

Abstract
Little is known about molecular changes occurring within retinal ganglion cells (RGCs) before their death in glaucoma. Taking advantage of the fact that γ-synuclein (Sncg) mRNA is expressed specifically and highly in adult mouse RGCs, we show in the DBA/2J mouse model of glaucoma that there is not only a loss of cells expressing this gene, but also a downregulation of gene expression of Sncg and many other genes within large numbers of RGCs. This downregulation of gene expression within RGCs occurs together with reductions in FluoroGold (FG) retrograde transport. Surprisingly, there are also large numbers of Sncg-expressing cells without any FG labeling, and among these many that have a marker previously associated with disconnected RGCs, accumulation of phosphorylated neurofilaments in their somas. These same diseased retinas also have large numbers of RGCs that maintain the intraocular portion while losing the optic nerve portion of their axons, and these disconnected axons terminate within the optic nerve head. Our data support the view that RGC degeneration in glaucoma has two separable stages: the first involves atrophy of RGCs, whereas the second involves an insult to axons, which causes the degeneration of axon portions distal to the optic nerve head but does not cause the immediate degeneration of intraretinal portions of axons or the immediate death of RGCs.
Keywords: retinal ganglion cell, axonal transport, axoplasmic transport, glaucoma, synuclein, axonopathy, DBA/2J
Introduction
Glaucoma, the second leading cause of blindness worldwide (Quigley and Broman, 2006), is a group of retinal neuropathies that usually occur late in life and involve the progressive degeneration of retinal ganglion cells (RGCs). The physical excavation of the optic nerve head (ONH), which is caused by the degeneration of intraocular RGC axons and the remodeling of glia and extracellular matrix, is the distinctive clinical characteristic and criteria for glaucoma diagnosis (Quigley, 1985). The increase in intraocular pressure (IOP) that is often associated with glaucoma usually begins years before the nerve fiber degeneration can be measured by current technologies. Visual field loss occurs later in the disease when large numbers of RGCs already have been lost (Kerrigan-Baumrind et al., 2000).
In the past few years, DBA/2J mice have become a popular animal model to study glaucoma because of some key similarities to the human disease (John, 2005). First, IOP elevation in these animals is not only spontaneous and progressive, but also age-related (John et al., 1998; Libby et al., 2005b). Second, the elevation of IOP triggers a progressive degeneration of retinal axons and atrophic excavation of the optic nerve head months after the increase in IOP (John et al., 1998; Libby et al., 2005b). Third, the degeneration of RGCs and their axons is not uniform across the retina but rather asynchronous and sectorial (Jakobs et al., 2005; Filippopoulos et al., 2006; Schlamp et al., 2006).
Most studies that have counted the number of RGCs in animal models of glaucoma have done so by injection of tracers such as FluoroGold (FG) into areas of the brain innervated by RGCs. However, because axonal transport has been shown to be affected in animal models of glaucoma (Anderson and Hendrickson, 1974; Minckler et al., 1976; Quigley and Anderson, 1977; Quigley et al., 1981; Pease et al., 2000), and retrograde tracers such as FG rely on active transport to get back to cell bodies (Kobbert et al., 2000), the use of FG cannot discriminate between loss of transport, loss of axons, and loss of cells. Here, we describe a molecular marker, γ-synuclein (Sncg) mRNA, that is suitable for quantitative studies of RGC numbers and gene expression and that identifies RGCs regardless of their connectivity to central targets. We also describe another marker, somatic accumulation of phosphorylated neurofilaments (pNFs), which identifies a subset of RGCs that are disconnected from their targets. With these markers, we show that the degeneration of RGCs in DBA/2J mice is characterized by two processes: widespread, progressive, and correlated decreases in gene expression and retrograde transport, as well as a later loss of distal but not proximal portions of axons, which does not result in the immediate loss of gene expression within RGCs. The extent of the gene downregulation and the presence of large numbers of disconnected RGCs are surprising findings that have important implications for the study and treatment of glaucoma.
Materials and Methods
Animals.
All experiments with mice were performed in accordance with the Association for Research in Vision and Ophthalmology statement for the use of animals in research under animal protocols approved by the Animal Care and Use Committees at The Johns Hopkins University and University of Washington. DBA/2J and C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed at either The Johns Hopkins University or housed and bred at the University of Washington in covered cages, fed with a standard rodent diet ad libitum, and maintained in a 12 h light/dark cycle.
Retrograde labeling of RGCs.
The mice were anesthetized with Avertin (1.3% tribromoethanol and 0.8% tert-amyl alcohol), and their heads were shaved and swabbed in Betadine. Using a Kopf (David Kopf Instruments, Tujunga, CA) stereotaxic device equipped with a mouse adapter, a midline incision was made in the scalp. Bilateral holes were drilled at −4.0 mm from bregma and ±0.5 mm from midline. Using a Hamilton (Reno, NV) syringe, 2 μl of 1% FG (hydroxystilbamidine; Invitrogen, Carlsbad, CA) in dH2O was injected 0.5 mm below the dura into both superior colliculi over the course of 2 min for each injection. After slow removal of the syringe, gelfoam pledgettes soaked in 5% FG were placed on the surface of the superior colliculi. After scalp suture with 4-0 silk, mice were placed on a warm blanket for recovery from anesthesia. One or 4 weeks after FG injection, mice were killed with 300 mg/kg sodium pentobarbitol followed by transcardial perfusion with 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer, pH 7.4. Retinas were dissected out immediately after perfusion, placed flat between glass coverslips, and immersion-fixed overnight in cold 4% PFA.
Quantitative reverse transcription-PCR.
Reverse transcriptase reactions were performed starting with 500 ng of total RNA for each sample and using the reverse transcription portion of a SuperScript III Platinum Two-Step quantitative reverse transcription-PCR (qRT-PCR) (Invitrogen) reaction to produce single-stranded cDNA. The real-time PCRs were completed using Platinum SYBR Green PCR qPCR SuperMix-UDG from the kit (Invitrogen) using the following primer set to amplify Sncg: GTCTCAACCTGGCACACTGAATG and AGAGGACCATAGGGTAAAAGGAGC. All qRT-PCR was performed in triplicate using an Applied Biosystems (Foster City, CA) Prism 7700 Sequence Detection System. Cycling conditions were as follows: 95°C for 10 min and cycles of 95°C for 15 s, 55°C for 20 s, and 72°C for 40 s. SDS2.1 software (Applied Biosystems) was used to visualize the data. The standard curve method described in Applied Biosystems User Bulletin no. 2 was used to determine relative changes in gene expression levels using β-actin as a reference, which did not change significantly in our samples.
In situ hybridization.
Digoxigenin (Dig) and FITC-labeled probes for Sncg (IMAGE 1448798) and GAD67 (IMAGE 5358787), and Dig-labeled probes for Nfl (IMAGE 4506903), Nf66 (IMAGE 4502421), tubulin β-III (IMAGE 5363050), Brn3b (IMAGE 5692570), Brn3a (IMAGE 1749761), Brn3c (IMAGE 40131244 and CX729528), AchRB2 (IMAGE 6838257), Hsp90 (IMAGE 6491307), FKB12 (IMAGE 3498682), PPIA (IMAGE 6442005), Thy1 (IMAGE 6312210), Tau (IMAGE 4504850), and TrkB (IMAGE 5707891) were synthesized and hydrolyzed by standard procedures. For in situ hybridization in retinal cryostat sections, animals were killed by CO2 inhalation or 300 mg/kg sodium pentobarbital, followed by transcardial perfusion with 4% PFA in PBS. Eyes were removed and postfixed in 4% PFA overnight, cryoprotected in 30% sucrose, and embedded in OCT. Frozen sections (10 μm) were subjected to digestion with proteinase K (20 μg/ml in PBS), postfixation (4% PFA; 10 min) and acetylation with 0.25% acetic anhydride in 0.1 m triethanolamine (TEA). Each of these steps was intercalated by PBS washes, and after the last wash the sections were incubated overnight at 65°C with hybridization solution [50% formamide, 1× Hybe solution (Sigma, St. Louis, MO), 1 mg/ml yeast RNA] containing 1 μg/ml Dig- and FITC-labeled riboprobes. After hybridization, slides were washed with 0.2× SSC at 72°C for 1 h, and endogenous peroxidases were quenched with a solution of 0.1% sodium azide and 0.3% hydrogen peroxide for 10 min. Bound probes were detected either with an AP-conjugated anti-Dig antibody followed by NBT/BCIP (nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate) reaction (Roche, Indianapolis, IN), or with peroxidase (POD)-conjugated anti-Dig and anti-FITC antibodies (Roche) followed by Cy-3 (PerkinElmer, Boston, MA) and Alexa 488 tyramides (Invitrogen). The antibody incubations and tyramides reactions were done separately for each probe, with an incubation of the sections with 0.1% sodium azide and 0.3% hydrogen peroxide for 25 min after the first tyramide reaction to inactivate the first POD enzyme. The sections were stained with 4′,6′-diamidino-2-phenylindole (DAPI) and coverslipped with Aqua PolyMount (Polysciences, Warrington, PA). For flat-mounted retinas, the eyes were enucleated and the retinas were dissected out from the globes, and flat-mounted after symmetrical incisions. The retinas were immediately fixed in buffered 4% PFA overnight at 4°C with gentle rocking, washed with PBT (PBS, 0.5% Tween 20) and stored in 100% methanol at −20°C until used for in situ hybridization. Flat-mounted retinas were rehydrated with descending series of graded methanol into PBT. After two PBT washes, the retinas were immobilized between two small heat-sealed sheets of 125 μm nylon mesh (Small Parts, Miami Lakes, FL). The tissue was incubated in 0.5% of acetic anhydride in TEA buffer, washed with PBT, and incubated for 1 h with hybridization solution [50% formamide, 1× Hybe solution (Sigma), 1 mg/ml yeast RNA, 0.5% Tween 20, 0.5% Chaps (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate)] at 65°C with gentle rocking. After prehybridization, the retinas were incubated in hybridization solution containing 1 μg/ml for each riboprobe overnight at 65°C with gentle rocking. The retinas were washed with descending series of graded hybridization solution into 2× SSC for 15 min each at 65°C. Also at this temperature, two additional 30 min washes of 0.2× SSC were done. Retinas were bleached in a solution containing 0.1% sodium azide and 0.3% of hydrogen peroxide for 45 min, rinsed with PBT, and preblocked with for 1 h in PBT plus 1% blocking reagent (Roche). Then the retinas were incubated 2 nights at 4°C with fresh blocking solution containing a 1:300 dilution of the POD-conjugated anti-Dig antibody. After PBT washes, Cy-3 or Alexa 488 tyramides were used for fluorescence signal detection. In double labeling in situ hybridizations of retina flat mounts, the retinas were incubated for 1 h in the 0.1% sodium azide plus 0.3% of hydrogen peroxide solution after the first tyramide detection to inactivate the first POD enzyme. After PBT washes, the tissues were preblocked again and incubated for two nights in the POD-conjugated anti-FITC antibody at 4°C. The Alexa 488 tyramide system was used to develop the second probe. For the detection of nuclei in tissues subjected to in situ hybridization, retinas were counterstained with DAPI or processed by a modified terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling procedure. For this, retinas were treated for 1 h at 37°C with 300 U/ml DNaseI, washed with PBT, and after preincubation in the terminal transferase (TdT) reaction buffer were incubated with 40 U/μl TdT enzyme and biotin-16-dUTP (Roche) for 4 h at 45°C. The reaction was stopped with a buffer containing 300 mm NaCl and 30 mm sodium citrate, and subsequently washed with PBT. For detection, the retinas were incubated with 7-amino-4-methylcoumarin-3-acetic acid (AMCA)-conjugated streptavidin (Jackson ImmunoResearch, West Grove, PA) overnight at 4°C.
Immunohistochemistry.
