

Abstract
We describe two new transgenic mouse lines for studying pathological changes of Tau protein related to Alzheimer's disease. They are based on the regulatable expression of the four-repeat domain of human Tau carrying the FTDP17 (frontotemporal dementia and parkinsonism linked to chromosome 17) mutation ΔK280 (TauRD/ΔK280), or the ΔK280 plus two proline mutations in the hexapeptide motifs (TauRD/ΔK280/I277P/I308P). The ΔK280 mutation accelerates aggregation (“proaggregation mutant”), whereas the proline mutations inhibit Tau aggregation in vitro and in cell models (“antiaggregation mutant”). The inducible transgene expression was driven by the forebrain-specific CaMKIIα (calcium/calmodulin-dependent protein kinase IIα) promoter. The proaggregation mutant leads to Tau aggregates and tangles as early as 2–3 months after gene expression, even at low expression (70% of endogenous mouse Tau). The antiaggregation mutant does not aggregate even after 22 months of gene expression. Both mutants show missorting of Tau in the somatodendritic compartment and hyperphosphorylation in the repeat domain [KXGS motifs, targets of the kinase MARK (microtubule affinity regulating kinase)]. This indicates that these changes are related to Tau expression rather than aggregation. The proaggregation mutant causes astrogliosis, loss of synapses and neurons from 5 months of gene expression onward, arguing that Tau toxicity is related to aggregation. Remarkably, the human proaggregation mutant TauRD coaggregates with mouse Tau, coupled with missorting and hyperphosphorylation at multiple sites. When expression of proaggregation TauRD is switched off, soluble and aggregated exogenous TauRD disappears within 1.5 months. However, tangles of mouse Tau, hyperphosphorylation, and missorting remain, suggesting an extended lifetime of aggregated wild-type Tau once a pathological conformation and aggregation is induced by a proaggregation Tau species.
Keywords: Alzheimer's disease, Tau, aggregation, reversibility, transgenic mouse models, tauopathy
Introduction
Tau is a microtubule (MT)-associated protein stabilizing microtubules as tracks for axonal transport (Garcia and Cleveland, 2001; Lee et al., 2001; Goedert et al., 2006; Mandelkow et al., 2007). Tau is highly soluble yet forms pathological “paired-helical filaments” (PHFs) in neurodegenerative diseases such as Alzheimer's disease (AD) or frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP17). The Tau–MT interaction is based on the “repeat-domain” of Tau, including three to four pseudorepeats of ∼31 residues, which also forms the core of PHFs, illustrating the close relationship between physiological and pathological functions.
The solubility of Tau has made it difficult to mimic its aggregation experimentally. This problem was solved in several ways: (1) The repeat domain aggregates faster than full-length Tau, and therefore removal of the flanking domains accelerates aggregation (Wille et al., 1992). (2) Polyanionic cofactors, such as heparin, acidic peptides, or RNA (Goedert et al., 1996; Kampers et al., 1996; Perez et al., 1996) promote aggregation by compensating the positive charges of Tau. (3) Some FTDP17 mutants show enhanced aggregation (Lewis et al., 2000; Lee et al., 2001; Brandt et al., 2005; LaFerla and Oddo, 2005; SantaCruz et al., 2005; McGowan et al., 2006; Götz et al., 2007). (4) Aggregation in transgenic mice is promoted by combining Tau mutations with APP (amyloid precursor protein) or presenilin mutations raising the level of β-amyloid (Aβ) (Götz et al., 2001; Lewis et al., 2001; Oddo et al., 2003; Roberson et al., 2007). Alternatively, Tau phosphorylation is enhanced by activating Tau kinases (Lucas et al., 2001; Cruz et al., 2003; Terwel et al., 2005; Tanemura et al., 2006). (5) Acceleration of aggregation by high protein concentrations was exploited in most mouse models in which exogenous Tau was strongly overexpressed (Andorfer et al., 2003; SantaCruz et al., 2005).
Here, we describe mouse models in which we combined Tau truncation and mutations at low Tau levels. PHF formation is based on hexapeptide motifs in the four-repeat domain (275VQIINK280, 306VQIVYK311) with a high propensity for β-structure (von Bergen et al., 2001), including the ΔK280 mutation known from FTDP17 (Rizzu et al., 1999), because this causes rapid aggregation (proaggregation mutant) (Barghorn et al., 2000). To test the toxicity of aggregation, we inserted proline mutations in the hexapeptide motifs that disrupt aggregation by destroying β-structure (antiaggregation mutant). The expression of TauRD/ΔK280 was restricted to the forebrain via the calcium/calmodulin-dependent protein kinase IIα (CaMKIIα)-promoter (Mayford et al., 1996) to avoid motor defects, which prevent analysis of cognitive functions because of inhibition of microtubule-based transport (Terwel et al., 2002; Mandelkow et al., 2004). The expression was regulated via the tetracycline-responsive transactivator (Gossen and Bujard, 1992) to study the recovery from pathology after switching off exogenous Tau (Ishihara et al., 1999).
The results show that aggregation properties of Tau in vitro are predictive of its behavior in mouse models. Strong propensity for β-structure leads to aggregation in neurons, but deleting the β-propensity by proline mutations does not. Aggregation causes neurotoxicity and synapse loss. Aggregates of exogenous Tau are reversed by discontinuing its expression. Unexpectedly, exogenous Tau prompts endogenous Tau to coaggregate as well, illustrating that the pathological properties of the foreign protein can be transmitted to the host protein.
Materials and Methods
Generation of TauRD transgenic mice
The proaggregation and antiaggregation transgenic mice were generated using the bidirectional transcription unit encoding the four-repeat domain of human tau/TauRD (244–372 aa) carrying the ΔK280 and ΔK280/I277P/I308P by PCR-based site-directed mutagenesis and PCR amplification. The cDNA was inserted into pBI5 bidirectional vector at ClaI and SalI restriction sites (Baron et al., 1995). The DNA plasmid was digested with XmnI and AseI enzymes and a fragment containing the Tet-operon-responsive element (tetO), bidirectional CMV promoter (Ptet-bi), TauRD, and luciferase sequences (5.75 kb) was microinjected into mouse fertilized eggs and reimplanted in pseudopregnant females. Founders were screened by PCR and gene reporter assay (luciferase assay). To develop an inducible Tet-OFF system, the Tau transgenic mouse lines were crossed with CaMKIIα-tTA transgenic mice (Mayford et al., 1996). The activator mice were a generous gift from Dr. E. Kandel (Columbia University, New York, NY).
All of the mouse strains have been conceived on an identical genetic background, C57BL/6, and all of the mice used in the present study were heterozygous. As negative control, nontransgenic littermates were used. During the gestation and in the first 6 weeks after birth, the mice were raised in presence of doxycycline hydrochloride (50 and 200 μg/ml, respectively) dissolved in 5% sucrose supplied in the drinking water to suppress the exogenous Tau during early development. For switch-off experiments, the proaggregation TauRD mice received 200 μg/ml doxycycline hydrochloride in the drinking water for 6 weeks. All animals were housed and tested according to standards of the German Animal Welfare Act.
The proaggregation and antiaggregation transgenic mice were identified by PCR of genomic mouse tail DNA (DNA isolation; Invitek, Berlin, Germany) using the primer pairs 5′-AAT GAG GTC GGA ATC GAA GG-3′ and 5′-TAG CTT GTC GTA ATA ATG GCG G-3′; 5′-CAT CGA TAT GCA GAC AGC CC-3′ and 5′-TCG ACT GGA CTC TGT CCT TG-3′, which amplified a 469 bp DNA fragment specific for the tTA trangene, and a 335 bp DNA fragment specific for the TauRD construct.
To evaluate the luciferase assay, fibroblast primary cultures were generated from ear biopsies. The fibroblasts were grown to 90% confluency and were transiently transfected with 0.4 μg of rtTA2-M2/pUHrT62–1 plasmid encoding the tetracycline transcriptional activators (tet-on) (Gossen and Bujard, 1992). The protein expression was induced by 1 μg/ml doxycycline, and the cell lysates were analyzed 24 h after gene expression. Luciferace activity was measured using the Lumat LB 9507 system (Berthold Technologies, Bad Wildbad, Germany).
