that an inflammatory response is mounted within the CNS after cerebral ischemia. Postischemic inflammation comprises the infiltration of polymorphonuclear granulocytes and monocytes/macrophages into the injured brain parenchyma, activation of microglia, and expression of proinflammatory cytokines, adhesion molecules, and other inflammatory mediators (Feuerstein et al., 1998; Dirnagl et al., 1999). Despite these well described inflammatory events after ischemia, the main impact of postischemic inflammation (beneficial or detrimental) is controversially discussed (del Zoppo et al., 2001; Feuerstein and Wang, 2001).
There is striking evidence that the infiltration of activated polymorphonuclear neutrophils (PMNs) (Kochanek and Hallenbeck, 1992; Jean et al., 1998; Prestigiacomo et al., 1999) into the injured parenchyma and the activation of microglia (Banati and Graeber, 1994; Kreutzberg, 1996; Minghetti and Levi, 1998) are playing an important role in the pathology of cerebral ischemia.
It is suggested that activated PMNs contribute to tissue damage by the release of oxygen radicals, proteases, and proinflammatory cytokines like TNFα (tumor necrosis factor α) (Barone et al., 1991; Jordan et al., 1999). As evidence, experimental strategies by avoiding PMN infiltration into the injured parenchyma are neuroprotective (Heinel et al., 1994; Beray-Berthat et al., 2003b; Miljkovic-Lolic et al., 2003). However, other studies failed to provide clear evidence of a cause–effect of PMN contribution to neuronal damage after ischemia (Hayward et al., 1996; Fassbender et al., 2002; Beray-Berthat et al., 2003a; Harris et al., 2005).
Similarly controversial findings exist regarding the role of microglia after ischemia. Several studies demonstrated the neurotoxic properties of activated microglia after ischemic or excitotoxic damage (Giulian et al., 1993; Kim and Ko, 1998; Rogove and Tsirka, 1998; Yrjanheikki et al., 1999), whereas considerable evidence shows that microglia triggered by injured/dying neurons mediate a reduction of neuronal damage and induction of tissue repair (Morioka et al., 1991; Rapalino et al., 1998; Streit, 2002; Kohl et al., 2003; Kitamura et al., 2004; Shaked et al., 2005; Hayashi et al., 2006; Lalancette-Hebert et al., 2007).
Considering these controversial findings on the role of PMNs and microglia during transient ischemia, strikingly few studies have evaluated the direct effect of these immune cells on neuronal survival/damage (Dinkel et al., 2004; Mitrasinovic et al., 2005). Despite that, although the combined recruitment of both cell types to tissue sites affected by cerebral ischemia is well established, a potential direct interaction of both immune cell types has not been investigated in depth. Two recent studies demonstrated a possible microglia–PMN interaction. By histology, microglia with engulfed PMNs were observed in zones of focal cerebral ischemia in vivo (Denes et al., 2007; Weston et al., 2007).
To better understand the complexity of cellular inflammation after experimental ischemia, we investigated the individual impact of PMNs, microglia, and macrophages on neuronal survival after ischemia and studied how these cells interact during the inflammatory response. To achieve this, we used an inflammation model that comprises oxygen–glucose deprivation (OGD) in organotypic hippocampal slice cultures (OHCs) as ischemic model and the application of immune cells onto the OHCs as simulation of the postischemic immune cell infiltration into the injured parenchyma.
Materials and Methods
Induction of focal cerebral ischemia by middle cerebral artery occlusion with endothelin-1
Focal cerebral ischemia was induced by occlusion of the left middle cerebral artery (MCA) via intracerebral microinjection of endothelin-1 (ET-1), following Sharkey and Butcher (1995) and Baldauf and Reymann (2005). Briefly, anesthesia was induced with halothane in a mixture of nitrous oxide and oxygen (50:50), and maintained with 2–3% halothane (Sigma-Aldrich) via a rat anesthetic mask (Stölting). A 29 gauge cannula was inserted through the brain close to the MCA. Ischemia was induced by injection of 376 pmol of ET-1 (Sigma-Aldrich). The animals were killed by an overdose of chloral hydrate (Sigma-Aldrich) after 1 d. Frozen brains were cut coronally and 30 μm slices were taken for additional staining.
Organotypic hippocampal slice cultures
Hippocampal interface organotypic cultures were prepared as previously described (Stoppini et al., 1991; Neumann et al., 2006) from postnatal day 7–9 Wistar rats (Harlan Winkelmann). For two-photon microscopy experiments, hippocampal slice cultures were prepared from transgenic B6.Cg-TgN(Thy1-YFP)16Jrs mice (The Jackson Laboratory; distributed by Charles River), which express enhanced yellow fluorescent protein (EYFP) at high levels in subsets of central neurons, including the pyramidal cells of the hippocampus (Feng et al., 2000). Hippocampi were dissected and transversely sliced at 350 μm thickness with a McIlwain tissue chopper (The Mickle Laboratory Engineering). Slices were transferred to Millicell membranes (Millipore). Cultures were maintained at 37°C in 1 ml of serum-based medium containing 50% MEM-Hanks, 25% HBSS, 17 mm HEPES, 5 mm glucose, pH 7.8 (Cell Concepts), 1 mm l-glutamine (Biochrom), 25% horse serum (Invitrogen), and 0.5% gentamycin (Biochrom) for 2–3 d. Cultures were then maintained in serum-free medium (Neurobasal A medium with B27 complement, 5 mm glucose, 1 mm l-glutamine). The OHCs were selected by adding a nontoxic concentration of propidium iodide (PI) (2 μg/ml; Sigma-Aldrich) 12 h before the experimental start. PI-negative slices were considered to be healthy, and only these slices were used for the experiments.
Isolation of polymorphonuclear granulocytes (PMNs)
Human PMNs were prepared from venous blood (8 ml) of healthy volunteers. The rat PMNs were prepared from blood of Wistar rats (Harlan Winkelmann). Briefly, anesthesia in rats was induced with chloral hydrate followed by opening the thorax. The left ventricle of the heart has been punctured under direct visualization, and 10–12 ml of blood was obtained per rat. The blood was collected in a vacutainer containing EDTA (BD Biosciences). The anticoagulated whole blood was carefully layered over 5 ml of the density gradient Polymorphprep (Axis-Shield). Samples were centrifuged for 35 min at 500 × g. After centrifugation, two leukocyte bands were visible. The top band contained the fraction of mononuclear cells, and the lower band contained the fraction of PMNs. The fraction of PMNs was collected by pipetting. PMNs were washed with PBS. Contaminating erythrocytes were removed by hypotonic lyses in plain H2O for 20 s followed by addition of the same volume of 2× PBS to restore osmolarity. PMNs were pelleted and resuspended in DMEM/10% fetal calf serum (FCS). Cell yield (1.5 × 107 cells per preparation) was determined by counting cells in a “Neubauer” hemocytometer, and viability (>99%) was assessed by trypan blue staining (0.4% trypan blue in PBS; Sigma-Aldrich). PMN preparations contained >95% neutrophils assessed by hematoxylin/eosin staining on smears, whereas only a few basophil and eosinophil granulocytes could be detected. No monocytes or lymphocytes contaminated the preparation.
