

Abstract
Disrupted-in-schizophrenia 1 (DISC1) and other genes have been identified recently as potential molecular players in chronic psychiatric diseases such as affective disorders and schizophrenia. A molecular mechanism of how these genes may be linked to the majority of sporadic cases of these diseases remains unclear. The chronic nature and irreversibility of clinical symptoms in a subgroup of these diseases prompted us to investigate whether proteins corresponding to candidate genes displayed subtle features of protein aggregation. Here, we show that in postmortem brain samples of a distinct group of patients with phenotypes of affective disorders or schizophrenia, but not healthy controls, significant fractions of DISC1 could be identified as cold Sarkosyl-insoluble protein aggregates. A loss-of-function phenotype could be demonstrated for insoluble DISC1 through abolished binding to a key DISC1 ligand, nuclear distribution element 1 (NDEL1): in human neuroblastoma cells, DISC1 formed expression-dependent, detergent-resistant aggregates that failed to interact with endogenous NDEL1. Recombinant (r) NDEL1 expressed in Escherichia coli selectively bound an octamer of an rDISC1 fragment but not dimers or high molecular weight multimers, suggesting an oligomerization optimum for molecular interactions of DISC1 with NDEL1. For DISC1-related sporadic psychiatric disease, we propose a mechanism whereby impaired cellular control over self-association of DISC1 leads to excessive multimerization and subsequent formation of detergent-resistant aggregates, culminating in loss of ligand binding, here exemplified by NDEL1. We conclude that the absence of oligomer-dependent ligand interactions of DISC1 can be associated with sporadic mental disease of mixed phenotypes.
Keywords: psychiatric disease, depression, bipolar disorder, multimerization, protein conformational disease, protein aggregation
Introduction
The neurobiology of chronic psychiatric diseases such as schizophrenia and affective disorders is in its infancy. The absence of specific and unequivocal biological correlates to these prevalent disorders has preserved a century-old clinical situation, where diagnosis still relies entirely on clinical interview. At the same time, huge progress has been made in many other chronic brain diseases, for example with the identification of key players and the elucidation of major molecular pathways in the classical neurodegenerative diseases Alzheimer's disease, Parkinson's disease, or polyglutamine diseases. Protein aggregation was discovered as a surprising common feature of neurodegenerative diseases, with the same proteins found aggregated in sporadic and familial forms of these diseases (Prusiner, 2001; Taylor et al., 2002).
Chronic psychiatric disorders present with considerable phenotypical heterogeneity, even within recognized clinical-diagnostic entities. In schizophrenia, chronicity of symptoms is mainly defined by progressive “negative symptoms” like avolition, lack of drive, and cognitive deficits that are serious and daily life-impacting in approximately one-third of cases and less serious but present in another one-third of cases (an der Heiden and Hafner, 2000). Furthermore, the lifelong recurrence of “positive” symptoms like acute disturbances of mood or cognition in untreated cases of affective disorders or schizophrenia, respectively, justifies terming these diseases chronic.
Genetic studies in families affected with chronic psychiatric disorders have hinted recently at an underlying molecular neurobiology. Investigations in a Scottish pedigree revealed that a familial mutation in the Disrupted-in-schizophrenia 1 (DISC1) gene, attributable to a balanced chromosome t(1;11) (q42.1;q14.3) translocation, segregated with a range of psychiatric phenotypes, including schizophrenia, bipolar disorder, and recurrent major depression (Millar et al., 2000; Blackwood et al., 2001) (for review, see Chubb et al., 2008). DISC1 plays a role in regulating neurodevelopment, neurosignaling, and cytoskeletal functions by interacting with a number of proteins, including phosphodiesterase-4B, fasciculation and elongation protein ζ 1, lissencephaly-1, and nuclear distribution element-like protein (NDEL1) (Kamiya et al., 2005; Millar et al., 2005; Ishizuka et al., 2006; Duan et al., 2007). Other genetic studies led to the identification of the neuregulin1 gene (Stefansson et al., 2002) and others (Harrison and Weinberger, 2005; Ross et al., 2006) as susceptibility factors for schizophrenia. These studies have established that under certain circumstances, the same genetic mutation can cause different psychiatric phenotypes in different affected family members. Shared risk loci of schizophrenia and bipolar disorder (Berrettini, 2003) as well as common endophenotypes (McDonald et al., 2004) have strengthened the notion of phenotypic variability caused by a single genetic defect in this group of diseases.
So far, no obvious and unequivocal neuropathology has been reproducibly reported in chronic psychiatric diseases, apart from enlarged third ventricles in cases of schizophrenia (Chua and McKenna, 2000). The chronic course in a significant subset of cases with schizophrenia and the affective disorders prompted us to investigate whether subtle signs of neuronal degeneration could be detected in affected brains. Because protein aggregation has been established as an unequivocal feature of the classical neurodegenerative diseases, we investigated whether the proteins of candidate genes associated with schizophrenia could be shown to be detergent insoluble or aggregated in sporadic forms of chronic psychiatric disorders.
Materials and Methods
Brain materials.
Huntington disease brains (BA9) were obtained from the Harvard Brain Bank (Harvard Brain Tissue and Resource Center, Belmont, MA). R6/2 transgenic mouse brains and littermate control brains (Mangiarini et al., 1996) were a generous gift from Gillian Bates (King's College London, London, UK). BA23 frozen cortex tissue from the Consortium Collection (Torrey et al., 2000) was obtained from the Stanley Medical Research Institute (SMRI) (Baltimore, MD).
Brain fractionation.
Snap-frozen brain samples were thawed on ice and homogenized 10% (w/v) in ice-cold 50 mm HEPES, pH 7.5, 100 mm KAc, 250 mm sucrose, 5 mm MgCl2, 1% Triton X-100, 5× protease inhibitor mixture (PI; Roche, Indianapolis, IN). Subsequently, the following sequential steps were performed twice: (1) centrifugation at 20,000 × g, 20 min at 4°C; (2) the pellet was taken up in buffer plus 1.6 m sucrose and layered on top of 2.3 m sucrose, followed by ultracentrifugation (130,000 × g, 45 min at 4°C; always in an MLS-55 rotor in an Optima ultracentrifuge; Beckman Coulter, Fullerton, CA); (3) the pellet was resuspended and digested with DNAseI (Roche) in 1 m NaCl (overnight at 4°C) and then centrifuged again (130,000 × g, 45 min at 4°C); (4) the pellet was washed with ice-cold 20 mm HEPES, pH 7.5, 5 mm EDTA, and 0.5% N-lauroylsarkosine (Sarkosyl) and then ultracentrifuged (130,000 × g, 45 min at 4°C). Brain samples were processed blinded; however, Western blot analysis was done unblinded.