Western blotting
To estimate total Tau levels, fresh brain tissue or tissue snap-frozen in liquid N2 and stored at −80°C was homogenized in 8 vol of lysis buffer [50 mm Tris-HCl, pH 7.4, 10% glycerol, 1% NP-40, 5 mm DTT, 1 mm EGTA, 20 mm NaF, 1 mm Na3VO4, 150 mm NaCl, protease inhibitors (Complete Mini; Roche, Indianapolis, IN), 5 mm CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), 100 U/ml benzonase, 5 μm okadaic acid]. Proteins were resolved by SDS-PAGE (17% polyacrylamide gels) and transferred to polyvinylidene difluoride membranes. The membranes were incubated in 5% nonfat milk in TBS-Tween for 1 h at room temperature. The blots were incubated at 4°C overnight in primary antibody followed by incubation 1 h at room temperature with secondary antibody. Ten micrograms of total protein was loaded per line for detection with pan-tau antibody K9JA (A-0024; 1:20,000; DakoCytomation, Carpinteria, CA) and analyzed by densitometry (LAS 3000; AIDA software; Raytest, Straubenhardt, Germany).
Extraction of Sarkosyl-insoluble Tau
A Sarkosyl-insoluble fraction of Tau was isolated from brain tissue as described previously (Greenberg and Davies, 1990). Briefly, the brain tissue was homogenized in 3 vol of cold buffer H (10 mm Tris-HCl, 1 mm EGTA, 0.8 m NaCl, 10% sucrose, pH 7.4) and centrifuged at 27,200 × g for 20 min at 4°C. The supernatant was collected and the resulting pellet was homogenized in buffer H and centrifuged at 27,200 × g for 20 min at 4°C. Both supernatants were combined, adjusted to 1% (w/v) N-lauroylsarcosine, and incubated at 37°C with shaking for 60–90 min. After centrifugation at 150,000 × g for 35 min at 20°C, the pellet was resuspended in 50 mm Tris-HCl, pH 7.4, using 0.5 μl of buffer for each milligram of initial weight of the brain tissue. Western blotting was used to analyze the supernatant and the insoluble pellet. The ratio between the volumes of the supernatant and the Sarkosyl-insoluble pellet loaded in 17% SDS gels was always 1:2 (the amount of protein in the insoluble fraction was doubled compared with soluble fraction). For quantification of Tau levels in each fraction, the Western blots were probed with pan-tau antibody K9JA and analyzed by densitometry (LAS 3000; AIDA software; Raytest).
Histology
Immunohistochemistry.
The samples for immunohistochemistry were prepared by paraffin embedding. Mice were anesthetized with 2-bromo-2-chloro-1,1,1-trifluoroethane and killed by decapitation. The brains were removed and weighed. Briefly, one of the hemispheres of the mouse brain was placed in 4% formaline for 24 h, and then dehydrated by sequential incubation in a series of ethanols and chloroform. The final embedding was in a mixture of paraffin and paraplast. Five micrometer coronal sections were obtained from each paraffin block. Endogenous peroxidase was quenched by treating the paraffin sections with 0.6% H2O2 and nonspecific binding of antibodies was eliminated by applying 10% horse serum for 30 min at room temperature. Primary antibody prepared in 1% horse serum was applied for 1 h at room temperature, followed by secondary antibody for 30 min incubation at room temperature. All of the antibody dilutions and the washing steps were performed in 10 mm Tris and 154 mm NaCl, pH 7.5. For secondary antibody and avidin-biotinylated peroxidase system, we used the Vectastain Universal Elite ABC kit (Vector Laboratories, Burlingame, CA). Antigen retrieval with citrate buffer was performed for an efficient epitope exposure to the antibodies. The following antibodies were used: 12E8 (1:500) for phosphorylated S262/S356 Tau (gift from Dr. P. Seubert, Elan Pharma, South San Francisco, CA); MC-1 (1:5; gift from Dr. P. Davies, Albert Einstein College, Bronx, NY); NeuN (1:1000) neuronal marker (Chemicon International, Temecula, CA); and GFAP (1:1000) glial marker (Sigma, St. Louis, MO). Rabbit polyclonal peptide antibodies were generated against pT231 (1:500; SA 5007), pS46 (1:50; SA 5010), first N-terminal insert (SA 4473), pS202 (1:60; Biosource, Camarillo, CA), and pS404 (1:100; Biosource).
Gallyas silver staining.
Five micrometer paraffin sections were silver stained according to published procedures (Braak and Braak, 1991).
Thioflavin S staining.
After removing the paraffin and rehydration, the autofluorescence was quenched (Sun et al., 2002), sections were incubated in 0.05% Thioflavin S (Sigma) for 8 min, and excess Thioflavin S was removed by two brief washes in 80% EtOH. After three washing steps in large volumes of tap water, the sections were mounted in Permafluor (Beckman Coulter, Paris, France).
Immunofluorescence.
Immunolabeling was performed as described previously (Rune et al., 2002; Kretz et al., 2004). Coronal frozen sections of transgenic and nontransgenic mouse brains were fixed in 4% paraformaldehyde and incubated overnight at 4°C with primary antibody against synaptophysin (1:1000; Boehringer, Bagnolet, France) and spinophilin (1:750; Upstate Biotechnology, Lake Placid, NY). The digitized images obtained with a laser-scanning microscope (SP2; Leica, Berlin, Germany) were analyzed by an imaging system (Openlab 2.2.5; Improvision, Conventry, UK). For evaluating the synaptophysin and spinophilin staining, defined areas were analyzed and the relative staining index was determined by multiplying the stained area (pixel number) with the intensity of staining (value on the gray scale). The result was divided by the number of measured areas. For each group, 20 fields in the stratum radiatum of the CA1 and CA3 were investigated. Means ± SD were calculated and statistical analyses were performed by ANOVA, followed by a post hoc test; p < 0.05 was considered to be significant.
Electron microscopy
Synapse count.
The electron microscopy analysis of the synapses was performed as described previously (Kretz et al., 2004). Briefly, 400 μm hippocampal slices after fixation in 1% glutaraldehyde, 1% paraformaldehyde in 0.1 m phosphate buffer were postfixed in 1% OsO4, dehydrated in ethanol, and embedded in Epon 820. Blocks were trimmed to contain only the stratum pyramidale and radiatum of the CA1 region. Ultrathin sections were cut on a Reichert-Jung OmU3 ultramicrotome. The sections were stained with uranyl acetate, followed by lead citrate. The spine synapse density was calculated using stereological methods as described previously (Prange-Kiel et al., 2004).
Immunogold negative stain electron microscopy.
To evaluate the presence of PHFs, the Sarkosyl-insoluble fractions were purified by iodixanol gradient and applied on 600-mesh carbon-coated copper grids for 1 min, washed twice with 1% albumin/PBS, and immunogold labeled with 10 nm gold particles (G1527; Sigma). The grids were incubated on a drop of antibody/gold solution for 1 h in a chamber with a water-saturated atmosphere. The grids were negatively stained with 2% uranyl acetate for 1 min, and the specimens were examined with a Philips CM12 electron microscope at 100 kV (Philips, Eindhoven, The Netherlands). The antibody dilutions were as follows: K9JA, 1:80 (DakoCytomation); Tau-1, 1:25 (gift from Dr. L. Binder, Northwestern University, Chicago, IL).
Results
Tau constructs and generation of transgenic mouse strains
We generated two inducible mouse strains that express mutant variants of the Tau repeat domain (TauRD: construct K18, with four repeats), which is essential for PHF aggregation (Fig. 1). One of the mutants is termed “proaggregation,” because it carries the Lys280 deletion, which was identified in a case with FTDP17 (Rizzu et al., 1999), and has a remarkable tendency to aggregate (Barghorn et al., 2000). As a negative counterpart, we generated a second “antiaggregation” mutant based on the TauRD/ΔK280 sequence with two additional proline mutations in the hexapeptide motifs (I277P, I308P) to inhibit the formation of β-structure and aggregation (von Bergen et al., 2000, 2001). Both mouse strains are double transgenic because we used the tetracycline controlled transactivator (tTA) to generate inducible transgenic mice that express TauRD under the control of the CaMKIIα promoter that drives protein expression in the forebrain (Mayford et al., 1996). In this Tet-OFF system, the administration of doxycycline in the drinking water represses the mutant TauRD gene expression. To avoid any potential developmental consequences from expression of mutant TauRD protein, all mice received doxycycline during the mating time and 6 weeks after birth.
Figure 1.

Constructs of Tau derived from Tau repeat domain. The top bar diagram represents the longest isoform of the human tau40 (441 residues). The bottom diagram shows the four-repeat construct TauRD derived from htau40. One of the constructs used here contains the FTDP17 mutation ΔK280 to accelerate aggregation by promoting β-structure (proaggregation mutant). The second mutant has additionally two proline mutations (I277P and I308P in the hexapeptide motifs) to inhibit the aggregation by disrupting β-structure (antiaggregation mutant).
Accumulation of Sarkosyl-insoluble Tau in proaggregation mutant
The mutant TauRD construct can be detected in Western blots as a band of 12–14 kDa. The clear difference between the molecular weight of the exogenous human and endogenous mouse Tau allowed us to distinguish these proteins and calculate their ratio (Fig. 2 A). The proaggregation and antiaggregation mouse lines had similar low levels of exogenous Tau, 70% or less of endogenous Tau (Fig. 2 B).
