

Abstract
Recent advance in the design of genetically encoded calcium indicators (GECIs) has further increased their potential for direct measurements of activity in intact neural circuits. However, a quantitative analysis of their fluorescence changes (ΔF) in vivo and the relationship to the underlying neural activity and changes in intracellular calcium concentration (Δ[Ca2+]i) has not been given. We used two-photon microscopy, microinjection of synthetic Ca2+ dyes and in vivo calibration of Oregon-Green-BAPTA-1 (OGB-1) to estimate [Ca2+]i at rest and Δ[Ca2+]i at different action potential frequencies in presynaptic motoneuron boutons of transgenic Drosophila larvae. We calibrated ΔF of eight different GECIs in vivo to neural activity, Δ[Ca2+]i, and ΔF of purified GECI protein at similar Δ[Ca2+] in vitro. Yellow Cameleon 3.60 (YC3.60), YC2.60, D3cpv, and TN-XL exhibited twofold higher maximum ΔF compared with YC3.3 and TN-L15 in vivo. Maximum ΔF of GCaMP2 and GCaMP1.6 were almost identical. Small Δ[Ca2+]i were reported best by YC3.60, D3cpv, and YC2.60. The kinetics of Δ[Ca2+]i was massively distorted by all GECIs, with YC2.60 showing the slowest kinetics, whereas TN-XL exhibited the fastest decay. Single spikes were only reported by OGB-1; all GECIs were blind for Δ[Ca2+]i associated with single action potentials. YC3.60 and D3cpv tentatively reported spike doublets. In vivo, the KD (dissociation constant) of all GECIs was shifted toward lower values, the Hill coefficient was changed, and the maximum ΔF was reduced. The latter could be attributed to resting [Ca2+]i and the optical filters of the equipment. These results suggest increased sensitivity of new GECIs but still slow on rates for calcium binding.
Keywords: GFP; calcium, neural activity; fluorescence; genetic probes; in vivo imaging
Introduction
The rapid development of fluorescent genetic probes [for review, see Miesenböck and Kevrekidis (2005) and Kleinfeld and Griesbeck (2005)] might soon allow routine recordings of activity in intact neural circuits and long-term studies in intact and behaving animals. Moreover, genetic probes promise to overcome the spatial limitations of electrode recordings by monitoring large ensembles of functionally related neurons (Fiala et al., 2002; Ng et al., 2002; Higashijima et al., 2003; Wang et al., 2003) as well as small subcellular compartments (Guerrero et al., 2005; Reiff et al., 2005). Whereas intracellular and extracellular electrode recordings report changes in membrane and field potential, respectively, genetic probes usually translate a change in ion concentration into changes in the fluorescence of green fluorescent protein (GFP) variants (Miyawaki, 2005). The kinetics of this translation process and the interplay with parameters of the cell at rest determine whether biological signals can be discerned from noise.
Direct assessment of membrane potential has been promised by several genetic probes (Siegel and Isacoff, 1997; Sakai et al., 2001; Ataka and Pieribone, 2002; Guerrero et al., 2002), but small fluorescence changes (ΔF) or slow kinetics prevented their successful application in vivo. On the other hand, synaptopHluorin (Miesenböck et al., 1998) successfully provided a fluorescent readout of slow changes in the net equilibrium of presynaptic vesicle release and recycling (Ng et al., 2002; Bozza et al., 2004; Shang et al., 2007). However, experiments in neurons of nematodes (Suzuki et al., 2003; Clark et al., 2007), fruit flies (Fiala et al., 2002; Reiff et al., 2002; Wang et al., 2003, 2004; Suh et al., 2004; Marella et al., 2006), zebrafish (Higashijima et al., 2003), and mice (Hasan et al., 2004; Díez-García et al., 2005; Heim et al., 2007) suggest that genetically encoded calcium indicators (GECIs) report spatial and temporal dynamics of neural activity with somewhat higher accuracy. Yet, they still suffer from rather small ΔF, poor signal-to-noise ratio (SNR), a capricious relationship to neural activity, slow kinetics, and changes in the dissociation constant (KD) in vivo (Hasan et al., 2004; Pologruto et al., 2004; Reiff et al., 2005). Most importantly, no faithful calibration of ΔF, the underlying neural activity, and change in intracellular calcium concentration (Δ[Ca2+]i) has been given for different GECIs in similar neurons in vivo so far.
We injected synthetic calcium indicators into subcellular compartments in Drosophila and calibrated ΔF of Oregon-Green-BAPTA-1 (OGB-1) in situ (Maravall et al., 2000). This allowed us to estimate [Ca2+]i at rest and to convert ΔF of OGB-1 to Δ[Ca2+]i at known rates of sustained neuronal activity when [Ca2+]i is at steady state. The amplitude of these fluorescence changes at steady state is not influenced by buffering of calcium by the indicator and thus independent of indicator concentration (see Discussion). Following this approach, ΔF of eight different GECIs was correlated to Δ[Ca2+]i at similar activity rates and to ΔF of purified GECI protein in vitro. In addition, the capacity of the GECIs to report transient changes in neural activity was analyzed. This way we analyzed Yellow Cameleon 3.3 (YC3.3) (Griesbeck et al., 2001), YC3.60, YC2.60 (Nagai et al., 2004), D3cpv (Palmer et al., 2006), TN-L15 (Heim and Griesbeck, 2004), TN-XL (Mank et al., 2006), GCaMP1.6 (GC1.6) (Ohkura et al., 2005), and GCaMP2 (Díez-García et al., 2005; Tallini et al., 2006) by two-photon laser-scanning microscopy (2PLSM) (Denk et al., 1990) in presynaptic boutons of transgenic Drosophila larvae and describe what these recent indicators can tell us about neural activity in vivo and what they still cannot.
Materials and Methods
Flies and genetics
Using the Gal4/UAS system (Brand and Perrimon, 1993), we directed expression of GECIs or monomeric DsRed (mDsRed) to the nervous system of flies. UAS-GECI flies carried cDNA for each GECI under UAS control as described by Reiff et al. (2005). Flies expressing YC3.3 (Griesbeck et al., 2001), GC1.6 (Ohkura et al., 2005), and TN-L15 (Heim and Griesbeck, 2004) were described by Reiff et al. (2005); TN-XL flies were described by Mank et al. (2006). For YC3.60, YC2.60 (Nagai et al., 2004), D3cpv (Palmer et al., 2006), and GCaMP2 (Tallini et al., 2006) new transgenic lines were generated.
We crossed 10 female, virgin elavC155-Gal4 (Lin and Goodman, 1994) flies to five male UAS-GECI flies. For experiments, we collected female, third-instar larval offspring. Where double heterozygous flies showed insufficient expression levels for imaging experiments, offspring was back-crossed to UAS-GECI or elavC155-Gal4 flies to yield flies homozygous for one or both transgenes. All animals were raised at 25°C on standard corn meal medium supplemented with yeast.
Transgenic flies were created by P-element-mediated germline transfection (Spradling and Rubin, 1982) in white− genetic background (w−, “Bayreuth” flies kindly provided by Christian Lehner, University of Bayreuth, Bayreuth, Germany). Stocks were established from hatching flies as described previously (Reiff et al., 2005). All fly strains used appeared normal in locomotion and neuromuscular junction (NMJ) gross morphology; however, this was not systematically addressed.
cDNAs for YC3.60 (provided by Atsushi Miyawaki, RIKEN Brain Science Institute, Wako City, Saitama, Japan) and TN-XL (provided by Marco Mank and Oliver Griesbeck, Max Planck Institute of Neurobiology, Martinsried, Germany) were amplified by PCR, adding NotI restriction sites to 3′ and 5′ ends, and inserted into the NotI site of the multicloning site of pUAST (Brand and Perrimon, 1993). Orientation of the insertion was controlled by sequencing.
D3cpv was provided in pcDNA3 in BamHI and EcoRI sites (by Maz Hasan, Max Planck Institute for Experimental Medicine, Heidelberg, Germany). An upstream His tag with NotI site at the 5′ end was inserted between HindIII and BamHI. A NotI/NotI fragment (including His tag and D3cpv) was then extracted and cloned into pUAST. Orientation and sequence were confirmed.
YC2.60 was generated by subcloning YC3.60 into pRSETb. PCR with primers GGCTACATCAGCGCTGCTGAATTACGTCACGTCATGACAAACC and GGTTTGTCATGACGTGACGTAATTCAGCAGCGCTGATGTAGCC were used to introduce two base exchanges (CAG to GAA), leading to the E104Q amino acid exchange in calmodulin as described by Nagai et al. (2004).
pUAST-GCaMP2 (Tallini et al., 2006) was generated by isolating a BglII/NotI fragment containing GCaMP2 preceded by His repeats from pN1 (provided by Junichi Nakai, RIKEN Brain Science Institute, Wako-shi, Saitama, Japan) and inserting this fragment between the BglII and NotI sites of pUAST.
UAS-mDsRed flies used for the injection of synthetic Ca2+ sensors were kindly provided by Gaia Tavosanis (Max Planck Institute of Neurobiology, Martinsreid, Germany).
GECIs
We compared ratiometric GECIs and single-chromophore indicators in their fluorescence responses to different activity levels in presynaptic boutons of the larval Drosophila NMJ. The GECIs are based on a Ca2+-binding protein fused to one or two different GFP variants. Ca2+ binding induces a conformational change in the binding protein(s) that leads either to enhanced fluorescent resonance energy transfer (FRET) (in ratiometric indicators) or to a higher ratio of deprotonated/protonated forms of the chromophore. The deprotonated and protonated forms are represented by the long-wavelength peak (490 nm) and the short-wavelength peak (375 nm) of GFP emission, respectively (Tsien, 1998). Thus, increase in Ca2+ increases the long-wavelength emission in GCaMPs. Specific constituents and basic properties of the GECIs included in this study are summarized in Table 1.
Table 1.
