

Abstract
The fast-activating/deactivating voltage-gated potassium channel Kv3.3 (Kcnc3) is expressed in various neuronal cell types involved in motor function, including cerebellar Purkinje cells. Spinocerebellar ataxia type 13 (SCA13) patients carrying dominant-negative mutations in Kcnc3 and Kcnc3-null mutant mice both display motor incoordination, suggested in mice by increased lateral deviation while ambulating and slips on a narrow beam. Motor skill learning, however, is spared. Mice lacking Kcnc3 also exhibit muscle twitches. In addition to broadened spikes, recordings of Kcnc3-null Purkinje cells revealed fewer spikelets in complex spikes and a lower intraburst frequency. Targeted reexpression of Kv3.3 channels exclusively in Purkinje cells in Kcnc3-null mice as well as in mice also heterozygous for Kv3.1 sufficed to restore simple spike brevity along with normal complex spikes and to rescue specifically coordination. Therefore, spike parameters requiring Kv3.3 function in Purkinje cells are involved in the ataxic null phenotype and motor coordination, but not motor learning.
Keywords: K channels, burst firing, motor dysfunction, cerebellum, Purkinje cell, complex spike
Introduction
Voltage-gated potassium (Kv) channels of the Kv3 subfamily enable neurons to fire narrow action potentials at high frequencies beyond ∼200 Hz. Four separate genes (Kcnc1–Kcnc4) encoding subunits Kv3.1–Kv3.4 are expressed in various combinations in CNS neurons in which they assemble into homotetrameric and heterotetrameric channels. Mice lacking the Kcnc3-encoded Kv3.3 subunit exhibit motor deficits manifested as increased lateral deviation while ambulating and increased slips while traversing a narrow beam (McMahon et al., 2004; Joho et al., 2006b). A role for Kv3.3 channels in motor coordination is also underscored by the recent finding that dominant-negative mutations found in the human KCNC3 gene correlate with the neurological disorder spinocerebellar ataxia 13 (Waters et al., 2006).
Kv3-type channels are distinguished by exceptionally rapid kinetics of activation and deactivation, rendering them ideally suited to drive repolarization that is rapid, both in its onset and relief, so that the next action potential upstroke can occur with minimal delay in the absence of lingering afterhyperpolarization. High-frequency spiking is facilitated by both minimizing sodium channel inactivation through brief action potentials and by promoting sodium channel deinactivation through the excursion to negative potentials during the fast afterhyperpolarization. Indeed, the loss of Kv3-type channels results in broadened spikes accompanied by decelerated firing (Rudy and McBain, 2001). Unlike most other neuron types in which Kv3.3 subunits are expressed with other Kv3-type subunits, Purkinje cells express high levels of Kv3.3 and lower levels of Kv3.4 (Weiser et al., 1994; Martina et al., 2003). Loss of Kv3.3 results in a doubling of action potential duration (McMahon et al., 2004). Furthermore, blockade with tetraethylammonium (TEA) or BDS-I at a concentration specific for Kv3 channels broadened spikes and decelerated spiking to a similar extent (McKay and Turner, 2004; Martina et al., 2007).
Purkinje cells serve as the sole output of the cerebellar cortex, providing frequency-modulated inhibitory input to the deep cerebellar and vestibular nuclei (DCN/VN) that, in turn, convey cerebellar output to other brain areas. Superimposed on the tonic firing of simple spikes are complex spikes that occur at a low frequency of ∼1 Hz (Schmolesky et al., 2002). Each complex spike consists of a brief burst of spikes at a very high frequency (>500 Hz). Postsynaptically to Purkinje cells, deep nuclear neurons are tonically inhibited (Telgkamp and Raman, 2002; Telgkamp et al., 2004) or temporally entrained by simple spike input (Gauck and Jaeger, 2000) and respond to complex spike input by strong inhibition followed by rebound excitation (Llinás and Mühlenthaler, 1988; Aizenman and Linden, 1999).
Given the putative roles of complex spikes in motor function, particularly in timing (Placantonakis et al., 2004), and the high frequency of sodium spikes within a complex spike, we hypothesized that altered complex spikes in Kcnc3-null mice underlie the observed motor deficits. To assess the role of Kv3.3 function in Purkinje cells, we restored Kv3.3 channels selectively in Purkinje cells of Kcnc3-null mutant mice and assessed the extent to which wild-type motor function was rescued under conditions in which Kv3.3 channel subunits remain absent in the rest of the brain.
Materials and Methods
Mouse breeding.
The generation and initial characterization of mice lacking Kv3.1 and Kv3.3 K+ channels has been described previously (Ho et al., 1997; Espinosa et al., 2001, 2004; Joho et al., 2006a). Mice were kept on a 12 h light/dark cycle, and all behavioral experiments were performed with age-matched mice (∼3–6 months of age) derived on a mixed genetic background (∼25% of the genome was derived from 129/SvJ, ∼25% from C57BL/6, and ∼50% from ICR). The introduction of the L7/Pcp2 promoter-driven tTA transgene (derived from FVB/N mice) and the bicistronic EGFP-Kv3.3 transgene (derived from B6SJLF1 mice) increased further the complexity of the genetic background. To avoid the introduction of genetic drift, no separate wild-type or mutant colonies were established, and to equalize the unavoidable genetic variability among individual mice of different genotypes, extended brother–sister mating was avoided. Using this breeding scheme, the genetic background varied among individual mice; however, the extent of variability was similar for wild-type mice, Kcnc3-null mutants, and Kv3.3-rescue mice. All experiments were approved by the Institutional Animal Care and Research Advisory Committee.
Measurement of lateral deviation.
For behavioral tests, naive mice were first tested on the force-plate actometer followed by the beam test on 5 consecutive days. Males and females were used because we did not observe gender differences in these tests (Joho et al., 2006b).
We used a sensitive force-plate actometer that allows the center of force of the mice to be determined with high spatial (<1 mm) and temporal (20 ms) resolution (Fowler et al., 2001). The walking trajectory is represented by linear connections of temporally consecutive centers of force (COFs), and the total distance traveled is the sum of the linear distances (Σdi) between consecutive COFs. The area measure corresponds to the sum of all triangular areas (Σai) defined by every three consecutive COF measurements (for details, see Joho et al., 2006b). For a quantitative measure of lateral deviation from the main walking trajectory, the summed areas (Σai) generated during the 6 min test are divided by the total distance traveled (Σdi) to obtain the lateral deviation index (LDI = Σai/Σdi). The LDI would equal zero if the walking trajectory were a straight line (area measure equals zero). The LDI (in millimeters) increases with increasing lateral deviation of the COFs from the main trajectory of movement and corresponds to the “relative amplitude” of deviation from the mean path, representing an operational measure for gait.
Beam test.
The beam test was performed as described previously (Joho et al., 2006b). Mice were placed at the end of a 100-cm-long, 1-cm- or 0.5-cm-wide Plexiglas beam suspended 10 cm above a surface and allowed to traverse the beam five times per day (no interval between trials) on 5 consecutive days. Forelimb and hindlimb slips on the 1 cm beam and sideway falls (loss of balance) on the 0.5 cm beam were counted by two observers blind to genotype (nearly all slips were by hind legs). The average numbers of slips and sideway falls per 100 cm traveled were used for data presentation and analysis.
Twitch score.
During the 5 d beam test, mice were observed every day for the presence of subtle twitches. Mice were rated as 0, 1, 2, or 3 (twitch score) according to frequency and strength of twitches by two observers that were blind to genotype. The averaged twitch scores for 5 d were used for data analysis.
Western blot analysis.
