

Abstract
DNA methylation is an epigenetic mechanism that plays a critical role in the repression of gene expression. Here, we show that DNA methyltransferase (DNMT) inhibition in hippocampal neurons results in activity-dependent demethylation of genomic DNA and a parallel decrease in the frequency of miniature EPSCs (mEPSCs), which in turn impacts neuronal excitability and network activity. Treatment with DNMT inhibitors reveals an activity-driven demethylation of brain-derived neurotrophic factor promoter I, which is mediated by synaptic activation of NMDA receptors, because it is susceptible to AP-5, a blocker of NMDA receptors. The specific effect of DNMT inhibition on spontaneous excitatory neurotransmission requires gene transcription and is occluded in the absence of the transcriptional repressor methyl-CpG-binding protein 2 (MeCP2). Interestingly, enhancing excitatory activity, in the absence of DNMT inhibitors, also produces similar decreases in DNA methylation and mEPSC frequency, suggesting a role for DNA methylation in the control of homeostatic synaptic plasticity. Furthermore, adding excess substrate for DNA methylation (S-adenosyl-l-methionine) rescues the suppression of mEPSCs by DNMT inhibitors in wild-type neurons, as well as the defect seen in MeCP2-deficient neurons. These results uncover a means by which NMDA receptor-mediated synaptic activity drives DNA demethylation within mature neurons and suppresses basal synaptic function.
Keywords: spontaneous neurotransmission, MeCP2, hippocampal neuron, FM1-43, S-adenosyl-l-methionine, homeostatic plasticity
Introduction
DNA methylation is a key cellular mechanism used to repress gene expression and promote genome stability in various species. In mammals, DNA methylation plays roles in many processes, such as X chromosome inactivation, genomic imprinting, and chromosome stability. DNA methyltransferases (DNMTs) are the enzymes responsible for adding methyl groups at the 5-position of cytosine residues within CpG dinucleotides. During development, widespread methylation changes occur in primordial germ cells and preimplantation embryos (Reik et al., 2001; Jaenisch and Bird, 2003). After cellular differentiation, DNA methylation changes are less numerous and thought to control the tissue-specific gene expression required to maintain the identity of cells from one generation to the next. Studies of DNA demethylation in differentiated cells suggests only a passive mechanism by which methylation patterns are lost during the DNA replication that occurs with each cellular division (Ehrlich, 2003). The idea that an active mechanism of demethylation can occur in a replication-independent manner is rather controversial. However, evidence for this type of demethylation does exist. For example, demethylation of transfected DNA into nonreplicating cells has been shown to occur (Paroush et al., 1990). More recently, the growth arrest and DNA damage protein Gadd45a was discovered to play a role in active demethylation of DNA in proliferating as well as in nondividing cells, by promoting DNA repair and thereby erasing methylation marks (Barreto et al., 2007).
The importance for DNA methylation in the brain is becoming apparent because of its association with a number of neurodevelopmental disorders. Both Fragile X and ICF (immuno-deficiency, centromeric region instability, facial anomalies) syndromes arise from malfunctions in the establishment of normal DNA methylation patterns (Turner et al., 1996; Hansen et al., 1999). Rett syndrome is caused by mutations in the DNA methyl-binding protein MeCP2 (methyl-CpG-binding protein 2), a protein important for interpreting DNA methylation and controlling the repression of gene transcription (Amir et al., 1999). Patients of all three syndromes manifest levels of mental retardation, demonstrating the importance for proper DNA methylation in the regulation of normal brain function.
DNMTs are highly expressed in neurons in the adult brain, suggesting they may have a functional role in postmitotic neurons (Goto et al., 1994; Brooks et al., 1996; Inano et al., 2000; Feng et al., 2005). Methylcytosine analogs, including 5-azacytidine (5azaC), can inhibit DNA methylation in many cell types (Robertson and Jones, 2000). Recent studies suggest that inhibiting DNA methylation in hippocampal slices blocks long-term memory and synaptic plasticity. Treatment with methylcytosine analogs blocks long-term potentiation and memory formation after contextual fear conditioning, a hippocampal-dependent associative learning and memory test (Levenson et al., 2006; Miller and Sweatt, 2007). In other studies, prolonged depolarization of cultured cortical neurons has been reported to result in a decrease in methylation in the promoter region of brain-derived neurotrophic factor (BDNF), a neurotrophin important for synaptic plasticity (Martinowich et al., 2003). These studies suggest that there may be a relationship between synaptic activity and DNA methylation in mature neurons. However, the mechanisms behind these changes in DNA methylation and synaptic function are unknown.
Materials and Methods
Cell culture.
Dissociated hippocampal cultures were prepared according to previously published protocols (Kavalali et al., 1999). Briefly, whole hippocampi were dissected from C57BL/6 mice on postnatal days 0–3, or from MeCP2 knock-out mice and littermate controls on postnatal day 0. Tissue was trypsinized for 10 min at 37°C, mechanically dissociated using siliconized glass pipettes, and plated onto Matrigel-coated coverslips. A concentration of 4 mm cytosine arabinoside (Sigma, St. Louis, MO) was added at 1 d in vitro (DIV). For DNA methylation and mRNA measurements, neurons were plated directly onto Matrigel-coated six-well plates. All experiments were performed on cultures 13–21 DIV.
Drug treatments and cell viability.
Twenty-four-hour treatment of hippocampal cultures was done with the following drugs: dimethylsulfoxide (DMSO; 1:1000), 5azaC (2.5 μm; Sigma), Zebularine (Zeb; 50 μm; Sigma), S-adenosyl-l-methionine chloride (100 μg/ml; Sigma), actinomycin D (Act D; 2.5 μm; Sigma), tetrodotoxin (TTX; 1 μm; Calbiochem, La Jolla, CA), or 2-amino-5-phosphonopentanoic acid (AP-5; 50 μm; Sigma). 5azaC (2.5 μm), S-adenosyl-l-methionine (SAM; 50 μg/ml), picrotoxin (PTX; 50 μm; Sigma), and TrkB-IgG antibody (10 μg/ml; R & D Systems, Minneapolis, MN) treatments were also done for 48 h where indicated. After treatment with DNMT inhibitors, cell viability was checked using trypan blue exclusion (Sigma). The percentages of dead cells were not significantly different between control cultures (7.4 ± 1.5% SEM, DMSO treated) and cultures treated with methylcytosine analogs (5azaC, 8.1 ± 2.4%; Zeb, 7.4 ± 2.1%).
Measurement of unmethylated DNA.
Genomic DNA was extracted from hippocampal cultures (DNeasy tissue kit; Qiagen, Valencia, CA), and bisulfite modification of 0.4 μg of DNA was performed (CpGenome DNA modification kit; Chemicon, Temecula, CA). Quantitative real-time PCR was used to determine the amount of unmethylated CpG island present in the BDNF promoter I according to previously published work (Levenson et al., 2006). Briefly, 2 μl of DNA was added to 10 μl of iQ SYBR Green Supermix (Bio-Rad, Hercules, CA), 7 μl of DEPC H2O, and 1 μl of each primer. The following primers were used at an 18 μm concentration: forward, 5′-GGGTAGTGATTTTGGGGAGGAAGTAT-3′; and reverse, 5′-CAACCTCTATACACAACTAAATCCACC-3′. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers were used as controls: forward, 5′-AGGTCGGTGTGAACGGATTTG-3′; and reverse, 5′-TGTAGACCATGTAGTTGAGGTCA-3′. Each sample was run in triplicate. Reactions were run on an Mx3000P real-time PCR machine (Stratagene, La Jolla, CA) with the following cycling program: 95°C for 3 min, 40 cycles of 95°C for 15 s, 60°C for 1 min, and 74°C for 15 s. Detection of fluorescent products was at the end of the last step. For each sample, a ΔCt value was determined (Ct BDNF − Ct GAPDH), followed by a ΔΔCt value relative to DMSO controls (ΔCt experimental treatment − ΔCt control treatment). Fold changes were determined by taking 2 to the power of ΔΔCt values. PCR products were run out on an agarose gel and visualized using ethidium bromide.
Measuring BDNF mRNA.
