

Abstract
The role of dopamine as a vulnerability factor and a toxic agent in Parkinson's disease (PD) is still controversial, yet the presumed dopamine toxicity is partly responsible for the “DOPA-sparing” clinical practice that avoids using l-3,4-dihydroxyphenylalanine (l-DOPA), a dopamine precursor, in early PD. There is a lack of studies on animal models that directly isolate dopamine as one determining factor in causing neurodegeneration. To address this, we have generated a novel transgenic mouse model in which striatal neurons are engineered to take up extracellular dopamine without acquiring regulatory mechanisms found in dopamine neurons. These mice developed motor dysfunctions and progressive neurodegeneration in the striatum within weeks. The neurodegeneration was accompanied by oxidative stress, evidenced by substantial oxidative protein modifications and decrease in glutathione. Ultrastructural morphologies of degenerative cells suggest necrotic neurodegeneration. Moreover, l-DOPA accelerated neurodegeneration and worsened motor dysfunction. In contrast, reducing dopamine input to striatum by lesioning the medial forebrain bundle attenuated motor dysfunction. These data suggest that pathology in genetically modified striatal neurons depends on their dopamine supply. These neurons were also supersensitive to neurotoxin. A very low dose of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (5 mg/kg) caused profound neurodegeneration of striatal neurons, but not midbrain dopamine neurons. Our results provide the first in vivo evidence that chronic exposure to unregulated cytosolic dopamine alone is sufficient to cause neurodegeneration. The present study has significant clinical implications, because dopamine replacement therapy is the mainstay of PD treatment. In addition, our model provides an efficient in vivo approach to test therapeutic agents for PD.
Keywords: cytosolic dopamine toxicity, neurodegeneration, l-DOPA, oxidative stress, glutathione, dopamine autoxidation
Introduction
The mechanism for the relatively selective loss of dopamine neurons in Parkinson's disease (PD) remains unknown. Dopamine has long been suspected as a toxic agent and a vulnerability factor for neurodegeneration in PD (Hastings et al., 1996; Barzilai et al., 2003). This presumed dopamine toxicity is responsible for the “DOPA-sparing” clinical practice that avoids using l-3,4-dihydroxyphenylalanine (l-DOPA), a dopamine precursor, in early PD. However, there is a lack of in vivo studies on animal models that directly isolate dopamine as the determining factor.
Dopamine is a very reactive molecule compared with other neurotransmitters, and dopamine degradation naturally produces oxidative species (Graham, 1978). More than 90% of dopamine in dopamine neurons is stored in abundant terminal vesicles and is protected from degradation (Eisenhofer et al., 2004). However, a small fraction of dopamine is cytosolic, and it is the major source of dopamine metabolism and presumed toxicity. Cytosolic dopamine undergoes degradation to form 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) as well as hydrogen peroxide via the monoamine oxidase pathway (Graham, 1978; Stokes et al., 1999). Alternatively, dopamine undergoes oxidation to form superoxide, hydrogen peroxide, and o-quinone or reacts with cysteine residues on glutathione or proteins to form cysteinyl-dopamine and cysteinyl-DOPAC conjugates (Hastings and Zigmond, 1994; Sulzer and Zecca, 2000; Barzilai et al., 2003).
The empirical evidence for dopamine as a toxic agent responsible for neurodegeneration in PD remains controversial (Agid et al., 1999; Fahn, 2005). Cell culture studies have shown that extracellular dopamine or l-DOPA is either toxic or protective against cell death (Walkinshaw and Waters, 1995; Simantov et al., 1996; Koshimura et al., 2000). In vivo, one of us demonstrated that a high dose of dopamine injected into the striatum caused selective dopamine terminal loss (Hastings et al., 1996). Sustained elevation of extracellular dopamine causes striatal neuron degeneration in the dopamine transporter (DAT) knock-out mice (Cyr et al., 2003). Both studies suggest that extracellular dopamine is toxic while leaving the role of cytosolic dopamine on neurodegeneration unanswered. In dealing with cytosolic dopamine, two recent studies examined mice with severely reduced vesicular monoamine transporter 2 (VMAT2) expression [5% of wild-type (WT) level]. Colebrooke et al. (2006) found age-related decline of striatal dopamine and motor performance in the absence of nigral cell loss. Using the same transgenic line, Caudle et al. (2007) reported mild substantia nigra dopamine neuron loss in aged mice, suggesting that cytosolic dopamine-mediated toxicity could be the potential cause. However, direct in vivo evidence that isolates cytosolic dopamine as the determining factor responsible for neurodegeneration is lacking.
In the present study, we have generated transgenic mice in which striatal neurons are engineered to take up extracellular dopamine released from dopaminergic terminals without acquiring the regulatory mechanisms typically found in dopamine neurons. These mice develop motor dysfunctions and progressive neurodegeneration in the striatum within weeks. Therefore, we were able to isolate cytosolic dopamine as the determining factor responsible for the neurodegeneration and provide definitive evidence that chronic exposure to unregulated cytosolic dopamine alone is sufficient to produce neurodegeneration in vivo.
Materials and Methods
Mice.
For most experiments, mice were kept on dietary doxycycline (DOX) rodent chow (200 mg/kg; Bio-Serv, Frenchtown, NJ) from gestation. Two- to four-month-old CaMKIIα-tTA; tetO-DAT double transgenic mice (CamDAT mice) were used after DOX withdrawal. When CamDAT mice maintained on DOX treatment were used as controls, sex- and age-matched littermates were used. When tetO-DAT single transgenic mice (TetDAT mice) were used as controls, hemizygous CaMKIIα-tTA mice were bred with homozygous tetO-DAT mice to generate CamDAT experimental group and TetDAT control group. All mice were kept on a 6:00 A.M. to 6:00 P.M. light cycle with ad libitum food and water. Behavioral tests were performed during the light period. All animal procedures were approved by the Institutional Animal Care and Usage Committee of The University of Chicago.
In situ hybridization and immunohistochemistry.
Mice were perfused with 4% paraformaldehyde. For DAT protein expression pattern, 40 μm sections were incubated with rat anti-mouse DAT antibody (1:5000, Millipore, Billerica, MA). For GFAP and α-synuclein immunostaining, sections from the same brains were incubated with rabbit anti-GFAP primary antibody (1:2500, DAKO Cytomation, Fort Collins, CO) or mouse anti-α-synuclein primary antibody (1:2000, BD Transduction, Lexington, KY). Sections were then incubated with secondary antibody (Jackson ImmunoResearch, West Grove, PA) and ABC (Vector Labs, Burlingame, CA). DAB was used for visualization.
