

Abstract
Oxidative stress may underlie age-dependent memory loss and cognitive decline. Toxic aldehydes, including 4-hydroxy-2-nonenal (HNE), an end product of lipid peroxides, are known to accumulate in the brain in neurodegenerative disease. We have previously shown that mitochondrial aldehyde dehydrogenase 2 (ALDH2) detoxifies HNE by oxidizing its aldehyde group. To investigate the role of such toxic aldehydes, we produced transgenic mice, which expressed a dominant-negative form of ALDH2 in the brain. The mice had decreased ability to detoxify HNE in their cortical neurons and accelerated accumulation of HNE in the brain. Consequently, their lifespan was shortened and age-dependent neurodegeneration and hyperphosphorylation of tau were observed. Object recognition and Morris water maze tests revealed that the onset of cognitive impairment correlated with the degeneration, which was further accelerated by APOE (apolipoprotein E) knock-out; therefore, the accumulation of toxic aldehydes is by itself critical in the progression of neurodegenerative disease, which could be suppressed by ALDH2.
Keywords: ALDH2, HNE, memory loss, mitochondria, oxidative stress, transgenic mice
Introduction
A decline in cognitive function is associated with aging, mainly as a result of oxidative stress in the brain (Mattson, 2004). Reactive oxygen species (ROS), strong oxidizing molecules including hydroxyl radicals, modify proteins, nucleic acids, and polyunsaturated fatty acids of the lipid membrane, and injure cells. Lipid peroxidation leads to the formation of highly reactive α,β-unsaturated aldehydes, primarily malondialdehyde and 4-hydroxy-2-nonenal (HNE) (Schneider et al., 2001). In particular, HNE is a strong electrophile and has the ability to readily adduct and damage proteins (Uchida, 2003). The accumulation of HNE-adducted proteins in pyramidal neurons has been observed in the brains of patients with Alzheimer's disease (AD) and Parkinson's disease (Yoritaka et al., 1996; Lovell et al., 1997; Montine et al., 1997; Sayre et al., 1997). HNE application in vitro mimics the pathological changes noted in AD, including modification of tau (Takeda et al., 2000; Liu et al., 2005) and β-amyloid (Murray et al., 2007), inhibition of the glucose transporter (Reagan et al., 2000), and microtubular disruption (Gadoni et al., 1993), and eventually leads to cell death (Mark et al., 1997).
HNE is adducted with glutathione via glutathione S-transferase (Xie et al., 1998), oxidized by aldehyde dehydrogenase (ALDH) to form 4-hydroxy-2-noneoate (Tjalkens et al., 1999), or reduced by aldo-keto reductase, alcohol dehydrogenase (ADH) or amyloid β-peptide-binding ADH (ABAD) to form 1,4-dihydroxynonene (Srivastava et al., 1998; Murakami et al., 2008). These multiple pathways are used to detoxify HNE in cells (Siems and Grune, 2003). We found previously that mitochondrial ALDH2-deficient neuronal cells exhibited increased vulnerability to HNE (Ohsawa et al., 2003a). The cells accumulated HNE and showed decreased resistance to oxidative insult, suggesting that ALDH2 functions as a protector against oxidative stress by oxidizing HNE. However, ALDH2 is well known to oxidize acetaldehyde produced from ethanol into acetate (Higuchi et al., 2004). A mutant allele, ALDH2*2, has a single point mutation (G→A) in exon 12 of the active ALDH2*1 gene and is confined to Asians. This mutation results in a substitution of glutamate 487 to lysine (E487K), acting in a dominant-negative manner (Bosron and Li, 1986). Individuals with the ALDH2*2 allele exhibit the alcohol-flushing syndrome attributable to an elevated blood acetaldehyde level.
We reported previously that a molecular epidemiological analysis revealed a higher concentration of lipid peroxides (LPOs) in the sera of ALDH2-deficient females than in those carrying an active ALDH2 (Ohsawa et al., 2003b), and that ALDH2 deficiency is a risk factor for late-onset AD, synergistically acting with the ε4 allele of the apolipoprotein E gene (APOE-ε4) (Kamino et al., 2000). This finding was recently confirmed by studies in China and Korea (Jo et al., 2007; Wang et al., 2008). To verify the role of ALDH2-deficiency in age-associated neurodegenerative diseases, we examined physiological changes, and learning and memory impairments by using transgenic (Tg) mice expressing ALDH2*2 in the brain. Here, we show that ALDH2-deficient mice present with age-dependent signs of neurodegeneration such as loss of pyramidal cells, activation of glial cells, and prominent deficits in cognition.