Figure 2.

Mutant TauRD protein levels in transgenic mice. Representative immunoblots probed with K9JA polyclonal antibody that recognizes the repeats plus C-terminal domain of Tau. A, Levels of Tau in cortex of mutant TauRD-inducible transgenic mice. Cortex brain lysates from nontransgenic littermate, proaggregation (TauRD/ΔK280) and antiaggregation (TauRD/ΔK280/2P) mutants at 9 months of gene expression. Ten micrograms of total protein is loaded per lane. B, Ratio of mutant TauRD versus endogenous Tau in transgenic mice at 9 months of gene expression (n = 3 mice/group) (mean ± SEM, 0.73 ± 0.02 for the proaggregation mutant; 0.68 ± 0.07 for the antiaggregation mutant). C, Immunoblots of Sarkosyl-soluble and -insoluble fractions from cortex lysates of proaggregation and antiaggregation mutant mice at 3 and 12 months of gene expression. Blot analysis with K9JA antibody. Sarkosyl-soluble fractions show the presence of endogenous (∼55 kDa) and exogenous (∼12–14 kDa) Tau protein in the case of proaggregation and antiaggregation mutants. The Sarkosyl-insoluble fractions indicate the aggregation of endogenous mouse Tau and exogenous mutant TauRD in the proaggregation mutant mice, but the absence of aggregation in the antiaggregation mutant mice.
The tendency of the mutant TauRD constructs to aggregate in the cortex was evaluated by Sarkosyl extraction (Greenberg and Davies, 1990), which reveals not only tangles but also pretangle aggregates. We compared two groups of animals, young mice (3 months of gene expression) and older mice (12 months of gene expression). The majority of exogenous TauRD protein as well as the three endogenous mouse Tau isoforms were found in the soluble fraction, both for proaggregation and antiaggregation mutants (Fig. 2 C). Sarkosyl-insoluble Tau was observed only for the case of the proaggregation mouse line. The ratio of mutant TauRD to endogenous mouse Tau was similar in the Sarkosyl-insoluble and -soluble fractions (0.4–0.5), showing that endogenous mouse Tau was incorporated into the aggregates. In contrast, the antiaggregation mouse line showed no Sarkosyl-insoluble pellet. Similarly, the wild-type control mice containing no human Tau also showed no Tau in the Sarkosyl-insoluble fraction, consistent with the highly soluble nature of native Tau.
The proaggregation mutant TauRD and mouse Tau coaggregate into neurofibrillary tangles
In the case of AD, increasing levels of Tau pathology and aggregation can be defined in terms of an early conformational change (MC1 antibody) (see Fig. 7 A–D) (details below), early stages of aggregation (Sarkosyl-insoluble Tau, “pretangles”), and fully developed tangles (visualized by Gallyas silver staining). We asked which of these stages are achieved in our mouse models. In the proaggregation mice, tangle formation started as early as 2–3 months of gene expression, and Gallyas-positive neurons were identified in a high number in the entorhinal cortex and amygdala of proaggregation mutants (Fig. 3 B,C) compared with the controls (Fig. 3 A). This result is remarkable considering that, in these mice, the expression of exogenous TauRD protein is low, less than that of mouse Tau (∼70%). At 12 months of gene expression, neurofibrillary tangle (NFT) pathology involved larger areas of entorhinal cortex (Fig. 3 D,E), and at 15 months, NFTs were present in the neocortex (Fig. 3 F,G). NFTs were morphologically heterogenous, showing flame-shaped structures (Fig. 3 B,E,G) and dystrophic neurites (Fig. 3 G, arrow). Some of the neurons that lacked NFTs showed a ballooned shape, indicative of incipient degeneration (Fig. 3 C, arrow). The elevated level of total Tau can be detected by the pan-tau antibody K9JA, which highlights the cells expressing the exogenous TauRD, despite the endogenous Tau background (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Figure 7.
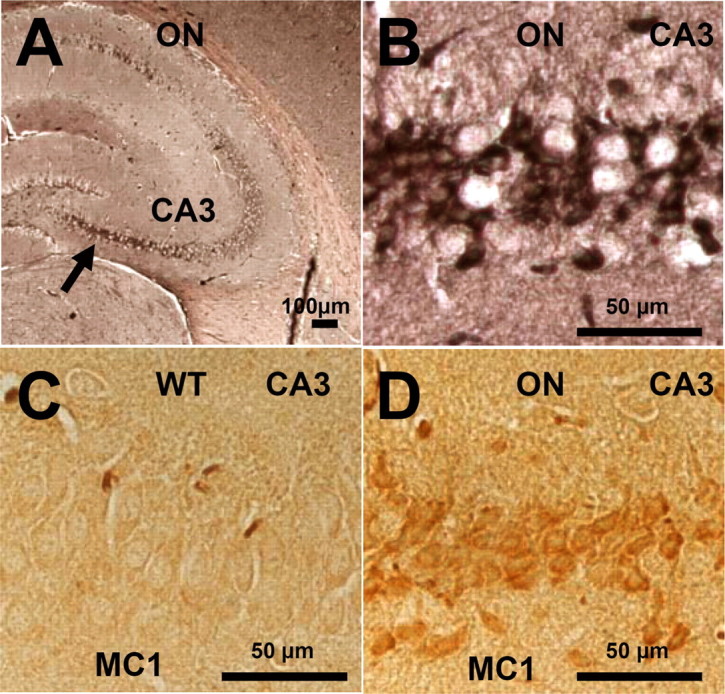
Neurofibrillary pathology and pathological conformation of mouse Tau in the hippocampus of TauRD/ΔK280 proaggregation mice. A, NFT pathology in the case of TauRD/ΔK280 proaggregation mice at 8 months after gene expression. The arrow indicates the CA3 hippocampal neurons stained by Gallyas silver staining. B, Higher magnification of hippocampal CA3 region, indicating formation of NFT pathology in the brain of proaggregation mice. C, D, Conformational changes of endogenous Tau identified by MC1 antibody in the brain of proaggregation mice at 8 months after gene expression (D). The same CA3 region in the wild-type mice is showing the absence of MC1 immunoreactivity (C). Scale bars: A, 100 μm; B–D, 50 μm.
Figure 3.
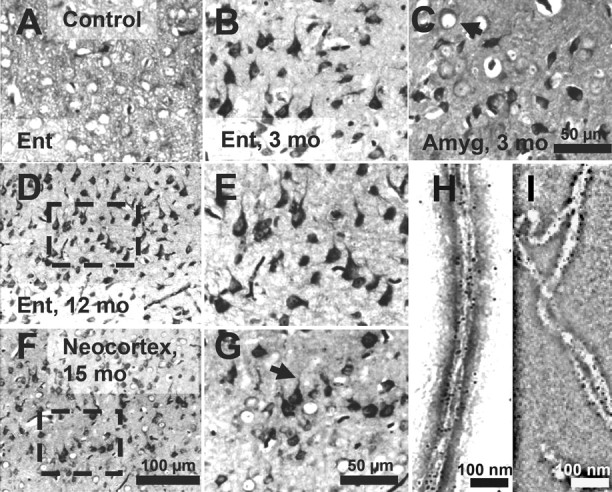
Tau aggregation in TauRD/ΔK280 transgenic mice by Gallyas silver staining. A, Entorhinal cortex of negative control (nontransgenic littermate) at 21 months. B, C, Gallyas-positive neurons in the entorhinal cortex (B) and in amygdala (C) in proaggregation mutant mice at 3 months after TauRD/ΔK280 gene expression. The arrow indicates a ballooned neuron in the amygdala. D, F, Accumulation of Gallyas-positive neurons in the entorhinal cortex at 12 months (D) and in neocortex at 15 months after TauRD/ΔK280 gene expression (F). E, G, Higher magnification of D and F; the arrow in G indicates dystrophic neurites. H, I, Negative stain electron microscopy of bundles of Tau filaments labeled with 10 nm immunogold, using antibodies K9JA (H) (against repeats plus C-terminal domain) and Tau-1 (I) (against region 189–207 aa before the repeats), demonstrating that mouse Tau and exogenous TauRD are present in the same filaments. The filaments are ∼10 nm wide and were isolated from proaggregation transgenic mice at 7 months of gene expression. Scale bars: A–C, E, G, 50 μm; D, F, 100 μm; H, I, 100 nm. Ent, Entorhinal cortex; Amyg, amygdala.