Hippocampal cultures were washed once with PBS. RNA was isolated by scraping neurons from six-well plates in 500 μl of RNA STAT-60 reagent (Tel-Test, Friendswood, TX) and transferred to 1.5 ml Eppendorf tubes. Tubes were incubated on ice for 5 min, and 100 μl of chloroform was added, mixed thoroughly, and incubated again on ice for 2 min. Tubes were spun for 15 min at 12,000 × g at 4°C. The upper, aqueous layer was transferred to new tubes, and 250 μl of isopropanol and 4 μl of linear acrylamide (Ambion, Austin, TX) were added, mixed, and incubated at −80°C for 1 h. Tubes were spun for 15 min at 12,000 × g at 4°C. Pellets were washed with 1 ml of 70% ethanol and resuspended in 20 μl of DEPC-treated H2O. To make cDNA, 0.8 μg of RNA was brought up to a total volume of 17 μl with DEPC H2O, treated with 1 μl of TURBO DNase (Ambion) in 2 μl of DNase buffer, mixed, and incubated at 37°C for 25 min. Five microliters of DNase inactivation reagent were mixed in for 2 min at 23°C and pelleted by spinning at 10,000 × g for 1 min, and supernatant was transferred to new tubes. Two microliters each of random hexamers (50 ng/μl), dNTPs (10 μm), and DEPC H2O were mixed in with the samples and incubated at 65°C for 5 min. Tubes were put on ice for 1 min, and 8 μl of 5× First Strand Buffer (Invitrogen, Carlsbad, CA) and 2 μl each of DTT (0.1 m; Invitrogen), RNase OUT (Invitrogen), and Superscript III Reverse Transcriptase (Invitrogen) were mixed in and incubated for 5 min at 25°C, 60 min at 50°C, and 15 min at 70°C. Real-time PCR was performed using 1 μl of cDNA added to 10.5 μl of iQ SYBR Green Supermix (Bio-Rad), 7.5 μl of H2O, and 1 μl of each primer. The following primers were used at a 10 μm concentration: BDNF forward, 5′-CCTGCATCTGTTGGGGAGAC-3′; and reverse, 5′-GCCTTGTCCGTGGACGTTTA-3′. GAPDH primers were used as controls: forward, 5′-AGGTCGGTGTGAACGGATTTG-3′; and reverse, 5′- TGTAGACCATGTAGTT-GAGGTCA-3′. Each sample was run in triplicate. Reactions were run on an Mx3000P real-time PCR machine (Stratagene) with the following cycling program: 95°C for 10 min, 40 cycles of 95°C for 20 s, 59°C for 30 s, and 72°C for 20 s. Detection of fluorescent products was at the end of the second step.
Immunocytochemistry.
Dissociated hippocampal neurons were fixed for 2 min at room temperature in PBS with 2% formaldehyde and 2% sucrose, followed by treatment with cold methanol for 10 min at −20°C. Then, the neurons were blocked in 2% goat serum for 1 h at room temperature. The cells were then incubated with primary antibodies, anti-postsynaptic density-95 (PSD-95) monoclonal (1:200; Affinity Bioreagents, Golden, CO) and anti-Synapsin polyclonal (1:1000; Synaptic Systems, Goettingen, Germany), added to 0.02% gelatin and 0.5% Triton X-100 in PBS overnight at 4°C. The next day, neurons were washed with PBS and incubated with fluorescent secondary antibodies, goat anti-rabbit (1:200; Invitrogen) and goat anti-mouse (1:200; Invitrogen). Coverslips were mounted on glass slides with Vectashield (Vector Laboratories, Burlingame, CA), and neurons were visualized on a Zeiss (Thornwood, NY) confocal microscope.
Electrophysiology.
Synaptic activity was recorded from hippocampal pyramidal neurons (from three or more different cultures for each experiment) using a whole-cell voltage-clamp technique. Data were acquired using an Axopatch 200B amplifier and Clampex 9.0 software (Molecular Devices, Sunnyvale, CA). Recordings were filtered at 2 kHz and sampled at 200 μs. A modified Tyrode's solution containing (in mm) 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, pH 7.4, was used as an external bath solution, with 50 μm PTX to isolate miniature EPSCs (mEPSCs) and EPSCs or with 10 μm 6-nitro-7-sulfamoylbenzo-(f)quinoxaline-2,3-dione (NBQX) to isolate miniature IPSCs (mIPSCs). For NMDA current recordings, the external bath solution also contained (in μm) 0 MgCl2, 10 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 50 PTX, 1 strychnine, and 15 glycine. The pipette internal solution for all voltage-clamp experiments contained (in mm) 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 tetraethylammonium-Cl, 4 Mg-ATP, 0.3 Na3GTP, pH 7.35, and 10 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide; 300 mOsm]. Field stimulation was applied through parallel platinum electrodes immersed in the perfusion chamber delivering 20 mA pulses. For whole-cell current-clamp recordings, the external solution contained 50 μm PTX, and the pipette internal solution contained (in mm) 110 K-gluconate, 20 KCl, 10 HEPES, 10 NaCl, 4 Mg-ATP, 0.3 Na3GTP, and 0.6 EGTA. Series resistances of recordings ranged between 7 and 25 MΩ and were not significantly different among drug treatments (DMSO, 15.1 ± 1.0; 5azaC, 13.8 ± 2.1; Zeb, 18.1 ± 2.1).
Fluorescence imaging.
For high potassium stimulation, synaptic boutons were loaded with FM1-43 during a 90 s incubation in Tyrode's solution containing 47 mm K+. After washing with a dye-free Tyrode's solution for 10 min, synaptic terminals were destained using a 90 mm K+ Tyrode's solution for 90 s, followed by three applications of 60 s (each separated by 60 s intervals). For spontaneous recycling pool experiments, boutons were loaded with FM2-10 during a 15 min incubation in Tyrode's solution containing TTX, washed for 10 min in dye-free Tyrode's solution, and destained with Tyrode's solution containing TTX for 20 min. This was followed by three applications of 90 mm K+ Tyrode's solution for 60 s (each separated by 60 s) to release all dye from synapses. All staining and washing solutions contained 10 μm CNQX and 50 μm AP-5 to prevent recurrent activity. Isolated boutons were selected during the wash, and fluorescence changes were measured during destaining. Images were obtained by a cooled, intensified digital CCD camera (Roper Scientific, Duluth, GA) during illumination at 480 nm via an optical switch (Sutter Instruments, Novato, CA). Images were acquired and analyzed using imaging software (Molecular Devices).
Data and statistical analyses.
mEPSCs (AMPA type) and mIPSCs were typically recorded for at least 3–5 min and analyzed using Mini Analysis Program version 6.0.3 (Synaptosoft, Decatur, GA). NMDA-type mEPSCs and all evoked EPSCs were analyzed using Clampfit 9.0 software (Molecular Devices). All data were tested for statistical significance by means of a two-tailed Student's t test, and all error bars represent the SEM.
Results
Effects of DNMT inhibition on miniature postsynaptic currents and synapse number
To examine whether alterations in DNA methylation influence synaptic transmission, hippocampal neurons were cultured from newborn C57BL/6 mice, matured (13–21 DIV) (Mozhayeva et al., 2002), and treated for 24 h with two different DNMT inhibitors, the methylcytosine analogs 5azaC or Zeb (Tawa et al., 1990). Whole-cell voltage-clamp recordings were performed on cultured hippocampal neurons immediately after 24 h chronic treatments. Spontaneous mEPSCs were measured in neurons treated with 5azaC, Zeb, or vehicle (DMSO). These recordings were done in the presence of TTX to block action potential (AP) firing and PTX to block inhibitory activity. We found a significant decrease in the frequency of mEPSCs in both the 5azaC- and Zeb-treated neurons compared with controls but no change in the amplitudes, or rise and decay times, of these events (Fig. 1 A–C and supplemental Fig. S1A–C, available at www.jneurosci.org as supplemental material). The changes in mEPSC frequency, but not kinetics or amplitudes, point to a possible presynaptic deficit or change in synapse number after DNMT inhibition. To test whether this effect on spontaneous activity is specific for excitatory synapses, mIPSCs were recorded in neurons in the presence of TTX and the AMPA-type glutamate receptor blocker NBQX. No changes were observed in the frequency, rise, and decay kinetics or amplitudes of mIPSCs in neurons with either 5azaC or Zeb treatments compared with controls, suggesting that DNMT inhibition is affecting only excitatory synaptic activity (Fig. 1 D–F and supplemental Fig. S1D–F, available at www.jneurosci.org as supplemental material). The fact that both DNMT inhibitors caused similar deficits in excitatory synaptic transmission suggests that inhibiting DNA methylation produces specific alterations in synapse function and not generalized, global nonspecific effects.