Cell studies.
Human neuroblastoma cells (NLF; Children's Hospital of Philadelphia, Philadelphia, PA) were grown to 70% confluency in 60 mm plates and transfected with 8 μg of pDISC1 or pcDNA-NDEL1 using Metafectene Reagent (Biontex, Martinsried, Germany). pDISC1 was a generous gift from Akira Sawa (Johns Hopkins Hospital, Baltimore, MD), and pcDNA-NDEL1 was a generous gift from Li-Huei Tsai (The Picower Institute, Massachusetts Institute of Technology, Cambridge, MA). After 14 and 24 h, cells were lyzed in 300 μl of lysis buffer [50 mm HEPES, pH 7.5, 300 mm NaCl, 250 mm sucrose, 5 mm EDTA, 5 mm glutathione (GSH), 1% NP-40, 0.2% Sarkosyl, 2× PIs (Roche, Basel, Switzerland), 1 mm PMSF] and centrifuged for 30 min at 1800 × g. The pellet was suspended in 300 μl of high-salt buffer (50 mm HEPES, pH 7.5, 1.5 m NaCl, 250 mm sucrose, 5 mm EDTA, 5 mm GSH, 1% NP-40, 0.2% Sarkosyl, 1 mm PMSF) and centrifuged for 30 min at 1800 × g. The resulting pellet was resuspended and subjected to a DNA digest for 30 min at 37°C in 50 mm Tris, pH 8, 250 mm sucrose, 5 mm MgCl2, 5 mm GSH, 1% NP-40, 2× PI (Roche), 1 mm PMSF, and DNase I 40 U/ml (Sigma, St. Louis, MO) and subsequently incubated at 4°C overnight. Samples were then centrifuged for 30 min at 1800 × g and resuspended in 300 μl of 50 mm HEPES and 0.2% Sarkosyl. Finally, probes underwent ultracentrifugation at 100,000 × g for 45 min at 4°C (TLA-55 rotor in Optima; Beckman Coulter), and the resulting pellets were solubilized in loading buffer. All steps were performed on ice unless otherwise stated.
Protein expression in Escherichia coli.
N-terminally His6-tagged human recombinant (r)DISC1(598–854), rDISC1(316–854), and NDEL1(1–345) were cloned into a modified pET-15b vector and expressed in E. coli BL21 (λDE3) Rosetta in medium plus 5 mm l-arginine, 5 mm MgSO4, 100 mg/ml carbenicillin, and 35 mg/ml chloramphenicol. Cells were lysed by lysozyme digestion, and after removal of the cytoplasmic fraction, inclusion bodies were solubilized in 50 mm Tris-HCl, pH 8, 2 mm imidazole, 500 mm NaCl, 6 m GuHCl, 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), and 20 mm β-mercaptoethanol. rNDEL1 was separated from the cytoplasmic fraction on Ni2+- nitrilotriacetic acid agarose while both rDISC1 fragments were separated from the inclusion body extract and refolded on-column in 20 mm Tris-HCl, pH 8, 10 mm imidazole, 500 mm NaCl, and 10 mm β-mercaptoethanol (1 h at 4°C). The His6-tag was removed from refolded rDISC1 protein by thrombin (Roche) digestion. Correct molecular weights of both DISC598 and rNDEL1 were verified by mass spectrometry (supplemental Fig. 11, available at www.jneurosci.org as supplemental material).
Antibodies.
Human DISC1: full-length recombinant DISC1(1–854) was purified and injected into two rabbits with 3 weekly boosts and RIBI adjuvant (Sigma). Antiserum was then pooled and affinity purified on immobilized DISC1(1–854) (for antiserum characterization, see supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Results were cross validated with affinity-purified antiserum generously supplied by Akira Sawa (supplemental Fig. 7, available at www.jneurosci.org as supplemental material) (Ozeki et al., 2003). Monoclonal antibody 3D4 was produced with a standard hybridoma cell fusion method after immunizing mice with full-length human recombinant DISC1 (P. Hendriks, S. R. Leliveld, C. Korth, unpublished observations). Neuregulin1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA) (s.c.-348); the antibody has been used successfully to characterize neuregulin1 in chronic psychiatric diseases (Law et al., 2004). NDEL1 antibody was purchased from Abcam (Cambridge, MA) (ab25959) (supplemental Fig. 9, available at www.jneurosci.org as supplemental material). NDEL1 antiserum was produced by immunizing a rabbit with full-length recombinant NDEL1 (see Fig. 2). Rabbit immunizations were performed in accordance with relevant guidelines and regulations; the animal experimentation protocol was authorized by the Regierungspräsidium Düsseldorf to C.K.
Figure 2.