Experiments were essentially performed as described previously (McMahon et al., 2004). Briefly, brains were dissected out and homogenized in cold lysis buffer (in mm: 1.5 KH2PO4, 8.1 Na2HPO4, 136.9 NaCl, 2.7 KCl, pH 7.4) containing 1% Triton X-100 and Complete protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN). Protein content was determined using the BCA protein assay kit (Pierce, Rockford, IL). Protein samples were separated by electrophoresis in 8.0% SDS-polyacrylamide and transferred to nitrocellulose membranes. Kv3.3 protein was detected with rabbit anti-Kv3.3 antibody (at 1:1200 dilution; Alomone Labs, Jerusalem, Israel) followed by peroxidase-conjugated goat anti-rabbit IgG antibody (Pierce). Enhanced chemiluminescence was used according to the manufacturer's instructions (Pierce).
Kv3.3 immunohistochemistry.
Anesthetized adult mice were transcardially perfused with cold PBS followed by 4% paraformaldehyde in PBS. Brains were dissected, postfixed for 1 h at 4°C, followed by cryoprotection in 40% sucrose in PBS. The tissue was embedded in OCT and 30 μm cryosections were mounted on Probe-On Slides (Thermo Fisher Scientific, Waltham, MA). Frozen sections were thawed and dried for 30 min followed by three 15 min washes with PBS. Sections were incubated for 1 h with blocking solution (4% goat serum, 0.3% Triton X-100, 0.05% Tween 20 in PBS). Rabbit anti-Kv3.3 (1:750; Alomone Labs) primary antibody was applied overnight at 4°C. Sections were then washed thrice for 15 min in PBS. Goat anti-rabbit Alexa 568-conjugated secondary antibody was applied in PBS (1:400; Invitrogen, Carlsbad, CA) at 4°C for 4 h followed by three 15 min rinses in PBS and coverslipping with Gel Mount (Biomeda, Foster City, CA) aqueous medium. To visualize fluorescence, a Carl Zeiss (Jena, Germany) LSM 510 confocal microscope with a krypton/argon-mixed gas laser was used. Images were collected digitally using a 63× oil (numerical aperture, 1.3) objective and processed with Adobe Photoshop 7.0.
Brain slice preparation and recording.
Artificial CSF (aCSF) was always the following (in mm): 125 NaCl, 3.25 KCl, 1.5 CaCl2, 1.5 MgCl2, 25 NaHCO3, 25 d-glucose, preoxygenated by carbogen (95% O2, 5% CO2) gas. Mice aged postnatal day 40 (P40) to P95 were anesthetized with ketamine/acepromazine before decapitation. The cerebellum was dissected to obtain 200-μm-thick parasagittal slices from the vermis in ice-cold aCSF using a vibratome. Slices were recovered at 35°C and submerged in an Edwards chamber for 30–45 min and thereafter at room temperature (22°C) until use in recording at least 45 min after slicing.
Slices were recorded in a submerged chamber maintained at 35 ± 0.5°C. Neurons were visualized at 40× with an Eclipse 6600FN epifluorescence microscope using differential interference contrast-infrared optics. Borosilicate glass recording pipettes (2–6 MΩ) were pulled on a Sutter P-87 puller. The internal solution consisted of the following (in mm): 130 K-methylsulfonate, 0.1 EGTA, 10 HEPES, 7 NaCl, 0.3 MgCl2, 5 di-Tris creatine-PO4, 2 Tris-ATP, and 0.5 Na-GTP. Whole-cell current-clamp recordings were obtained using an Axoclamp 2B amplifier filtering at 10 kHz in bridge mode. Pipette resistance was kept at 15–25 MΩ for spike waveform measures by monitoring the bridge balance. Data were collected at 40 kHz with pCLAMP 9.2 software operating on a Dell (Round Rock, TX) Dimension 8250 interfaced with a Digidata 1322A. Purkinje cells were easily identified by morphology and position. Only neurons capable of stable spontaneous spiking overshooting 0 mV were recorded.
Climbing fibers were stimulated by 100 μs stimuli from an A365 stimulus isolator (World Precision Instruments, Sarasota, FL) triggered by pClamp delivered via a theta pipette in the granule layer pulled as above from 1.5 mm theta glass broke to ∼10 μm filled with HEPES-aCSF (in mm: 150 NaCl, 3.35 KCl, 1.5 MgCl2, 20 d-glucose, 10 HEPES, 1.5 CaCl2, pH 7.4 with NaOH). Climbing fiber responses were distinguished by their all-or-none nature, were free from antidromic contamination and parallel-fiber evoked responses. F–I plots were compiled from 1.2 s depolarizing steps from a holding potential of −70 mV.
Data analysis and presentation.
Complex and spontaneous simple spike analysis was performed in Clampfit (Molecular Devices, Sunnyvale, CA), and spike rates for F–I plots were measured in Minianalysis (Synaptosoft, Decatur, GA). All measurements except spike rates for F–I plots were taken from the resting potential. The simple spike rate was measured from the interspike interval immediately preceding the complex spikes, and the pause was taken as the interval between the last spikelet and first simple spike after the complex spike. For data presentations, means and SEM were used. For statistical analyses, two-tailed Student's t tests, one-factor ANOVAs followed by multiple comparisons (Tukey's tests), and Kruskal–Wallis tests were used as appropriate. For repeated measures, genotype was the between factor and days on beam was the within factor for repeated-measures ANOVAs (OriginLab, Northampton, MA).
Results
Lack of Kv3.3 channels results in loss of spikelets within complex spikes
A climbing fiber-triggered complex spike consists of a large-amplitude sodium spike followed by several spikelets of smaller amplitudes (Schmolesky et al., 2002). The frequency of spikelets within a complex spike may reach >500 Hz, a discharge rate that requires fast-repolarizing potassium currents likely mediated by Kv3-type channels (Rudy and McBain, 2001). Because simple spike duration was doubled and the fast afterhyperpolarization reduced in Kv3.3-deficient Purkinje cells (McMahon et al., 2004), we hypothesized that the increased refractory period in the absence of Kv3.3 channels may lower the probability of spikelet generation within complex spikes. Hence, because of the increased interspikelet interval, Kv3.3-deficient Purkinje cells should have fewer spikelets within complex spikes compared with wild-type mice. If complex spikes are involved in some aspects of the timing function in the cerebellum, perturbed complex spikes with fewer spikelets may lead to imprecise timing and be responsible for some of the motor deficits observed in Kcnc3-null mutants.
To assess potential alterations in complex spikes in the absence of Kv3.3 channels, we performed whole-cell current-clamp recordings of Purkinje cells in cerebellar slices from wild-type and Kv3.3-deficient mice (Kcnc3 mutants). Most recordings were obtained from the same adult mice that had previously been subjected to behavioral tests to assess motor function (see below). Threshold stimuli to climbing fibers were delivered from a stimulating electrode located in the granule cell layer. The complex spike parameters measured included the number of spikelets after the initial large-amplitude spike and the first interspike interval (the time between the initial large sodium spike and the first spikelet).
At the resting potential, wild-type Purkinje cells fired spontaneous simple spikes with an action potential width (at half-height) at 35°C of 0.15 ± 0.004 ms (number of cells, n = 31) (Table 1). Climbing fiber-evoked complex spikes in wild-type mice comprised the initial large Na+ spike followed on average by 2.61 ± 0.29 spikelets (n = 15) with a first interspike interval corresponding to an instantaneous frequency of 625 ± 39 Hz (Fig. 1A). In contrast to wild type, the number of spikelets that follow the initial large sodium spike in complex spikes of Kv3.3-deficient Purkinje cells was significantly reduced to 1.17 ± 0.17 spikelets (n = 10; ANOVA, p < 0.001), and the first interspike interval was prolonged corresponding to an instantaneous frequency of 214 ± 9 Hz (Fig. 1B). As expected, simple spikes were nearly doubled in duration to 0.26 ± 0.005 ms (n = 33; ANOVA, p < 0.001). Therefore, in the absence of Kv3.3, complex spikes are generated but they contain fewer spikelets and the duration of simple spikes is approximately doubled.
Table 1.