After the in situ hybridization procedure, the retinas were removed from the nylon mesh and incubated for three nights at least in one or two of the following primary antibodies: one of two mouse monoclonal antibodies against phosphorylated neurofilaments, SMI31 (Covance, Berkeley, CA) or 2F11 (DakoCytomation, Carpinteria, CA), together with rabbit polyclonal antibodies against tubulin β-III (TUJ1; Sigma), GFAP (Advanced Immunochemical, Long Beach, CA), FluoroGold (Chemicon International, Temecula, CA), or Brn3 (C-13; Santa Cruz Biotechnology, Santa Cruz, CA). The antibodies were diluted in PBT (1% Tween 20) containing 10% of normal goat serum. After incubation with the primary antibodies, the retinas were washed with PBT and incubated in the respective secondary antibodies for 2 h each. After washes with PBT, the flat-mount retinas were mounted in PolyMount. For optic nerve immunolabeling, optic nerves from perfused animals were dissected and fixed overnight in 4% PFA. After cryoprotection in 30% sucrose, the optic nerves were frozen and 10 μm cryostat sections were cut. Optic nerve sections were incubated for 2 h in either mouse monoclonal antibodies 2F11 (DakoCytomation) or 3A10 (Developmental Studies Hybridoma Bank, Iowa City, IA) and in the rabbit polyclonal antibody TUJ1 at room temperature. After washes with PBT, the optic nerve sections were incubated in secondary antibodies for 1 h, washed in PBT, counterstained with DAPI, and mounted with PolyMount.
Quantitative imaging.
Automated counting of RGCs in retinas was performed using protocols similar to those described by others (Filippopoulos et al., 2006; Li et al., 2007; Zhong et al., 2007), except that we used Sncg expression to identify RGCs. Flat-mounted retinas were imaged using a 0.4 numerical aperture Zeiss 20× objective and a motorized Zeiss 200M inverted microscope (Carl Zeiss, Oberkochen, Germany) equipped with a Linear Encoded Flat Top Zeiss 200 Ludl motorized stage (Ludl, Hawthorne, NY), Roper Scientific Coolsnap FX digital camera (Photometrics, Tucson, AZ), and Excite fluorescence source (EXFO Burleigh, Victor, NY). A custom script written for IPlab (Becton Dickinson, Rockville, MD) was used to control the microscope, camera, and stage. Retinas were imaged in their entirety by adjacent nonoverlapping frames, with autofocusing on the ganglion cell layer based on the expression of Sncg. Exposure times were nonsaturating and held constant for retina sets that would be the subject of internal comparisons. The fluorophores and fluorescence Zeiss filter cubes used were as follows: DAPI and AMCA detected with Zeiss filter set 49 [excitation (Ex.), G365; split, FT395; emission (Em.), BP445/50], Alexa 488 detected with filter Zeiss set 38 (Ex., BP470/40; split, FT495; Em., BP525/50), Cy-3 detected with Zeiss filter set 43 HE (Ex., BP550/25; split, FT570; Em., BP605/70), and Cy-5 detected with Chroma Technology filter set HQ:Cy5 (Ex., HQ620/60; split, Q660lp; Em., HQ700/75). Before image quantification, composite images for each of the four fluorescence channels were generated from half-sized images (10× equivalent). The composites were used to identify the position of the optic nerve, the contour of the retina, as well as the contour of the retina that could be measured (excluding curled edges), and used to develop a region of interest (ROI) mask to exclude internal regions in each of the four channels that might produce quantification artifacts, such as rips or other distortions in the tissue and an occasional highly fluorescent nonbiological material. The composites, overlaid with a grid, also were used to mark the position and category (weak expressing or strong expressing) of each RGC with somatic accumulation of pNFs. The ROI and pNF masks were magnified to twice their size before being applied to the original unprocessed images for subsequent quantification steps. Each set of original ∼200 20× image files, containing the same field imaged in all four channels, was opened and quantified sequentially. RGCs were identified based on the expression of Sncg mRNA in all cases except the retinas shown in Figure 4, B and E, which used FG to identify RGCs. Objects considered RGCs were identified by threshold criteria: intensity over background within that same image after local background subtraction and smoothening functions. Optimal identification of cells was obtained by applying higher threshold values in the periphery and a calculated graded value for images between retina periphery and center. All retinas belonging to the same experiment used identical threshold values. Because of the overlap of RGC somas, particularly in the center of the retina, it was necessary to break up large objects using the IPlab erode function; a useful criteria for separating overlapping cells was empirically determined to be an area of ∼136 μm2 as too large to be single cells. An additional cutoff value of ∼2 μm was applied to exclude objects (e.g., axons and background) that were too small to be RGC somas. Using these settings, the program reproducibly identified ∼60–80,000 RGCs in young retinas. For each cell, the area and centroid position were measured, as well as mean intensity values. The mean fluorescence values for each cell that are presented here represent the mean intensity of pixels within the cell after subtraction of the mean fluorescence of pixels within the ROI of that image that did not contain cells (local background). Total fluorescence values of each cell, a measurement that more closely represents the amount of mRNA per cell, were calculated as the mean fluorescence value multiplied by the number of pixels for each cell. Quantification results for each set of images were saved as sets of tabulated data that were subsequently combined and further processed using IPlab scripts. For example, scripts were used to calculate the xy coordinates of each cell within the retina, and to calculate the z-scores: the observed intensity for each cell in the retina minus the mean retina intensity, divided by the SD of the retina intensities. The calculation of nearest neighbor distance (NND) was done using a sequence of custom scripts written for Perl, R, and C programming languages. All data were analyzed statistically using Partek Software (Partek, St. Louis, MO) or Excel (Microsoft, Redmond, WA), and graphed using Parkek, IPlab, Excel, and CA-CricketGraphIII (Computer Associates, Islandia, NY) software.
Figure 4.
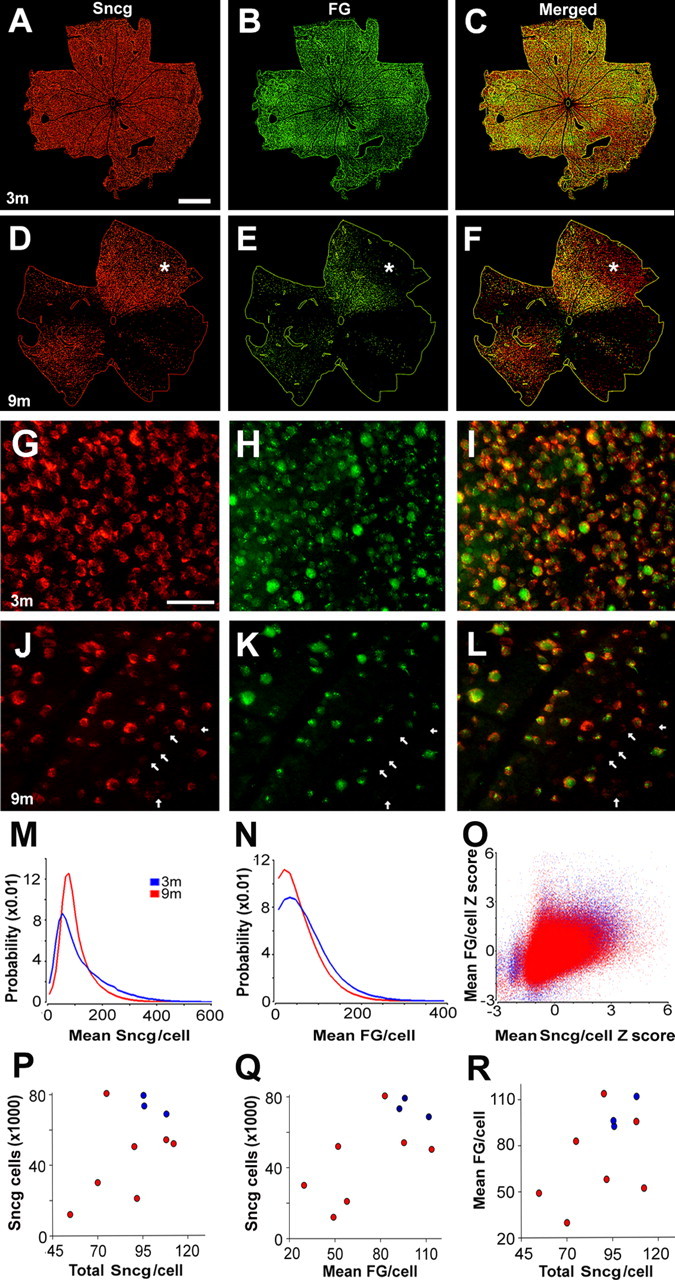
Sectorial loss of Sncg-expressing cells and decreased Sncg expression occur together with decreases in FG retrograde labeling. A–F, Binarized images show RGCs identified either by their Sncg expression (red) or their FG labeling (green) in 3 month (A–C) and 9 month (D–F) DBA/2J mice. The asterisks in the retina from the 9 month DBA/2J (D–F) show an area with fewer FG-labeled cells than Sncg-expressing cells. The traces in the binarized images mark the outer edges of retina flat mounts used for analyses as well as position of the ONH. G–L, Sncg expression (G, J) and FG labeling (H, K) shown alone, and together (I, L), show variable intensities in different RGCs of 3 month (G–I) and 9 month (J–L) DBA/2J mice. The arrows in the 9 month retinas (J–L) mark RGCs with minimal expression of Sncg mRNA and little or no FG labeling. M, Probability distribution of mean Sncg expression per cell in retinas from 3 month (blue; n = 183,334 cells; n = 3 retinas) and 9 month (n = 256,837 cells; n = 7 retinas) DBA/2J mice. N, Probability distribution of mean FG labeling per cell (using the same cells shown in M) in the retinas from 3 month (blue) and 9 month (red) DBA/2J mice. O, Positive correlation between Sncg mRNA expression and FG retrograde labeling per cell (using the same cells shown in M and N), expressed as Z-scores (value minus the mean for that retina, divided by the retina SD) in the retinas of 3 month (blue) and 9 month (red) DBA/2J mice. P, Correlation between the retina mean value of total Sncg per cell and the total number of Sncg-expressing cells per retina of 3 month (blue dots) and 9 month (red dots) DBA/2J mice. Q, Correlation between the retina mean value of mean FG fluorescence per cell and the total number of Sncg-expressing cells per retina of 3 month (blue dots) and 9 month (red dots) DBA/2J mice. R, Correlation between the retina mean value of the total Sncg per cell and retina mean value of the mean FG per cell in the retinas of 3 month (blue dots) and 9 month (red dots) DBA/2J mice. m, Month. Scale bars: A–F, 500 μm; G–L, 50 μm.
Results
Sncg mRNA is expressed specifically in the majority or all adult mouse RGCs
A candidate-based search for mRNAs expressed highly and specifically in RGCs of adult mice, conducted by in situ hybridization of cryostat sections of retinas from control C57BL/6J mice, identified Sncg (also known as Persyn) as a gene with robust and specific mRNA expression in adult mouse RGCs. As evident by low (Fig. 1A)- and high (Fig. 1B)-magnification views of retinas from 3 month DBA/2J mice, Sncg mRNA was found only in the ganglion cell layer (GCL) and a very small number of cells in the inner nuclear layer, consistent with the known small number of displaced RGCs in the mouse retina (Dräger and Olsen, 1981). To determine whether Sncg was expressed only in RGCs and not in displaced amacrine cells within the GCL, double in situ hybridization experiments using fluorescence-based detection were performed on retina cryostat sections (Fig. 1 C–E) and flat mounts (Fig. 1 F–H), using probes for Sncg and the GABA synthetic enzyme 67 kDa glutamic acid decarboxylase (GAD67; Gad1), a gene expressed in a fraction of amacrine cells found in the GCL (May et al., 2007). In retina sections, the GAD67 probe labeled cells in the inner one-third of the inner nuclear layer, the known location of the majority of amacrine cells, as well as cells within the GCL, which were smaller than the cells labeled by the Sncg probe (Fig. 1 C–E). In retina flat mounts focusing on the GCL, comparable numbers of the larger Sncg-expressing cells and the smaller GAD67-expressing cells were observed intermingled (Fig. 1 F–H). Quantification of retina flat-mount images showed that Sncg and GAD67 were expressed in similar numbers of cells, that no cells coexpressed both markers, and that only 20% of GCL cells expressed neither marker (Fig. 1I). The fraction of GCL cells expressing Sncg (45%) matched well with previous estimates of the fraction of mouse GCL cells that are RGCs (41–44%) (Jeon et al., 1998). To confirm that the great majority of RGCs express Sncg, similar analyses were performed using retina sections (data not shown) and flat mounts from 3 month DBA/2J mice in which RGCs had been labeled by injection of FG into both superior colliculi. Both visual inspection (Fig. 1J) and quantification (Fig. 1K) showed a near-perfect correspondence between the cells labeled by FG and the cells that expressed Sncg. Sncg mRNA stands in contrast to another RGC marker, Brn3b, which is only expressed in about one-third of RGCs in the retina of adult mice (Senatorov et al., 2006). Whereas an antibody that recognizes all Brn3s labels a greater fraction of RGCs, these are still only a subset of Sncg-positive cells in the retinas of both young and old DBA/2J including many large cells that can only be RGCs (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Similarly, Sncg mRNA labels a greater proportion of RGCs than Brn3a mRNA (data not shown), Brn3b mRNA (Fig. 2C), and Brn3c mRNA (no cells) (data not shown). Thus, in stark contrast to Brn3 mRNA or protein reagents, in addition to providing a much higher signal-to-noise, Sncg mRNA also identifies more RGCs within the adult mouse retina. Together, these results demonstrate that Sncg mRNA is an optimal Nissl-like marker for the majority if not all RGCs within the adult mouse retina.