Figure 1.
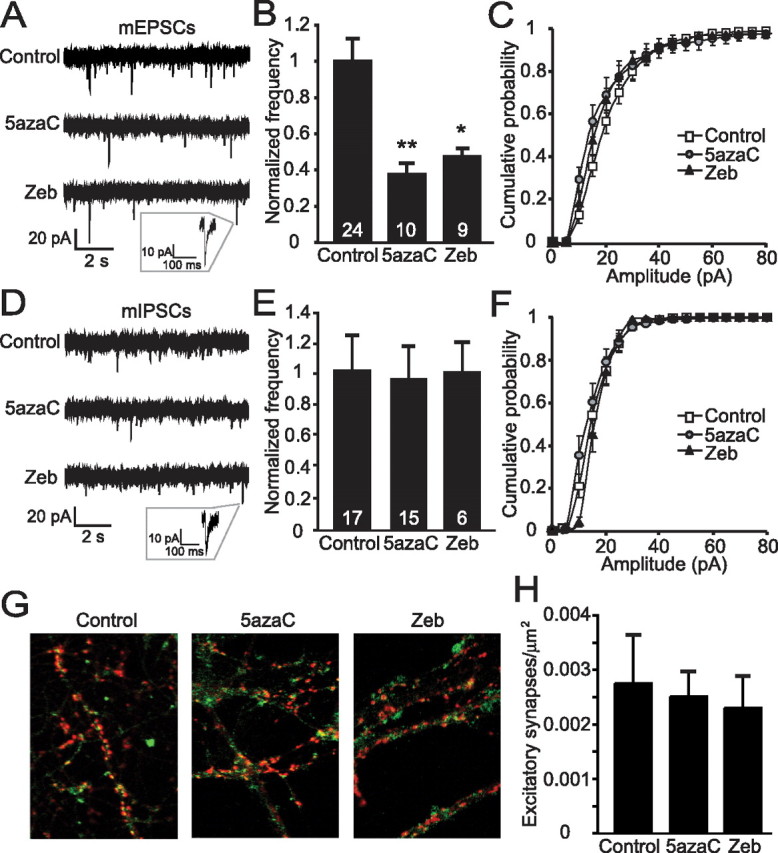
Inhibiting DNMT activity in neurons causes a deficit in excitatory synaptic transmission. A, Sample traces of mEPSCs recorded from mature hippocampal cultures treated 24 h with DNMT inhibitors 5azaC (2.5 μm) and Zeb (50 μm). B, Bar graph showing a decrease in the frequencies of mEPSCs recorded from 5azaC- and Zeb-treated neurons compared with controls (**p < 0.01; *p < 0.05). The numbers on the bars indicate the number of experiments (B, E). C, Histograms of mEPSC amplitudes show no differences among control, 5azaC, or Zeb treatments. D, Sample recordings of mIPSCs from cultures treated with inhibitors of DNMT activity. E, Bar graph depicts no change in mIPSC frequencies from 5azaC- or Zeb-treated neurons compared with controls. F, Cumulative histograms of mIPSC amplitudes show no differences between control and DNMT inhibitor-treated neurons. G, Dissociated hippocampal cultures were immunostained with antibodies to Synapsin (red) and PSD-95 (green) to determine the number of excitatory synapses. H, The bar graph shows no alterations in excitatory synapse number among control, 5azaC-treated, or Zeb-treated cultures.
The deficits in mEPSC frequency produced by DNMT inhibition could be produced by decreases in excitatory synapse number and/or release probability from excitatory presynaptic terminals (Chao et al., 2007). Therefore, we assessed the number of functional excitatory synapses formed onto cultured hippocampal neurons after 24 h of DNMT inhibition. Neurons were immunostained for Synapsin, a presynaptic vesicle protein, and PSD-95, a postsynaptic scaffolding protein found specifically at excitatory synapses. The analysis of colocalized Synapsin and PSD-95 revealed that the number of excitatory synapses was unaffected by treatment with either 5azaC or Zeb, indicating that the decreases in spontaneous mEPSCs are not the result of a decreased number of excitatory inputs (Fig. 1G,H).
Short-term synaptic plasticity and FM dye imaging after DNMT inhibition
We also examined the properties of evoked excitatory neurotransmission in response to AP stimulation to determine whether there were deficits in release probability after inhibition of DNMT activity. After treatment with 5azaC or Zeb for 24 h, there were no changes in excitatory postsynaptic response depression during a 10 Hz field stimulation (Fig. 2A,B) or in the paired-pulse ratios of these responses recorded at various stimulation frequencies (Fig. 2C). In addition, no changes were seen in evoked EPSC amplitudes, and no changes were seen in any of these measurements after a longer, 48 h exposure to these DNMT inhibitors (data not shown). These observations are consistent with a previous study that did not find effects of these drugs on basal synaptic transmission or short-term synaptic plasticity in hippocampal slices (Levenson et al., 2006). These findings suggest that inhibition of DNMTs by methylcytosine analogs specifically affects the release probability of spontaneous miniature excitatory events.
Figure 2.

DNMT inhibition for 24 h specifically affects spontaneous presynaptic function. A, Normalized sample traces of the first 20 evoked EPSCs, recorded in the presence of PTX, in response to 10 Hz field stimulation from neurons treated with inhibitors of DNMTs. B, Average normalized EPSC amplitudes from treated neurons measured during 20 s of 10 Hz stimulation show no alterations in response depression after treatment with DNMT inhibitors compared with controls (control, n = 15; 5azaC, n = 14; Zeb, n = 12). C, Paired-pulse ratios of the first two evoked EPSCs in response to various stimulation frequencies were not significantly different in DNMT inhibitor-treated neurons compared with controls. D, Total synaptic vesicle pools were loaded with FM1-43 by 47 mm K+-induced depolarization and destained using 90 mm K+. The kinetics of destaining was not different between control and 5azaC-treated neurons. Inset, The bar graph depicts no change in total recycling synaptic vesicles from control and 5azaC-treated synapses measured by the total change in fluorescence during 90 mm K+ destaining (control, n = 8 coverslips; 5azaC, n = 8 coverslips). E, Spontaneously recycling synaptic vesicles were loaded for 15 min in the presence of TTX and destained for 20 min in the same manner. Destaining of spontaneous vesicles was slower in 5azaC-treated synapses compared with controls (**p < 0.01). Inset, The bar graph indicates no difference in the numbers of spontaneously recycling synaptic vesicles between control and DNMT inhibitor-treated neurons (control, n = 6; 5azaC, n = 5).
To further explore the presynaptic properties of these neurons, we used FM dye imaging to measure aspects of both evoked and spontaneous synaptic vesicle recycling. To label all recycling vesicles within presynaptic terminals, cultures were stimulated with 47 mm K+ solution for 90 s in the presence of the styryl dye FM1-43 (Betz et al., 1996; Harata et al., 2001). After a brief wash, the kinetics of release of these vesicles was measured by destaining synapses with 90 mm K+ solution for 90 s. The rates of dye loss from total vesicle pools in control and DNMT inhibitor-treated synapses was similar (Fig. 2D), as were the total amounts of dye trapped within individual synapses (Fig. 2D, inset), indicating that the sizes of the total vesicle pools were unchanged. The properties of spontaneously recycling vesicles from neurons treated with inhibitors of DNMT activity were also assessed. Vesicles were loaded for 15 min in the presence of TTX to block activity and washed, and the destaining of individual synapses was measured for 20 min, again in the presence of TTX to block synaptic activity. Treatment with 5azaC resulted in slower destaining kinetics compared with controls (Fig. 2E), suggesting a specific deficit in spontaneous fusion after DNMT inhibition. The total amount of dye released during destaining was similar between control and 5azaC-treated synapses (Fig. 2E, inset), implying that the number of vesicles that recycle spontaneously was unaffected. Together, these results support a specific role for DNA methylation in the control of spontaneous vesicle release from presynaptic terminals.
