

Abstract
Alzheimer's disease is characterized by the accumulation of neurotoxic amyloidogenic peptide Aβ, degeneration of the cholinergic innervation to the hippocampus (the septohippocampal pathway), and progressive impairment of cognitive function, particularly memory. Aβ is a ligand for the p75 neurotrophin receptor (p75NTR), which is best known for mediating neuronal death and has been consistently linked to the pathology of Alzheimer's disease. Here we examined whether p75NTR is required for Aβ-mediated effects. Treatment of wild-type but not p75NTR-deficient embryonic mouse hippocampal neurons with human Aβ1–42 peptide induced significant cell death. Furthermore, injection of Aβ1–42 into the hippocampus of adult mice resulted in significant degeneration of wild-type but not p75NTR-deficient cholinergic basal forebrain neurons, indicating that the latter are resistant to Aβ-induced toxicity. We also found that neuronal death correlated with Aβ1–42 peptide-stimulated accumulation of the death-inducing p75NTR C-terminal fragment generated by extracellular metalloprotease cleavage of full-length p75NTR. Although neuronal death was prevented in the presence of the metalloprotease inhibitor TAPI-2 (tumor necrosis factor-α protease inhibitor-2), Aβ1–42-induced accumulation of the C-terminal fragment resulted from inhibition of γ-secretase activity. These results provide a novel mechanism to explain the early and characteristic loss of cholinergic neurons in the septohippocampal pathway that occurs in Alzheimer's disease.
Keywords: p75NTR, Alzheimer's disease, basal forebrain, cholinergic neurons, regulated intramembrane proteolysis (RIP), apoptosis, neurodegeneration
Introduction
Alzheimer's disease is a neurodegenerative disorder characterized by senile plaque pathology, neurofibrillary tangles, and neuronal death, with resultant impairment of memory and cognitive function. Amyloid β protein (Aβ), the major component of senile plaques, is a 39–43 aa peptide produced by cleavage of the amyloid protein precursor. According to the “amyloid hypothesis,” accumulation of soluble Aβ in the brain is the primary cause of the pathogenesis of Alzheimer's disease (Hardy and Selkoe, 2002), but with Aβ oligomers rather than amyloid deposits being the cause of the neurotoxicity associated with this condition (Dahlgren et al., 2002; Klein et al., 2004; Gandy, 2005).
Loss of basal forebrain cholinergic neurons that innervate the hippocampus and neocortex is an early and key feature of Alzheimer's disease (Yan and Feng, 2004; Wu et al., 2005). These cholinergic neurons express high levels of the p75 neurotrophin receptor (p75NTR), which mediates cell death in a wide range of neuronal subtypes (Dechant and Barde, 2002). p75NTR expression has been consistently linked to changes occurring in Alzheimer's disease (Schliebs, 2005), including the death of basal forebrain neurons (Yeo et al., 1997; Volosin et al., 2006). Like the amyloid protein precursor (APP), the extracellular domain of p75NTR is cleaved by a metalloprotease, generating a transmembrane-linked C-terminal fragment. This is then cleaved by γ-secretase, liberating a soluble intracellular domain (Jung et al., 2003). Ligand binding can initiate this process, and both fragments of p75NTR have been shown to mediate death signaling (Kenchappa et al., 2006; Podlesniy et al., 2006; Coulson et al., 2008; Underwood et al., 2008).
The classic p75NTR ligands are the neurotrophins nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3. These are secreted as immature forms (proneurotrophins), which are sometimes cleaved to produce the mature forms (Fahnestock et al., 2001). It has previously been proposed that increased levels of proneurotrophins, such as occur in Alzheimer's disease, may underlie cholinergic neuronal degeneration (Fahnestock et al., 2001; Volosin et al., 2006). However, it is also possible that Aβ could be directly responsible for activating p75NTR-mediated cell death in Alzheimer's disease, given that it is a ligand for the receptor (Yaar et al., 1997) and can stimulate p75NTR-mediated death signaling cascades in cell lines (Costantini et al., 2005; Coulson, 2006). In this study, we provide evidence of a direct link between p75NTR signaling and Aβ-induced toxicity in hippocampal neurons in vitro and in cholinergic basal forebrain neurons in vivo.
Materials and Methods
Aβ preparation.
Human Aβ1–42 peptide and control human Aβ1–16 peptide were purchased from Dr. J Elliot (Yale University, New Haven, CT). Unless otherwise stated, peptides were dissolved in sterile water at a concentration of 200 μm and stored at 4°C (Yaar et al., 1997). Peptide solutions were incubated at 37°C for 1 h before experimental use and were found to occur in a predominantly oligomeric form (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), which was more toxic to hippocampal neurons than the fibrillar form (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material).
