

Abstract
The role of presenilin-1 (PS1) in neuronal phosphatidylinositol 3-kinase (PI3K)/Akt signaling was investigated in primary neuronal cultures from wild-type (WT) and PS1 null (PS1−/−) embryonic mouse brains. Here we show that in PS1−/− cultures, the onset of neuronal maturation coincides with a decrease in the PI3K-dependent phosphorylation-activation of Akt and phosphorylation-inactivation of glycogen synthase kinase-3 (GSK-3). Mature PS1−/− neurons show increased activation of apoptotic caspase-3 and progressive degeneration preceded by dendritic retraction. Expression of exogenous WT PS1 or constitutively active Akt in PS1−/− neurons stimulates PI3K signaling and suppresses both caspase-3 activity and dendrite retraction. The survival effects of PS1 are sensitive to inhibitors of PI3K kinase but insensitive to γ-secretase inhibitors. Familial Alzheimer disease (FAD) mutations suppress the ability of PS1 to promote PI3K/AKT signaling, prevent phosphorylation/inactivation of GSK-3 and promote activation of caspase-3. These mutation effects are reversed upon coexpression of constitutively active Akt. Together, our data indicate that the neuroprotective role of PS1 depends on its ability to activate the PI3K/Akt signaling pathway and that PS1 FAD mutations increase GSK-3 activity and promote neuronal apoptosis by inhibiting the function of PS1 in this pathway. These observations suggest that stimulation of PI3K/Akt signaling may be beneficial to FAD patients.
Keywords: Alzheimer's disease, apoptosis, neuronal death, Akt, phosphorylation, signaling
Introduction
The phosphatidylinositol-3 kinase (PI3K) cell survival pathway is activated at the plasma membrane in response to growth factors (Datta et al., 1999; van der Heide et al., 2006) or cell-cell adhesion (Pece et al., 1999; Ditlevsen et al., 2003; Loers et al., 2005). The survival effects of PI3K signaling are mediated by its downstream effector protein kinase B/Akt which is activated by phosphorylation at serine 473 and threonine 308 (Fayard et al., 2005). Akt inactivates components of the apoptotic machinery, including the Bcl-2 family protein Bad, the apoptotic executor caspase-9, and members of the Forkhead family of transcription factors while it activates the NF-κB and CREB transcription factors (for review, see Datta et al., 1997; Brunet et al., 2001). In addition, PI3K/Akt signaling regulates translation by activating mTOR (mammalian target of rapamycin) (Ruggero and Sonenberg, 2005), and suppresses the activity of glycogen synthase kinase-3 (GSK-3) (Cross et al., 1995) that has been implicated in neuronal cell death, tau overphosphorylation and production of Aβ peptides (for review, see Kaytor and Orr, 2002; Bhat et al., 2004). The importance of GSK-3β in brain function is underlined by previous data showing increased neurodegeneration, Alzheimer's disease (AD)-like tau hyperphosphorylation, and behavioral abnormalities in GSK-3β conditional knock-out mice, all of which were reversed after restoration of normal GSK-3β levels (Engel et al., 2006).
PI3K/Akt signaling is of central importance for neuronal physiology. In addition to its role in neuronal survival (Brunet et al., 2001), this signaling has been implicated in dendritic morphogenesis (Jaworski et al., 2005), establishment of neuronal polarity (Jiang et al., 2005), synaptic potentiation (Opazo et al., 2003; Wang et al., 2003) and memory formation (Lin et al., 2001; Mizuno et al., 2003). Previous reports link abnormalities of this pathway with an increasing number of neurodegenerative disorders including Huntington's disease (Humbert et al., 2002), and Parkinson's disease (Chen et al., 2004; Ries et al., 2006).
Presenilin1 (PS1) is a ubiquitously expressed transmembrane protein that plays critical roles in development (Shen et al., 1997; Wong et al., 1997) and in early onset familial Alzheimer's disease (FAD) (Sherrington et al., 1995). Recent studies in fibroblast cells implicate PS1 in the regulation of the PI3K/Akt signaling pathway (Baki et al., 2004; Kang et al., 2005; Uemura et al., 2007). Here we used primary neuronal cultures to investigate the role of PS1 and its FAD mutants in the neuronal PI3K signaling, as such studies may yield information on the mechanism by which PS1 FAD mutations promote neurodegeneration. Our data show that the onset of neuronal maturation coincides with a decline in the ability of PS1 null neurons to support PI3K/Akt signaling resulting in impaired phosphorylation/inactivation of GSK-3β, increased activation of apoptotic caspase-3 and progressive neurodegeneration. Exogenous PS1 or constitutively active Akt restore PI3K signaling and GSK-3β phosphorylation and suppress neuronal death. Importantly, PS1 FAD mutations inhibit the functions of PS1 in PI3K/Akt signaling and promote apoptosis. Our data indicate that the neuroprotective role of PS1 depends on its ability to promote the PI3K/Akt signaling and suggest a mechanism by which PS1 FAD mutations contribute to neurodegeneration.
Materials and Methods
Reagents and antibodies.
Common reagents were from Sigma (St. Louis, MO). Neurobasal medium and B-27 supplement were from Invitrogen (Eugene, OR) PI3K inhibitor LY294002, anti-Akt, anti-phospho-Akt (Ser473), anti-phospho-GSK-3β (Ser9), anti-activated caspase-3 antibodies were from Cell Signaling Technology (Beverly, MA). Anti-GSK-3β antibody was from BD Biosciences (Franklin Lakes, NJ). Antibodies against the N-terminal and C-terminal fragments of PS1 (PS1/NTF and PS1/CTF, respectively) have been described previously (Baki et al., 2004). γ-secretase inhibitors (L-685,458 and compound E) were from Calbiochem (La Jolla, CA). Anti MAP2 and anti NeuN antibodies were from Millipore (Temecula, CA). Alexa Fluor secondary antibody conjugates and ProLong Gold Antifade mounting reagent were from Invitrogen.