Isolation of rat primary microglia
Primary microglia cultures were prepared from postnatal day 2–3 Wistar rats (Harlan Winkelmann) and cultured in DMEM supplemented with 10% FCS (Biochrom), 1% Pen/Strep (Biochrom), and 1% l-glutamine (Biochrom). After 6 or 7 d of primary cultivation, microglia cells were separated from other cell types by shaking, placed in a 25 ml flask (TPP) at a density not exceeding 5 × 105 cells/ml and maintained in 5% CO2 at 37°C for 2 d. Cultures consisted of 95% microglia cells as determined by staining with Alexa 568-conjugated Griffonia simplicifolia isolectin B4 (Invitrogen).
Culture of macrophage cell line RAW 264.7
The macrophage cell line RAW 264.7 was cultured in DMEM supplemented with 10% FCS (Biochrom), 1% Pen/Strep (Biochrom), and 1% l-glutamine (Biochrom) at a density not exceeding 1 × 106 cells/ml and maintained in 5% CO2 at 37°C.
Application of cells onto OHCs
Isolated primary microglia or macrophages were trypsinated (trypsin/EDTA; Biochrom), centrifuged at 500 × g for 2 min, and finally resuspended in Neurobasal medium (Invitrogen). PMNs were freshly prepared shortly before the experimental start. PMNs were also resuspended in Neurobasal medium. The cells were applied directly onto 10-d-old OHCs in a volume of 1 μl of Neurobasal medium containing 8 × 104 microglia, macrophages, or 1 × 105 PMNs. Viability of microglia, macrophages, and PMNs after application onto the OHC was confirmed in initial experiments by previous staining with 5-chloromethylfluorescein diacetate (CMFDA) (Invitrogen) and by time-lapse microscopy of these cells (data not shown). As indicated, OHCs were fixed with 4% paraformaldehyde (PFA) and mounted with 3:1 PBS/glycerol. OHCs were then further analyzed with the indicated microscopy approach.
Oxygen–glucose deprivation
The membrane inserts carrying up to three OHCs were placed into 1 ml of glucose-free medium in sterile six-well culture plates (TPP) that had previously been saturated with 5% CO2/95% N2 for 10 min. Then OHC were subjected to the OGD [40 min of OGD in a temperature-controlled hypoxic chamber (Billups-Rothenberg); no glucose medium; N2/CO2 atmosphere] before retransfer into normal conditions. Control cultures were kept in regular medium (plus glucose) under normoxic conditions. The cultures were analyzed as indicated by individual experiments 24 or 48 h after OGD.
Analysis of cell death
Cell death was evaluated by cellular incorporation of PI at 24 or 48 h after OGD. Cultures were incubated with PI-containing medium (10 μm) for 2 h at 33°C. Fluorescent images were acquired in a semiautomatic manner (Nikon motorized stage; LUCIA software) and analyzed by densitometry to quantify necrotic cell death (LUCIA Image analysis software). Based on transmission light images, the area of analysis was determined such that only the CA area (CA1–3) was analyzed, whereas the dentate gyrus was excluded. Background correction was performed automatically by a control square (150 × 150 μm) in the stratum moleculare outside the pyramidal cell layer. To combine data from individual experiments, the densitometric mean value of the respective insult of an individual experiment was set to the value of 1 and given as relative fluorescence intensity. All other data are given as relative fluorescence intensity of the insult damage.
Electrophysiology
Wistar rats (25 d old) (Harlan Winkelmann) were killed by a blow on the neck. After decapitation, the brain was quickly removed and placed into ice-cold artificial CSF (ACSF) having the following composition (in mm): 124 NaCl, 4.9 KCl, 1.3 MgSO4, 2.5 CaCl2, 1.2 KH2PO4, 25.6 NaHCO3, 10 d-glucose, saturated with 95% O2 and 5% CO2, pH 7.4. Transverse hippocampal slices (350 μm thickness) with adjacent subicular and entorhinal cortices were prepared using a vibratome (microm HM 650V). The slices were placed on Millicell membranes in a six-well cluster dish (Sigma-Aldrich) with 1 ml of high K+ culture media [25% horse serum (Invitrogen); 40% Eagle's Basal Essential Media (BME) (Sigma-Aldrich); 25% Earle's balanced salt solution (Sigma-Aldrich); 10% 250 mm NaHEPES in BME, pH 7.3; 0.5 mm l-glutamine (Sigma-Aldrich); 28 mm glucose, pH 7.32]. One hour after preparation, 2 μl of high K+ culture media containing 4 × 105 PMNs and 2 μl of cell-free media for control experiments, respectively, were applied on top of the slices and incubated overnight at 33°C in a humidified carbogen atmosphere (95% O2/5% CO2). Then, the slices were transferred into an interface-type recording chamber saturated with carbogen at 33 ± 1°C and constantly superfused with ACSF. Synaptic responses were elicited by stimulation of the Schaffer collateral–commissural fibers in the stratum radiatum of the CA1 region using lacquer-coated stainless-steel stimulating electrodes. Glass electrodes (filled with ACSF, 1–4 MΩ) were placed in the apical dendritic layer to record field EPSPs (fEPSPs). The initial slope of the fEPSP was used as a measure of this potential. The stimulus strength of the test pulses was adjusted to 30% of the EPSP maximum. During baseline recording, three single stimuli (10 s interval) were averaged every 5 min. After tetanization, recordings were taken as indicated in Figure 1 C. Once a stable baseline had been established, long-term potentiation was induced by application of four times two-paired pulses in intervals of 200 ms (theta burst). The interval between the paired pulses was 10 ms, and the width of a single pulse was 0.2 ms.
Figure 1.

PMNs infiltrate the brain parenchyma 1 d after in vivo focal ischemia. A1, Hematoxylin/eosin staining of a coronal section shows an ischemic lesion in the cortex 1 d after focal ischemia. A2, Higher magnification from the edge of the ischemic lesion shows polymorphonuclear cells pointed by black arrows. A3, The black box in A2 is enlarged in A3 and demonstrates the occurrence of PMNs shown by the polymorph nucleus. Scale bar: A1, 200 μm; A2, 20 μm; A3, 10 μm. B, A total of 2 × 105 PMNs was applied in 1 μl onto untreated OHCs. Quantification of neuronal death in CA1–3 was determined by PI incorporation after 24 h [control (CTRL) vs PMNs, nonsignificant (n.s.); n = 9/bar]. In the panel next to the bar chart are representative PI fluorescent images showing neuronal death in CA1–3 after 24 h. C, Because of the nearly twice greater surface of 25-d-old slices, we applied 4 × 105 PMNs in 2 μl onto 1 DIV slices. The recording shows that applied PMNs did not influence EPSP signal nor the amount and persistence of LTP (CTRL vs PMNs, n.s.; CTRL = 11; PMNs, n = 6). Analogous traces represent typical recordings of single experiments taken 10 min before tetanization and 60 min after tetanization. Error bars indicate SEM.
Two-photon microscopy
For two-photon microscopy, PMNs were labeled with Cell Tracker Orange [5-(and-6)-(((4-chloromethyl)benzoyl)-amino)tetramethylrhodamine (CMTMR)] (7.5 μm in PBS, 10 min, room temperature; Invitrogen). At different time points after OGD induction, the OHCs were fixed with 4% PFA, subsequently mounted and subjected to three-dimensional two-photon microscopy. In other experiments, viable OHCs were analyzed. PMNs were labeled with CMFDA and microglia with CMTMR and both applied onto OHCs. Two hours after experimental start, the OHCs were placed in a custom-built chamber supplied with 37°C and 5% CO2 directly under the microscope. The two-photon microscope setup was used exactly as previously described (Neumann et al., 2006). OHCs prepared from transgenic B6.Cg-TgN(Thy1-YFP)16Jrs mice (Feng et al., 2000) were imaged in a modus at 800 and 920 nm wavelength of the laser (MaiTai; Spectra Physics) using a scanning window of 90 × 150 μm size. With a resolution in Z of 1 μm, the entire thickness of the OHC was scanned, first at 920 nm and then at 800 nm wavelength with no filter. The emission of EYFP at 800 nm was negligible as was the emission of Cell Tracker Orange at 920 nm. Image stacks were exported as two independent 16-bit multilayer TIFF stacks and subsequently reconstructed using the Volocity software package (Improvision).