Hippocampal cultures.
All animal experiments were approved by the University of Queensland Animal Ethics Committee. When examining the effects of genotype, experiments were performed in parallel. Hippocampi were dissected from embryonic day 16 (E16) wild-type and p75NTR-knock-out (Lee et al., 1992) C57BL/6 mice. They were then dissociated in 0.05% trypsin (Invitrogen, Melbourne, Australia), plated on 35 mm dishes coated with poly-l-lysine (0.1 mg/ml; Sigma-Aldrich, Sydney, Australia), and cultured for 24 h in a 1:1 mixture of minimal essential medium (Invitrogen) and Ham's F-12 medium (Invitrogen) supplemented with NeuroCult (StemCell Technologies, Vancouver, British Columbia, Canada). Approximately 95% of cells within the cultures were p75NTR-positive neurons (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). Live cells within a defined grid were then counted (t = 0) under phase microscopy (n = 4 wells per condition per experiment). Aβ peptides or inhibitors were dissolved in fresh culture medium and applied after the t = 0 cell count. The final concentrations were 5 or 20 μm Aβ peptides, a 1:100 dilution of anti-extracellular p75NTR antibody (Ab1554; Millipore Bioscience Research Reagents, Melbourne, Australia), 200 nm compound E γ-secretase inhibitor (Calbiochem, Melbourne, Australia), and 20 μm tumor necrosis factor-α protease inhibitor-2 (TAPI-2; Peptides International, Louisville, KY). In some experiments using the metalloprotease inhibitor TAPI-2, a second 20 μm dose of TAPI-2 was added into the culture medium at t = 6 h. Twenty-four hours after Aβ treatment, the viability of the same neurons was again assessed, and the percentage survival was determined.
p75NTR cleavage in hippocampal neurons.
To determine the effect of Aβ treatment on the proteolysis of p75NTR, hippocampal neurons plated in six-well plates for 24 h were pretreated with 5 μm β-clasto-lactacystin (Calbiochem) proteasome inhibitor for 90 min, after which they were treated for 3 h with a mixture of 20 μm Aβ1–42, 200 nm compound E, and/or 200 nm phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich). Cell lysates were separated using 4–20% Tris-glycine gradient gels (Invitrogen) and Western blotted using rabbit anti-human p75NTR intracellular domain (1:2000; Promega, Sydney, Australia). Immunoreactive bands were quantified using ImageJ software. The amount of C-terminal fragment was determined as a ratio to full-length p75NTR within each lane.
DNA constructs.
Full-length p75NTR rat cDNA was as previously described (Coulson et al., 2000). A p75NTR C-terminal fragment mimic that is constitutively cleaved (ΔE14) was made as described by Jung et al. (2003). The C99 APP plasmid contained Gal4 DNA binding and VP16 transactivation domains fused to the carboxyl end (C99-Gal4) (Sernee et al., 2003). A ΔE14 construct was similarly modified (ΔE14-Gal4) using standard methods. The luciferase reporter gene construct (pUAS) and the constitutively active plasmid (pCMV-Gal) were as previously described (Karlstrom et al., 2002).
p75NTR cleavage in HEK293 cells.
The effect of Aβ treatment on the proteolysis of p75NTR was determined using human embryonic kidney 293 (HEK293) cells grown in RPMI (Roswell Park Memorial Institute) medium and 10% fetal bovine serum. Cells were plated at 7.5 × 104 in 24-well plates and transiently transfected with 1 μg of plasmid DNA using Fugene6 (Roche, Basel, Switzerland) per well. Aβ1–42 (16 μm) was added to the cells either 48 (overnight treatment) or 52 (3 h treatment) hours after transfection. Before cell harvest, cells were treated with β-clasto-lactacystin, compound E, and PMA as described above. Cell lysates were Western blotted and the results were analyzed as described above.
Luciferase cleavage assay.
For luciferase experiments, HEK293 cells were plated and transfected with the pUAS plasmid together with the ΔE14-Gal4, C99-Gal4, or pCMV-Gal plasmid. The cells were then incubated overnight and/or for 3 h in Aβ or compound E, before being harvested in reporter lysis buffer. Lysis supernatant (20 μl) was assessed using the Luciferase Activity System according to the manufacturer's instructions (Promega). In each experiment, three to six replica lysates were analyzed. The total protein in each lysate was also quantified using a BCA assay kit (Pierce, Rockford, IL).