Neuronal cultures.
Primary neuronal cultures were prepared separately from E15.5 PS1+/+, PS1−/−, and PS1+/− embryonic brains and genotypes were determined as described previously (Shen et al., 1997). The neocortices and hippocampi were dissected out, treated with 0.025% of trypsin in Krebs-Ringer buffer [containing (in mm) 120 NaCl, 5.0 KCl, 25 NaHCO3, 1.2 KH2PO4, 1.5 MgCl2, 10 glucose, pH 7.4] for 10 min at 37 C and, subsequently, were mechanically dissociated in DNase I-containing (0.008%) Krebs-Ringer buffer. After filtration through a 40 μm nylon mesh and centrifugation, the dissociated cells were resuspended in Neurobasal medium containing B27 and plated on poly-d-lysine coated 12-well dishes at 4 × 105 cells/well. On culture day 1, the medium was replaced with fresh Neurobasal-B27 and 20% of the culture medium was renewed on 7 or 8 d in vitro (DIV).
Constructs, viruses and infections.
Constructs for constitutively active (N-myristoylated HA-Akt) and dominant-negative (HA-Akt K179M) Akt (Franke et al., 1995) were a generous gift from Dr T. F. Franke (Columbia University, New York, NY). cDNAs for wild-type and mutant PS1 or for constitutively active and dominant negative Akt were subcloned into either pHSVPrPUC or p1005 herpes simplex virus (HSV) (for EGFP expression) vectors (McPhie et al., 1997; Chen et al., 2007) and replication-defective, recombinant HSV viruses were prepared as described previously (McPhie et al., 1997; Chen et al., 2007). For preparation of lysates used in Western blots, cultures were infected at ∼1 multiplicity of infection (moi). Under these conditions, expression of exogenous PS1 in PS1−/− neuronal cultures was comparable to the levels of PS1 in WT mouse neurons. For immunofluorescence studies, a moi of 0.4 was used. Infected cells were harvested either 2 or 3 d after infection.
Treatment with inhibitors and cell lysates.
Treatment with PI3K inhibitor LY294002 (50 μm), γ-secretase inhibitor l-685,458 (0.5 μm), and γ-secretase inhibitor compound E (1 μm) was for 20–24 h. Cell lysates for Western blotting were prepared in SDS lysis buffer [containing (in mm) 100 Tris-HCl, 20 NaCl, 10 EGTA, 10 EDTA, 1% SDS, 20 NaF, 5 sodium orthovanadate, 1 sodium pyrophosphate, and 200 microcystin] containing complete protease inhibitor mixture (Boehringer Mannheim, Indianapolis, IN). In all experiments, the same amount of total protein was loaded for each sample.
Immunocytochemistry.
Primary neurons grown on poly-d-lysine coated glass coverslips were fixed in 2.5% paraformaldehyde for 10 min at room temperature, washed with 0.3 m glycine in TBS, permeabilized in 0.1% Triton X-100 for 5 min on ice, washed and blocked in Superbock blocking Buffer (Pierce, Rockford, IL) for 1–2 h. Incubation with primary antibodies (in Superbock blocking Buffer) was overnight and with Alexa Fluor secondary antibody conjugates (in Superbock) was for 1 h. For visualization of nuclei, 4,6,diamidino-2-phenylindole (DAPI) was added in the last wash after secondary antibody incubation for 5 min. For detection of apoptosis, the APO-AC Annexin V-C3 (Sigma) was used according to the manufacturer's instructions.
Statistical analysis.
Statistical analysis was based on densitometric data from several independent experiments. Results are expressed as mean ± SEM. Statistical significance was determined with one tail Student's t test. One asterisk indicates p ≤ 0.05, and two asterisks indicate p ≤ 0.005.
Results
PS1 null neurons display impaired PI3K/Akt signaling and increased apoptosis at the end of their second week in culture
To investigate the role of PS1 in neuronal PI3K/Akt signaling, we prepared primary cortical cultures from PS1−/− embryonic brains or from brains from their wild-type or PS1+/− littermates. Based on nuclear DAPI staining together with double immunostaining with the neuronal marker NeuN and the dendritic marker MAP2, the purity of our neuronal cultures was higher than 95% (data not shown). It is well known that at the end of their first week in vitro primary neuronal cultures enter a maturation period characterized by extensive dendritic branching and synaptogenesis that is most prominent during the second week (Dotti et al., 1988; Ichikawa et al., 1993; Lesuisse and Martin, 2002). Examination of lysates from PS1+/+, PS1+/−, and PS1−/− neurons approaching the end of their second week in culture (13 DIV) revealed that PS1−/− neurons are significantly impaired in their ability to activate PI3K/Akt signaling, as indicated by the decreased phosphorylation of both, the PI3K effector protein kinase Akt (Fig. 1Aa,Ab,B) and the Akt target GSK-3β kinase (Fig. 1Aa,Ad,C). In contrast to PS1−/− neurons, PS1+/− neurons were as effective as the wild-type ones in terms of phosphorylation of both Akt (Fig. 1B) and GSK-3β (Fig. 1C), indicating that one copy of PS1 is sufficient for activating the pathway. In agreement with a critical role of the PI3K/Akt signaling in the suppression of apoptosis (Datta et al., 1999; Brunet et al., 2001), PS1−/− neurons showed consistently increased activation of the apoptotic executor caspase 3 (Fig. 1Ae,D). Increased apoptosis in the absence of PS1 was also confirmed by the increase in nuclear fragmentation observed in 13 DIV PS1−/− neuronal cultures (Fig. 1E) and the increased staining of PS1−/− neurons with the early apoptotic marker annexin V (Fig. 1F). These data indicated that absence of PS1 results in decreased activation of the neuronal PI3K/Akt signaling and increased neuronal apoptosis.