Kcnc-null allele dependence of electrophysiological and behavioral parameters
| Genotype (Kv3.1; Kv3.3) |
||||||
|---|---|---|---|---|---|---|
| +/+; +/+ | +/+; −/− | +/+; −/− rescue | +/+; +/− | +/−; −/− | +/−; −/− rescue | |
| Input resistance (MΩ) | 23.3 ± 0.83 (29) | 24.2 ± 0.75 (28) | 23.6 ± 1.23 (17) | ND | ND | ND |
| Simple spike width (ms) | 0.15 ± 0.004 (31) | 0.26 ± 0.005*** (33) | 0.14 ± 0.006 (21) | ND | ND | ND |
| Interspike interval (ms) | 9.56 ± 1.02 (14) | 14.2 ± 1.30* (10) | 7.31 ± 0.72 (7) | ND | ND | ND |
| Slope (Hz/200 pA) | 29.3 ± 3.19 (9) | 11.8 ± 2.55** (7) | 25.8 ± 3.42 (6) | ND | ND | ND |
| Spikelets in complex spikes | 2.61 ± 0.29 (15) | 1.17 ± 0.17*** (10) | 2.30 ± 0.25 (7) | ND | ND | ND |
| Instantaneous frequency (Hz) | 625 ± 39 (15) | 214 ± 9*** (10) | 694 ± 85 (7) | ND | ND | ND |
| Pause after complex spike (ms) | 12.4 ± 1.29 (14) | 23.4 ± 3.65** (10) | 13.0 ± 1.68 (7) | ND | ND | ND |
| Lateral deviation index (mm) | 0.52 ± 0.03 (23) | 0.71 ± 0.04*** (33) | 0.59 ± 0.03 (27) | 0.55 ± 0.02 (29) | 0.84 ± 0.03*** (12) | 0.72 ± 0.05** (7) |
| Slips on 1 cm beam, day 1 | 1.79 ± 0.26 (23) | 6.18 ± 0.85** (32) | 4.12 ± 0.51# (27) | 4.97 ± 0.93 (29) | 11.1 ± 1.45** (12) | 9.14 ± 2.37 (7) |
| Slips on 1 cm beam, day 5 | 0.38 ± 0.07 (23) | 1.27 ± 0.26** (32) | 0.48 ± 0.08 (27) | 0.31 ± 0.06 (29) | 1.30 ± 0.34*** (12) | 0.66 ± 0.28 (7) |
For several electrophysiological and behavioral measures, the Kcnc-null allele dependence is shown. The numbers of independent data points are listed in parentheses (cells for electrophysiological measures and mice for behavior). ND, Not determined.
Significant differences from wild type are highlighted in bold (#p = 0.051; *p < 0.05; **p < 0.01; ***p < 0.001).
Figure 1.

Purkinje-cell-targeted Kv3.3 expression restores normal action potentials and complex spikes in Kcnc3-null mutants. Complex spikes triggered by climbing fiber stimulation (arrow) in Purkinje cells at the resting potential in wild-type mice (A), Kcnc3-null mutant (B), and on restoration of Kv3.3b expression (C). In Kcnc3-null mutants, both the number of spikelets after the initial large sodium spike and the instantaneous frequency derived from the first interspike interval are reduced (Table 1). Purkinje-cell-targeted Kv3.3 expression is sufficient to restore these parameters to those of wild-type mice. On the right, individual action potentials of wild type, Kcnc3-null mutant, and on restoration of Kv3.3 expression show action potential broadening and loss of fast afterhyperpolarization in Kcnc3-null mutants. The thin traces show spike broadening in the presence of 1 mm TEA. Compared with the Kcnc3-null mutant, TEA results in more substantial spike broadening and abolishes the fast afterhyperpolarization in wild-type and rescue mice.
When we compared the spontaneous firing frequency of simple spikes and the pauses after complex spikes between genotypes, we found significant differences for both parameters (Fig. 2A,B). Both the interspike interval (n = 10; ANOVA, p < 0.05) and pause duration (p < 0.01) were increased in Kv3.3-deficient Purkinje cells. In addition, in Kcnc3-null mice, the spike frequency as a function of injected current (F–I relationship) was significantly reduced (repeated-measures ANOVA, p < 10−6) (Fig. 2C) and the gain of the relationship was significantly decreased in Kv3.3-lacking Purkinje cells (11.8 ± 2.6 Hz/200 pA; n = 7) compared with wild-type cells (29.3 ± 3.2 Hz/200 pA; n = 9; ANOVA, p < 0.01), indicating that spiking at higher frequencies was more affected in Kv3.3-deficient Purkinje cells.
Figure 2.
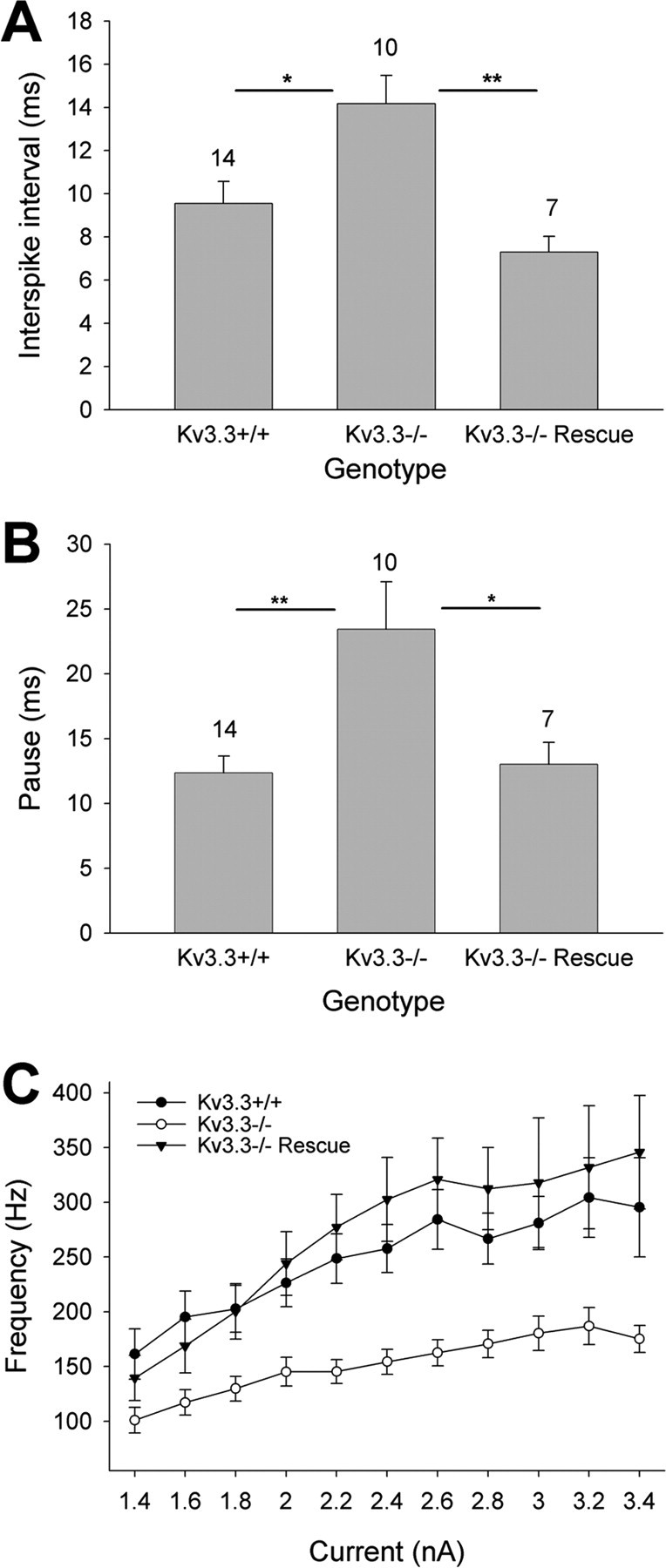
Purkinje-cell-targeted Kv3.3 expression restores normal simple spike frequency, post-complex spike pause duration, and high-frequency firing in Kcnc3-null mutants. A, The interspike interval of spontaneous simple spiking at the resting potential was increased in Kv3.3-deficient Purkinje cells and restored to that of wild type in rescue Purkinje cells (ANOVA). B, The duration of pauses in simple spiking that follow complex spikes is longer in Purkinje cells of Kcnc3-null mice and restored to that of wild type in rescue mice (ANOVA) (sample sizes are indicated above bars; *p < 0.05; **p < 0.01). C, In Kcnc3-null mice, the spike frequency as a function of injected current (F–I relationship) was significantly reduced (repeated-measures ANOVA, p < 10−6) and restored to wild-type values in rescue mice (p < 0.43). Error bars indicate SEM.