Frozen sections (20 μm) were used for in situ hybridization protocol. In situ probe for mouse DAT corresponds to the sequence from 1053 to 1801 nt (GenBank accession number NM_010020). In situ probes for mouse preprodynorphin (Pdyn) and mouse preproenkephalin (PPE) correspond to the sequence from 120 to 614 nt (GenBank accession number U64968) and the sequence from 342 to 1025 nt (GenBank accession number M13227), respectively. Digoxigenin nonradioactive in situ hybridization was used to detect DAT expression (Chen et al., 2005), and 35S radioactive in situ hybridization was used to detect Pdyn and PPE expression (Ding et al., 2007).
Western blot.
For quantification of DAT protein expression levels, brains were lysed in NaCl-Tris-EDTA buffer, and 50 μg of protein was separated on SDS-polyacrylamide gel. The membrane was then blocked and incubated with rat anti-mouse DAT antibody (1:2000, Millipore) and mouse anti-mouse glyceraldehyde-3-phosphate dehydrogenase (1:200,000, Abcam, Cambridge, MA) sequentially. Signals were then detected by horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) and enhanced chemiluminescence (Fisher, Hanover Park, IL).
Unbiased stereological cell counting and brain volume estimation.
Mice were perfused with 4% paraformaldehyde. Brains were sectioned into 40 μm serial sections. One of six sections were stained with mouse anti-mouse NeuN antibody (1:1000, Millipore) and visualized with DAB. The estimation of the total number of NeuN+ neurons in the dorsal striatum was determined using the optical fractionator probe (Stereo Investigator 6 from MicroBrightField, Williston, VT) under 100× oil lens. The 300 × 300 μm grid and 18 × 18 μm counting frame were used. The Gundersun's coefficient of error was 0.05 (m = 1) in this study. To quantify dopamine neurons in substantia nigra pars compacta (SNc), one of three sections was used. The detailed method was described in our previous publication (Chen et al., 2005).
Slides were scanned, and the areas of cerebral cortex, striatum, and olfactory bulb were measured using ImageJ (NIH). Volumes of these structures were estimated by Cavalier principles.
Transmission electron microscopy.
Mice (n = 3 for DOX group and n = 3 for no DOX group) were perfused with 4% paraformaldehyde and 2% glutaraldehyde. Striatum was dissected and postfixed in the same fixative. Then blocks were processed by osmium tetroxide, dehydrated, and embedded in epoxy resin. Ninety nanometer sections were cut and examined under Tecnai F30 scanning transmission electron microscope.
HPLC.
Tissue dopamine, metabolites, and protein cysteinyl-catechols were measured according to previously published methods (LaVoie and Hastings, 1999). Briefly, tissues were homogenized in 1 ml of cold 0.1N perchloric acid and centrifuged (30,000 × g, 20 min), and the resulting supernatant was separated from the protein pellet. An aliquot of the supernatant was filtered before analysis for dopamine, DOPAC, and HVA levels on HPLC with electrochemical detection. The precipitated protein pellet was hydrolyzed in 6N HCl (100°C, under vacuum, 24 h), dried, and alumina extracted before analysis by HPLC with electrochemical detection for cysteinyl-catechol conjugates. All catechols were identified and quantified by comparison to standards.
Open field locomotor activity and food consumption.
Each mouse was placed in an open field chamber (40 cm long × 40 cm wide × 37 cm high, Med Associates, Georgia, VT). Illumination of open filed was set to 20 lux. Locomotor activity was monitored by infrared beams that record the animal's location and path. Data were collected in 20 min sessions with a preceding 10 min habituation period.
For home cage food consumption, mice were individually housed. Preweighed rodent chow was given, and the amount of consumed rodent chow was recorded after 24 h. For small pellet consumption, mice were tested weekly in transparent cylinder with small pellets (20 mg of each) for 5 min. The number of pellets consumed was recorded.
Unilateral lesion of dopamine neurons and forepaw adjust stepping test.
Mice were anesthetized and put into stereotaxics. Desipramine (25 mg/kg, Sigma-Aldrich, St. Louis, MO) was injected intraperitoneally to protect the norepinephrine neurons. Thirty minutes later, 6-OHDA (1 μl of 3 mg/ml dissolved in 0.01% ascorbate in saline, Sigma-Aldrich) was injected into left medial forebrain bundle (coordinates: anteroposterior, −1.3 mm from bregma; lateral, 1.3 mm from bregma; dorsoventral, −4.9 to −5.2 mm from the skull surface) via a 28-gauge stainless-steel cannula that stayed in the brain for 5 min after the injection before being taken out.
The hindpaws of mice were held up and the front paws put on a treadmill as the belt moved forward for a complete cycle. Each mouse was tested in three nonconsecutive cycles. Stepping was videotaped and scored by a blind observer afterward. The numbers of left and right paw steps were counted.
MPTP treatment and silver staining.
Two weeks after DOX withdrawal, mice received saline or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Sigma-Aldrich) at dosages of 4 × 2 mg/kg or 4 × 5 mg/kg intraperitoneally at 2 h intervals (n = 5). One week later, mice were perfused with 4% paraformaldehyde and cryoprotected with 30% sucrose. Brains were sliced on a cryostat. Silver staining was done with FD NeuroSilver Kit (FD NeuroTechnologies, Ellicott City, MD).
l-DOPA treatment. Four weeks after DOX withdrawal, mice were divided into two groups to receive daily intraperitoneal injections of l-DOPA (300 mg/kg, Sigma-Aldrich) plus benserazide (100 mg/kg, Sigma-Aldrich) or saline for 4 weeks. Benserazide is an inhibitor of peripheral DOPA decarboxylase to prevent the conversion of l-DOPA in periphery. Body weight was measured daily during treatment. By the end of fourth week of l-DOPA treatment, mice were perfused, and unbiased stereological cell counting was performed to assess neuron number in striatum.
Glutathione measurement.
Four weeks after DOX withdrawal, mice were killed, and striatum, cerebral cortex, and cerebellum were dissected on ice and then stored in a −80°C freezer. Reduced glutathione (GSH) and oxidized glutathione (GSSG) content were measured using a glutathione assay kit (Cayman Chemical, Ann Arbor, MI) (n = 5). GSH and GSSG contents were normalized to protein content and presented as nmol/mg protein. Protein was quantified with BCA protein quantification kit (Fisher).
Statistical analysis.
Data were analyzed using StatView 5.0.1. Unpaired two-tailed Student's t tests were used when genotype or DOX treatment was the only grouping variable. ANOVA were used when additional factors were considered. Repeated-measures ANOVA were used when data were collected in multiple trials. In figures, * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001. The exact p value (if it is not <0.001) is indicated in figure legends.