Characteristics of GECIs and OGB-1 in vivo and in vitro
| Probe | GFP variantsa | Functional sensorb | Hill coefficient |
KD |
Max. change |
τ risei (40 Hz)k | τ decayj (40 Hz)k | |||
|---|---|---|---|---|---|---|---|---|---|---|
| In vitroc | In vivod | In vitroe | In vivof | In vitrog | In vivoh | |||||
| YC3.3 | ECFP/Citrine | CaM/M13 | — | 2.50 | — | 0.47 μm | — | 67% | 1.41 s | 1.05 s |
| — | (1.5 μm) | 38 Hz | (100%) | |||||||
| YC3.60 | ECFP/cpVenus | CaM/M13 E104Q | 2.24 | 1.67 | 0.63 μm | 0.36 μm | 346% | 136% | 0.82 s | 0.73 s |
| (1.7) | (0.25 μm) | 30 Hz | (560%) | |||||||
| YC2.60 | ECFP/cpVenus | CaM/M13 | — | 1.84 | — | 0.40 μm | — | 194% | —k | 5.24 sk |
| (2.4) | (0.04 μm) | 32 Hz | (560%) | |||||||
| D3cpv | ECFP/cpVenus | CaM/M13 redesigned | 0.63 | 0.96 | 0.66 μm | 0.49 μm | 190% | 90% | 0.68 sk | 1.96 sk |
| (0.74) | (0.53 μm) | 41 Hz | (530%) | |||||||
| TN-L15 | ECFP/Citrine | csTnC | 1.29 | 2.59 | 0.72 μm | 0.36 μm | 82% | 60% | 0.81 s | 1.49 s |
| (0.89) | (1.2 μm) | 30 Hz | (140%) | |||||||
| TN-XL | ECFP/cpCitrine | csTnC mutl | 1.90 | 2.74 | 2.20 μm | 0.77 μm | 252% | 106% | 0.59 s | 0.20 s |
| (1.7) | (2.5 μm) | 65 Hz | (400%) | |||||||
| GC1.6 | cpEGFP | CaM/M13 | — | 2.34 | 1.23 μm | 0.64 μm | — | 162% | 1.38 s | 0.45 s |
| (3.8) | (0.15 μm) | 54 Hz | (490%) | |||||||
| GC2 | cpEGFP | CaM/M13 | — | 2.30 | — | 0.75 μm | — | 156% | 0.63 s | 0.38 s |
| (3.8) | (0.15 μm) | 63 Hz | (490%) | |||||||
| OGB-1 | — | BAPTA | 0.97 | 1.48 | 0.24 μm | 0.26 μm | 11 | 316% | 0.24 sk | 0.38 sk |
| (0.17 μm) | 21 Hz | (14) | ||||||||
a“GFP variants” lists the chromophores used in each indicator. cp, Circular permuted variant; EGFP, enhanced YFP; ECFP, enhanced CFP. b“Functional sensor” indicates the Ca2+ binding and interaction moieties. CaM, Calmodulin; csTnC, chicken skeletal muscle troponin C. Hill coefficients are listed for ccuvette measurements (Fig. 4) and din vivo measurements (Fig. 3). KD values shown represent ecuvette measurements (Fig. 4) and fin vivo data (Fig. 3), respectively. Max. change represents (ΔR/R)max or (ΔF/F)max in gcuvette (uncorrected spectrophotometer measurements) for [Ca2+] = 39.8 μm and hamplitudes at 160 Hz in vivo (2PLSM measurements) as shown in Figure 4. iτ rise and jτ decay were determined from single-exponential fits to rise and decay phases of indicator responses to 40 Hz. k20 Hz for YC2.60, D3cpv, and OGB-1. These values represent the properties of both the indicator and the specific fly lines. Values in parentheses represent literature values from original publications as cited in the text. lFour mutations within the C-terminal lobe of troponin C altered Mg2+ and Ca2+ binding properties.
Physiology and optical imaging
The larval preparation and solutions have been described previously (Macleod et al., 2002; Reiff et al., 2005). Unless stated otherwise, HL6 with 7 mm l-glutamate and 1.5 mm Ca2+ at pH 7.2 was used for superfusion of preparations and filling of electrodes. l-Glutamate effectively blocks postsynaptic muscle contractions at concentrations ≥5 mm without influencing presynaptic Ca2+ dynamics. In brief, late third-instar larvae were pinned to the bottom of a recording chamber with Sylgard lining and cut open along the dorsal midline using buckled scissors (Frohnhäuser); fat body, gut, and big trachea were removed. Segmental nerves were severed, and the ventral nerve cord (vnc) and brain were removed. Larval NMJs were recorded at muscle 6/7 in abdominal segments 2, 3, or 4. Presynaptic boutons were stimulated by placing the cut end of a segmental nerve into a suction electrode. We induced action potentials (APs) by applying voltage pulses (5.5 V, 0.3 ms) to the nerve (Iso-Stim 01-D; NPI Electronic). Pulses were applied at frequencies of 160, 80, 40, 20, 10, and 0 Hz, respectively, over a period of 2 s. Sequence of stimulus frequencies was altered pseudorandomly. Recording time was 8 s (12 s for YC2.60 because of slow decay time constant of the Ca2+ response) (see Figs. 2, 3), and stimulus period was 2 s starting after 2 s. The interval between individual recordings was 1 min at least.
Figure 2.
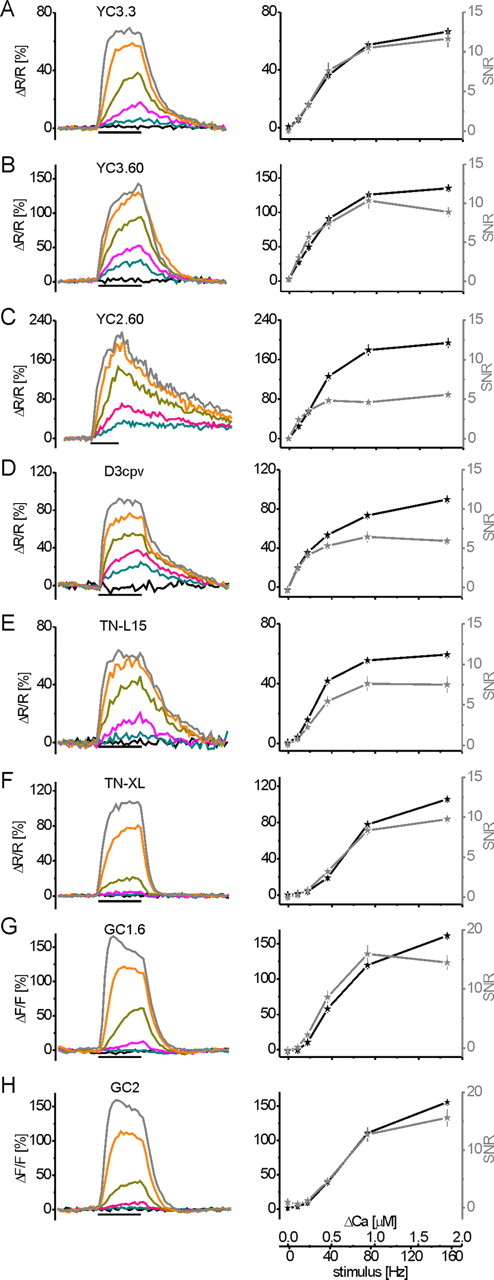
GECI fluorescence changes at sustained neural activity and Δ[Ca2+]i. Transgenic flies were engineered to allow the expression of eight different GECIs in vivo. Fluorescence changes were analyzed at presynaptic terminals of the larval NMJ, similar to the experiment in Figure 1 [2 s stimulus period indicated by the black bar underneath the recording traces; fAP = 0 (black), 10 (blue), 20 (purple), 40 (green), 80 (orange), and 160 (gray) Hz]. A, YC3.3; B, YC3.60; C, YC2.60; D, D3cpv; E, TN-L15; F, TN-XL; G, GC1.6; H, GC2. To the left, mean fractional fluorescence changes (ΔR/R for all dual-chromophore indicators and ΔF/F for GC1.6 and GC2) of a large number of experiments are plotted as a function of time (for n, see Results). To the right, the maximum fluorescence change (black trace) and SNR (gray trace) calculated at steady state are plotted as a function of the stimulus frequency and the corresponding Δ[Ca2+]i, respectively (for conversion of fluorescence changes to Δ[Ca2+]i via OGB-1, see Fig. 1). Images were recorded at 8 Hz frame rate for 8 s (12 s for YC2.60; note different time scale of stimulus bar).
Figure 3.

Summary of the in vivo quantification of GECI and OGB-1 fluorescence changes at steady state. A, B, Side-by-side comparison of maximum fractional fluorescence changes (A) and SNR (B) plotted as a function of Δ[Ca2+]i (logarithmic x- and y-axes). C, Δ[Ca2+]i necessary to elicit a fractional fluorescence change with SNR of 2 under our recording conditions. D, Time constants for the decay of the fluorescence change, determined after 40 Hz stimulation (20 Hz for YC2.60, D3cpv, and OGB-1). Parameters in A–C were analyzed at steady state; OGB-1 data are from Figure 1, and GECI data are from Figure 2.
We imaged individual boutons using a custom-built 2P microscope [design kindly provided by Winfried Denk, Max Planck Institute for Medical Research, Heidelberg, Germany (Wachowiak et al., 2004)], which allows for wide-field or 2P imaging through the same objective [63×/0.90 numerical aperture (NA) for GECIs, 40×/0.80 NA for OGB-1 and Magnesium Green (Invitrogen), water-immersion, IR Achroplan; Zeiss]. Wide-field illumination used a 150 W mercury arc lamp housed in a lamp house and power supply from Opti-Quip (models 770 and 1600) with optical filters [450/50 excitation, 480 long-pass (LP) dichroic, and 510/50 emission for yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP); 565/30 excitation, 585 LP dichroic, and 620/60 emission for mDsRed; all optical filters and dichroic mirrors by AHF]. The epifluorescence condenser was coupled to the microscope head containing tube lens, mirrors, and step motors to move the objective in three dimensions while the optical path was kept nearly constant (steering: Sutter MP285). Emitted light was projected onto the chip (1040 × 1392 pixels) of a CCD camera (Cool Snap HQ and MetaView software; Visitron Systems). Switch from wide-field to 2P microscopy involved moving of two mirrors. 2P fluorescence was excited by a mode-locked Ti:Sapphire laser (<100 fs, 80 MHz, 700–1000 nm; pumped by a 10 W Millenia laser; both Tsunami; Spectraphysics). Laser intensity was held constant at 6–15 mW for GECIs and at 45 mW for OGB-1 and Magnesium Green to minimize photobleaching and allow sufficient SNR (Patterson and Piston, 2000). Ratiometric GECIs were excited at 830 nm, GCaMPs at 920–930 nm, and OGB-1 and Magnesium Green at 950 nm. An emission filter (700 SP) was inserted in front of a cassette of two photomultiplier tubes, allowing simultaneous recording of different wavelengths of light. The photomultiplier tubes were equipped with bandpass dichroic mirrors (485/40 for CFP; 535/30 for YFP; 510/50 for GCaMPs, OGB-1, and Magnesium Green; and 620/60 for DsRed). Image acquisition was controlled by custom software [CfNT, written by R. Stepnoski (Bell Labs, Murray Hill, NJ) and M. Müller (Max Planck Institute for Medical Research, Heidelberg, Germany)].
Ca2+ signals were recorded at 64 × 64 pixel resolution at 8 Hz frame rate or at 1 × 64 pixel line scans at 500 Hz, respectively. Experiments were controlled by custom software [written in Delphi (Borland) by J. Haag (Max Planck Institute of Neurobiology, Martinsried, Germany)] using an analog-to-digital converter (DAS-1602/12; Computerboards). All data were analyzed using custom software written in IDL (RSI).
Data analysis
Further data evaluation and signal processing was done in Matlab R2006b (MathWorks) software and Origin 7.5 (Additive).
Single-chromophore indicators.
For background subtraction, a homogeneous region neighboring individual boutons was selected, and its intensity was subtracted from the intensity of the bouton. (For line scans, a time-averaged mean intensity next to each bouton was subtracted as background.) Bleach correction of individual bouton intensity traces was done by deleting the stimulation period before fitting a single-exponential function to each trace and subtracting the resulting function from the original fluorescence trace. From the corrected data for each bouton, fractional fluorescence changes (ΔF/F) were calculated by subtracting the average intensity measured before stimulus onset (average of nine control frames = Fcntrl) from the fluorescence in each image Ft of a series and subsequently dividing the difference by Fcntrl. (For line scans, the average of the first 240 lines was used as Fcntrl.)