Western blots demonstrating that molecular interactions of DISC1 and NDEL1 in NLF cells depend on solubility of DISC1. After transient transfection of NLF cells with DISC1, cells were lyzed and subjected to a fractionation protocol, including ultracentrifugation in the presence of cold Sarkosyl (see Materials and Methods). a, b, The supernatant after the first low-speed spin in 0.2% cold Sarkosyl (a) and the pellet purified, including ultracentrifugation (b), were analyzed by Western blot. The Western blot was horizontally cut at ∼55 kDa and stained with mouse α-DISC1 monoclonal antibody 3D4 (top) or polyclonal NDEL1 antiserum (bottom); before film exposure, cut blot parts stained with different antibodies were reassembled. U, Untransfected; D14 or N14, transient transfection with DISC1 or NDEL1, respectively, for 14 h; D24 or N24, transient transfection with DISC1 or NDEL1, respectively, for 24 h. Baseline expression of DISC1 was not detectable; DISC1 immunoreactivity increased after prolonged transfection time (a, lanes U-D24, top); there was baseline NDEL1 expression detectable, which increased after prolonged transient transfection of NDEL1 but not DISC1 (a, lanes U-N24). DISC1 pelleted as Sarkosyl-insoluble in an expression-dependent manner (b, top, compare D14 with D24), without pulling down NDEL1 (b, bottom, D14-D24); NDEL1 itself did not pellet under the same conditions used for DISC1 (b, bottom, N14-N24). The asterisk indicates a 72 kDa immunoreactive band that is strongly enriched in the Sarkosyl-insoluble fraction, as is a 55 kDa band. c, Soluble fractions of DISC1 transfected lysates were further processed by size exclusion chromatography and analyzed by Western blot. Although overexpression of DISC1 did not qualitatively change the size distribution of DISC1 multimers, NDEL1 was redistributed dramatically toward high-molecular size complexes in an expression level-dependent manner as evidence for a sensitive regulation of NDEL1 by soluble DISC1 (bottom three panels). The DISC1-positive bands >200 kDa reflect SDS-resistant oligomers, similar to the ∼180 kDa bands we observed for rDISC316 (supplemental Fig. 10c, available at www.jneurosci.org as supplemental material). Vertical gridlines are shown to facilitate comparisons of aligned lanes corresponding to the same eluted fractions.
Size exclusion chromatography.
Protein samples and NLF cell lysates (0.2–0.5 ml/run) were fractionated on a Superdex 200 10/300 GL (GE Healthcare Bio-Sciences, Little Chalfont, Buckinghamshire, UK) at 0.5 ml/min in 20 mm Tris-HCl, pH 8, 1 mm EDTA, 5 mm dithiothreitol (DTT), 500 mm NaCl, and 0.5% CHAPS using an Ákta HPLC (GE Healthcare). For binding studies, rDISC598 and rNDEL1 were labeled cysteine specifically with 5-iodoactetyl-fluorescein (Invitrogen, Carlsbad, CA). NLF cells transfected with pDISC1 (14 or 24 h) plus nontransfected controls were lysed in 50 mm HEPES, pH 7.5, 500 mm NaCl, 5 mm DTT, 5 mm EDTA, 1% CHAPS, 20 mm β-glycerolphosphate, 5 mm NaF, and 5× PIs. Cell debris was removed by centrifugation (5 min at 10,000 × g). Size exclusion chromatography (SEC) lysate fractions were concentrated by protein precipitation (four volumes, 50/50 methanol/acetone, 1 h at −80°C, then centrifugation at 20,000 × g for 30 min at 4°C). Pellets were taken up in loading buffer.
Dynamic light scattering.
rDISC598, rDISC316 (after thrombin digestion), and rNDEL1 (after SEC) were measured at 0.2–2 mg/ml in 20 mm Tris-HCl, pH 8, 500 mm NaCl, 1 mm EDTA, and 5 mm DTT at 16°C with a DynaPro MS/X running DYNAMICS V6 (Wyatt Technology Corporation, Santa Barbara, CA).
Statistical testing was performed with SPSS (11.0) Software (SPSS, Chicago, IL). ANOVA corrected for unequal variances, different group case numbers, and multiple testing was used to compare means of control versus disease cases. Pearson's two-tailed correlation test was used to analyze covariance of parametric case variables with P/H ratios for DISC1, and Spearman's correlation test for nonparametric case variables.
Results
Purifying the insoluble proteome
For identifying subtle protein aggregates in postmortem brains of patients with chronic psychiatric disease, a procedure was developed to isolate the “insoluble proteome” of brain tissue by means of a sequential biochemical fractionation (see Materials and Methods). The protocol was validated using mutant, polyglutamine huntingtin aggregates, which are the smallest known pathogenic protein inclusions, visible only after immunostaining (Scherzinger et al., 1997). In this context, Huntington disease is a well-suited model, because the polyglutamine protein aggregates are also present in cortical tissue, an anatomic site remote from the caudate nucleus, the primary site of neuropathology (Gutekunst et al., 1999; Kuemmerle et al., 1999).
Sequential biochemical fractionation was performed both with postmortem brains from patients with Huntington disease as well as with R6/2 mice (supplemental Fig. 4a–c, available at www.jneurosci.org as supplemental material), a transgenic mouse model of Huntington disease, where the exon 1, including an extended CAG repeat domain, is expressed (Mangiarini et al., 1996). The final cold (0.5%) Sarkosyl-resistant pellet showed greatly enriched huntingtin immunoreactivity, which we estimated to be ∼100-fold after comparing the total protein content of starting material and pellet (supplemental Fig. 4a, available at www.jneurosci.org as supplemental material).
The purification protocol for the insoluble proteome protocol was transferred to brains from patients with schizophrenia, bipolar disorder, and monopolar depression, as well as normal controls. The SMRI Consortium Collection was used, an established collection of 60 well-diagnosed postmortem cases (Torrey et al., 2000) that has been validated frequently and used to investigate chronic psychiatric disease-associated proteins (for publication overview, see http://www.stanleyresearch.org/publications/consortiumall.asp). Frozen brain pieces from cingulum cortex (BA23), a brain region that is considered an anatomical region associated with psychiatric disease pathology (Hulshoff Pol et al., 2001; Katsel et al., 2005; Narr et al., 2005), were used. Lack of protein degradation in these samples was confirmed by intactness of degradation-sensitive marker dihydropyrimidinase-related protein-2 (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) (Franzen et al., 2003).