Confocal microscopy
Cultures were examined with a confocal microscope equipped with a 40× magnification Plan Neofluar 0.75 objective, an argon laser emitting at 488 nm, and a helium/neon laser emitting at 543 nm. Multitracking was used to avoid cross talk between channels. Images were analyzed with Carl Zeiss software (Pascal; Carl Zeiss).
Time-lapse video microscopy
The cellular dynamics of PMN–microglia interaction were investigated using OHCs or an in vitro PMN–microglia coculture.
Time-lapse microscopy of OHCs.
Fluorescently labeled PMNs [7-amino-4-chloromethylcoumarin(CMAC)] and microglia (CMTMR) were applied onto OHCs. Two hours after start of the experiment, the OHCs were placed in a custom-made chamber (Incubator S-M; Pecon) adjusted to 37°C and 5% CO2 (CTI-Controller 3700 digital; Tempcontrol 37–2 digital; Pecon).
Time-lapse microscopy of PMN–microglia coculture.
Primary microglia were cultured onto Matrigel-coated surfaces in a 12-well plate (Falcon; BD Biosciences Discovery Labware). Matrigel basement membrane matrix (BD Biosciences), containing laminin as a major component, was diluted in ice-cold DMEM (ratio, 1:20) and polymerized at 37°C for 30 min. Thereafter, 2 × 105 microglia/well were placed and recovered for 1 d. Freshly prepared human PMNs were applied to the primary microglia culture. If indicated, PMNs were CMFDA labeled prior to application. Thirty minutes after the experimental start, the coculture was placed into the chamber as described above. The time-lapse microscope was based on an Axiovert 200M (Carl Zeiss) stage equipped with a 10×, numerical aperture (NA) 0.3 lens or a 32×, NA 0.5 lens (Carl Zeiss) and a CCD camera (Axiocam MRm; Carl Zeiss). Images were recorded at defined time intervals. The data were subsequently analyzed with the Carl Zeiss software (AxioVs40 V4.5).
Statistical analysis
All data are given as mean ± SEM. Statistical analysis was performed by one-way ANOVA followed by post hoc comparison (Tukey's test). A value of p < 0.05 was considered statistically significant.
Results
PMNs infiltrate the brain parenchyma after focal ischemia
Given the heterogeneity of the data concerning the relevance of peripheral immune cells for ischemia-induced neuronal damage, we first wanted to investigate which type of peripheral immune cell was initially recruited into the injured brain parenchyma after transient ischemia. To this end, endothelin was injected directly above the MCA. Endothelin as vasoconstrictor occluded the MCA for ∼30 min. We found that, 1 d after injection, endothelin-mediated transient focal ischemia induced a cellular infiltrate that was mainly composed of PMNs as detectable from the characteristic lobulated nuclei of infiltrating cells in immunohistological sections of the damaged area (Fig. 1 A). We found no marked infiltration with other peripheral immune cells such as macrophages or lymphocytes. Thus, the acute cellular infiltrate of the ischemia-injured brain in vivo was dominated by PMNs, suggesting a prominent role of this immune cell type for the additional development of neuronal damage.
Application of human PMNs onto organotypic hippocampal cultures does not influence neuronal survival
Following the results of the in vivo ischemia-induced recruitment of PMNs (Fig. 1 A), one goal of this study was to clarify the role of PMNs for the development of neuronal viability. To this end, we chose a well established model of ischemic injury in situ using OHCs that maintain many characteristics of true brain–parenchyma, especially the complex three-dimensional structure of neuronal circuits, yet allow for precise control of cellular or humoral factors impacting on neuronal viability as described previously (Neumann et al., 2006). The first step was to evaluate whether PMNs as such had a detrimental effect on neuronal viability in healthy OHCs. Because of the very limited availability of primary rat PMNs, we chose to use human PMNs throughout this study and performed only key experiments with primary rat PMNs. Direct application of up to 2 × 105 human PMNs onto control OHCs had no effect on neuronal viability in the cornu ammonis (CA1–CA3) area after 24 h (Fig. 1 B). Additionally, we investigated the effect of PMNs on neuronal function by electrophysiological EPSP recording with subsequent LTP induction in 1 d in vitro (DIV) hippocampal slices. This approach provides two critical parameters of neuronal health. First, the shape and value of the synaptic signal reflects the quantity and integrity of the involved synapses. Second, the amount and persistence of the synaptic potentiation represents a highly sensitive marker for neuronal viability and functionality. According to the EPSP signal, neither the persistence nor the amount of the LTP was disturbed by application of 4 × 105 PMNs onto the 1 DIV hippocampal slices (Fig. 1 C). These experiments indicated that PMNs derived from healthy human volunteers did not cause neuronal cell loss or neuronal electrophysiological disturbance in rat OHCs.
PMNs exacerbate neuronal damage after oxygen–glucose deprivation
To examine the effect of PMNs on CNS cells after ischemia, we simulated the PMN infiltration into the brain parenchyma by direct application of PMNs onto post-OGD OHCs (Fig. 2 A). Application of increasing numbers of PMNs onto the OHCs after OGD resulted in a significant exacerbation of neuronal damage compared with OGD-induced neuronal damage alone (Fig. 2 B). Representative fluorescence images of the densitometric quantification (Fig. 2 B) are shown in Figure 2 C. These data suggest that PMNs can aggravate the outcome of ischemic neurological insults after 24 h. For all the following experiments involving direct application of PMNs onto OHCs, 1 × 105 PMNs were used.
Figure 2.

Polymorphonuclear granulocytes (PMNs) exacerbate neuronal damage after OGD. A, Different numbers of PMNs (0.5 × 105, 1 × 105, 2 × 105) were applied onto OHCs after OGD. B, Quantification of neuronal death in CA1–3 was determined by PI incorporation after 24 h (***p < 0.001 vs OGD; n = 7/bar). Error bars indicate SEM. CTRL, Control. C, Representative PI fluorescent images showing neuronal death in CA1–3 after 24 h.
PMNs migrate rapidly into hippocampal slices independent of OGD-induced neuronal damage
We had previously shown that microglia can migrate deeply into OGD-damaged OHCs to provide neuroprotection (Neumann et al., 2006). Thus, we speculated that also PMNs might immigrate into OHCs, albeit with a neurotoxic effect after OGD. To test this assumption directly, we subjected cocultures of OGD-treated OHCs and PMNs to two-photon microscopy as previously described (Neumann et al., 2006). In this model, OHCs are made from mice that express EYFP under a specific Thy-1 promoter that leads to strong expression of the transgene in hippocampal neurons (Feng et al., 2000). We observed the rapid migration of PMNs into the brain tissue after 1 h under basal conditions. However, the PMN immigration rate into OGD-damaged slices was indistinguishable from that observed under control conditions (Fig. 3 A,B). Also, at later time points, we detected a uniform distribution of PMNs within the slice and no differences between OGD conditions and basal conditions were observed (Fig. 3 A–F). Additionally, we studied the morphology of EYFP-positive neurons within the OHCs after PMN application. The application of PMNs onto hippocampal slices without OGD showed morphologically intact neurons (neuronal body, dendrites, axons) at all analyzed time points (Fig. 3 A,C,E). In contrast, OGD slices containing PMNs were characterized by a severe loss of axons and dendrites after 6 h (Fig. 3 D) and an almost complete loss of EYFP-positive neurons associated with the appearance of subcellular granular material, presumably stemming from neuronal apoptosis and necrosis, in the CA1 area after 24 h (Fig. 3 F). We also noted the appearance of holes in the otherwise homogenous layer of intact neuronal somata (Fig. 3 F, arrowheads), which probably showed the loss of cell bodies in the respective areas. Thus, although PMNs have the ability to invade OHCs under normal conditions, they do not cause neuronal damage, unless this process is initiated by OGD.