Double-chromophore indicators.
The ratio of fluorescence values from both channels was calculated after background subtraction and bleach correction. The resulting trace was processed as described above to yield relative changes in the fluorescence ratio (ΔR/R).
SNR.
SNR was calculated for each bouton as the ratio of the mean ΔR/R or ΔF/F from five frames around peak amplitude and the corresponding SD. Presented values represent mean SNRs of all boutons measured at one stimulus protocol. (For line scans, amplitudes and SNR were calculated separately for each stimulus event within individual recordings. Then all SNR values from all four trials were averaged). The Δ[Ca2+]i and frequency of action potentials (fAP) leading to SNR = 2 were interpolated by fitting experimental results to sigmoidal functions in semilogarithmic plots (concentration log-scaled, SNR linear).
Two-photon guided dye injection
Sharp electrodes (R ∼ 100 MΩ; Science Products) were made on a standard puller (P-97; Sutter Instruments) and backfilled with OGB-1 or Magnesium Green, respectively (10 mm dye, in 0.5 m KCl and 2 m KAc). Photomultipliers of the 2PLSM were equipped with red (620/60 nm) and green (535/50 nm; AHF) bandpass filters for simultaneous dual-channel recording of monomeric DsRed and synthetic calcium dyes (red channel: axon terminals at larval NMJs; green channel: dye-filled electrode tip/loaded boutons). Excitation was adjusted to 980 nm for dye injection and 950 nm for recordings. Pressure was applied to the electrode to provide a minimal efflux from the tip and to avoid Ca2+ diffusion into the electrode. Injections were mostly done into boutons, occasionally into axons. The injection electrode was steered with electronic manipulator units (Luigs and Neumann Feinmechanik). Membrane potentials were between −20 and −55 mV; NMJs displaying lower membrane potentials did not show Ca2+ responses after dye injection. Dye concentration reached a plateau after 20 min (data not shown). Concentration in NMJs was unknown. However, ΔF/F amplitudes at steady-state Ca2+ are independent of indicator concentration, and thus different experiments were pooled. In contrast, time constants and SNR of fluorescence signals are affected by dye concentration. The presented time constants and SNRs represent an average over different concentrations in an experimentally realistic range.
Calcium measurements in NMJs
We used a method that allows intracellular Ca2+ measurements without wavelength ratioing described by Maravall et al. (2000). Briefly this method relies on a known KD for the given indicator, in situ measurements of Rf = Fmax/Fmin, and linear [Ca2+]/stimulus relationship of the system under observation. It provides resting [Ca2+] by
 |
and Δ[Ca2+] for a given steady-state response as follows:
 |
where [Ca2+]0 is intracellular [Ca2+] at rest, F0 is fluorescence at rest, and
 |
See Maravall et al. (2000) for details. KD of OGB-1 was determined in the cuvette as 240 nm, in good accordance with values in the literature.
Fmin was determined by bath applying BAPTA-AM (200 μm; Invitrogen) in HL6 with zero [Ca2+]. Using transgenic animals expressing YC2.60, we determined effective concentrations and exposure times for in situ Ca2+ buffering with BAPTA-AM (data not shown). Standard HL6 with glutamate and 1.5 mm [Ca2+] was exchanged with HL6 with glutamate, zero [Ca2+], 1 mm EGTA, and 130 μm BAPTA-AM. Diffusion of AM esters was allowed for 30 min before washout with HL6. Earlier washout led to incomplete intracellular Ca2+ buffering. After washout, residual Ca2+ responses were recorded that were markedly slowed down (data not shown), representing an effect of BAPTA-AM at low concentration. After 30 min, no response to any stimulus was noticeable, and Fmin could be determined.
In vivo KD values represent the [Ca2+] and stimulus frequency at half-maximum fluorescence changes. Hill coefficients were derived from fitting experimental results to a model for the Hill coefficient:
 |
where r is the experimental response, f is the stimulus frequency, n is the Hill coefficient, and KD is the half-maximum stimulus frequency.
Protein expression and purification
For cuvette determination of Ca2+ binding curves, GECI cDNAs were subcloned into the pRSETB vector (Invitrogen), which is optimized for protein expression using the T7 expression system and carries a 6xhis-tag 5′ of the multiple cloning site. Protein expression was achieved using the Escherichia coli strain BL-21 (Invitrogen). Induction of protein expression took place at an OD600 of 0.6–0.8 with 0.5 mm IPTG (isopropyl-β-d-thiogalactoside) for 2–3 h at 37°C. His-tagged protein was bound to a Ni-NTA-Sepharose column by shaking for 2 h at 4°C. After washing the column with 10 ml of protein wash buffer (containing 10 mm imidazole), GECI protein was eluted by competitively displacing it with a high concentration of imidazole (150 mm).
Spectroscopic determination of KD values
For determination of KD values, freshly purified protein was used. For titration, a prewarmed (room temperature) titration kit (Calcium Calibration Buffer Kit with Magnesium #1, C3721; Invitrogen) was applied as follows. Two stock solutions were prepared: zero calcium: mix 1 ml of zero calcium buffer with 1 volume of protein solution [∼0.2–1 μm protein, directly into the cuvette (Hellma Precision Cells Quartz Suprasil, type 101-QS/10 mm path)]. High calcium: mix 5.4 ml of 39.8 μm free calcium buffer with 5.4 volumes of protein solution.
Subsequently, the zero calcium stock was put into the fluorescence spectrophotometer (Cary Eclipse fluorometer; Varian) to determine a baseline spectrum. Excitation wavelength for a CFP/YFP-FRET pair was 432 nm. Emission was determined in the range from 450 to 600 nm (all bandwidths 5 nm). Excitation wavelength for GC1.6 was 470 nm. Emission was determined in the range from 470 to 600 nm (all bandwidths, 5 nm). Excitation wavelength for OGB-1 was 475 nm (bandwidth, 10 nm). Emission was determined in the range from 490 to 600 nm (bandwidths, 5 nm).
Adjustment of free [Ca2+] was achieved by reciprocal dilution (replacing same amount of zero [Ca2+] buffer with the [Ca2+] stock) to the desired concentrations. Zero, 0.065, 0.100, 0.225, 0.350, 0.600, 0.850, 1.35, 1.73, 2.85, 4.87, 7.37, 14.9, 29.9, and 39.8 μm free [Ca2+] were used as reference points to determine the KD value for GECIs (for OGB-1 concentrations were 0, 0.017, 0.038, 0.065, 0.1, 0.15, 0.225, 0.351, 0.602, 1.35, and 39.8 μm free [Ca2+]). Calculation of the volumes that had to be replaced was according to the manufacturer's manual.
After measuring the spectra, the ΔR/R at distinct [Ca2+] was calculated as follows:
 |
with Rx = Ratio of YFP/CFP intensities at [Ca2+] = x and RRest = 0.065 μm. For OGB-1 and GC1.6, ΔF/F was determined analogously. To make cuvette measurements comparable with in vivo data acquired at the 2PLSM, two corrections were made: (1) we calculated ΔR/R using RRest at 0.065 μm. This deviation from real resting [Ca2+]i (∼31 nm) has negligible influence on resulting ΔR/R for GC1.6 and TN-XL. For YC3.60, D3cpv, and OGB-1, however, this influence had to be considered (because of the steep slope of the titration curve close to zero [Ca2+]i). Rx for a hypothetical Ca2+ level of 31 nm was interpolated for the titration of these GECIs, and this value was used as RRest. For OGB-1, a titration value was measured at 38 nm and used for RRest. (2) We mimicked bandpass filters by integrating the emission intensities from 465 to 505 nm for CFP, from 520 to 550 nm for YFP and 510–560 nm for OGB-1 and GC1.6. We measured a titration curve of a purified GECI protein at the two-photon microscope in aqueous solution as described for cuvette measurements and found that this bandpass correction leads to a maximum difference in ΔR/R under both imaging conditions of only 3.7%. Extraction of the KD values was done by sigmoidal fits to the dose–response curves (logarithmic [Ca2+] vs ΔR/R, normalized to 39.8 μm [Ca2+]).
Results
In a first series of experiments, neural activity was evoked at known rates (fAP), and the relationship to ΔF exhibited by OGB-1 and Δ[Ca2+]i was quantified. These recordings as well as all other live recording experiments were done in presynaptic boutons of transgenic Drosophila larvae. Thus, we had to develop a protocol for the injection of synthetic calcium indicators into targeted neuronal compartments in Drosophila.
Two-photon guided dye injection into genetically labeled presynaptic boutons
We used sharp quartz electrodes to inject the green synthetic Ca2+ indicators OGB-1 and Magnesium Green into presynaptic boutons labeled in red by transgenic expression of mDsRed (see Materials and Methods). Injected boutons and axons were of 2–5 and 1 μm diameter, respectively (Fig. 1A–C). Fluorescence was excited at 980 nm and visualized by dual-channel 2PLSM (Denk et al., 1990; Wachowiak et al., 2004). This allowed guidance of the dye-filled electrode tip toward boutons and axons and dye loading by current injection (Fig. 1B,C). A similar strategy has previously been described for patch electrodes and large cell bodies (Margrie et al., 2003; Komai et al., 2006). Twenty minutes after removal of the electrode dye concentration reached a plateau (data not shown). At that point, we began our experiments. The axon was stimulated with trains of action potentials at different frequencies (0–160 Hz). Figure 1D shows intensity changes of OGB-1 (raw data) in several boutons stimulated at 160 Hz (stimulation from t = 2–4 s). Baseline fluorescence was reached again ∼2 s after the offset of the stimulus. ΔF/F of OGB-1 at all stimulation frequencies is summarized in Figure 1, E and F. The OGB-1 response saturates with increasing fAP (Fig. 1F, black triangles), whereas responses recorded with Magnesium Green exhibited an almost linear increase in ΔF/F over the same stimulus range (Fig. 1G). The fast kinetics of the Magnesium Green responses (data not shown) and linear increase in amplitude can be explained by low Ca2+ affinity (KD ∼ 6 μm; Invitrogen), whereas the saturation curve of OGB-1 (Fig. 1E,F) reflects high Ca2+ affinity (KD = 170 nm; Invitrogen) (Table 1). Notably, the approximately linear increase of the Magnesium Green response suggests that the calcium concentration reached in presynaptic boutons is a linear function of fAP up to 160 Hz.
Figure 1.