Insoluble DISC1 in schizophrenia and affective disorders
The cold Sarkosyl-insoluble pellets and the starting brain homogenate from each single case of the 60 fractionated SMRI brain samples were generated, immunoblotted, and probed with antiserum against DISC1 (Fig. 1). We identified a ∼72 kDa band as the major immunoreactive band using affinity-purified DISC1 antiserum both in the homogenate and in the Sarkosyl-insoluble fraction (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), similar to what has been described previously (James et al., 2004; Ishizuka et al., 2006); this band has also been shown to be enriched in nuclear fractions (Sawamura et al., 2005). The DISC1 antiserum used was potentially able to also recognize higher molecular weight isoforms of DISC1 (supplemental Fig. 6a, available at www.jneurosci.org as supplemental material). A subset of postmortem cases with chronic psychiatric disease but not normal controls exhibited strong DISC1 immunoreactivity in the insoluble fraction (Fig. 1 a). The mean value of the fraction of insoluble DISC1 with respect to total DISC1 present in the starting brain homogenate (P/H), was in fact higher in the disease versus normal group (1.6 vs 0.4, respectively; p = 0.002, ANOVA), although the significance of a different mean value should be considered with reservations attributable to the likely heterogeneity of diseases. More revealing was a considerable scatter of the data points (Fig. 1 b), clearly defining a subgroup of cases with high P/H values (7 of 45), whereas the rest overlap with the controls. Covariation of DISC1 P/H ratios with variables lifetime fluphenazine dosage, postmortem interval, brain mass, storage time, substance or alcohol abuse, or pH was excluded by a correlation analysis (Pearson's). In contrast, we could not detect immunoreactivity to neuregulin1 in the insoluble proteome of our samples (supplemental Fig. 8, available at www.jneurosci.org as supplemental material).
Figure 1.
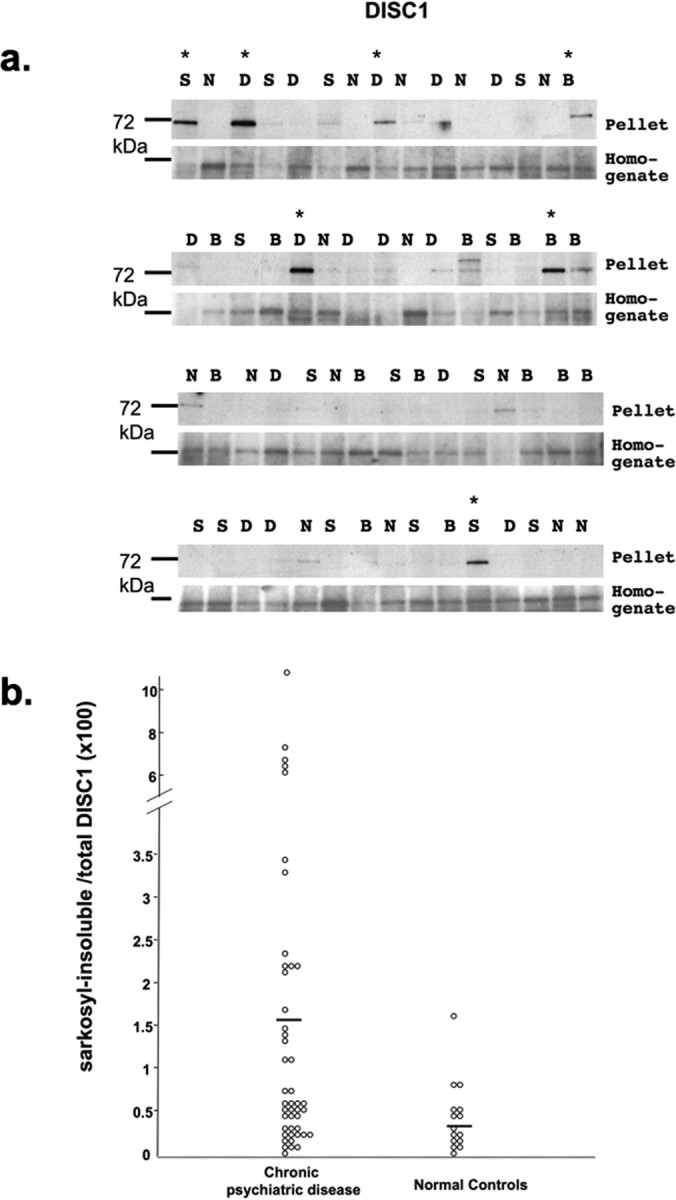
a, Western blot for DISC1 of cold Sarkosyl-insoluble pellets (top) and starting brain homogenates (bottom) of 60 individual cases (SMRI Consortium Collection) of schizophrenia (S), bipolar disorder (B), depression (D), and normal controls (N). Each lane corresponds to one case. Asterisks indicate immunoreactivity above threshold (defined as strongest background immunoreactivity in normal controls) in the insoluble pellet fraction. b, Scatter plot of the ratio of Sarkosyl-insoluble pelleted material (P) by the starting material (H) of DISC1 from diseased cases versus normal controls, quantified by densitometry of Western blots from a. Eighty-six percent of positive cases from a were also found to have a high P/H ratio. Calculation of P/H ratio leads to slight adjustments of positive cases relative to a, when the homogenate concentration was relatively abundant. The solid bold lines mark the arithmetic means. A pronounced scatter of P/H values is evident across the three disease groups; current clinical interview-based diagnostic protocols cannot avoid the grouping of pathological entities with different molecular underpinnings under a similar broad diagnostic category. Therefore, mean values for any of these groups, although of interest, should be relativized. Means of diseased (n = 45) versus normal (n = 15) brains was 1.6 versus 0.4, respectively (p = 0.002; ANOVA).
Detergent-insoluble DISC1 does not interact with NDEL1 in vivo
To investigate whether detergent insolubility of DISC1 was associated with impaired functioning in vivo, we transfected NLF human neuroblastoma cells with full-length DISC1. Lysis of transiently transfected cells at different time points correlated with low (14 h) or high (24 h) expression levels, paralleled by an expression-dependent pelleting of cold-Sarkosyl-resistant DISC1 (Fig. 2 b). This assay was used to investigate the molecular interaction of DISC1 with one of its key physiological ligands. We chose NDEL1, a known interaction partner of DISC1, the interactions of which have been well investigated, and that is involved in neurofilament assembly, neurogenesis, and neurite outgrowth during neuronal development (Ozeki et al., 2003; Nguyen et al., 2004; Duan et al., 2007).
The major immunoreactive DISC1 band in human postmortem brains is at ∼72 kDa, whereas in the cell transfection studies, it is at 100 kDa (Figs. 1, 2) (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). A point could be raised that these two DISC1-immunoreactive proteins are substantially different. However, a ∼72 kDa also appears as an immunoreactive band in NLF cells transfected with full-length DISC1 that is enriched in the Sarkosyl-insoluble fraction even relatively more than the 100 kDa band (Fig. 2, asterisks). Although we concede that the artificial system of overexpressing full-length DISC1 may be substantially different from the fine regulation of DISC1 transcript expression in human brains, we believe that the phenotype of acquisition of insolubility may be comparable, in particular for DISC1 species with an electrophoretic mobility at ∼72 kDa.