Purkinje-cell-specific restoration of Kv3.3 channels restores complex spikes
To express Kv3.3 channels in a Purkinje-cell-specific manner on the Kcnc3-null mutant background, we created a transgene in which both enhanced green fluorescent protein (EGFP) and Kv3.3b (the major splice variant in Purkinje cells) are expressed together from a bidirectional promoter that requires expression of the tetracycline transactivator protein (tTA). To confer Purkinje-cell-specific expression, we introduced the transactivator selectively to Purkinje cells by breeding with another transgenic mouse in which the transactivator was under the control of the Purkinje-cell-specific L7/Pcp2 promoter (Zu et al., 2004). Several bitransgenic lines were obtained that expressed Kv3.3b and EGFP as a fluorescent reporter, and two lines (313 and 396) were used for the experiments described here.
In both bitransgenic lines, Kv3.3 expression was only detectable by Western blots in cerebellum but not in brainstem or forebrain/midbrain (Fig. 3A). Mice carrying the Kv3.3 transgene alone without the tTA transgene lacked Kv3.3 expression indicating that leaky expression of the Kv3.3 transgene was absent (Fig. 3A). Compared with wild type, Kv3.3 expression levels in the cerebellum of rescue mice were lower, presumably reflecting the lack of Kv3.3 that is normally expressed in cerebellar granule cells and parallel fibers (Fig. 3A). In both bitransgenic lines, Kv3.3 immunoreactivity and EGFP fluorescence was colocalized and restricted to Purkinje cells (Fig. 3B). On the Kcnc3-null background, Kv3.3 transgene expression was detected throughout the soma, proximal dendrites, and axons (Fig. 3B, arrows) of Purkinje cells.
Figure 3.
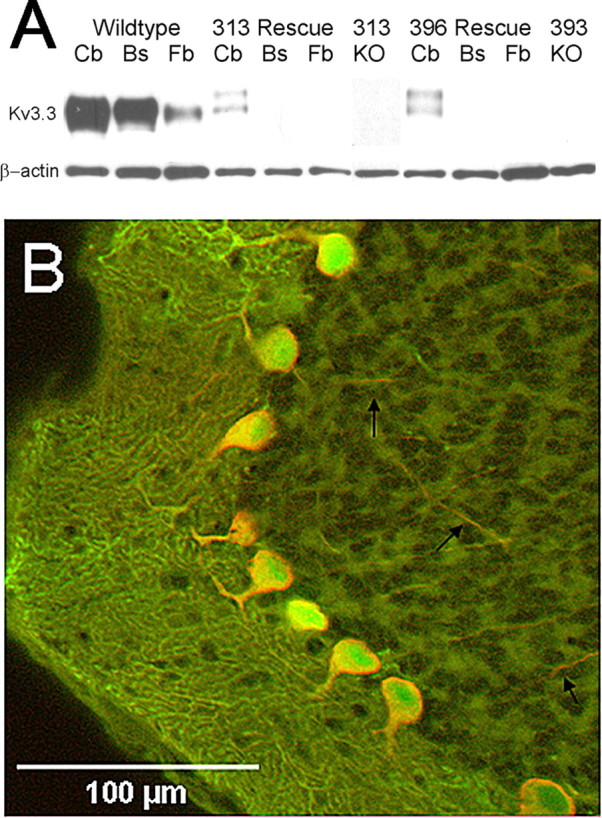
Purkinje-cell-targeted expression of Kv3.3 channel subunits. A, Western blot analysis shows Kv3.3 expression in wild-type cerebellum (Cb), brainstem (Bs), and forebrain/midbrain (Fb). In contrast, in both bitransgenic lines of rescue mice (313 and 396), targeted Kv3.3 expression is restricted to the cerebellum and absent from brainstem and forebrain/midbrain. In the absence of the L7-tTA transgene, both monotransgenic lines (KO) do not show detectable Kv3.3 expression in the cerebellum, indicating that leaky transcription of the Kv3.3 transgene is absent. β-Actin served as a loading control. B, The majority of Purkinje cells coexpress green fluorescent protein (GFP) (green) and Kv3.3b (red). Cytoplasmic GFP expression extends throughout the dendrites of the molecular layer. Kv3.3b expression is mostly restricted to somatic membranes, proximal dendrites, and is visible in axons traversing the granule cell layer (black arrows).
Cerebellar slices were prepared from bitransgenic mice (line 313), and climbing fiber-evoked complex spikes and spontaneous simple spikes in EGFP-positive somata were measured and compared with those of wild type and Kcnc3-null mutants. Input resistances were comparable across genotypes and the values were consistent with that expected in adult rodents (McKay and Turner, 2005). Complex spikes from bitransgenic rescue mice were similar to those of wild-type mice (Fig. 1C). The initial large sodium spike was followed by 2.30 ± 0.25 spikelets (n = 7), and the instantaneous frequency was 694 ± 85 Hz. These values are not different from those observed in wild-type Purkinje cells, but they are significantly different from those in Kcnc3-null mutants (ANOVA, p < 0.01). In bitransgenic Kcnc3-null mutants in which Kv3.3 channels were expressed selectively in Purkinje cells (referred to as rescue mice), the short duration of simple spikes was restored to 0.14 ± 0.006 ms (n = 21), a value not different from wild-type mice. A Purkinje cell recording from the 396 line was also consonant with rescue of all the above spike parameters (data not shown). Moreover, the interspike interval of simple spikes, pause duration after complex spikes, and F–I relationship were also restored to wild-type values (Fig. 2) (data are summarized in Table 1), in agreement with the notion that the Kv3.3 current enables neurons to fire narrow action potentials at high frequencies.
Genetic restoration of spike brevity could be reversed by bath application of 1 mm TEA (Fig. 1C) to an extent comparable with the effect on wild-type Purkinje cells (Fig. 1A). The TEA effect was mostly occluded in Kcnc3-null mice, suggesting that the rescue was indeed mediated by restoring Kv3.3 current (Fig. 1B). Based on the aforementioned rescue of spike parameters in Purkinje cells altered in Kcnc3-null mice in acute slices, we inferred that spiking was similarly rescued in vivo and next tested whether behavioral parameters were rescued concurrently.
Purkinje-cell-restricted Kv3.3 reexpression rescues motor function
We used the force-plate actometer to assess quantitatively the motor performance of wild-type mice, Kcnc3-null mutants, and rescue mice. The LDI corresponds to the relative amplitude of lateral deviation from the mean path while ambulating and represents an operational measure for gait and force exertion (for details, see Materials and Methods). The LDI of Kcnc3-null mice was significantly increased compared with that of wild-type mice in agreement with previous work (WT, 0.522 ± 0.026, n = 23; Kcnc3-null, 0.712 ± 0.037, n = 33; ANOVA, p = 0.0002). The LDI of rescue mice was, however, significantly smaller than that of Kcnc3-null mutants (rescue, 0.588 ± 0.025; n = 27; p = 0.015) and not different from wild-type mice (p = 0.35) (Fig. 4A). These data indicate that rescue mice, wherein Kv3.3 channels are exclusively expressed in Purkinje cells but still absent from the rest of the brain, display significantly less lateral deviation from their mean walking trajectory than Kcnc3-null mutants and are not different from wild-type mice.
Figure 4.

