

Abstract
In the subthalamic nucleus (STN) of Parkinson's disease (PD) patients, a pronounced synchronization of oscillatory activity at beta frequencies (15–30 Hz) accompanies movement difficulties. Abnormal beta oscillations and motor symptoms are concomitantly and acutely suppressed by dopaminergic therapies, suggesting that these inappropriate rhythms might also emerge acutely from disrupted dopamine transmission. The neural basis of these abnormal beta oscillations is unclear, and how they might compromise information processing, or how they arise, is unknown. Using a 6-hydroxydopamine-lesioned rodent model of PD, we demonstrate that beta oscillations are inappropriately exaggerated, compared with controls, in a brain-state-dependent manner after chronic dopamine loss. Exaggerated beta oscillations are expressed at the levels of single neurons and small neuronal ensembles, and are focally present and spatially distributed within STN. They are also expressed in synchronous population activities, as evinced by oscillatory local field potentials, in STN and cortex. Excessively synchronized beta oscillations reduce the information coding capacity of STN neuronal ensembles, which may contribute to parkinsonian motor impairment. Acute disruption of dopamine transmission in control animals with antagonists of D1/D2 receptors did not exaggerate STN or cortical beta oscillations. Moreover, beta oscillations were not exaggerated until several days after 6-hydroxydopamine injections. Thus, contrary to predictions, abnormally amplified beta oscillations in cortico-STN circuits do not result simply from an acute absence of dopamine receptor stimulation, but are instead delayed sequelae of chronic dopamine depletion. Targeting the plastic processes underlying the delayed emergence of pathological beta oscillations after continuing dopaminergic dysfunction may offer considerable therapeutic promise.
Keywords: subthalamic nucleus, cerebral cortex, basal ganglia, Parkinson's disease, dopamine, 6-hydroxydopamine
Introduction
The frequency characteristics of brain activity vary predictably with behavior (Steriade, 2000; Buzsaki and Draguhn, 2004). The neurotransmitter dopamine is particularly important in coordinating neuronal activity across different frequencies during executive processes. Acute disruption of dopamine transmission, through blockade of receptors or synthesis, rapidly restructures the spectral pattern of activity within and between the basal ganglia and cerebral cortex (Degos et al., 2005; Costa et al., 2006; Burkhardt et al., 2007). Behavior changes accordingly, with acute disruptions producing severe motor deficits such as catalepsy (Ayd, 1961).
Chronic disruption of dopamine transmission also greatly affects frequency-domain interactions, as exemplified in Parkinson's disease (PD), in which midbrain dopamine neurons progressively die. Excessive synchronization of neuronal activity, and particularly oscillations, is a critical functional change accompanying Parkinsonism (Bergman et al., 1998; Boraud et al., 2005; Gatev et al., 2006; Hammond et al., 2007). In PD patients, synchronization within and between the rhythmic activities of single neurons and/or neuronal populations in the basal ganglia, particularly the subthalamic nucleus (STN), and cortex preferentially occurs at beta (15–30 Hz) frequencies (Brown et al., 2001; Levy et al., 2002; Amirnovin et al., 2004; Kuhn et al., 2005; Weinberger et al., 2006). These exaggerated beta oscillations are reduced during voluntary movements (Amirnovin et al., 2004; Kuhn et al., 2004; Williams et al., 2005) and are attenuated, together with motor symptoms, by dopamine-replacement therapies (Brown et al., 2001; Levy et al., 2002; Williams et al., 2002; Priori et al., 2004; Kuhn et al., 2006). Furthermore, electrical stimulation of STN at beta frequencies, which artificially induces excessive beta activity synchronization, promotes bradykinesia in patients (Chen et al., 2007). These clinical studies suggest that, by inappropriately coordinating neuronal activities, exaggerated beta oscillations play pathological “antikinetic” roles in PD (Brown, 2006).
Once established, excessive beta oscillations can be effectively “filtered out” by acute pharmacological therapies, but this does not necessarily mean they arise from acute loss of dopaminergic frequency filtering rather than slower long-term adaptations. This issue is not just important for understanding PD pathophysiology; any dependence on continuing plasticity might offer crucial new targets for disease-modifying interventions. However, dissecting the mechanisms underlying pathologically synchronized beta oscillations is challenged by clinical constraints, and because these activities have not been particularly forthcoming in animal models. Indeed, although animal models of PD have established the central tenet that dopamine modulates basal ganglia and cortical activities in the frequency domain, most studies emphasize the emergence of pathologically synchronized oscillations with frequencies of ≤15 Hz (Bergman et al., 1998; Boraud et al., 2005; Gatev et al., 2006; Hammond et al., 2007), i.e., substantially lower frequencies than those recorded clinically. We have shown previously, however, that 6-hydroxydopamine (6-OHDA) lesions of dopamine neurons, a well established rat model of PD (Schwarting and Huston, 1996a,b), significantly increases beta oscillations in local field potentials (LFPs) recorded from the STN and cortex (Sharott et al., 2005). Here, we define the neural basis of abnormal beta oscillations using this clinically relevant chronic PD model, and test whether acute disruption of dopamine transmission provokes their emergence.
Materials and Methods
Experimental procedures were performed on adult male Sprague Dawley rats (Charles River, Margate, UK), and were conducted in accordance with the Animals (Scientific Procedures) Act, 1986 (United Kingdom), and with Society for Neuroscience Policies on the Use of Animals in Neuroscience Research.
Electrophysiological recordings in anesthetized rats.
Recordings were made in eight dopamine-intact control rats (288–366 g) and nine chronically 6-OHDA-lesioned rats (309–411 g at the time of recording). Anesthesia was induced with 4% v/v isoflurane (Isoflo; Schering-Plough, Welwyn Garden City, UK) in O2, and maintained with urethane (1.3 g/kg, i.p.; ethyl carbamate; Sigma, Poole, UK) and supplemental doses of ketamine (30 mg/kg, i.p.; Ketaset; Willows Francis, Crawley, UK) and xylazine (3 mg/kg, i.p.; Rompun; Bayer, Leverkusen, Germany) as described previously (Magill et al., 2001, 2006). All wound margins were infiltrated with the local anesthetic, bupivacaine (0.75% w/v; Astra, Kings Langley, UK). Animals were then placed in a stereotaxic frame (Kopf, Tujunga, CA). Body temperature was maintained at 37 ± 0.5°C using a homeothermic heating device (Harvard Apparatus, Edenbridge, UK). Electrocorticograms (ECoGs), electrocardiographic activity, and respiration rate were monitored constantly to ensure the animals' well being (Magill et al., 2006). The ECoG was recorded via a 1-mm-diameter steel screw juxtaposed to the dura mater above the right frontal (somatic sensory–motor) cortex (4.5 mm anterior and 2.0 mm lateral of bregma) (Paxinos and Watson, 1986), and was referenced against another screw implanted in the skull above the ipsilateral cerebellar hemisphere. The raw ECoG was bandpass filtered (0.3–1500 Hz, −3 dB limits) and amplified (2000×; DPA-2FS filter/amplifier; Scientifica, Harpenden, UK) before acquisition. Extracellular recordings of unit activity and LFPs in and around the STN were simultaneously made using “silicon probes” (NeuroNexus Technologies, Ann Arbor, MI). Each probe had 16 recording contacts arranged in a single vertical plane, with a contact separation of 100 μm. Each contact had an impedance of 0.9–1.3 MΩ (measured at 1000 Hz) and an area of ∼400 μm2. The same probe was used throughout, but it was cleaned after each experiment in a proteolytic enzyme solution (Magill et al., 2006). This was sufficient to ensure that contact impedances and recording performance were not altered by probe use and reuse. Monopolar probe signals were recorded using high-impedance operational amplifiers (Advanced LinCMOS; Texas Instruments, Dallas, TX) and were referenced against a screw implanted above the contralateral cerebellum. After preamplification, extracellular signals were further amplified (1000–2000×) and low-pass filtered (0–6000 Hz) using programmable differential amplifiers (Lynx-8; Neuralynx, Tucson, AZ). In another two untreated control rats (325 and 350 g) and three chronically 6-OHDA-lesioned rats (370–433 g at time of recording), recordings of unit activity in STN (but not LFPs) were made using glass electrodes instead of silicon probes, according to standard methods (Magill et al., 2001). Briefly, extracellular recordings of the action potentials of STN neurons were made using 15–25 MΩ glass electrodes (tip diameter, ∼1.5 μm), which contained saline solution (0.5 m NaCl) and Neurobiotin (1.5% w/v; Vector Laboratories, Peterborough, UK). Glass electrode signals were amplified (10×) through the active bridge circuitry of an Axoprobe-1A amplifier (Molecular Devices, Sunnyvale, CA), AC-coupled, amplified an additional 100×, and then bandpass filtered at 300–5000 Hz (DPA-2FS; Scientifica). The ECoG and probe/glass electrode signals were each sampled at 17.5 kHz on-line using a Power1401 Analog-Digital converter and a PC running Spike2 acquisition and analysis software (Cambridge Electronic Design, Cambridge, UK). The recording strategy was to place three probe contacts within the STN. The STN was identified on-line by comparison of recorded unit activity with the known characteristic discharges of STN neurons under slow-wave activity (SWA) in urethane anesthesia (Magill et al., 2001). Neurons are more active and more densely packed in STN compared with neighboring structures. Thus, probe contacts within STN tended to register more intense multiunit activity (see Figs. 1F, 2E). The recording of LFPs evoked by bipolar electrical stimulation of the ipsilateral frontal cortex allowed unequivocal targeting of the STN on-line (Magill et al., 2004). Finally, all recording locations were additionally verified after the experiments using standard histological procedures (Magill et al., 2006). Activity was recorded first during SWA, which accompanies deep anesthesia and is similar to activity observed during natural sleep, and second during episodes of “global activation,” which contain patterns of activity that are more analogous to those observed during the awake, behaving state (Steriade, 2000). Transition from SWA to activation is exemplified by obliteration of the cortical slow oscillation (∼1 Hz), as well as δ (1–4 Hz) and spindle (7–14 Hz) oscillations (Steriade, 2000). Notably, the neuronal activity patterns present under this anesthetic regimen may only be qualitatively similar to those present in the unanesthetized brain. Nevertheless, the urethane-anesthetized animal still serves as a useful model for assessing the impact of extremes of brain state on functional connectivity within and between the basal ganglia and cortex (Magill et al., 2006). Sensory stimulation and subsequent global activation were elicited by pinching the hindpaw for 10–15 s with serrated forceps that were driven by a standard pneumatic pressure, as described previously (Magill et al., 2006). The animals did not exhibit either a marked change in ECG or respiration rate, and did not exhibit a hindpaw withdrawal reflex, in response to the pinch. Moreover, withdrawal reflexes were not present during episodes of spontaneous and/or prolonged global activation, thus indicating anesthesia was adequate throughout recordings.