It has been shown experimentally that artificial, exogenously expressed C-terminally truncated DISC1 (codons 1–597) (Millar et al., 2000) no longer binds NDEL1 and exhibits dominant-negative cellular phenotypes (Ozeki et al., 2003; Kamiya et al., 2005). Haploinsufficiency of the DISC1 gene with decreased DISC1 expression levels could also lead to disturbed DISC1-NDEL1 interactions and account for disease phenotypes (Millar et al., 2005). We therefore investigated whether DISC1 insolubility caused an impairment of its NDEL1 interaction.
NDEL1 overexpression did not lead to a visible, detergent-insoluble pellet under the conditions in which overexpressed DISC1 was pelleting (Fig. 2 b), indicating that there was an aggregation propensity of DISC1 but not for NDEL1. Furthermore, insoluble, pelleting DISC1 did not pull down endogenous NDEL1, indicating that their interaction was abolished (Fig. 2 b, bottom). In contrast, overexpression of soluble DISC1 led to a dose-dependent shift of NDEL1 to soluble, high-molecular weight complexes demonstrating the profound influence of DISC1 on NDEL1 interactions (Fig. 2 c). These data paralleled our findings that in postmortem brains of cases with insoluble DISC1, NDEL1 was not pulled down or copelleting (supplemental Fig. 9, available at www.jneurosci.org as supplemental material).
A cell-free in vitro assay of oligomer-dependent DISC1-NDEL1 interaction
We investigated DISC1 multimerization-dependent molecular interactions in a cell-free in vitro system by expressing DISC1 and NDEL1 recombinantly in E. coli.
Because we could not obtain stably folded full-length rDISC1 expressed in E. coli, we expressed two fragments of rDISC1 to serve as an in vitro model for DISC1 aggregation, namely rDISC598 and rDISC316, corresponding to codons 598–854 and 316–854, respectively (supplemental Fig. 10, available at www.jneurosci.org as supplemental material). The rationale for choosing these fragments was to include known NDEL1-binding and DISC1 homomerization domains to best model the multimerization-dependent DISC1-NDEL1 interaction. Although rDISC598 contains the domain involved in NDEL1 binding, rDISC316 also contains all five predicted coiled-coil/leucine zipper domains and a central self-association domain (codons 403–504) (Ozeki et al., 2003; Kamiya et al., 2005).
Using SEC to characterize multimerization, rDISC598 was found to exist as dimeric (60–70 kDa), octameric (∼250 kDa), and multimeric (>300 kDa) species under physiologically relevant conditions (Fig. 3 a). rDISC598 dimer displayed essentially the same far-UV circular dichroism (CD) spectrum as the combined oligomer plus multimer fraction, indicating that rDISC598 self-associates as folded subunits (supplemental Fig. 10b, available at www.jneurosci.org as supplemental material). rDISC598 gradually formed a Sarkosyl-resistant precipitate when stored at 80 μm, suggesting that the range from rDISC598 dimers up to Sarkosyl-insoluble aggregates represents a self-association continuum that parallels aggregation of endogenous 72 kDa DISC1 (supplemental Fig. 10c, available at www.jneurosci.org as supplemental material). Accordingly, it has been reported that C-terminally truncated DISC1 perturbs neuronal development by directly binding to and redistributing wild-type DISC1, indicating that the C-terminal domain controls proper DISC1 self-association (Ozeki et al., 2003; Kamiya et al., 2005).
Figure 3.
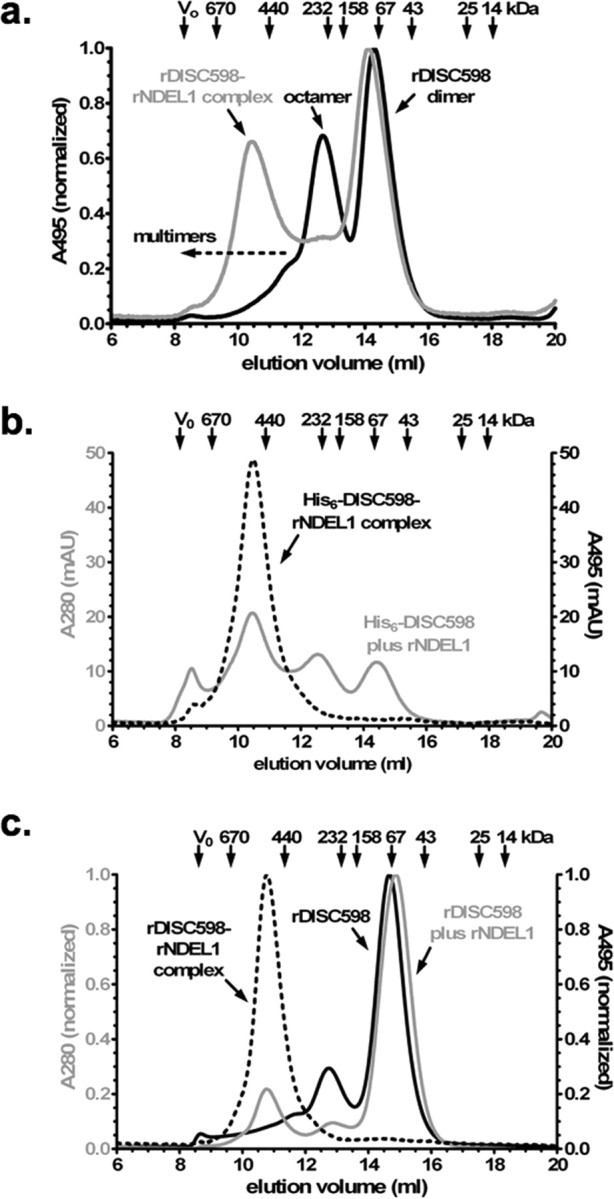
Analysis of interaction between rDISC598 and rNDEL1 by SEC. a, Normalized SEC profile at 495 nm of fluorescein-labeled rDISC598 alone (black) and combined with nonlabeled rNDEL1 (gray; 1:4 as monomer). The resulting complex peak (∼500 kDa) reflects a homogenous species, whereby the disappearance of the rDISC598 octamer peak indicates that NDEL1 preferentially binds the octamer and not the dimer (∼65 kDa) and is unlikely to bind multimers (>300 kDa). b, SEC profile at 280 nm (total protein; gray) and 495 nm of His6-tagged rDISC598 plus fluorescein-tagged NDEL1 (dashed black; 1:4), confirming that NDEL1 has little or no affinity for multimeric rDISC598. The presence of the hexahistidine tag increases favors multimerization without affecting overall structure, as verified by CD spectroscopy. c, Normalized SEC profile at 280 and 495 nm of nonlabeled DISC598 without (solid black) or with (gray; 10:1) fluorescein-labeled rNDEL1. Again, the rDISC598-rNDEL1 complex (495 nm; dashed black) is homogenous, even with an excess of rDISC598. The rDISC598 used here is the isolated and then concentrated dimer fraction, verifying that previously non-NDEL1-binding dimers can form NDEL1-binding octamers.