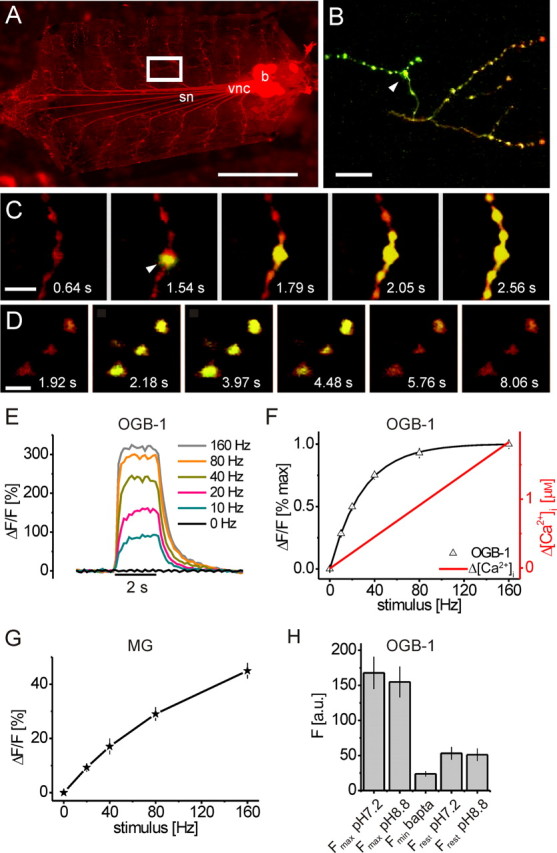
In vivo cross-calibration of OGB-1 fluorescence changes, neural activity, and Δ[Ca2+]i. A, A transgenic Drosophila larva expressing monomeric DsRed (elavC155-gal4→UAS-mDsRed) in all neurons has been prepared for recording. For imaging experiments, the brain was removed and severed nerve ends were placed into a suction electrode for electrical stimulation. OGB-1 was injected into presynaptic boutons using sharp electrodes (white box in A, close-up in B–D) and 2PLSM imaging [image: incident fluorescence light microscopy; brain (b), vnc, segmental nerves (sn)]. B–D, 2P excitation and graphic overlay of simultaneously recorded mDsRed (red) and OGB-1 (green) fluorescence. B, One minute after injection of the synthetic calcium indicator OGB-1 (green), intense green fluorescence is visible near the injection site (white arrowhead). C, Zoom-in during the 2PLSM-controlled injection of OGB-1 into a single bouton (white arrowhead): rapid filling and initial diffusion of OGB-1 along the axonal branch is visible (electrode barely visible). D, Fluorescence changes of OGB-1 (raw data) in response to a train of APs. Time points of frames are indicated; stimulation: 80 Hz from t = 2 to t = 4 s. E, ΔF/F of OGB-1 in boutons evoked by trains of APs at different frequencies plotted as a function of time (fAP = 0–160 Hz; n = 17, 30, 32, 32, 31, and 30 boutons). F, Cross-calibration of OGB-1 fluorescence changes (black triangles, extracted from the data in E; black line represents a hyperbolic fit) to neural activity (fAP) and Δ[Ca2+]i (red trace) in situ. ΔF/F and Δ[Ca2+]i are plotted as a function of fAP. OGB-1 exhibited fluorescence saturation at 160 Hz stimulation, and Δ[Ca2+]i increased linearly up to 160 Hz. Determination of Fmax and Fmin (see H) allowed conversion of the fluorescence changes into an estimate of changes in [Ca2+]i. G, Calcium influx increases approximately linearly with the frequency of action potentials in presynaptic boutons. Steady-state amplitude and SNR of fractional fluorescence changes are plotted as a function of stimulus frequency. Magnesium Green (MG) responses increased almost linearly with stimulus intensity (15 < n < 24). H, Determination of Fmax and Fmin of OGB-1 in situ. Fmax was reached at 160 Hz stimulation; there was no further increase when raising the extracellular pH from 7.2 to 8.8 (p ∼ 0.5; n = 11). Fmin was reached in HL6 with zero [Ca2+] by buffering of [Ca2+]i by excess BAPTA-AM, which reduced the resting fluorescence Frest to Fmin (FminBAPTA vs Frest pH7.2, p < 0.01; n = 11). Frest was unaffected by the extracellular pH (Frest pH7.2 vs Frest pH8.8, p ∼ 0.5; n = 11). Imaging frame rate was 8 Hz throughout. Scale bars: A, 1 mm; B, 20 μm; C, 10 μm; D, 5 μm. Calibration: E, 2 s.
Estimation of intracellular calcium
We used in situ fluorescence saturation of OGB-1 to estimate [Ca2+]i (Maravall et al., 2000). Assuming a linear stimulus (fAP)–[Ca2+]i relationship, as suggested by the response of the low-affinity Ca2+ indicator Magnesium Green (see above), [Ca2+]i at rest and Δ[Ca2+]i at steady state during stimulus trains can be calculated from the fluorescence response of OGB-1. This translation of OGB-1 fluorescence changes into [Ca2+]i after Equations 1 and 2 requires knowledge of the following parameters in situ: (1) KD, (2) maximum ΔF/F evoked by neural activity, (3) Fmax, and (4) Fmin. (1) The KD for OGB-1 was determined as 240 nm at 22°C in the cuvette, which is in good accordance with our in vivo OGB-1 measurements (see Fig. 4F, Table 1). (2) For (ΔF/F)max in situ, complete saturation of the OGB-1 fluorescence response was estimated from fitting a hyperbolic function y = y0 × [Ca]/([Ca] + KD) to the data points (Fig. 1F), where y0 is the asymptotic fluorescence value. The measured response at 160 Hz stimulation was ∼99% of this value (318% ΔF/F) (Figs. 1F, 4F). (3) For Fmax, we confirmed that OGB-1 fluorescence in situ cannot be further increased by stimulating at 160 Hz and extracellular pH 8.8 (Fig. 1H). Increasing the extracellular pH increases the intracellular Ca2+ accumulation at steady state (Lnenicka et al., 2006) because Ca2+ extrusion from presynaptic boutons in Drosophila NMJs is mainly achieved by a Ca2+/H+ exchanger. In supplemental Figure 1A (available at www.jneurosci.org as supplemental material), we demonstrate this effect using GC1.6-expressing boutons. Stimulation at 40 Hz reversibly increased ΔF/F to ∼80% ΔF/F at pH 8.8, which is a threefold increase compared with pH 7.2. The baseline fluorescence showed no significant increase (data not shown), suggesting that increased ΔF/F of GC1.6 is not caused by a shift in the intracellular pH (Ohkura et al., 2005). For OGB-1, however, ΔF/F at 160 Hz could not be further increased at pH 8.8 (Fig. 1H), indicating that OGB-1 was saturated with Ca2+ at this fAP in situ and extracellular pH 7.2, as suggested by the saturation curve (Fig. 1F). Thus, the fluorescence at 160 Hz represents the true Fmax and could be used to calculate (ΔF/F)max. (4) Fmin was determined by creating an intracellular environment of effectively zero [Ca2+]i using extracellular solution of zero [Ca2+]i together with the cell-permeable Ca2+ chelator BAPTA-AM (130 μm) (Fig. 1H) (see Materials and Methods). Using Equation 1, we determined [Ca2+]i Rest = 31 nm. Equation 2 gave Δ[Ca2+]i 10Hz = 92 nm and Δ[Ca2+]i 20Hz = 229 nm. A linear fit (Fig. 1F, red line) resulted in estimates of Δ[Ca2+]i at steady state for each of the applied stimuli: 0.11 μm/10 Hz, 0.22 μm/20 Hz, 0.45 μm/40 Hz, 0.91 μm/80 Hz, and 1.82 μm/160 Hz. The obtained high Δ[Ca2+]i at high action potential frequencies is in accordance with the slightly sublinear increase of the Magnesium Green response (Fig. 1G). At these concentrations, a slightly sublinear increase of the Magnesium Green response is predicted by the law of mass action and can be explained by beginning sublinear binding of Ca2+ to the indicator as [Ca2+]i approaches 0.5 × KD (Yasuda et al., 2004).
Figure 4.

In vivo versus in vitro: quantitative comparison of fractional fluorescence changes of GECIs and OGB-1 at steady state. GECI and OGB-1 fluorescence changes were analyzed at the end of stimulus trains in presynaptic boutons of transgenic larvae (black traces) and compared with the purified protein and OGB-1 in solution in the cuvette. A, YC3.60; B, TN-L15; C, D3cpv; D, TN-XL; E, GC1.6; F, OGB-1. Fractional fluorescence changes (ΔR/R and ΔF/F) in vivo (black traces) are compared with in vitro ΔR/R and ΔF/F calculated from spectrophotometer data either directly (light gray traces) or after applying corrections (dark gray traces; see Materials and Methods). Correction was done for the resting calcium concentration and the width of the bandpass filters in the detection pathway of the 2PLSM. Both corrections significantly reduce Fmax, which is most obvious for the synthetic indicator OGB-1 (F). Applying these corrections, the fluorescence change of OGB-1 in vivo matches perfectly the one observed in vitro. All GECIs show significant deviations of in vivo from in vitro (A–E), the origin of which remains to be investigated. However, the applied corrections cancel out effects of the applied imaging conditions, and the remaining differences between GECIs probably represent interactions with the chemical environment.
GECIs in vivo: cross-calibration of steady-state fluorescence changes, neural activity, and Δ[Ca2+]i
Experimental evidence (Pologruto et al., 2004; Reiff et al., 2005) suggests that, in general, available GECIs suffer from low-sensitivity, low-SNR, nonlinear concentration dependence with unusual supralinear and sublinear regimes and slow binding kinetics. These features influence whether and how a given GECI reports transient and fast Δ[Ca2+]i and whether Δ[Ca2+]i falls into the dynamic range of the indicator.
In the following experiments, nerves were stimulated at 0–160 Hz as described for OGB-1 (Fig. 1). Fluorescence changes of eight different GECIs were recorded in presynaptic boutons of transgenic animals and plotted as a function of time (Fig. 2, left). The stimulus-evoked fractional fluorescence changes at the end of 2 s stimulus trains represent steady-state conditions. Other than during dynamic calcium concentration changes, at steady state the amplitude of an indicator's fluorescence change is independent of indicator concentration (see Discussion). Thus, amplitudes and SNR of fractional fluorescence changes were extracted at steady state and related to neural activity and Δ[Ca2+]i (Fig. 2, right) as determined in the OGB-1 experiments (Fig. 1F). Time constants for the rise and decay were derived from single-exponential functions fitted to the respective phase of the 40 Hz response unless a given indicator showed saturation at this frequency. In these cases (OGB-1, D3cpv, and YC2.60), the 20 Hz responses were analyzed (see Discussion). Data represent mean ± SEM. The eight GECIs performed as follows [summaries are given in Table 1 and supplemental Table 1 (available at www.jneurosci.org as supplemental material)].
YC3.3 (Fig. 2A): The dependence of SNR on [Ca2+]i is shown in Figure 3B. The lowest fAP that still yielded fluorescence changes with an SNR > 2 was 20 Hz. Fitting these data allowed us to interpolate the minimum Δ[Ca2+]i or fAP at which a SNR of 2 would be reached. For YC3.3, this was the case at Δ[Ca2+]i ∼140 nm/13 Hz (Fig. 3C). We observed a linear increase in the response up to 40 Hz and beginning saturation above. (ΔR/R)max was 66.6 ± 0.9%, and the half-maximum ΔR/R, corresponding to the KD, was reached at ∼38 Hz or 0.47 μm [Ca2+]i in vivo (Fig. 3A). Rise and decay displayed time constants of 1.41 and 1.05 s, respectively (Fig. 3D, Table 1) (48 < n < 63).