Figure 1.

Exaggerated beta oscillations are present in the cortex and subthalamic nucleus of 6-OHDA-lesioned anesthetized rats. A, Frontal ECoGs and time-evolving power spectrogram of ECoG activity. Exaggerated beta oscillations emerge during spontaneous activation (2) but not SWA (1). Calibration: 0.2 mV, 500 ms. a.u., Arbitrary units. B, Spectrograms of simultaneously recorded activity in cortex (ECoG) and subthalamic nucleus (STN-LFP) during spontaneous activation. Weak power at 50 Hz is line noise. C, Power spectra (Power) of ECoGs, as well as LFPs simultaneously recorded from within, immediately above and below the STN, in a representative lesioned and control animal during activation. Gray boxes indicate the classic beta band (15–30 Hz). D, E, Beta oscillations are evident in the discharges of single STN units, as shown in autocorrelograms (AC) and periodograms (Lomb). The dashed line in periodogram indicates p = 0.05. STN neurons fire either spike bursts or single spikes in time with most (but not all; arrowhead in E) beta cycles in oscillatory LFPs, as shown in spike-triggered LFP averages (STAv). Power spectra of ECoG and STN-LFP simultaneously recorded with neuron in D are also shown. Calibration: D, 0.2 mV (ECoG), 0.5 mV (unit), 0.1 mV (LFP), 100 ms; E, 0.125 mV, 100 ms. F, Beta oscillations are also reflected in rhythmic multiunit activity in STN (contacts 2–4), but not immediately above or below (contacts 1 and 5). Traces 1–5 of units and LFPs were recorded simultaneously. Calibration: 0.1 mV, 100 ms. G, Plot of STN neurons with significant oscillations in their spike trains (those recorded with silicon probes or glass electrodes are represented by diamonds and circles, respectively). H, Coherence (left) and total beta coherence at 17–23 Hz (right) between single units and LFPs in the STN as recorded with probes (n = 14 and 8 unit/LFP pairs in lesioned and control, respectively). The dashed line is the 95% confidence limit. Data are means ±1 SEM. *p = 0.005. I, Mean firing rates of STN neurons during activated brain state. Data are means ±1 SEM. *p < 0.001. Data in A–F are from different lesioned animals.
Figure 2.

Beta oscillations are not prevalent in the cortex and subthalamic nucleus of dopamine-intact anesthetized rats. A, Frontal ECoGs and time-evolving power spectrogram of ECoG activity in a control animal. Neither global activation (2) nor SWA (1) brain states were associated with prominent beta oscillations. B, Spectrograms of simultaneously recorded activity in the cortex (ECoG) and subthalamic nucleus (STN-LFP) during spontaneous activation. Weak power at 50 Hz is line noise. C, Power spectra (Power) of ECoGs as well as LFPs simultaneously recorded from within, immediately above, and below STN, in a representative control animal before and after systemic treatment with D1 and D2 receptor antagonists (SCH-23390 at 0.5 mg/kg and raclopride at 2 mg/kg). Gray boxes indicate the classic beta band (15–30 Hz). D, During activation in control animals, the discharges of single STN units were not clearly periodic, as shown by flat autocorrelograms (AC) and periodograms (Lomb). Spike-triggered LFP averages (STAv) were correspondingly flat. The power spectra of ECoG and STN-LFP simultaneously recorded with the neuron in D are also shown. E, Simultaneous recordings of multiunit activity and LFPs in the STN (contacts 2–5), as well as immediately above and below the STN (contacts 1 and 6, respectively) of a control rat. Calibrations are the same as in Figure 1, except in D: 0.1 mV (unit).
Dopamine receptor antagonists.
To achieve acute and widespread pharmacological disruption of dopamine transmission, rats received systemic injections of potent and selective antagonists of dopamine D1-like receptors [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SCH-23390), 0.5 mg/kg, s.c.; Sigma] and D2-like receptors [3,5-dichloro-N-(1-ethylpyrrolidin-2-ylmethyl)-2-hydroxy-6-methoxybenzamide [S(−)-raclopride], 2 mg/kg, i.p.; Sigma]. Both drugs were dissolved in physiological saline solution and administered within 10 s of each other. Antagonists and doses were selected according to previous behavioral observations (Degos et al., 2005).
6-Hydroxydopamine lesions of dopamine neurons.
Unilateral 6-OHDA lesions were performed on 213–293 g rats, as described previously (Magill et al., 2001). Twenty-five min before the injection of 6-OHDA, all animals received a bolus of desipramine (25 mg/kg, i.p.; Sigma) to minimize the uptake of 6-OHDA by noradrenergic neurons (Schwarting and Huston, 1996a). Anesthesia was induced and maintained with isoflurane (as above). The neurotoxin 6-OHDA (hydrochloride salt; Sigma) was dissolved immediately before use in ice-cold 0.9% w/v NaCl solution containing 0.02% w/v ascorbate to a final concentration of 4 mg/ml. Then, 3 μl of 6-OHDA solution was injected into the region adjacent to the medial substantia nigra (4.5 mm posterior and 1.2 mm lateral of bregma, and 7.9 mm ventral to the dura) (Paxinos and Watson, 1986). The extent of the dopamine lesion was assessed 14 or 15 d after 6-OHDA injection by challenge with apomorphine (0.05 mg/kg, s.c.; Sigma) (Schwarting and Huston, 1996b). The lesion was considered successful in those animals that made ≥100 net contraversive rotations in 20 min (Magill et al., 2001). Note that the emergence of exaggerated beta oscillations is not dependent on apomorphine (Sharott et al., 2005). Electrophysiological recordings were performed ipsilateral to 6-OHDA lesions in anesthetized rats 21–35 d after surgery, when pathophysiological changes in the basal ganglia are likely to have leveled out near their maxima (Vila et al., 2000).
Electrophysiological recordings in behaving rats.
Dopamine-intact rats (260–373 g; n = 5) were anesthetized with isoflurane and implanted with a screw above the frontal cortex for subsequent ECoG recordings (as above). Behavioral monitoring, electrophysiological recordings, and antagonist administration were performed 8–10 d after implantation surgery. The ECoGs were referenced against a screw implanted centrally above the cerebellum, and bandpass filtered (0.1–3000 Hz) and amplified (1000×; EPA-6 amplifier; Sensorium, Charlotte, VT) before acquisition at 20 kHz. The induction of catalepsy, defined as the delayed or absent correction of an abnormal positioning of the extremities, was tested 10–15 min after administration of SCH-23390 and raclopride using a standard bar test (Degos et al., 2005). None of the animals were able to retract their forepaws from the raised horizontal bar (10 cm above the floor) and correct their unusual posture within 1 min of paw placement; thus, all were considered fully cataleptic (Degos et al., 2005). For recordings in behaving lesioned subjects, rats (215–246 g; n = 5) were anesthetized with isoflurane, unilaterally injected with 6-OHDA, and implanted with a screw above the ipsilateral frontal cortex in a single surgical session (as above). Recordings of ECoGs were performed between 3:00 P.M. and 6:00 P.M. (reversed light/dark cycle: lights on 8:00 A.M. to 8:00 P.M.) on postoperative days 1, 4, and 15, and care was taken to record when the animals were fully alert but immobile, behavior known to favor the emergence of abnormal beta oscillations (Sharott et al., 2005). Lesioned rats were confirmed as such by apomorphine challenge (see above).