Figure 3.

The migration of PMNs into the OHCs under basal and OGD conditions. PMNs (1 × 105) were labeled with CMTMR (red) and then directly applied onto OHCs prepared from B6.Cg-TgN(Thy1-YFP)16Jrs mice (neurons: green). At indicated time points (basal, 1, 6, 24 h; OGD, 1, 6, 24 h), slices were fixed with 4% PFA and subsequently Z-stacks through the whole OHCs were performed by using two-photon microscopy. A–F, Images show three-dimensional reconstruction of the OHCs and the representative focal plane (middle of OHCs) in the CA1 neuronal layer at the indicated time points after PMN application under basal conditions (A, C, E) or after OGD (B, D, F). Scale bars, 20 μm.
Exogenous microglia counteract the neurotoxicity of PMNs
We previously showed that microglia can have a neuroprotective function in the OGD-induced neuronal damage within OHCs (Neumann et al., 2006). We next asked whether this might also hold true for the PMN-induced neurotoxicity. The direct application of PMNs (Fig. 4 A), microglia, or macrophages (RAW264.7) to control OHCs had no effect on neuronal viability in the CA area after 48 h (Fig. 4 B,C). However, although the presence of PMNs in OGD-treated OHCs showed a strong exacerbation of OGD-induced neuronal damage after 24 and 48 h (Fig. 4 B,C), microglia application resulted in a significant reduction of OGD-induced neuronal damage after 48 h but not after 24 h (Fig. 4 B,C). No significant effect on neuronal death was observed after application of macrophages (Fig. 4 B,C). We next wanted to analyze whether microglia were also able to counteract the massive neurotoxicity induced by PMNs. Therefore, we applied PMNs (1 × 105) in combination with microglia or macrophages (0.8 × 105 each) directly onto the OHCs (Fig. 4 D). The combined application of PMNs and microglia resulted in a significant reduction of PMN-caused exacerbation of neuronal damage after OGD (Fig. 4 E). In contrast, no significant effect was detected by simultaneous application of PMNs and macrophages. These data suggested that there might be a direct interaction between microglia and PMNs, which significantly attenuated the deleterious effects of PMNs on neuronal survival.
Figure 4.

Exogenous microglia counteract PMN neurotoxicity. In A–C, the effects of PMNs, microglia, and macrophages (RAW264.7) were examined individually. A, PMNs (1 × 105), microglia (0.8 × 105), and macrophages (0.8 × 105) were applied directly onto the OHCs after OGD. B, Quantification of neuronal death in CA1–3 was determined by PI incorporation after 24 and 48 h (24 h: ***p < 0.01 vs PMNs; 48 h: ***p < 0.001 PMNs vs OGD, **p < 0.01 MIC vs OGD; n = 9/bar). C, Representative PI fluorescent images showing neuronal death in CA1–3 48 h after OGD. In D–F, the effect of combined application of PMNs/microglia or PMNs/RAW264.7 was studied. D, PMNs were applied directly onto the OHCs together with microglia or macrophages after OGD. E, Quantification of neuronal death in CA1–3 was determined by PI incorporation after 24 h (***p < 0.001 PMNs vs PMNs/MIC; n = 9/bar). Error bars indicate SEM. F, Representative PI fluorescent images showing neuronal death in CA1–3 24 h after OGD. MIC, Microglia; RAW, RAW264.7.
Microglia engulf PMNs within the OHCs
Based on these findings, we investigated a possible direct microglia–PMN interaction in OGD-damaged OHCs. Therefore, we applied fluorescently labeled PMNs and microglia onto the OHCs. Confocal microscopy analyses revealed that exogenous microglia had engulfed PMNs in OGD-treated OHCs (Fig. 5 A). By means of two-photon microscopy and three-dimensional reconstruction of individual microglia, we found that endogenous as well as newly applied microglia were able to fully incorporate single or multiple PMNs within the OHCs (Fig. 5 B).To investigate the cellular dynamics of such an engulfing process in more detail, we applied fluorescently labeled PMNs and microglia onto OHCs and recorded their migratory behavior by time-lapse microscopy. Surprisingly, we observed that, in addition to inactive (immotile and presumably apoptotic) PMNs, microglia were able to also engulf active (motile and viable) PMNs (Fig. 5 C; supplemental movie 1, available at www.jneurosci.org as supplemental material).
Figure 5.

Microglia phagocytose PMNs within the OHCs. PMNs (1 × 105) were labeled with CMFDA (green) (A, B) or CMAC (blue) (C) and microglia with CMTMR (red) and then directly applied onto OHCs. A, OHC was fixed with 4% PFA and subsequently investigated using confocal microscopy. B, The images from a living slice show the three-dimensional reconstruction of one microglia that had already phagocytosed two PMNs (arrows) and was in progress to phagocytose another PMN (asterisk). The dotted line indicates the contact between microglia and PMNs. C, Time-lapse video microscopy was performed 4 h after experimental onset. Images show the engulfing of a motile PMN by the microglia. The white line indicates the migration pathway of the PMN. The microglia contacted the PMN at the time (3 min after start of time-lapse imaging) at which the PMN showed a velocity of 7.6 μm/min. The white dotted line shows the contact point between microglia and PMN (supplemental movie 1, available at www.jneurosci.org as supplemental material). Scale bars: A, 20 μm; B, 5 μm; C, 10 μm.
Microglia engulf viable, motile, nonapoptotic PMNs
The previous experiments suggested that the microglia was able to engulf fully viable PMNs. However, although the phagocytosis of apoptotic cellular material by microglia is a relatively common process (Stolzing and Grune, 2004; Takahashi et al., 2005; Chan et al., 2006), the capture of active and viable cells by microglia has not been observed before. Phototoxicity and bleaching prevented us from performing hour-long time-lapse sequences on individual OHCs. However, in several instances, we were able to document the uptake of live PMNs by parenchymal microglia within the brain slices in a manner very similar to the one observed before for isolated microglia (supplemental movie 6, available at www.jneurosci.org as supplemental material), which suggests that the behavior of the externally added microglia was similar to the parenchymal cells. The related low frequency of phagocytosis events in individual image sequences did not allow performing a thorough quantitative analysis of PMN phagocytosis in this ex vivo model system. We therefore performed cell culture experiments in vitro. PMNs (3 × 105) were cocultured with primary microglia (0.75 × 105) in vitro. These experiments allowed to clearly visualize the engulfing process of motile PMNs by individual microglia (Fig. 6 A,B) and demonstrated that before being engulfed and phagocytosed PMNs could exhibit profound motility over long time periods (Fig. 6 A,A*).