Figure 1.
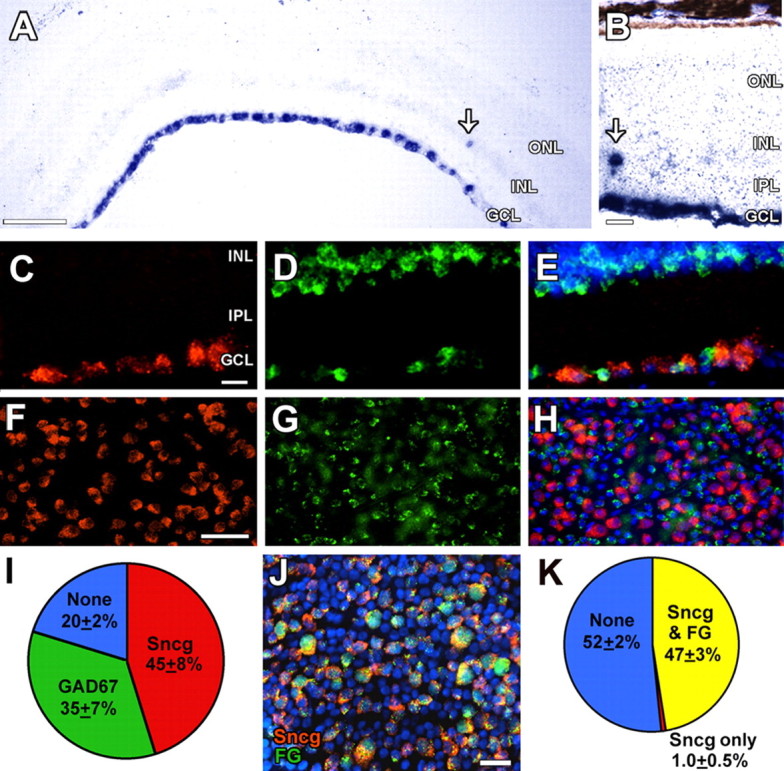
Sncg mRNA is expressed specifically and abundantly in adult mouse RGCs. A, Low-power view of a retina from a 3 month DBA/2J mouse after in situ hybridization detection of Sncg mRNA shows expression in the GCL. B, Higher-power view of an overdeveloped in situ hybridization shows Sncg mRNA only in the GCL and an occasional cell in the inner nuclear layer. C–H, Expression of Sncg (C, F), GAD67 (D, G) mRNAs and merged views together with nuclei in blue (E, H) in cryostat sections (C–E) and flat mounts (F–H) of retinas from C57BL/6J mice. I, Pie chart showing the percentage of GCL cells that express Sncg mRNA (red), GAD67 mRNA (green), and cells that express neither marker (blue), as determined from retina flat-mount images (n = 2260 cells; n = 3 retinas). J, Colocalization of Sncg mRNA (red) and FG retrograde tracer labeling (green), together with nuclei (blue). K, Pie chart of the percentage of GCL cells with both Sncg expression and FG labeling (yellow), Sncg expression only (red), and cells without either marker (blue), as determined from retina flat-mount images (n = 1465 cells; n = 4 retinas). The arrows (A, B) mark displaced ganglion cells. ONL, Outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer. Scale bars: A, 100 μm; B–E, J, 20 μm; F–H, 50 μm.
Figure 2.
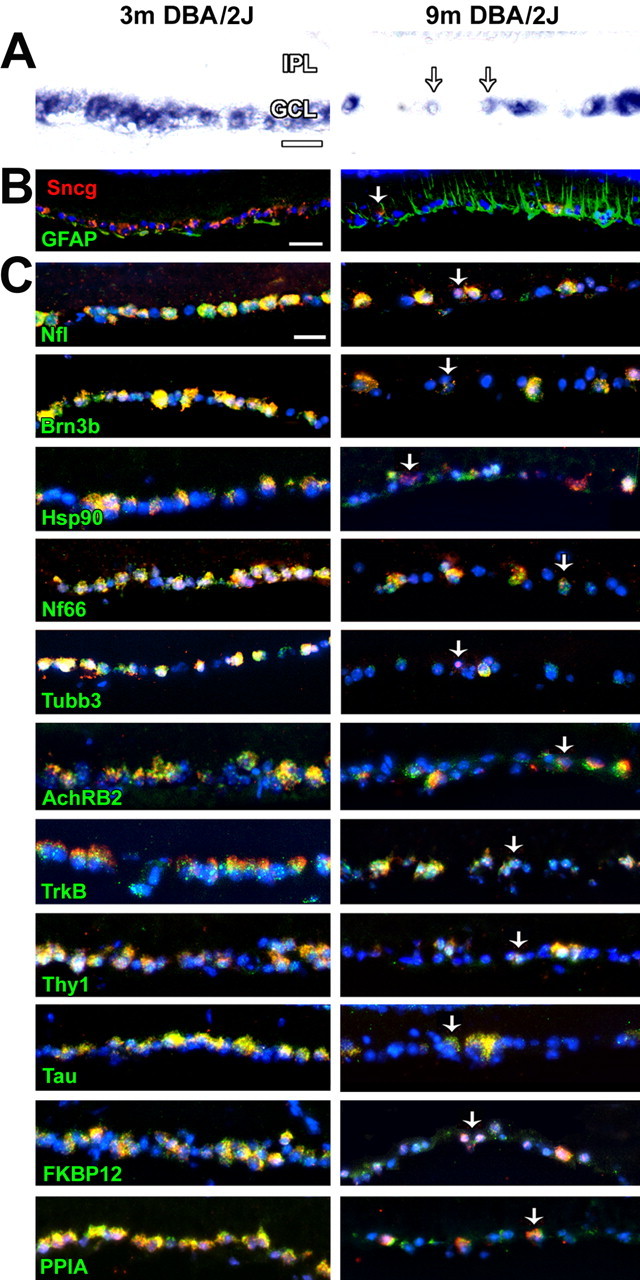
Gene expression is downregulated within RGCs in affected regions of DBA/2J retinas. A, Sncg mRNA expression in retina sections of 3 and 9 month DBA/2J mice. B, Sncg mRNA (red), GFAP immunoreactivity (green), and nuclei staining (DAPI; blue) in retina sections of 3 and 9 month DBA/2J mice. C, Colocalization of Sncg mRNA (red) with RGC-specific genes (green) Nfl, Brn3b, and Hsp90; genes that are enriched in but not specific to RGCs, Nf66, tubulin β-III (Tubb3), AchRB2, TrkB, Thy1, and Tau; and genes that have widespread or ubiquitous expression, FKBP12 and PPIA, in retina sections of 3 and 9 month DBA/2J mice shown together with nuclei staining (DAPI; blue). The arrows (A–C) mark RGCs with reduced expression of Sncg. m, Month; IPL, inner plexiform layer. Scale bars: A–C, 20 μm.
We next sought to determine whether Sncg mRNA could be used to identify retinas with RGC degeneration by quantitative PCR (qRT-PCR). For this purpose, we compared retinas from 3 and 9 month DBA/2J mice (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Sncg mRNA was downregulated (p < 0.03) in the group of 9 month DBA/2J retinas (n = 17) when compared with the 3 month DBA/2J retinas (n = 6). However, the variability in the amount of Sncg expression in the 9 month retinas was very large, with most retinas having values that overlap the range of values seen in the 3 month retinas. Such a large range in the measurement of individual variables is common in DBA/2J mice because of the variable phenotype, and particularly between 9 and 12 months of age presumably because this is the most dynamic phase of degeneration (Libby et al., 2005b). These qRT-PCR results show that, even with a truly specific RGC marker that is expressed at very high levels in RGCs, qRT-PCR is an insensitive tool at measuring RGC degeneration in DBA/2J unless very large sample sizes are used. In addition, qRT-PCR cannot identify retinas with small sectors of degeneration, nor can it determine whether the reduction of Sncg mRNA observed in some 9 month retinas is attributable to a loss of RGCs, a decline in the expression of Sncg per cell, or a combination of both factors.
Gene expression is decreased within RGCs in glaucomatous regions of DBA/2J retinas
In situ hybridization studies in cryostat sections of 3 and 9 month DBA/2J retinas demonstrated not only a significant loss of Sncg-expressing cells in some retina sections of affected old animals but also the downregulation of Sncg mRNA in some of the remaining RGCs (Fig. 2A). Regions of the GCL without Sncg-expressing cells or with RGCs expressing low levels of Sncg were found in patches, consistent with the reported sectorial pattern of degeneration in DBA/2J mice (Jakobs et al., 2005; Filippopoulos et al., 2006; Schlamp et al., 2006). To determine whether the patches of lost and reduced Sncg expression coincided with regions undergoing the reactive gliosis recently reported in the DBA/2J mouse (Inman and Horner, 2007), retina sections subjected to in situ hybridization were analyzed for GFAP immunoreactivity. Whereas GFAP protein expression was confined to astrocytes in the retinas of 3 month DBA/2J, strong GFAP was observed also in Müller cells in the regions of 9 month DBA/2J retinas with reduced Sncg expression (Fig. 2B). Increases in the expression of GFAP within Müller cells also were observed at the level of mRNA with in situ hybridization (data not shown). To determine whether the downregulation of Sncg expression within individual RGCs was specific to this one gene, or whether it heralded a more global downregulation of gene expression in affected RGCs, the mRNA expression of other genes expressed in RGCs were also analyzed together with Sncg in the retinas of 3 and 9 month DBA/2J mice. Double probe in situ hybridization experiments (Fig. 2C) demonstrated that RGCs with Sncg mRNA downregulation also had downregulation of mRNAs for genes that in the adult mouse retina are specific to RGCs, such as neurofilament light (Nfl), the transcription factor Brn3b (Pou4F2), and heat shock protein 90 (Hsp90), genes that are enriched in RGCs but also expressed in other retina cells, such as neurofilament-66 (Ina or NF66), tubulin β-III (Tubb3), nicotinic acetylcholine receptor β2 (Chrnb2 or AChRB2), tyrosine kinase receptor B (Ntrk2 or TrkB), thymus cell antigen 1 θ (Thy1), and microtubule-associated protein tau (Mapt or Tau), as well as genes with widespread or ubiquitous expression, such as the FK506 binding protein FKB12 (Fkbp1b or FKBP12) and the cyclosporine A binding protein PPIA (PPIA). To determine whether gene expression might be affected also in displaced amacrine cells that were interspersed with RGCs with such pronounced downregulation of gene expression, Scng and GAD67 probes were used together in both retina sections (data not shown) and flat mounts of 3 and 9 month DBA/2J mice (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Consistent with the results of some others who reported no change in several amacrine cell markers (Jakobs et al., 2005), we observed no change in GAD67 expression in amacrine cells, including those that are among regions with greatly reduced numbers of RGCs. These studies also show that Sncg mRNA remains confined to RGCs and is not expressed in displaced amacrine cells even after dramatic loss of RGCs. Together, these results show that RGCs but not displaced amacrine cells downregulate gene expression in DBA/2J and show that this transcriptional downregulation is widespread if not general within RGCs.