Data analysis.
Epochs of robust cortical slow-wave activity or global activation were selected according to the previously described characteristics of these brain states (Magill et al., 2001, 2006). Frequency analyses were performed on 100.1 ± 0.1 s (mean ± SEM) and 94.8 ± 2.3 s of data, 100.1 ± 0.1 and 97.2 ± 2.8 s of data, and 100.1 ± 0.1 and 88.8 ± 7.4 s of data recorded during robust SWA and during activation in chronically lesioned, untreated control, and antagonist-treated control animals, respectively. Recordings of LFPs and ECoGs were low-pass filtered at 100 Hz and down-sampled to 250 Hz off-line in Spike2 for analysis purposes. Frequency-domain (spectral) analyses were performed in Matlab v6.0 (The MathWorks, Natick, MA) using a mixed-radix algorithm. Spectra were estimated by dividing the data epochs into a number of disjoint sections of 1 s duration. Frequency resolution was 1 Hz. Data were windowed with a Hanning filter to control “spectral leakage.” In the main analysis of STN-LFPs, we averaged the power from each of three probe contacts in STN, and compared this across brain states and treatment groups. In a subsidiary analysis, this average of STN-LFP power was compared with the LFP power separately averaged across the two nearest probe contacts above and below STN in each animal. We also analyzed the linear phase/amplitude relationships (as defined by coherence) and mutual information between STN signals that were maximally representative of activity in local regions of the STN in the different groups. Our original monopolar LFPs were not suitable for this, and we could not extract a sufficient number of bipolar LFPs because, on average, we had only three probe contacts in each STN. Accordingly, we sought evidence of elevated synchronization across regions of the STN using multiunit signals from each probe contact (see Figs. 1F, 2E). The multiunit activity was derived by high-pass filtering the wideband probe signals at 500 Hz off-line (Spike2), and converting the analog signal into a point process by considering those data points that exceeded the mean +3 SD of the analog signal. The coherence between point process data representing multiunit activity at each probe contact in the STN was then determined using previously published methods (Halliday et al., 1995) and software (Neurospec version 1.0; www.neurospec.org). Data were divided into blocks of 8192 data points, affording a frequency resolution of 2.18 Hz. Lower-bound estimates of the mutual information between STN sites were derived from the coherence, as described previously (Borst and Theunissen, 1999). Before performing our analyses of coherence and mutual information between multiunit activities recorded at different STN sites, we first assessed the analog signals (see above) for the possibility of coregistration of the same unit(s) across contact pairs because such contamination might confound estimates of coherence. Coregistration of the same unit(s) would be reflected as large, but narrow, symmetrical and centrally aligned peaks in cross-correlograms (1 ms bins) of the analog signals. Thus, in the absence of signal contamination, activity counts in the central cross-correlogram bin should be similar to counts in flanking bins. We rejected 4 of 27, 3 of 24, and 3 of 24 signal pairs from lesioned animals, untreated controls, and antagonist-treated controls, respectively, where the cross-correlogram bin centered at t = 0 ms had ≥35% of the mean count in the flanking two bins on either side. This narrow total sampling period of 5 ms was necessary to ensure relative local stationarity, even in the presence of the biological synchronization predicted in lesioned animals (where oscillatory temporal interactions were likely to occur with a period of ∼50 ms). The mean excess counts in the central bins of the remaining cross-correlograms did not differ from unity, suggesting negligible contamination (4 ± 3% and 6 ± 3% above flanking bins for lesioned animals and untreated controls, respectively, and 6 ± 4% below flanking bins for antagonist-treated controls; all p > 0.05, one-sample t tests; median of all excess counts was 1% above mean count in flanking bins). Finally, autocorrelograms (2 ms bins) and spike-triggered LFP averages were constructed using standard procedures in Spike2, and Lomb periodograms of spike trains (0.5–100 Hz range) according to previous publications (Magill et al., 2001).
Statistical testing.
For analysis of recordings in anesthetized animals, power values were normalized by logarithmic transformation before statistical testing (Halliday et al., 1995). Transformed power data were normally distributed, as judged by the single-sample Kolmogorov–Smirnov test and p ≤ 0.05 to reject (SPSS; SPSS, Chicago, IL). To determine whether power in the beta band was greatest in the STN-LFPs and ECoGs after 6-OHDA lesions, in control animals or after antagonist treatment of control animals, we used combinations of two-tailed unpaired and paired t tests (corrected for multiple comparisons) (see below). Because analyzing data from these three groups involved a comparison of paired data (i.e., untreated controls vs treated controls) and comparison of these data with a third group that was not paired (lesioned animals), this combination of t tests was more suitable than either a standard ANOVA or repeated-measures ANOVA. Equality of variances was assessed using Levene's test (p ≤ 0.05 to reject). Where ANOVAs were performed for other multiple group comparisons, Mauchly's test was used to confirm the sphericity of the data and, when nonspherical, a Greenhouse-Geisser correction performed. All tests specified in the text remained significant at p ≤ 0.05 after corrections for multiple testing using the false discovery rate procedure (Curran-Everett, 2000), unless otherwise stated. Means and corresponding SEMs are given in the text and illustrations. For comparisons of ECoG data from behaving control animals before and after saline or antagonist administration (100 s per treatment per animal), we used a one-way repeated-measures ANOVA, with post hoc Bonferroni's t tests for multiple comparisons with the control group (SigmaStat; Systat Software, London, UK). For comparisons of ECoG data from behaving lesioned animals (100 s per day per animal), which had smaller sample sizes, we used nonparametric statistical testing because some data sets were not normally distributed (p < 0.05; Kolmogorov–Smirnov test). We thus used Friedman's repeated-measures ANOVA on ranks (more suitable than standard ANOVAs for assessing nonparametric data), with Student–Newman–Keuls tests for post hoc pairwise multiple comparisons (SigmaStat). The Mann–Whitney rank sum test was used for comparisons with unpaired nonparametric data, i.e., between control and lesioned animals. Medians and corresponding quartiles are shown in the figures where appropriate.
Results
Pathological beta oscillations are dependent on dopamine loss and brain state
A key aim of this study was to define the neural basis of abnormal beta oscillations in cortico-STN circuits. It was thus important to simultaneously detect the synchronized activity of local populations of neurons in and around STN. Our strategy was to sample both single-unit and multiunit activity and LFPs using linear arrays with multiple, spatially defined recording contacts (silicon probes) (Magill et al., 2006). The STN is not only small (< 0.1 mm3) but it is also located deep (7.5–8 mm) in the adult rat brain (Oorschot, 1996), which greatly challenges the use of these recording arrays in behaving rodents. We thus chose to make our initial analyses in anesthetized animals, which had the additional important benefit of allowing us to record with the probes during two well defined and controlled brain states, slow-wave activity and global activation (Magill et al., 2006). The former state is dominated by low-frequency rhythms (≤10 Hz) and is qualitatively similar to natural sleep, whereas the latter contains patterns of activity that are more analogous to those observed during waking behavior (Steriade, 2000). Hence, we could define any interaction between gross brain state and the emergence of pathological synchrony. However, because pathological beta oscillations are expressed in alert PD patients, but have not been studied in anesthetized subjects, our first priority was to establish meaningful parallels between clinical data and our experimental paradigm.
We recorded epidural ECoGs from the frontal cortex, as well as activity at multiple sites in and around the STN, in anesthetized dopamine-intact control rats (n = 8) and unilaterally 6-OHDA-lesioned rats (n = 9) during SWA and global activation. Spontaneous or sensory-driven transitions from SWA to global activation were accompanied by the emergence of prominent beta oscillations (15–25 Hz) in ECoGs and STN-LFPs recorded from 6-OHDA-lesioned animals (Fig. 1A–C), but not those recorded from control animals (Figs. 1C, 2). The prevalent frequencies of these abnormal beta oscillations in cortex and STN were similar across all 6-OHDA-lesioned animals, with an average peak frequency of around 20 Hz (see Figs. 1, 4A, 5A). Thus, chronic 6-OHDA lesions of dopamine neurons profoundly exaggerate beta oscillations in field activity recorded from cortex and STN during the activated brain state but not during SWA. Moreover, the beta oscillations evident in cortical and STN field activities in our rat model of PD compare favorably with previous findings in PD patients.
Figure 4.