Kcnc-null allele dependence of lateral deviation. A, The lateral deviation indices (LDI in millimeters) for wild-type (Kv3.3+/+), Kcnc3-null mutant (Kv3.3−/−), and rescue mice are shown (number of animals tested is shown above the bars). The LDI is significantly increased in Kcnc3-null mice compared with wild-type and rescue mice. Rescue mice are not different from wild-type (ANOVA). B, The lateral deviation indices for controls (+/+; +/−), heterozygous Kv3.1/homozygous Kv3.3 mutants (+/−; −/−), and the corresponding Kv3.3-rescue mice are shown (number of animals tested is shown above the bars). Compared with controls, the LDI is significantly increased in +/−; −/− mice and in Kv3.3-rescue mice on the +/−; −/− background (ANOVA). Error bars indicate SEM.
Kcnc3-null mutants not only show increased lateral deviation while ambulating but also increased slips, presumably because of reduced motor coordination, when traversing a narrow beam (Joho et al., 2006b). To assess motor coordination, mice from the same groups used to measure lateral deviation were subjected to the 1 cm beam test. Mice of all three genotypes improved their performance on the 1 cm beam during the 5 d test (repeated-measures ANOVA, p < 10−6). The three genotypes were, however, significantly different from each other (wt/mutant, p < 10−6; mutant/rescue, p < 10−4; wt/rescue, p = 0.026) (Fig. 5A). On the first day, Kcnc3-null mutants made significantly more slips on the beam than wild-type mice (wt/mutant, p < 10−4, ANOVA) confirming previous work (Joho et al., 2006b). Rescue mice made more slips than wild type but fewer slips than Kcnc3 mutants on day 1, although these differences were only marginally significant (wt/rescue, p = 0.051; mutant/rescue, p = 0.063, ANOVA). On days 2–5 of testing, however, the performances of wild-type and rescue mice were indistinguishable (wt/rescue, p > 0.5, ANOVA). In stark contrast, even on day 5, Kcnc3 mutants did not reach the performance levels of wild-type or rescue mice (wt/mutant, p = 0.004; rescue/mutant, p = 0.007), although their performance had improved at a rate not slower than that of wild-type mice. We had previously shown that the beam performance of Kcnc3 mutants did not improve further even after 10 d of training (Joho et al., 2006b). Hence, it appears that Kcnc3-null mutants are fully capable of motor skill learning but are impaired in the execution of the learned motor skill.
Figure 5.
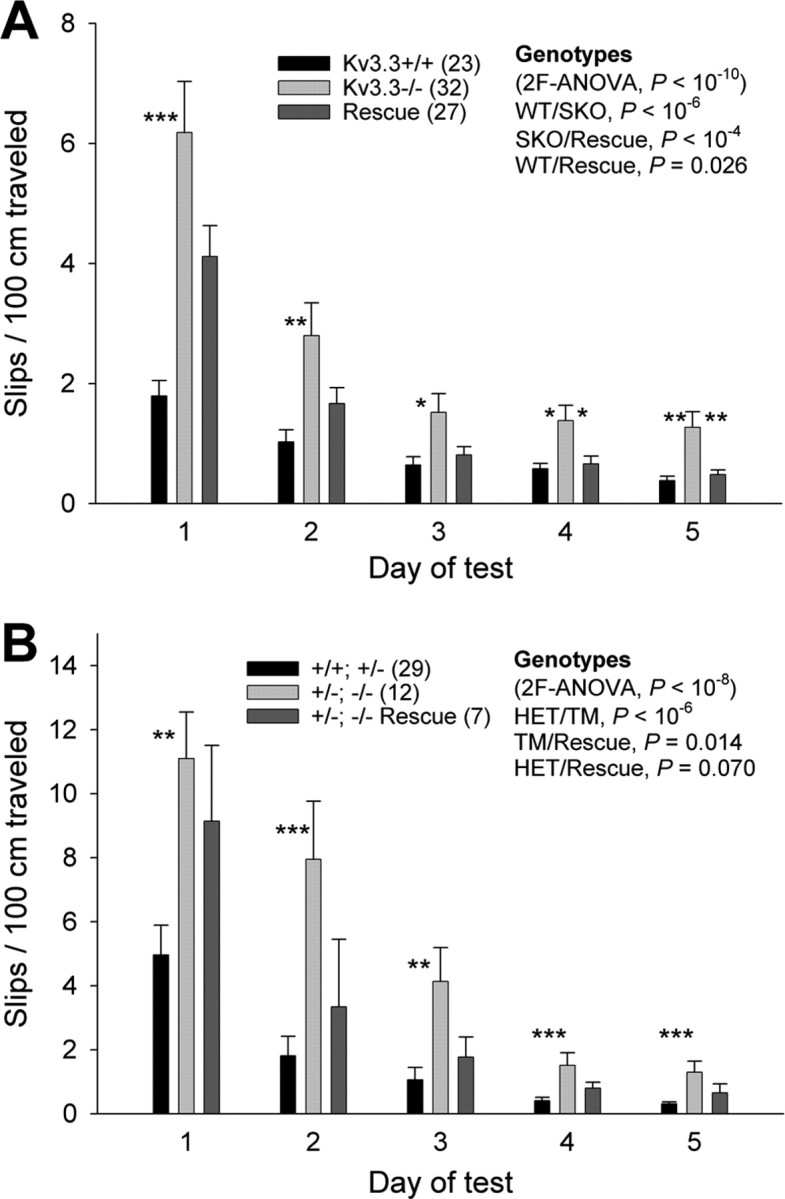
Purkinje-cell-specific expression of Kv3.3 rescues motor task performance. A, During the 5 d test on the 1 cm beam, Kcnc3-single mutants (SKO) perform worse than wild-type (p < 10−6) and rescue mice (repeated-measures ANOVA, p < 10−4). Although rescue mice perform better than Kcnc3-null mutants do, their performance is worse than that of wild type (repeated-measures ANOVA, p = 0.026) during the 5 d test, probably because of the impaired beam performance on day 1 (ANOVA; wt/rescue, p = 0.051). Importantly, all genotypes significantly improve their performance during the 5 d test (repeated-measures ANOVA, p < 10−6). Significant differences between SKO and wild-type or rescue mice for each day are indicated (ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001). B, Mice carrying three Kcnc-null alleles [i.e., heterozygous Kv3.1/homozygous Kv3.3 mutants (+/−; −/−)] perform worse than +/+; +/− controls (repeated-measures ANOVA, p < 10−6) and rescue mice on the +/−; −/− background (p = 0.014). Although rescue mice perform better than +/−; −/− mutants, their performance appears worse than that of controls (p = 0.070). Importantly, all genotypes significantly improve their performance during the 5 d test (repeated-measures ANOVA, p < 10−6). Significant differences between +/−; −/− mice and +/+; +/− or rescue mice for each day are indicated (ANOVA, **p < 0.01, ***p < 0.001). Error bars indicate SEM.
Kv3.3-deficient mice in which also a Kv3.1 allele is missing (+/−; −/−) show an even greater impairment of motor function (i.e., an additional increase in LDI and more slips on the 1 cm beam compared with +/+; −/− single-mutant mice) (Joho et al., 2006b). Thus, we asked whether expression of Kv3.3 in Purkinje cells of +/−; −/− mice would still rescue some aspects of motor function. As expected, the LDI of +/−; −/− mice (0.838 ± 0.027; n = 12) was significantly increased compared with that of +/+; +/− control mice (0.553 ± 0.019; n = 29; ANOVA, p < 10−6). In contrast to the restoration of LDI to wild-type levels on the +/+; −/− background, Purkinje cell-targeted Kv3.3 expression on the +/−; −/− background did not restore the LDI to control levels (0.718 ± 0.049; n = 7; p = 0.002) (Fig. 4B). The LDI of rescue mice was somewhat smaller than that of +/−; −/− mutants (p = 0.057). These data indicate that Purkinje-cell-restricted restoration of Kv3.3 channel function on the Kv3.1+/−Kv3.3−/− background does not rescue lateral deviation, whereas it does rescue it on the Kv3.1+/+Kv3.3−/− background.