Reductions in Sncg expression per cell occur in old DBA/2J but not old C57BL/6J mice
To determine whether the decreases in Sncg expression detected by qRT-PCR were attributable mainly to decreases in the number of Sncg-expressing cells, decreases in the expression of Sncg per cell, or a combination of both factors, as well as to confirm that decreases in Sncg expression were not the effect of age, the expression of Sncg was studied in flat-mounted retinas from 2 and 15 month DBA/2J mice, the latter being selected because of extreme anterior segment dysplasia, and compared with 2 and 12 month C57BL/6J mice, a strain that does not develop glaucoma. On visual inspection, no obvious changes in either the number of Sncg-expressing cells or the amount of Sncg expression per cell were observed between the young mice from both strains, C57BL/6J (Fig. 3A) and DBA/2J (Fig. 3B), and the old mice from the C57BL/6J strain (Fig. 3C). Only in the retinas of the severely affected old DBA/2J mice (Fig. 3D) were there regions with apparent reductions in the number of Sncg-expressing cells as well as the presence of cells with greatly reduced Sncg expression (Fig. 3D, arrows). To quantify the reduction of Sncg expression per cell in retinas with different number of Sncg-expressing cells, we developed a quantitative imaging protocol for flat-mounted retinas that identified RGCs based on the expression of Sncg (see Materials and Methods). In C57BL/6J mice, there were no significant age-dependent changes in either the number of Sncg-expressing cells (Fig. 3E) or the amount of Sncg expression per cell (Fig. 3F). In contrast, in the DBA/2J mice, both the number of Sncg-expressing cells (Fig. 3G) and the Sncg expression per cell (Fig. 3H) were significantly reduced in the severely affected old mice (p < 0.002). Thus, the decreases of Sncg detected in aged DBA/2J animals at the level of whole retina qRT-PCR reflect the combined effect of a decrease in the number of Sncg-expressing cells and a decrease in the amount of Sncg expression per cell, and neither of these changes are the result of a normal aging process.
Figure 3.

Decreases in the number of Sncg-expressing cells and the amount of Sncg expression per cell occur in RGCs of old DBA/2J mice but not old C57BL/6J mice. Sncg expression (red), and nuclei (blue) in flat-mounted retinas of 2 month (A) and 12 month (C) C57BL/6J, and 2 month (B) and 15 month (D) DBA/2J mice. Scale bar, 20 μm. The arrows (D) mark RGCs with reduced expression of Sncg mRNA. Number of Sncg-expressing cells (RGCs) (E, G) and total Sncg expression per cell (F, H) in retinas of young and old C57BL/6J (E, F) and DBA/2J (G, H) mice. Retina numbers were as follows: 2 month C57BL/6J (n = 3), 12 month C57BL/6J (n = 3), 2 month DBA/2J (n = 5), and 15 month DBA/2J (n = 3). m, Month. Error bars represent the SD. **p < 0.002.
Progressive reductions in Sncg expression correlate with reductions in FluoroGold retrograde labeling
To determine whether RGCs with diminished gene expression were compromised functionally, we examined the ability of those cells to be labeled by retrograde transport of FG injected into the superior colliculus. For this purpose, Sncg expression and FG retrograde labeling were compared in retina flat mounts of 3 and 9 month DBA/2J mice. In the retinas of 3 month DBA/2J mice, there was uniform labeling throughout the retina with both Sncg mRNA and FG (Fig. 4 A–C). Generally concordant labeling of Sncg mRNA and FG also was observed in the retinas of 9 month DBA/2J mice as well, even in retinas with sectorial degeneration patterns (Fig. 4 D–F). However, in some retinas of 9 month DBA/2J mice, there were regions evident in low-power views (Fig. 4 D–F, asterisks) that had reduced or absent FG in regions with Sncg expression. Higher magnification views of unaffected retinas (Fig. 4 G–I) confirmed that there was general concordance between Sncg expression and FG labeling, with larger cells tending to have higher levels of Sncg expression and higher FG labeling. In separate experiments, large RGCs labeled with antibodies to the nonphosphorylated form of neurofilament heavy (e.g., SMI32) consistently had higher expressions of Sncg expression relative to other nearby RGCs (data not shown). In high-magnification views of affected DBA/2J retinas (Fig. 4 J–L), particularly in boundary regions between affected and unaffected sectors, three subpopulations of RGCs were evident: (1) RGCs with levels of both Sncg expression and FG labeling similar to RGCs observed in the retinas from young and nonaffected old mice, (2) RGCs that appeared smaller but that still had low-to-moderate levels of Sncg expression and FG labeling, and (3) very small RGCs with extremely low expression of Sncg and little or no FG labeling (Fig. 4 J–L, arrows). This concordant lowering of both Sncg expression and FG labeling in the retinas of old DBA/2J mice was evident when analyzing the whole population of RGCs (Fig. 4M,N). The shift to lower values in the distribution of mean Sncg expression per cell (Fig. 4M) was matched by a similar shift to lower values in the distribution of mean FG per cell (Fig. 4N). When the values of mean Sncg expression and FG labeling for all cells were normalized to the mean of their retina as a way of comparing all cells in different retinas, Sncg expression and FG labeling were shown to be correlated (r = 0.46; n = 440,171 cells; p < 0.0001) (Fig. 4O). Interestingly, the correlation for the 3 month retinas (r = 0.57; n = 183,334 cells; p < 0.0001) was higher than for the 9 month retinas (r = 0.39; n = 256,837 cells; p < 0.0001). Similar degrees of correlation between levels of Sncg expression and FG labeling were found among every individual 9 month retina (r = 0.32–0.56; n = 12,108–80,534 cells; all p < 0.0001), with lower correlations being found in the retinas with lower numbers of Sncg-positive cells (data not shown). Thus, within all retinas, there is a tight correlation between RGC gene expression and FG retrograde transport, but this correlation becomes weaker as the retinas have less RGCs remaining. When analyzing these same retinas for the mean values for each retina, as opposed to the mean values for individual cells, weak correlations were apparent between the number of remaining Sncg-expressing cells, which is a measure of disease severity, and Sncg expression per cell (Fig. 4P) (r = 0.49; n = 10), between the number of remaining Sncg-expressing cells and amount of FG labeling per cell (Fig. 4Q) (r = 0.69; n = 10), and also between Sncg mRNA per cell and FG labeling per cell (Fig. 4R) (r = 0.51; n = 10). These correlations showed obvious trends but lacked significant p values because of the small number of retinas in which both FG and Sncg could be analyzed together. However, when also using other retinas that had not been labeled with FG, statistical significance was reached for the correlation between Sncg expression per cell and number of Sncg-expressing cells (r = 0.56; n = 20; p < 0.02). These results show that both at the level of whole retinas as well as cells within those retinas, the expression of Sncg and labeling with FG are tightly correlated and both decline together in the more affected DBA/2J, and that in addition, at the level of individual RGCs the correlation between Sncg expression and FG becomes weaker as the retinas become more diseased. One possible mechanism that would explain a declining correlation between RGC gene expression and FG labeling is if diseased retinas had large numbers of RGCs with highly dysfunctional or absent retrograde transport. To explore this possibility, we sought additional molecular markers and performed more detailed analyses of the pattern of degeneration observed within individual retinas.
Retina regions that contain RGCs with somatic phosphorylated neurofilament accumulation have many disconnected RGCs
To determine whether there were any RGCs in the retinas of affected DBA/2J mice that were completely disconnected from the brain, we examined a marker previously reported to accumulate in the soma of only RGCs that are disconnected after partial optic nerve crush, phosphorylated forms of neurofilaments (pNF) (Dieterich et al., 2002). In healthy mouse RGCs, pNF is not found in RGC somas or dendrites, but rather only in axons and only starting 200–1000 μm away from the cell body (Nixon et al., 1989). Two different antibodies that recognize pNF were used for these studies and provided similar results (data not shown). The SMI31 antibody, which recognizes phosphorylated heavy neurofilament, labeled distal portions of RGC axons as well as horizontal cells, but also had widespread nuclear labeling throughout the mouse retina that was presumed to be nonspecific (data not shown). The 2F11 antibody, which recognizes a phosphorylated epitope in middle- and high-molecular weight forms of neurofilaments (Luider et al., 1992) and was the antibody used to label pNF in rat RGCs disconnected after partial optic nerve crush (Dieterich et al., 2002), lacked the nonspecific nuclear labeling. Neither antibody had labeling in soma or dendrites of RGCs in young DBA/2J or young or old C57BL/6J mice, except for an occasional cell in an occasional retina (data not shown). However, in retinas with loss of Sncg-expressing cells, numerous RGCs were found with accumulation of pNF within their somas (Fig. 5A,D). Some cells had either weak somatic or weak somatic and dendritic accumulation (Fig. 5 A–D, arrowheads), whereas other cells (Fig. 5 A–D, arrow) had stronger accumulation of pNF throughout both soma and dendrites. Both groups of cells had considerable Sncg expression (Fig. 5B), showing them to be transcriptionally active RGCs, although often lower than other nearby comparably sized RGCs (Fig. 5B, stars). Although these pNF-positive RGCs (pNF+ RGCs) occur in a continuum of intensities and morphologies, for the purpose of analysis we categorized them as two groups: those having weak pNF somatic or somatodendritic labeling but normal RGC morphologies, and those having strong pNF somatodendritic labeling and abnormal morphologies with processes resembling neurites (additional examples provided in supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 5.
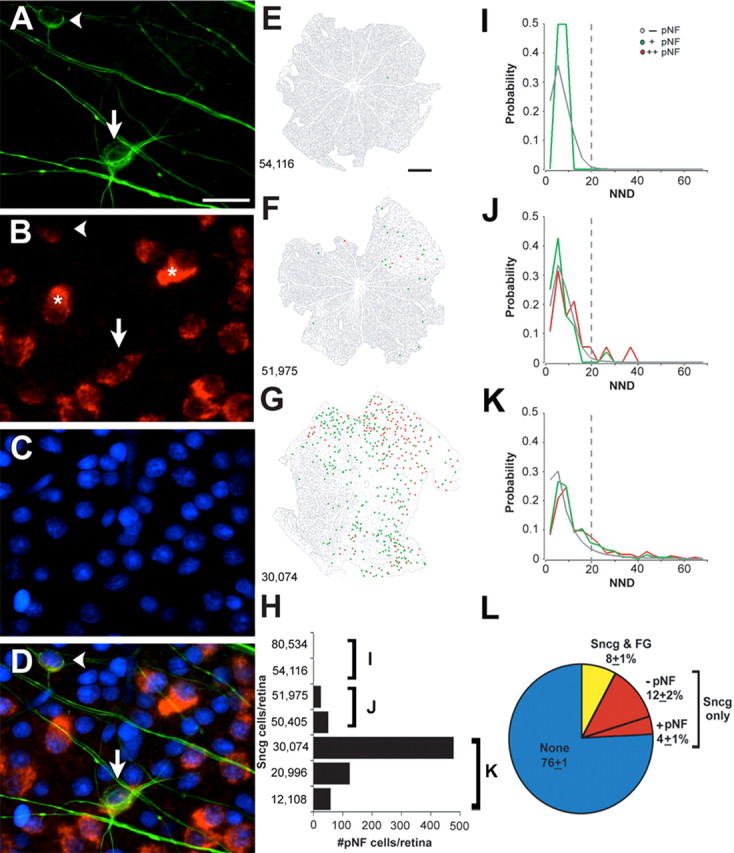
RGCs with weak and strong accumulation of pNF (pNF+ RGCs) are found in sectors of RGC degeneration and mark the position of other RGCs that also lack FG transport. A–D, RGCs with faint (arrowhead) and strong (arrow) pNF immunoreactivity in their cell bodies and dendrites (A) express Sncg mRNA (B), but at levels that are relatively low when compared with some nearby RGCs (asterisks). E–G, Traces of retinas from 9 month DBA/2J mice representing three extents of disease progression: unaffected (E), mild (F), and severe (G), categorized as described in the text and supplemental Figure 3 (available at www.jneurosci.org as supplemental material). H, Number of pNF+ RGCs per retina in unaffected (bracket I), mild (bracket J), or severe (bracket K) retinas, relative to the number of remaining Sncg-expressing cells. I–K, Probability distribution of NND for both Sncg-expressing cells without pNF accumulation (gray), as well as pNF+ RGCs with weak (green) and strong (red) pNF accumulation, graphed for retinas that are unaffected (I) (n = 114,019 cells; n = 2 retinas), mild (J) (n = 87,146 cells; n = 2 retinas), or severe (K) (n = 55,672 cells; n = 3 retinas). The dashed line marks the value 3 SDs above the mean value observed in retinas of 3 month DBA/2J mice (n = 183,334 cells; n = 3 retinas). L, Pie chart of the percentage of GCL cells within a ∼35 μm radius of pNF+ RGCs that have both Sncg expression and FG labeling (yellow), Sncg mRNA only (red) with or without pNF accumulation, and cells without either marker (blue) (n = 419 cells; n = 4 retinas). Scale bars: A–D, 20 μm; E–G, 500 μm.