Aβ injections.
Eight-week-old wild-type and p75NTR knock-out C57BL/6 mice were anesthetized with 8 mg/kg xylazine and 80 mg/kg ketamine by intraperitoneal injection and mounted in a stereotaxic frame. The skull was exposed and a burr hole made at stereotaxic coordinates measured from bregma (anteroposterior, −2.0 mm; mediolateral, −1.3 mm; ventral, −2.2 mm), through which 0.5 μl of saline or 100 μg/ml Aβ was injected into the CA1 region of the hippocampus at a rate of 0.5 μl/min. The needle was kept in this configuration for 2 min to prevent reflux of the injected material along the injection track. In cases in which mice were administered bilateral injections, they received Aβ1–16 ipsilaterally and saline contralaterally, or Aβ1–42 ipsilaterally and Aβ1–16 contralaterally. Animals were perfused with 4% paraformaldehyde 14 d after injection.
Histology.
To determine cell survival of cholinergic basal forebrain neurons, the rostral half of each brain between −1.4 and −0.5 mm from bregma (the medial septum and diagonal band of Broca) was cut by vibratome into 15 60-μm-thick serial coronal sections. Floating sections were stained with a rabbit anti-choline acetyltransferase (ChAT) polyclonal antibody (1:500; Ab143; Millipore Bioscience Research Reagents), and all ChAT-positive neurons in the area of interest were counted based on the method of Greferath et al. (2000).
Statistical analysis.
The data were analyzed by ANOVA using the Newman–Keuls multiple-comparison test. All results are expressed as mean ± SD.
Results
To investigate whether p75NTR is required for Aβ-induced neuronal death, we first established an in vitro model of toxicity in E16 embryonic hippocampal neuron cultures, which have been found to be independently sensitive to both Aβ- (Ueda et al., 1994) and p75NTR-mediated death (Troy et al., 2002). We found that 24 h after plating in serum-free medium, neurons were sensitive to an overnight incubation with oligomeric 5 or 20 μm Aβ1–42 but not 20 μm Aβ1–16 or vehicle (Fig. 1A).
Figure 1.

Aβ1–42-induced toxicity of hippocampal neurons requires p75NTR. A, The percentage survival of cells after treatment with Aβ1–42 or control peptide Aβ1–16. Wild-type (WT) but not p75NTR-deficient (KO) neurons were sensitive to Aβ1–42-induced toxicity. Aβ1–16 had no effect on neuronal survival. B, Treatment of wild-type hippocampal neuronal cultures with a p75NTR-blocking antibody (Ab1554) significantly inhibited Aβ1–42 toxicity. n = 4 experiments. *p < 0.05; ***p < 0.001.
To test whether p75NTR was required for the observed neuronal death, the effect of Aβ1–42 on hippocampal neurons isolated from p75NTR-deficient mice was determined. In contrast to the results obtained with wild-type neurons, Aβ1–42 failed to induce the death of p75NTR-deficient neurons even at 20 μm (Fig. 1A), indicating that p75NTR is required for Aβ-induced death in vitro. An extracellular antibody to p75NTR also significantly inhibited Aβ1–42-induced hippocampal cell death (Fig. 1B).
Given that Aβ is a known ligand for p75NTR, the effect of Aβ1–42 on p75NTR proteolytic processing was next examined. Hippocampal neurons were treated for 3 h with 20 μm Aβ1–42 before analysis by Western blot (Fig. 2A). This analysis revealed that neurons exposed to Aβ1–42 contained significantly more p75NTR C-terminal fragment than control neurons (Fig. 2B), the amount being equivalent to that found when intracellular cleavage was blocked with compound E, a γ-secretase inhibitor, for the same period of time (Fig. 2A,B). An intracellular domain fragment of p75NTR was not apparent. To determine whether cleavage was required for death signaling, extracellular cleavage was blocked with the metalloprotease inhibitor TAPI-2 (Jung et al., 2003). This completely inhibited Aβ1–42- but not staurosporine-induced neuronal death in a dose-dependent manner (Fig. 2C,D). In contrast, compound E treatment resulted in a slight increase in Aβ-induced death (Fig. 2E).
Figure 2.