YC3.60 (Fig. 2B) showed a fivefold higher ΔR/R at 10 Hz stimulation compared with YC3.3 and the highest SNR of all GECIs (SNR > 3) at this frequency. SNR of 2 was reached at Δ[Ca2+]i ∼80 nm/8 Hz (Fig. 3B,C). ΔR/R increased linearly up to 40 Hz, and fAP > 40 Hz led to a sublinear increase in the signal amplitude with (ΔR/R)max = 135.8 ± 4.4%. The half-maximum response corresponds to ∼30 Hz stimulation or a KD of 0.36 μm in vivo (Fig. 3A). Rise and decay displayed time constants of 0.82 and 0.73 s, respectively (Fig. 3D, Table 1) (33 < n < 71).
YC2.60 (Fig. 2C): A SNR of 2 was reached at Δ[Ca2+]i ∼100 nm/9 Hz (Fig. 3B,C). ΔR/R increased linearly up to 40 Hz. At 80 and 160 Hz, amplitudes increased sublinearly with (ΔR/R)max = 193.8 ± 9.7%, and the half-maximum response corresponds to ∼32 Hz or a KD of 0.40 μm in vivo (Fig. 3A). Responses of comparable size had previously not been reported for any ratiometric GECI in vivo. The response amplitudes may still be slightly underestimated, because a plateau was not fully reached after 2 s stimulation. However, SNR did not increase linearly (see Discussion), and the slow kinetics is disadvantageous for most experiments. A fit to the decay of the 20 Hz response showed a time constant of 5.24 s for the decay (single exponentials could not be fit to the rise) (Fig. 3D, Table 1) (29 < n < 30).
D3cpv (Fig. 2D) is a reengineered variant of YC, whose sites of interaction with wild-type calmodulin were eliminated (see Discussion). SNR of 2 was reached at Δ[Ca2+]i ∼100 nm/9 Hz (Fig. 3B,C). ΔR/R increased sublinearly when frequencies exceeded 20 Hz and reached (ΔR/R)max = 89.7 ± 3.9%. The half-maximum response is reached at ∼41 Hz, reflecting a KD of 0.49 μm in vivo (Fig. 3A). Rise and decay of the 20 Hz responses displayed time constants of 0.68 and 1.96 s, respectively (Fig. 3D, Table 1) (48 < n < 56).
TN-L15 (Fig. 2E) uses the same chromophores as YC3.3 but troponin C as calcium binding moiety. SNR of 2 was reached at Δ[Ca2+]i ∼200 nm/18 Hz (Fig. 3B,C). ΔR/R increased about linearly up to 40 Hz with (ΔR/R)max = 59.5 ± 2.3%. The half-maximum response is reached at ∼30 Hz or a KD of 0.36 μm [Ca2+] in vivo (Fig. 3A). Rise and decay displayed time constants 0.81 and 1.49 s, respectively (Fig. 3D, Table 1) (27 < n < 30).
TN-XL (Fig. 2F) exhibited pronounced supralinear behavior at lower activity rates. SNR of 2 was reached at Δ[Ca2+]i ∼350 nm/31 Hz (Fig. 3B,C). Higher activity rates rapidly evoked a sublinear increase. (ΔR/R)max was 105.9 ± 2.7%, and the half-maximum response corresponds to ∼65 Hz or a KD of 0.77 μm in vivo (Fig. 3A). TN-XL responses showed time constants of 0.59 s for the rise and the fastest decay time constant of all GECIs with 0.20 s (Fig. 3D, Table 1) (48 < n < 87).
GC1.6 (Fig. 2G): This single-chromophore GECI in our study exhibited ΔF/F with SNR of 2 at Δ[Ca2+]i ∼220 nm/20 Hz (Fig. 3B,C). ΔF/F increased supralinearly with stimulus intensity up to 40 Hz and sublinearly above. (ΔF/F)max was 161.6 ± 13.1%, and the highest maximum SNR of all GECIs in this study was reached at 80 Hz (15.9 ± 1.5) (Fig. 3B). The half-maximum response was reached at ∼54 Hz or a KD of 0.64 μm in vivo (Fig. 3A). Time constants of 40 Hz responses were 1.38 s for the rise and 0.45 s for the decay (Fig. 3D, Table 1) (42 < n < 52).
Finally, we generated UAS-GCaMP2 (Tallini et al., 2006; Díez-García et al., 2007) flies that carried the full cDNA (including his-tag) under control of the UAS. Live imaging with these GCaMP2-expressing flies was hardly possible because of the low baseline fluorescence of the indicator (Mao et al., 2008). Only 3 of ∼50 fly strains gave rise to high enough expression that it could be detected in vivo when animals were homozygous for both Gal4 and UAS-GCaMP2. The fractional fluorescence changes recorded with GC2 mostly mimicked those exhibited by GC1.6 (Fig. 2G). However, an SNR of 2 was only reached at stimulus frequencies >20 Hz (Δ[Ca2+]i ∼300 nm/27 Hz) (Fig. 3B,C). ΔF/F increased supralinearly with stimulus intensity up to 20 Hz, then linearly up to 80 Hz. (ΔF/F)max was 155.7 ± 4.2% with an SNR of 15.6 ± 1.5 (Fig. 3A,B). This corresponds to a KD of 0.75 μm in vivo (∼63 Hz), which is close to the KD of GC1.6. Time constants of 40 Hz responses were 0.63 s for the rise and 0.38 s for the decay (Fig. 3D, Table 1) (30 < n < 58).
In vitro versus in vivo response characteristics of GECIs at steady state
We assessed the influence of the cellular environment on the signaling properties of the GECIs and OGB-1. We measured fluorescence changes at defined Δ[Ca2+] exhibited by purified GECI protein in the cuvette of a spectrophotometer and compared it to our measurements in vivo (Fig. 4). Two corrections were applied to the cuvette data to allow this comparison: first, the bandpass filters in the detection pathway of the 2PLSM were mimicked; and second, baseline [Ca2+] in vivo was taken into consideration (see Materials and Methods). Because fluorescence changes of single and dual wavelength indicators are both expressed as changes relative to baseline fluorescence, [Ca2+] at baseline limits the maximum fluorescence change and the signaling capacity of an indicator, in particular for low-KD indicators (see Discussion). Thus, both corrections reduce the magnitude of the fluorescence responses (Fig. 4). This effect can be seen for all GECIs (Fig. 4A–E) and OGB-1 (Fig. 4F) by comparing uncorrected (light gray traces) with corrected (dark gray traces) spectrophotometer data. Most importantly, deviations of the corrected spectrophotometer data from the data acquired in presynaptic boutons (Fig. 4, black traces) can likely be attributed to interactions of the indicator with the intracellular environment (see Discussion).
At low Δ[Ca2+], YC3.60 showed rather similar response properties in vitro and in vivo (compare Figs. 2B, 4A). YC3.60 was identified as most promising GECI in this study for the detection of low rates of activity or small Δ[Ca2+]i. However, above 0.25 μm Δ[Ca2+] (fAP > 20 Hz), the large fluorescence changes in vitro were not retained in vivo (Fig. 4A). However, Figure 4A indicates a relatively large dynamic range (notice the linear scaling of the x-axis in Fig. 4) over which neural activity is reported about linearly (up to Δ[Ca2+] of 0.4 μm/40 Hz). Such linear regimes with high signaling capacity were less pronounced in D3cpv, TN-L15, and TN-XL (Fig. 4B–D): D3cpv saturated more quickly, whereas the troponin-C-based GECIs exhibited more complex binding curves in vivo with supralinear and sublinear regimes. Interestingly, these three indicators showed larger ΔR/R in vivo (black curves) than in vitro (dark gray curves) when Δ[Ca2+] exceeded 0.25 μm (Fig. 4B–D). Also, GC1.6 responses exhibited complex Ca2+-binding dynamics (Fig. 4E), and at Δ[Ca2+] > 1 μm, the observed GC1.6 responses were smaller in vivo. However, in vivo responses higher than in vitro were observed within a certain range (0.2 < Δ[Ca2+] < 1 μm). The mechanistic interpretations of these findings remain to be given. The comparison of OGB-1 between the cuvette and in vivo (Fig. 4F) convincingly demonstrates that the applied methods are suitable to make significant statements on calcium indicator properties in vivo and in vitro: the applied corrections reduced the original in vitro ΔF/F of OGB-1 (light gray trace) as expected, and the obtained curve (dark gray trace) matches the in vivo data (black trace) over the full range of Δ[Ca2+]. The maximum fluorescence change is thus identical under both conditions. However, because the calibration was done using OGB-1 in vitro, effects of the cellular environment on the KD of OGB-1 may be hidden (see Discussion).
From the data described so far, the KD value and the Hill coefficient of the different GECIs were calculated (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material; Table 1). For all GECIs that we measured under both conditions, the KD was significantly decreased in vivo (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Moreover, the differences in the KD values of the individual GECIs were less pronounced in vivo. Thus, as a rule of thumb, the in vivo KD of GECIs appears to be shifted toward higher affinity compared with our in vitro calibration [with YC2.60 and GC2 being the only exception, because they were not calibrated in cuvettes in our lab, and the in vitro KD values were taken from literature (Nagai et al., 2004; Tallini et al., 2006)]. For the Hill coefficient (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material), the picture is slightly more complex. However, there is a systematic change too. In vivo, the Hill coefficient of GECIs based on wild-type calmodulin (GC1.6, YC2.60, and YC3.60; no data available on YC3.3) is decreased. This finding is in line with [Ca2+]-dependent interactions of GECI proteins with native wild-type proteins: at higher [Ca2+] concentrations, such interactions are favored, interfering with GECI function. In contrast, D3cpv and the troponin-C-based indicators TN-L15 and TN-XL showed higher Hill coefficients in vivo, indicative of increased cooperativity of calcium binding to the indicator.
Fluorescence changes of GECIs and OGB-1 in response to short Ca2+ transients in vivo
The combination of concentration, sensitivity, and speed of the fluorescence transition kinetics determines how well transient calcium fluctuations can be resolved by a fluorescent indicator. The favorable combination of backward binding rate (Kb) and forward binding rate (Kf) allows some synthetic Ca2+ indicators to report neuronal activity with single action potential resolution (Schiller et al., 1995; Helmchen et al., 1996, 1997; Maravall et al., 2000). In contrast, cytosolic GECIs were reported to lack sufficient SNR and Ca2+ binding kinetics to report individual APs (Pologruto et al., 2004; Reiff et al., 2005) (but see Díez-García et al., 2007). We assessed the capacity of cytosolic GECIs and OGB-1 to signal a transient increase in [Ca2+]i in presynaptic boutons of transgenic Drosophila by 2PLSM fluorescence recordings at high temporal resolution.
In a first set of experiments, we chose TN-XL for its fast decay time constant (Fig. 3D) and evoked APs at 10, 20, and 40 Hz over a period of 4 s. The power spectra of the fractional fluorescence changes did in no case reveal significant peaks corresponding to the action potential frequency (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Similar measurements in OGB-1 filled boutons revealed clear stimulus-related peaks for 10 and 20 Hz experiments (supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material), whereas individual APs at 40 Hz and higher activity rates were masked by the rather slow time constant for the fluorescence decay of OGB-1 (supplemental Fig. 2D, available at www.jneurosci.org as supplemental material).