To confirm that these pNF+ RGCs were disconnected, to determine their number, and to determine whether their presence might be used as a marker to assess the state of disease progression, we undertook quantitative studies of pNF+ RGCs within flat-mounted retinas. In the retinas used for this particular analysis, the mean number pNF+ RGCs per retina in 9 month DBA/2J mice (105 ± 169 pNF+ RGCs/retina; n = 7 retinas) was 100 times higher than in the retinas of 3 month DBA/2J mice (1 ± 2 pNF+ RGCs/retina; n = 3 retinas), and the few observed in the young animals all had only weak somatic expression (data not shown). As with other measures in 9 month DBA/2J mice, there was the expected large variability between retinas in terms of the number of pNF+ RGCs, with some retinas with no such RGCs and others with several hundred (a representative set of retinas are shown in Fig. 5 E–G, and the full set of seven retinas was used for graphs Fig. 5 H–L and are shown in supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In terms of disease severity, we use a categorization scheme similar to that previously used in DBA/2J mice based on the appearance of the optic nerve (Anderson et al., 2005, 2006; Jakobs et al., 2005; Libby et al., 2005a), except that our analysis of the optic nerve involved antibodies to three separate axonal markers, pNF, TUJ1, and 3A10, that label phosphorylated neurofilaments, tubulin β-III, and a neurofilament-associated protein, respectively. Our categorization scheme was also based on the number and pattern of remaining RGCs within the retina. Although we did not attempt to distinguish between moderate and severe phenotypes, we were able to separate mild from unaffected retinas based on the presence or absence of small sectors with reduced RGC densities. Through this combined analysis of the optic nerve and retina, we were able to categorize all the retinas used in this study into unaffected, mild, or severe groups (see supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Two 9 month DBA/2J retinas were classified as unaffected based on there being no evidence of sectorial loss of Sncg cells (Fig. 5E, gray cells; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These unaffected retinas, which also had no obvious loss of FG labeling as well as immunoreactivity in the optic nerve for three axonal markers that was comparable with that found in 3 month DBA/2J optic nerves (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), had no or very few pNF+ RGCs (Fig. 5E, green point; H, columns labeled I). Two retinas were classified as having mild degeneration because only a small sector had reductions in Sncg-expressing cells (Fig. 5F, sparse gray cells in upper right of the retina; supplemental Fig. 3, retina sectors marked with asterisk, available at www.jneurosci.org as supplemental material). These mildly affected retinas, which also had sectors lacking FG that matched the sectors lacking Sncg expression (data not shown) as well as optic nerves indistinguishable from control nerves (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), had both weak and strong (Fig. 5F, green and red points, respectively) pNF+ RGCs that were in or near sectors with reduced RGC densities. In these retinas, the total number of pNF+ RGCs (Fig. 5H, columns labeled J) and the percentage of pNF+ RGCs relative to the total number of RGCs (0.05–0.1%) were small. The three remaining retinas were classified as severe because of the large sectors with loss of Sncg-expressing RGCs (Fig. 5G, reduced gray cells; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These severely affected retinas, which also had large loss of FG label that generally matched the loss of RGCs (Fig. 4 D–F) as well as optic nerves with greatly reduced number of axons (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), had both weak and strong (Fig. 5G, green and red points, respectively) pNF+ RGCs in large numbers (Fig. 5H, columns labeled K), and in a greater percentage of remaining RGCs (0.5–1.8%). Because pNF+ RGCs are found principally in sectors of RGC degeneration, and in numbers and percentages that increase with disease severity, somatodendritic accumulation of phosphorylated neurofilaments must be a stage in the degeneration of all RGCs or at least a subset of RGCs.
Visual inspection of retinas showing the position of all weak and strong pNF+ RGCs in the context of all remaining RGCs (Fig. 5 E–G) gives the impression that strong pNF+ RGCs (red points) may be a later stage of weak pNF+ RGCs (green points). To test this model, we performed NND analyses (Fig. 5 I–K). NND, which already has been used to study RGC degeneration in DBA/2 retinas (Filippopoulos et al., 2006), calculates the distance from the centroid of every RGC to the nearest RGC centroid. Within unaffected 9 month retinas, no RGCs were found with strong accumulation of pNF, and the mean NND for the cells with weak accumulation of pNF, 7.2 μm (n = 2), was similar to that of cells without pNF accumulation, 7.3 μm (n = 114,017), and also similar to the mean NND in 3 month DBA/2J retinas, 7.8 μm (n = 183,334) (not shown in Fig. 5). In retinas with mild degeneration, the mean NND for RGCs with strong accumulation of pNF, 12 μm (n = 19), but not the mean NND for RGCs with weak accumulation of pNF, 7.2 μm (n = 56), was greater than the mean NND for RGCs without pNF accumulation, 8.0 μm (n = 87,071) (one-way ANOVA, p < 0.0001). In retinas with severe degeneration, the mean NND for RGCs with strong accumulation of pNF, 15 μm (n = 207), exceeded the mean NND for RGCs with weak accumulation of pNF, 12 μm (n = 451), which in turn exceeded the mean NND for RGCs with no pNF accumulation, 9 μm (n = 55,014) (one-way ANOVA, p < 0.003). Because RGCs with strong accumulation of pNF were not present in the unaffected retinas, they were the only ones with increased NND in the mildly affected retinas, and had the greatest NND in the severely affected retinas, these analyses show that RGCs with weak and strong accumulation of pNF are different from one another and that the ones with strong pNF occur later in the disease progression. The simplest explanation, then, is that weak and strong pNF+ RGCs represent early and late stages of degeneration within the same population of RGCs. A similar analysis based on percentage of RGCs with abnormally large NNDs, defined as NNDs >3 SDs above the mean NND of control retinas (Fig. 5 I–K, dotted lines) supports the same conclusions (data not shown). However, all these analyses also showed that pNF+ RGCs are only a subset of the RGCs within affected regions: for example, in severely affected retinas, they are only 2.3% of RGCs with NNDs >3 SDs above the mean NND of control retinas. At this point, it is unclear whether all RGCs go through a transient stage including pNF accumulation or whether this accumulation occurs only in some RGCs.
To confirm that pNF accumulation in RGC soma meant that these RGCs were disconnected, as proposed for the case of partial optic nerve crush (Dieterich et al., 2002), as well as to determine whether any other RGCs might be disconnected, the fraction of GCL cells with Sncg expression and FG labeling was determined for cells that were within a ∼35 μm radius of pNF+ RGCs (Fig. 5L). In this analysis, all pNF+ RGCs lacked FG labeling, supporting that they were disconnected. However, 60% of nearby RGCs lacking pNF accumulation also lacked FG, suggesting that they too were disconnected. Based on the fraction of GCL cells that have Sncg expression and the fraction of these RGCs that have FG labeling in healthy young retinas (Fig. 1K) and severely affected regions of old retinas (Fig. 5L), it is evident that the retinas of 9 month DBA/2J amid sectorial degeneration have large numbers of RGCs that are either completely disconnected or totally deficient in FG transport and yet maintain RGC gene expression.
Disconnected RGCs maintain intraocular portions of their axons and these axons terminate within the optic nerve head
To confirm that accumulation of phosphorylated neurofilament in the somas of these RGCs was attributable to their being disconnected rather than just dysfunctional, retinas from 9 month (n = 5) and 15 month (n = 6) DBA/2J mice, as well as control retinas from 2 month DBA/2J (n = 28), 2 month C57BL/6J (n = 3), and 12 month C57BL/6J (n = 3) mice, all previously subjected to Sncg in situ hybridization, were immunostained with antibodies against tubulin β-III and pNF (Fig. 6). Low-power views of flat-mounted retinas from all control retinas, including the 12 month C57BL/6J retina shown in Figure 6, showed normal levels of Sncg expression (Fig. 6A), and strong immunoreactivity of tubulin β-III along intraretinal axons in the central (Fig. 6C) and peripheral (data not shown) retina, whereas strong pNF immunostaining was observed only in the central retina (Fig. 6E). In low-power views of the retinas of old DBA/2J mice that had a large loss of Sncg-expressing cells, such as the 15 month DBA/2J retina shown in Figure 6, the dramatic loss of Sncg-expressing cells was paralleled by a corresponding dramatic loss of tubulin β-III immunoreactivity in intraretinal axons (Fig. 6D). However, in these retinas depleted of both Sncg-expressing cells and tubulin β-III axonal labeling, strong pNF immunoreactivity was still observed within large numbers of intraretinal axons (Fig. 6F). Higher magnification views of the retina periphery of control retinas, including the 12 month C57BL/6J retina shown in Figure 7, demonstrated that weak pNF immunoreactivity was detectable within some intraretinal axons that also had tubulin β-III immunoreactivity (Fig. 7 A–C). In contrast, in the retina periphery of DBA/2J that had few remaining cells expressing Sncg, including the retina from a 15 month DBA/2J mouse shown in Figure 7, strong pNF immunostaining was observed in axons with weak tubulin β-III immunoreactivity (Fig. 7 D–F). The peripheral axons labeled by pNF antibodies were thick and exhibited large varicosities that were not observed with the tubulin β-III antibody (Fig. 7 D–F). In most retinas depleted of Sncg-expressing cells, it was possible to follow single axons stained with the pNF antibodies from the ONH to the far periphery where RGC cell bodies often had accumulation of pNF (Fig. 7G). These single axons had increased width and/or pNF immunofluorescence nearer to the ONH (Fig. 7I), relative to the retina periphery (Fig. 7H). These data support the view that the pNF+ RGC axons are largely intact within the retina but must have a break or dramatic impairment in axonal transport at or beyond the ONH.
Figure 6.