Figure 1.

Impaired PI3K/Akt signaling and increased apoptosis in 13DIV PS1−/− neurons. A, Lysates from13DIV PS1+/+, PS1−/− and PS1+/− mouse primary neurons were analyzed for phosphorylated (Ser473) and total Akt (a, b) phosphorylated (Ser9) and total GSK-3β (c, d), cleaved (activated) caspase 3 (cap3) and PS1-CTF (33B10). B–D, Statistical analysis with each bar representing data from five to seven embryos. E, DAPI nuclear staining of PS1+/+ and PS1−/− neurons cultured on glass coverslips for 13 d. Fragmented nuclei are indicated by circles, nuclear fragments are indicated by arrows. F, Annexin V-CY3 staining of 13 DIV PS1+/+ and PS1−/− neurons. Asterisks are described in Materials and Methods.
PS1−/− neurons lose their ability to activate PI3K/Akt signaling at the onset of the neuronal maturation period
To examine whether the reduced activity of PI3K/Akt signaling in 13 DIV PS1−/− neurons is the result of apoptosis, we followed the phosphorylation of Akt and GSK-3 as well as the activation of caspase 3 at different developmental stages in the presence and absence of PS1. Because WT and PS1+/− neurons show similar levels of PI3K/Akt signaling (Fig. 1 A–C), we used combined cultures of PS1+/+ and PS1+/− neurons as controls for the PS1−/− cultures. Figure 2A shows that the ability of PS1−/− neurons to activate PI3K/Akt signaling depends on their developmental stage. Although during the first 4 d in vitro, PS1−/− neurons are as effective as the WT ones in activating the PI3K/Akt signaling (Fig. 2 Aa–Ad, 2 and 4 DIV), a decrease in the phosphorylation/activation of Akt is observed at about the end of the first week in culture, a time that coincides with the onset of maturation period (Dotti et al., 1988). This decrease is followed by a drop in the phosphorylation/inactivation of the Akt target GSK3-β (Fig. 2Ac,Ad). Statistical analysis of the data show that a drop in the ratio of phospho to total Akt is first observed at 6–7 DIV (Fig. 2B) and by day 10, this ratio becomes less than half of the control. In addition, Akt-dependent phosphorylation of GSK-3β decreases (Fig. 2A,C) and accumulation of cleaved (active) caspase-3 increases (Fig. 2Af,D). No differences in the levels of β-tubulin were observed between PS1−/− and control cultures (Fig. 2Ae,E).
Figure 2.

Downregulation of PI3K/Akt signaling in PS1−/− neurons at the onset of neuronal maturation period. A, PS1−/− and PS1+/− mouse primary neurons were maintained in culture for the indicated periods. Lysates were analyzed as indicated. B–E, Statistical analysis with each bar representing data from at least three independent experiments (dark gray, PS1−/−; light gray, PS1+/−). Levels for ph-Akt and ph-GSK were normalized to the levels of total Akt and GSK respectively and data are presented as ratio of phosphorylated total protein. All values are expressed relative to the respective values of the PS1+/− samples. F, Phase contrast pictures of PS1+/+ and PS1−/− primary neurons cultured for 7, 10, and 13 d in vitro. G, MAP2 (red) immunostaining of PS1+/+ and PS1−/− mouse primary neurons cultured on coverslips for 7, 9, and 13 d. Nuclei (blue) were stained with DAPI.
Morphological examination of our cultures shows that PS1−/− neurons are similar to PS1+/+ until about the middle of the second week in culture (Fig. 2F). After this period, PS1−/− neurons display signs of degeneration evident by the reduced complexity of the neuronal network and the progressively increasing number of shrunk and/or fragmented cells (Fig. 2F). In most of our kinetic experiments, the first signs of degeneration in PS1−/− cultures were evident between days 11 and 13 and degeneration extended to >50% of the population was evident at about the beginning of the third week in culture. No morphological differences were observed between PS1+/+ and PS1+/− neurons until the end of their third week in vitro (data not shown). Immunostaining with the dendritic marker MAP2 indicates that PS1+/+ and PS1−/− cultures have similar dendritic complexities and are indistinguishable until ∼9 DIV (Fig. 2G). At the end of the second week however, when a mature network has been established in PS1+/+ cultures, PS1−/− neurons show dendrite retraction and increased nuclear fragmentation (Fig. 1E), suggesting increased neurodegeneration. Together, our data suggest that at the onset of the maturation period, PS1 becomes necessary for activation of neuronal PI3K/Akt signaling and suppression of cell death. Importantly, abnormalities in Akt phosphorylation of PS1−/− neurons precede both, the increase in apoptotic caspase 3 (Fig. 2D) and the appearance of neurodegenerative changes (Figs. 2F,G), suggesting that increased apoptosis and neurodegeneration may result from downregulation of PI3K/Akt cell survival signaling.