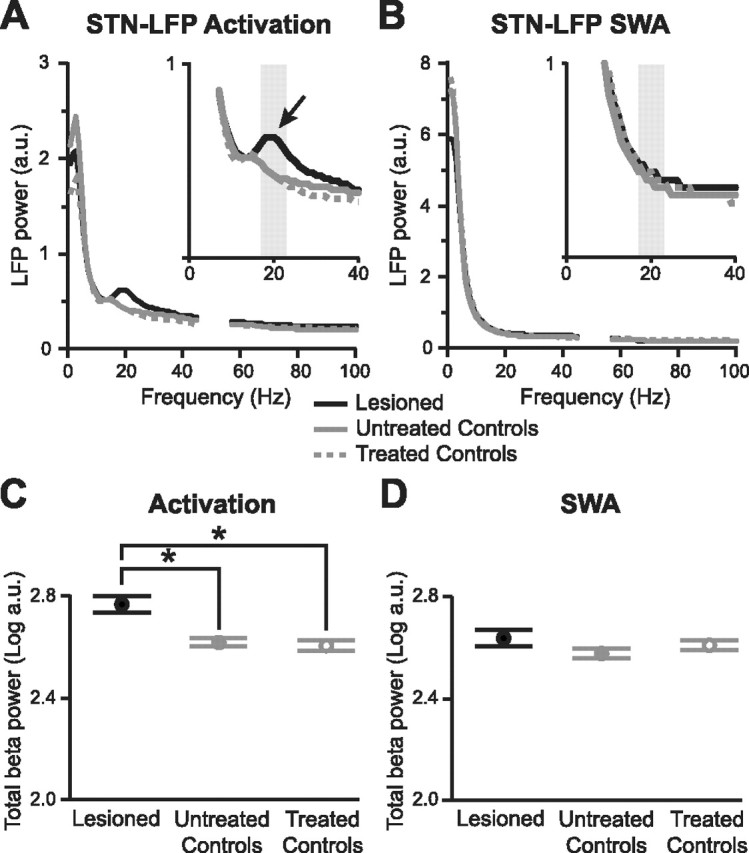
Chronic 6-OHDA lesions, but not acute antagonist treatment, exaggerate beta oscillations in the subthalamic nucleus. A, Average power spectra of STN-LFPs recorded in untreated control animals (n = 8), controls after acute antagonist treatment (n = 8, “treated controls”), and chronically lesioned animals (n = 9) during the activated brain state. The gray boxes indicate the peak beta band (17–23 Hz) used in quantitative analyses in C and D. Spectral power at ∼50 Hz (line noise) was removed for clarity. a.u., Arbitrary units. B, Average power spectra of STN-LFPs recorded in the same animals during slow-wave activity. There are no distinct peaks in power at beta frequencies. C, D, Average log power of peak beta oscillations (17–23 Hz) in STN-LFPs across experimental groups and brain states. Power values in A and B were normalized by logarithmic transformation before statistical testing. Data are means ±1 SEM. *p < 0.01. Note the small error bars (SEM) indicating limited variability between beta oscillations in different animals.
Figure 5.
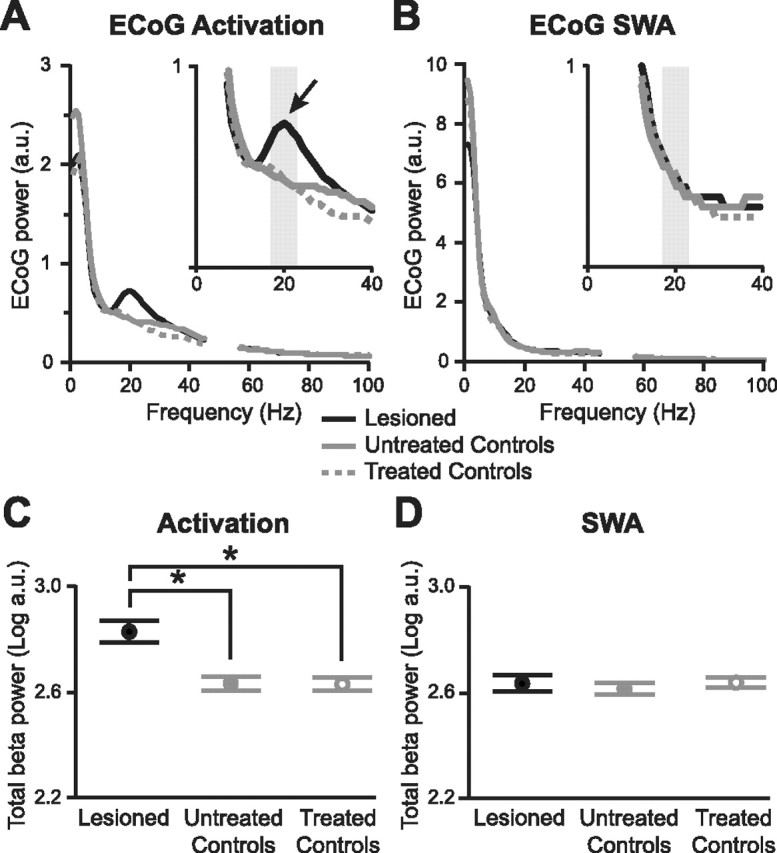
Chronic 6-OHDA lesions, but not acute antagonist treatment, exaggerate beta oscillations in the cerebral cortex. A, Average power spectra of ECoGs recorded in untreated control animals (n = 8), controls after acute antagonist treatment (n = 8, “treated controls”), and chronically lesioned animals (n = 9) during the activated brain state. The gray boxes indicate the peak beta band (17–23 Hz) used in quantitative analyses in C and D. Spectral power at ∼50 Hz (line noise) was removed for clarity. a.u., Arbitrary units. B, Average power spectra of ECoGs recorded in same animals during slow-wave activity. C, D, Average log power of peak beta oscillations (17–23 Hz) in ECoGs across experimental groups and brain states. The power values in A and B were normalized by logarithmic transformation before statistical testing. Data are means ±1 SEM. *p < 0.01. Note the small error bars (SEM) indicating limited variability between beta oscillations in different animals.
Having validated our experimental paradigm, we next addressed a key question: what is the neural basis and functional significance of these pathological beta oscillations? Cortical field activity, such as the ECoG, likely reflects the synchronized subthreshold activities and, to a lesser extent, suprathreshold activities of populations of principal neurons. Similar inferences have been made for LFPs recorded in the basal ganglia (Boraud et al., 2005), but the irregular, nonlaminated architecture of the STN makes interpretation of STN-LFPs more difficult. Prominent beta oscillations in LFPs were largely restricted to STN, compared with immediately above or below STN (Fig. 1C), suggesting a direct link between the recorded LFPs and STN neuron activity. We quantitatively tested for the focal expression of beta oscillations in STN (compared with the neighboring ventral zona incerta and cerebral peduncle) after averaging LFP power across frequencies, probe recording contacts in or around STN, and across lesioned animals during the activated brain state. Here, we averaged power across 17–23 Hz, thus concentrating on those frequency bins underlying the peak of beta activity in lesioned animals (see Figs. 1, 4A). Henceforth, for convenience, we will refer to this frequency range as the “peak beta” (βp) band. A one-way ANOVA confirmed position [three levels; “above STN” (in the ventral zona incerta, <200 μm from dorsal STN border), “within STN,” and “below STN” (in the cerebral peduncle, <200 μm from the ventral STN border)] as a main effect (p = 0.002). Post hoc paired t tests confirmed that the LFP power in the βp band in STN (mean log power, 2.766 ± 0.033) significantly exceeded that immediately above STN (2.617 ± 0.022; p = 0.002) and below STN (2.643 ± 0.023; p = 0.001).
The focality of the exaggerated beta oscillations in STN-LFPs suggests they are generated by events in local ensembles of STN neurons. To gain further insights into the neural basis of these pathological rhythms, we also studied the simultaneously recorded single-unit and multiunit activities. In lesioned rats, single STN neurons (n = 24; of which 14 were recorded with probes, and 10 with glass electrodes) typically fired either bursts of spikes or single spikes in time with many, but not necessarily all, cycles of the beta oscillations in LFPs (Fig. 1D,E). Accordingly, the spike trains of most STN neurons (14 of 24) contained a significant oscillatory component at ∼20 Hz (Fig. 1G), and significant coherence between single units and STN-LFPs peaked at similar beta frequencies (Fig. 1H). Rhythmic beta activity was not restricted to single STN neurons and, indeed, beta LFP oscillations were also reflected in periodic grouped discharges of multiunits within STN, but not immediately above or below STN (Fig. 1F). Importantly, and in keeping with LFP data, these characteristic single-unit and multiunit activities were not seen in the STN of control rats (Fig. 2). The spike trains of 6 of 18 STN neurons recorded in control rats (eight neurons recorded with probes, 10 with glass electrodes) contained significant oscillations, but these oscillations were outside the beta band (Fig. 1G). Coherence between single units and STN-LFPs in control rats did not peak at beta frequencies, and unit/LFP coherence in the βp band was significantly higher in lesioned rats compared with control rats (Fig. 1H). Moreover, on average, STN neurons in lesioned rats fired significantly faster than those in control rats (Fig. 1I). In lesioned animals, the mean firing rate of STN neurons that exhibited significant beta oscillations in their spike trains was similar to the mean firing rate of STN neurons that did not oscillate (34.0 ± 3.4 Hz and 29.5 ± 3.7 Hz, respectively; p = 0.4). For oscillating neurons, there was no clear relationship between the power of the spike train oscillation (as measured in the Lomb periodogram) and firing rate (R2 = 0.13; p = 0.19). In control animals, oscillatory STN neurons exhibited similar firing rates to nonoscillatory STN neurons (13.8 ± 2.7 Hz and 14.6 ± 2.0 Hz, respectively; p = 0.8). Together, these data suggest that, after 6-OHDA lesions, populations of STN neurons tend to engage in synchronized oscillatory firing at beta frequencies during the activated state, and that the postsynaptic membrane currents underlying this striking activity also summate in time and space to generate beta oscillations in STN-LFPs.