As mentioned above, the aggregation starts in the entorhinal cortex of proaggregation mutants at 3 months of gene expression (Fig. 4 A). In contrast, in the case of the antiaggregation Tau mice, we observed no NFTs in the entorhinal cortex at 3 months of gene expression, and not even after 22 months (Fig. 4 B). The presence of NFTs in proaggregation mice and their absence in the antiaggregation mice were confirmed by Thioflavin S staining (Fig. 4 C,D).
Figure 4.

NFTs in proaggregation but not in antiaggregation mice. A, Gallyas-positive neurons in the entorhinal cortex of proaggregation mutant mice at 3 months after TauRD/ΔK280 gene expression. B, Absence of Gallyas-positive neurons in the entorhinal cortex of antiaggregation mice at 22 months after TauRD/ΔK280/2P gene expression. C, Thioflavin S staining of the entorhinal cortex confirms the presence of NFTs in proaggregation mice at 3 months after TauRD/ΔK280 gene expression and absence of NFTs in the antiaggregation mutant (D) at 3 months after TauRD/ΔK280/2P gene expression. Scale bars: A–D, 20 μm. Ent, Entorhinal cortex.
We searched for Tau filaments in the Sarkosyl-insoluble material from the entorhinal cortex of proaggregation mice at 7 months of gene expression by negative stain electron microscopy. Bundles of filaments resembling those of AD were identified after enrichment by iodixanol density gradient centrifugation and immunogold labeling with a pan-tau antibody (K9JA, recognizing the repeats and the C-terminal tail) and antibody Tau-1 (Fig. 3 H,I). Antibody Tau-1 (Binder et al., 1985) recognizes an unphosphorylated epitope around residue 200 in the flanking region before the repeats that is not present in the exogenous TauRD. This confirms that endogenous mouse Tau contributes to the filaments formed from the proaggregation Tau mutant.
Early phosphorylation of the KXGS motifs and conformational changes in proaggregation TauRD, but not with antiaggregation TauRD
In AD, Tau occurs in an aggregated and hyperphosphorylated form. We therefore investigated the relationship between these two properties in our mouse models. The Tau repeat domain lacks most of the phosphorylation sites that are characteristic of AD Tau, but it contains the KXGS motifs that are targets of the microtubule affinity regulating kinase (MARK). There are four such motifs in TauRD, one per repeat, and phosphorylation can be detected by the antibody 12E8 (Fig. 5 A), which recognizes phosphorylated S262 and S356 (Seubert et al., 1995). In the case of the proaggregation mutant, the hippocampal pyramidal neurons in the CA1 region showed an increased phosphorylation at S262/S356 after 2 months of gene expression (Fig. 5 B). The phosphorylated TauRD/ΔK280 was pathological, relocated to the somatic compartment at 2 months of gene expression, and showed somatodendritic missorting in the neurons of the subiculum at 12 months (Fig. 5 D). In the case of the antiaggregation mutant, there was no phosphorylation at KXGS motifs in a similar hippocampal region at 2 months of gene expression (Fig. 5 C). After 12 months of gene expression, the subicular neurons showed only a low level of KXGS phosphorylation (Fig. 5 E), whereas the control mice showed no 12E8 staining.
Figure 5.
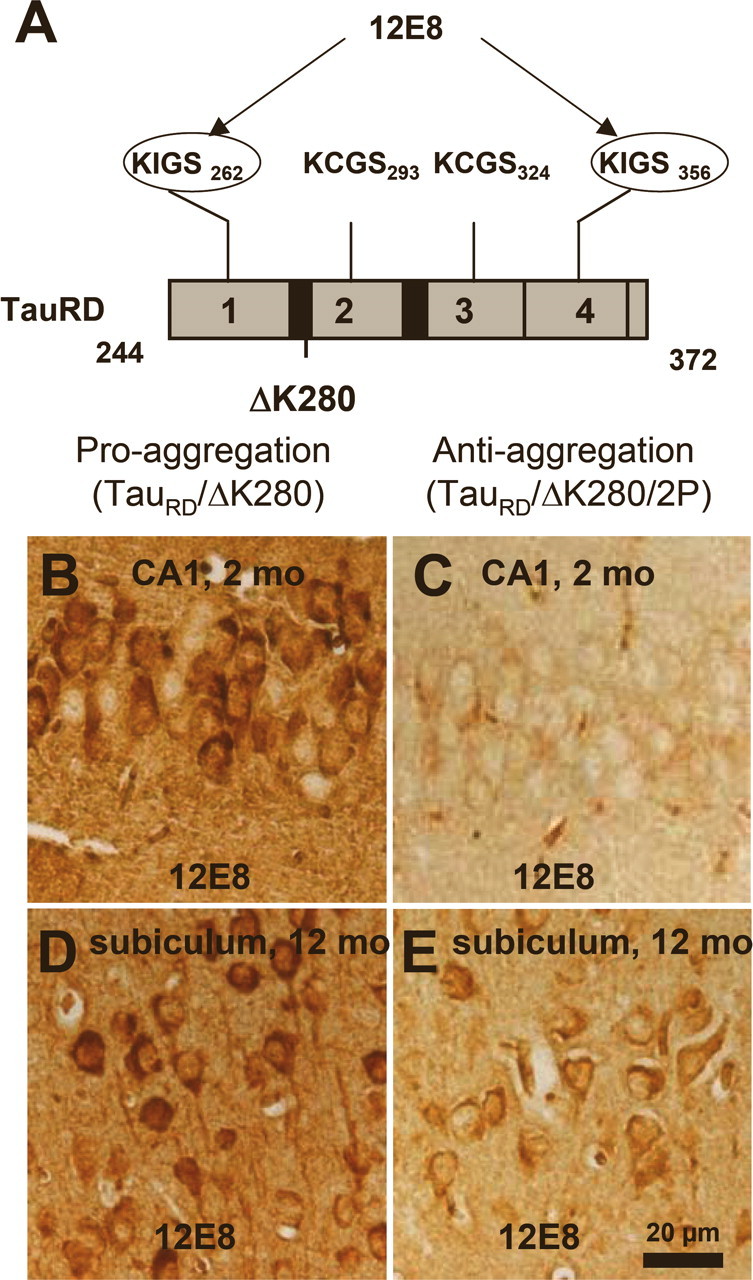
Phosphorylation at KXGS motifs and mislocalization of TauRD/ΔK280 to the somatodendritic compartment. A, Bar diagram of Tau repeat domain and 12E8 epitopes, the phosphorylation sites of MARK. B, Somatic relocalization of hyperphosphorylated TauRD/ΔK280 in hippocampal, CA1 neurons at 2 months after gene expression of the proaggregation mutant. C, Absence of hyperphosphorylated TauRD/ΔK280/2P in hippocampal CA1 region at 2 months after gene expression of the antiaggregation mutant. D, Immunohistochemistry of proaggregation mouse brains at 12 months of TauRD/ΔK280 gene expression showed phosphorylation and translocation of Tau protein in the somatodendritic compartment of subiculum and (E) only mild phosphorylation of Tau protein in the antiaggregation mutant at the same time point. Scale bar: B–E, 20 μm.
Because the endogenous mouse Tau coaggregates with exogenous TauRD, we asked whether mouse Tau is phosphorylated at sites diagnostic for AD outside the repeat domain (mostly Ser-Pro or Thr-Pro motifs). Indeed, in the case of the proaggregation mice, the aggregated endogenous mouse Tau was phosphorylated in the domains flanking the repeats (e.g., at Thr231 and Ser46) (Fig. 6 F,G). Several antibodies diagnostic for Alzheimer-like Tau phosphorylation [e.g., AT8 (pS202/pT205) or PHF-1 (pS396/pS404)] were checked but did not show any reactivity (in both cases, only the first of the two residues was phosphorylated). Interestingly, strong immunoreactivity of phosphorylated Tau at single sites (S404, T231, S202, S46, and S262 or S356) was identified in the CA3 region, neurons that bear heavy NFT pathology at 5 months of gene expression (Fig. 6 B,D,F–I). Similarly, neurons in the amygdala exhibiting pronounced tangle pathology by Gallyas staining (Fig. 6 C) showed phosphorylation at KXGS motifs (Fig. 6 E). These data support the above results from Sarkosyl-insoluble fractions and electron microscopy that both human and mouse Tau became missorted and coaggregated in the brains of the transgenic animals. Neurons with aggregated Tau in the hippocampal region were also positive for staining with antibody MC-1, confirming that aggregation was accompanied by a pathological conformational in mouse Tau in the proaggregation mutant mice (note that the MC-1 epitopes is present only in mouse Tau, not in the truncated human Tau) (Fig. 7 A,B,D). MC-1 staining was not observed with antiaggregation mice (Fig. 7 C).
Figure 6.