Reintroduction of PS1 in PS1−/− neurons restores PI3K/Akt signaling and suppresses neuronal degeneration
To examine whether exogenous PS1 is able to restore PI3K/Akt signaling and rescue PS1−/− neurons from apoptosis, we used recombinant HSV viruses carrying the human PS1-cDNA PS1−/− neurons were infected the 10th or 11th DIV when the pathway is already suppressed and cultures show increased activation of caspase-3. Figure 3A shows that reintroduction of PS1 in PS1−/− neurons was sufficient to stimulate phosphorylation of both Akt (Fig. 3Aa,Ab,B) and GSK-3β (Fig. 3Ac,Ad,B) and to suppress activation of apoptotic caspase3 (Fig. 3Af,B). Thus, PS1 is necessary and sufficient for the activation of PI3K signaling and suppression of apoptosis. To further investigate the effect of PS1 at the single cell level, PS1−/− neurons were infected with HSV-EGFP-vector (V) and HSV-EGFP-PS1 (PS1) recombinant viruses at a low moi, so that noninfected cells would serve as an internal control. Figure 3C shows that, compared with either noninfected (Fig. 3C, EGFP-negative in left and right panels) or vector-infected neurons (Fig. 3C, left, EGFP-positive), neurons expressing HSV-EGFP-PS1 (Fig. 3C, right, EGFP-positive) retain their processes suggesting that exogenous PS1 was able to prevent the dendrite retraction characteristic of PS1−/− neurons at this developmental stage (Fig. 2F).
Figure 3.

Re-introduction of PS1 into PS1−/− neurons restores PI3K/Akt signaling and suppresses neuronal degeneration. A, PS1−/− primary neurons were infected with either HSV-vector virus (V), or HSV-PS1 (PS1) recombinant virus on their 10th day in culture. Lysates were analyzed as indicated. B, Statistical analysis of the results from four independent experiments. C, MAP2 (red) and DAPI (blue) staining of 13 DIV PS1−/− mouse primary neurons, infected with EGFP-expressing (green) HSV-EGFP-vector and HSV-EGFP-PS1 recombinant viruses.
The survival effects of PS1 are mediated by PI3K/Akt signaling
Our data indicate that exogenous PS1 is able to restore PI3K/Akt signaling and to suppress apoptosis and dendrite retraction. To examine whether the survival effects of PS1 are mediated by PI3K/Akt signaling, we first asked whether pharmacological inhibition of PI3K blocks the survival effects of exogenous PS1 in PS1 null neurons. Figure 4A shows that the PI3K inhibitor LY294002, but not the gamma-secretase inhibitor l-685,458, blocks the PS1-induced phosphorylation/inactivation of GSK-3β and the suppression of apoptotic caspase-3, indicating that PI3K signaling is necessary for both the antiapoptotic and the GSK3-suppressing effects of PS1. We next asked whether stimulation of PI3K/Akt signaling is sufficient to increase survival of PS1 null neurons. Figure 4B shows that expression of a constitutively active (CA) Akt in PS1−/− neurons induces phosphorylation/inactivation of GSK-3β whereas it suppresses apoptotic caspase-3. Importantly, similar to the effects of exogenous PS1, constitutively active Akt increased neuronal survival and prevented dendritic retraction. Figure 4C shows that although vector expressing PS1−/− neurons (top left, EGFP-positive) have either already lost their MAP2 staining or have retracted their dendrites, neurons expressing CA Akt retain their dendrites. Together, our data indicate that the survival effects of PS1 are mediated by PI3K/Akt signaling.
Figure 4.

PS1 rescues via PI3K/Akt signaling. A, PS1−/− primary neurons, infected with either HSV-vector virus (V), or HSV-PS1 (PS1) recombinant viruses were incubated in the absence (NT) or presence of LY294002 (LY) or the γ-secretase inhibitor l-685,458 (γ-inh). Lysates were analyzed as indicated. B, Lysates from PS1−/−, PS1+/+ and PS1+/− primary neurons, infected with HSV-EGFP-vector, HSV-EGFP-constitutively active Akt or HSV-EGFP dominant-negative Akt(DN) viruses were analyzed as indicated. C, MAP2 (red) and DAPI (blue) staining of 14 DIV PS1−/− mouse primary neurons, infected with EGFP-expressing (green) HSV-EGFP-vector and HSV-EGFP CA-Akt recombinant viruses.
PS1 FAD mutations impair neuronal PI3K/Akt signaling, leading to increased GSK-3 activity and apoptosis
To determine the effects of PS1 FAD mutations on neuronal PI3K/Akt signaling, we used recombinant HSV viruses carrying either WT or mutant human PS1 to infect PS1+/− neurons, as expression of mutant PS1 on top of one copy of wild-type PS1 is a condition most closely resembling that of FAD. Figure 5A shows that whereas exogenous WT PS1 stimulates phosphorylation of both Akt and GSK-3β above vector levels, PS1 FAD mutants appear ineffective (Fig. 5A). Statistical analysis of our data revealed that, in contrast to wild-type PS1, PS1 FAD mutants were consistently impaired in their ability to stimulate phosphorylation/activation of Akt and phosphorylation/inactivation of the Akt target GSK-3β (Figs. 5B,C), indicating that FAD mutations suppress the ability of PS1 to activate the PI3K/Akt pathway. Furthermore, all PS1 FAD mutants induced a statistically significant activation of apoptotic caspase-3 above basal levels (Figs. 5A,D). Collectively, our data support the conclusion that PS1 promotes neuronal survival and inactivation of GSK-3β by stimulating the PI3K/Akt signaling and that inhibition of this signaling by PS1 FAD mutations results in increased GSK-3 activity and neuronal apoptosis.
Figure 5.

PS1 FAD mutations impair neuronal PI3K/Akt signaling, leading to increased GSK-3 activity and apoptosis. A, PS1+/− primary neurons were infected with HSV-vector (V) or HSV recombinant viruses encoding WT PS1 (wt) or one of the PS1 FAD mutations shown on the top of the lanes. Lysates were analyzed as indicated. B–D, Statistical analysis with each mutation being analyzed in at least four (B, C) or at least three (D) independent experiments.