Figure 6.

Microglia engulf motile PMNs. The panels show CMFDA (green)-labeled PMNs (3 × 105) cocultured with primary microglia (0.75 × 105). Cell movements were recorded by time-lapse microscopy. A, The panel displays a microglia that engulfed a motile CMFDA-labeled PMN (arrow). A*, The image depicts the PMN migration path (white line) before the microglia contacted and engulfed the PMN (red circle). The integrated white graph shows the PMN velocity of 5 μm/min at the time the microglia had touched the PMN (supplemental movie 2, available at www.jneurosci.org as supplemental material). B, Images show the engulfment of an immotile CMFDA-labeled PMN (arrow) (supplemental movie 4, available at www.jneurosci.org as supplemental material). Scale bars, 10 μm.
We frequently observed that microglia adopted a “chasing behavior” while attempting to engulf PMNs, either including cellular protrusions (supplemental movie 2, available at www.jneurosci.org as supplemental material) or the whole cell bodies (Fig. 6 B; supplemental movie 3, available at www.jneurosci.org as supplemental material). Because also immotile (presumably apoptotic or preapoptotic) PMNs were engulfed by microglia (Fig. 6 B; supplemental movie 4, available at www.jneurosci.org as supplemental material), we next addressed the question of whether engulfed PMNs always exhibited signs of proapoptosis. Therefore, we transferred PMNs to primary microglia and added FITC-labeled Annexin V to the coculture. Annexin V binds to phosphatidylserine on the outer side of the membrane of cells that have already initiated the apoptotic cascade (Fadok et al., 1992). Thus, in our coculture system, proapoptotic cells (especially PMNs) were detectable by an increased FITC signal (Fig. 7). This set of experiments revealed that microglia indeed engulfed both proapoptotic PMNs and healthy PMNs (Fig. 7; supplemental movie 5, available at www.jneurosci.org as supplemental material). Our experimental setup required a 48 h resting period of primary microglia before use. Attempts to apply microglia directly shaken from the culture were not successful because these cells were not very active and partially died during the first hour of investigation. Thus, it could not be excluded that microglia were activated by the necessary culture conditions. Microscopic analysis of the cells showed that untreated microglia were very adhesive to plastic but, in the presence of supernatant of OGD-treated OHC or PMNs, detached from the plastic surface and obtained a motile morphology (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), which suggested, that indeed the microglia was activated by the presence of PMNs or the supernatant of OGD-treated OHC. Thus, it was possible that the observed phagocytosis behavior was a general phenomenon of activated macrophage-like cells and not specific for microglia. To test this assumption, we compared the ability of primary microglia with peritoneal macrophages freshly isolated from rats by peritoneal lavage with PBS. Analysis showed that despite being motile and frequently touching or dragging PMNs in the culture peritoneal macrophages never engulfed PMNs, whether apoptotic or alive (supplemental movie 7, available at www.jneurosci.org as supplemental material), whereas microglia cocultured in the same experiment phagocytosed on average 2.6 PMNs per cell (supplemental Fig. 2 and movie 8, available at www.jneurosci.org as supplemental material). Thus, the ability to phagocytose live or dead PMNs seemed to be specific for microglia activated by the presence of PMNs or the supernatant of OGD-treated OHC and was not observed with peripheral macrophages, at least from the peritoneum.
Figure 7.
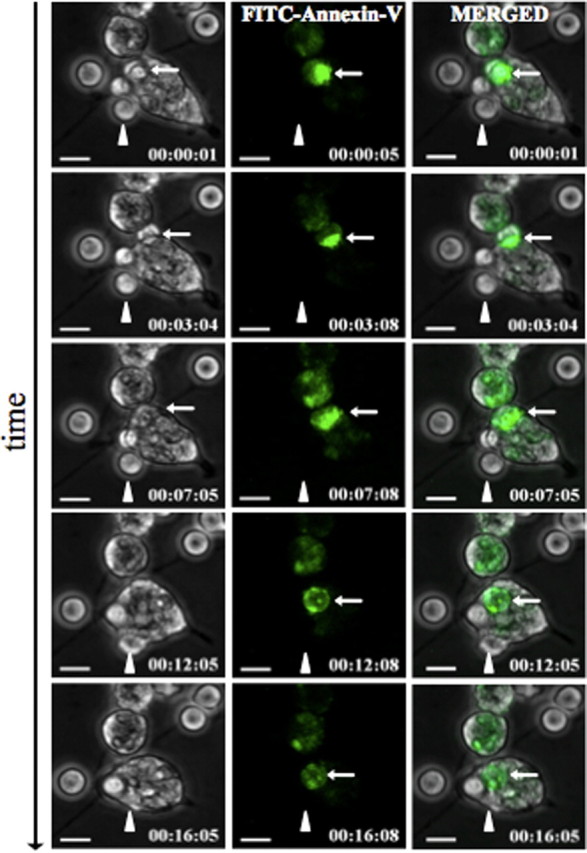
Microglia engulf preapoptotic and nonapoptotic PMNs. FITC-conjugated Annexin V was added to the microglia–PMN coculture, and thus proapoptotic cells were visualized by an increased FITC signal. The white thin arrow points on an Annexin V-positive PMN that was fully engulfed after 12 min. The arrowhead shows a PMN that was engulfed without exhibiting a FITC signal at any time point of the engulfment process (supplemental movie 5, available at www.jneurosci.org as supplemental material). Scale bars, 10 μm.
Blocking the engulfment process of PMNs by microglia worsens the outcome of neuronal viability after OGD
Next, we investigated whether the engulfment of PMNs by microglia had any consequence on neuronal viability after OGD. It has been shown that apoptotic PMN cells can be internalized by binding to the αvβ3-integrin receptor on macrophages. In addition, lectin-like receptors have been shown to be involved in this process (Fadok et al., 1998; Meszaros et al., 1999). Thus, we evaluated the potential of the integrin-blocking tetrapeptide RGDS (Arg-Gly-Asp-Ser) and the lectin inhibitor N-acetyl glucosamine (GlcNAc) to block the engulfing process by preincubating primary microglia with the reagents before adding PMNs to the culture. We distinguished between engulfment of motile or immotile PMNs by time-lapse microscopy. RGDS and GlcNAc blocked the engulfing of both immotile and, interestingly, also motile PMNs, highly significantly (Fig. 8 A). Thereby GlcNAc was more efficient than RGDS (Fig. 8 A). We also noted a slight synergistic effect on the blockade of the engulfment process when both substances were combined. This synergistic effect was more pronounced in the engulfment of nonmotile cells (Fig. 8 A). Additionally, we always observed altered microglia–PMN interaction patterns in the presence of RGDS and GlcNAc. Whereas untreated microglia bound and engulfed PMNs, treated microglia bound several PMNs but mostly failed to ingest them (Fig. 8 B).
Figure 8.

Blockage of the engulfment process of PMNs by microglia worsens the outcome of neuronal viability after OGD. A, Microglia were preincubated with RGDS (1 mm) or GlcNAc (20 mm) or both for 20 min before PMNs were added to the microglia. Time-lapse microscopy was performed to distinguish the engulfment of immotile and motile PMNs by the microglia. Column scatterplots show the number of PMNs engulfed by microglia under the indicated conditions. Significance values are shown above the columns. B, The images show representative microglia that were exposed to PMNs. The arrows on the top image display the engulfed PMN within the microglia. The bottom image shows PMNs that adhere to the microglia without being internalized. C, Microglia were preincubated with RGDS (1 mm) and GlcNAc (20 mm) or both for 20 min before they were applied together with either human-derived (hPMN) or rat-derived (rPMN) PMN onto OGD-treated OHCs. Quantification of neuronal death in CA1–3 was determined by PI incorporation after 24 h [**p < 0.01, hPMN plus MIC (OGD) vs hPMN plus MIC plus RGDS/GlcNAc (OGD); ***p < 0.001, rPMN plus MIC (OGC) vs rPMN plus MIC plus RGDS/GlcNAc (OGD); n = 5–7/bar]. Error bars indicate SEM. CTRL, Control; MIC, microglia.