Materials and Methods
Transgenic mice.
To generate Tg mice that express ALDH2*2, a transgene containing a mouse version of ALDH2*2 under the control of the human elongation factor-1α (EF1α) promoter was constructed. A DNA fragment containing ALDH2*2 in plasmid pBluescript SK (Ohsawa et al., 2003a) was digested with XbaI and inserted into the XbaI site of plasmid pEF1-BOS (Mizushima and Nagata, 1990). The 4.2 kb ApaLI–PvuI fragment of EF1α promoter-ALDH2*2 derived from the resultant plasmid was purified and dissolved in TE buffer at a concentration of 3 μg/ml. Tg mice were produced in accordance with a standard procedure. In brief, the DNA fragment was microinjected into 800 fertilized eggs taken from superovulated C57BL/6 females. The injected eggs were surgically transferred to the oviducts of C57BL/6 pseudopregnant females to produce male chimeric mice. Sixteen mice carrying the transgene were selected by PCR of tail DNA with primers 5′-NNTCTAGAGCCACCATGGCTCAGGGCGCCCATA and 5′-GAAGGGTTGACGGTGGGAAATGTT. F1 heterozygous animals were backcrossed to C57BL/6 mice for more than five generations, and homozygous mice (ALDH2*2+/+) were finally obtained. Next, three independent lines (DAL101, DAL102, and DAL103) were selected according to the expression of ALDH2*2 in the brain determined by the following method. Poly(A)+ RNA was extracted from the brain and mismatch reverse transcription (RT)-PCR was performed with ALDH2-specific primers 5′-AGATGTCAGGGAGTGGCAGG (see Fig. 1, primer A) and 5′-GTGGCACTTTGACAGTAACCGTCTTAACGT (see Fig. 1, primer B). The amplified cDNA fragment was incubated with AclI and subjected to PAGE. Two digested fragments (54 and 30 bp) were derived from exogenous ALDH2*2, whereas the fragment not digested with AclI (84 bp) was from endogenous ALDH2. To quantify the expression of ALDH2 and ALDH2*2 transcripts in each region of the brain, their cDNA was qualified by ABI7700 with real-time RT-PCR primers and probe 5′-ACCTGTCCCAAGCTCTGCAG, 5′-GGAGACTGGGCCCCAAAC, and 5′-CTGGCACTGTGTGGATCAACTGCTACG for endogenous ALDH2, and 5′-AAAACCCAACAAGATATACTGAGAAAAAC, 5′-GACCTGATACTCTCAAGGGTCCC, and 5′-CACTGTCTAGAGTGAGGGTCCCCACCTG for exogenous ALDH2*2. DAL101 was further mated with ApoE−/− mice (purchased from Taconic Farms) and offspring carrying the genotype ALDH2*2+/+/ApoE−/− were obtained.
Figure 1.
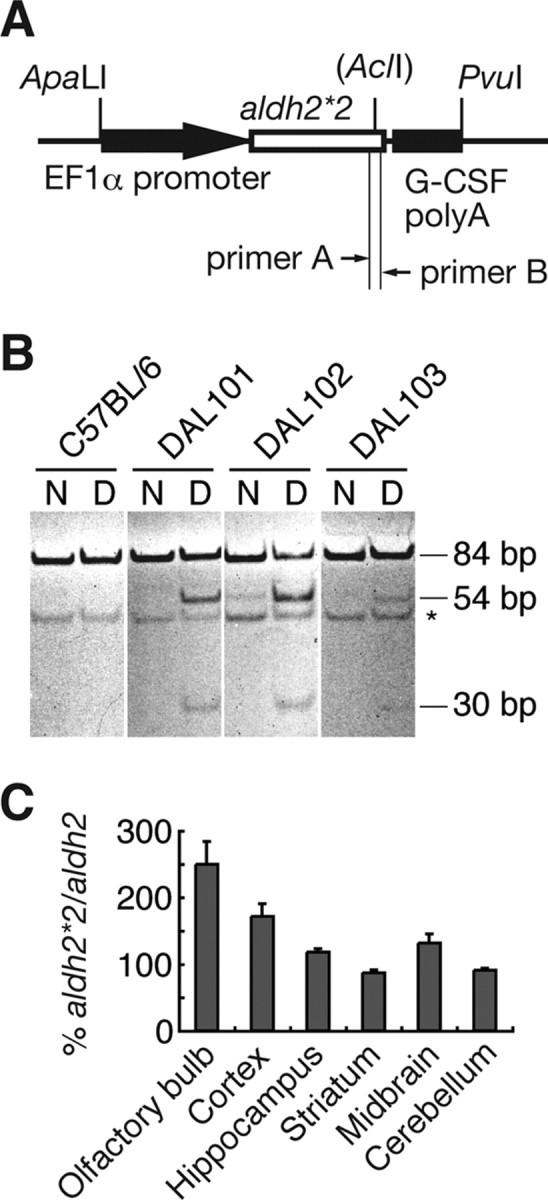
Generation of DAL mice. A, Schematic drawing of the transfected DNA fragment. DAL mice were generated by the injection of 4.2 kb ApaLI–PvuI fragment into fertilized eggs. B, RT-PCR analysis revealed that three transgenic founders expressed exogenous ALDH2*2 in the brain. For PCR, primers shown in A were used (see Materials and Methods). The obtained PCR fragments (N) were digested with AclI (D). Two short fragments (54 and 30 bp) were derived from exogenous ALDH2*2, whereas the nondigested fragment was from endogenous ALDH2. *Nonspecific bands. C, In the brain of DAL101, real-time RT-PCR analysis revealed more exogenous ALDH2*2 transcripts relative to endogenous ALDH2 transcripts in the olfactory bulb, cortex, hippocampus, and midbrain. Data are mean ± SEM (n = 4).