To determine whether the proapoptotic effects of mutant PS1 are related to their inability to support proper PI3K/Akt survival signaling, we asked whether expression of a constitutively active form of Akt would be able to reverse the effects of PS1-FAD mutations. Figure 6 shows that coexpression of constitutively active Akt together with mutant PS1 was able to restore phosphorylation of GSK-3β and suppress activation of caspase-3, indicating that the proapoptotic effects of PS1 FAD mutations largely depend on mutation-induced dysregulation of PS1/PI3K/Akt signaling. Collectively, our data support the conclusion that PS1 promotes neuronal survival and inactivation of GSK-3β by stimulating the PI3K/Akt signaling and that inhibition of this signaling by PS1 FAD mutations results in increased GSK-3 activity and neuronal apoptosis.
Figure 6.
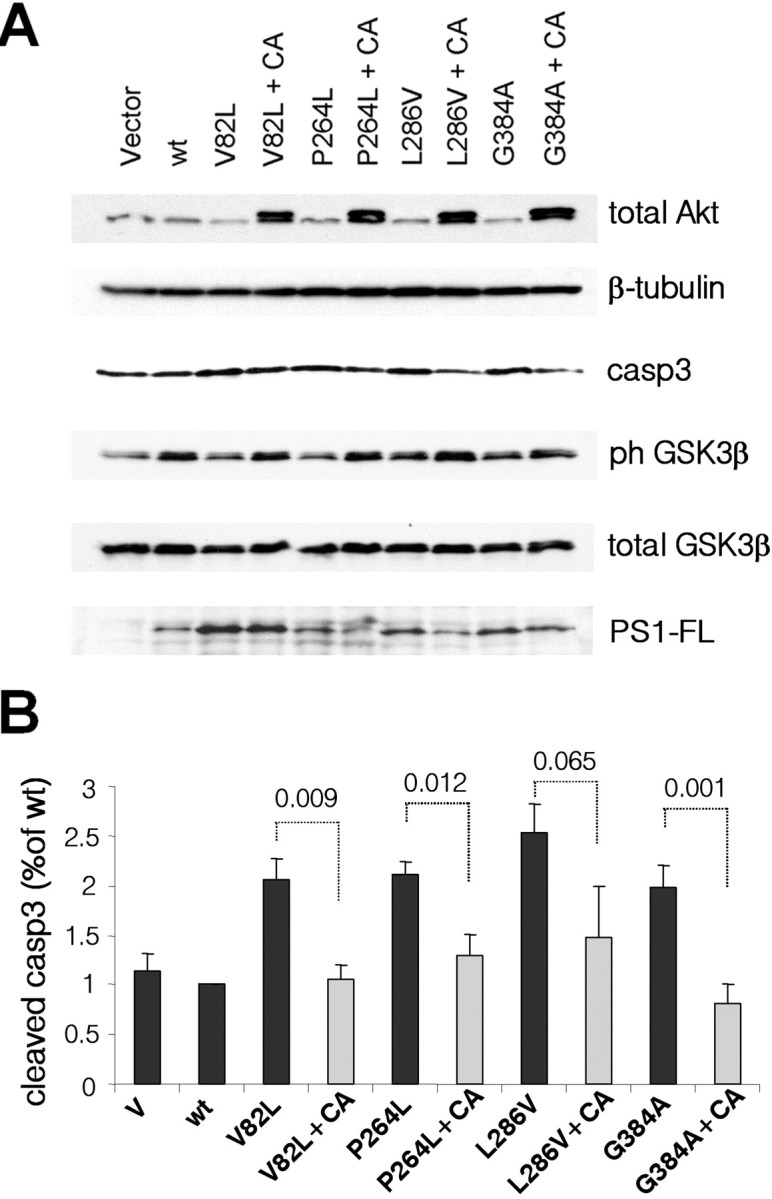
Constitutively active Akt reverses the effects of PS1 FAD mutations. A, PS1+/− primary neurons were infected with HSV-vector (V) or HSV recombinant viruses encoding WT PS1 (wt) or one of the PS1 FAD mutations shown. CA refers to coinfection with HSV-mutant-PS1 viruses plus HSV-CA-Akt viruses (+CA). Lysates were analyzed as indicated. B, Statistical analysis of the data from three independent experiments. The numbers indicate statistical significance.