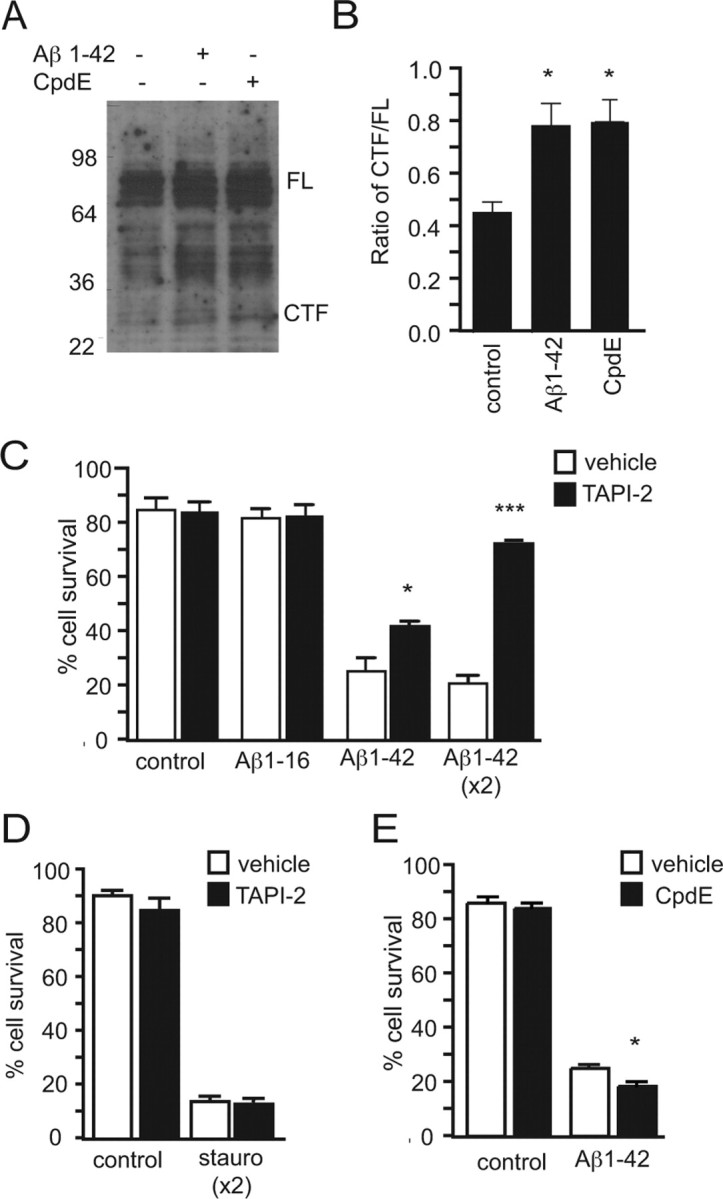
Aβ1–42 toxicity regulates and requires extracellular cleavage of p75NTR. A, Anti-p75NTR Western blots of hippocampal neuronal cultures treated for 3 h with Aβ1–42 and the γ-secretase inhibitor compound E (CpdE). All conditions contain β-clasto-lactacystin. B, These treatments result in a significant increase in the amount of C-terminal fragment (CTF) as a ratio of full-length (FL) p75NTR as quantified by densitometry. C, D, Aβ1–42 toxicity (C) but not staurosporine (stauro) toxicity (D) is significantly inhibited after treatment with the metalloprotease inhibitor TAPI-2. x2, Double TAPI-2 treatment. n = 3 experiments. E, Aβ1–42 toxicity is promoted in the presence of CpdE. n = 2 experiments. *p < 0.05; ***p < 0.001.
To further examine the effect of Aβ on p75NTR cleavage, p75NTR was expressed in HEK293 fibroblast cells. Overnight treatment [but not 3 h treatment (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material)] with Aβ1–42 resulted in a significant increase in the amount of C-terminal fragment (Fig. 3A,B). However, in contrast to phorbol ester (PMA) treatment, there was less generation of the intracellular domain fragment of p75NTR (Fig. 3C). Interestingly, when the two treatments were combined, the resulting cleavage appeared cumulative (Fig. 3 A–C; supplemental Fig. 3A, available at www.jneurosci.org as supplemental material), suggesting that their actions were independent, possibly acting on different subcellular pools of p75NTR. Because increased amounts of C-terminal fragment could result from either increased metalloprotease activity or reduced γ-secretase activity, and coexpression of an APP γ-secretase substrate (C99) together with p75NTR also significantly promoted accumulation of the p75NTR C-terminal fragment (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material) (Urra et al., 2007), we hypothesized that Aβ1–42 might be competing with p75NTR for γ-secretase activity, rather than, or as well as, acting as a p75NTR ligand to promote extracellular cleavage.