An important question was whether interfering with the engulfment process affected the neuronal viability after OGD. To test this, we applied PMNs and microglia simultaneously onto the OHCs after OGD. Microglia and OHCs were also preincubated with RGDS/GlcNAc before application. Indeed, the presence of RGDS or GlcNAc alone, and, more efficiently, in combination strongly reduced the neuroprotective function of coapplied microglia in this model (Fig. 8 C). In addition, we obtained similar results by using primary rat PMNs, confirming that the observed behavior was specific for PMNs (Fig. 8 C).
Thus, blocking the engulfment of PMNs by microglia severely compromised the neuroprotective function that microglia exerted on neurons on OGD exposure pointing to a physiological role of this cellular function of microglia.
Discussion
This study was designed to determine the role of individual cell types of the innate immune system that contribute to the postischemic inflammation after cerebral ischemia. Taking advantage of our neuroinflammation model, we were able to simulate the migration and infiltration of these cells to the site of damage in the neuronal tissue. It is generally accepted that the main immune cells involved in the inflammation-induced secondary neuronal damage are PMNs, microglia, and macrophages, which all are recruited as early as the postischemic inflammation is initiated. However, PMNs and local microglia are the first cells present on site, followed by peripheral microglia and monocytes/macrophages. Although all three cell types potentially exhibit cytotoxicity by releasing noxious substances such as cytokines, oxygen radicals, and proteases (Hallenbeck et al., 1986; Barone et al., 1991; Minghetti and Levi, 1998), their individual contribution for the overall damage remains unclear.
There is still an ongoing debate as to the importance of the individual cell types in brain ischemia. In an attempt to overcome this limitation, we chose to use a well established ex vivo model of neuroinflammation (Ullrich et al., 2001; Mitrasinovic et al., 2005; Neumann et al., 2006). Using this model, we were now able to demonstrate that indeed only PMNs sharply increased neuronal damage associated with transient ischemia. This was not a general phenomenon of infiltrating phagocytes, because neither local nor externally added microglia nor macrophages appeared to be neurotoxic after cerebral ischemia. In fact, we and others previously showed that local (Kohl et al., 2003) or externally added (Neumann et al., 2006) microglia may even be neuroprotective in this model. Our data provide strong evidence that PMNs are a critical innate immune cell type that is responsible for the increase of neuronal damage associated with ischemia. This finding is well in line with previous studies showing enhanced neuronal death after ischemia in a dissociated neuronal culture (Dinkel et al., 2004). Moreover, in vivo studies strongly correlate the degree of PMN infiltration to the size of the neuronal damage (Beray-Berthat et al., 2003b; Weston et al., 2007). Another study found a positive correlation between the intensity of myeloperoxidase staining, a hallmark of PMN activity, and the degree of neuronal damage in the area of ischemia (Matsuo et al., 1994). This finding also strongly supports the view that PMNs are critically involved in the progress of postischemic neuronal damage.
Interestingly, the inhibition of reactive oxygen species (ROS) production by NAD(P)H-oxidases, one of the key actions of inflammatory PMNs (Segal, 2005), results in strong neuroprotective effects in rodent models of ischemic stroke (Wang et al., 2006; L. L. Tang et al., 2007). Moreover, interfering with the homing of immune cells by an antiadhesive therapy has been shown to be neuroprotective (Connolly et al., 1996; Yanaka et al., 1996). However, because this approach affects indiscriminately all immune cell types, it did not provide any suggestions as to whether PMNs might play a specific role in this context. In addition, it is assumed that also cells of the adaptive immune system can invade areas of ischemia-induced neuroinflammation (Arumugam et al., 2005). Because their contribution is still unclear, our model is well suited to examine this matter in the future. In summary, in the line of these previous data and our study, it seems likely that PMNs constitute an important neurotoxic cell type under ischemic conditions. Because we did not find evidence that microglia and macrophages were neurotoxic, at least 48 h after ischemia, we assume that interfering with the function of PMNs might be a particularly promising option to limit the extent of neuronal damage after stroke.
A major conclusion from our study is that there seems to be a natural mechanism aiming precisely at this goal. After coapplying microglia and PMNs onto ischemically damaged OHCs, we found a strong decrease of neuronal damage compared with the application of PMNs alone. This was a rather unexpected finding, because numerous studies demonstrated that microglia preactivated by inflammatory mediators are strongly neurotoxic (Arai et al., 2004; Minghetti et al., 2004; Qin et al., 2004; Fordyce et al., 2005; Huang et al., 2005). However, a close inspection of these studies shows that microglial activation was achieved by exposure to microbial products (Arai et al., 2004; Qin et al., 2004; Huang et al., 2005) or viral particles (Lim et al., 2003; Minghetti et al., 2004). Lipopolysaccharide (LPS) or viral particles are well known to trigger a massive immune cell reaction via Toll-like receptors (TLRs) (Kawai and Akira, 2006). Recent evidence suggests that, even in the situation of stroke, which constitutes a sterile inflammation, Toll-like mediated pathways can be triggered on microglia (Lehnardt et al., 2007) or even directly on neurons (S. C. Tang et al., 2007) and that this response increases neuronal damage after stroke. However, the signaling induced by bacterial TLR triggers might well be different from the one induced by endogenous inflammatory mediators because neurons, which express both TLR-2 and -4 do not respond to classical triggers of these receptors such as peptidoglycan or LPS (S. C. Tang et al., 2007). It might, therefore, be possible that also microglia are able to distinguish between microbial and other inflammatory stimuli of the TLR system. In this concept, microglia activated by nonmicrobial inflammation might exert neuroprotective effects, whereas microbial activation of microglia preferentially triggers neurotoxicity.
Our study also provides insights into the mechanism whereby this protection is accomplished. We demonstrate that microglia were very effective in phagocytosing invading PMNs. Macrophage-like cells are well known to clear the inflamed tissue from cell debris, such as apoptotic PMNs. Two recent histological studies have also demonstrated brain-resident microglia associated with PMN-related debris in areas of ischemia-induced neuronal damage (Denes et al., 2007; Weston et al., 2007). However, our data show that, in addition to proapoptotic PMNs, also fully viable, Annexin V-negative, PMNs were effectively engulfed by microglia.
In contrast, we have never observed microglia engulfing other types of immune cells such as freshly prepared lymphocytes and monocytes, and in addition we did not find engulfing of PMNs by macrophage lines or freshly prepared peritoneal macrophages. These findings underscore the specificity of the microglia–PMN engagement. By time-lapse microscopy, we observed microglia exerting a PMN chasing behavior, either including cellular protrusions or the whole cell bodies. To the best of our knowledge, the phagocytosis of active and viable immune cells by other immune cells has not been described so far and therefore might represent an entirely novel way of immune control. This is probably attributable to the fact that such a mechanism can only be revealed by life cell imaging, which is technically challenging, especially with the cell types investigated in the current study.