Discussion
Several studies indicate that PS1 has neuroprotective activities. Data from animal models show that PS1 deficiency results in perinatal death (Shen et al., 1997; Wong et al., 1997) with neuronal loss prominent on embryonic days 17–18 (Shen et al., 1997) and that conditional inactivation of both presenilins in the adult forebrain results in age-dependent neurodegeneration (Saura et al., 2004) tissue shrinkage and apoptosis (Feng et al., 2004). Furthermore, downregulation of PS1 in NT2 cells induces apoptosis (Hong et al., 1999), and primary neuronal cultures from PS1 null embryos show increased vulnerability to oxidative stress (Nakajima et al., 2001). The current study provides evidence that the neuroprotective role of PS1 depends on its ability to promote PI3K/Akt signaling, a cellular pathway critical to cell survival (Datta et al., 1999; Brunet et al., 2001). Mature primary PS1−/− neurons display impaired PI3K/Akt signaling and increased activation of proapoptotic caspase-3 followed by neurodegenerative changes indicated by extensive dendritic retraction and increased pyknotic or fragmented nuclei. These data are in agreement with the critical role of PI3K/Akt signaling in suppressing both, neuronal apoptosis (Brunet et al., 2001) and dendritic retraction (Jaworski et al., 2005). Introduction of exogenous human PS1 in PS1−/− neurons restores PI3K signaling, suppresses apoptotic caspase-3 and prevents retraction of dendrites. Importantly, these PS1 effects are blocked by PI3K inhibitors showing that the effects of PS1 on the phosphorylation of both Akt and GSK-3β are mediated by PI3K activity. Furthermore, our data suggest that the survival effects of PS1 are mediated by the PI3k/Akt cell signaling pathway. In accordance with this suggestion, a constitutively active form of Akt suppressed apoptosis and prevented dendritic retraction in PS1−/− cultures (Fig. 5). Thus, although contribution of other pathways cannot be excluded, our data show that PS1/PI3K/Akt signaling is both necessary and sufficient to suppress apoptosis and prevent neuronal degeneration. In accordance with previous studies showing that the ability of PS1 to activate PI3K/Akt signaling is independent of γ-secterase activity (Baki et al., 2004; Kang et al., 2005), the survival effects of PS1 were not affected by γ-secretase inhibitors. It should be noted that the PS1 homologous protein presenilin 2 (PS2) which has also been implicated in the regulation of PI3K/Akt signaling (Kang et al., 2005), is present in our PS1−/− cultures. Currently, it is unclear what the contribution of PS2 is in our system and whether absence of both presenilins would result in accelerated neuronal degeneration.
It has been shown that after the initial stages of neurite outgrowth and establishment of neuronal polarity, primary neuronal cultures enter a maturation period characterized by extensive branching and formation of synaptic contacts. These maturation activities start at about the end of the first week in vitro and peak at the end of the second week (Dotti et al., 1988; Ichikawa et al., 1993; Lesuisse and Martin, 2002). Neuronal PI3K signaling is activated in response to a variety of stimuli including growth factors (van der Heide et al., 2006), neurotrophins (Mizuno et al., 2003), cell–cell adhesion molecules (Ditlevsen et al., 2003; Loers et al., 2005), and synaptic activity (Perkinton et al., 2002; Sutton and Chandler, 2002; Man et al., 2003). It is expected that cell-contact-dependent mechanisms of PI3K activation (Sutton and Chandler, 2002; Ditlevsen et al., 2003; Man et al., 2003; Loers et al., 2005) are mostly inactive in immature neuronal cultures and that these mechanisms become important during the maturation period when extensive cell-cell contacts are established. Interestingly, our kinetic analysis reveals that PS1 becomes necessary for maintaining PI3K signaling during this developmental stage, suggesting that PS1 may be necessary for contact-dependent activation of neuronal PI3K/Akt signaling. This suggestion is supported by recent data that PS1 regulates the cadherin-dependent activation of PI3K (Baki et al., 2004; Uemura et al., 2007).
Several lines of evidence suggest that PS1 FAD mutations may compromise the neuroprotective effect of PS1. For example, neurons from PS1 M146L knock-in mice show increased vulnerability to glucose deprivation and chemical hypoxia of (Mattson et al., 2000) whereas overexpression of PS1 FAD mutants A246E and C410Y in primary neurons inhibits Akt and increases apoptosis (Weihl et al., 1999). In addition, mutant L286V increases susceptibility to apoptosis induced by trophic factor withdrawal or amyloid β-peptides (Guo et al., 1997). Here we used primary neuronal cultures heterozygous for WT PS1 to examine the effects of eight PS1 FAD mutants on PI3K/Akt signaling and apoptosis in the presence of a WT PS1 allele, a situation closely resembling the in vivo FAD condition. Our data show that compared with WT PS1, all FAD mutants tested were impaired in their ability to stimulate PI3K/Akt signaling and all promoted activation of apoptotic caspase-3. Three of the mutants examined (V82L, L286V, G384A) suppressed PI3K/Akt signaling below the vector control levels indicating these mutants may have dominant negative effects on this signaling and may stimulate apoptosis by suppressing the survival pathway. Other mutants, however, activated apoptotic caspase-3 (Fig. 6D) in the absence of any significant dominant negative effects on PI3K/Akt signaling (Fig. 6B), suggesting that some mutants may have pleiotropic effects that may contribute to the observed apoptosis independently of PI3K/Akt signaling. This may include loss-of-function effects on γ-secretase transcriptional signaling (Marambaud and Robakis, 2005). Regardless of the potential contribution of PI3K-independent mechanisms to mutant-induced apoptosis however, the suppression of neuronal apoptosis by constitutively active Akt (Fig. 4) suggests that the inability of mutant PS1 to support appropriate PS1/PI3K/Akt signaling is a major contributor to the apoptotic effects of these mutants. In support to this notion, coexpression of constitutively active Akt with PS1 FAD mutants reversed the mutant-induced apoptosis (Fig. 6) (Weihl et al., 1999)). Interestingly, in vivo administration of constitutively active Akt was recently shown to rescue dopamine neurons of the substantia nigra in a potent neurotoxin model of Parkinson's disease (Ries et al., 2006).