Figure 3.

Aβ1–42 inhibits γ-secretase cleavage of p75NTR. A–C, Western blot (A) and quantification of the amount of the C-terminal fragment (CTF; B) and intracellular domain fragments (ICD; C) of p75NTR in lysates of HEK293 cells transfected with full-length (FL) p75NTR before overnight treatment with Aβ1–42 and/or 3 h treatment with PMA and/or compound E (CpdE). All cultures were treated with β-clasto-lactacystin. D, Western blot of p75NTR C-terminal fragment mimic ΔE14 in lysates of transfected HEK293 cells after overnight treatment with Aβ1–42 or 3 h treatment with CpdE or PMA. E, F, Luciferase activity in lysates of HEK293 cells coexpressing ΔE14-Gal4 (E) or C99-Gal4 (F) together with pUAS (black bars) after treatment with Aβ1–16, Aβ1–42, or CpdE. Gray bars indicate positive-control (pCMV-Gal- and pUAS-transfected cells) and negative-control (cells transfected with pUAS alone) conditions. The total amount of total protein in lysates (white bars; in micrograms per microliter) is also shown in E (n = 6 samples from 2 experiments). *p < 0.05; **p < 0.01; ***p < 0.001. n = 4 experiments for all other conditions.
Therefore, we next tested whether Aβ could alter the rate of intracellular cleavage of a p75NTR protein lacking the extracellular domain (ΔE14) (Jung et al., 2003). Although not as efficient as treatment with a γ-secretase inhibitor, cells treated overnight with Aβ had discernibly less intracellular domain fragment than untreated cells (Fig. 3D). To quantify this, we used a ΔE14 construct and a similar APP construct (C99-Gal4) in which the Gal4 transactivation domains had been fused to their carboxyl ends (Sernee et al., 2003). Only when released after γ-secretase cleavage of the C-terminal protein can the activator domain promote transcription of a cotransfected luciferase reporter gene (pUAS), the activity of which can be fluorometrically quantified in cell lysates (Karlstrom et al., 2002). Using this method, we found that overnight treatment of ΔE14-Gal4-expressing HEK293 cells with Aβ1–42 significantly inhibited the resultant luciferase activity, whereas the control peptide Aβ1–16 had no significant effect (Fig. 3E). Importantly, our results using the C99-Gal4 construct also showed a significant effect after an overnight Aβ treatment regimen, which was similar to the level of inhibition seen when the γ-secretase inhibitor was applied overnight (Fig. 3F). Given that the assay is based on a transcriptional readout, it was not surprising that no significant effects were seen after the 3 h treatments. These results indicate that Aβ1–42 can generically inhibit γ-secretase activity, and suggest that the increased level of p75NTR C-terminal fragment in cells treated with Aβ is, at least partially, caused by this effect.
Loss of basal forebrain cholinergic neurons through degeneration of the septohippocampal pathway is a major feature of Alzheimer's disease. To determine whether Aβ1–42-induced toxicity in cholinergic neurons innervating the hippocampus was mediated by p75NTR, Aβ1–42 or Aβ1–16 peptide solution was injected into the hippocampus of wild-type and p75NTR-deficient mice. Fourteen days after the injection, the septohippocampal pathway was examined histologically. Regardless of the injected solution or mouse genotype, the needle track through the CA1 region of the hippocampus was identified, revealing no obvious difference in the amount of tissue damage in this area (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). In contrast, the number of ChAT-positive neurons in the medial septum and diagonal band of Broca after Aβ1–42 exposure was affected by genotype. First, the number of ChAT-positive neurons in the diagonal band of Broca of the ipsilateral hemisphere of wild-type mice injected with saline (n = 4, data not shown) or Aβ1–16 was not significantly different from that found in uninjected hemispheres (Fig. 4A). However, the number of ChAT-positive neurons in the same region of mice injected with Aβ1–42 was significantly reduced compared with that of uninjected, saline-injected, or Aβ1–16-injected animals (Fig. 4A,B). Most importantly, however, the number of ChAT-positive neurons in p75NTR-deficient mice after injection with Aβ1–16 or Aβ1–42 did not differ significantly, and was similar to that found in uninjected wild-type mice (Fig. 4A,C). These results demonstrate that p75NTR is also required for Aβ1–42-mediated neuronal toxicity in vivo.
Figure 4.