Miniature NMDA currents are decreased after treatment with DNMT inhibitors
Another possible explanation for the previously observed decrease in mEPSC frequency is an increase in the number of silent, or only NMDA receptor-containing, synapses after 5azaC and Zeb treatments. To assess the number of silent synapses in our cultures, we used whole-cell voltage clamp to measure spontaneous mEPSCs carried via NMDA receptors. To isolate these events, the recordings were done in 0 mm Mg2+ to allow current flow through NMDA receptors without depolarization, and in the presence of the NMDA coagonist glycine, the AMPA receptor blocker CNQX, and two blockers of inhibitory activity, PTX and strychnine. After 3–5 min of recording, the NMDA antagonist AP-5 was then washed on to establish a baseline noise level for each recording. The charge values [pA*ms (Q)] per 10 s intervals of NMDA mEPSCs were averaged and then divided by the baseline Q determined after the addition of AP-5. This charge ratio was significantly smaller in neurons treated with DNMT inhibitors compared with controls (Fig. 3A,B), indicating that treatment with DNMT inhibitors triggers a parallel decrease in AMPA receptor-mediated and NMDA receptor-mediated mEPSCs, the opposite effect from what would be expected as a result of an increase in silent synapses. Nevertheless, individual NMDA mEPSC events are difficult to discern, therefore this decrease could be the result of a decrease in either the frequency of these events or in their amplitudes. Changes in current amplitudes are typically the result of a postsynaptic alteration in receptor number or responsiveness, a change that should also be apparent when measuring evoked NMDA EPSCs. However, the average evoked NMDA EPSC amplitudes were similar among 5azaC-, Zeb-, and control-treated cultures (Fig. 3C,D). As a result, these data further support a specific deficit in presynaptic release probability from excitatory synapses after chronic DNMT inhibition.
Figure 3.

Spontaneous NMDA mEPSCs are decreased after inhibition of DNMTs. A, Sample traces of whole-cell voltage-clamp recordings of NMDA mEPSCs done in the presence of CNQX, PTX, strychnine, glycine, and TTX. AP-5 was then washed on to determine baseline noise levels. B, Bar graph demonstrating that both 5azaC and Zeb treatments resulted in significant decreases in the amount of mEPSC charge (Q) per 10 s intervals divided by the baseline amount of Q after AP-5 was added. **p < 0.01; ***p < 0.001. C, Sample traces of evoked NMDA EPSCs recorded from treated neurons. D, Bar graph showing no changes in average EPSC amplitudes after treatment with DNMT inhibitors. The numbers on the bars indicate the number of experiments (B, D).
Mechanistic insight into the control of mEPSC frequency by DNA methylation
The molecular substrate for the DNMT methylation reaction is SAM. The interaction of methylcytosine analogs, such as 5azaC, with DNMTs interferes with the abilities of the enzymes to transfer methyl groups from this endogenous substrate to cytosines in CpG dinucleotides. However, in the presence of excess SAM, DNMTs can actually methylate 5azaC-containing DNA (Gabbara and Bhagwat, 1995). Therefore, we surmised that the addition of excess SAM may reverse the DNMT inhibitor-induced deficits in spontaneous synaptic transmission. The previously seen decrease in mEPSC frequency after 24 h 5azaC and Zeb treatments was no longer seen after the addition of SAM (Fig. 4A), indicating a rescuing effect of the drug. Furthermore, the proposed functions of 5azaC and Zeb are to inhibit DNMT-dependent methylation of appropriate CpG dinucleotides, thus resulting in the improper transcription of specific genes. Therefore, the effects of these two drugs should be dependent on active gene expression. To investigate whether the deficit in mEPSC frequency caused by inhibition of DNMT activity is dependent on transcription, we treated cultures with the RNA polymerase inhibitor Act D, in combination with either 5azaC or Zeb. Act D treatment was able to block the decrease in mEPSC frequency seen in neurons treated with DNMT inhibitors (Fig. 4B). These results demonstrate that the defects in miniature excitatory event frequency seen after blocking the ability of DNMTs to methylate DNA is, in fact, a result of increased gene transcription and not some nonspecific side effect of the drugs.
Figure 4.
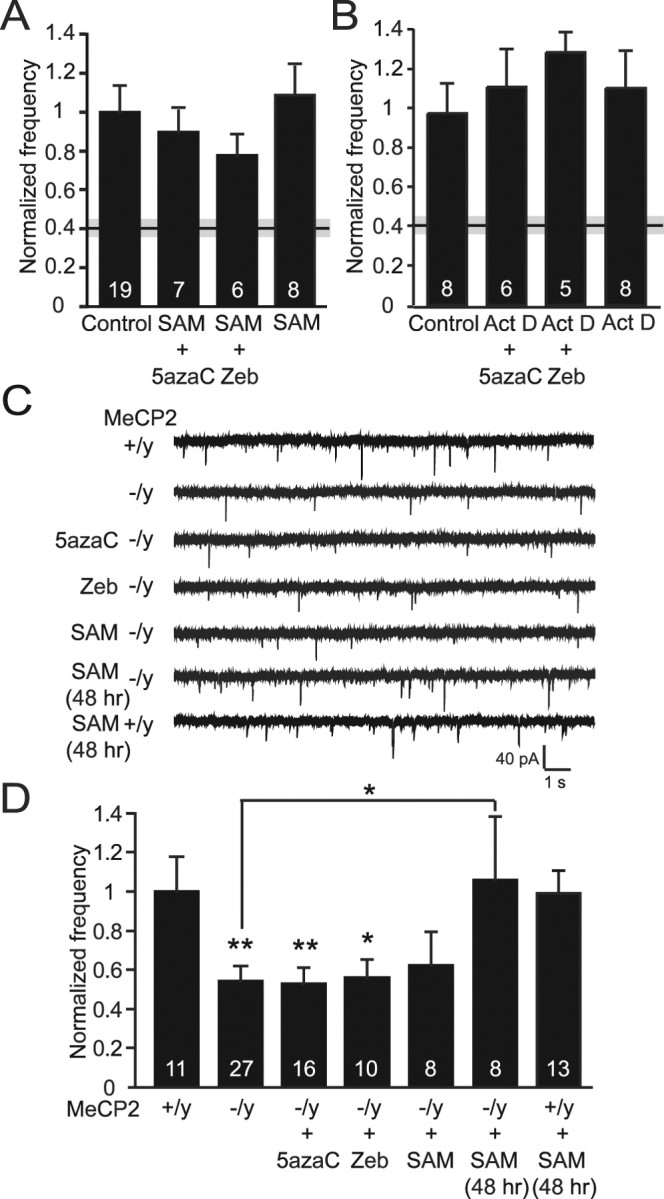
Changes in spontaneous excitatory neurotransmission after DNMT inhibition are mediated by the loss of function of the transcriptional repressor MeCP2. A, Bar graph demonstrating that cotreatment with DNMT inhibitors and the methyl donor, SAM, or treatment with SAM alone, results in no alterations in mEPSC frequency compared with controls. The black and gray bar indicating the mean ± SEM for 5azaC treatment is shown for comparison. B, Bar graph revealing no changes in mEPSC frequencies when neurons were treated with DNMT inhibitors in combination with the inhibitor of transcriptional activation, Act D, suggesting that gene transcription is required for the deficit seen with DNMT inhibitor treatment alone. The black and gray bar indicating the mean ± SEM depicts the 5azaC result. C, Representative traces of mEPSCs recorded from MeCP2-deficient neurons. D, Bar graph showing a significant decrease in mEPSC frequency in MeCP2 knock-out (−/y) neurons compared with wild-type (+/y) littermate controls. A decrease in frequency is also seen after 24 h treatments of knock-out neurons with 5azaC, Zeb, or SAM. However, a 48 h application of SAM onto MeCP2-deficient neurons was able to significantly reverse this frequency deficit compared with control-treated knock-out neurons (**p < 0.01; *p < 0.05). The numbers on the bars indicate the number of experiments (A, B, D).