Activation of PI3K/Akt signaling promotes phosphorylation/inactivation of GSK-3 (Cross et al., 1995). Accordingly, our data show that phosphorylation of GSK-3β closely parallels that of Akt and that downregulation of PS1/PI3K/Akt signaling by FAD mutations results in reduced phosphorylation of GSK-3 indicating that FAD mutations increase GSK-3 activity by inactivating Akt. Over-activation of GSK-3 in response to PS1 FAD mutations has been reported in several cell culture systems (Weihl et al., 1999; Pigino et al., 2003; Baki et al., 2004; Kang et al., 2005) and in brains of knock-in mice (Baki et al., 2004; Tanemura et al., 2006). Because GSK-3 is a major tau kinase (for review, see Kaytor and Orr, 2002; Bhat et al., 2004), it is expected that increased GSK-3 activity would result in increased phosphorylation of tau at GSK-3 target residues. Indeed, we and others have previously shown that presenilin suppresses tau phosphorylation via PI3K/Akt/GSK-3 signaling (Baki et al., 2004; Kang et al., 2005) and that PS1 FAD mutants unable to inactivate GSK-3 fail to suppress GSK-3-dependent phosphorylation of exogenous tau (Baki et al., 2004). In primary neurons however, we observed no consistent difference between WT and mutant PS1 in terms of tau phosphorylation at GSK-3 target residues, although we did observe such a tendency in several of our experiments (data not shown). One possible explanation for this discrepancy is that unlike the exogenous four-repeat tau analyzed in our previous work (Baki et al., 2004), embryonic tau present in primary neurons is already heavily phosphorylated (Mawal-Dewan et al., 1994) and short-term changes in GSK-3 activity, within the timeframe of exogenous PS1 expression, may not be sufficient to produce consistently detectable effects on the phosphorylation of embryonic tau. In agreement with this interpretation, we and others reported hyperphosphorylated tau in the brains of adult PS1 I213T knock-in mice (Baki et al., 2004; Tanemura et al., 2006).
Our data, combined with those from previous studies on the role of WT and mutant presenilins on PI3K/Akt signaling (Weihl et al., 1999; Baki et al., 2004; Kang et al., 2005), suggest that this pathway may be downregulated in FAD. In agreement with this notion, decreased phosphorylation of Akt and increased GSK-3 activity were found in lymphoblasts derived from FAD patients carrying PS1 or PS2 mutations and in HEK293 cells expressing the Swedish APP FAD mutation (Ryder et al., 2004). Conflicting results, however, have been reported for the activity of this pathway in sporadic AD. Zunbenco et al. (1999) reported reduced PI3K activity in AD brain tissue, whereas Griffin et al. (2005) reported increased Akt phosphorylation in AD brain. Postmortem delays in the removal and processing of human brain tissue have been shown to affect both the stability and phosphorylation of many proteins (Ferrer et al., 2007). Accordingly, kinetic studies in our laboratory using mouse brain tissue showed a rapid and significant reduction in the phosphorylation of several phosphoproteins, including Akt, GSK-3 and CREB, most prominent during the first postmortem hour (our unpublished observation). It is therefore possible that the variable postmortem intervals, in combination with different methodologies used in the above studies, contribute to the conflicting data.
In summary, our data indicate that PS1 prevents neurodegeneration by activating the PI3K/Akt signaling of mature neurons. PS1 FAD mutations inhibit this PS1 function and promote deregulation of GSK-3 activity and neuronal apoptosis. These data indicate a mechanism by which PS1 FAD mutations may contribute to neurodegeneration and suggest that stimulation of the PI3K/Akt signaling pathway may be used as a strategy for the treatment of at least the familial cases of Alzheimer disease.
Footnotes
This work was supported by National Institutes of Health Grants NS047229-04 and AG008200-19 to N.K.R., and AG021185 to R.L.N., and by Alzheimer's Association Grant NIRG 04 1011 to L.B. We thank Dr. T. F. Franke, Columbia University, for the constructs for constitutively active and dominant negative Akt.
References
- Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J. 2004;23:2586–2596. doi: 10.1038/sj.emboj.7600251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem. 2004;89:1313–1317. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3β (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18:1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- Chen Y, Bodles AM, McPhie DL, Neve RL, Mrak RE, Griffin WS. APP-BP1 inhibits Abeta42 levels by interacting with Presenilin-1. Mol Neurodegener. 2007;2:3. doi: 10.1186/1750-1326-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Ditlevsen DK, Kohler LB, Pedersen MV, Risell M, Kolkova K, Meyer M, Berezin V, Bock E. The role of phosphatidylinositol 3-kinase in neural cell adhesion molecule-mediated neuronal differentiation and survival. J Neurochem. 2003;84:546–556. doi: 10.1046/j.1471-4159.2003.01538.x. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Hernandez F, Avila J, Lucas JJ. Full reversal of Alzheimer's disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci. 2006;26:5083–5090. doi: 10.1523/JNEUROSCI.0604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase B/Akt at a glance. J Cell Sci. 2005;118:5675–5678. doi: 10.1242/jcs.02724. [DOI] [PubMed] [Google Scholar]