Aβ1–42 toxicity of basal forebrain cholinergic neurons in vivo is prevented by p75NTR deficiency. The hippocampi of wild-type (WT) and p75NTR knock-out (KO) mice were injected with Aβ, and after 14 d the number of ChAT-positive cells in each hemisphere was counted. A, The number of wild-type ChAT-positive neurons in the hemisphere ipsilateral to the Aβ1–42 injection site was significantly reduced compared with that in uninjected or Aβ1–16-injected animals. The numbers of ChAT-positive neurons in p75NTR-deficient mice after injection with Aβ1–16 or Aβ1–42 were not significantly different, and were similar also to that found in uninjected wild-type mice. n ≥ 4 per condition; ***p < 0.001. B, C, Photomicrographs of ChAT-positive cells in the basal forebrain of wild-type (B) and p75NTR-deficient (C) mice 14 d after injection with Aβ.
Discussion
Here we provide evidence that p75NTR is required for oligomeric Aβ1–42-mediated neuronal death in vitro and in vivo, further strengthening the case that p75NTR plays a role in the etiology of Alzheimer's disease.
We found that embryonic hippocampal neurons, which, unlike their adult counterparts, express high levels of p75NTR, were killed by bathing the cultures with oligomeric Aβ1–42 peptides, but were unaffected by Aβ1–16 peptides. This finding is in agreement with many other studies showing that the carboxyl amino acids of the amyloid peptide are important for neuronal toxicity (Klein et al., 2004; Costantini et al., 2005), and supports the more recent idea that the soluble oligomeric protofibril form of Aβ1–42 mediates amyloid neurotoxicity (Dahlgren et al., 2002; Gandy, 2005). Moreover, we demonstrated that Aβ1–42-mediated neurotoxicity is abolished in the absence of p75NTR expression. This confirms the involvement of p75NTR-mediated signaling pathways in neuronal death reported in previous studies using nonprimary cell cultures treated with amyloid peptides (Coulson, 2006).
We found that Aβ-mediated death signaling resulted in, and required, increased production of the p75NTR C-terminal fragment. It has been demonstrated that Aβ binds to the extracellular domain of p75NTR (Yaar et al., 1997), and that p75NTR mediates death signaling in hippocampal neurons after neurotrophin application (Troy et al., 2002). We found that an antibody to the p75NTR extracellular domain had a significant effect on Aβ1–42-induced death. It is therefore possible that the interaction of Aβ1–42 with p75NTR promotes extracellular cleavage, such as occurs in response to other p75NTR ligands (Kenchappa et al., 2006; Podlesniy et al., 2006; Coulson et al., 2008). Although it is also a possibility that the antibody was preventing the binding and activation of p75NTR by endogenously produced neurotrophins (Troy et al., 2002; Volosin et al., 2006), the fact that γ-secretase inhibitor treatment failed to promote cell death in the absence of Aβ1–42 suggests that additional Aβ1–42 effects may play a role in the promotion of cell death.
Our experiments using HEK293 cells indicate that inhibition of γ-secretase activity is a significant factor in promoting the accumulation of the C-terminal fragment. Although the exact mechanism by which this occurs is not known, our finding that APP processing is also affected by Aβ, together with the demonstration that increased expression of the APP C-terminal fragment has a similar effect on p75NTR processing, suggests that Aβ1–42 could act as a competitive γ-secretase substrate. This mode of action, in agreement with our finding that γ-secretase inhibitor treatment promoted rather than inhibited Aβ-induced neuronal death, would preclude Aβ-induced cell death being mediated via the intracellular domain pathway described by others (Kenchappa et al., 2006; Podlesniy et al., 2006). However, we have previously shown that the C-terminal fragment of p75NTR is constitutively active in signaling neuronal death, and have recently defined a signaling cascade by which it results in caspase activity (Coulson et al., 2000, 2008; Underwood et al., 2008).
In support of p75NTR playing a key role in Aβ1–42-induced neurodegeneration, we found that injection of Aβ1–42 into the hippocampus of wild-type mice, a model of Alzheimer's disease, resulted in the degeneration of ChAT-positive forebrain neurons in vivo. Although our analysis did not specifically determine that ChAT-positive cells had died after Aβ1–42 exposure at axonal terminals, it did demonstrate the loss of cellular neurotransmitter production and hence loss of function of these neurons. It has also been shown in aged animals that ChAT downregulation correlates with loss of hippocampal function (Yeo et al., 1997) and that this precedes the death of basal forebrain neurons (Greferath et al., 2000).