In mammalian cells, one primary role of DNA methylation is to promote the binding of transcriptional repressor proteins to the promoters of specific genes. One such DNA methyl-binding protein is MeCP2. Mutations in the MeCP2 gene are associated with the neurological disorder Rett syndrome. We previously reported a defect in excitatory neurotransmission in cultured hippocampal neurons from MeCP2-deficient mice, of which included a specific decrease in the frequency of mEPSCs (Nelson et al., 2006). We were intrigued by the similar decrease in mEPSC frequency seen after DNMT inhibition, suggesting a shared mechanism for the control of excitatory spontaneous transmission by DNA methylation and MeCP2. To investigate this possibility, primary hippocampal cultures were isolated from newborn MeCP2 knock-out mice, and mature cultures were treated with DNMT inhibitors. No further reduction in the frequency of mEPSCs from these neurons was detected after the addition of either 5azaC or Zeb (Fig. 4C,D), effectively denoting that the loss of MeCP2 function, either in the knock-out or by eliminating DNA methylation, is the primary cause of this deficit. These results also suggest that the effects of DNMT inhibition occur in postmitotic neurons because MeCP2 is not expressed in glial cells (Shahbazian et al., 2002).
Next, the possibility that the methyl donor, SAM, may also alleviate the synaptic defects seen in MeCP2-deficient neurons was assessed. A decrease in mEPSC frequency was still seen after a 24 h treatment with the DNMT substrate; however, after 48 h, SAM was able to rescue the defect in spontaneous event frequency seen in MeCP2 knock-out neurons (Fig. 4C,D). The need for a longer exposure to SAM is not surprising because previous work has shown that an increase in DNA methylation does not occur until after a >36 h treatment with SAM (Noh et al., 2005). The transcriptional repressor MeCP2 is the only methyl-binding protein that can bind to singly methylated CpG dinucleotides (Lewis et al., 1992), whereas other proteins containing a methyl-binding domain (MBD) require additional sites (Hendrich and Bird, 1998). It is possible that the addition of excess SAM leads to an increase in the number of methylated CpGs on specific promoters, allowing for other MBDs to bind in the absence of MeCP2 (modeled in supplemental Fig. S2, available at www.jneurosci.org as supplemental material). We saw no effect of SAM treatment, for either 24 or 48 h, on wild-type neurons (Fig. 4A,C,D), suggesting that the binding of MeCP2 to sufficiently methylated promoter regions has already repressed the expression of genes responsible for suppressing synaptic function. The ability of SAM to rescue the synaptic deficits in the MeCP2 knock-out is intriguing, and future studies will be important to determine whether SAM can rescue the behavioral deficits in mouse models of Rett syndrome.
Synaptic activity drives DNA methylation-dependent effects on mEPSCs
Our data, thus far, support a role for DNA methylation in synaptic function, which overlaps with the role of MeCP2 as a transcriptional repressor. Recently, the function of MeCP2 was shown to be susceptible to activity-dependent regulation via phosphorylation in response to Ca2+ (Chen et al., 2003; Zhou et al., 2006). We hypothesized that DNA methylation may be an additional synaptic activity-dependent mechanism by which neurons can control gene expression. To determine whether the deficit in synaptic transmission caused by DNMT inhibitors is dependent on activity, mEPSCs were recorded from cultures silenced with TTX during the entire 24 h they were exposed to either 5azaC or Zeb. In the absence of AP firing, treatment with these DNMT inhibitors no longer resulted in decreased frequency of spontaneous miniature excitatory events (Fig. 5A). Thus, the ability of DNA methylation to influence synaptic transmission requires neuronal activity, suggesting that the methylation status of genes that play a role in controlling synapse function may be regulated by synaptic activity.
Figure 5.

Treatment of hippocampal cultures with DNMT inhibitors reveals activity-dependent alterations in synaptic transmission. A, Bar graph shows no changes in mEPSC frequency when cultures were treated with DNMT inhibitors in the presence of TTX, suggesting that neuronal activity is required for the decrease in frequency seen with DNMT inhibitor treatment alone (black and gray bar indicates mean ± SEM). B, Bar graph indicates no difference in the frequencies of mEPSCs when 5azaC and Zeb treatments included the NDMA antagonist AP-5. The black and gray bar indicating the mean ± SEM is the result of 5azaC treatment alone. C, Representative traces of mEPSCs recorded after 48 h treatments with PTX and 5azaC. D, Bar graph depicts a decrease in mEPSC frequency after PTX treatment that mimics that seen after treatment with 5azaC. There was no further decrease in mEPSC frequency when the treatments were combined (***p < 0.001; *p < 0.05). The numbers on the bars indicate the number of experiments (A, B, D).
A well known mechanism by which synaptic activity can drive changes in gene expression is by calcium influx through postsynaptic NMDA-type glutamate receptors (Bito et al., 1997). Because we observed a requirement of synaptic activity for DNA methylation-induced alterations in synaptic function, we tested the importance of NMDA receptor-dependent calcium signaling in this regulation. We exposed our cultures to the NMDA receptor antagonist AP-5 and measured the effects of DNMT inhibition on mEPSCs. The previously seen decrease in mEPSC frequency after treatments with either 5azaC or Zeb was blocked in the presence of AP-5 (Fig. 5B), suggesting that calcium influx through NMDA receptors is, at least in part, responsible for the effects DNMT inhibition has on excitatory synaptic transmission. AP-5 treatment alone had no effect on mEPSC properties (Fig. 5B and data not shown). Together, these data indicate that the regulation of DNA methylation in mature neurons is dependent on neuronal activity and possibly NMDA receptor-dependent calcium signaling and that these activity-dependent changes in DNA methylation play a fundamental role in controlling basal synaptic function.
To further support the importance of synaptic activity in the control of DNA demethylation, we increased the network activity within our cultures by treating them for 48 h with the GABAA receptor antagonist PTX and/or 5azaC. These experiments were done in the absence of DNMT inhibitors to determine the effects of activity alone on synaptic transmission. Increasing excitatory activity with 48 h PTX treatment caused a decrease in mEPSC frequency, similar to that seen with 5azaC alone (Fig. 5C,D). When cultures were treated with both PTX and 5azaC, we found no additional decrease in mEPSC frequency compared with either treatment alone (Fig. 5C,D). Interestingly, previous studies found a decrease in spontaneous firing after an increase in excitatory drive in cultured cortical neurons using bicuculline, another GABAA antagonist (Turrigiano et al., 1998). This decreased firing in response to chronic increased network activity is considered an example of homeostatic plasticity. Our findings are similar in that a chronic increase in excitatory activity, using PTX, causes a homeostatic decrease in mEPSC frequency in hippocampal neurons. The similar decrease seen with 5azaC treatment, and the occlusion of the DNMT inhibitor-mediated mEPSC frequency deficit by PTX, suggests a shared, possibly homeostatic, mechanism between the two in the control of spontaneous synaptic transmission.
Activity dependence of BDNF promoter demethylation and mRNA expression
To examine whether changes in DNA methylation indeed occur in response to DNMT inhibition, we focused on the BDNF gene, because of its earlier association of high-potassium depolarization-mediated demethylation in its promoter region (Martinowich et al., 2003). After treating dissociated hippocampal cultures with the DNMT inhibitor 5azaC, we isolated genomic DNA and processed it for bisulfite modification. Using quantitative real-time PCR with primers specific for a CpG island located in the promoter region for exon I of the BDNF gene, we found a significant increase in unmethylated DNA after 5azaC treatment compared with vehicle controls (Fig. 6A,B), suggesting that demethylation of this BDNF promoter occurs in mature hippocampal cultures. Our electrophysiology data suggested an activity-dependent effect on mEPSCs after changes in DNA methylation. Therefore, we hypothesized that the spontaneous network activity present in these mature cultures (Virmani et al., 2006) may exert control over BDNF expression by regulating methylation at the BDNF promoter I. When we measured the methylation status of the CpG island after cotreatment with 5azaC and either TTX or AP-5, the increase in demethylation after 5azaC treatment was prevented when either synaptic activity was blocked with TTX or when NMDA receptor activity was blocked using AP-5 (Fig. 6A,B). Blocking synaptic activity using TTX treatment alone had no effect on the amount of unmethylated DNA (data not shown). These results demonstrate that spontaneous network activity present in these cultures is sufficient to render the BDNF promoter susceptible to demethylation and likely alter BDNF expression. We also increased the network activity in our cultures by treating them with PTX alone to demonstrate that decreases in DNA methylation can be seen without incubation with DNMT inhibitors. After a 48 h increase in network excitability, we found an increase in demethylation of the BDNF promoter, again reminiscent of the effects seen after DNMT inhibition (Fig. 6A,B). The mechanism by which neuronal activity regulates the methylation status of genes, such as BDNF, may be that activity makes DNA susceptible to demethylation by causing damage/repair or perhaps facilitates putative demethylation activity.