Pathological beta synchronization is spatially distributed and affects information coding within STN
The focal increases in βp power of STN-LFPs in chronically lesioned animals during global activation could arise through increased oscillatory activity of STN neurons in a local network of fixed size (together with a tighter temporal locking of oscillatory activity across that network) and/or through a spatial extension of an oscillating network to recruit more distributed neurons within STN. We have demonstrated increased oscillatory activity at the level of single STN neurons (Fig. 1G), thus providing some evidence supporting the former. Whether the latter also occurred was explicitly addressed by testing for increased coherence in the βp band between sites within STN in the lesioned animals relative to controls. To this end, we wanted electrical signals that were maximally representative of activity in local (restricted to <100 μm) regions of the STN, so we used multiunit activity rather than the monopolar LFPs recorded from each probe contact. There was a significant increase in coherence between multiunit activities recorded across STN sites (up to 300 μm apart) in chronically lesioned animals, compared with controls, and again, this coherence peaked at ∼20 Hz, and was limited to the βp band (Fig. 3A,B). The functional significance of this increased beta synchronization within STN may lie in the accompanying increase in the shared or mutual information coded by neurons in different regions of STN. Within the information theory framework, the total information capacity of a set of processing elements is related to the sum of the entropies of the individual elements less an amount that corresponds to the mutual information between elements. To quantify this, we derived lower-bound estimates of mutual information from the coherence between sites in the STN. There was an increase in the mutual information between sites in the STN of chronically lesioned animals compared with untreated controls (Fig. 3C). Thus, in 6-OHDA-lesioned rats, pathological beta oscillations in STN-LFPs reflect excessive and distributed synchronization of activity at similar frequencies between neurons, which may diminish the information coding capacity of processing channels within STN.
Figure 3.

Temporal coupling and mutual information of multiunit activities in the subthalamic nucleus increase after 6-OHDA lesions. A, Average coherence spectra of STN multiunit activities recorded in untreated control animals (n = 21 contact pairs), controls after acute antagonist treatment (n = 21 contact pairs, “treated controls”), and chronically lesioned animals (n = 23 contact pairs). The gray box indicates the beta band (17–23 Hz) used in quantitative analyses. B, Average coherence of STN activities at beta frequencies (17–23 Hz) across experimental groups. Data are means ±1 SEM. *p < 0.05. C, Mutual information between sites within the STN. Data are means ±1 SEM. *p < 0.05.
Only chronic disruption of dopamine transmission increases beta oscillations in STN-LFPs
How then does this pathological network state arise and become established? Are exaggerated beta oscillations the product of slowly evolving chronic adaptations to dopaminergic denervation, or is the acute disruption of dopamine transmission a sufficient cause? To address these questions, we recorded from dopamine-intact anesthetized control rats before and after systemic treatment with selective antagonists of dopamine D1-like receptors (SCH-23390; 0.5 mg/kg, s.c.) and D2-like receptors (raclopride; 2 mg/kg, i.p.). Group power spectra of STNLFPs averaged across recording contacts within STN and across animals showed that, during the activated brain state, a discrete peak in beta activity was only present in chronically lesioned animals, and that this peak centered around 20 Hz (Fig. 4A). Accordingly, during global activation, chronically lesioned animals had significantly more βp LFP power in STN than untreated controls (Fig. 4C) (p = 0.002, unpaired t test) or antagonist-treated controls (p = 0.001). Acute blockade of dopamine transmission in control animals did not result in the emergence of a distinct peak in beta activity, and there was no difference between STN-LFPs recorded before and after antagonist treatment in control animals (Figs. 2C, 4A,C) (p = 0.322). Moreover, during SWA, distinct beta peaks were not present in STN-LFPs, and βp LFP power was similar between the three experimental groups (Fig. 4B,D) (p > 0.1 for all comparisons). In summary, chronic but not acute disruption of dopamine transmission significantly potentiates beta oscillations in the STN, and this effect is restricted to the activated brain state. However, dopamine receptor blockade did affect activities at other frequencies. During global activation, acute disruption of dopamine transmission suppressed STN-LFP power across 26–48 Hz, and consequently, in this low gamma frequency band, treated control animals had significantly less activity than lesioned and untreated control animals (Fig. 4A) (log power suppressed to 96 and 97% of lesioned and untreated controls; both p < 0.02, unpaired and paired t tests, respectively).
Only chronic disruption of dopamine transmission increases beta oscillations in ECoGs
Group power spectra of frontal ECoGs averaged across animals showed that, during the activated brain state, a discrete peak in beta activity was again confined to chronically lesioned animals, and that this peak also centered around 20 Hz (Fig. 5A). During global activation, chronically lesioned animals had significantly more βp ECoG power than untreated controls (Fig. 5C) (p = 0.002, unpaired t test) or antagonist-treated animals (p = 0.001). There was no difference between ECoGs recorded before and after antagonist treatment in control animals (Figs. 2C, 5A,C) (p > 0.5, paired t test). Furthermore, during SWA, distinct beta peaks were not present, and ECoG power was similar between the three experimental groups (Fig. 5B,D) (p > 0.38 for all comparisons). In short, potentiation of cortical beta oscillations was restricted to chronically lesioned animals in the activated state, and therefore paralleled changes in the STN. Yet, as in the STN, dopamine receptor blockade was not without some effects. During activation, acute disruption of dopamine transmission suppressed ECoG activity across 26–48 Hz, such that both lesioned animals and untreated controls had significantly more activity than treated controls within this low gamma frequency band (Fig. 5A) (log power suppressed to 95% of lesioned and untreated controls, both p = 0.01, paired and unpaired t tests, respectively). Thus, dopamine receptor blockade induced similar changes in cortical and STN gamma activity.
Differences in βp power of STN-LFPs and ECoGs between experimental groups were not caused by relatively greater brain activation in chronically lesioned animals. Indeed, reductions of slow wave (1–2 Hz) ECoG power during global activation were similar in chronically lesioned animals, untreated controls, and antagonist-treated controls (mean log powers, −0.585 ± 0.080, −0.515 ± 0.037, and −0.520 ± 0.055; p > 0.5 for all comparisons). In summary, the exaggeration of beta oscillations in STN-LFPs and ECoGs in the activated state in chronically 6-OHDA-lesioned animals was unlikely to have simply resulted from an acute response to reduced dopamine transmission because it was absent in control animals treated with receptor antagonists during comparable degrees of brain activation.
Predictability of D1/D2 antagonist actions in anesthetized animals
To ensure that the failure of the receptor antagonists to induce exaggerated beta oscillations did not result from unpredicted or compromised drug actions under the anesthetic regimen used here, we tested the effects of antagonist administration on midbrain dopamine neurons. The activity of these neurons is partly controlled by local and extrinsic dopamine receptors, so they act as robust “biosensors” of agonist/antagonist actions. Indeed, activation of receptors with the mixed D1/D2 receptor agonist apomorphine decreases the firing of dopamine neurons (Bunney et al., 1973), whereas acute blockade of D1 or D2 receptors increases their activity (Carlson et al., 1986; Pucak and Grace, 1994; Mereu et al., 1995). We performed in vivo extracellular recordings of identified dopamine neurons (n = 3) in anesthetized animals (n = 3) during acute drug treatments. As expected, systemic administration of apomorphine (0.05 mg/kg, s.c.) led to a prolonged reduction in the activity of all dopamine neurons, which was consistently and rapidly attenuated by coadministration of SCH-23390 and raclopride (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Antagonist treatment also led to an increase in spontaneous activity, and the effects of a subsequent dose of apomorphine were blocked (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Together, these data indicate that, under this anesthetic regimen, SCH-23390 and raclopride effectively blocked dopamine receptors in the brain, and thus, acted as predicted.