Results
Generation of transgenic mice with ectopic DAT overexpression in the forebrain
The transgenic mice with inducible forebrain DAT overexpression were engineered using the tetracycline inducible system (Gossen and Bujard, 1992) (Fig. 1 A). A tetO-DAT knock-in line was generated by inserting the tetO promoter into the 5′ untranslated region of DAT, placing DAT under the transcriptional control of the tetO promoter. In homozygous tetO-DAT mice, DAT expression is reduced to ∼5% of WT expression levels (data not shown). The tetO-DAT mice were then crossed with the well characterized forebrain-specific CaMKIIα-tTA mice (Mayford et al., 1996) (from The Jackson Laboratory, Bar Harbor, ME) to generate CamDAT mice. In these mice, ectopic DAT expression is driven by tTA-dependent tetO activity in a forebrain-specific manner. In addition, DAT is also expressed in dopamine neurons from the remaining wild-type allele.
Figure 1.

Generation of transgenic mice with forebrain expression of DAT (CamDAT mice). A, Inducible forebrain DAT expression mice were generated by breeding forebrain-specific CaMKIIα-tTA mice and tetO-DAT mice. B, DAT mRNA expression was detected in the striatum (STR), cerebral cortex (CTX), hippocampus (HIP), olfactory bulb (not shown), and olfactory tubercle (not shown). Endogenous DAT mRNA was detected in SNc. In control mice (CTL), no DAT mRNA expression was detected in the forebrain. C, DAT proteins were detected in regions with mRNA expression as well as in globus pallidus (arrow) and substantia nigra pars reticulata (arrowhead) that are main striatum efferent nuclei. Top, Forebrain region shown in more rostral sections. Bottom, Midbrain region shown in more caudal sections.
To characterize DAT expression, homozygous tetO-DAT mice were mated with hemizygous CaMKIIα-tTA mice to generate CamDAT mice and TetDAT mice. All mice were kept on dietary DOX (200 mg/kg rodent chow, from Bio-Serv) from gestation until the experiments started, inhibiting ectopic DAT expression. DOX was then removed to induce ectopic DAT expression in adult mice. After 1 week of DOX withdrawal, ectopic DAT mRNA expression was detected in the dorsal striatum, nucleus accumbens, olfactory tubercle, olfactory bulb, cortex, and hippocampus (for lower-power photomicrograph, see Fig. 1 B; for a high-magnification image at the cellular level, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Ectopically expressed DAT protein was highly expressed in the striatum and moderately expressed in the cortex and hippocampus. As expected for a protein enriched in axon terminals, DAT was most abundantly present in two main striatum efferent nuclei, globus pallidus and substantia nigra pars reticulata, that receive inputs from striatal D1-positive and D2-positive medium spiny neurons, respectively (Fig. 1 C). DAT protein was also expressed on cell bodies of neurons in striatum of CaMDAT mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We used Western blot analysis to quantify DAT protein levels. In the forebrain, DAT protein levels in the CamDAT mice after 3 weeks of DOX withdrawal were approximately three times the levels of DOX-treated mice (n = 4 per group, unpaired t test, p < 0.001).
Ectopic DAT overexpression in the forebrain significantly altered dopamine turnover in the striatum
Figure 2 A illustrates a schematic of the predicted dopamine turnover in the CamDAT mice and WT mice, which is supported by our HPLC data (Fig. 2 B). In CamDAT mice after 3 weeks of DOX withdraw, striatal tissue dopamine content decreased by 76%, whereas DOPAC and HVA increased by 68 and 368%, respectively. Similarly, in cerebral cortex, tissue dopamine decreased by 66%, whereas DOPAC and HVA increased by 38 and 192%, respectively. In contrast, in the control tissue cerebellum, where dopamine levels were 0.2% of the striatal level because of the lack of dopamine input or DAT expression, dopamine levels were not changed by DOX withdrawal, whereas DOPAC and HVA levels increased by 31 and 47%, respectively. To exclude the possibility that decreased dopamine level results from loss of dopamine neurons in midbrain, we quantified dopamine neurons by unbiased stereological cell counting. The dopamine neuron numbers in SNc were comparable between the DOX group and no DOX group of CamDAT mice (5938 ± 535 and 6300 ± 364, respectively; data are given as mean ± SEM).
Figure 2.
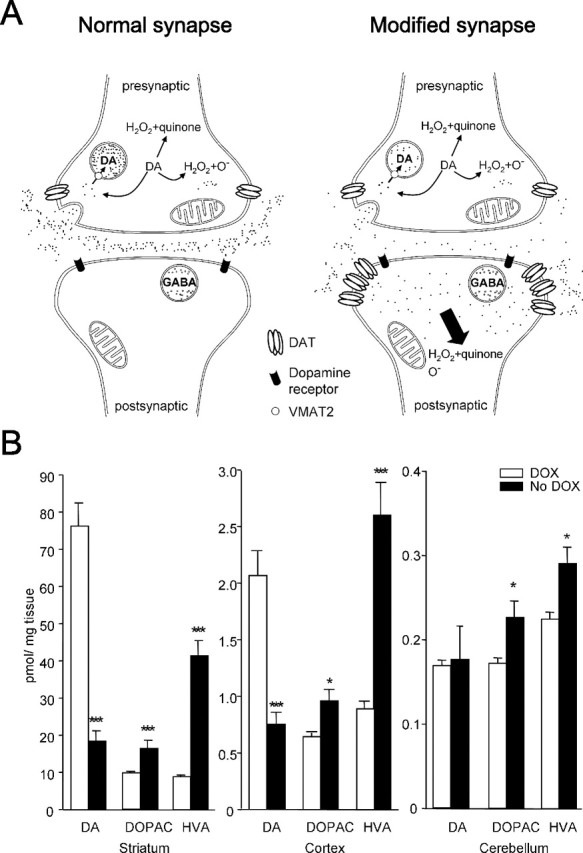
Alternation of dopamine turnover in CamDAT mice. A, A schematic illustration of genetically engineered synapse in the striatum. Most dopamine released by presynaptic dopamine terminal is taken up by postsynaptic neurons expressing ectopic DAT. There is no VMAT2 expression in postsynaptic neurons. Uptaken dopamine is degraded in cytoplasm and produces quinine, H2O2, and other reactive species in postsynaptic neurons. B, After 3 weeks of DOX withdrawal, striatum and cortex dopamine content decreased and its metabolites, DOPAC and HVA, increased. In cerebellum, dopamine level was unchanged, whereas HVA and DOPAC levels increased. Note the differences in y-axis scales (n = 8 per group, unpaired t test, p < 0.001 for dopamine in striatum, p < 0.001 for DOPAC in striatum, p = 0.01 for HVA in striatum, p < 0.001 for dopamine in cortex, p = 0.018 for DOPAC in cortex, p < 0.001 for HVA in cortex, p = 0.015 for DOPAC in cerebellum, p = 0.013 for HVA in cerebellum).
The sharp decrease of dopamine and increases of DOPAC and HVA in CamDAT mice suggest that postsynaptic neurons act as a “sink” that quickly sequesters presynaptic dopamine (Fig. 2 A) and that dopamine is turned over at a high rate, a phenomenon that has been described in the DAT knock-out mice (Jones et al., 1998). The increased dopamine metabolites in the cerebellum suggest diffusions from dopamine terminals. This is consistent with the observed increases of HVA and DOPAC in the cerebellum of l-DOPA-treated animals (el Gemayel et al., 1986).