All mice were kept on a 12 h light/dark cycle with ad libitum access to food and water. The studies were approved by the Animal Care and Use Committee of Nippon Medical School. All experiments were performed by examiners blinded to the genotypes or treatments of the mice.
Primary culture of neurons.
Primary cultures of cortical neurons were prepared from 17 d mouse embryos by the method described previously (Ohsawa et al., 2007). In brief, neocortical tissue was cleaned of meninges, minced, and treated with trypsin. After mechanical dissociation by pipetting, cells were resuspended in DMEM (Invitrogen) supplemented with insulin (5 μg/ml), transferrin (5 μg/ml), putrescine (100 μm), selenium (5 ng/ml), and penicillin–streptomycin, and then plated onto poly-l-lysine-coated plates at a density of 5 × 104 cells/cm2. After 3 d of the culture, we used neurons for analysis of cell viability. For preparing hippocampal cells, hippocampus of 17 d mouse embryo was cleaned of meninges, minced, and treated with protease mixture (SUMILON). After mechanical dissociation by pipetting, we resuspended cells in nerve cell culture medium (SUMILON), and then plated onto poly-l-lysine-coated plates at a density of 5 × 104 cells/cm2, changed one-half of the medium once every 2 d, and then used neurons at day 1 and day 9 for analysis of neurite outgrowth and cell viability, respectively. Neuronal identity was confirmed by immunostaining with the neuron marker anti-TUJ-1 (Covance) and/or anti-MAP2 (Sigma-Aldrich) and astrocyte marker anti-glial fibrillary acidic protein (GFAP) (ThermoImmunon) antibodies. After incubation with BODIPY FL goat anti-mouse IgG (Invitrogen), cells were imaged by confocal scanning microscopy (Olympus) using excitation and emission filters of 488 and 510 nm, respectively. Preparations containing >90% neurons were used for experiments. Apoptosis was detected by DNA strand breaks using terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end-labeling (TUNEL) according to the procedure of the manufacturers (MBL).
Preliminary observation and neurological reflexes.
Mice were first observed in their home cages for general activity and levels of aggression. Each mouse was then observed in a bare cage for general health, presence of whiskers, and appearance of fur. Mice were challenged to evaluate reflex responses including eye blink, ear twitch, whisker touch, and righting reflexes (Miyakawa et al., 2001).
Open-field test.
Locomotor activity was measured using an open-field apparatus (45 × 45 × 65 cm) (Kim et al., 2006). Animals were placed in the center of the apparatus in standard indoor-lighting conditions and total distance traveled for 10 min was recorded. Between sessions, the apparatus was wiped clean with 70% ethanol.
Wire hanging test.
The neuromuscular strength was tested by the wire hanging test (Hamann et al., 2003). In brief, mice were placed on a wire netting, which was lightly shaken, causing the mouse to grip the wire. After a 5 s cutoff time, the wire netting was turned upside down (180°) and the latency to fall was recorded for the maximum time.