Delayed exaggeration of beta oscillations after disruption of dopamine transmission in behaving animals
Chronic 6-OHDA lesions, but not acute treatment with selective D1 and D2 antagonists, reliably exaggerated beta oscillations in the frontal cortex of anesthetized animals. To further test the possibility that anesthesia might have occluded antagonist actions on beta activity, we recorded frontal ECoGs in behaving dopamine-intact animals before and after antagonist treatment (n = 5 rats) (Fig. 6A–C). As shown previously (Degos et al., 2005), systemic coadministration of SCH-23390 (0.5 mg/kg) and raclopride (2.0 mg/kg) induced severe catalepsy, with an onset of 10–15 min to a full-blown cataleptic state (Fig. 6C). However, despite this profound behavioral effect, antagonist treatment did not produce an exaggerated peak in ECoG power at beta frequencies (Fig. 6B). The peak frequency of abnormal beta oscillations previously recorded in behaving lesioned animals was 25–26 Hz (Sharott et al., 2005) (Fig. 6D), which is slightly higher than oscillations present during anesthesia. Thus, for statistical testing, the frequency band analyzed was shifted higher and wider (19–31 Hz) than that analyzed in anesthetized animals to safely accommodate the predicted peak frequency of beta oscillations during behavior. Quantitative analyses confirmed that neither antagonist nor saline vehicle treatment augmented cortical beta oscillations above untreated control levels (Fig. 6B,C) [two control periods combined, two saline periods combined, and two antagonist periods (at ∼20 and ∼25 min) were combined for analysis]. Instead, during catalepsy, there was a small but significant reduction in beta power compared with controls (power in controls and antagonist treated, 3.03 ± 0.15 × 10−4 mV2 and 2.77 ± 0.24 × 10−4 mV2, respectively; p = 0.02, one-way repeated-measures ANOVA and Bonferroni's t test). Together with our control studies of dopamine neurons, these experiments in behaving animals demonstrate that the inability of acute dopamine receptor blockade to exaggerate beta oscillations is unlikely to result from the possible confounding effects of anesthesia.
Figure 6.

Cortical beta oscillations are not augmented by acute antagonist treatments in behaving control animals, and pathological beta oscillations are delayed after 6-OHDA injections in lesioned animals. A, Right frontal ECoGs recorded in a representative dopamine-intact rat before (control) and after systemic administration of vehicle (saline) or a combination of selective D1 and D2 receptor antagonists (SCH-23390 at 0.5 mg/kg and raclopride at 2 mg/kg). Recording times after injections are indicated. Calibration: 0.5 mV, 250 ms. B, Power spectra of ECoGs recorded from the single animal in A, and average power spectra for the group of dopamine-intact rats (n = 5). Recording times after injections are indicated in parentheses (in minutes). The gray boxes and dashed lines indicate the peak beta band (22–28 Hz) and wider beta band (19–31 Hz), respectively, as used in quantitative analyses in C and D. Note that antagonist treatment did not augment beta oscillations. C, Total power of peak beta oscillations (22–28 Hz, gray) and wider beta oscillations (19–31 Hz, black) averaged across all dopamine-intact rats and treatments. Data are means ±1 SEM. Animals were fully cataleptic 10–15 min after administration of D1/D2 antagonists. D, Left, Average power spectra of ECoGs recorded ipsilateral to 6-OHDA lesions in behaving rats (n = 5) on postoperative days 1, 4, and 15. Right, Comparison of peak beta power (22–28 Hz) in ECoGs recorded from lesioned, behaving rats. Data are medians (black lines) and 25th/75th percentiles (boxes). *p < 0.05. E, Peak beta power (22–28 Hz) in ECoGs recorded from control behaving rats.
Our data are also important in implying that the emergence of pathological beta oscillations is appreciably delayed after lesion induction by 6-OHDA injection. To test this idea directly, we recorded frontal ECoGs on postoperative days 1, 4, and 15 from behaving rats (n = 5) that had received a 6-OHDA injection on day 0. The appearance of exaggerated beta oscillations was indeed delayed, insofar that a distinct spectral peak at beta frequencies was not evident in ECoGs recorded on postoperative days 1 and 4, but was clearly present on day 15 (Fig. 6D). The peak frequency of the exaggerated beta oscillations in these behaving lesioned rats was ∼25 Hz (Fig. 6D). As before, we then focused on ECoG activity around this peak and compared power in the peak beta band (22–28 Hz) across lesioned and control animals. Peak beta power in lesioned animals was significantly increased on day 15 compared with days 1 and 4 (Friedman's ANOVA and Student–Newman–Keuls tests) (Fig. 6D), as well compared with control dopamine-intact animals (Mann–Whitney test) (Fig. 6, compare D, E). Note that peak beta power (22–28 Hz) in control animals was unchanged by saline or antagonist administration (Fig. 6B,C). However, peak beta power in lesioned animals on day 1 was significantly reduced compared with control animals (Mann–Whitney test) (Fig. 6, compare D, E). Together, these data show that beta oscillations in behaving animals only became exaggerated several days after chronic disruption of dopamine transmission was initiated by 6-OHDA injections.
Discussion
We addressed the critical issue of how inappropriately synchronized beta-frequency (15–30 Hz) oscillations arise in corticobasal ganglia circuits in PD. Although exaggerated beta oscillations are attenuated by acute dopaminergic therapies, we found that chronic disruption of dopamine transmission was a prerequisite for their emergence. Thus, pathological amplification of beta oscillations in these circuits in PD is likely a consequence of plasticity induced by long-term progressive dopamine depletion rather than an acute network response to loss of dopamine transmission.
Clinical evidence suggests a functional association between exaggerated beta oscillations and disease symptoms. Moreover, these abnormal oscillations are acutely suppressed by dopamine-replacement therapies (Brown, 2006). As such, one might predict that they arise in PD through an acute pharmacological imbalance, that is, they rapidly appear because they are no longer “filtered out” by reduced dopamine. Here, we tested this important prediction, with notable results. We used a combination of potent and selective antagonists of D1-like and D2-like receptors to acutely disrupt dopamine transmission in intact animals. These are the principal receptor classes in the cortex and basal ganglia (Seamans and Yang, 2004; Surmeier et al., 2007) and, thus, it is expected that neuronal activity and behavior would be significantly altered after their blockade. Indeed, profound catalepsy developed shortly after antagonist administration (Degos et al., 2005). Importantly, however, acute disruption of dopamine transmission did not exaggerate cortical and STN beta oscillations, although still affecting activity at other frequencies. This finding has two major implications. First, although excessive beta synchronization might cause paucity or slowing of movement in PD (see below), these phenomena are not inextricably linked, insofar that an arguably similar motor deficit may occur in catalepsy without abnormal beta rhythms. Second, because acute antagonist treatment was ineffective at inducing excessive beta oscillations, they must instead arise through relatively slow and long-term adaptive and/or compensatory processes. Our recordings of cortical activity at several time points after 6-OHDA injections showed that beta oscillations are not exaggerated until >4 d after lesion. This agrees with data from our antagonist experiments, but is remarkable because striatal extracellular dopamine levels decrease to <20% within 1 h of midbrain 6-OHDA injections (Svenningsson et al., 1999). Yet, the relatively slow evolution of excessive beta synchronization is consistent with the gradual development of other pathophysiological hallmarks, e.g., significant losses of dopaminergic fibers from striatum or alterations in STN metabolic activity are only detectable 3 d after toxin injection (Vila et al., 2000). In short, the delayed emergence of pathological beta oscillations after 6-OHDA injections, or their absence after acute receptor blockade, does not parallel the rapid disruption of dopamine transmission that occurs in both cases, but rather correlates with the genetic, neurochemical, and structural changes that slowly ensue as compensatory or adaptive sequelae in PD and its chronic animal models (Zigmond et al., 1990; Surmeier et al., 2007). The delayed onset of abnormal beta oscillations in the 6-OHDA-lesioned rat model of PD may have behavioral parallels in other PD models, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated monkeys. Early motor deficits after acute MPTP toxicity are often dystonic in nature, whereas classic Parkinsonism may be delayed by one or more weeks after intoxication (Tabbal et al., 2006). This natural history is consistent with clinical experience: acute exposure to dopamine receptor antagonists may cause dystonia and related dyskinesias, but Parkinsonism requires more chronic neuroleptic use (Ayd, 1961).
Guided by clinical findings, we demonstrate here that chronic disruption of dopamine transmission after 6-OHDA lesions of midbrain dopamine neurons leads to a significant increase in the power of beta oscillations in the sensory–motor cortex and STN of anesthetized rats. The overt synchronization of oscillations was effectively limited to beta frequencies, although most other studies in animal models of PD have emphasized the emergence of abnormal oscillations with frequencies of ≤15 Hz (Bergman et al., 1998; Boraud et al., 2005; Gatev et al., 2006). Exaggerated beta oscillations were highly dynamic, and their emergence during anesthesia was brain-state dependent; they arose during global brain activity analogous to that accompanying the waking state. Our recordings in alert lesioned animals support this. Thus, the predominant pattern of oscillatory activity in this rodent model of PD accurately reflects that seen in unmedicated PD patients. Although the neural basis of cortical beta oscillations, as measured in LFPs, is reasonably well established (Rubino et al., 2006), this is not the case for STN-LFP oscillations, and particularly those in this rodent model. We found that abnormal beta oscillations were expressed in the STN of 6-OHDA-lesioned rats at the levels of single-units, multiunit ensembles, and LFPs. Individual STN neurons could fire a single spike or a burst of spikes in a phase-locked manner during every oscillatory LFP cycle, or just a proportion of cycles. These rodent data extend and agree with previous findings in PD patients (Kuhn et al., 2005; Weinberger et al., 2006). Thus, after chronic 6-OHDA lesions, populations of STN neurons tend to engage in synchronized oscillatory firing at beta frequencies. The underlying postsynaptic currents most likely summate and generate beta oscillations in STN-LFPs. Our simultaneous recordings of activity in and around STN directly demonstrate that pathological beta oscillations are focally expressed within STN (compared with adjacent structures), thus supporting inferences from previous clinical studies (Chen et al., 2006; Weinberger et al., 2006). Subthalamic nucleus neurons are intrinsic pacemakers, but the stereotypical unit activity we recorded in lesioned rats has not been described ex vivo (Bevan et al., 2002). Although adaptive processes within STN cannot be disregarded, it thus seems likely that intact STN afferents are critical for orchestrating excessive beta oscillations generated therein. Our ECoG data highlight a role for direct cortical inputs (Magill et al., 2004), but trans-striatal influences via the external globus pallidus cannot be discounted (Bevan et al., 2002), and future studies should investigate these candidate mechanisms.