Selective binding of DISC1 octamers to NDEL1
N-terminally His6-tagged rNDEL1 was expressed in soluble form as an oligomer of 400 ± 100 kDa according to SEC and in accordance with its endogenous expression in NLF cells (Fig. 2 c). In dynamic light scattering (DLS), it displayed a hydrodynamic diameter (D H) of 13.4 ± 0.6 nm (300 ± 30 kDa). We attributed any discrepancies between SEC and DLS data to the possibly nonglobular nature of the complexes studied here and the use of detergents for SEC. When we combined cysteine-selectively fluorescein-labeled rDISC598 with nonlabeled rNDEL1 and fractionated the resulting complex by SEC, we found that the octamers but not the dimers bound rNDEL1, resulting in a selective shift of only the octameric rDISC598-rNDEL1 complex (Fig. 3 a,b). Formation of a single rDISC598-rNDEL1 complex (∼500 kDa) was confirmed using fluorescein-labeled rNDEL1 and nonlabeled rDISC598 (Fig. 3 c). Even in the presence of a large excess of rDISC598 (by monomer), including a relative excess of multimers, we could not detect significant amounts of rNDEL1-rDISC598 complexes >500 kDa (Fig. 3 c), suggesting that multimer affinity for NDEL1 is greatly reduced compared with the octamers.
These data were further corroborated by DLS analyses: refolded rDISC316 was measured to be an oligomer with a D H of 15.0 ± 1.0 nm (370 ± 60 kDa), and rNDEL1-rDISC316 formed a monodisperse 1:1 complex with a D H of 19.2 ± 1.0 nm (670 ± 80 kDa) without producing any significant population of larger complexes. In line with these biophysical findings, we have not been able to detect NDEL1 in detergent-insoluble fractions in the postmortem SMRI brains (supplemental Fig. 9, available at www.jneurosci.org as supplemental material) or as copelleting with insoluble DISC1 (Fig. 2 b), indicating that high-molecular weight complexes of DISC1 outside an oligomerization optimum do not bind NDEL1.
Discussion
Our study demonstrates a link between sporadic and familial cases of DISC1-related chronic psychiatric disease by showing that insoluble DISC1 present in brains of a subset of sporadic cases with mental disease leads to dysfunctional molecular interactions similar to what has been described for mutant DISC1 from familial cases. Our findings provide a mechanism for DISC1 dysfunction in a group of sporadic cases of affective disorders or schizophrenia. Using two independent experimental models, we show how rDISC1 multimerization mimicking the starting point for the formation of Sarkosyl-resistant aggregates, disrupts NDEL1 binding and leads to a loss-of-function phenotype. Although DISC1 multimerization does not appear to result from extensive misfolding, it might be triggered by subtle conformational changes and/or other posttranslational modifications that are not easily detected by the cellular protein quality control machinery.
It is possible that loss of interaction of DISC1 with proteins other than NDEL1 contributes to the disease phenotype (Millar et al., 2005). However, the disturbed interaction of mutant DISC1 with NDEL1 was reported to accompany profound changes in cellular distribution and functions (Kamiya et al., 2005) and to induce a severe behavioral phenotype in a transgenic mouse model (Hikida et al., 2007; Li et al., 2007; Pletnikov et al., 2008). We therefore believe that the in vitro dynamics of the DISC1-NDEL1 interaction can serve as an excellent model for DISC1 conformational functionality.
The classical neurodegenerative diseases are grouped together as protein misfolding diseases, because they are characterized by misfolded proteins that are aggregated in different sizes and in different subcellular compartments. Although it has been established that the aggregates have a role in the pathophysiological process, it is currently disputed whether the aggregates themselves are toxic. In fact, extensive aggregation might actually have a protective role by sequestering of toxic species (Taylor et al., 2002; Cohen et al., 2006). We do not think that the type of Sarkosyl-resistant DISC1 aggregates described here are directly comparable with those found in classic misfolding diseases, because we do not have evidence of extensive conformational misfolding of multimeric DISC1, and we do not have evidence of a toxic DISC1 species corresponding to a gain-of-function conformation, as for instance the case with polyglutamine aggregates (Li et al., 1995; Trottier et al., 1995). Our findings, in analogy to familial DISC1 cases, can be sufficiently explained by a loss-of-function phenotype. However, for many aggregated proteins characteristic of degenerative brain conditions, toxic gain-of -function and loss-of-function cannot strictly be separated. In the present case, for example, it cannot be excluded that aggregated DISC1 acquires novel binding to other molecules and thereby could indirectly exert additional influence on DISC1-NDEL1 interactions through a gain-of-function. Whether such toxic gain-of-function of insoluble DISC1 would eventually lead to a subtle degenerative mechanism in selected neurons remains speculative at this point.
Furthermore, it is very likely that compartment-specific DISC1 aggregation can cause different cellular phenotypes and thus clinical outcomes, although the level of overall cellular DISC1 may remain mostly unaffected. Because the possibly uncontrolled multimerization of DISC1 has functional consequences in impairing NDEL1 interaction, the here-described subset of insoluble-DISC1 “positive” cases may nevertheless be categorized as part of a broad spectrum of protein conformational disorders.