Given the fact that phagocytosis was inhibitable by RGDS peptides and GlcNAc, we assumed that integrins such as vitronectin receptors (Ruoslahti, 1996) or lectins mediated the engulfment process. Although this is well known for the uptake of apoptotic cells (Fadok et al., 1998; Meszaros et al., 1999), this surprisingly also held true for the uptake of presumably live cells. We cannot formally exclude that even Annexin V-negative cells were already proapoptotic thereby exposing vitronectin or lectin ligands. However, the fact that the cells were very motile argued against this hypothesis, because one of the earliest events of apoptosis is loss of migration (Savill et al., 2002).
Our study also indicates that interference with the phagocytosis process by RGDS and GlcNAc abolished the neuroprotective function of microglia. It is well conceivable that dying PMNs mediate neurotoxicity by the release of toxic intracellular compounds (Denes et al., 2007; Weston et al., 2007), and that, consequently, prompt phagocytosis of apoptotic PMNs by microglia might prevent the secretion of toxic compounds. In the light of these findings, we propose that clearance of PMNs from the nervous tissue might be an effective strategy to protect neurons from PMN neurotoxicity. Whether the engulfment of viable PMNs contributes to this protection remains to be determined and requires to be examined in additional studies. In our system, phagocytosis of motile (presumably viable) PMNs was even more frequent than the uptake of nonmotile cells. Therefore, it appears unlikely that this process is irrelevant for the protection we observed. How microglia turn off the neurotoxicity of a live PMN currently remains enigmatic. A possible mechanism might include active interference with cytokine secretion and production of ROS. This might be mediated by the rapid destruction of engulfed PMNs, which appears to be happening to PMNs after phagocytosis by microglia in our system. Finally, it has also been shown that macrophages release immune-regulatory cytokines such as TGF-β (Savill et al., 2002) after uptake of apoptotic PMNs. A similar process might also be exhibited by microglia after uptake of PMNs, with TGF-β mediating neuroprotection by reducing the general inflammatory responses. Future studies are required to verify whether the processes observed in our ex vivo model are truly active and physiologically relevant in vivo. If so, future therapies might aim achieving neuroprotection by supporting microglia activation, thereby rapidly eliminating infiltrating PMNs from the ischemic lesion site.
Footnotes
This work was supported by grants from the State of Saxony-Anhalt (3594M/0405M) (K.G.R., O.U.), the German Research Community (Deutsche Forschungsgemeinschaft) (GU 769/2-1) (M.G.), and the German Federal Ministry for Science and Education (01GO0504-CAI). We thank Susanne v. Kenne and Cornelia Garz for excellent technical assistance and Gabriella Orlando for critical reading of this manuscript. We also thank Roland Hartig for help with differential interference contrast microscopy.
References
- Arai H, Furuya T, Yasuda T, Miura M, Mizuno Y, Mochizuki H. Neurotoxic effects of lipopolysaccharide on nigral dopaminergic neurons are mediated by microglial activation, interleukin-1beta, and expression of caspase-11 in mice. J Biol Chem. 2004;279:51647–51653. doi: 10.1074/jbc.M407328200. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Granger DN, Mattson MP. Stroke and T-cells. Neuromol Med. 2005;7:229–242. doi: 10.1385/NMM:7:3:229. [DOI] [PubMed] [Google Scholar]
- Baldauf K, Reymann KG. Influence of EGF/bFGF treatment on proliferation, early neurogenesis and infarct volume after transient focal ischemia. Brain Res. 2005;1056:158–167. doi: 10.1016/j.brainres.2005.07.035. [DOI] [PubMed] [Google Scholar]
- Banati RB, Graeber MB. Surveillance, intervention and cytotoxicity: is there a protective role of microglia? Dev Neurosci. 1994;16:114–127. doi: 10.1159/000112098. [DOI] [PubMed] [Google Scholar]
- Barone FC, Hillegass LM, Price WJ, White RF, Lee EV, Feuerstein GZ, Sarau HM, Clark RK, Griswold DE. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: myeloperoxidase activity assay and histologic verification. J Neurosci Res. 1991;29:336–345. doi: 10.1002/jnr.490290309. [DOI] [PubMed] [Google Scholar]
- Beray-Berthat V, Palmier B, Plotkine M, Margaill I. Neutrophils do not contribute to infarction, oxidative stress, and NO synthase activity in severe brain ischemia. Exp Neurol. 2003a;182:446–454. doi: 10.1016/s0014-4886(03)00106-7. [DOI] [PubMed] [Google Scholar]
- Beray-Berthat V, Croci N, Plotkine M, Margaill I. Polymorphonuclear neutrophils contribute to infarction and oxidative stress in the cortex but not in the striatum after ischemia-reperfusion in rats. Brain Res. 2003b;987:32–38. doi: 10.1016/s0006-8993(03)03224-4. [DOI] [PubMed] [Google Scholar]
- Chan A, Hummel V, Weilbach FX, Kieseier BC, Gold R. Phagocytosis of apoptotic inflammatory cells downregulates microglial chemoattractive function and migration of encephalitogenic T cells. J Neurosci Res. 2006;84:1217–1224. doi: 10.1002/jnr.21029. [DOI] [PubMed] [Google Scholar]
- Connolly ES, Jr, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, Stern DM, Solomon RA, Gutierrez-Ramos JC, Pinsky DJ. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996;97:209–216. doi: 10.1172/JCI118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Becker KJ, Hallenbeck JM. Inflammation after stroke: is it harmful? Arch Neurol. 2001;58:669–672. doi: 10.1001/archneur.58.4.669. [DOI] [PubMed] [Google Scholar]
- Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, Allan SM. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab. 2007;27:1941–1953. doi: 10.1038/sj.jcbfm.9600495. [DOI] [PubMed] [Google Scholar]
- Dinkel K, Dhabhar FS, Sapolsky RM. Neurotoxic effects of polymorphonuclear granulocytes on hippocampal primary cultures. Proc Natl Acad Sci USA. 2004;101:331–336. doi: 10.1073/pnas.0303510101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- Fadok VA, Warner ML, Bratton DL, Henson PM. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3) J Immunol. 1998;161:6250–6257. [PubMed] [Google Scholar]
- Fassbender K, Ragoschke A, Kuhl S, Szabo K, Fatar M, Back W, Bertsch T, Kreisel S, Hennerici M. Inflammatory leukocyte infiltration in focal cerebral ischemia: unrelated to infarct size. Cerebrovasc Dis. 2002;13:198–203. doi: 10.1159/000047776. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Feuerstein G, Wang X, Barone F. Oxford: Blackwell Scientific; 1998. Cerebrovascular disease: pathophysiology, diagnosis and management. [Google Scholar]
- Feuerstein GZ, Wang X. Inflammation and stroke: benefits without harm? Arch Neurol. 2001;58:672–674. doi: 10.1001/archneur.58.4.672. [DOI] [PubMed] [Google Scholar]
- Fordyce CB, Jagasia R, Zhu X, Schlichter LC. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci. 2005;25:7139–7149. doi: 10.1523/JNEUROSCI.1251-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Vaca K, Corpuz M. Brain glia release factors with opposing actions upon neuronal survival. J Neurosci. 1993;13:29–37. doi: 10.1523/JNEUROSCI.13-01-00029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallenbeck JM, Dutka AJ, Tanishima T, Kochanek PM, Kumaroo KK, Thompson CB, Obrenovitch TP, Contreras TJ. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke. 1986;17:246–253. doi: 10.1161/01.str.17.2.246. [DOI] [PubMed] [Google Scholar]