This functional loss of cholinergic forebrain neurons in vivo was also prevented by p75NTR deficiency, supporting the hypothesis that p75NTR-mediated degeneration of basal forebrain neurons is an early consequence of increased oligomeric Aβ levels and is tightly linked to the loss of septohippocampal function observed in both animal models and human patients with Alzheimer's disease (Klein et al., 2004; Gimenez-Llort et al., 2007). Although a significant difference in the numbers of ChAT-positive basal forebrain neurons correlated directly with Aβ1–42 exposure, we found no obvious difference in the amount of damage in the hippocampus after injections into the CA1 region, regardless of Aβ peptide length or genotype. In the adult hippocampus, unlike the embryonic hippocampus and the adult basal forebrain, p75NTR is downregulated, although it can be upregulated in response to injury (Dechant and Barde, 2002). This observation further highlights the connection between the degeneration of neurons that express high levels of p75NTR and Aβ1–42 peptide exposure. Although our in vitro results demonstrate that Aβ1–42-induced, p75NTR-mediated neuronal death is likely to result from inhibition of γ-secretase activity, other injury-induced factors, such as modulation of synaptic plasticity (Klein et al., 2004), and/or neurotrophins released by reactive astrocytes (Pehar et al., 2004), could also contribute to, and indeed may be required to, promote cell death in vivo via a p75NTR-dependent mechanism.
In conclusion, we have shown that Aβ1–42-induced neuronal degeneration of embryonic hippocampal neurons in vitro and basal forebrain neurons in vivo requires p75NTR. Furthermore, the activation of neuronal loss strongly correlates with increased accumulation of the constitutively active death-signaling C-terminal fragment of p75NTR generated by inhibition of γ-secretase activity. Although p75NTR has been causally linked to the progression of Alzheimer's disease for over a decade, the current results provide a novel mechanism to explain the early and characteristic degeneration of the septohippocampal pathway that occurs in this disease.
Footnotes
This work was supported by the National Health and Medical Research Council of Australia. A.S. is the recipient of a Development and Promotion of Science and Technology Talent Scholarship (Mahidol University, Salaya, Thailand). We thank Rowan Tweedale for editorial assistance and Dr. G. Evin (Department of Pathology, University of Melbourne, Australia; C99-Gal4) and Prof. U. Lendal (Karolinska Institute, Stockholm, Sweden; pUAS and pCMV-Gal) for DNA constructs.
References
- Costantini C, Rossi F, Formaggio E, Bernardoni R, Cecconi D, Della-Bianca V. Characterization of the signaling pathway downstream p75 neurotrophin receptor involved in β-amyloid peptide-dependent cell death. J Mol Neurosci. 2005;25:141–156. doi: 10.1385/JMN:25:2:141. [DOI] [PubMed] [Google Scholar]
- Coulson EJ. Does the p75 neurotrophin receptor mediate Aβ-induced toxicity in Alzheimer's disease? J Neurochem. 2006;98:654–660. doi: 10.1111/j.1471-4159.2006.03905.x. [DOI] [PubMed] [Google Scholar]
- Coulson EJ, Reid K, Baca M, Shipham K, Hulett SM, Kilpatrick TJ, Bartlett PF. Chopper, a new death domain of the p75 neurotrophin receptor which mediates rapid neuronal cell death. J Biol Chem. 2000;275:30537–30545. doi: 10.1074/jbc.M005214200. [DOI] [PubMed] [Google Scholar]
- Coulson EJ, May LM, Osborne SL, Reid K, Underwood CK, Meunier FA, Bartlett PF, Sah P. p75 neurotrophin receptor mediates neuronal cell death by activating GIRK channels through phosphatidylinositol 4,5-bisphosphate. J Neurosci. 2008;28:315–324. doi: 10.1523/JNEUROSCI.2699-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- Dechant G, Barde YA. The neurotrophin receptor p75NTR: novel functions and implications for diseases of the nervous system. Nat Neurosci. 2002;5:1131–1136. doi: 10.1038/nn1102-1131. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer's disease. Mol Cell Neurosci. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- Gandy S. The role of cerebral amyloid β accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez-Llort L, Blazquez G, Canete T, Johansson B, Oddo S, Tobena A, LaFerla FM, Fernandez-Teruel A. Modeling behavioral and neuronal symptoms of Alzheimer's disease in mice: a role for intraneuronal amyloid. Neurosci Biobehav Rev. 2007;31:125–147. doi: 10.1016/j.neubiorev.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Greferath U, Bennie A, Kourakis A, Bartlett PF, Murphy M, Barrett GL. Enlarged cholinergic forebrain neurons and improved spatial learning in p75 knockout mice. Eur J Neurosci. 2000;12:885–893. doi: 10.1046/j.1460-9568.2000.00976.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Jung KM, Tan S, Landman N, Petrova K, Murray S, Lewis R, Kim PK, Kim DS, Ryu SH, Chao MV, Kim TW. Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J Biol Chem. 2003;278:42161–42169. doi: 10.1074/jbc.M306028200. [DOI] [PubMed] [Google Scholar]