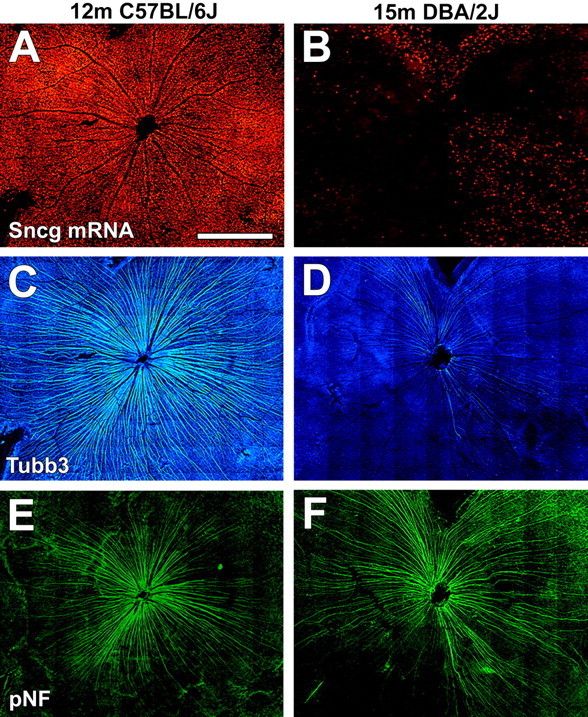
The loss of Sncg-expressing cells is matched by a reduction in tubulin β-III but not pNF labeling within intraretinal axons. Two retina flat mounts, from a 12 month C57BL/6J mouse (A, C, E) and a 15 month DBA/2J mouse with severe degeneration (B, D, F) showing the relationship between Sncg mRNA expression (red) (A, B), tubulin β-III (Tubb3 or TUJ1) immunoreactivity (blue) (C, D), and pNF immunoreactivity (green) (E, F). m, Month. Scale bar, 500 μm.
Figure 7.
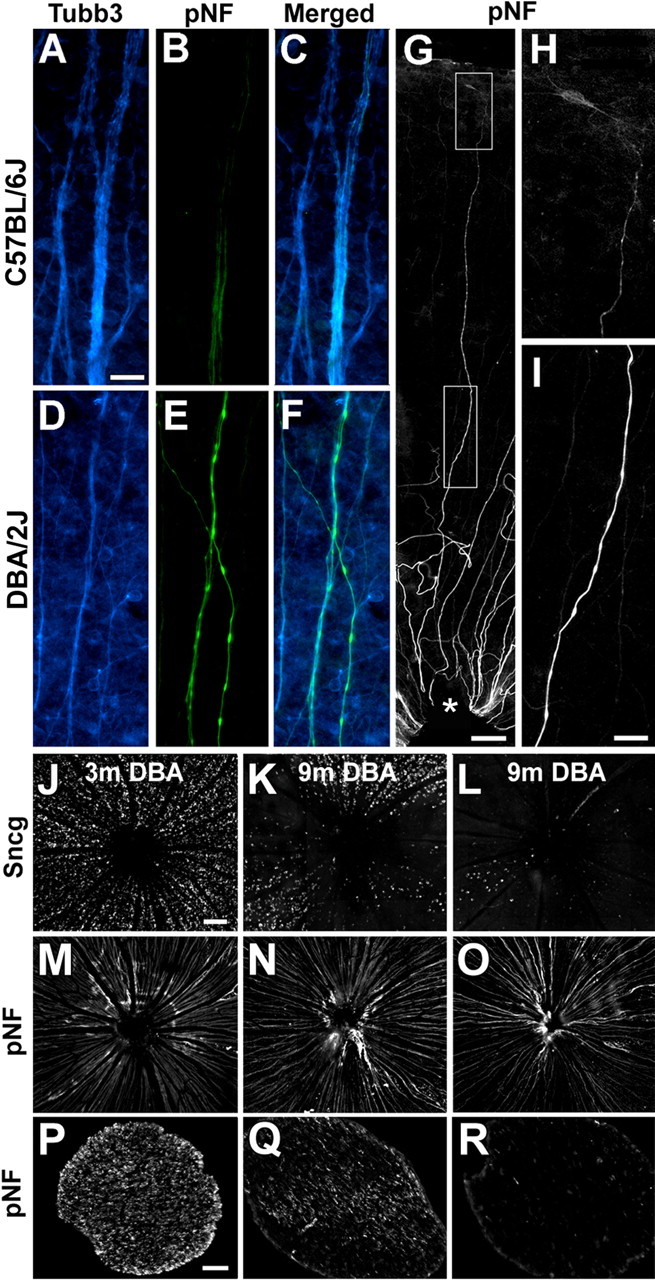
Severely affected DBA/2J mice have accumulation of pNF within intraretinal axons and have loss of optic nerve axons. Peripheral retina axons from 12 month C57BL/6J (A–C) and 15 month DBA/2J mice (D–F) labeled with antibodies to tubulin β-III (Tubb3) (A, D) and pNF (B, E), shown next to the merged views (C, F). G, Retina from a 15 month DBA/2J mouse in which a single pNF-positive axon can be followed from the ONH (asterisk) to the most peripheral part of the retina in which an RGC soma also has pNF labeling. H, I, Higher power views of two regions of the same pNF-labeled axon shown in G are shown magnified so that the axon near the soma (H) and ONH (I) can be compared. J–R, Sncg mRNA expression (J–L), and pNF staining of axons inside the retina (M–O) and in the proximal optic nerve (P–R) for one 3 month DBA/2J mouse (J, M, P) and two 9 month DBA/2J mice with lesser (K, N, Q) and greater (L, O, R) loss of Sncg-expressing cells. Note the correspondence between the number of Sncg-expressing cells within the retina and the number of pNF axons in the proximal optic nerve, but the lack of correspondence of the pNF labeling within the retina. m, Month. Scale bars: A–F, H, I, 20 μm; J–O, 100 μm; G, P–R, 50 μm.
To examine whether these pNF+ axons extended into the optic nerve, we examined the retinas and nerves with the same pNF antibodies. Unaffected retinas, including the 3 month DBA/2J retina shown in Figure 7, but also 9 and 15 month DBA/2J retinas without degeneration sectors, had uniform distribution or RGCs around the ONH (Fig. 7J), and strong pNF immunoreactivity in axons both within the retina (Fig. 7M) and optic nerve (Fig. 7P). In contrast, in retinas depleted of RGCs, such as the two 9 month DBA/2J retinas shown in Figure 7, sectors depleted of Sncg-expressing cells extended all the way to the ONH (Fig. 7K,L). Interestingly, in these affected retinas, pNF immunoreactivity within the retina was strong and widespread throughout the retina (Fig. 7N,O), similar to what was observed in the control retina (compare Fig. 7M), despite being mostly lost from the optic nerve (Fig. 7Q,R, and compare P). The lack of labeling with pNF within the nerve was not attributable to a lack of transport or expression of this one marker but rather attributable to a loss of axons because the severe loss of axons within the optic nerve was also observed with two additional antibodies, TUJ1 and 3A10, that label integral axonal proteins tubulin β-III and a neurofilament-associated protein, respectively (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Qualitatively, the labeling of optic nerves with the three axonal markers, pNF, TUJ1, and 3A10, always matched the degree and pattern of loss of Sncg-expressing cells within the retina (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Thus, the finding that did not match was the presence of large number of intraretinal axons labeled with pNF.
To determine whether the discordance between the labeling of axons seen inside the retina and in the optic nerve was attributable to large number of axons terminating within the ONH, punches were taken containing a small piece of the central retina, the entire ONH, as well as a proximal portion of the optic nerve, and these were sectioned longitudinally and immunolabeled with pNF antibodies (Fig. 8 A–G). Unaffected retinas, including the 9 month DBA/2J retina shown, had the expected uniform labeling of pNF axons within the retina (Fig. 8A), and the expected uniform labeling with pNF and TUJ1 in the optic nerve (Fig. 8C). The sum projection of longitudinal sections of the optic nerve punch also showed strong and uniform labeling with pNF (Fig. 8E). In contrast, in severely affected retinas such as the 9 month DBA/2J retina shown, where pNF axons are still present in the retina (Fig. 8B) but mostly lost from the optic nerve (Fig. 8D), the sum projection of longitudinal sections of the ONH punches showed that the majority of pNF axons end just outside the retina. Fluorescence intensity measurements of these sum projections demonstrated that, in this affected retina (Fig. 8G, red line), most pNF-labeled axons ended within 500 μm from the retina, in stark contrast to the pNF axons of the unaffected retina (Fig. 8G, blue line). Thus, because myelination begins ∼600–800 μm behind the globe in the mouse (May and Lutjen-Drecoll, 2002), we show here that large numbers of pNF axons in affected retinas end within the optic nerve head itself.
Figure 8.
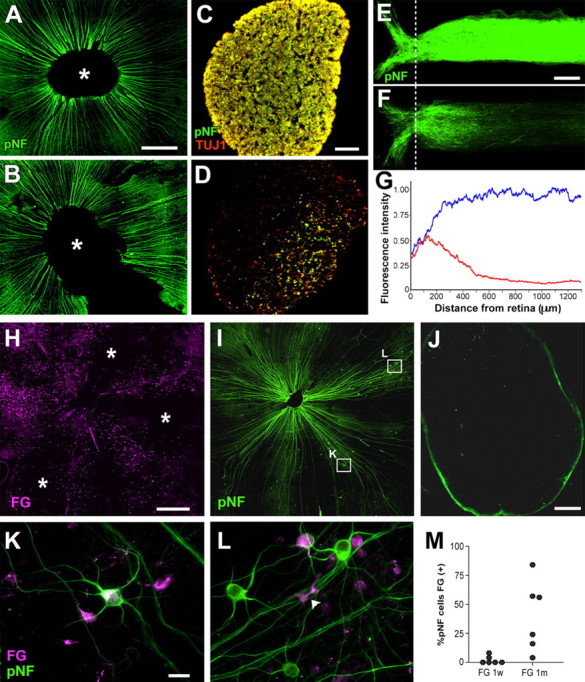
pNF-positive axons in glaucomatous retinas of DBA/2J mice end within the ONH, and pNF+ RGCs survive disconnected for ∼1 month. A, B, pNF-positive axons are found within the retina in unaffected (A) and highly affected (B) 9 month DBA/2J retinas. The asterisk indicates the missing retina surrounding the ONH, analyzed in longitudinal sections of ONH punches in E and F. C, D, Cross sections of the optic nerves, at ∼2 mm from the retina, from the unaffected (C) and affected (D) retinas presented in A and B, respectively, show uniform labeling of optic nerve axons with pNF (green) and TUJ1 (red) in the unaffected DBA/2J mouse (C) and significant loss of both pNF- and TUJ1-positive axons in the affected DBA/2J mouse (D). E, F, Sum projection images of longitudinal sections of ONH and proximal ON region dissected from the unaffected (E) and affected (F) retinas presented in A and B, respectively. Fluorescence intensity measurements (G) of the unaffected (blue) and affected (red) retinas shown in A and B, respectively, show that the majority of pNF-positive axons from the affected retina end within 500 μm of the retina. The dotted line in E and F indicate the position of the choroid, from which distances were measured in G. H, I, Retina flat mount of a 9 month DBA/2J mouse shows the relationship between FG labeling 1 month after injection in the superior colliculi (magenta) (H) and pNF immunoreactivity (green) (I). J, Cross section of the optic nerve from retina presented in H and I immunolabeled with pNF, showing the near complete loss of axons by the time of analysis. K, L, pNF+ RGCs (green) with (K) and without (L) FG retrograde labeling (magenta) 1 month after FG injection, found in the retina shown in H and I. M, Percentage of pNF+ RGCs that have FG labeling in 9 month DBA/2J 1 week (n = 25 cells in each of 6 retinas) and 1 month (n = 25 cells in each of 5 retinas; n = 19 in 1 retina) after FG injection. Note that, 1 week after FG injection, a very small number of pNF+ RGCs have FG labeling, whereas about one-half of pNF+ RGCs have FG 1 month after FG injection. w, Week; m, month. Scale bars: A, B, H, I, 500 μm; C, D, J, 50 μm; E, F, 200 μm; K, L, 20 μm.