The same mice were tested on the 1 cm beam. All genotypes improved their performance during the 5 d test (repeated-measures ANOVA, p < 10−6). However, the three genotypes differed from each other (control/mutant, p < 10−6; mutant/rescue, p = 0.014; control/rescue, p = 0.070) (Fig. 5B). On all days of the test, +/−; −/− mutants made significantly more slips than +/+; +/− control mice (days 1–3, p < 0.01; days 4 and 5, p < 0.001, ANOVA), again, in agreement with previous work (Joho et al., 2006b). Although rescue mice performed better than +/−; −/− mutants during the course of the 5 d test, the actual differences on individual days did not reach statistical significance.
Mice carrying three Kcnc-null alleles (−/−; +/− and +/−; −/− mice) exhibit sideways falls when traversing a 0.5 cm beam (Joho et al., 2006b). We therefore tested whether restoration of Kv3.3 function in Purkinje cells would prevent falls in +/−; −/− mutants (Fig. 6). All three genotypes improved their performance during the 5 d test (p < 10−8). The +/−; −/− mutant performed worse than control (+/+; +/−) or rescue (+/−; −/−) mice (control/mutant, p < 10−6; mutant/rescue, p < 0.001); however, there was no difference between control and rescue mice (control/rescue, p = 0.30). These data suggest that heterozygous Kv3.1 (+/−), homozygous Kv3.3 (−/−) mice in which Kv3.3 expression has been targeted to Purkinje cells but is still absent in the rest of the brain perform like wild-type-like mice (Kv3.1+/+Kv3.3+/−) in terms of falls while traversing the narrow 0.5 cm beam.
Figure 6.

Purkinje-cell-specific expression of Kv3.3 restores balance. On the 0.5 cm beam, mice of all genotypes improve their performance during the 5 d test (repeated-measures ANOVA, p < 10−8). Mutants carrying three Kcnc-null alleles (+/−; −/−) lose balance more often than +/+; +/− control mice (repeated-measures ANOVA, p < 10−6) and rescue mice (p = 0.001). Control mice do not differ from rescue mice (p = 0.30). Significant differences between +/−; −/− mice and +/+; +/− or rescue mice for each day are indicated (ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001). Error bars indicate SEM.
The data thus far indicate that reduced motor performance correlates directly with altered Purkinje cell firing. Because Kv3.3 subunits are coexpressed with Kv3.4 in axon terminals of motor neurons at the neuromuscular junction in which the absence of Kv3.3 may affect neurotransmitter release (Brooke et al., 2004), we determined whether changes in muscle strength might have contributed to the observed motor phenotype. We placed individual mice on an inverted cage top and measured the latency to fall (maximal time, 180 s). There were no differences between control (+/+; +/−, 154.0 ± 9.6 s; n = 29), Kv3.3-deficient (+/−; −/−, 158.7 ± 14.9 s; n = 12) and the corresponding rescue mice (176.4 ± 3.6 s; n = 7; ANOVA, p = 0.55), suggesting that no obvious changes in muscle strength were responsible for the motor dysfunction in Kcnc3-deficient mice.
Purkinje-cell-restricted Kv3.3 expression does not reduce muscle twitches
In the rescue mice, Kv3.3 function is only restored in Purkinje cells but is still absent from the rest of the brain in which Kv3.3 is normally expressed (e.g., in parallel fibers of cerebellar granule cells, deep nuclear neurons, red nucleus, motor neurons). Kcnc3-null mice (+/+; −/−) and particularly +/−; −/− mutants exhibit extensive, frequent twitches involving discrete subsets of muscles throughout the body. We used a semiquantitative scoring system to assess whether restoration of Kv3.3 expression in Purkinje cells might abolish twitches. Compared with control mice (+/+; +/−), mutant mice carrying three Kcnc-null alleles (+/−; −/−) showed a significantly increased twitch score [+/+; +/−, 0.62 ± 0.10, n = 29; +/−; −/−, 1.67 ± 0.11, n = 12; p = 0.001, Kruskal–Wallis (p < 10−4)]. The twitch score of rescue mice was not different from that of +/−; −/− mutants (1.51 ± 0.17; n = 7) but significantly different from control mice (p < 0.01). Therefore, Purkinje-cell-specific restoration of Kv3.3 function does not abolish twitchiness in Kcnc3-deficient mice.
Discussion
Here, we showed that in addition to maintaining narrow simple spikes, the presence of Kv3.3 channels in Purkinje cells is also required for a complete set of high-frequency spikelets in complex spikes. We determined that Purkinje cells represent an important cell type in which Kv3.3 function is involved in motor coordination and the ataxic phenotype of the Kcnc3-null mouse. Specifically, the execution of coordinated movement, but not motor skill learning or the muscle twitching phenotype, is related to Kv3.3-sensitive spike parameters.
Motor task execution but not motor skill learning requires Kv3.3 function in Purkinje cells
In addition to Purkinje cells, Kv3.3 is expressed in several neuronal populations known to be involved in motor function, such as α-motor neurons, the subthalamic nucleus, deep cerebellar, vestibular, and red nuclei. It is remarkable that reexpression of Kv3.3 channels exclusively in Purkinje cells of Kcnc3-single mutants (+/+; −/−) not only fully restored the properties of complex and simple spikes (Fig. 1) but also reduced lateral deviation to wild-type levels and restored motor performance on the 1 cm beam on days 2–5 (Figs. 4A, 5A). Rescue mice show intermediate beam performance on day 1 (Table 1) before learning and habituation, perhaps because loss of Kv3.3 expression in other types of neurons may be anxiogenic, particularly when the test is novel. It has been reported that anxiety decreases performance in a variant of the beam test (Lepicard et al., 2003). Alternatively, parallel fiber input to Purkinje cells, which is affected when Kcnc1 and Kcnc3 alleles are absent (Matsukawa et al., 2003), is initially abnormal but undergoes plastic changes on training that compensate for the augmented glutamatergic drive.
Even in Kv3.1+/−Kv3.3−/− mice, which show very poor beam performance, Purkinje-cell-restricted Kv3.3 reexpression resulted in significantly improved motor performance on the 1 cm beam (Fig. 5B) and decreased falls on the 0.5 cm beam (Fig. 6). These data highlight the pivotal role of Kv3.3-sensitive spike parameters in Purkinje cells in motor task execution. On the Kv3.1+/−Kv3.3−/− background, Purkinje-cell-targeted expression of Kv3.3 did not restore lateral deviation to wild-type levels (Fig. 4B). We believe that part of the increase of the LDI in Kv3.3-deficient mice, particularly in +/−; −/− mice, results from the underlying twitchiness that is present in these mice. This is in agreement with the observation that twitchiness was still present in rescue mice (no improved twitch score) and the LDI was not improved to control level on the +/−; −/− background. The persistence of twitches in rescue mice suggests that the twitches observed in Kcnc3-null mutant mice is not caused by altered Purkinje cell firing.
A remarkable finding is the observation that lack of Kv3.3 channels from Purkinje cells results in a deficit of motor task execution but that the actual process of motor skill learning is not negatively affected (Figs. 5, 6). Kcnc3-null mice (+/+; −/− and +/−; −/−) improve their performance on the 1 cm and 0.5 cm beams just as rapidly as wild-type mice during the 5 d test, suggesting that altered simple and complex spike firing does not affect this type of motor skill learning. This is in agreement with theories of cerebellar learning stipulating a role for parallel fiber-Purkinje cell long-term depression (LTD) and the previous observation that LTD is intact at these synapses in the absence of Kv3.3 (Matsukawa et al., 2003). The contribution of climbing fiber input to LTD is thought to arise from calcium influx in the dendrite, whereas the alterations here affect the sodium spikes of resulting complex spikes at the soma.