Beta oscillations may subserve intracortical and intercortical information processing during normal movement preparation and execution (Rubino et al., 2006). Given that the cortex is the primary extrinsic afferent of the basal ganglia, the same may hold true for information processing within and between corticobasal ganglia circuits. In primates, beta power in the cortex and STN decreases during movement preparation (or might increase, depending on the postural configuration), attenuates further at self-paced or externally paced movement initiation, and increases with movement termination (Amirnovin et al., 2004; Kuhn et al., 2004; Doyle et al., 2005; Williams et al., 2005; Devos et al., 2006; Rubino et al., 2006). Under normal conditions, with dopaminergic innervation intact, appropriately synchronized beta oscillations (at low levels) may thus be advantageous for sensory–motor information processing and task performance (MacKay, 1997; Baker et al., 1999; Uhlhaas and Singer, 2006). However, in the case of PD and associated motor symptoms, particularly akinesia/bradykinesia, excessive beta synchronization may be counterproductive or truly pathological (Brown, 2006; Uhlhaas and Singer, 2006). But how might pathological beta synchronization actually impair motor processing? Assuming that the cortex (and basal ganglia) uses a neural code in which the precise timing of outputs is, or can be, fully used by targeted neurons (Boraud et al., 2005), then during synchronized activity, the potential information encoded by a neuronal population will be smaller than the sum of information encoded by its single elements because of the mutual information shared between neurons (Averbeck et al., 2006). In extreme cases, such as that of the STN in PD, synchronization across a neuronal population can reduce distinct information carrying channels to unity. We found that the mutual information between STN neuronal ensembles was significantly increased in lesioned animals compared with controls. This reduced capacity for information encoding in the STN could underlie dysfunction. Attenuation of information through pathological synchrony will also affect STN targets, because synchronized activity is preferentially propagated at postsynaptic sites through temporal summation. In line with this, beta oscillations in the STN are coherent with those in the output nuclei and cortex in both the 6-OHDA-lesioned rat model (Sharott et al., 2005) and PD patients (Brown, 2006). The net result is that the pathological beta synchronization evident in our rodent model of PD, and PD patients, will impair information coding capacity in cortical-basal ganglia circuits during motor processing. However, the firing rates of STN neurons also significantly increased after 6-OHDA lesions, suggesting that rate changes may also be of great importance in Parkinsonism.
To conclude, the 6-OHDA-lesioned rat model of PD is now well placed to give further insights into the mechanisms underlying the exaggerated beta oscillations seen in idiopathic PD, and will further aid the identification of novel therapeutic targets. When pathological beta oscillations become established through chronic dopamine loss, acute drug therapies can attenuate them, with concomitant relief from movement symptoms (Brown, 2006). The abnormal beta activity exhibited by this model thus offers a clinically relevant biomarker that can be exploited in drug discovery. Moreover, because the emergence of pathological beta oscillations is appreciably delayed after the initial insult to dopamine neurons, there is a window of opportunity to prevent the underling plasticity through alternative disease-modifying strategies and counteract the evolving beta oscillations for symptomatic benefit.
Footnotes
This work was supported by the Medical Research Council (United Kingdom) and The Dana Foundation. A.S. was supported by the European Union Marie Curie Program. We are grateful to N. Maurice for advice on the use of receptor antagonists, to B. Averbeck for insightful comments on mutual information analysis, and to T. Klausberger, M. Brown, T. Senior, and K. Allen for their valuable input throughout. We also thank E. Norman, C. Francis, K. Whitworth, and B. Micklem ARPS for expert technical assistance.
References
- Amirnovin R, Williams ZM, Cosgrove GR, Eskandar EN. Visually guided movements suppress subthalamic oscillations in Parkinson's disease patients. J Neurosci. 2004;24:11302–11306. doi: 10.1523/JNEUROSCI.3242-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averbeck BB, Latham PE, Pouget A. Neural correlations, population coding and computation. Nat Rev Neurosci. 2006;7:358–366. doi: 10.1038/nrn1888. [DOI] [PubMed] [Google Scholar]
- Ayd FJ., Jr A survey of drug-induced extrapyramidal reactions. JAMA. 1961;175:1054–1060. doi: 10.1001/jama.1961.03040120016004. [DOI] [PubMed] [Google Scholar]
- Baker SN, Kilner JM, Pinches EM, Lemon RN. The role of synchrony and oscillations in the motor output. Exp Brain Res. 1999;128:109–117. doi: 10.1007/s002210050825. [DOI] [PubMed] [Google Scholar]
- Bergman H, Feingold A, Nini A, Raz A, Slovin H, Abeles M, Vaadia E. Physiological aspects of information processing in the basal ganglia of normal and parkinsonian primates. Trends Neurosci. 1998;21:32–38. doi: 10.1016/s0166-2236(97)01151-x. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ. Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci. 2002;25:525–531. doi: 10.1016/s0166-2236(02)02235-x. [DOI] [PubMed] [Google Scholar]
- Boraud T, Brown P, Goldberg JA, Graybiel AM, Magill PJ. Oscillations in the basal ganglia: The good, the bad and the unexpected. In: Bolam JP, Ingham CA, Magill PJ, editors. The basal ganglia Vol VIII. New York: Springer Science and Business Media; 2005. pp. 3–24. [Google Scholar]
- Borst A, Theunissen FE. Information theory and neural coding. Nat Neurosci. 1999;2:947–957. doi: 10.1038/14731. [DOI] [PubMed] [Google Scholar]
- Brown P. Bad oscillations in Parkinson's disease. J Neural Transm. 2006;70(Suppl):27–30. doi: 10.1007/978-3-211-45295-0_6. [DOI] [PubMed] [Google Scholar]
- Brown P, Oliviero A, Mazzone P, Insola A, Tonali P, Di Lazzaro V. Dopamine dependency of oscillations between subthalamic nucleus and pallidum in Parkinson's disease. J Neurosci. 2001;21:1033–1038. doi: 10.1523/JNEUROSCI.21-03-01033.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunney BS, Aghajanian GK, Roth RH. Comparison of effects of L-dopa, amphetamine and apomorphine on firing rate of rat dopaminergic neurones. Nat New Biol. 1973;245:123–125. doi: 10.1038/newbio245123a0. [DOI] [PubMed] [Google Scholar]
- Burkhardt JM, Constantinidis C, Anstrom KK, Roberts DC, Woodward DJ. Synchronous oscillations and phase reorganization in the basal ganglia during akinesia induced by high-dose haloperidol. Eur J Neurosci. 2007;26:1912–1924. doi: 10.1111/j.1460-9568.2007.05813.x. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Carlson JH, Bergstrom DA, Walters JR. Neurophysiological evidence that D-1 dopamine receptor blockade attenuates postsynaptic but not autoreceptor-mediated effects of dopamine agonists. Eur J Pharmacol. 1986;123:237–251. doi: 10.1016/0014-2999(86)90665-5. [DOI] [PubMed] [Google Scholar]
- Chen CC, Pogosyan A, Zrinzo LU, Tisch S, Limousin P, Ashkan K, Yousry T, Hariz MI, Brown P. Intra-operative recordings of local field potentials can help localize the subthalamic nucleus in Parkinson's disease surgery. Exp Neurol. 2006;198:214–221. doi: 10.1016/j.expneurol.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Chen CC, Litvak V, Gilbertson T, Kuhn A, Lu CS, Lee ST, Tsai CH, Tisch S, Limousin P, Hariz M, Brown P. Excessive synchronization of basal ganglia neurons at 20 Hz slows movement in Parkinson's disease. Exp Neurol. 2007;205:214–221. doi: 10.1016/j.expneurol.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Costa RM, Lin SC, Sotnikova TD, Cyr M, Gainetdinov RR, Caron MG, Nicolelis MA. Rapid alterations in corticostriatal ensemble coordination during acute dopamine-dependent motor dysfunction. Neuron. 2006;52:359–369. doi: 10.1016/j.neuron.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Curran-Everett D. Multiple comparisons: philosophies and illustrations. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1–R8. doi: 10.1152/ajpregu.2000.279.1.R1. [DOI] [PubMed] [Google Scholar]