The presence of a large number of axons ending within the ONH implies that RGCs that are disconnected do not immediately die, but must be capable of surviving long periods of time. To test this possibility, we examined 9 month DBA/2J retinas with pNF antibodies 1 week and 1 month after injection of FG into the superior colliculus (Fig. 8 H–M). In the animals labeled with FG 1 week before fixation and analysis, the grand majority of pNF+ RGCs lacked FG labeling. However, in the animals labeled with FG 1 month before fixation and analysis, both FG-positive (Fig. 8K) and FG-negative (Fig. 8L) pNF+ RGCs were found. Any pNF+ RGCs that were positive for FG must have become disconnected during that 1-month-long interval. However, any pNF+ RGCs that were negative for FG must have been disconnected at the time of FG label 1 month before and must have survived a full month disconnected. In the samples labeled with FG 1 month before but not the samples labeled 1 week before, smaller cells that are presumed to be microglial cells also were labeled with FG in addition to RGCs. Labeling of microglia with FG would imply phagocytosis of degenerating RGCs by microglial cells, as has been shown before in an experimental rat glaucoma model after a long-term retrograde labeling (Naskar et al., 2002). Counts of FG-positive and -negative pNF+ RGCs (Fig. 8M) confirmed that 1 week after FG label nearly all pNF+ RGCs in all retinas lacked FG label. However, 1 month after FG labeling, different retinas had very different fractions of FG-positive and -negative pNF+ RGCs. Whereas the average fraction of FG-positive pNF+ RGCs in these 9 month DBA/2J retinas labeled with FG 1 month before is about one-half, some retinas had nearly all pNF+ RGCs connected whereas other retinas had nearly all pNF+ RGCs disconnected at the time of FG label, and yet other retinas, such as the one shown in Figure 8, H–L, had a mix of FG-labeled and -unlabeled pNF+RGCs. A low-power view of one such retina (Fig. 8H) shows sectors with and without FG labeling, indicating that only some sectors were connected to the brain at the time of the FG injection, although most axons had degenerated in the optic nerve by the time the of analysis (Fig. 8J). Interestingly, the sectors lacking FG also generally lacked pNF axons, showing that the half-life of disconnected pNF axons cannot be much longer than 1 month. Thus, we conclude the half-life of pNF+ RGCs and their intraretinal axons is ∼1 month.
Discussion
Sncg (also known as Persyn) is a small protein expressed in subsets of neurons and in carcinogenic breast and ovarian tissue believed to perform chaperone-like functions (Buchman et al., 1998; Ninkina et al., 1998; Bruening et al., 2000; Surguchov et al., 2001; George, 2002; Surgucheva et al., 2005). In the mouse retina, during development Sncg is regulated by the transcription factor Brn3b and expressed specifically in RGCs (Mu et al., 2004), and in the adult remains expressed in RGCs and not other retina cell types (Trimarchi et al., 2007). Its specificity coupled to the fact that Sncg is one of the most highly expressed mRNAs in purified rat RGCs (Farkas et al., 2004), made it a particularly attractive choice for an mRNA marker of adult mouse RGCs. In our study, we first confirmed that Sncg mRNA is expressed in most if not all RGCs and no other cells within the adult mouse retina. Then, we showed that there is a highly significant correlation between Sncg expression per cell and the amount of FG retrograde transport per cell, and that both Sncg expression and FG labeling decrease together in affected DBA/2J retinas. Others have shown previously in other RGC injury models including more acute animal models of glaucoma that a different gene expressed in RGCs, Thy1, also is downregulated before cell loss (Schlamp et al., 2001; Huang et al., 2006). Here, we show that the downregulation of gene expression within RGCs is not specific to Sncg and Thy1, but rather is very widespread if not global because it is observed among genes that are specific to or enriched in RGCs as well as genes that have more widespread expression. Because the genes downregulated within RGCs are involved in many critical functions of RGCs, such as transcription, axonal and dendritic structure, synaptic transmission, growth factor response, and stress response, RGCs with gene downregulation likely have a multifaceted or even general alterations of structure and function that should be considered in the design of therapeutic strategies.
A challenge in studying DBA/2J mice has been the phenotypic variability that necessitates the use of large sample sizes in analyses that are performed at the level of whole retinas. Here, we demonstrate that analyses comparing cells rather than retinas can be performed on smaller numbers of retinas, including retinas that have very small regions of RGC degeneration. These small regions, as well as the larger fan-shaped sectors of degeneration that have been reported by others (Danias et al., 2003; Jakobs et al., 2005; Filippopoulos et al., 2006; Schlamp et al., 2006), have large proportions of RGCs that retain Sncg gene expression but are not labeled by FG injected into the superior colliculus, and among these RGCs an additional subset that have somatic accumulation of pNFs, a marker expressed in RGCs that are disconnected from the brain after partial optic nerve crush (Dieterich et al., 2002). Our results confirm previous studies showing that pNF+ RGCs are extremely rare within healthy retinas, because under normal circumstances pNF is found only in axons starting at 200–1000 μm from RGC soma (Nixon et al., 1989), consistent with the function of pNF in maintaining axon caliber in myelinated axons (Hoffman et al., 1984; Shea et al., 1993). In contrast, in diseased DBA/2J retinas, pNF+ RGCs can be found in large numbers and in positions consistent with the extent of loss of RGCs. Thus, the number and position of pNF+ RGCs are sensitive monitors of the extent and location of the glaucomatous damage. A previous study (Jakobs et al., 2005) had shown that affected DBA/2J retinas have RGCs with aberrant distribution of nonphosphorylated heavy neurofilaments within soma and dendrites. It is likely that the cells they described represent either the same or an overlapping population to the ones we describe. However, because many RGCs normally have heavy neurofilaments in their somas, the determination as to whether any one RGC is diseased based on the appearance of labeling is problematic. In contrast, using somatic accumulation of pNF as a marker, the determination as to whether these RGCs are abnormal is unambiguous.
Aberrant accumulation of pNF in axonal swellings and somas has been a hallmark of axonal transport disruption in neurodegenerative diseases (Manetto et al., 1988; Munoz et al., 1988; Mizusawa et al., 1989; Sobue et al., 1990; Rouleau et al., 1996; Stokin et al., 2005). Because phosphorylation of neurofilaments has been shown to decrease active transport of neurofilaments through retinal axons (Jung and Shea, 1999), it is possible that excess pNF within intraocular axons may itself cause progressive declines in RGC gene expression and retrograde transport. Whereas in other cases axonal and even somatic accumulation of pNF may be indicative of an impairment rather than loss of axonal transport, there are two lines of evidence that argue that in this animal model of glaucoma RGCs accumulate pNF in their somas because they are physically disconnected from their targets. First, we show that pNF+ RGCs cannot be labeled by FG injection into the superior colliculus 7 d before. Second, in and only in retinas with the largest proportions of pNF+ RGCs, there is a large discrepancy between the numbers of axons labeled by pNF inside the retina versus in the optic nerve. This discrepancy is not attributable to a specific impairment of transport of pNF or a defective phosphorylation of neurofilaments within the optic nerve, because those axons are absent from the optic nerve according to two other integral axonal proteins, tubulin β-III and a neurofilament-associated protein. Furthermore, we show that these axons end within the optic nerve head itself.
The preservation of RGCs within the retina but loss of axons within the nerve had been reported before in DBA/2J but only when apoptotic pathways were inhibited, in Bax−/− DBA/2J mice (Libby et al., 2005a). We demonstrate here that, in otherwise unmodified DBA/2J mice, large numbers of RGCs have axons that end within the optic nerve head. These results are consistent with findings in human glaucoma and other glaucoma animal models in which most profound pathological changes occur at the level of the lamina cribosa (Quigley, 2005; Nickells, 2007). Whereas such a loss might be the result of an acute insult at the optic nerve head, it is also possible that the insult is elsewhere in the retinal ganglion cell, its axon, or its terminal, leading to axonal die-back, as recently reported in DBA/2J mice (Schlamp et al., 2006), or be the result of an acute insult elsewhere in the retinal ganglion cell axon that results in Wallerian degeneration of only myelinated portions of the axons [as discussed in reviews by Coleman (2005) and Whitmore et al. (2005)]. Surprisingly, these RGCs with severed axons remain alive in the retina, as evident by still transcribing Sncg mRNA, for as long as a month. Such longevity of pNF+ RGCs in this animal model of glaucoma match the long life of pNF+ RGCs after partial optic nerve crush (Dieterich et al., 2002). Based on the large number of pNF+ RGCs, the long survival of these cells, the fact that among RGCs that cannot be labeled by FG only a subset are pNF+ RGCs, and the observation that in severely affected retinas the discrepancy in the number of axons inside versus outside far exceeds the number of pNF+ RGCs (data not shown), we conclude that glaucomatous retinas that have begun to lose RGCs have large proportions of RGCs with axons that are disconnected at the level of the ONH. We estimate that disconnected RGCs represent at least 5–10% of all remaining RGCs at middle and late stages of the disease. It seems likely that large numbers of disconnected RGCs and axons are important factors in the progression of glaucoma and may be important therapeutic targets.
Our results lend new support to an emerging view of how glaucoma progresses (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). There are two distinct degenerative processes that contribute to the ultimate loss of the majority of RGCs. The first process involves linked declines in RGC gene expression and retrograde transport that are likely chronic and widespread across the retina. Whether the decreases in gene expression cause the decreases in transport or vice versa, and whether these are attributable to RGC intrinsic or rather extrinsic factors, remain to be determined. However, there must be a second acute degenerative process because there are also large numbers of RGCs that are near one another with no detectable retrograde transport but that retain RGC gene expression. This second process likely involves damage to the axon that results in the rapid degeneration of the distal portion while there is still preservation of the proximal portion of the axon for as long as a month. Separate study of these two degeneration processes in DBA/2J and other promising mouse models of glaucoma (Mabuchi et al., 2003; Nakazawa et al., 2006; Senatorov et al., 2006) especially after various treatments that have been reported to slow disease progression in DBA/2J mice (Anderson et al., 2005; Ward et al., 2007; Zhong et al., 2007) will likely lead to both a better understanding of disease mechanisms as well as the discovery of additional promising interventions.
Footnotes
This work was supported by a “Catalyst for a Cure” grant from the Glaucoma Research Foundation (N.M.-A., M.L.V., P.J.H.), Postdoctoral Fellowship Grant NEI 5 T32 EY07143-12 (I.S.), and Extramural Research Facilities Construction Project Grant 1 C06 RR-15488-01 from the National Center for Research Resources–National Institutes of Health. We thank Jeremy Nathans, Harry Quigley, and Alejandra Bosco for valuable comments on this manuscript, and Robert Scharpf for advice on statistical analyses.