Only a subset of clinical cases were rated positive for insoluble DISC1. Insoluble DISC1 was seen in cases of depression, bipolar disorder, and schizophrenia, suggesting that insolublility of specific proteins like DISC1 may be a generalized phenomenon that cuts across traditional diagnostic boundaries. Although our data suggest that ∼20% of sporadic cases of chronic psychiatric disease may involve DISC1 insolubility-associated pathology, the remainder of cases may involve other genes or proteins reflecting the heterogeneity of these phenotypic disease categories. Whether the possible insolubility of such proteins would also be a phenotypic marker remains to be determined. Molecular scenarios of how this causal heterogeneity on the genetic or posttranslational level may converge into the known, broad phenotypical and diagnostic disease categories have been proposed to be common endpoints of disturbed glutamate metabolism (Harrison and Weinberger, 2005), for example.
Our findings parallel the phenotypical heterogeneity of familial DISC1 cases in the original Scottish pedigree (St Clair et al., 1990; Blackwood et al., 2001; Blackwood and Muir, 2004). We find it remarkable that a single mutation can result in such diverse phenotypes as schizophrenia, bipolar disorder, and recurrent depression. In an American family with a DISC1 frameshift mutation, an overlap of schizophrenic and affective symptoms was also reported (Sachs et al., 2005); however, this mutation does only partially segregate with severe mental illness (Green et al., 2006). A direct conclusion from these familial cases is that yet unidentified factors modulate the final specification of the behavioral phenotype independent from the upstream molecular cause. Our findings that insoluble DISC1 is associated with both schizophrenic and affective phenotypes are thus in line with the phenotypic diversity observed in familial DISC1-associated mental disease and add to the mounting evidence that alterations in candidate genes or proteins have clinical phenotypes that cross traditional clinical diagnostic boundaries (Ross et al., 2006; Craddock and Owen, 2007; Owen et al., 2007). Because translocation mutations tend to influence more than just one gene, additional evidence for DISC1-related association with psychiatric phenotypes has been sought (Owen et al., 2007); we believe that our data, for a subset of sporadic cases, contribute to DISC1-associated pathology of psychiatric diseases. A wealth of genetic data has been quoted in support of a novel classification of chronic psychotic disorders dissolving the strict Kraepelinian dichotomy to account for the linkage of single genotypes with mixed phenotypes (Craddock and Owen, 2007; Owen et al., 2007). Our findings now link a subset of sporadic cases with mental disease of mixed phenotypes to DISC1-associated pathology beyond the genetic level in that posttranslationally modified, insoluble DISC1, can serve as a distinguishing biomarker.
In how far is a mere fraction of insoluble DISC1 sufficient to cause a disease phenotype? Patients from the Scottish pedigree are heterozygous for the DISC1 mutation, and given the high penetrance of the phenotype (Blackwood et al., 2001; Blackwood and Muir, 2004), it seems that a loss of 50% of the gene dose is pathogenic (by haploinsufficiency). However, no data have been reported on DISC expression levels in these patients, and compensatory mechanisms may increase gene expression of the healthy allele. The insoluble DISC1 fractions in our investigations represent an average of all insoluble DISC of millions of cells that end up in one Western blot lane. Although our method is extremely sensitive in detecting these aggregates, disease-causing loss of DISC1-NDEL1 interaction may hinge on specific cells that happen to contribute only a few percent of the whole protein content of the homogenate used. If such a scenario would be the case, our method would even underestimate the number of DISC1-“positive” cases of chronic psychiatric disease.
In summary, we demonstrate an association of insoluble DISC1 with sporadic cases of affective disorders and schizophrenia, thus establishing a unique biological signature for a subset of phenotypically diverse cases. Our findings may now lead to a more detailed characterization of insoluble DISC1-positive cases not restricted to phenotypical psychiatric categories. By defining these patient subgroups, specific causal pharmacotherapies aimed at restoring or compensating DISC1-insolublilty related dysfunction are becoming a possibility.
Footnotes
This work was supported by the Stanley Medical Research Institute (Baltimore, MD) (C.K.). S.R.L. was supported by a grant from the German Research Foundation (Deutsche Forschungsgemeinschaft). Postmortem brain tissue was donated by The Stanley Medical Research Institute brain collection, courtesy of Drs. Michael B. Knable, E. Fuller Torrey, Maree J. Webster, and Robert H. Yolken.