- Degos B, Deniau JM, Thierry AM, Glowinski J, Pezard L, Maurice N. Neuroleptic-induced catalepsy: electrophysiological mechanisms of functional recovery induced by high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2005;25:7687–7696. doi: 10.1523/JNEUROSCI.1056-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Szurhaj W, Reyns N, Labyt E, Houdayer E, Bourriez JL, Cassim F, Krystkowiak P, Blond S, Destee A, Derambure P, Defebvre L. Predominance of the contralateral movement-related activity in the subthalamo-cortical loop. Clin Neurophysiol. 2006;117:2315–2327. doi: 10.1016/j.clinph.2006.06.719. [DOI] [PubMed] [Google Scholar]
- Doyle LM, Kuhn AA, Hariz M, Kupsch A, Schneider GH, Brown P. Levodopa-induced modulation of subthalamic beta oscillations during self-paced movements in patients with Parkinson's disease. Eur J Neurosci. 2005;21:1403–1412. doi: 10.1111/j.1460-9568.2005.03969.x. [DOI] [PubMed] [Google Scholar]
- Gatev P, Darbin O, Wichmann T. Oscillations in the basal ganglia under normal conditions and in movement disorders. Mov Disord. 2006;21:1566–1577. doi: 10.1002/mds.21033. [DOI] [PubMed] [Google Scholar]
- Halliday DM, Rosenberg JR, Amjad AM, Breeze P, Conway BA, Farmer SF. A framework for the analysis of mixed time series/point process data–theory and application to the study of physiological tremor, single motor unit discharges and electromyograms. Prog Biophys Mol Biol. 1995;64:237–278. doi: 10.1016/s0079-6107(96)00009-0. [DOI] [PubMed] [Google Scholar]
- Hammond C, Bergman H, Brown P. Pathological synchronization in Parkinson's disease: networks, models and treatments. Trends Neurosci. 2007;30:357–364. doi: 10.1016/j.tins.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Kuhn AA, Williams D, Kupsch A, Limousin P, Hariz M, Schneider GH, Yarrow K, Brown P. Event-related beta desynchronization in human subthalamic nucleus correlates with motor performance. Brain. 2004;127:735–746. doi: 10.1093/brain/awh106. [DOI] [PubMed] [Google Scholar]
- Kuhn AA, Trottenberg T, Kivi A, Kupsch A, Schneider GH, Brown P. The relationship between local field potential and neuronal discharge in the subthalamic nucleus of patients with Parkinson's disease. Exp Neurol. 2005;194:212–220. doi: 10.1016/j.expneurol.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Kuhn AA, Kupsch A, Schneider GH, Brown P. Reduction in subthalamic 8–35 Hz oscillatory activity correlates with clinical improvement in Parkinson's disease. Eur J Neurosci. 2006;23:1956–1960. doi: 10.1111/j.1460-9568.2006.04717.x. [DOI] [PubMed] [Google Scholar]
- Levy R, Ashby P, Hutchison WD, Lang AE, Lozano AM, Dostrovsky JO. Dependence of subthalamic nucleus oscillations on movement and dopamine in Parkinson's disease. Brain. 2002;125:1196–1209. doi: 10.1093/brain/awf128. [DOI] [PubMed] [Google Scholar]
- MacKay WA. Synchronised neuronal oscillations and their role in motor processes. Trends Cogn Sci. 1997;1:176–183. doi: 10.1016/S1364-6613(97)01059-0. [DOI] [PubMed] [Google Scholar]
- Magill PJ, Bolam JP, Bevan MD. Dopamine regulates the impact of the cerebral cortex on the subthalamic nucleus-globus pallidus network. Neuroscience. 2001;106:313–330. doi: 10.1016/s0306-4522(01)00281-0. [DOI] [PubMed] [Google Scholar]
- Magill PJ, Sharott A, Bevan MD, Brown P, Bolam JP. Synchronous unit activity and local field potentials evoked in the subthalamic nucleus by cortical stimulation. J Neurophysiol. 2004;92:700–714. doi: 10.1152/jn.00134.2004. [DOI] [PubMed] [Google Scholar]
- Magill PJ, Pogosyan A, Sharott A, Csicsvari J, Bolam JP, Brown P. Changes in functional connectivity within the rat striatopallidal axis during global brain activation in vivo. J Neurosci. 2006;26:6318–6329. doi: 10.1523/JNEUROSCI.0620-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu G, Lilliu V, Vargiu P, Muntoni AL, Diana M, Gessa GL. Depolarization inactivation of dopamine neurons: an artifact? J Neurosci. 1995;15:1144–1149. doi: 10.1523/JNEUROSCI.15-02-01144.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oorschot DE. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the cavalieri and optical disector methods. J Comp Neurol. 1996;366:580–599. doi: 10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 2. Sydney: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- Priori A, Foffani G, Pesenti A, Tamma F, Bianchi AM, Pellegrini M, Locatelli M, Moxon KA, Villani RM. Rhythm-specific pharmacological modulation of subthalamic activity in Parkinson's disease. Exp Neurol. 2004;189:369–379. doi: 10.1016/j.expneurol.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Pucak ML, Grace AA. Evidence that systemically administered dopamine antagonists activate dopamine neuron firing primarily by blockade of somatodendritic autoreceptors. J Pharmacol Exp Ther. 1994;271:1181–1192. [PubMed] [Google Scholar]
- Rubino D, Robbins KA, Hatsopoulos NG. Propagating waves mediate information transfer in the motor cortex. Nat Neurosci. 2006;9:1549–1557. doi: 10.1038/nn1802. [DOI] [PubMed] [Google Scholar]
- Schwarting RK, Huston JP. Unilateral 6-hydroxydopamine lesions of meso-striatal dopamine neurons and their physiological sequelae. Prog Neurobiol. 1996a;49:215–266. doi: 10.1016/s0301-0082(96)00015-9. [DOI] [PubMed] [Google Scholar]
- Schwarting RK, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol. 1996b;50:275–331. doi: 10.1016/s0301-0082(96)00040-8. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Sharott A, Magill PJ, Harnack D, Kupsch A, Meissner W, Brown P. Dopamine depletion increases the power and coherence of beta-oscillations in the cerebral cortex and subthalamic nucleus of the awake rat. Eur J Neurosci. 2005;21:1413–1422. doi: 10.1111/j.1460-9568.2005.03973.x. [DOI] [PubMed] [Google Scholar]
- Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 2000;101:243–276. doi: 10.1016/s0306-4522(00)00353-5. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Fourreau L, Bloch B, Fredholm BB, Gonon F, Le Moine C. Opposite tonic modulation of dopamine and adenosine on c-fos gene expression in striatopallidal neurons. Neuroscience. 1999;89:827–837. doi: 10.1016/s0306-4522(98)00403-5. [DOI] [PubMed] [Google Scholar]
- Tabbal SD, Mink JW, Antenor JA, Carl JL, Moerlein SM, Perlmutter JS. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced acute transient dystonia in monkeys associated with low striatal dopamine. Neuroscience. 2006;141:1281–1287. doi: 10.1016/j.neuroscience.2006.04.072. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006;52:155–168. doi: 10.1016/j.neuron.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Vila M, Perier C, Feger J, Yelnik J, Faucheux B, Ruberg M, Raisman-Vozari R, Agid Y, Hirsch EC. Evolution of changes in neuronal activity in the subthalamic nucleus of rats with unilateral lesion of the substantia nigra assessed by metabolic and electrophysiological measurements. Eur J Neurosci. 2000;12:337–344. doi: 10.1046/j.1460-9568.2000.00901.x. [DOI] [PubMed] [Google Scholar]
- Weinberger M, Mahant N, Hutchison WD, Lozano AM, Moro E, Hodaie M, Lang AE, Dostrovsky JO. Beta oscillatory activity in the subthalamic nucleus and its relation to dopaminergic response in Parkinson's disease. J Neurophysiol. 2006;96:3248–3256. doi: 10.1152/jn.00697.2006. [DOI] [PubMed] [Google Scholar]
- Williams D, Tijssen M, Van Bruggen G, Bosch A, Insola A, Di Lazzaro V, Mazzone P, Oliviero A, Quartarone A, Speelman H, Brown P. Dopamine-dependent changes in the functional connectivity between basal ganglia and cerebral cortex in humans. Brain. 2002;125:1558–1569. doi: 10.1093/brain/awf156. [DOI] [PubMed] [Google Scholar]
- Williams D, Kuhn A, Kupsch A, Tijssen M, van Bruggen G, Speelman H, Hotton G, Loukas C, Brown P. The relationship between oscillatory activity and motor reaction time in the parkinsonian subthalamic nucleus. Eur J Neurosci. 2005;21:249–258. doi: 10.1111/j.1460-9568.2004.03817.x. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]