Ectopic DAT overexpression in the forebrain led to motor dysfunction, body weight loss, and lethality
Ectopic DAT expression in the forebrain had severe behavioral consequences (Fig. 3 A). If not treated with dietary DOX, CamDAT pups were smaller, and most of them died ∼1 week after weaning. If CamDAT mice were treated with DOX from gestation stage on, they were indistinguishable from TetDAT mice. However, if DOX treatment was withdrawn at any time point, most mice died 4–8 weeks afterward (Fig. 3 A).
Figure 3.

Behavioral phenotypes of CamDAT mice. A, DOX treatment was required for the survival of CamDAT mice. B, In the open field, locomotor activity of CamDAT mice was normal 1 week after DOX withdrawal. However, their locomotor activity sharply declined in the second week of DOX withdrawal (n = 4 per group, repeated-measures ANOVA, p < 0.001). C, Progressive body weight loss in CamDAT mice after DOX withdrawal (n = 6, repeated-measures ANOVA, p < 0.001 for group × week interaction). D, Four weeks after DOX withdrawal, CamDAT mice in their home cages consumed very little regular rodent chow (n = 5 per group, individually housed, unpaired t test, p < 0.001). E, Two weeks after DOX withdrawal, when provided with easily accessible chocolate flavored pellets (20 mg each) on the cage floor, CamDAT mice ate more than TetDAT mice (n = 6 per group, repeated-measures ANOVA, p < 0.001 for group × week interaction).
We noted that lethality in CamDAT mice after DOX withdrawal was preceded by severe hypolocomotion and lack of feeding. CamDAT mice developed severely impaired locomotor activity after 2 weeks of DOX withdrawal (Fig. 3 B). We monitored animals' body weight and food intake throughout DOX withdrawal period. Significant body weight loss was obvious in CamDAT mice but not in TetDAT (Fig. 3 C) or CamDAT mice on DOX (data not shown). Body weight loss was apparently caused by lack of feeding (Fig. 3 D). However, when food intake was assessed using easily accessible food (20 mg pellets on the floor), CamDAT mice after 2 weeks of DOX withdrawal (already underweight) consumed more than the control group (Fig. 3 E). Therefore, the lack of feeding is most likely a result of motor dysfunction rather than the lack of appetite.
Neuropathology in forebrain DAT overexpression mice
To explore whether neuropathological changes might underlie the behavioral abnormalities of CamDAT mice, mice were perfused, and brain sections were immunostained with a neuron-specific marker (NeuN) after 4 weeks of DOX withdrawal. We first measured the volume of striatum and cortex as well as olfactory bulb. We found significant striatal and cortical atrophy in CamDAT mice after DOX withdrawal compared with those kept on DOX (Fig. 4 A,C). These areas have both ectopic DAT expression and dopaminergic input. In contrast, there was no change in the olfactory bulb, which also has ectopic DAT expression but has no significant dopamine afferents (Fig. 4 C). The dopamine neurons in the glomerular layer do not project to the granule cell layer, where ectopic DAT expressing neurons are localized.
Figure 4.
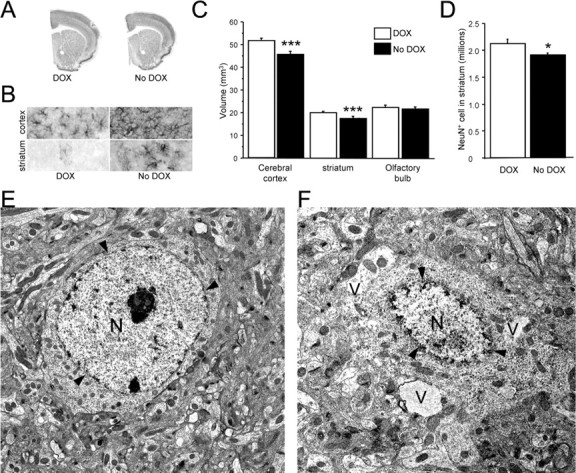
Neuropathology in the forebrain of CamDAT mice. A, C, Four weeks after DOX withdrawal, the volumes of cerebral cortex and striatum decreased by 15 and 10% respectively, whereas volume of olfactory bulb was unchanged (n = 4 per group, unpaired t test, p < 0.001 for both striatum and cortex). B, Astrogliosis was detected by GFAP staining in the cerebral cortex and striatum. n = 6 per group. D, NeuN-positive cells in the striatum decreased by 10% (n = 4, unpaired t test, p = 0.04). E, Transmission electron microscopic image of a normal striatal neuron from a CamDAT mouse under DOX treatment. F, Abnormal morphologies were found in striatum of CamDAT mice after 4 weeks of DOX withdrawal. A severely affected neuron displayed vacuolization (V) and disintegration of the nucleus (N). Arrowheads indicate the nuclear envelope. Scale bar, 2 μm.
We analyzed the striatal neuropathology further by unbiased stereology counting of NeuN-positive cells. We found 10% neuron loss in the striatum of CamDAT mice after DOX withdrawal compared with those kept on DOX (Fig. 4 D). Although the neuronal loss strongly suggested dopamine toxicity, the degree of the cell loss was not sufficient to account for the behavioral changes. Therefore, we sought further evidence of the neuronal dysfunction that precedes actual cell loss.
We investigated ultrastructural evidence for neurodegeneration by transmission electron microscopy. Striatal neurons showed various degrees of abnormal morphologies. Some affected neurons only displayed dilated organelles, such as Golgi apparatus and mitochondria. Other affected neurons displayed prominent cytoplasmic organelle disintegration, although the morphology of their nucleus remained relatively normal. The more severely affected neurons displayed vacuolization and disintegration of the nucleus (Fig. 4 F). The latter two types of pathology account for ∼50% of striatal neurons. The typical apoptotic morphology, such as cell shrinkage, chromatin condensation, membrane blebbing, and phagocytosis, was lacking. We also used apoptotic markers to examine potential apoptosis. Likewise, we found no increase of active caspase-3 or terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-positive neurons in CamDAT mice with ectopic DAT expression (data not shown).
To explore further potential functional changes in striatal neurons, we measured dynorphin and enkephalin mRNA expression, which are neuropeptides specifically found in D1-positive and D2-positive neurons, respectively. Semiquantitative radioactive in situ hybridization for dynorphin and enkephalin mRNA in striatum did not reveal differences in expression levels 4 weeks after DOX removal in CamDAT mice compared with control group (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), suggesting that these peptidergic phenotypes in D1-positive and D2-positive neurons were preserved, although there were profound changes in morphology.