References
- an der Heiden W, Hafner H. The epidemiology of onset and course of schizophrenia. Eur Arch Psych Clin Neurosci. 2000;250:292–303. doi: 10.1007/s004060070004. [DOI] [PubMed] [Google Scholar]
- Berrettini W. Evidence for shared susceptibility in bipolar disorder and schizophrenia. Am J Med Genet. 2003;123:59–64. doi: 10.1002/ajmg.c.20014. [DOI] [PubMed] [Google Scholar]
- Blackwood DH, Muir WJ. Clinical phenotypes associated with DISC1, a candidate gene for schizophrenia. Neurotox Res. 2004;6:35–41. doi: 10.1007/BF03033294. [DOI] [PubMed] [Google Scholar]
- Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ. Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet. 2001;69:428–433. doi: 10.1086/321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua SE, McKenna PJ. A skeptical view of the neuropathology of schizophrenia. In: Harrison PJ, Roberts GW, editors. The neuropathology of schizophrenia. Oxford: Oxford UP; 2000. pp. 291–337. [Google Scholar]
- Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK. The DISC locus in psychiatric illness. Mol Psychiatry. 2008;13:36–64. doi: 10.1038/sj.mp.4002106. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Craddock N, Owen MJ. Rethinking psychosis: the disadvantages of a dichotomous classification now outweigh the advantages. World Psychiatry. 2007;6:20–27. [PMC free article] [PubMed] [Google Scholar]
- Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, Kitabatake Y, Liu XB, Yang CH, Jordan JD, Ma DK, Liu CY, Ganesan S, Cheng HJ, Ming GL, Lu B, Song H. Disrupted-in-schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007;130:1146–1158. doi: 10.1016/j.cell.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzen B, Yang Y, Sunnemark D, Wickman M, Ottervald J, Oppermann M, Sandberg K. Dihydropyrimidinase related protein-2 as a biomarker for temperature and time dependent post mortem changes in the mouse brain proteome. Proteomics. 2003;3:1920–1929. doi: 10.1002/pmic.200300535. [DOI] [PubMed] [Google Scholar]
- Green EK, Norton N, Peirce T, Grozeva D, Kirov G, Owen MJ, O'Donovan MC, Craddock N. Evidence that a DISC1 frame-shift deletion associated with psychosis in a single family may not be a pathogenic mutation. Mol Psychiatry. 2006;11:798–799. doi: 10.1038/sj.mp.4001853. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. image 45. [DOI] [PubMed] [Google Scholar]
- Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, Wu D, Xue R, Andrade M, Tankou S, Mori S, Gallagher M, Ishizuka K, Pletnikov M, Kida S, Sawa A. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci USA. 2007;104:14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulshoff Pol HE, Schnack HG, Mandl RC, van Haren NE, Koning H, Collins DL, Evans AC, Kahn RS. Focal gray matter density changes in schizophrenia. Arch Gen Psychiatry. 2001;58:1118–1125. doi: 10.1001/archpsyc.58.12.1118. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Paek M, Kamiya A, Sawa A. A review of disrupted-in-schizophrenia-1 (DISC1): neurodevelopment, cognition, and mental conditions. Biol Psychiatry. 2006;59:1189–1197. doi: 10.1016/j.biopsych.2006.03.065. [DOI] [PubMed] [Google Scholar]
- James R, Adams RR, Christie S, Buchanan SR, Porteous DJ, Millar JK. Disrupted in schizophrenia 1 (DISC1) is a multicompartmentalized protein that predominantly localizes to mitochondria. Mol Cell Neurosci. 2004;26:112–122. doi: 10.1016/j.mcn.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, Sawamura N, Park U, Kudo C, Okawa M, Ross CA, Hatten ME, Nakajima K, Sawa A. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- Katsel P, Davis KL, Gorman JM, Haroutunian V. Variations in differential gene expression patterns across multiple brain regions in schizophrenia. Schizophr Res. 2005;77:241–252. doi: 10.1016/j.schres.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- Law AJ, Shannon Weickert C, Hyde TM, Kleinman JE, Harrison PJ. Neuregulin-1 (NRG-1) mRNA and protein in the adult human brain. Neuroscience. 2004;127:125–136. doi: 10.1016/j.neuroscience.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Li W, Zhou Y, Jentsch JD, Brown RA, Tian X, Ehninger D, Hennah W, Peltonen L, Lonnqvist J, Huttunen MO, Kaprio J, Trachtenberg JT, Silva AJ, Cannon TD. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci USA. 2007;104:18280–18285. doi: 10.1073/pnas.0706900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XJ, Li SH, Sharp AH, Nucifora FC, Jr, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- McDonald C, Bullmore ET, Sham PC, Chitnis X, Wickham H, Bramon E, Murray RM. Association of genetic risks for schizophrenia and bipolar disorder with specific and generic brain structural endophenotypes. Arch Gen Psychiatry. 2004;61:974–984. doi: 10.1001/archpsyc.61.10.974. [DOI] [PubMed] [Google Scholar]
- Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, Clair DM, Muir WJ, Blackwood DH, Porteous DJ. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, Malloy MP, Chubb JE, Huston E, Baillie GS, Thomson PA, Hill EV, Brandon NJ, Rain JC, Camargo LM, Whiting PJ, Houslay MD, Blackwood DH, Muir WJ, Porteous DJ. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- Narr KL, Toga AW, Szeszko P, Thompson PM, Woods RP, Robinson D, Sevy S, Wang Y, Schrock K, Bilder RM. Cortical thinning in cingulate and occipital cortices in first episode schizophrenia. Biol Psychiatry. 2005;58:32–40. doi: 10.1016/j.biopsych.2005.03.043. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Shu T, Sanada K, Lariviere RC, Tseng HC, Park SK, Julien JP, Tsai LH. A NUDEL-dependent mechanism of neurofilament assembly regulates the integrity of CNS neurons. Nat Cell Biol. 2004;6:595–608. doi: 10.1038/ncb1139. [DOI] [PubMed] [Google Scholar]
- Owen MJ, Craddock N, Jablensky A. The genetic deconstruction of psychosis. Schizophr Bull. 2007;33:905–911. doi: 10.1093/schbul/sbm053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozeki Y, Tomoda T, Kleiderlein J, Kamiya A, Bord L, Fujii K, Okawa M, Yamada N, Hatten ME, Snyder SH, Ross CA, Sawa A. Disrupted-in-schizophrenia-1 (DISC-1): mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc Natl Acad Sci USA. 2003;100:289–294. doi: 10.1073/pnas.0136913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH, Ross CA. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:173–186. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Shattuck lecture–neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;52:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Sachs NA, Sawa A, Holmes SE, Ross CA, DeLisi LE, Margolis RL. A frameshift mutation in disrupted in schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol Psychiatry. 2005;10:758–764. doi: 10.1038/sj.mp.4001667. [DOI] [PubMed] [Google Scholar]
- Sawamura N, Sawamura-Yamamoto T, Ozeki Y, Ross CA, Sawa A. A form of DISC1 enriched in nucleus: altered subcellular distribution in orbitofrontal cortex in psychosis and substance/alcohol abuse. Proc Natl Acad Sci USA. 2005;102:1187–1192. doi: 10.1073/pnas.0406543102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, Gosden C, Evans HJ. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, Gunnarsdottir S, Ivarsson O, Chou TT, Hjaltason O, Birgisdottir B, Jonsson H, Gudnadottir VG, Gudmundsdottir E, Bjornsson A, Ingvarsson B, Ingason A, Sigfusson S, Hardardottir H, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- Torrey EF, Webster M, Knable M, Johnston N, Yolken RH. The Stanley foundation brain collection and neuropathology consortium. Schizophr Res. 2000;44:151–155. doi: 10.1016/S0920-9964(99)00192-9. [DOI] [PubMed] [Google Scholar]
- Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, Agid Y, Brice A, Mandel J-L. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]