- Feng R, Wang H, Wang J, Shrom D, Zeng X, Tsien JZ. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer's presenilin-1 and presenilin-2. Proc Natl Acad Sci USA. 2004;101:8162–8167. doi: 10.1073/pnas.0402733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Santpere G, Arzberger T, Bell J, Blanco R, Boluda S, Budka H, Carmona M, Giaccone G, Krebs B, Limido L, Parchi P, Puig B, Strammiello R, Strobel T, Kretzschmar H. Brain protein preservation largely depends on the postmortem storage temperature: implications for study of proteins in human neurologic diseases and management of brain banks: a BrainNet Eure Study. J Neuropathol Exp Neurol. 2007;66:35–46. doi: 10.1097/nen.0b013e31802c3e7d. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem. 2005;93:105–117. doi: 10.1111/j.1471-4159.2004.02949.x. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sopher BL, Furukawa K, Pham DG, Robinson N, Martin GM, Mattson MP. Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–4222. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CS, Caromile L, Nomata Y, Mori H, Bredesen DE, Koo EH. Contrasting role of presenilin-1 and presenilin-2 in neuronal differentiation in vitro. J Neurosci. 1999;19:637–643. doi: 10.1523/JNEUROSCI.19-02-00637.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Ichikawa M, Muramoto K, Kobayashi K, Kawahara M, Kuroda Y. Formation and maturation of synapses in primary cultures of rat cerebral cortical cells: an electron microscopic study. Neurosci Res. 1993;16:95–103. doi: 10.1016/0168-0102(93)90076-3. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kang DE, Yoon IS, Repetto E, Busse T, Yermian N, Ie L, Koo EH. Presenilins mediate phosphatidylinositol 3-kinase/AKT and ERK activation via select signaling receptors. Selectivity of PS2 in platelet-derived growth factor signaling. J Biol Chem. 2005;280:31537–31547. doi: 10.1074/jbc.M500833200. [DOI] [PubMed] [Google Scholar]
- Kaytor MD, Orr HT. The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol. 2002;12:275–278. doi: 10.1016/s0959-4388(02)00320-3. [DOI] [PubMed] [Google Scholar]
- Lesuisse C, Martin LJ. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol. 2002;51:9–23. doi: 10.1002/neu.10037. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- Loers G, Chen S, Grumet M, Schachner M. Signal transduction pathways implicated in neural recognition molecule L1 triggered neuroprotection and neuritogenesis. J Neurochem. 2005;92:1463–1476. doi: 10.1111/j.1471-4159.2004.02983.x. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D'Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Robakis NK. Genetic and molecular aspects of Alzheimer's disease shed light on new mechanisms of transcriptional regulation. Genes Brain Behav. 2005;4:134–146. doi: 10.1111/j.1601-183X.2005.00086.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Zhu H, Yu J, Kindy MS. Presenilin-1 mutation increases neuronal vulnerability to focal ischemia in vivo and to hypoxia and glucose deprivation in cell culture: involvement of perturbed calcium homeostasis. J Neurosci. 2000;20:1358–1364. doi: 10.1523/JNEUROSCI.20-04-01358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawal-Dewan M, Henley J, Van de Voorde A, Trojanowski JQ, Lee VM. The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J Biol Chem. 1994;269:30981–30987. [PubMed] [Google Scholar]
- McPhie DL, Lee RK, Eckman CB, Olstein DH, Durham SP, Yager D, Younkin SG, Wurtman RJ, Neve RL. Neuronal expression of beta-amyloid precursor protein Alzheimer mutations causes intracellular accumulation of a C-terminal fragment containing both the amyloid beta and cytoplasmic domains. J Biol Chem. 1997;272:24743–24746. doi: 10.1074/jbc.272.40.24743. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, Takei N, Tran MH, He J, Nakajima A, Nawa H, Nabeshima T. Phosphatidylinositol 3-kinase: a molecule mediating BDNF-dependent spatial memory formation. Mol Psychiatry. 2003;8:217–224. doi: 10.1038/sj.mp.4001215. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Miura M, Aosaki T, Shirasawa T. Deficiency of presenilin-1 increases calcium-dependent vulnerability of neurons to oxidative stress in vitro. J Neurochem. 2001;78:807–814. doi: 10.1046/j.1471-4159.2001.00478.x. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O'Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece S, Chiariello M, Murga C, Gutkind JS. Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell-cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J Biol Chem. 1999;274:19347–19351. doi: 10.1074/jbc.274.27.19347. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries V, Henchcliffe C, Kareva T, Rzhetskaya M, Bland R, During MJ, Kholodilov N, Burke RE. Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson's disease. Proc Natl Acad Sci USA. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D, Sonenberg N. The Akt of translational control. Oncogene. 2005;24:7426–7434. doi: 10.1038/sj.onc.1209098. [DOI] [PubMed] [Google Scholar]
- Ryder J, Su Y, Ni B. Akt/GSK3beta serine/threonine kinases: evidence for a signalling pathway mediated by familial Alzheimer's disease mutations. Cell Signal. 2004;16:187–200. doi: 10.1016/j.cellsig.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, III, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Sutton G, Chandler LJ. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem. 2002;82:1097–1105. doi: 10.1046/j.1471-4159.2002.01031.x. [DOI] [PubMed] [Google Scholar]
- Tanemura K, Chui DH, Fukuda T, Murayama M, Park JM, Akagi T, Tatebayashi Y, Miyasaka T, Kimura T, Hashikawa T, Nakano Y, Kudo T, Takeda M, Takashima A. Formation of tau inclusions in knock-in mice with familial Alzheimer disease (FAD) mutation of presenilin 1 (PS1) J Biol Chem. 2006;281:5037–5041. doi: 10.1074/jbc.M509145200. [DOI] [PubMed] [Google Scholar]
- Uemura K, Kuzuya A, Shimozono Y, Aoyagi N, Ando K, Shimohama S, Kinoshita A. GSK3beta activity modifies the localization and function of Presenilin 1. J Biol Chem. 2007 doi: 10.1074/jbc.M610708200. [DOI] [PubMed] [Google Scholar]
- van der Heide LP, Ramakers GM, Smidt MP. Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol. 2006;79:205–221. doi: 10.1016/j.pneurobio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/s0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP. Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci. 1999;19:5360–5369. doi: 10.1523/JNEUROSCI.19-13-05360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Zubenko GS, Stiffler JS, Hughes HB, Martinez AJ. Reductions in brain phosphatidylinositol kinase activities in Alzheimer's disease. Biol Psychiatry. 1999;45:731–736. doi: 10.1016/s0006-3223(98)00073-0. [DOI] [PubMed] [Google Scholar]