Perturbed complex spikes: potential impact on deep nuclear neurons
The changes in complex and simple spikes in Kv3.3-deficient Purkinje cells may affect neurons in the deep cerebellar and vestibular nuclei and, therefore, cerebellar output. Less characterized, but potentially relevant, are recurrent collaterals to inhibitory interneurons in the cerebellar cortex. Action potentials within both complex and simple spikes are broadened suggesting that GABA release may be augmented. Unless postsynaptic neurons adapt, tonic inhibition by simple spikes and compound IPSPs triggered by complex spikes, despite the reduced spikelet number and intraburst frequency, might be augmented overall. Alternatively, the net effect might be decreased inhibition, for the frequencies of simple spikes (Akemann and Knöpfel, 2006) as well as spikes within complex spike bursts are both diminished, and complex spikes contain fewer spikelets. Interestingly, the magnitude of the effect of Kv3.3 loss on simple spike frequency, a ∼40% reduction, is less than that on the intraburst frequency of complex spikes, a ∼70% reduction, consistent with higher frequency firing being more acutely sensitive to the absence of Kv3.3, as reflected in the gain of the F–I relationship. Thus, although all these parameters are rescued and therefore potential candidates, complex spikes seem relatively more affected.
The synapse made by Purkinje cells onto large glutamatergic projection neurons of the DCN is in a powerful position to determine cerebellar output. Rebound excitation is robust because each deep nuclear neuron receives synaptic input from up to ∼30 Purkinje cells (Chan-Palay, 1977) during synchronized complex spike firing that is expected to exert powerful inhibitory effects through temporal summation. As a primarily depressing synapse, the Purkinje cell-to-DCN synapse is considerably more sensitive to sudden changes in Purkinje cell firing rate than tonic input at a constant frequency (Telgkamp and Raman, 2002; Pedroarena and Schwarz, 2003; Telgkamp et al., 2004). Given the enhancement of parallel fiber input to Purkinje cells (Matsukawa et al., 2003), it is unclear whether the decrease in spontaneous simple spike frequency described in slices generalize to Purkinje cells in vivo with intact parallel fibers. Complex spikes, and the pauses that follow, are poised to have a greater impact on the DCN, for they involve sudden changes in input. Because inhibitory drive onto the glutamatergic neurons of the DCN undergoes compensatory upregulation in mutant mice lacking Purkinje cells (Garin et al., 2002; Linnemann et al., 2004), it may be possible that DCN neurons can adapt to altered tonic inhibition, whereas there is no evidence that these neurons can adapt to altered temporal summation of burst input with a decelerated intraburst frequency comprised of fewer spikes. These properties suggest that the complex spike alterations reported here are liable to have especially pronounced behavioral ramifications and be less subject to adaptation or compensation in the Kcnc3-null mutant.
Innervation of single DCN neurons by multiple Purkinje neurons is likely crucial for the alterations reported here to be consequential for the DCN firing given that, in individual neurons, axonal propagation of action potentials, particularly of spikelets, which are more often of stout stature, is unreliable at the high frequencies attained within complex spikes (Khaliq and Raman, 2005; Monsivais et al., 2005). Intriguingly, Kv3.3 channels thus far appear not to be enriched at nodes of Ranvier, in contrast to Kv3.1 (Chang et al., 2007), offering a potential explanation for the conduction failures and suggesting this is not the subcellular locus of Kv3.3 function.
After the complex spike itself, the ensuing pause in Purkinje cell firing allows DCN neurons to fire unimpeded during, and shortly after, the rebound burst. That Kcnc3-null Purkinje cells exhibit longer pauses in slices suggests that, regardless of whether the initial rebound burst is weaker, the duration over which the DCN can fire without inhibition from Purkinje cells is longer. Short pause durations are restored along with the complex spike burst. Thus, the rebound burst proper, longer spike trains thereafter, or both in the DCN may contribute to the motor phenotype.
The exact functional roles of complex spikes have mostly remained enigmatic. Aside from their posited role in learning, correlations between complex spikes and motor behavior implicate complex spikes in acute motor function. Synchronous complex spike firing of discrete subsets of Purkinje cells is thought to orchestrate coherent muscle contraction with high temporal precision (Welsh et al., 1995; Fukuda et al., 2001). More precisely, complex spikes have been posited to correct for ongoing adjustments of movement velocity during acute motor execution even after a task has been learned (Ebner et al., 2002; Pasalar et al., 2006). Aberrant movement velocity, such as the sudden acceleration seen in human patients with cerebellar pathology, could plausibly cause increased lateral deviation through increased exertion of force when each step is initiated and cause slips on the 1 cm beam. Indeed, we noted that Kcnc3-null mice, especially those additionally lacking a Kcnc1 allele, frequently appear to kick abruptly as they try to traverse the beam when slips are made.
Mice with mutations in P/Q-type calcium channels exhibit ataxia and an increase in the variance of the interspike interval of simple spikes (Walter et al., 2006). Potential concomitant alterations in complex spikes, however, were not explored despite the fact both channels affect complex spikes. The interspike interval variability is normal in Kcnc3-null mice (Akemann and Knöpfel, 2006). Purkinje-cell-specific ablation of the sodium channel Nav1.6 caused ataxia and virtually abrogated spontaneous simple spikes (Levin et al., 2006). Although complex spikes were not assessed, this directly corroborates the notion of the general involvement of Purkinje cell spike parameters in motor coordination.
Large glutamatergic neurons of the DCN express Kv3.1 and Kv3.3 (McMahon et al., 2004; Chang et al., 2007), suggesting functional redundancy. The cell types that express Kv3.2 in the DCN remain unidentified. We do not yet know whether Kcnc1/Kcnc3 double-null mutant mice are rescued behaviorally or whether rebound bursts, which also have a high intraburst frequency, become intrinsically impaired in these mice. It is intriguing that restoration of Kv3.3 in Purkinje cells is sufficient to rescue motor performance of Kv3.1+/−Kv3.3−/− mice. These observations further reinforce the importance of Kv3.3 function in Purkinje cells.
Footnotes
This work was supported by National Institutes of Health Grant NS042210 (R.H.J.). We are indebted to Drs. Ray Turner and Bruce McKay for their valuable help and advice on recording from cerebellar slices and to Dr. Harry Orr for providing L7/Pcp-2-tTA mice. We thank Mitali Bose for expert technical assistance in the final stages of this work.