References
- Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol. 1974;13:771–783. [PubMed] [Google Scholar]
- Anderson MG, Libby RT, Gould DB, Smith RS, John SW. High-dose radiation with bone marrow transfer prevents neurodegeneration in an inherited glaucoma. Proc Natl Acad Sci USA. 2005;102:4566–4571. doi: 10.1073/pnas.0407357102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MG, Libby RT, Mao M, Cosma IM, Wilson LA, Smith RS, John SW. Genetic context determines susceptibility to intraocular pressure elevation in a mouse pigmentary glaucoma. BMC Biol. 2006;4:20. doi: 10.1186/1741-7007-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruening W, Giasson BI, Klein-Szanto AJ, Lee VM, Trojanowski JQ, Godwin AK. Synucleins are expressed in the majority of breast and ovarian carcinomas and in preneoplastic lesions of the ovary. Cancer. 2000;88:2154–2163. [PubMed] [Google Scholar]
- Buchman VL, Adu J, Pinon LG, Ninkina NN, Davies AM. Persyn, a member of the synuclein family, influences neurofilament network integrity. Nat Neurosci. 1998;1:101–103. doi: 10.1038/349. [DOI] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Danias J, Lee KC, Zamora MF, Chen B, Shen F, Filippopoulos T, Su Y, Goldblum D, Podos SM, Mittag T. Quantitative analysis of retinal ganglion cell (RGC) loss in aging DBA/2NNia glaucomatous mice: comparison with RGC loss in aging C57/BL6 mice. Invest Ophthalmol Vis Sci. 2003;44:5151–5162. doi: 10.1167/iovs.02-1101. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Trivedi N, Engelmann R, Gundelfinger ED, Gordon-Weeks PR, Kreutz MR. Partial regeneration and long-term survival of rat retinal ganglion cells after optic nerve crush is accompanied by altered expression, phosphorylation and distribution of cytoskeletal proteins. Eur J Neurosci. 2002;15:1433–1443. doi: 10.1046/j.1460-9568.2002.01977.x. [DOI] [PubMed] [Google Scholar]
- Dräger UC, Olsen JF. Ganglion cell distribution in the retina of the mouse. Invest Ophthalmol Vis Sci. 1981;20:285–293. [PubMed] [Google Scholar]
- Farkas RH, Qian J, Goldberg JL, Quigley HA, Zack DJ. Gene expression profiling of purified rat retinal ganglion cells. Invest Ophthalmol Vis Sci. 2004;45:2503–2513. doi: 10.1167/iovs.03-1391. [DOI] [PubMed] [Google Scholar]
- Filippopoulos T, Danias J, Chen B, Podos SM, Mittag TW. Topographic and morphologic analyses of retinal ganglion cell loss in old DBA/2NNia mice. Invest Ophthalmol Vis Sci. 2006;47:1968–1974. doi: 10.1167/iovs.05-0955. [DOI] [PubMed] [Google Scholar]
- George JM. The synucleins. Genome Biol. 2002;3:REVIEWS3002. doi: 10.1186/gb-2001-3-1-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PN, Griffin JW, Price DL. Control of axonal caliber by neurofilament transport. J Cell Biol. 1984;99:705–714. doi: 10.1083/jcb.99.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Fileta J, Guo Y, Grosskreutz CL. Downregulation of Thy1 in retinal ganglion cells in experimental glaucoma. Curr Eye Res. 2006;31:265–271. doi: 10.1080/02713680500545671. [DOI] [PubMed] [Google Scholar]
- Inman DM, Horner PJ. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia. 2007;55:942–953. doi: 10.1002/glia.20516. [DOI] [PubMed] [Google Scholar]
- Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005;171:313–325. doi: 10.1083/jcb.200506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John SW. Mechanistic insights into glaucoma provided by experimental genetics the cogan lecture. Invest Ophthalmol Vis Sci. 2005;46:2649–2661. doi: 10.1167/iovs.05-0205. [DOI] [PubMed] [Google Scholar]
- John SW, Smith RS, Savinova OV, Hawes NL, Chang B, Turnbull D, Davisson M, Roderick TH, Heckenlively JR. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Invest Ophthalmol Vis Sci. 1998;39:951–962. [PubMed] [Google Scholar]
- Jung C, Shea TB. Regulation of neurofilament axonal transport by phosphorylation in optic axons in situ. Cell Motil Cytoskeleton. 1999;42:230–240. doi: 10.1002/(SICI)1097-0169(1999)42:3<230::AID-CM6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Kerrigan-Baumrind LA, Quigley HA, Pease ME, Kerrigan DF, Mitchell RS. Number of ganglion cells in glaucoma eyes compared with threshold visual field tests in the same persons. Invest Ophthalmol Vis Sci. 2000;41:741–748. [PubMed] [Google Scholar]
- Kobbert C, Apps R, Bechmann I, Lanciego JL, Mey J, Thanos S. Current concepts in neuroanatomical tracing. Prog Neurobiol. 2000;62:327–351. doi: 10.1016/s0301-0082(00)00019-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Semaan SJ, Schlamp CL, Nickells RW. Dominant inheritance of retinal ganglion cell resistance to optic nerve crush in mice. BMC Neurosci. 2007;8:19. doi: 10.1186/1471-2202-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005a;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, Cosma IM, Snow A, Wilson LA, Smith RS, Clark AF, John SW. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005b;22:637–648. doi: 10.1017/S0952523805225130. [DOI] [PubMed] [Google Scholar]
- Luider TM, van Dommelen MW, Tibboel D, Meijers JH, Ten Kate FJ, Trojanowski JQ, Molenaar JC. Differences in phosphorylation state of neurofilament proteins in ganglionic and aganglionic bowel segments of children with Hirschsprung's disease. J Pediatr Surg. 1992;27:815–819. doi: 10.1016/0022-3468(92)90372-e. [DOI] [PubMed] [Google Scholar]
- Mabuchi F, Aihara M, Mackey MR, Lindsey JD, Weinreb RN. Optic nerve damage in experimental mouse ocular hypertension. Invest Ophthalmol Vis Sci. 2003;44:4321–4330. doi: 10.1167/iovs.03-0138. [DOI] [PubMed] [Google Scholar]
- Manetto V, Sternberger NH, Perry G, Sternberger LA, Gambetti P. Phosphorylation of neurofilaments is altered in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1988;47:642–653. doi: 10.1097/00005072-198811000-00007. [DOI] [PubMed] [Google Scholar]
- May CA, Lutjen-Drecoll E. Morphology of the murine optic nerve. Invest Ophthalmol Vis Sci. 2002;43:2206–2212. [PubMed] [Google Scholar]
- May CA, Nakamura K, Fujiyama F, Komatsu Y, Yanagawa Y. Homozygous GAD65 and heterozygous GAD67 knock-out mice reveal normal retinal development and maintenance despite reduced amounts of GABA. Acta Neuropathol (Berl) 2007;113:101–103. doi: 10.1007/s00401-006-0157-6. [DOI] [PubMed] [Google Scholar]
- Minckler DS, Tso MO, Zimmerman LE. A light microscopic, autoradiographic study of axoplasmic transport in the optic nerve head during ocular hypotony, increased intraocular pressure, and papilledema. Am J Ophthalmol. 1976;82:741–757. doi: 10.1016/0002-9394(76)90012-x. [DOI] [PubMed] [Google Scholar]
- Mizusawa H, Matsumoto S, Yen SH, Hirano A, Rojas-Corona RR, Donnenfeld H. Focal accumulation of phosphorylated neurofilaments within anterior horn cell in familial amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 1989;79:37–43. doi: 10.1007/BF00308955. [DOI] [PubMed] [Google Scholar]
- Mu X, Beremand PD, Zhao S, Pershad R, Sun H, Scarpa A, Liang S, Thomas TL, Klein WH. Discrete gene sets depend on POU domain transcription factor Brn3b/Brn-3.2/POU4f2 for their expression in the mouse embryonic retina. Development. 2004;131:1197–1210. doi: 10.1242/dev.01010. [DOI] [PubMed] [Google Scholar]
- Munoz DG, Greene C, Perl DP, Selkoe DJ. Accumulation of phosphorylated neurofilaments in anterior horn motoneurons of amyotrophic lateral sclerosis patients. J Neuropathol Exp Neurol. 1988;47:9–18. doi: 10.1097/00005072-198801000-00002. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, Michaud N, Hafezi-Moghadam A, Miller JW, Benowitz LI. Tumor necrosis factor-α mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006;26:12633–12641. doi: 10.1523/JNEUROSCI.2801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naskar R, Wissing M, Thanos S. Detection of early neuron degeneration and accompanying microglial responses in the retina of a rat model of glaucoma. Invest Ophthalmol Vis Sci. 2002;43:2962–2968. [PubMed] [Google Scholar]
- Nickells RW. From ocular hypertension to ganglion cell death: a theoretical sequence of events leading to glaucoma. Can J Ophthalmol. 2007;42:278–287. [PubMed] [Google Scholar]
- Ninkina NN, Alimova-Kost MV, Paterson JW, Delaney L, Cohen BB, Imreh S, Gnuchev NV, Davies AM, Buchman VL. Organization, expression and polymorphism of the human persyn gene. Hum Mol Genet. 1998;7:1417–1424. doi: 10.1093/hmg/7.9.1417. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Lewis SE, Dahl D, Marotta CA, Dräger UC. Early posttranslational modifications of the three neurofilament subunits in mouse retinal ganglion cells: neuronal sites and time course in relation to subunit polymerization and axonal transport. Brain Res Mol Brain Res. 1989;5:93–108. doi: 10.1016/0169-328x(89)90001-6. [DOI] [PubMed] [Google Scholar]
- Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000;41:764–774. [PubMed] [Google Scholar]
- Quigley HA. Early detection of glaucomatous damage. II. Changes in the appearance of the optic disk. Surv Ophthalmol. 1985;30:111, 117–126. doi: 10.1016/0039-6257(85)90080-3. [DOI] [PubMed] [Google Scholar]
- Quigley HA. Glaucoma: macrocosm to microcosm the Friedenwald lecture. Invest Ophthalmol Vis Sci. 2005;46:2662–2670. doi: 10.1167/iovs.04-1070. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Anderson DR. Distribution of axonal transport blockade by acute intraocular pressure elevation in the primate optic nerve head. Invest Ophthalmol Vis Sci. 1977;16:640–644. [PubMed] [Google Scholar]
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Addicks EM, Green WR, Maumenee AE. Optic nerve damage in human glaucoma. II. The site of injury and susceptibility to damage. Arch Ophthalmol. 1981;99:635–649. doi: 10.1001/archopht.1981.03930010635009. [DOI] [PubMed] [Google Scholar]
- Rouleau GA, Clark AW, Rooke K, Pramatarova A, Krizus A, Suchowersky O, Julien JP, Figlewicz D. SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann Neurol. 1996;39:128–131. doi: 10.1002/ana.410390119. [DOI] [PubMed] [Google Scholar]
- Schlamp CL, Johnson EC, Li Y, Morrison JC, Nickells RW. Changes in Thy1 gene expression associated with damaged retinal ganglion cells. Mol Vis. 2001;7:192–201. [PubMed] [Google Scholar]
- Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006;7:66. doi: 10.1186/1471-2202-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatorov V, Malyukova I, Fariss R, Wawrousek EF, Swaminathan S, Sharan SK, Tomarev S. Expression of mutated mouse myocilin induces open-angle glaucoma in transgenic mice. J Neurosci. 2006;26:11903–11914. doi: 10.1523/JNEUROSCI.3020-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea TB, Paskevich PA, Beermann ML. The protein phosphatase inhibitor okadaic acid increases axonal neurofilaments and neurite caliber, and decreases axonal microtubules in NB2a/d1 cells. J Neurosci Res. 1993;35:507–521. doi: 10.1002/jnr.490350507. [DOI] [PubMed] [Google Scholar]
- Sobue G, Hashizume Y, Yasuda T, Mukai E, Kumagai T, Mitsuma T, Trojanowski JQ. Phosphorylated high molecular weight neurofilament protein in lower motor neurons in amyotrophic lateral sclerosis and other neurodegenerative diseases involving ventral horn cells. Acta Neuropathol (Berl) 1990;79:402–408. doi: 10.1007/BF00308716. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Surgucheva I, Ninkina N, Buchman VL, Grasing K, Surguchov A. Protein aggregation in retinal cells and approaches to cell protection. Cell Mol Neurobiol. 2005;25:1051–1066. doi: 10.1007/s10571-005-8474-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surguchov A, McMahan B, Masliah E, Surgucheva I. Synucleins in ocular tissues. J Neurosci Res. 2001;65:68–77. doi: 10.1002/jnr.1129. [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Stadler MB, Roska B, Billings N, Sun B, Bartch B, Cepko CL. Molecular heterogeneity of developing retinal ganglion and amacrine cells revealed through single cell gene expression profiling. J Comp Neurol. 2007;502:1047–1065. doi: 10.1002/cne.21368. [DOI] [PubMed] [Google Scholar]
- Ward MS, Khoobehi A, Lavik EB, Langer R, Young MJ. Neuroprotection of retinal ganglion cells in DBA/2J mice with GDNF-loaded biodegradable microspheres. J Pharm Sci. 2007;96:558–568. doi: 10.1002/jps.20629. [DOI] [PubMed] [Google Scholar]
- Whitmore AV, Libby RT, John SW. Glaucoma: thinking in new ways—a role for autonomous axonal self-destruction and other compartmentalised processes? Prog Retin Eye Res. 2005;24:639–662. doi: 10.1016/j.preteyeres.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Zhong L, Bradley J, Schubert W, Ahmed E, Adamis AP, Shima DT, Robinson GS, Ng YS. Erythropoietin promotes survival of retinal ganglion cells in DBA/2J glaucoma mice. Invest Ophthalmol Vis Sci. 2007;48:1212–1218. doi: 10.1167/iovs.06-0757. [DOI] [PubMed] [Google Scholar]