Next, we evoked brief volleys of 2, 5, and 10 APs (Fig. 5) (APs separated by 10 ms, volleys by 500 ms). OGB-1 reported even single action potentials with 43.6 ± 2.5% ΔF/F and SNR > 15 (Fig. 6A). Volleys of 2, 5, and 10 APs were reported with increasing ΔF/F and SNR (Fig. 5B). SNRs were calculated for each individual burst and given as the average of all bursts from the four traces. In contrast to OGB-1, no GECI responded to transient Δ[Ca2+]i associated with single spikes. Spike doublets were reported with SNR > 3 only by YC3.60 and YC2.60. D3cpv provided SNR of 2.2. Only YC3.3 and GC2 failed to report volleys of five APs. Volleys of 10 APs were reported by all GECIs, although with widely differing amplitudes, SNRs, and rise times (Figs. 5C–J, 6B; supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 5.
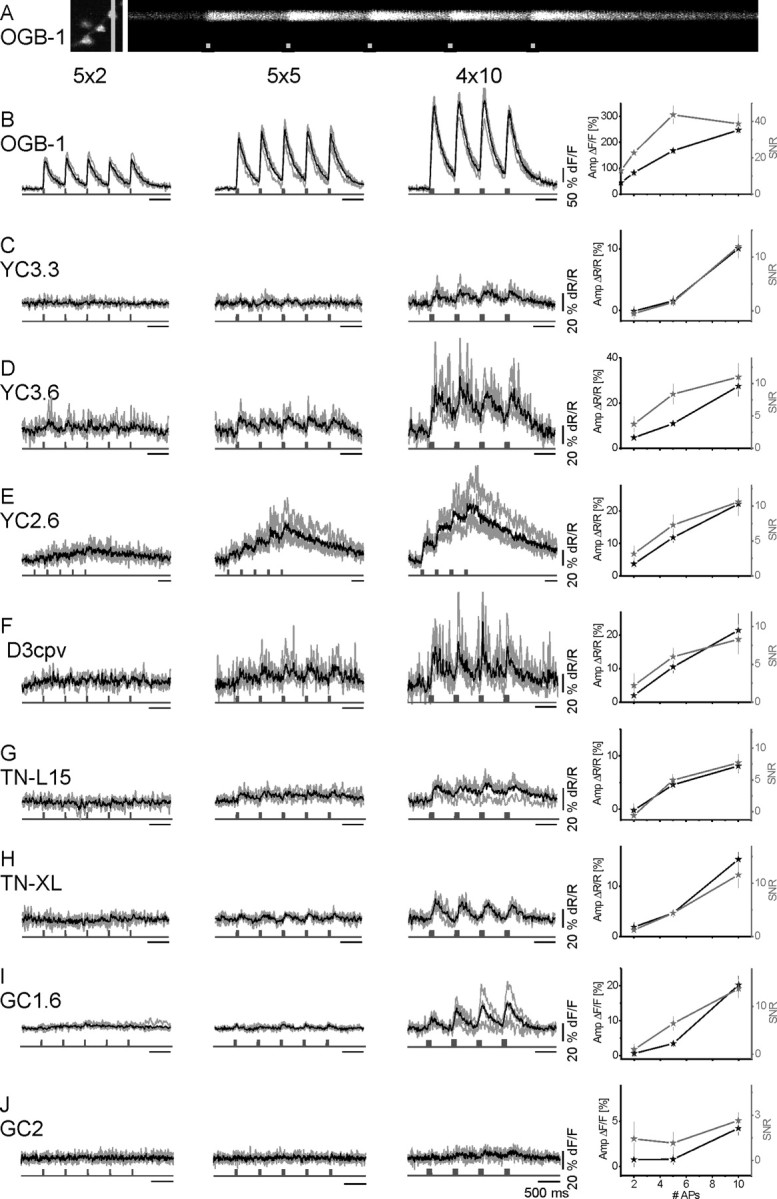
In vivo comparison of fluorescence transients of GECIs and OGB-1 in response to short AP volleys. GECIs and OGB-1 fluorescence changes were recorded in response to biologically more realistic stimuli. Nerves were stimulated with short AP sequences. A, 2PLSM recording in line-scan mode at an acquisition rate of 500 Hz. Left, Close-up of a small branch of a larval NMJ showing several boutons labeled with OGB-1. Position of the line scan is indicated by the light gray line. Right, Raw line-scan data show transient fluorescence increases evoked by calcium influx during AP volleys (light gray squares, 5 APs at 100 Hz/volley; x-axis represents time). Calibration, 200 ms. B–J, OGB-1 (B) and GECI (C–J) recording traces. AP volleys were spaced by 500 ms. Within a volley, APs were elicited at 100 Hz in packs of 2, 5, and 10 APs per volley (first, second, and third columns, respectively). Shown are four individual recording traces from different boutons (gray) and their mean (black). The maximum amplitude of the mean fluorescence response is plotted against the number of APs per volley (mean ± SEM). In addition, the average SNR of single responses to single AP volleys is plotted. Only OGB-1 reliably reported single APs (SNR of >15; see B and supplemental Fig. 3A, supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 6.

A, OGB-1 fluorescence transients in response to single action potentials in vivo. OGB-1 reliably reports individual APs in individual recorded traces with SNR > 15. A single AP was elicited every 500 ms (experimental details as in Fig. 5). Traces represent four individual measurements from different boutons (gray) and their mean (black). B, Time constants for the rise of fluorescence changes in response to fast and transient Ca2+ fluctuations. Time constants were determined for the fluorescence signals plotted in Figure 5 (volleys of 10 APs at 100 Hz, first volley analyzed). In general, GECIs with a low KD display fast rise times with the exception of YC2.60. The latter may be because of very high protein concentration. Time constants were determined for data from t = 0 to the peak of the response to the first volley fit by a single-exponential function.
In a last series of experiments, we expressed GECIs in tangential neurons of the optic lobe of adult flies and recorded fluorescence changes in response to bath-applied high-KCl solution (Fig. 7). All GECIs exhibited fluorescence changes under these conditions. However, the fluorescence responses elicited by the rather strong stimulation with 100 mm KCl were relatively small. This might be explained by only small Δ[Ca2+]i in the analyzed neurons and/or high rates of neural activity at rest and resulting high levels of baseline [Ca2+]i before KCl application. Nevertheless, the result suggests that our experimental findings in larval motoneurons can be generalized to neurons in the CNS of adult flies (Jayaraman and Laurent, 2007). However, these experiments do not allow a more quantitative analysis of GECI signaling properties in terms of amplitudes and kinetics.
Figure 7.

GECI responses in the adult CNS in vivo. All GECIs reproducibly exhibited fluorescence changes in the CNS of adult Drosophila flies. GECIs were expressed in a subset of large tangential neurons (and few unknown interneurons) of the optic lobe (genotype, DB331-Gal4→ UAS-GECI). Details of the preparation were given by Joesch et al. (2008). In short, a young female fly was glued to a holder and its head was bent down such that its back side faced upward. The back side of the head was opened up to expose the caudal optic lobes with GECI-expressing neurons. The preparation was covered with Ringer's solution and imaged for 33 s using wide-field epifluorescence microscopy. During the experiment, the external potassium concentration was raised to 100 mm. Fluorescence responses were recorded at somata and axons of the large tangential cells. Each data point represents the maximum fluorescence change recorded in one animal. Before calculation of the fractional fluorescence changes, motion artifacts were reduced by alignment of the sequential images. DB331-Gal4 flies were a generous gift from Reinhard Stocker, University of Fribourg, Fribourg, Switzerland.
Discussion
The advent of optical recording with genetic probes triggered a renaissance for systemic neuroscience in invertebrates and in particular in Drosophila, in which genetic probes can be combined with circuit-breaking genetic tools (Marella et al., 2006; Holmes et al., 2007). Hence, we used Drosophila neurons as a testing ground to compare eight different GECIs in vivo that can similarly be applied to neurons in vertebrates.
Meaningful optical recordings rely on the faithful interpretation of fluorescence changes with respect to the underlying neural activity and [Ca2+]i. This relationship has been reported to be notoriously difficult to interpret when using GECIs (Hasan et al., 2004; Pologruto et al., 2004; Reiff et al., 2005; Jayaraman and Laurent, 2007; Tay et al., 2007). At best, fluctuations in [Ca2+]i represent a low-pass filtered version of fluctuations in the membrane potential. The Δ[Ca2+]i is then reported by GECI signals with rather small amplitude and low SNR that, depending on the GECI concentration and KD value, become further distorted. Thus, the spatial and temporal relationship between neural activity and GECI signals is far from trivial, which will be discussed here.
Steady state
In calcium imaging studies, fluorescence changes are commonly expressed as changes relative to baseline (Ft − F0)/F0 (F0 is fluorescence at baseline, and Ft is fluorescence at time t). Thus, [Ca2+]i at baseline reduces (ΔF/F)max and the signaling capacity of the indicator by increasing F0. The signaling capacity is highest within the dynamic range of an indicator where Δ[Ca2+]i is reported by a linear increase in ΔF of maximum magnitude. Thus, the dynamic range of the indicator should ideally match the regime of expected Δ[Ca2+]i that are superimposed on a given [Ca2+]i at baseline. Furthermore, the rate constants for binding and unbinding of calcium to the indicator must be fast enough to allow detection of short Δ[Ca2+]i transients [Borst and Abarbanel (2007), their Equation 10], and indicators should exhibit bright fluorescence and large ΔF to provide a good SNR. Following these guidelines, we can rank the eight GECIs for their capability to report Δ[Ca2+]i at steady state and thus to report different rates of sustained neural activity (Yasuda et al., 2004).
In presynaptic boutons of Drosophila, Δ[Ca2+]i of 10–11 nm/AP were superimposed on [Ca2+]i at rest of 31 nm (Fig. 1). Small Δ[Ca2+]i were reported best by YC3.60 (Fig. 3C), and higher Δ[Ca2+]i fell within the broad dynamic range of this indicator (Fig. 4A, black trace). Also D3cpv reported small Δ[Ca2+]i quite well but showed a narrow dynamic range. YC2.60 provided fluorescence changes of magnitude similar to YC3.60 (Figs. 2, 4), but lower SNR (for discussion of SNR, see below). YC3.3, TN-L15, TN-XL, GC1.6, and GC2 did not allow detection of small Δ[Ca2+]i from baseline. Nevertheless, the large maximum fluorescence change and high SNR in GCaMPs and TN-XL and their KD makes these GECIs valuable indicators to address large Δ[Ca2+]i or medium Δ[Ca2+]i superimposed on high [Ca2+]i at rest. However, their capricious Ca2+-binding dynamics (Fig. 4), with sublinear and supralinear regimes (Pologruto et al., 2004; Reiff et al., 2005), and the increase in ΔF/F with repetition of AP volleys (Fig. 5I) have to be taken into account.
SNR
The calculated SNR values do only partially represent an endogenous property of the indicator (see below). The SNR is also dependent on all parameters of the imaging setup (e.g., laser intensity, pixel dwell time, photon noise, and collection efficiency), on the imaged volume, and on the indicator concentration (SNR depends on the square root of the number of detected photons). The only of these variables that we could neither hold constant nor measure was the concentration of the GECIs in boutons in vivo, a problem that will be faced by most experimenters, too. Instead we aimed for a high expression level of each GECI in vivo using constant copy numbers for Gal4 and the UAS-GECI (see Materials and Methods), which probably comes closest to the strategy typically used in in vivo imaging experiments that involve GECIs. Thus, the given SNR values represent useful landmarks that reflect the mixed properties of the GECI (see below) and a given fly strain, i.e., expression level.