Coaggregation and phosphorylation of endogenous and exogenous Tau in proaggregation mutant mice. A, Coronal section from the brain of proaggregation mice at 5 months of gene expression by Gallyas silver staining. The frames indicate NFTs in the hippocampus and amygdala of proaggregation mutant mice. B, C, Higher magnification of the hippocampus and amygdala. D, E, The same areas as B and C show positive 12E8 staining. F–I, Pyramidal neurons, CA3 region positive for pT231, pS46, pS202, and pS404. These sites lie outside the repeats and demonstrate the coaggregation of mouse Tau. Scale bars: A, 1 mm; B–I, 100 μm. amyg, Amygdala.
Age-dependent neuronal loss and astrogliosis in the dentate gyrus of the proaggregation transgenic mice
To assess whether tangle pathology was associated with neurodegeneration, the neuronal loss was evaluated in the brain of the transgenic mice. Coronal brain sections of wild-type, proaggregation, and antiaggregation mice were immunostained with the neuronal-specific marker NeuN for different time points, young mice at 5 months and old mice at 24 months of gene expression. For proaggregation mutants, neuronal loss was detected in the granule cells of the dentate gyrus starting at 5 months of gene expression (Fig. 8 B, arrows). In aged proaggregation mice, at 24 months of gene expression there was a remarkable neuronal loss of the granule cells in the dentate gyrus and shrinkage of the molecular layer (Fig. 8 E). In contrast, in the case of the antiaggregation mice, both young and old, the layers of granule neurons in the dentate gyrus were comparable with those in the wild-type mice (Fig. 8 A,C,D,F).
Figure 8.

Neuron loss and astrogliosis in hippocampus of proaggregation transgenic mice. Coronal brain sections were immunostained with NeuN (neuron-specific marker). A–C, Comparison between the layers of granule cells in the dentate gyrus of control, proaggregation, and antiaggregation mice at 5 months of gene expression. Arrows indicate the loss of neurons in the dentate gyrus of the proaggregation mutant. D–F, Control, proaggregation mutant, and antiaggregation mutant at 24 months of gene expression. Note the reduction of the granule cell layer in the dentate gyrus in the case of the proaggregation mutant compared with antiaggregation mutant and control mice. The double arrows indicate the shrinkage of the molecular layer for proaggregation mutant and no changes in the case of antiaggregation mutant and wild-type mice. G–I, Paraffin brain sections immunostained for GFAP. Note increased GFAP immunoreactivity in the hilus of the proaggregation mutant at 21 months of gene expression compared with antiaggregation mutant and wild-type mice, indicating that aggregation of TauRD induces inflammatory reactions. Scale bars: A–F, 100 μm; G–I, 50 μm.
Reactive changes after Tau accumulation and neuronal loss in the brains of the transgenic mice were evaluated by GFAP immunohistochemical staining. Astrogliosis was present in the hilus region of proaggregation mutant mice after 21 months of TauRD/ΔK280 gene expression (Fig. 8 H), but not with antiaggregation mutants and nontransgenic littermates for a similar time point (Fig. 8 G,I). An increased number of GFAP-positive cells were noticed in the entorhinal and piriform cortex of the proaggregation mutant mice (data not shown).
The level of presynaptic proteins and spine synapses decreases in the hippocampus of proaggregation transgenic mice
Because loss of synapses is known as one of the earliest changes in AD (Terry et al., 1991; Coleman and Yao, 2003), we asked how synapses and synaptic proteins were influenced by the presence of Tau in the brains of proaggregation and antiaggregation transgenic mice. Synaptophysin, a constituent of the synaptic vesicles, was chosen as a presynaptic marker (Rune et al., 2002), and spinophilin, a protein present in the dendritic spines, was selected as a postsynaptic marker (Muly et al., 2004). Frozen brain sections of the proaggregation and antiaggregation TauRD transgenic mice at 9.5 months of gene expression were quantitatively evaluated by immunohistochemistry for the levels of the synaptic proteins (Fig. 9 A). Significantly, the immunoreactivity of synaptophysin in the stratum radiatum of the CA1 hippocampal region in the case of the proaggregation mice was reduced by ∼70% compared with antiaggregation mutants and wild-type littermates. Similarly, in the CA3 region of the proaggregation mutant, the synaptophysin immunoreactivity decreased to ∼50% compared with control and antiaggregation mutant (Fig. 9 B). There was also a moderate decrease of spinophilin immunoreactivity in the proaggregation mutant (26% in CA1 region) (Fig. 9 C).
Figure 9.

TauRD/ΔK280 reduces spine-synapse density and synaptic markers. A, Stratum radiatum of CA1 region of hippocampus. Coronal brain sections from control, proaggregation, and antiaggregation mice were immunostained for the presynaptic marker synaptophysin. Immunohistochemistry staining was performed on animals after 9 months of TauRD gene expression and age-matched nontransgenic littermates. B, Quantification of staining shows that synaptophysin is strongly decreased in proaggregation mice [e.g., by ∼70% in the CA1 region (n = 9–16; mean ± SE; p < 0.001) and by ∼56% in the CA3 region (n = 9–16; mean ± SE; * p < 0.05)]. C, Quantification of spinophilin immunostaining in the CA1 and CA3 region shows no significant differences between proaggregation and antiaggregation mice, and a slight decrease compared with nontransgenic littermates (n = 9–16; mean ± SE). D, E, Brain sections from proaggregation and antiaggregation (9.5 months ON) mice were imaged by electron microscopy (stratum radiatum of the CA1 region). F, Quantitative evaluation of spine-synapses in the stratum radiatum of the CA1 hippocampal region. Note the decrease by 27% of the spine synapse number in the proaggregation mutant compared with antiaggregation and control mice (proaggregation mice had 6.7 ± 0.7 spine-synapses per 6.4 μm3; antiaggregation mice had 9.7 ± 0.5 spine synapses per 6.4 μm3; wild-type mice had 9.2 ± 0.6 spine synapses per 6.4 μm3) (n = 10; mean ± SE; *** p < 0.001). Scale bars: A, 40 μm; D, E, 600 nm.
Using thin sectioning electron microscopy with unbiased stereological evaluation, the number of spine synapses was measured in the stratum radiatum of the CA1 region of the proaggregation and antiaggregation mutants at 9.5 months of TauRD expression (Fig. 9 D,E). The spine synapses were distinguished from shaft synapses and a synapse was defined as the presence of (1) presynaptic, postsynaptic parts and synaptic cleft, (2) vesicles in the presynaptic region, and (3) a postsynaptic density plaque. We found a 27% decrease in spine-synapse count in the proaggregation mutant (from 9.2 to 6.7 per 6.4 μm3), compared with wild-type control. Antiaggregation mutant showed no reduction in spine synapses.
Missorting, phosphorylation, and aggregation of TauRD/ΔK280 protein are reversible after switching off the expression; only mouse Tau tangles tend to persist
An important question in the AD field is whether pathological changes and aggregation of Tau protein are reversible. The inducible TauRD mouse model allowed us to address this question by switching off the Tau expression through addition of 200 μg/ml doxycycline to the drinking water. Groups of mice were induced for 3 or 9 months, and for each group the transgene expression was switched off for 1.5 months, followed by biochemical and immunohistochemical analysis.
Switching the expression on for 3 months and then off for 1.5 months revealed that the level of exogenous TauRD/ΔK280 Sarkosyl-soluble and -insoluble protein almost disappeared (>90%) (Fig. 10 A). Interestingly, the aggregated endogenous mouse Tau in the Sarkosyl-insoluble fractions showed only a mild decrease, indicating that the tangle fraction of mouse Tau, once formed, is more persistent (presumably because of continued synthesis and nucleated aggregation) (see Discussion). Similar results were obtained in the case of older mice (9 months ON, and then 1.5 months OFF) (Fig. 10 B).
Figure 10.