In addition to neurodegeneration, immunohistochemical staining with an antibody against GFAP revealed obvious astrogliosis in striatum and cortex of CamDAT mice after 4 weeks of DOX withdrawal but not in control mice (Fig. 4 B). Although α-synuclein accumulation was observed in several PD models, we did not find α-synuclein accumulation or upregulation in striatal neurons by immunostaining and Western blot (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), suggesting that neurodegeneration in CamDAT mice is likely to be α-synuclein independent.
Neurodegeneration was accompanied by oxidative protein modifications and decreased tissue glutathione content
To further investigate the mechanism of neurodegeneration, we examined evidence for oxidative stress. Dopamine undergoes oxidation to dopamine-quinone, which reacts with cysteine residues on proteins to form cysteinyl-dopamine and cysteinyl-DOPAC conjugates (Hastings and Zigmond, 1994; Sulzer and Zecca, 2000; Barzilai et al., 2003). To assess the abundance of quinone-modified proteins, HPLC analyses of 5-cysteinyl-dopamine and 5-cysteinyl-DOPAC levels were used to assess for quinone modified protein (Hastings et al., 1996; LaVoie and Hastings, 1999). We found significant increases of dopamine-quinone by 527% and DOPAC-quinone by 149% evidenced by modified cysteine residues in CamDAT mice after 3 weeks of DOX withdrawal compared with animals kept on DOX. DOPA-quinone modified cysteine residues were unchanged (Fig. 5 A). Similarly, in cerebral cortex, we found increases of dopamine-quinone by 138% and DOPAC-quinone by 88% noted by modified cysteine residues. DOPA-quinone modified cysteine residues were unchanged (Fig. 5 A). In contrast, in the cerebellum, no change was found for dopamine, DOPA, or DOPAC-quinone modified cysteine residues (Fig. 5 A).
Figure 5.

Oxidative stress in transgenic mice. A, Oxidation-induced protein modification by dopamine. Using HPLC, we found significant increases of cys-dopamine and cys-DOPAC in CamDAT mice after 3 weeks of DOX withdrawal. Similar increases were found in cerebral cortex. In contrast, no change was found in the cerebellum (n = 8 per group, unpaired t test, ***p < 0.001). B, Decreased glutathione content in brain tissues. Total glutathione (GSH+GSSG), GSH, and GSSG were 26% (p < 0.01), 29% (p < 0.01), and 27% (p < 0.05) lower, respectively, in striatum of CamDAT mice after 4 weeks of DOX withdrawal compared with control mice (n = 5 per group, unpaired t test, *p < 0.05, **p < 0.01). No change was found either in the cortex or cerebellum (p > 0.05, n = 5 per group).
In addition to reactive quinones, dopamine metabolism also produces H2O2 and superoxide, which may contribute to oxidative stress. Therefore, glutathione content was examined. Compared with control mice, total glutathione (GSH+GSSG), GSH, and GSSG content in striatum decreased by 26% (p < 0.01), 29% (p < 0.01), and 27% (p < 0.05), respectively, in CamDAT mice 4 weeks after DOX withdrawal (n = 5), which alone could be sufficient to cause mitochondria damage in brain (Jain et al., 1991). In cerebral cortex and cerebellum, total glutathione (GSH+GSSG), GSH, and GSSG content appeared normal (Fig. 5 B).
l-DOPA accelerated neurodegeneration and body weight loss
If cytosolic dopamine is responsible for the aforementioned neuropathology, l-DOPA would be predicted to accelerate such a process by boosting dopamine supply. Four weeks after DOX withdrawal, we treated CamDAT mice with another 4 weeks of daily intraperitoneal injections of 300 mg/kg l-DOPA plus 100 mg/kg benserazide, and body weight was monitored. l-DOPA-treated CamDAT mice lost more body weight than saline control (41.5 vs 25% by the end of fourth week, respectively, n = 5, p < 0.001) (Fig. 6 A). Unbiased stereological cell counting in striatum revealed significant NeuN+ cell loss in l-DOPA-treated CamDAT mice compared with saline-treated CamDAT mice (n = 4, 12% loss, p < 0.05) (Fig. 6 B).
Figure 6.

l-DOPA accelerates neurodegeneration and body weight loss. Four weeks after DOX withdrawal, CamDAT mice were injected daily with 300 mg/kg l-DOPA plus 100 mg/kg benserazide, and body weight was monitored. A, l-DOPA-treated mice gradually lost more body weight than saline control (41.5% vs 25%, respectively, n = 5 per group, unpaired t test, p < 0.001). Day 0 indicates the first day of l-DOPA treatment. Blank data points indicate the days without treatment. B, l-DOPA accelerated neurodegeneration in striatum assayed by unbiased stereological cell counting (12% more loss of NeuN+ neurons in l-DOPA-treated mice, n = 4 per group, unpaired t test, p = 0.03). *p < 0.05, **p < 0.01, ***p < 0.001.
Removing dopamine supply stopped progression of motor dysfunction
Striatal neurons in CamDAT mice express ectopic DAT without the capacity to synthesize dopamine, and therefore the accumulation of cytoplasmic dopamine in these mice depends on supply of extracellular dopamine. To assess whether the behavioral impairment is dopamine dependent (therefore a putative dopamine toxicity effect), we performed unilateral lesions of the medial forebrain bundle by 6-hydroxydopamine before DOX withdrawal to remove the major source of dopamine supply for striatal neurons.
One month after the lesion, CamDAT mice's usage of individual forelimbs was assessed weekly by stepping behavior on a motorized treadmill. Stepping of the contralateral forelimb controlled by the lesion side was significantly impaired (16% of that of intact side), consistent with our previous observation of motor deficit resulting from dopaminergic deafferentation (Chang et al., 1999). The 6-OHDA-lesion paradigm used in current study damages >90% dopamine neurons, which is confirmed by immunostaining with tyrosine hydroxylase antibody (data not shown). After DOX withdrawal, stepping of the ipsilateral forelimb controlled by the intact side declined drastically (Fig. 7) (stepping of the ipsilateral forelimb controlled by the intact side before DOX withdrawal was used as baseline). In contrast, there was no further deterioration of the stepping of the contralateral limb. In fact, a gradual partial improvement of stepping of the contralateral forelimb controlled by the lesion side was noted (Fig. 7). These data suggest that the stepping dysfunction of the intact side after DOX withdrawal was caused by uptake of released dopamine from presynaptic terminals, and such toxicity was spared in the lesion side, where the source of dopamine was removed. The partial improvement of the contralateral limb is intriguing. There are at least two possibilities. One is gradual behavioral adaptation after acute damage. The second possibility is that forced use of this particular limb after drastic deterioration of the other limb induced functional recovery from the lesion. Functional and anatomical recovery after 6-OHDA lesion through exercise or forced use in rodents has been reported by other groups (Tillerson et al., 2001; Dobrossy and Dunnett, 2003). Together, the above data strongly suggest that the behavioral abnormality in CamDAT mice is dependent on dopamine supply.