Figure 6.
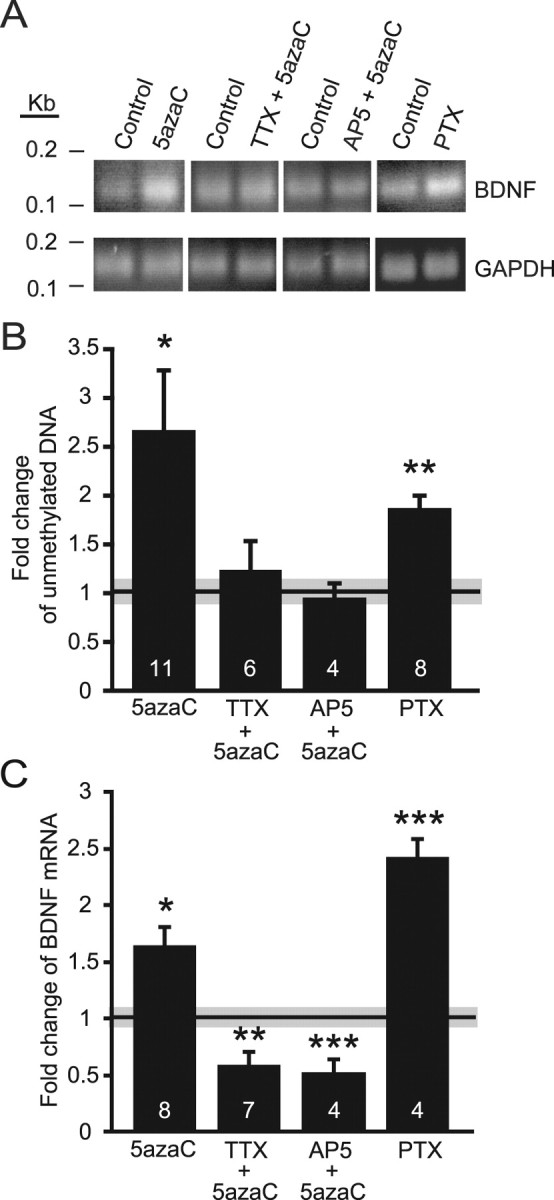
Demethylation of BDNF promoter I and increased BDNF mRNA expression occurs after DNMT inhibition and both are dependent on synaptic activity. A, Bisulfite modification of genomic DNA followed by quantitative PCR (Q-PCR) to measure levels of unmethylated BNDF promoter I. Representative gel electrophoresis of Q-PCR results after control, PTX, and 5azaC treatments with and without TTX or AP-5. B, Bar graph shows a significant increase in the level of unmethylated BDNF promoter I after 5azaC and PTX treatments compared with controls (black and gray bar indicates mean ± SEM) but no changes in the level of unmethylation when 5azaC treatments were in combination with TTX or AP-5 (*p < 0.05; **p < 0.01). C, Bar graph showing increased BDNF mRNA in response to both 5azaC and PTX treatments and decreased expression with 5azaC plus TTX or AP-5 cotreatments (*p < 0.05; **p < 0.01; ***p < 0.001). The numbers on the bars indicate the number of experiments (B, C).
We next assayed whether the changes in methylation of the BDNF promoter I resulted in changes in BDNF mRNA expression. Using quantitative real-time PCR with primers specific to BDNF exon VI, the mRNA coding exon of the gene, we determined the levels of BDNF expression in our cultures after treatment with the DNMT inhibitor 5azaC. Treatment with 5azaC leads to a significant increase in BDNF mRNA levels compared with controls (Fig. 6C), which is the expected result of the decrease in methylation of the BDNF promoter I. We also tested the role of synaptic activity in controlling changes in BDNF expression in response to alterations in DNA methylation by either decreasing the network activity in our cultures, with either TTX or AP-5 in the presence of 5azaC, or increasing activity, using PTX alone. Increasing network activity, using PTX, resulted in a similar increase in BDNF mRNA expression, which also corresponds with the previous changes in demethylation (Fig. 6C). Interestingly, both the blocker of APs and the antagonist of NMDA receptors significantly decreased the expression of BDNF (Fig. 6C). These results were expected given that there is ample evidence indicating an activity-dependent regulation of BDNF gene expression through the regulation of promoter III or IV (Tao et al., 1998). The expression of BDNF mRNA is likely being effected by the regulation of additional promoter regions, such as III or IV, in response to decreased activity. In conclusion, our studies suggest a novel mechanism by which synaptic activity can drive an increase in BDNF transcription through the demethylation of BDNF promoters.
DNMT inhibition reduces spontaneous network activity
To demonstrate the functional significance of the effects of DNMT inhibitors on basal synaptic function, we measured spontaneous firing rates of neurons treated with 5azaC using whole-cell current-clamp electrophysiology. Spontaneous neurotransmission is thought to have effects on numerous aspects of synaptic function, including synapse maturation, dendritic protein synthesis, and homeostatic plasticity (Sutton et al., 2004; Frank et al., 2006). After 24 h DNMT inhibition, we found a fivefold decrease in the mean spontaneous firing rates compared with controls (Fig. 7 A–C). This deficit was not attributable to changes in intrinsic properties of the neurons, because average input resistance (control, 177.5 ± 15.6 MΩ; 5azaC, 193.8 ± 23.1 MΩ) and resting membrane potential (control, 65 ± 2.1 mV; 5azaC, 62.6 ± 1.6 mV) were unaffected by DNMT inhibitor treatment. Furthermore, both AP amplitudes (control, 100.7 ± 31 mV; 5azaC, 100.1 ± 4.0 mV) and half-widths (control, 2.8 ± 0.3 ms; 5azaC, 2.8 ± 0.3 ms) were unchanged. Finally, current step injections were used to determine AP firing threshold and the number of APs fired with each increasing 10 pA current step amplitudes. AP firing thresholds were similar between control and 5azaC-treated neurons (Fig. 7D,E), and the numbers of APs fired with each increasing current step amplitude (plotted as a measure of voltage change) were unaltered after 5azaC treatment (Fig. 7F). Therefore, our data suggest that the decrease in mEPSC frequency, after treatment with DNMT inhibitors, is likely leading to an even larger deficit in overall network activity, without altering the intrinsic or firing properties of individual neurons.
Figure 7.

Spontaneous synaptic firing is reduced after inhibition of DNMT activity. A, Sample current-clamp recordings from 5azaC- and control-treated hippocampal cultures. B, Bar graph depicting a fivefold decrease in mean spontaneous firing rates after DNMT inhibition (*p < 0.05). C, Cumulative probability graph reveals decreased firing rates in all recorded neurons after 5azaC treatment compared with controls. D, Sample recordings showing similar increases in voltage and firing of APs in response to increasing current injections. E, Bar graph showing no difference in AP firing threshold between neurons treated with DNMT inhibitor and controls. F, Histogram demonstrating no significant differences between control and 5azaC-treated neurons in the numbers of APs fired in response to depolarizing voltage changes. The numbers on the bars indicate the number of experiments (B, E).