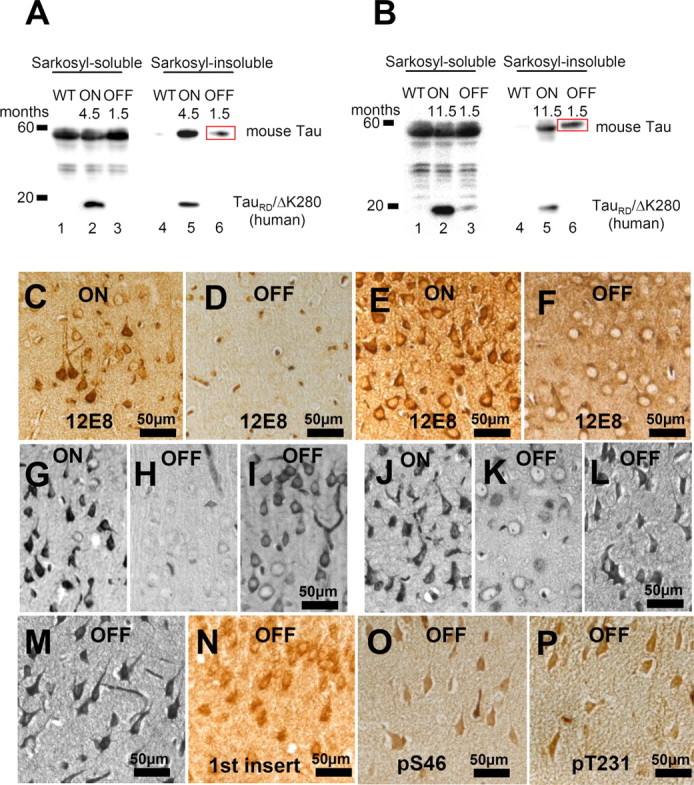
Phosphorylation and aggregation of Tau is reduced after switching off the expression of TauRD/ΔK280. A, Representative immunoblots of Sarkosyl-soluble and Sarkosyl-insoluble fractions from cortex homogenates of young proaggregation mice: lanes 1–3, Sarkosyl-soluble fractions; and lanes 4–6, Sarkosyl-insoluble fractions of wild-type mice; proaggregation mutant mice 4.5 months ON; or proaggregation mutant mice 3 months ON and 1.5 months OFF. B, Immunoblot analysis of Sarkosyl-soluble fractions (lanes 1–3) and Sarkosyl-insoluble fractions (lanes 4–6) from cortex homogenates in the case of older proaggregation mice. Lanes 1 and 4 show wild type; lanes 2 and 5 show proaggregation mutant, 11.5 months expression ON; and lanes 3 and 6 show proaggregation mutant, 10 months ON and 1.5 months OFF. Note that after switch-off for 1.5 months, the insoluble human TauRD disappears, whereas the insoluble mouse Tau stays aggregated (lane 6 in A and B; red boxes). C, Phosphorylation at KXGS motifs in Tau repeats seen by the 12E8 antibody in the cortex of proaggregation mutant at 4.5 months TauRD/ΔK280 gene expression and (D) loss of 12E8 immunoreactivity after 1.5 months switching off gene expression (3 months ON and 1.5 months OFF). E, F, 12E8 immunoreactivity in proaggregation transgenic mice at 11.5 months gene expression (E) was reversible after suppression of TauRD/ΔK280 gene for 1.5 months (10 months ON and 1.5 months OFF) (F). G, Tangles at 4.5 months of TauRD/ΔK280 gene expression in entorhinal cortex (Gallyas staining). H, I, Two examples of reduction (H) or persistence (I) of tangles in the entorhinal cortex after switching off TauRD/ΔK280 gene expression for 1.5 months in mice with initial 3 months gene expression (3 months ON and 1.5 months OFF). J, NFTs in entorhinal cortex of proaggregation mutant at 11.5 months TauRD/ΔK280 expression. K, L, Two examples of switch off experiments: after 1.5 month suppression of TauRD/ΔK280 gene expression, the tangle pathology decreased (K) or partly persisted (L) in proaggregation mice initially induced for 10 months (10 months ON and 1.5 months OFF). M–P, Entorhinal neurons in the brain of proaggregation mice after 1.5 months of switch off of the TauRD/ΔK280 transgene (10 months ON and 1.5 months OFF) were positive for Gallyas staining (M), and at the same time were stained by antibodies against the first N-terminal insert of Tau with antibody SA4473 (N), against Tau phosphorylated at S46 (O) and at T231 (P). Note that full-length mouse Tau is present in the persisting tangles after switching off exogenous TauRD. Scale bars: C–P, 50 μm.
The pathological somatodentritic localization of Tau phosphorylated at KXGS motifs showed complete reversibility in the somatosensory and piriform cortex after 1.5 months of switching off the TauRD/ΔK280 transgene (Fig. 10 C,D). Similar results were obtained in the case of older mice at 9–10 months of gene expression and 1.5 months of switch-off (Fig. 10 E,F). With regard to phosphorylation, the switching-off of TauRD/ΔK280 for 1.5 months after 10 months of expression lead to complete reversal of MARK type phosphorylation, independently of age and localization, whereas the neurofibrillary tangles were only partly reversed. This is in agreement with the above biochemical analysis, arguing that aggregates of exogenous TauRD are reversible, but aggregates of endogenous mouse Tau, once formed, are difficult to reverse (Fig. 10 G–L). Similar data were obtained by switching on for 3 months and then switching off the Tau transgene for 3 months (data not shown). To check the persistence of endogenous mouse Tau in tangles after switch-off experiments (10 months ON and 1.5 months OFF), coronal paraffin brain sections were probed by antibodies recognizing only endogenous mouse tau, for example against the first insert (Ab SA4473), against pS46 and pT231 (proline-rich domain before the repeats) (Fig. 10 N–P). All regions positive for Gallyas silver staining showed positive immunoreactivity, highlighting the persistence of endogenous Tau aggregates and of enhanced phosphorylation at SP or TP motifs.
To evaluate the toxicity of the aggregated mouse tau, we investigated the neuronal cell loss and astrogliosis in proaggregation mice after switching off the TauRD/ΔK280 transgene for 1.5 months. The hippocampal pyramidal areas that retained NFT pathology after switch-off displayed altered neuronal morphology and neuronal loss (Fig. 11 A–D). Phosphorylation at four phosphoepitopes located outside of the exogenous TauRD/ΔK280 construct, namely S46, S202, S404, and T231, confirmed that the Tau protein involved in the persisting NFTs and altered neuronal morphology was indeed endogenous mouse Tau (Fig. 11 E–H). The astrocytosis that was visible in the hilus area of the proaggregation mutant mouse at 6 months of gene expression, became clearly reduced after 1.5 months of switching off the transgene (Fig. 11 I,J).
Figure 11.
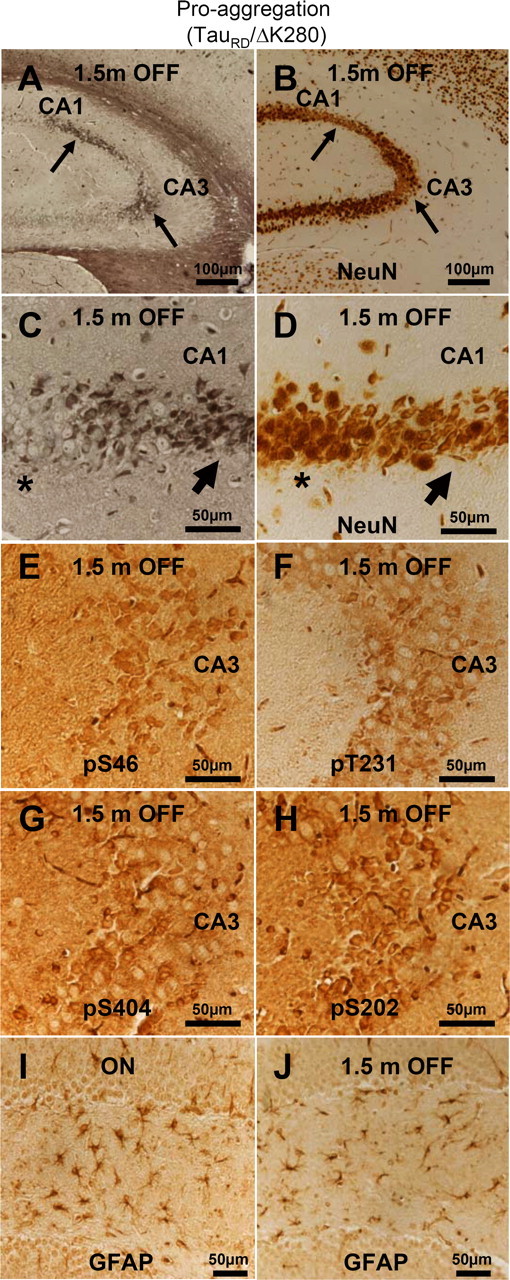
Aggregated mouse Tau is toxic to hippocampal neurons. A, NFT pathology (Gallyas silver staining) in the hippocampus of proaggregation mutant mice after switching off the TauRD/ΔK280 transgene expression for 1.5 months (10 months ON and 1.5 months OFF). B, NeuN staining of CA1 and CA3 hippocampal area with NFT pathology displays neuronal loss (arrows) after switching off the transgene for 1.5 months (10 months ON and 1.5 months OFF). C, Higher magnification of A showing NFT pathology by Gallyas silver staining. D, Higher magnification of B showing altered neuronal morphology and loss of neurons in the areas affected by NFT pathology after 1.5 months of switching off the transgene. Note that region of higher silver staining in C corresponds to lower neuron count in D (arrows). In contrast, the areas not affected by NFT pathology do not display altered neuronal morphology (stars). E–H, Phosphorylation of endogenous mouse Tau at S46, T231, S202, and S404 in the CA3 region of proaggregation mutant mice after 1.5 months of switching off gene expression (10 months ON and 1.5 months OFF). Note hyperphosphorylation of aggregated mouse Tau. I, J, The astrocytosis identified by GFAP was reduced in the proaggregation mutant after 1.5 months switching off (J) (4.5 months ON and 1.5 months OFF), compared with the proaggregation mutant with continuous transgene expression (I) (6 months ON). Scale bars: A, B, 100 μm; C–J, 50 μm.