- Karlstrom H, Bergman A, Lendahl U, Naslund J, Lundkvist J. A sensitive and quantitative assay for measuring cleavage of presenilin substrates. J Biol Chem. 2002;277:6763–6766. doi: 10.1074/jbc.C100649200. [DOI] [PubMed] [Google Scholar]
- Kenchappa RS, Zampieri N, Chao MV, Barker PA, Teng HK, Hempstead BL, Carter BD. Ligand-dependent cleavage of the p75 neurotrophin receptor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neurons. Neuron. 2006;50:219–232. doi: 10.1016/j.neuron.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, Estevez AG, Barbeito L. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004;89:464–473. doi: 10.1111/j.1471-4159.2004.02357.x. [DOI] [PubMed] [Google Scholar]
- Podlesniy P, Kichev A, Pedraza C, Saurat J, Encinas M, Perez B, Ferrer I, Espinet C. Pro-NGF from Alzheimer's disease and normal human brain displays distinctive abilities to induce processing and nuclear translocation of intracellular domain of p75NTR and apoptosis. Am J Pathol. 2006;169:119–131. doi: 10.2353/ajpath.2006.050787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliebs R. Basal forebrain cholinergic dysfunction in Alzheimer's disease—interrelationship with β-amyloid, inflammation and neurotrophin signaling. Neurochem Res. 2005;30:895–908. doi: 10.1007/s11064-005-6962-9. [DOI] [PubMed] [Google Scholar]
- Sernee MF, Evin G, Culvenor JG, Villadangos JA, Beyreuther K, Masters CL, Cappai R. Selecting cells with different Alzheimer's disease gamma-secretase activity using FACS. Differential effect on presenilin exon 9 γ- and ε-cleavage. Eur J Biochem. 2003;270:495–506. doi: 10.1046/j.1432-1033.2003.03405.x. [DOI] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ. Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J Biol Chem. 2002;277:34295–34302. doi: 10.1074/jbc.M205167200. [DOI] [PubMed] [Google Scholar]
- Ueda K, Fukui Y, Kageyama H. Amyloid β protein-induced neuronal cell death: neurotoxic properties of aggregated amyloid β protein. Brain Res. 1994;639:240–244. doi: 10.1016/0006-8993(94)91736-1. [DOI] [PubMed] [Google Scholar]
- Underwood CK, Reid K, May LM, Bartlett PF, Coulson EJ. Palmitoylation of the C-terminal fragment of p75NTR promotes death signaling and is required for subsequent cleavage by γ-secretase. Mol Cell Neurosci. 2008;37:346–358. doi: 10.1016/j.mcn.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Urra S, Escudero CA, Ramos P, Lisbona F, Allende E, Covarrubias P, Parraguez JI, Zampieri N, Chao MV, Annaert W, Bronfman FC. TrkA receptor activation by nerve growth factor induces shedding of the p75 neurotrophin receptor followed by endosomal γ-secretase-mediated release of the p75 intracellular domain. J Biol Chem. 2007;282:7606–7615. doi: 10.1074/jbc.M610458200. [DOI] [PubMed] [Google Scholar]
- Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ. Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci. 2006;26:7756–7766. doi: 10.1523/JNEUROSCI.1560-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CK, Thal L, Pizzo D, Hansen L, Masliah E, Geula C. Apoptotic signals within the basal forebrain cholinergic neurons in Alzheimer's disease. Exp Neurol. 2005;195:484–496. doi: 10.1016/j.expneurol.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA. Binding of β-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J Clin Invest. 1997;100:2333–2340. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Feng J. Alzheimer's disease: interactions between cholinergic functions and β-amyloid. Curr Alzheimer Res. 2004;1:241–248. doi: 10.2174/1567205043331992. [DOI] [PubMed] [Google Scholar]
- Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, Mobley WC, Longo FM. Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci. 1997;17:7594–7605. doi: 10.1523/JNEUROSCI.17-20-07594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]