References
- Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol. 1999;82:1697–1709. doi: 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- Akemann W, Knöpfel T. Sodium current influences the rate of spontaneous firing of Purkinje neurons. J Neurosci. 2006;26:4602–4612. doi: 10.1523/JNEUROSCI.5204-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke RE, Moores TS, Morris NP, Parson SH, Deuchars J. Kv3 voltage-gated potassium channels regulate neurotransmitter release from mouse motor nerve terminals. Eur J Neurosci. 2004;20:3313–3321. doi: 10.1111/j.1460-9568.2004.03730.x. [DOI] [PubMed] [Google Scholar]
- Chang SY, Zagha E, Kwon ES, Ozaita A, Bobik M, Martone ME, Ellisman MH, Heintz N, Rudy B. Distribution of Kv3.3 potassium channel subunits in distinct neuronal populations of mouse brain. J Comp Neurol. 2007;502:953–972. doi: 10.1002/cne.21353. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V. Berlin: Springer; 1977. Cerebellar dentate nucleus: organization, cytology and transmitters. [Google Scholar]
- Ebner TJ, Johnson MT, Roitman A, Fu Q. What do complex spikes signal about limb movements? Ann NY Acad Sci. 2002;978:205–218. doi: 10.1111/j.1749-6632.2002.tb07568.x. [DOI] [PubMed] [Google Scholar]
- Espinosa F, McMahon A, Chan E, Wang S, Ho CS, Heintz N, Joho RH. Alcohol hypersensitivity, increased locomotion, and spontaneous myoclonus in mice lacking the potassium channels Kv3.1 and Kv3.3. J Neurosci. 2001;21:6657–6665. doi: 10.1523/JNEUROSCI.21-17-06657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Marks GA, Heintz N, Joho RH. Increased motor drive and sleep loss in mice lacking Kv3-type potassium channels. Genes Brain Behav. 2004;3:90–100. doi: 10.1046/j.1601-183x.2003.00054.x. [DOI] [PubMed] [Google Scholar]
- Fowler SC, Birkestrand BR, Chen R, Moss SJ, Vorontsova E, Wang G, Zarcone TJ. A force-plate actometer for quantitating rodent behaviors: illustrative data on locomotion, rotation, spatial pattering, stereotypies, and tremor. J Neurosci Methods. 2001;107:107–124. doi: 10.1016/s0165-0270(01)00359-4. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Yamamoto T, Llinás R. The isochronic band hypothesis and climbing fibre regulation of motricity: an experimental study. Eur J Neurosci. 2001;13:315–326. doi: 10.1046/j.0953-816x.2000.01394.x. [DOI] [PubMed] [Google Scholar]
- Garin N, Hornung JP, Escher G. Distribution of postsynaptic GABAA receptor aggregates in the deep cerebellar nuclei of normal and mutant mice. J Comp Neurol. 2002;447:210–217. doi: 10.1002/cne.10226. [DOI] [PubMed] [Google Scholar]
- Gauck V, Jaeger D. The control of rate and timing of spikes in the deep cerebellar nuclei by inhibition. J Neurosci. 2000;15:3006–3016. doi: 10.1523/JNEUROSCI.20-08-03006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CS, Grange RW, Joho RH. Pleiotropic effects of a disrupted K+ channel: reduced body weight, impaired motor skill and muscle contraction, but no seizures. Proc Natl Acad Sci USA. 1997;94:1533–1538. doi: 10.1073/pnas.94.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joho RH, Marks GA, Espinosa F. Kv3 potassium channels control the duration of different arousal states by distinct stochastic and clock-like mechanisms. Eur J Neurosci. 2006a;23:1567–1574. doi: 10.1111/j.1460-9568.2006.04672.x. [DOI] [PubMed] [Google Scholar]
- Joho RH, Street C, Matsushita S, Knöpfel T. Behavioral motor dysfunction in Kv3-type potassium channel-deficient mice. Genes Brain Behav. 2006b;5:472–482. doi: 10.1111/j.1601-183X.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Axonal propagation of simple and complex spikes in cerebellar Purkinje neurons. J Neurosci. 2005;25:454–463. doi: 10.1523/JNEUROSCI.3045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepicard EM, Venault P, Negroni J, Perez-Diaz F, Joubert C, Nosten-Bertrand M, Berthoz A, Chapouthier G. Posture and balance responses to a sensory challenge are related to anxiety in mice. Psychiatr Res. 2003;118:273–284. doi: 10.1016/s0165-1781(03)00069-6. [DOI] [PubMed] [Google Scholar]
- Levin SI, Khaliq ZM, Aman TK, Grieco TM, Kearney JA, Raman IM, Meisler MH. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a1 (Nav1.6) in cerebellar Purkinje neurons and granule cells. J Neurophysiol. 2006;96:785–793. doi: 10.1152/jn.01193.2005. [DOI] [PubMed] [Google Scholar]
- Linnemann C, Sultan F, Pedroarena CM, Schwarz C, Thier P. Lurcher mice exhibit potentiation of GABAA-receptor-mediated conductance in cerebellar nuclei neurons in close temporal relationship to Purkinje cell death. J Neurophysiol. 2004;91:1102–1107. doi: 10.1152/jn.00163.2003. [DOI] [PubMed] [Google Scholar]
- Llinás R, Mühlenthaler M. Electrophysiology of guinea pig cerebellar nuclear cells in the in vitro brainstem-cerebellar preparation. J Physiol (Lond) 1988;404:241–258. doi: 10.1113/jphysiol.1988.sp017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Yao GL, Bean BP. Properties and functional role of voltage-dependent potassium channels in dendrites of rat cerebellar Purkinje neurons. J Neurosci. 2003;23:5698–5707. doi: 10.1523/JNEUROSCI.23-13-05698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Metz AE, Bean BP. Voltage-dependent potassium currents during fast spikes of rat cerebellar Purkinje neurons: inhibition by BDS-I toxin. J Neurophysiol. 2007;97:563–571. doi: 10.1152/jn.00269.2006. [DOI] [PubMed] [Google Scholar]
- Matsukawa H, Wolf AM, Matsushita S, Joho RH, Knöpfel T. Motor dysfunction and altered synaptic transmission at the parallel fiber-Purkinje cell synapse in mice lacking the potassium channels Kv3.1 and Kv3.3. J Neurosci. 2003;23:7677–7684. doi: 10.1523/JNEUROSCI.23-20-07677.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BE, Turner RW. Kv3 K+ channels enable burst output in rat cerebellar Purkinje cells. Eur J Neurosci. 2004;20:729–739. doi: 10.1111/j.1460-9568.2004.03539.x. [DOI] [PubMed] [Google Scholar]
- McKay BE, Turner RW. Physiological and morphological development of the rat cerebellar Purkinje cell. J Physiol (Lond) 2005;567:829–850. doi: 10.1113/jphysiol.2005.089383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon A, Fowler SC, Perney T, Akemann W, Knöpfel T, Joho RH. Altered olivocerebellar circuit properties in the absence of the voltage-gated potassium channels Kv3.1 and Kv3.3. Eur J Neurosci. 2004;19:3317–3327. doi: 10.1111/j.0953-816X.2004.03385.x. [DOI] [PubMed] [Google Scholar]
- Monsivais P, Clark BA, Roth A, Häusser M. Determinants of action potential propagation in cerebellar Purkinje cell axons. J Neurosci. 2005;25:464–472. doi: 10.1523/JNEUROSCI.3871-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasalar S, Roitman AV, Durfee WK, Ebner TJ. Force field effects on cerebellar Purkinje cell discharge with implications for internal models. Nat Neurosci. 2006;9:1404–1411. doi: 10.1038/nn1783. [DOI] [PubMed] [Google Scholar]
- Pedroarena CM, Schwarz C. Efficacy and short-term plasticity at GABAergic synapses between Purkinje and cerebellar nuclei neurons. J Neurophysiol. 2003;89:704–715. doi: 10.1152/jn.00558.2002. [DOI] [PubMed] [Google Scholar]
- Placantonakis DG, Bukovsky AA, Zeng XH, Kiem HP, Welsh JP. Fundamental role of inferior olive connexin 36 in muscle coherence during tremor. Proc Natl Acad Sci USA. 2004;101:7164–7169. doi: 10.1073/pnas.0400322101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Schmolesky MT, Weber J, De Zeeuw CI, Hansel C. The making of a complex spike: ionic composition and plasticity. Ann NY Acad Sci. 2002;978:359–390. doi: 10.1111/j.1749-6632.2002.tb07581.x. [DOI] [PubMed] [Google Scholar]
- Telgkamp P, Raman IM. Depression of inhibitory synaptic transmission between Purkinje cells and neurons of the cerebellar nuclei. J Neurosci. 2002;22:8447–8457. doi: 10.1523/JNEUROSCI.22-19-08447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telgkamp P, Padgett DE, Ledoux VA, Woolley CS, Raman IM. Maintenance of high-frequency transmission at Purkinje to cerebellar nuclear synapses by spillover from boutons with multiple release sites. Neuron. 2004;41:113–126. doi: 10.1016/s0896-6273(03)00802-x. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JPA, Nolte D, Mock AF, Evidente VG, Fee D, Müller U, Dürr A, Brice A, Papazian DM, Pulst SM. Mutations in the voltage-gated potassium channel KCNC3 cause degenerative and developmental CNS phenotypes. Nat Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Lang EJ, Sugihara I, Llinás R. Dynamic organization of motor control within the olivocerebellar system. Nature. 1995;374:453–457. doi: 10.1038/374453a0. [DOI] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]