Discussion
Neurofibrillary lesions are hallmarks of AD and other tauopathies, and mice expressing different Tau variants were developed for modeling the pathology (Lee et al., 2001; Brandt et al., 2005; LaFerla and Oddo, 2005; McGowan et al., 2006; Götz et al., 2007). Important questions are whether Tau hyperphosphorylation is necessary for aggregation, how endogenous Tau contributes to pathology, whether changes are reversible, and how Tau affects synapses and neurons. We designed mouse models in two variants in which the Tau repeat domain is expressed as proaggregation or antiaggregation mutants. The proaggregation mutation ΔK280 (identified in FTDP17) (Rizzu et al., 1999), strongly induces aggregation by enhancing β-structure (von Bergen et al., 2001; Khlistunova et al., 2006), which can be blocked by proline mutations in the hexapeptide motifs. The TauRD expression was driven in the forebrain by a CaMKIIα-promoter (Mayford et al., 1996). This allowed the development of models resembling AD in that pathology appears first in the entorhinal region and hippocampus, but not in the spinal cord, as in some previous mouse models (Götz et al., 1995; Spittaels et al., 1999). Another advantage was the regulatable Tau expression (Tet-off system) (Gossen and Bujard, 1992), which enabled analysis of the reversibility of Tau pathology.
The analysis of Tau effects is complicated by the multidomain structure of Tau. The repeat domain harbors the microtubule interactions and PHF aggregation, but these are modulated by other domains (hairpin or paperclip folding) (Jicha et al., 1999; Binder et al., 2005; Jeganathan et al., 2006). The repeats contain few phosphorylation sites (KXGS motifs) that strongly inhibit Tau–Tau or Tau–MT interactions (Schneider et al., 1999), whereas other domains contain multiple sites whose roles are uncertain (Stoothoff and Johnson, 2005). These problems are circumvented by using only the repeat domain for transfection.
With these tools, we addressed several issues: (1) What determines Tau aggregation in mice? Does the β-propensity predispose to aggregation? (2) Does Tau aggregation in mice affect similar regions as in AD? (3) Does Tau expression or aggregation correlate with neuronal toxicity? (4) What is the influence of Tau phosphorylation sites outside the repeats that are diagnostic of AD? (5) Can Tau aggregation, phosphorylation, and toxicity be reversed? (6) Does mouse Tau cooperate with exogenous Tau to generate pathology?
High β-propensity of TauRD/ΔK280 promotes aggregation even at low concentrations
Previous mouse models overexpressed human Tau under the Thy-1 promoter, resulting in hyperphosphorylation and somatodendritic localization but not tangle formation (Götz et al., 1995; Duff et al., 2000). The discovery of Tau mutations in FTDP17 helped in generating transgenic models for tauopathy. Thus, expressing Tau/P301L under the prion promoter caused NFTs in spinal cord, brainstem, and pretangles in cortex (Lewis et al., 2000), resulting in the loss of motor neurons. Other models using Tau/P301L under control of the CaMKIIα-promoter resulted in cortical NFTs at 3–5 months (Ramsden et al., 2005; SantaCruz el al., 2005). The rate of aggregation depended on the expression level so that only mice with high overexpression (13-fold) formed NFTs at an early age. In our proaggregation mice, NFTs appeared early (2–3 months) in the entorhinal cortex even at low expression (∼70% of endogenous tau). Aggregation occurred only with proaggregation but not with antiaggregation TauRD. Because these variants only differ in amyloidogenicity, they argue that the β-propensity drives aggregation in vitro and in brains. If the β-propensity is weaker (as in full-length mutant Tau), one requires higher Tau concentrations for aggregation within the animal's life span. If the β-propensity is destroyed (as in antiaggregation Tau), tangle formation does not occur. Another way to enhance β-propensity is the deletion of flanking regions outside the repeats because these can shield against aggregation (Wille et al., 1992; Binder et al., 2005). This explains why tangles develop faster with the proaggregation repeat domain than with full-length Tau (Eckermann et al., 2007).
Coaggregation of TauRD/ΔK280 with endogenous mouse Tau
There is a debate on whether transgenic human Tau interacts with endogenous mouse Tau. The uncertainty arises partly because mouse Tau does not normally form aggregates, although its assembly in vitro is indistinguishable from human Tau (Kampers et al., 1999). Secondly, full-length mouse and human Tau isoforms are difficult to discern because of their similarity and heterogeneity (several isoforms, multiple states of phosphorylation). Some experiments suggested that mouse Tau might protect transgenic mice from developing NFTs, and to overcome this impediment, several mouse models were generated on a knock-out background of endogenous mouse Tau (Andorfer et al., 2003; Terwel et al., 2005). Others demonstrated that lack of the prolyl-isomerase Pin1 caused NFTs from endogenous mouse Tau (Liou et al., 2003). Similarly, overexpression of p25, an activator of cdk5, caused NFT-like Tau aggregation (Cruz et al., 2003; Fischer et al., 2005). Thus, the role of mouse Tau in PHF formation remained elusive. In our transgenic mice, the analysis was simplified because the exogenous repeat domain is easily discerned from endogenous Tau, and in addition it aggregates rapidly. The unexpected observation was that human TauRD induced pathological changes even in mouse Tau, including missorting, pathological conformation, phosphorylation, and coaggregation. This is consistent with the fact that different human and mouse Tau isoforms can coassemble in vitro (Kampers et al., 1999). The abnormal distribution and phosphorylation of mouse Tau argues that aggregation of the exogenous Tau can trigger abnormal changes in the total Tau pool (i.e., foreign Tau can “poison” host Tau).
Loss of neurons and astrogliosis
Apart from aggregation, our transgenic mice displayed other features of AD, namely, loss of neurons and astrogliosis, analogous to other Tau models (Lewis et al., 2000; Allen et al., 2002; Ramsden et al., 2005). In proaggregation mice, neuronal loss started in the dentate gyrus at 5 months. This neurodegeneration shows that neuronal loss stems from neurotoxicity induced by aggregation because it is absent in antiaggregation mice. Neuronal loss and altered morphology remained after switching off the TauRD/ΔK280 transgene for 1.5 months, particularly in regions showing NFT pathology. Recent reports on Tau/P301L mice demonstrated that brief transgene suppression (1.5–2 months) sufficed to stop neuronal loss in the CA1 region, but the number of neurons did not recover (SantaCruz et al., 2005). The GFAP immunoreactivity is normally limited to white matter, whereas in AD brains reactive astrocytes occur around plaques (Dickson et al., 1988). Astrogliosis was detected in other Tau models resulting from neurofibrillary pathology (Schindowski et al., 2006; Yoshiyama et al., 2007). Our mice displayed GFAP-positive cells in the hilus region of proaggregation but not antiaggregation mutants, demonstrating that aggregation induces an inflammatory response. However, switching off the transgene for 1.5 months in proaggregation mice resulted in reduced inflammation and Gallyas-stained tangles.
Amyloidogenic proaggregation TauRD/ΔK280 induces loss of synapses
Memory impairment in AD correlates best with synaptic loss (Terry et al., 1991; Selkoe, 2002). Indeed, wild-type Tau can damage synapses in cell models by inhibiting intraneuronal transport (Stamer et al., 2002; Thies and Mandelkow, 2007). In the present study, synaptophysin was downregulated in the CA1–CA3 regions in proaggregation mice. Consistent with this, a P301S tauopathy model showed that hippocampal synaptic pathology appears early in neurodegeneration, accompanied by decreased presynaptic synaptophysin and postsynaptic GluR2/3 (Yoshiyama et al., 2007). We observed only a moderate decrease of spinophilin in the CA1–CA3 regions. Gallyas-positive neurons in the CA3 region and hyperphosphorylated Tau in the hippocampus of the proaggregation mice could influence the CA3–CA1 connections through Schaffer collaterals. This might provide a mechanism whereby TauRD/ΔK280 affects the function of hippocampal neurons. Interestingly, the number of spine-synapses was significantly downregulated in the CA1 region of proaggregation but not in antiaggregation or control mice.