Deficit in mEPSC frequency after DNMT inhibition is not mediated by the DNA methylation-induced increase in BDNF expression
The previously observed decrease in mEPSC frequency after treatment with DNMT inhibitors was occluded when synaptic activity was increased using PTX, indicating a possible interaction between homeostatic plasticity and alterations in DNA methylation. Homeostatic plasticity is intended to constrain the level of activity within a neuronal network so as not to overload the system with either synapse-strengthening or synapse-weakening forms of synaptic Hebbian plasticity. Interestingly, previous work has established roles for BDNF in mediating aspects of homeostatic plasticity. For example, increased BDNF signaling, which typically enhances excitatory synaptic transmission (Kang and Schuman, 1995; Akaneya et al., 1997), causes an increase in inhibitory activity when exogenous BDNF is applied chronically to cortical cultures (Rutherford et al., 1998). Because we have shown that synaptic activity can drive an increase in BDNF transcription through the demethylation of BDNF promoters, it is possible that enhanced BDNF expression may be mediating the decrease in mEPSC frequency seen after DNMT inhibition. To test this, we treated our hippocampal cultures with an antibody to the high-affinity BDNF receptor TrkB (TrkB-IgG) to block BDNF signaling while still inhibiting DNMTs using 5azaC. Neurons treated for 48 h with TrkB-IgG (10 μg/ml) showed no change in mEPSC frequency compared with controls (Fig. 8A,B). TrkB-IgG treatment also had no effect on the decrease in mEPSC frequency seen with 5azaC treatment (Fig. 8A,B), suggesting that increased BDNF signaling is not causing this deficit in basal synaptic transmission. Treatment with TrkB-IgG antibody did, however, cause a significant increase in mEPSC amplitudes compared with controls (Fig. 8A,C), a result similar to that seen in a previous study (Rutherford et al., 1998). This significant increase in amplitudes was also seen when 5azaC was included with the TrkB-IgG treatment, although to a lesser extent (Fig. 8A,C). These data show that increased BDNF expression is not causing a homeostatic decrease in mEPSC frequency and suggest some additional unknown mechanism by which activity-dependent changes in DNA methylation are causing deficits in excitatory synapse function.
Figure 8.

The decrease in mEPSC frequency seen after DNMT inhibition is not mediated by enhanced BDNF signaling. A, Sample traces of mEPSCs recorded from neurons treated with TrkB-IgG antibodies in the presence and absence of DNMT inhibitors. B, Bar graph shows a significant decrease in mEPSC frequency with both 5azaC alone and TrkB-IgG plus 5azaC treatments (*p < 0.05). The number on the bars indicate the number of experiments. C, Cumulative histogram of mEPSC amplitudes reveals a significant increase after TrkB-IgG treatment and a somewhat smaller, but still significant, increase when neurons were exposed to TrkB-IgG in the presence of the DNMT inhibitor 5azaC (*p < 0.05; **p < 0.01).
Discussion
Together, our findings suggest that spontaneous synaptic transmission between postmitotic neurons is regulated by alterations in DNA methylation that occur in response to synaptic activity. In response to DNMT inhibition, we found a significant decrease in the frequency of mEPSCs as well as the rate of spontaneous synaptic vesicle fusion as detected by FM dye release, which correlates with a decrease in BDNF promoter I methylation and an increase in BDNF expression. Our results demonstrate that both effects on synaptic transmission and BDNF promoter methylation are dependent on the background neuronal activity occurring in our cultures. This activity-dependent regulation is partially attributable to the calcium influx through NMDA receptors and presumably leads to the demethylation of specific gene promoters. This demethylation of specific genes may be mediated by an active demethylating enzyme in neurons; however, the existence of such an enzymatic activity remains an open question. Alternatively, activity may drive demethylation through a process that takes advantage of the DNA damage and repair machinery (Brooks et al., 1996).
Activity-dependent regulation of DNA methylation
Regardless of the underlying mechanism(s), there is increasing support for activity-driven changes in DNA methylation in postmitotic neurons. For instance, prolonged depolarization of cortical cultures results in decreased methylation of the BDNF promoter IV (Martinowich et al., 2003). In addition, both increases and decreases in the DNA methylation patterns of two genes involved in synaptic plasticity, protein phosphatase 1 and reelin, respectively, were found in the hippocampus after fear conditioning (Miller and Sweatt, 2007). The current study explores the implications of these previous observations at the cellular level and uncovers two new facets. First, synaptic network activity can drive DNA demethylation in neurons by activating NMDA receptor-dependent signaling. Second, decreased DNA methylation, in turn, may selectively suppress spontaneous neurotransmission and reduce spontaneous network activity in dissociated neuronal cultures.
Activity-dependent DNA demethylation contributes to homeostatic synaptic plasticity
In this study, we also observed a large deficit in spontaneous network activity in response to DNMT inhibition, which was not associated with changes in the intrinsic electrical properties of the neurons, suggesting that the decrease in spontaneous mEPSCs is likely having a significant effect on the overall network excitability of our cultures. This observation is consistent with previous evidence that the activity of cells with high membrane resistance can be regulated by unitary quantal events (Carter and Regehr, 2002) as well as the finding that asynchronous neurotransmitter release may trigger persistent reverberatory activity in neuronal networks (Lau and Bi, 2005). In addition to regulation of network activity, alterations in spontaneous neurotransmission can influence multiple neuronal processes that include synapse maturation and the stability of neuronal networks (McKinney et al., 1999; Zucker, 2005) as well as regulation of local protein synthesis machinery in dendrites (Sutton et al., 2004; Sutton and Schuman, 2006). Furthermore, spontaneous neurotransmission is a robust readout for homeostatic mechanisms such as synaptic scaling (Turrigiano et al., 1998; Kilman et al., 2002). Our data demonstrated a significant decrease in mEPSC frequency after an increase in network activity, performed by chronically treating our cultures with PTX. This result is reminiscent of the homeostatic changes in synaptic transmission observed previously in response to chronic alterations in network activity levels (Turrigiano et al., 1998). The decrease in mEPSC frequency we observed was correlated with demethylation of the BDNF promoter I and an increase in BDNF expression. Previous work has established roles for BDNF in mediating some forms of homeostatic plasticity. A chronic decrease in endogenous BDNF signaling in cortical cultures increases the excitability of pyramidal neurons, similar to what occurs after chronic activity blockade (Desai et al., 1999). Conversely, exogenous BDNF application increases the firing rates of inhibitory interneurons causing an overall decrease in network activity (Rutherford et al., 1998). In our hands, increased BDNF signaling does not appear to be mediating the homeostatic decrease in mEPSC frequency after DNMT inhibition because blocking BDNF signaling with TrkB-IgG did not rescue this effect. Previous work also found no effect of increased BDNF on mEPSC properties, but they found that increased BDNF was able to reverse the augmentation in mEPSC amplitudes caused by chronic activity blockade (Rutherford et al., 1998). Interestingly, we found an increase in mEPSC amplitudes as a result of decreased BDNF signaling, an effect that is partially rescued by DNMT inhibitors, presumably through the enhancement in BDNF expression. The fact that DNMT inhibitors alone had no effect on mEPSC amplitudes suggests that, in our cultures, baseline levels of BDNF expression under control conditions may saturate the impact of BDNF. Thus, a reduction in BDNF signaling after TrkB-IgG treatment leads to an increase in mEPSC amplitudes uncovering an effect of BDNF similar to previous observations by Rutherford et al. (1998). Furthermore, an increase over normal BDNF levels caused by promoter demethylation could partially rescue the increase in mEPSC amplitudes. Together, our data suggest two mechanisms by which DNA methylation can regulate forms of homeostatic plasticity: by counteracting the effects of decreased BDNF signaling on mEPSC amplitude and by causing a decrease in mEPSC and firing frequency via alterations in the methylation status of some currently unidentified gene(s). Therefore, the increase in levels of synaptic activity, DNA demethylation, and BDNF expression during development of synaptic networks may underlie the dependence of the expression locus of homeostatic plasticity on the age of neuronal cultures (Wierenga et al., 2006).
Implications for MeCP2 function and the synaptic basis of Rett syndrome
Our results demonstrate an intimate relationship between DNA methylation in neurons and the function of the transcriptional repressor MeCP2. The deficit seen in spontaneous synaptic transmission after 5azaC and Zeb treatments was occluded in the absence of MeCP2, the loss of which causes a similar defect in synapse function (Nelson et al., 2006). In addition, treatment of MeCP2 knock-out neurons with the methyl donor SAM was able to reverse this decrease in miniature neurotransmission. Supplemental Figure S2 (available at www.jneurosci.org as supplemental material) depicts a model that may reconcile the impact of synaptic activity on DNA methylation and the function of MeCP2. According to this model, in the absence of activity, MeCP2 is able to bind to methylated CpG dinucleotides and repress the transcription of target genes (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). In the presence of synaptic activity, however, demethylation occurs (possibly through DNA damage/repair), and MeCP2 can no longer bind to the promoter unless remethylation occurs via DNMT activity. In the presence of activity and 5azaC, DNMTs become covalently bound to the methylcytosine analog and can no longer methylate the target promoters. Therefore, the coincidence of activity and 5azaC uncovers the activity-driven demethylation process and its impact on neurotransmission. In MeCP2 knock-out neurons, however, MeCP2 is not present to bind to methylated promoters and repress gene transcription (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). Here, the addition of excess SAM may result in multiple methylated sites at target promoter regions (Noh et al., 2005). The increase in the number of methylated cytosines, in turn, may facilitate the binding of other MBD transcriptional repressors to these sites, thus partially compensating for the loss of MeCP2. This scenario is supported by the previous findings that although MeCP2 binds to singly methylated promoters, other MBDs may require multiple methylation sites for binding (Lewis et al., 1992; Meehan et al., 1992).