Figure 7.

Motor dysfunction in CamDAT mice is dopamine supply dependent. DOX treatment was removed 1 month after unilateral 6-OHDA lesion, and stepping of the forelimb controlled by the intact side worsened progressively, dropping from 100 to 15% of baseline level during 4 weeks DOX withdrawal, whereas stepping of the forelimb controlled by the lesion side improved from 16 to 50% of baseline level (n = 5 per group, repeated-measures ANOVA, p < 0.001 for side × week interaction).
Low-dose MPTP causes neurodegeneration in the striatum
MPTP-treated mice usually develop dopamine neuron degeneration and are commonly used as PD models (Przedborski et al., 2004). The selective loss of dopamine neurons in the MPTP model depends on DAT expression in dopamine neurons (Gainetdinov et al., 1997; Donovan et al., 1999). Our data suggest that CamDAT mice are especially vulnerable to dopamine-induced neurodegeneration. To examine whether they are also especially vulnerable to MPTP and to confirm that their ectopically expressed DAT is functional, MPTP was administrated to mice, and neurodegeneration in striatum, cortex, and hippocampus was examined. Two weeks after DOX withdrawal, a single dose of MPTP (30 mg/kg, i.p.) killed all seven CamDAT mice within 24 h, whereas CamDAT mice kept on DOX remained active. Therefore, we lowered the dose of MPTP to 2 mg/kg and 5 mg/kg. All mice were alive 1 week after MPTP treatment. Using silver staining, degenerative neurons in striatum were found in CamDAT mice treated with both dosages of MPTP (Fig. 8). This type of degenerating neuron was not found in saline-treated mice or in low-dose MPTP-treated CamDAT mice kept on DOX. At this low dose of MPTP, no neurodegeneration was observed in the substantia nigra. These data suggest that ectopically expressed DAT in striatal neurons are functional, and these neurons are more susceptible to MPTP treatment than dopamine neurons in substantia nigra, which have protective mechanisms including the presence of VMAT2.
Figure 8.

Low-dose MPTP causes neurodegeneration in forebrain. Using FD silver staining kit, degenerative neurons in striatum were found in an MPTP dose-dependent manner (2 mg/kg, 5 mg/kg). The degenerative neurons (arrows) are characterized by dense, dark silver precipitates in their cell bodies. This type of neuron is absent in saline and DOX groups. n = 3 per group. Scale bar, 20 μm.
Discussion
One important feature of the present in vivo model is that we were able to isolate dopamine as the determining factor in causing neurodegeneration. In our model, dopamine supply determines the progression of neurodegeneration, which is accelerated by l-DOPA treatment and attenuated by removing dopaminergic afferents. Therefore, we provide definitive evidence that chronic exposure to unregulated cytosolic dopamine alone is sufficient to produce neurodegeneration in vivo. Moreover, severe neuropathology and motor dysfunctions develop within weeks, making it a practical model for studying mechanisms and testing therapeutic agents.
In the transgenic mice, striatal neurons were engineered to take up extracellular dopamine released from dopaminergic terminals without acquiring intrinsic regulatory mechanisms typically found in dopamine neurons. Therefore, we were able to isolate the contribution of dopamine without introducing other confounding factors. In normal dopamine neurons, there are major regulatory mechanisms that include the VMAT2 and regulated dopamine synthesis and activity. Those mechanisms maintain cytosolic dopamine homeostasis and keep cytosolic dopamine within relatively nontoxic levels (Zigmond et al., 1989; Kumer and Vrana, 1996; Eisenhofer et al., 2004). Therefore, it may require decades of exposure to low levels of cytosolic dopamine or may require impaired protective mechanisms for neuropathology to take place.
How does cytosolic dopamine cause toxicity? Our data suggest that the detrimental effects of dopamine in the present animal model are mediated via dopamine metabolism and oxidation. H2O2 and other reactive oxidative species produced naturally in dopamine degradation pathways may oxidize proteins, lipids, and DNA, which ultimately cause cell dysfunction and neurodegeneration (Stokes et al., 1999). Of particular importance is the modification of cysteinyl residues by dopamine- and DOPAC-quinones. Cysteinyl residues, the prime target of electrophilic quinones, often reside at the active sites of proteins, and thus, their covalent modifications by quinones could inactivate function, alter conformation, and promote aggregation of proteins (Graham, 1978). One of the main defense mechanisms for such detrimental effects is glutathione. Glutathione is ready to bind dopamine- and DOPAC-quinones by their thiol group so that other proteins are spared of their detrimental modifications. In agreement with cytosolic dopamine toxicity in our transgenic model, there are dramatic increases of 5-cysteinyl-dopamine and 5-cysteinyl-DOPAC and decrease of total glutathione, similar to what have been found in PD patients (Schulz et al., 2000).
Dopamine toxicity-induced neurodegeneration may involve α-synuclein, including its oxidative modification. It has been found that dopamine and other catechols inhibit α-synuclein fibrillization and lead to accumulation of α-synuclein protofibrils. These include studies in both cell culture (Montine et al., 1997; Conway et al., 2001; Cappai et al., 2005; Norris et al., 2005) and in animal models (Li et al., 2004; Mazzulli et al., 2006). It has been further shown that dopamine-autoxidized products but not dopamine itself are responsible for modifying α-synuclein, mainly by noncovalent modification on 125YEMPS129 motif by dopaminochrome, DA-quinone, DOPA-quinone, and DOPAC-quinone. Therefore, we investigated the possibility that α-synuclein is a major mediator of dopamine toxicity, but did not find elevated α-synuclein expression or accumulation, suggesting that neurodegeneration in CamDAT mice is likely to be α-synuclein independent.
Our data show severe motor impairment after DOX removal in CamDAT mice. Because striatal neuron loss is modest, striatal neuron dysfunction is more likely to be the cause of their motor impairment. Our EM ultrastructural data indicate that ∼50% striatal neurons have degenerative morphologies. Low striatal dopamine content might be another contributor. However, our unilateral lesion data clearly indicate that dopamine toxicity is a much more significant contributor than dopamine deficiency to the motor impairment phenotype in CamDAT mice.
The major pathological phenotype of our mice bears some similarities to that of models of Huntington's disease, which is characterized by neuropathology in striatal medium spiny neurons. Interestingly, extracellular dopamine has been shown to worsen neuronal loss caused by mutant huntingtin in both cultured cells and mice models (Hastings et al., 1996; Jakel and Maragos, 2000; Cyr et al., 2003, 2006; Charvin et al., 2005; Tang et al., 2007). In comparison, the CamDAT mouse is a cytosolic dopamine toxicity model. Therefore dopamine is likely to be a toxic agent, whether elevated inside or outside of neurons.