It has to be noted that the indicator concentration influences in addition how fast steady state is achieved inside a bouton (see below). But it does not influence the level of this steady state (including [Ca2+]i Rest), which is an exclusive function of the driving forces for calcium influx and efflux (Borst and Abarbanel, 2007).
Signal kinetics
So far, we have mostly considered the nondynamic features of ΔF when calcium influx and efflux are in equilibrium and the concentration of calcium-bound-indicator is constant. However, GECIs should ideally report individual APs with sufficient SNR in a quantitative manner (Wallace et al., 2008; Mao et al., 2008). This property is limited by the concentration of the indicator and the forward and backward binding rates that define the KD as KD = Kb/Kf (Schiller et al., 1995; Tank et al., 1995; Helmchen et al., 1996, 1997; Maravall et al., 2000; Borst and Abarbanel, 2007). A high indicator concentration slows down the time constant of the fluorescence signal and thus reduces the magnitude of the fluorescence response during a single AP. Also, in experiments using long stimulus trains (Figs. 2, 3D), the indicator concentration can affect the time constants of the rise and decay. However, exemplary calculations for the two GECIs TN-L15 and TN-XL show that their kinetics are dominated by their binding rates and are largely independent of the indictor concentration (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Whatever the reason for the given kinetics might be, i.e., binding rates or indicator concentration, time constants close to those presented here can be expected if GECIs are expressed at a level that enables in vivo imaging.
In addition, the rise and decay time in these experiments is affected by indicator saturation. If the steady state [Ca2+] is far below the KD of an indicator, the system is linear and the time constants for the rise and the decay are equal. If the steady-state calcium concentration approaches the KD of an indicator or even exceeds it, the rise will appear faster and the decay slower compared with the linear case (Borst and Abarbanel, 2007). We thus analyzed time constants from steady-state responses in the linear regime of the indicators (20 and 40 Hz stimuli).
Transient neural activity
OGB-1 allowed the detection of a single AP with an average SNR > 15 (Figs. 5B, 6A). No GECI allowed the detection of single spikes. Volleys of two APs were tentatively reported by YC3.60, YC2.60, and D3cpv (Fig. 5; supplemental Table 1, available at www.jneurosci.org as supplemental material). Significant further improvements leading to GECIs with faster reaction kinetics and very high KD are required, in particular when tagged GECIs and the assessment of short-lived, large Δ[Ca2+]i in spatially restricted microdomains are considered (Schneggenburger and Neher, 2005). The recently achieved rapid fluorescence decay of TN-XL in vivo (τTN-XL = 200 ms; τOGB-1 = 380 ms) (Fig. 3D) and differences in Kf of two to three orders of magnitude (Miyawaki et al., 1997; Naraghi, 1997) suggest that calcium binding can be further modified in future GECIs. The low sensitivity and short decay time of TN-XL and the failure of the high-sensitivity GECIs YC3.60, YC2.60, and D3cpv to report Δ[Ca2+]i associated with single spikes suggest that the on rate of calcium binding to GECIs is the current bottleneck for the detection of fast calcium transients.
A further difficulty in the detection of Δ[Ca2+]i associated with a single AP arises from the large volume (∼2–70 μm3) of type 1b boutons (Hoang and Chiba, 2001) that causes small-volume-averaged Δ[Ca2+]i compared with boutons of ∼0.04 μm3 in the mouse brain (Schikorski and Stevens, 1997). Such differences might explain why GC2 came close to the detection of single APs when large populations of fibers and boutons were imaged simultaneously in the mouse cerebellum (Díez-García et al., 2007).
Ratiometric and single-chromophore GECIs
Ratiometric analysis reduces motion artifacts and eliminates correlated noise in the two monitored signals, whereas uncorrelated noise can be increased, in particular under photon-limited conditions (then, a single-chromophore GECI might provide lower noise). In CFP/YFP pairs, the FRET efficiency can reach up to 98% (Shimozono et al., 2006). Then, the denominator in the emission ratio r = YFP/CFP approaches zero, which gives rise to noise and low SNR as in YC2.60 (Figs. 2, 3). Single-chromophore GCaMPs showed the highest SNR of all GECIs at high Δ[Ca2+]i (SNR > 15).
In vivo versus in vitro
Shortcomings of GECIs in vivo have been attributed to interactions of the GECIs calmodulin and the calmodulin-binding peptide (Hasan et al., 2004) with cellular calmodulin [10–100 μm (Xia and Storm, 2005)] and calmodulin-regulated proteins (Mori et al., 2004). Complementary modification of the relevant interaction sites within the binding interface of calmodulin and M13 (Palmer et al., 2006), or replacement of calmodulin-M13 by troponin C (Heim and Griesbeck, 2004; Mank et al., 2006), should reduce such interactions. Our comparison of in vivo and in vitro responses supports the relevance of such interactions. GECIs that use wild-type calmodulin-M13 (YC3.60 and GC1.6) showed reduced ΔFmax and a reduced Hill coefficient in vivo, whereas ΔFmax of troponin-C-based GECIs and D3cpv was retained, and the Hill coefficients were increased in vivo (Fig. 4; supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material).
Injection of synthetic dyes in Drosophila and [Ca2+]i estimation
Drosophila is a classic model organism for studies on learning and memory (Quinn et al., 1974) and their underlying molecular mechanisms (Lin and Goodman, 1994). More recently, GECIs are increasingly used to study information processing in intact neural circuits of the Drosophila brain. Thus, we expect that the described dye injection into genetically labeled neurons is highly useful for the calibration of other neurons too.
Our calibration produced results that are in good accordance with previous work at the Drosophila NMJ (Macleod et al., 2004). Because we took the KD of OGB-1 from our in vitro calibration, factors that influence indicator performance, such as the ionic strength, osmolarity, pH, and protein environment differed from the in vivo situation. This may lead to an underestimation of the in vivo KD for OGB-1 (Thomas et al., 2000). Assuming a twofold higher in vivo KD of OGB-1 compared with our measurement in the cuvette would result in a twofold increase of [Ca2+]i both at rest and at steady state during prolonged stimulation, as well as twofold higher in vivo KD values of the GECIs. Earlier measurements of [Ca2+]i Rest suggest that this is rather unlikely (Macleod et al., 2004).
Concluding remarks
Since the first prototypic GECIs were published (Miyawaki et al., 1997; Nakai et al., 2001), GECIs have been improved with respect to bright fluorescence, increased FRET efficiency, decreased FRET at resting [Ca2+]i, reduced magnesium sensitivity, KD, and response time constants. Still all GECIs exhibit low fluorescence and quantum yield compared with synthetic indicators. Thus, to yield sufficient SNR, more molecules per volume are needed, which increases the external buffer capacity. This is even more the case because a single GECI molecule binds up to four Ca2+ ions, which in addition may cause higher-order reaction kinetics. The latter becomes evident when ΔF is rather “loosely” related to activity and [Ca2+]i. Some of these aspects were improved in YC3.60 and D3cpv, making these indicators most sensitive for the detection of small [Ca2+]i and low rates of activity. Most importantly, those improvements were largely retained in vivo. GECIs such as GCaMPs and TN-XL, with their particular KD, Hill coefficient, and fluorescence time course, are suitable for different experimental demands. Thus, selecting a particular GECI by matching its in vivo properties to the expected [Ca2+]i of the experimental system provides a promising way to decipher changes in neuronal activity in intact organisms.
Footnotes
This work was supported by the Max Planck Society and a Human Frontier Science Program grant to K. Ito, A.B., and B. Nelson. We thank Atsushi Miyawaki and Roger Tsien for providing DNA; Katrin Deininger, Wolfgang Essbauer, Alexandra Ihring, Maximilian Joesch, and Christian Theile for experimental help; and Winfried Denk for the design and initial help on the 2P microscope.
The authors declare no competing financial interests.