These findings suggest that MeCP2 is the methyl-binding protein responsible for mediating the effects of DNA methylation on neuronal function. An interesting future avenue to pursue is the possible affects of SAM treatment on many of the behavioral deficits seen in MeCP2-deficient mice. Therefore, our results also have implications toward treatment of Rett syndrome patients with SAM in an attempt to ameliorate some of their neurological symptoms. Interestingly, there has already been a related study in human Rett syndrome (RTT) patients that received folinic acid treatments (Ormazabal et al., 2005). Folinic acid is the molecular precursor to methionine, among other reaction products, and is used to convert homocysteine to methionine, which is then activated by methionine adenosyltransferase to make SAM. RTT patients treated with folinic acid showed improvements in social behavior and presented less stereotypical “hand-washing” movements and fewer seizures (Ormazabal et al., 2005), signifying that folinic acid, and possibly its reaction product SAM, are viable treatments for patients with Rett syndrome.
In conclusion, our data indicate a role for DNA methylation in the control of synaptic function, which shares a common pathway with the methyl-binding protein MeCP2. Furthermore, our data suggest that neuronal activity can drive the transcription of genes important for controlling spontaneous neurotransmitter release by regulating their methylation status. Previous work has demonstrated that activity can also induce the phosphorylation of MeCP2, causing its dissociation with target genes and relieving its repression of transcription (Chen et al., 2003). Activity-dependent loss of DNA methylation may be an additional mechanism by which MeCP2 is released from the promoters of target genes. Our results concerning DNA methylation and its impact on neurotransmission may suggest a homeostatic mechanism by which neuronal nuclei can monitor alterations in activity levels and adjust neurotransmitter output via altering gene expression and thus impact network excitability.
Footnotes
This work was supported by grants from the National Institute of Mental Health (E.T.K., L.M.M.) and the Division of Basic Sciences Training Program at the University of Texas Southwestern Medical Center (E.D.N.). E.T.K. is an Established Investigator of the American Heart Association. We thank Drs. R. Bassel-Duby, M. Colledge, S. McKnight, E. Nestler, and E. Olson for critically reading this manuscript and Drs. M. Adachi, M. W. Akhtar, and S. Ashimi for helpful discussions.
References
- Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J Neurosci. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, Niehrs C. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Smith CB. Imaging exocytosis and endocytosis. Curr Opin Neurobiol. 1996;6:365–371. doi: 10.1016/s0959-4388(96)80121-8. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. Ca2+-dependent regulation in neuronal gene expression. Curr Opin Neurobiol. 1997;7:419–429. doi: 10.1016/s0959-4388(97)80072-4. [DOI] [PubMed] [Google Scholar]
- Brooks PJ, Marietta C, Goldman D. DNA mismatch repair and DNA methylation in adult brain neurons. J Neurosci. 1996;16:939–945. doi: 10.1523/JNEUROSCI.16-03-00939.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nat Neurosci. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. 1999;6:284–291. [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M. Expression of various genes is controlled by DNA methylation during mammalian development. J Cell Biochem. 2003;88:899–910. doi: 10.1002/jcb.10464. [DOI] [PubMed] [Google Scholar]
- Feng J, Chang H, Li E, Fan G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J Neurosci Res. 2005;79:734–746. doi: 10.1002/jnr.20404. [DOI] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbara S, Bhagwat AS. The mechanism of inhibition of DNA (cytosine-5-)-methyltransferases by 5-azacytosine is likely to involve methyl transfer to the inhibitor. Biochem J. 1995;307:87–92. doi: 10.1042/bj3070087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Numata M, Komura JI, Ono T, Bestor TH, Kondo H. Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation. 1994;56:39–44. doi: 10.1046/j.1432-0436.1994.56120039.x. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harata N, Pyle JL, Aravanis AM, Mozhayeva M, Kavalali ET, Tsien RW. Limited numbers of recycling vesicles in small CNS nerve terminals: implications for neural signaling and vesicular cycling. Trends Neurosci. 2001;24:637–643. doi: 10.1016/s0166-2236(00)02030-0. [DOI] [PubMed] [Google Scholar]
- Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inano K, Suetake I, Ueda T, Miyake Y, Nakamura M, Okada M, Tajima S. Maintenance-type DNA methyltransferase is highly expressed in post-mitotic neurons and localized in the cytoplasmic compartment. J Biochem (Tokyo) 2000;128:315–321. doi: 10.1093/oxfordjournals.jbchem.a022755. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Klingauf J, Tsien RW. Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci USA. 1999;96:12893–12900. doi: 10.1073/pnas.96.22.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABAA receptors clustered at neocortical synapses. J Neurosci. 2002;22:1328–1337. doi: 10.1523/JNEUROSCI.22-04-01328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau PM, Bi GQ. Synaptic mechanisms of persistent reverberatory activity in neuronal networks. Proc Natl Acad Sci USA. 2005;102:10333–10338. doi: 10.1073/pnas.0500717102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20:5085–5092. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Mozhayeva MG, Sara Y, Liu X, Kavalali ET. Development of vesicle pools during maturation of hippocampal synapses. J Neurosci. 2002;22:654–665. doi: 10.1523/JNEUROSCI.22-03-00654.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Noh JS, Sharma RP, Veldic M, Salvacion AA, Jia X, Chen Y, Costa E, Guidotti A, Grayson DR. DNA methyltransferase 1 regulates reelin mRNA expression in mouse primary cortical cultures. Proc Natl Acad Sci USA. 2005;102:1749–1754. doi: 10.1073/pnas.0409648102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormazabal A, Artuch R, Vilaseca MA, Aracil A, Pineda M. Cerebrospinal fluid concentrations of folate, biogenic amines and pterins in Rett syndrome: treatment with folinic acid. Neuropediatrics. 2005;36:380–385. doi: 10.1055/s-2005-873078. [DOI] [PubMed] [Google Scholar]
- Paroush Z, Keshet I, Yisraeli J, Cedar H. Dynamics of demethylation and activation of the alpha-actin gene in myoblasts. Cell. 1990;63:1229–1237. doi: 10.1016/0092-8674(90)90418-e. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Robertson KD, Jones PA. DNA methylation: past, present and future directions. Carcinogenesis. 2000;21:461–467. doi: 10.1093/carcin/21.3.461. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Wall NR, Aakalu GN, Schuman EM. Regulation of dendritic protein synthesis by miniature synaptic events. Science. 2004;304:1979–1983. doi: 10.1126/science.1096202. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Tawa R, Ono T, Kurishita A, Okada S, Hirose S. Changes of DNA methylation level during pre- and postnatal periods in mice. Differentiation. 1990;45:44–48. doi: 10.1111/j.1432-0436.1990.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Turner G, Webb T, Wake S, Robinson H. Prevalence of fragile X syndrome. Am J Med Genet. 1996;64:196–197. doi: 10.1002/(SICI)1096-8628(19960712)64:1<196::AID-AJMG35>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Virmani T, Atasoy D, Kavalali ET. Synaptic vesicle recycling adapts to chronic changes in activity. J Neurosci. 2006;26:2197–2206. doi: 10.1523/JNEUROSCI.4500-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Walsh MF, Turrigiano GG. Temporal regulation of the expression locus of homeostatic plasticity. J Neurophysiol. 2006;96:2127–2133. doi: 10.1152/jn.00107.2006. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Minis: whence and wherefore? Neuron. 2005;45:482–484. doi: 10.1016/j.neuron.2005.02.003. [DOI] [PubMed] [Google Scholar]