Our data highlight the importance of cellular regulatory mechanisms for cytosolic dopamine homeostasis in preventing dopamine induced neurotoxicity and the importance of protective biochemical pathways such as glutathione. Under normal conditions, those protective mechanisms along with others such as the ubiquitin-proteasome system might work in concert to prevent dopamine neurons from degeneration. However, the detrimental effects of dopamine could be exaggerated under conditions of genetic defects or environmental challenges. For example, the disruption of dopamine vesicular storage by α-synuclein has been suggested (Conway et al., 2001; Lotharius and Brundin, 2002; Cooper et al., 2006). DJ-1-deficient mice have been shown to exhibit elevated DAT function and total tissue dopamine content (Chen et al., 2005). Parkin-deficient mice have been reported to have increased extracellular dopamine (Bonifati et al., 2003; Goldberg et al., 2003). Conversely, parkin has been reported to be one of the targets modified by dopamine quinone (LaVoie et al., 2005). Synuclein protofibrils can also be modified and stabilized by catechols related to dopamine but not other compounds (Conway et al., 2001).
Our results have significant clinical implications for the potential detrimental effects of dopamine replacement therapies for PD, including l-DOPA and some of the gene therapy approaches. Results from the recent large-scale clinical trials are mixed. l-DOPA improved the signs and symptoms of the disease, suggesting neuroprotection. However, the same trial showed reduced uptake of DAT substrates, suggesting more rapid dopamine terminal loss in l-DOPA-treated patients (Rascol et al., 2000; Parkinson Study Group, 2002; Whone et al., 2003; Holloway et al., 2004; Fahn, 2005). Our l-DOPA treatment data indicate that under conditions of compromised defense mechanisms, dopamine replacement therapies will accelerate neurodegeneration. This point is also relevant for many of the PD gene therapy approaches that involve the expression of tyrosine hydroxylase and aromatic l-amino acid decarboxylase either via direct in vivo viral vector delivery or via implantation of ex vivo engineered cells (Kang et al., 2001; Liste et al., 2004). Virus-infected neurons (mostly striatal neurons) and most engineered cells (e.g., fibroblasts) do not have the complete cellular machinery to protect them from cytosolic dopamine toxicity. Therefore, reducing cytosolic dopamine toxicity might be necessary to prevent any potential damages associated with such therapies.
Footnotes
This work was supported in part by the Parkinson's Disease Foundation (X.Z.); the American Parkinson Disease Association (U.J.K.); a grant from Brain Research Center of the 21st Century Frontier Research Program funded by the Ministry of Science and Technology, Republic of Korea (U.J.K.); National Institute of Neurological Disorders and Stroke (NINDS) Grant NS043286 (U.J.K.); and NINDS Grant NS044076 (T.G.H.). We thank Manuel Utset and Robert Burke for their help with neuropathology; William Dauer for critical reading of this manuscript; and Nancy Tian, Sandra Rokosik, Khalid Fakhro, Hyun Ah Yoon, Ali Hussain, and Sarah Manning for technical assistance.
References
- Agid Y, Ahlskog E, Albanese A, Calne D, Chase T, De Yebenes J, Factor S, Fahn S, Gershanik O, Goetz C, Koller W, Kurth M, Lang A, Lees A, Lewitt P, Marsden D, Melamed E, Michel PP, Mizuno Y, Obeso J, et al. Levodopa in the treatment of Parkinson's disease: a consensus meeting. Mov Disord. 1999;14:911–913. doi: 10.1002/1531-8257(199911)14:6<911::aid-mds1001>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Barzilai A, Daily D, Zilkha-Falb R, Ziv I, Offen D, Melamed E, Shirvan A. The molecular mechanisms of dopamine toxicity. Adv Neurol. 2003;91:73–82. [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Cappai R, Leck SL, Tew DJ, Williamson NA, Smith DP, Galatis D, Sharples RA, Curtain CC, Ali FE, Cherny RA, Culvenor JG, Bottomley SP, Masters CL, Barnham KJ, Hill AF. Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 2005;19:1377–1379. doi: 10.1096/fj.04-3437fje. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, Colebrooke RE, Di Monte DA, Emson PC, Miller GW. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JW, Wachtel SR, Young D, Kang UJ. Biochemical and anatomical characterization of forepaw adjusting steps in rat models of Parkinson's disease: studies on medial forebrain bundle and striatal lesions. Neuroscience. 1999;88:617–628. doi: 10.1016/s0306-4522(98)00217-6. [DOI] [PubMed] [Google Scholar]
- Charvin D, Vanhoutte P, Pages C, Borrelli E, Caboche J. Unraveling a role for dopamine in Huntington's disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc Natl Acad Sci USA. 2005;102:12218–12223. doi: 10.1073/pnas.0502698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Cagniard B, Mathews T, Jones S, Koh HC, Ding Y, Carvey PM, Ling Z, Kang UJ, Zhuang X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280:21418–21426. doi: 10.1074/jbc.M413955200. [DOI] [PubMed] [Google Scholar]
- Colebrooke RE, Humby T, Lynch PJ, McGowan DP, Xia J, Emson PC. Age-related decline in striatal dopamine content and motor performance occurs in the absence of nigral cell loss in a genetic mouse model of Parkinson's disease. Eur J Neurosci. 2006;24:2622–2630. doi: 10.1111/j.1460-9568.2006.05143.x. [DOI] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr M, Beaulieu JM, Laakso A, Sotnikova TD, Yao WD, Bohn LM, Gainetdinov RR, Caron MG. Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc Natl Acad Sci USA. 2003;100:11035–11040. doi: 10.1073/pnas.1831768100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr M, Sotnikova TD, Gainetdinov RR, Caron MG. Dopamine enhances motor and neuropathological consequences of polyglutamine expanded huntingtin. FASEB J. 2006;20:2541–2543. doi: 10.1096/fj.06-6533fje. [DOI] [PubMed] [Google Scholar]
- Ding Y, Restrepo J, Won L, Hwang DY, Kim KS, Kang UJ. Chronic 3,4-dihydroxyphenylalanine treatment induces dyskinesia in aphakia mice, a novel genetic model of Parkinson's disease. Neurobiol Dis. 2007;27:11–23. doi: 10.1016/j.nbd.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrossy MD, Dunnett SB. Motor training effects on recovery of function after striatal lesions and striatal grafts. Exp Neurol. 2003;184:274–284. doi: 10.1016/s0014-4886(03)00028-1. [DOI] [PubMed] [Google Scholar]