- Harris AK, Ergul A, Kozak A, Machado LS, Johnson MH, Fagan SC. Effect of neutrophil depletion on gelatinase expression, edema formation and hemorrhagic transformation after focal ischemic stroke. BMC Neurosci. 2005;6:49. doi: 10.1186/1471-2202-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Tomimatsu Y, Suzuki H, Yamada J, Wu Z, Yao H, Kagamiishi Y, Tateishi N, Sawada M, Nakanishi H. The intra-arterial injection of microglia protects hippocampal CA1 neurons against global ischemia-induced functional deficits in rats. Neuroscience. 2006;142:87–96. doi: 10.1016/j.neuroscience.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Hayward NJ, Elliott PJ, Sawyer SD, Bronson RT, Bartus RT. Lack of evidence for neutrophil participation during infarct formation following focal cerebral ischemia in the rat. Exp Neurol. 1996;139:188–202. doi: 10.1006/exnr.1996.0093. [DOI] [PubMed] [Google Scholar]
- Heinel LA, Rubin S, Rosenwasser RH, Vasthare US, Tuma RF. Leukocyte involvement in cerebral infarct generation after ischemia and reperfusion. Brain Res Bull. 1994;34:137–141. doi: 10.1016/0361-9230(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Huang Y, Liu J, Wang LZ, Zhang WY, Zhu XZ. Neuroprotective effects of cyclooxygenase-2 inhibitor celecoxib against toxicity of LPS-stimulated macrophages toward motor neurons. Acta Pharmacol Sin. 2005;26:952–958. doi: 10.1111/j.1745-7254.2005.00136.x. [DOI] [PubMed] [Google Scholar]
- Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery. 1998;43:1382–1397. doi: 10.1097/00006123-199812000-00076. [DOI] [PubMed] [Google Scholar]
- Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res. 1999;43:860–878. doi: 10.1016/s0008-6363(99)00187-x. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Kim WK, Ko KH. Potentiation of N-methyl-d-aspartate-mediated neurotoxicity by immunostimulated murine microglia. J Neurosci Res. 1998;54:17–26. doi: 10.1002/(SICI)1097-4547(19981001)54:1<17::AID-JNR3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Takata K, Inden M, Tsuchiya D, Yanagisawa D, Nakata J, Taniguchi T. Intracerebroventricular injection of microglia protects against focal brain ischemia. J Pharmacol Sci. 2004;94:203–206. doi: 10.1254/jphs.94.203. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23:1367–1379. doi: 10.1161/01.str.23.9.1367. [DOI] [PubMed] [Google Scholar]
- Kohl A, Dehghani F, Korf HW, Hailer NP. The bisphosphonate clodronate depletes microglial cells in excitotoxically injured organotypic hippocampal slice cultures. Exp Neurol. 2003;181:1–11. doi: 10.1016/s0014-4886(02)00049-3. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Lim MC, Brooke SM, Sapolsky RM. gp120 neurotoxicity fails to induce heat shock defenses, while the overexpression of hsp70 protects against gp120. Brain Res Bull. 2003;61:183–188. doi: 10.1016/s0361-9230(03)00113-8. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Onodera H, Shiga Y, Nakamura M, Ninomiya M, Kihara T, Kogure K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994;25:1469–1475. doi: 10.1161/01.str.25.7.1469. [DOI] [PubMed] [Google Scholar]
- Meszaros AJ, Reichner JS, Albina JE. Macrophage phagocytosis of wound neutrophils. J Leukoc Biol. 1999;65:35–42. doi: 10.1002/jlb.65.1.35. [DOI] [PubMed] [Google Scholar]
- Miljkovic-Lolic M, Silbergleit R, Fiskum G, Rosenthal RE. Neuroprotective effects of hyperbaric oxygen treatment in experimental focal cerebral ischemia are associated with reduced brain leukocyte myeloperoxidase activity. Brain Res. 2003;971:90–94. doi: 10.1016/s0006-8993(03)02364-3. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol. 1998;54:99–125. doi: 10.1016/s0301-0082(97)00052-x. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Visentin S, Patrizio M, Franchini L, Ajmone-Cat MA, Levi G. Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions. Neurochem Res. 2004;29:965–978. doi: 10.1023/b:nere.0000021241.90133.89. [DOI] [PubMed] [Google Scholar]
- Mitrasinovic OM, Grattan A, Robinson CC, Lapustea NB, Poon C, Ryan H, Phong C, Murphy GM., Jr Microglia overexpressing the macrophage colony-stimulating factor receptor are neuroprotective in a microglial-hippocampal organotypic coculture system. J Neurosci. 2005;25:4442–4451. doi: 10.1523/JNEUROSCI.0514-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morioka T, Kalehua AN, Streit WJ. The microglial reaction in the rat dorsal hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab. 1991;11:966–973. doi: 10.1038/jcbfm.1991.162. [DOI] [PubMed] [Google Scholar]
- Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. 2006;20:714–716. doi: 10.1096/fj.05-4882fje. [DOI] [PubMed] [Google Scholar]
- Prestigiacomo CJ, Kim SC, Connolly ES, Jr, Liao H, Yan SF, Pinsky DJ. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke. 1999;30:1110–1117. doi: 10.1161/01.str.30.5.1110. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Rapalino O, Lazarov-Spiegler O, Agranov E, Velan GJ, Yoles E, Fraidakis M, Solomon A, Gepstein R, Katz A, Belkin M, Hadani M, Schwartz M. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med. 1998;4:814–821. doi: 10.1038/nm0798-814. [DOI] [PubMed] [Google Scholar]
- Rogove AD, Tsirka SE. Neurotoxic responses by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol. 1998;8:19–25. doi: 10.1016/s0960-9822(98)70016-8. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, Hart RP, Schwartz M. Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem. 2005;92:997–1009. doi: 10.1111/j.1471-4159.2004.02954.x. [DOI] [PubMed] [Google Scholar]
- Sharkey J, Butcher SP. Characterisation of an experimental model of stroke produced by intracerebral microinjection of endothelin-1 adjacent to the rat middle cerebral artery. J Neurosci Methods. 1995;60:125–131. doi: 10.1016/0165-0270(95)00003-d. [DOI] [PubMed] [Google Scholar]
- Stolzing A, Grune T. Neuronal apoptotic bodies: phagocytosis and degradation by primary microglial cells. FASEB J. 2004;18:743–745. doi: 10.1096/fj.03-0374fje. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang LL, Ye K, Yang XF, Zheng JS. Apocynin attenuates cerebral infarction after transient focal ischaemia in rats. J Int Med Res. 2007;35:517–522. doi: 10.1177/147323000703500411. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Diestel A, Eyupoglu IY, Nitsch R. Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat Cell Biol. 2001;3:1035–1042. doi: 10.1038/ncb1201-1035. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–189. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- Weston RM, Jones NM, Jarrott B, Callaway JK. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab. 2007;27:100–114. doi: 10.1038/sj.jcbfm.9600324. [DOI] [PubMed] [Google Scholar]
- Yanaka K, Camarata PJ, Spellman SR, McCarthy JB, Furcht LT, Low WC, Heros RC. Neuronal protection from cerebral ischemia by synthetic fibronectin peptides to leukocyte adhesion molecules. J Cereb Blood Flow Metab. 1996;16:1120–1125. doi: 10.1097/00004647-199611000-00005. [DOI] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]