Reversibility of aggregation
A major discussion point in AD research is the reversibility of Tau missorting, pathological conformation, hyperphosphorylation, and aggregation, especially considering that Aβ-induced toxicity is mediated by Tau (Santa Cruz et al., 2005; Roberson et al., 2007). Our proaggregation mice displayed complete reversibility of the changes involving TauRD/ΔK280 [i.e., once the expression ceased, the protein disappeared in soluble and aggregated form within 6 weeks, consistent with proteasomal degradation (Petrucelli et al., 2004)]. The result agrees with our N2a cell model in which aggregation of TauRD/ΔK280 and toxicity was reversible after switch-off (Wang et al., 2007). Larger aggregates (tangles) may be cleared by autophagy (Rubinsztein et al., 2007), although even tangles are in dynamic equilibrium with Tau subunits, judging by the intrinsic lability of PHFs (Li et al., 2002), and could therefore disappear via the proteasome pathway. The unexpected feature was, however, the persistence of pathological changes of endogenous mouse Tau after switching off the exogenous TauRD (Fig. 10 M–P). Our interpretation is that these aggregates are continuously replenished from the pool of newly synthesized mouse Tau, which elongates existing seeds of filaments, once they have been generated with the previous help of exogenous mutant TauRD. This would also explain the results of SantaCruz et al. (2005) in which the levels of exogenous Tau mRNA, neuronal loss, and memory were reversible, but tangle pathology was not. Thus, the persistence of aggregated mouse Tau illustrates how pathological properties of a foreign protein (human TauRD) can be transmitted to the host protein.
Footnotes
This work was supported by the Max Planck Society and Deutsche Forschungsgemeinschaft. We thank Anne Hofmann for excellent technical assistance, and Dr. A. Haemisch and his team of the animal facility at University of Hamburg Medical School for their continuous help in mouse breeding. We gratefully acknowledge reagents from Dr. E. Kandel (Columbia University, New York, NY) (CaMKIIα-tTA activator transgenic mice), Dr. P. Seubert (Elan Pharma, South San Francisco, CA) (12E8 antibody), Dr. L. I. Binder (Northwestern University, Chicago, IL) (Tau-1 antibody), and Dr. P. Davies (Albert Einstein College, Bronx, NY) (MC-1 antibody).
References
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Zheng-Fischhofer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow EM, Mandelkow E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–11721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- Baron U, Freundlieb S, Gossen M, Bujard H. Co-regulation of two gene activities by tetracycline via a bidirectional promoter. Nucleic Acids Res. 1995;23:3605–3606. doi: 10.1093/nar/23.17.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- Brandt R, Hundelt M, Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta. 2005;1739:331–354. doi: 10.1016/j.bbadis.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer's disease. Neurobiol Aging. 2003;24:1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH. Alzheimer's disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol. 1988;132:86–101. [PMC free article] [PubMed] [Google Scholar]
- Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Burki K, Davies P. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A, Schönig K, Bujard H, Haemisch A, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of Alzheimer tauopathy. J Biol Chem. 2007;282:31755–31765. doi: 10.1074/jbc.M705282200. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron. 2005;48:825–838. doi: 10.1016/j.neuron.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Cleveland DW. Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr Opin Cell Biol. 2001;13:41–48. doi: 10.1016/s0955-0674(00)00172-1. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- Goedert M, Klug A, Crowther RA. Tau protein, the paired helical filament and Alzheimer's disease. J Alzheimers Dis. 2006;9:195–207. doi: 10.3233/jad-2006-9s323. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J, Probst A, Spillantini MG, Schafer T, Jakes R, Burki K, Goedert M. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995;14:1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Götz J, Deters N, Doldissen A, Bokhari L, Ke Y, Wiesner A, Schonrock N, Ittner LM. A decade of tau transgenic animal models and beyond. Brain Pathol. 2007;17:91–103. doi: 10.1111/j.1750-3639.2007.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Berenfeld B, Davies P. Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer's disease. J Neurosci Res. 1999;55:713–723. doi: 10.1002/(SICI)1097-4547(19990315)55:6<713::AID-JNR6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Kampers T, Friedhoff P, Biernat J, Mandelkow EM. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–349. doi: 10.1016/s0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- Kampers T, Pangalos M, Geerts H, Wiech H, Mandelkow E. Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer's disease. FEBS Lett. 1999;451:39–44. doi: 10.1016/s0014-5793(99)00522-0. [DOI] [PubMed] [Google Scholar]
- Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- Kretz O, Fester L, Wehrenberg U, Zhou L, Brauckmann S, Zhao S, Prange-Kiel J, Naumann T, Jarry H, Frotscher M, Rune GM. Hippocampal synapses depend on hippocampal estrogen synthesis. J Neurosci. 2004;24:5913–5921. doi: 10.1523/JNEUROSCI.5186-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Oddo S. Alzheimer's disease: Abeta, tau and synaptic dysfunction. Trends Mol Med. 2005;11:170–176. doi: 10.1016/j.molmed.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Li L, von Bergen M, Mandelkow EM, Mandelkow E. Structure, stability, and aggregation of paired helical filaments from tau protein and FTDP-17 mutants probed by tryptophan scanning mutagenesis. J Biol Chem. 2002;277:41390–41400. doi: 10.1074/jbc.M206334200. [DOI] [PubMed] [Google Scholar]
- Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E, von Bergen M, Biernat J, Mandelkow E-M. Structural principles of Tau and the paired helical filaments of Alzheimer's disease. Brain Pathol. 2007;17:83–90. doi: 10.1111/j.1750-3639.2007.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Thies E, Trinczek B, Biernat J, Mandelkow E. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol. 2004;167:99–110. doi: 10.1083/jcb.200401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Muly EC, Smith Y, Allen P, Greengard P. Subcellular distribution of spinophilin immunolabeling in primate prefrontal cortex: localization to and within dendritic spines. J Comp Neurol. 2004;469:185–197. doi: 10.1002/cne.11001. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Perez M, Valpuesta JM, Medina M, Montejo de Garcini E, Avila J. Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J Neurochem. 1996;67:1183–1190. doi: 10.1046/j.1471-4159.1996.67031183.x. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Prange-Kiel J, Rune GM, Leranth C. Median raphe mediates estrogenic effects to the hippocampus in female rats. Eur J Neurosci. 2004;19:309–317. doi: 10.1111/j.0953-816x.2003.03124.x. [DOI] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in The Netherlands. Am J Hum Genet. 1999;64:414–421. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- Rune GM, Wehrenberg U, Prange-Kiel J, Zhou L, Adelmann G, Frotscher M. Estrogen up-regulates estrogen receptor alpha and synaptophysin in slice cultures of rat hippocampus. Neuroscience. 2002;113:167–175. doi: 10.1016/s0306-4522(02)00152-5. [DOI] [PubMed] [Google Scholar]
- SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, Buee L. Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, Lee VM-Y. Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem. 1995;270:18917–18922. doi: 10.1074/jbc.270.32.18917. [DOI] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739:280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Sun A, Nguyen XV, Bing G. Comparative analysis of an improved thioflavin-s stain, Gallyas silver stain, and immunohistochemistry for neurofibrillary tangle demonstration on the same sections. J Histochem Cytochem. 2002;50:463–472. doi: 10.1177/002215540205000403. [DOI] [PubMed] [Google Scholar]
- Tanemura K, Chui DH, Fukuda T, Murayama M, Park JM, Akagi T, Tatebayashi Y, Miyasaka T, Kimura T, Hashikawa T, Nakano Y, Kudo T, Takeda M, Takashima A. Formation of tau inclusions in knock-in mice with familial Alzheimer disease (FAD) mutation of presenilin 1 (PS1) J Biol Chem. 2006;281:5037–5041. doi: 10.1074/jbc.M509145200. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Terwel D, Dewachter I, Van Leuven F. Axonal transport, tau protein, and neurodegeneration in Alzheimer's disease. Neuromolecular Med. 2002;2:151–165. doi: 10.1385/NMM:2:2:151. [DOI] [PubMed] [Google Scholar]
- Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem. 2005;280:3963–3973. doi: 10.1074/jbc.M409876200. [DOI] [PubMed] [Google Scholar]
- Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci USA. 2000;97:5129–5134. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, Mandelkow E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J Biol Chem. 2001;276:48165–48174. doi: 10.1074/jbc.M105196200. [DOI] [PubMed] [Google Scholar]
- Wang YP, Biernat J, Pickhardt M, Mandelkow E, Mandelkow EM. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci USA. 2007;104:10252–10257. doi: 10.1073/pnas.0703676104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille H, Drewes G, Biernat J, Mandelkow EM, Mandelkow E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J Cell Biol. 1992;118:573–584. doi: 10.1083/jcb.118.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]