References
- Ataka K, Pieribone VA. A genetically targetable fluorescent probe of channel gating with rapid kinetics. Biophys J. 2002;82:509–516. doi: 10.1016/S0006-3495(02)75415-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst A, Abarbanel HD. Relating a calcium indicator signal to the unperturbed calcium concentration time-course. Theor Biol Med Model. 2007;4:7. doi: 10.1186/1742-4682-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza T, McGann JP, Mombaerts P, Wachowiak M. In vivo imaging of neuronal activity by targeted expression of a genetically encoded probe in the mouse. Neuron. 2004;42:9–21. doi: 10.1016/s0896-6273(04)00144-8. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Clark DA, Gabel CV, Gabel H, Samuel ADT. Temporal activity patterns in thermosensory neurons of freely moving Caenorhabditis elegans encode spatial thermal gradients. J Neurosci. 2007;27:6083–6090. doi: 10.1523/JNEUROSCI.1032-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Díez-García J, Matsushita S, Mutoh H, Nakai J, Ohkura M, Yokoyama J, Dimitrov D, Knöpfel T. Activation of cerebellar parallel fibers monitored in transgenic mice expressing a fluorescent Ca2+ indicator protein. Eur J Neurosci. 2005;22:627–635. doi: 10.1111/j.1460-9568.2005.04250.x. [DOI] [PubMed] [Google Scholar]
- Díez-García J, Akemann W, Knöpfel T. In vivo calcium imaging from genetically specified target cells in mouse cerebellum. Neuroimage. 2007;34:859–869. doi: 10.1016/j.neuroimage.2006.10.021. [DOI] [PubMed] [Google Scholar]
- Fiala A, Spall T, Diegelmann S, Eisermann B, Sachse S, Devaud JM, Buchner E, Galizia CG. Genetically expressed cameleon in Drosophila melanogaster is used to visualize olfactory information in projection neurons. Curr Biol. 2002;12:1877–1884. doi: 10.1016/s0960-9822(02)01239-3. [DOI] [PubMed] [Google Scholar]
- Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- Guerrero G, Siegel MS, Roska B, Loots E, Isacoff EY. Tuning FlaSh: redesign of the dynamics, voltage range, and color of the genetically encoded optical sensor of membrane potential. Biophys J. 2002;83:3607–3618. doi: 10.1016/S0006-3495(02)75361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero G, Reiff DF, Agarwal G, Ball RW, Borst A, Goodman CS, Isacoff EY. Heterogeneity in synaptic transmission along a Drosophila larval motor axon. Nat Neurosci. 2005;8:1188–1196. doi: 10.1038/nn1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MT, Friedrich RW, Euler T, Larkum ME, Giese G, Both M, Duebel J, Waters J, Bujard H, Griesbeck O, Tsien RY, Nagai T, Miyawaki A, Denk W. Functional fluorescent Ca2+ indicator proteins in transgenic mice under TET control. PLoS Biol. 2004;2:e163. doi: 10.1371/journal.pbio.0020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim N, Griesbeck O. Genetically encoded indicators of cellular calcium dynamics based on troponin C and green fluorescent protein. J Biol Chem. 2004;279:14280–14286. doi: 10.1074/jbc.M312751200. [DOI] [PubMed] [Google Scholar]
- Heim N, Garaschuk O, Friedrich MW, Mank M, Milos RI, Kovalchuk Y, Konnerth A, Griesbeck O. Improved calcium imaging in transgenic mice expressing a troponin C-based biosensor. Nat Methods. 2007;4:127–129. doi: 10.1038/nmeth1009. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B. Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Borst JG, Sakmann B. Calcium dynamics associated with a single action potential in a CNS presynaptic terminal. Biophys J. 1997;72:1458–1471. doi: 10.1016/S0006-3495(97)78792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima S, Masino MA, Mandel G, Fetcho JR. Imaging neuronal activity during zebrafish behavior with a genetically encoded calcium indicator. J Neurophysiol. 2003;90:3986–3997. doi: 10.1152/jn.00576.2003. [DOI] [PubMed] [Google Scholar]
- Hoang B, Chiba A. Single-cell analysis of Drosophila larval neuromuscular synapses. Dev Biol. 2001;229:55–70. doi: 10.1006/dbio.2000.9983. [DOI] [PubMed] [Google Scholar]
- Holmes TC, Sheeba V, Mizrak D, Rubovszky B, Dahdal D. Circuit-breaking and behavioral analysis by molecular genetic manipulation of neural activity in Drosophila. In: North G, Greenspan RJ, editors. Invertebrate neurobiology. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; 2007. pp. 19–52. [Google Scholar]
- Jayaraman V, Laurent G. Evaluating a genetically encoded optical sensor of neural activity using electrophysiology in intact adult fruit flies. Front Neural Circuits. 2007;1:3. doi: 10.3389/neuro.04.003.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joesch M, Plett J, Borst A, Reiff DF. Response properties of motion-sensitive visual interneurons in the lobula plate of Drosophila melanogaster. Curr Biol. 2008;18:368–374. doi: 10.1016/j.cub.2008.02.022. [DOI] [PubMed] [Google Scholar]
- Kleinfeld D, Griesbeck O. From art to engineering? The rise of in vivo mammalian electrophysiology via genetically targeted labeling and nonlinear imaging. PLoS Biol. 2005;3:e355. doi: 10.1371/journal.pbio.0030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai S, Denk W, Osten P, Brecht M, Margrie TW. Two-photon targeted patching (TPTP) in vivo. Nat Protoc. 2006;1:647–652. doi: 10.1038/nprot.2006.100. [DOI] [PubMed] [Google Scholar]
- Lin DM, Goodman CS. Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Lnenicka GA, Grizzaffi J, Lee B, Rumpal N. Ca2+ dynamics along identified synaptic terminals in Drosophila larvae. J Neurosci. 2006;26:12283–12293. doi: 10.1523/JNEUROSCI.2665-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod GT, Hegström-Wojtowicz M, Charlton MP, Atwood HL. Fast calcium signals in Drosophila motor neuron terminals. J Neurophysiol. 2002;88:2659–2663. doi: 10.1152/jn.00515.2002. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Marin L, Charlton MP, Atwood HL. Synaptic vesicles: test for a role in presynaptic calcium regulation. J Neurosci. 2004;24:2496–2505. doi: 10.1523/JNEUROSCI.5372-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank M, Reiff DF, Heim N, Friedrich MW, Borst A, Griesbeck O. A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change. Biophys J. 2006;90:1790–1796. doi: 10.1529/biophysj.105.073536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao T, O'Connor DH, Scheuss V, Nakai J, Svoboda K. Characterization and subcellular targeting of GCaMP-type genetically-encoded calcium indicators. PLoS ONE. 2008;3:e1796. doi: 10.1371/journal.pone.0001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J. 2000;78:2655–2667. doi: 10.1016/S0006-3495(00)76809-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella S, Fischler W, Kong P, Asgarian S, Rueckert E, Scott K. Imaging taste responses in the fly brain reveals a functional map of taste category and behavior. Neuron. 2006;49:285–295. doi: 10.1016/j.neuron.2005.11.037. [DOI] [PubMed] [Google Scholar]
- Margrie TW, Meyer AH, Caputi A, Monyer H, Hasan MT, Schaefer AT, Denk W, Brecht M. Targeted whole-cell recordings in the mammalian brain in vivo. Neuron. 2003;39:911–918. doi: 10.1016/j.neuron.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, Kevrekidis IG. Optical imaging and control of genetically designated neurons in functioning circuits. Annu Rev Neurosci. 2005;28:533–563. doi: 10.1146/annurev.neuro.28.051804.101610. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Miyawaki A. Innovations in the imaging of brain functions using fluorescent proteins. Neuron. 2005;48:189–199. doi: 10.1016/j.neuron.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Mori MX, Erickson MG, Yue DT. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 2004;304:432–435. doi: 10.1126/science.1093490. [DOI] [PubMed] [Google Scholar]
- Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:137–141. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–268. doi: 10.1016/s0143-4160(97)90064-6. [DOI] [PubMed] [Google Scholar]
- Ng M, Roorda RD, Lima SQ, Zemelman BV, Morcillo P, Miesenböck G. Transmission of olfactory information between three populations of neurons in the antennal lobe of the fly. Neuron. 2002;36:463–474. doi: 10.1016/s0896-6273(02)00975-3. [DOI] [PubMed] [Google Scholar]
- Ohkura M, Matsuzaki M, Kasai H, Imoto K, Nakai J. Genetically encoded bright Ca2+ probe applicable for dynamic Ca2+ imaging of dendritic spines. Anal Chem. 2005;77:5861–5869. doi: 10.1021/ac0506837. [DOI] [PubMed] [Google Scholar]
- Palmer AE, Giacomello M, Kortemme T, Hires SA, Lev-Ram V, Baker D, Tsien RY. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol. 2006;13:521–530. doi: 10.1016/j.chembiol.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Piston DW. Photobleaching in two-photon excitation microscopy. Biophys J. 2000;78:2159–2162. doi: 10.1016/S0006-3495(00)76762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Yasuda R, Svoboda K. Monitoring neural activity and [Ca2+] with genetically encoded Ca2+ indicators. J Neurosci. 2004;24:9572–9579. doi: 10.1523/JNEUROSCI.2854-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn WG, Harris WA, Benzer S. Conditioned behavior in Drosophila melanogaster. Proc Natl Acad Sci U S A. 1974;71:708–712. doi: 10.1073/pnas.71.3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff DF, Thiel PR, Schuster CM. Differential regulation of active zone density during long-term strengthening of Drosophila neuromuscular junctions. J Neurosci. 2002;22:9399–9409. doi: 10.1523/JNEUROSCI.22-21-09399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff DF, Ihring A, Guerrero G, Isacoff EY, Joesch M, Nakai J, Borst A. In vivo performance of genetically encoded indicators of neural activity in flies. J Neurosci. 2005;25:4766–4778. doi: 10.1523/JNEUROSCI.4900-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai R, Repunte-Canonigo V, Raj CD, Knöpfel T. Design and characterization of a DNA-encoded, voltage-sensitive fluorescent protein. Eur J Neurosci. 2001;13:2314–2318. doi: 10.1046/j.0953-816x.2001.01617.x. [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Helmchen F, Sakmann B. Spatial profile of dendritic calcium transients evoked by action potentials in rat neocortical pyramidal neurones. J Physiol. 1995;487:583–600. doi: 10.1113/jphysiol.1995.sp020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Shang Y, Claridge-Chang A, Sjulson L, Pypaert M, Miesenböck G. Excitatory local circuits and their implications for olfactory processing in the fly antennal lobe. Cell. 2007;128:601–612. doi: 10.1016/j.cell.2006.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimozono S, Hosoi H, Mizuno H, Fukano T, Tahara T, Miyawaki A. Concatenation of cyan and yellow fluorescent proteins for efficient resonance energy transfer. Biochemistry. 2006;45:6267–6271. doi: 10.1021/bi060093i. [DOI] [PubMed] [Google Scholar]
- Siegel MS, Isacoff EY. A genetically encoded optical probe of membrane voltage. Neuron. 1997;19:735–741. doi: 10.1016/s0896-6273(00)80955-1. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM. Transposition of cloned P elements into Drosophila germ line chromosomes. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- Suh GS, Wong AM, Hergarden AC, Wang JW, Simon AF, Benzer S, Axel R, Anderson DJ. A single population of olfactory sensory neurons mediates an innate avoidance behaviour in Drosophila. Nature. 2004;431:854–859. doi: 10.1038/nature02980. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Kerr R, Bianchi L, Frøkjaer-Jensen C, Slone D, Xue J, Gerstbrein B, Driscoll M, Schafer WR. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron. 2003;39:1005–1017. doi: 10.1016/j.neuron.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Tallini YN, Ohkura M, Choi BR, Ji G, Imoto K, Doran R, Lee J, Plan P, Wilson J, Xin HB, Sanbe A, Gulick J, Mathai J, Robbins J, Salama G, Nakai J, Kotlikoff MI. Imaging cellular signals in the heart in vivo: cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc Natl Acad Sci U S A. 2006;103:4753–4758. doi: 10.1073/pnas.0509378103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank DW, Regehr WG, Delaney KR. A quantitative analysis of presynaptic calcium dynamics that contribute to short-term enhancement. J Neurosci. 1995;15:7940–7952. doi: 10.1523/JNEUROSCI.15-12-07940.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay LH, Griesbeck O, Yue DT. Live-cell transforms between Ca2+ transients and FRET responses for a troponin-C-based Ca2+ sensor. Biophys J. 2007;93:4031–4040. doi: 10.1529/biophysj.107.109629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Tovey SC, Collins TJ, Bootman MD, Berridge MJ, Lipp P. A comparison of fluorescent Ca2+ indicator properties and their use in measuring elementary and global Ca2+ signals. Cell Calcium. 2000;28:213–223. doi: 10.1054/ceca.2000.0152. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Wachowiak M, Denk W, Friedrich RW. Functional organization of sensory input to the olfactory bulb glomerulus analyzed by two-photon calcium imaging. Proc Natl Acad Sci U S A. 2004;101:9097–9102. doi: 10.1073/pnas.0400438101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Wong AM, Flores J, Vosshall LB, Axel R. Two-photon calcium imaging reveals an odor-evoked map of activity in the fly brain. Cell. 2003;112:271–282. doi: 10.1016/s0092-8674(03)00004-7. [DOI] [PubMed] [Google Scholar]
- Wang Y, Guo HF, Pologruto TA, Hannan F, Hakker I, Svoboda K, Zhong Y. Stereotyped odor-evoked activity in the mushroom body of Drosophila revealed by green fluorescent protein-based Ca2+ imaging. J Neurosci. 2004;24:6507–6514. doi: 10.1523/JNEUROSCI.3727-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DJ, Meyer zum Alten Borgloh S, Astori S, Yang Y, Bausen M, Kügler S, Palmer AE, Tsien RY, Sprengel R, Kerr JN, Denk W, Hasan MT. Single-spike detection in vitro and in vivo with a genetic Ca2+ sensor. Nat Methods. 2008 doi: 10.1038/nmeth.1242. in press. [DOI] [PubMed] [Google Scholar]
- Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE. 2004;2004:pl5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]