- Donovan DM, Miner LL, Perry MP, Revay RS, Sharpe LG, Przedborski S, Kostic V, Philpot RM, Kirstein CL, Rothman RB, Schindler CW, Uhl GR. Cocaine reward and MPTP toxicity: alteration by regional variant dopamine transporter overexpression. Brain Res Mol Brain Res. 1999;73:37–49. doi: 10.1016/s0169-328x(99)00235-1. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56:331–349. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- el Gemayel G, Trouvin JH, Prioux-Guyonneau M, Jacquot C, Cohen Y. Dopaminergic metabolism in various rat brain areas after L-dopa loading. J Pharm Pharmacol. 1986;38:691–694. doi: 10.1111/j.2042-7158.1986.tb03112.x. [DOI] [PubMed] [Google Scholar]
- Fahn S. Does levodopa slow or hasten the rate of progression of Parkinson's disease? J Neurol. 2005;252(Suppl 4):IV37–IV42. doi: 10.1007/s00415-005-4008-5. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Fumagalli F, Jones SR, Caron MG. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J Neurochem. 1997;69:1322–1325. doi: 10.1046/j.1471-4159.1997.69031322.x. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- Hastings TG, Zigmond MJ. Identification of catechol-protein conjugates in neostriatal slices incubated with [3H]dopamine: impact of ascorbic acid and glutathione. J Neurochem. 1994;63:1126–1132. doi: 10.1046/j.1471-4159.1994.63031126.x. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway RG, Shoulson I, Fahn S, Kieburtz K, Lang A, Marek K, McDermott M, Seibyl J, Weiner W, Musch B, Kamp C, Welsh M, Shinaman A, Pahwa R, Barclay L, Hubble J, LeWitt P, Miyasaki J, Suchowersky O, Stacy M, et al. Pramipexole vs levodopa as initial treatment for Parkinson disease: a 4-year randomized controlled trial. Arch Neurol. 2004;61:1044–1053. doi: 10.1001/archneur.61.7.1044. [DOI] [PubMed] [Google Scholar]
- Jain A, Martensson J, Stole E, Auld PA, Meister A. Glutathione deficiency leads to mitochondrial damage in brain. Proc Natl Acad Sci USA. 1991;88:1913–1917. doi: 10.1073/pnas.88.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakel RJ, Maragos WF. Neuronal cell death in Huntington's disease: a potential role for dopamine. Trends Neurosci. 2000;23:239–245. doi: 10.1016/s0166-2236(00)01568-x. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang UJ, Lee WY, Chang JW. Gene therapy for Parkinson's disease: determining the genes necessary for optimal dopamine replacement in rat models. Hum Cell. 2001;14:39–48. [PubMed] [Google Scholar]
- Koshimura K, Tanaka J, Murakami Y, Kato Y. Effects of dopamine and L-DOPA on survival of PC12 cells. J. Neurosci Res. 2000;62:112–119. doi: 10.1002/1097-4547(20001001)62:1<112::AID-JNR12>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Kumer SC, Vrana KE. Intricate regulation of tyrosine hydroxylase activity and gene expression. J Neurochem. 1996;67:443–462. doi: 10.1046/j.1471-4159.1996.67020443.x. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Peroxynitrite- and nitrite-induced oxidation of dopamine: implications for nitric oxide in dopaminergic cell loss. J Neurochem. 1999;73:2546–2554. doi: 10.1046/j.1471-4159.1999.0732546.x. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Li J, Zhu M, Manning-Bog AB, Di Monte DA, Fink AL. Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson's and Alzheimer's disease. FASEB J. 2004;18:962–964. doi: 10.1096/fj.03-0770fje. [DOI] [PubMed] [Google Scholar]
- Liste I, Navarro B, Johansen J, Bueno C, Villa A, Johansen TE, Martinez-Serrano A. Low-level tyrosine hydroxylase (TH) expression allows for the generation of stable TH+ cell lines of human neural stem cells. Hum Gene Ther. 2004;15:13–20. doi: 10.1089/10430340460732427. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson's disease. Hum Mol Genet. 2002;11:2395–2407. doi: 10.1093/hmg/11.20.2395. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Mishizen AJ, Giasson BI, Lynch DR, Thomas SA, Nakashima A, Nagatsu T, Ota A, Ischiropoulos H. Cytosolic catechols inhibit α-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J Neurosci. 2006;26:10068–10078. doi: 10.1523/JNEUROSCI.0896-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Picklo MJ, Amarnath V, Whetsell WO, Jr, Graham DG. Neurotoxicity of endogenous cysteinylcatechols. Exp Neurol. 1997;148:26–33. doi: 10.1006/exnr.1997.6662. [DOI] [PubMed] [Google Scholar]
- Norris EH, Giasson BI, Hodara R, Xu S, Trojanowski JQ, Ischiropoulos H, Lee VM. Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J Biol Chem. 2005;280:21212–21219. doi: 10.1074/jbc.M412621200. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA. 2002;287:1653–1661. doi: 10.1001/jama.287.13.1653. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Tieu K, Perier C, Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson's disease. J Bioenerg Biomembr. 2004;36:375–379. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med. 2000;342:1484–1491. doi: 10.1056/NEJM200005183422004. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- Simantov R, Blinder E, Ratovitski T, Tauber M, Gabbay M, Porat S. Dopamine-induced apoptosis in human neuronal cells: inhibition by nucleic acids antisense to the dopamine transporter. Neuroscience. 1996;74:39–50. doi: 10.1016/0306-4522(96)00102-9. [DOI] [PubMed] [Google Scholar]
- Stokes AH, Hastings TG, Vrana KE. Cytotoxic and genotoxic potential of dopamine. J Neurosci Res. 1999;55:659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res. 2000;1:181–195. doi: 10.1007/BF03033289. [DOI] [PubMed] [Google Scholar]
- Tang TS, Chen X, Liu J, Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington's disease. J Neurosci. 2007;27:7899–7910. doi: 10.1523/JNEUROSCI.1396-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillerson JL, Cohen AD, Philhower J, Miller GW, Zigmond MJ, Schallert T. Forced limb-use effects on the behavioral and neurochemical effects of 6-hydroxydopamine. J Neurosci. 2001;21:4427–4435. doi: 10.1523/JNEUROSCI.21-12-04427.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkinshaw G, Waters CM. Induction of apoptosis in catecholaminergic PC12 cells by L-DOPA. Implications for the treatment of Parkinson's disease. J Clin Invest. 1995;95:2458–2464. doi: 10.1172/JCI117946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whone AL, Watts RL, Stoessl AJ, Davis M, Reske S, Nahmias C, Lang AE, Rascol O, Ribeiro MJ, Remy P, Poewe WH, Hauser RA, Brooks DJ. Slower progression of Parkinson's disease with ropinirole versus levodopa: the REAL-PET study. Ann Neurol. 2003;54:93–101. doi: 10.1002/ana.10609. [DOI] [PubMed] [Google Scholar]
- Zigmond RE, Schwarzschild MA, Rittenhouse AR. Acute regulation of tyrosine hydroxylase by nerve activity and by neurotransmitters via phosphorylation. Annu Rev Neurosci. 1989;12:415–461. doi: 10.1146/annurev.ne.12.030189.002215. [DOI] [PubMed] [Google Scholar]