Immunohistochemistry.
The brain was removed and left one was kept at −80°C for biochemical assay. The right brain was further fixed for 4 h at room temperature by using 4% paraformaldehyde in PBS and subsequently cryoprotected with 30% sucrose in PBS. Frozen brain was sectioned at 8 μm thickness using a cryostat (Leica), mounted on slides, and air-dried. To stain sections with antibodies against NeuN (clone A60; Millipore), GFAP, and phospho-Tau (clone AT8; Pierce), the M.O.M. kit (Vector Laboratories) was used by the method according to the manufacturer. For double staining with anti-NeuN, antimonoclonal antibody and rabbit anti-GFAP polyclonal antibody (Abcam), we used Texas Red goat anti-mouse IgG (Invitrogen) and BODIPY FL goat anti-rabbit IgG (Invitrogen) as secondary antibodies and visualized signals with a laser-scanning confocal microscope.
Immunoblot analysis.
The left hippocampus was dissected and Dounce-homogenized in cold lysate buffer [10 mm Tris, pH 7.4, 320 mm sucrose, 1% Triton X-100, 1% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), 0.025% NaN3, 1 mm EDTA, 1 mm EGTA, 50 mm sodium fluoride, 2 mm sodium orthovanadate, and protease inhibitor Complete tablets EDTA-free (Roche)] and stored at −80°C. Protein concentration was determined with the BCA protein assay kit (Pierce). Equal amounts of protein (100 μg) were separated on 10–20% polyacrylamide gels (Daiichi), transferred onto PolyScreen transfer membranes (PerkinElmer), and incubated in blocking buffer consisting of TBS (10 mm Tris, pH 7.6, and 150 mm NaCl) with 5% bovine serum albumin. For immunodetection of specific bands on the blots, Can Get Signal immunoreaction enhancer solution (Toyobo) was used by the method according to the manufacturer. Blots were incubated with primary antibodies to tau phospho-Ser-202/205 (AT-8; 1:500; Pierce), pan-tau (TAU-5; 1:1000; Millipore), p35 and p25 C terminus (C-19; 1:200; Santa Cruz), and cyclin-dependent kinase 5 (Cdk5) (C-8; 1:100; Santa Cruz) and further incubated with alkaline phosphatase-conjugated secondary antibodies (1:10,000; Cappel). Signals were visualized with AttoPhos substrate (Roche). Blots were stripped with stripping buffer (62.5 mm Tris, pH 6.8, 100 mm 2-mercaptoethanol, 2% SDS) and reprobed with antibody to β-actin (1:10,000; Sigma-Aldrich) to normalizing the amounts of loaded proteins.
HNE concentration.
Concentration of HNE in the brain was determined using a colorimetric assay kit (HAE-586; OXIS International) according to the manufacturer's instructions. The protein concentration of each sample was measured using a BCA protein assay reagent (Pierce).
Visual object recognition test.
The visual object recognition test (vORT) was used to test recognition memory (Wang et al., 2004). Mice were first habituated in a 25 × 40 cm cage for 6 h before training. During 10 min of training, two plastic blocks with different shapes and colors were presented. Object recognition was scored by the number of approaches to and/or sniffs of the object and was reported as percentage preference (recognition index). One day after training, one of the conditioned blocks was replaced with a novel object to test for memory retention, and recognition index was scored during 5 min of testing.
Water maze test.
The apparatus used for water maze tasks was a circular pool (1.5 m diameter) painted white and filled with water maintained at 25°C, and rendered opaque by the addition of nontoxic white paint. The maze is located in a room containing several simple visual, extramaze cues. Mice were trained in the morning to swim to a 10-cm-diameter circular clear acryl resin platform submerged 1 cm beneath the surface of the water and invisible to the mice while swimming. Mice were monitored by a camera mounted above the pool, and all trials were stored on PC for subsequent analysis. In each trial, the mouse was placed and released into the pool at one of four designated start points in a pseudorandom order and allowed to find and escape to the submerged platform and remain there for 20 s. If a mouse failed to find the platform within 60 s, it was guided carefully to the platform and allowed to remain there for 20 s. After this, each mouse was dried gently with a towel and placed into a holding cage for 30 min until the start of next trial. Mice were given four trials a day for 5 d. Retention of the spatial training was assessed 1 h after the last training trial. A single probe trial consisted of a 60 s free swim in the pool without the platform. Mice were placed and released at the location opposite the site where the platform had been located and the time spent in each quadrant was recorded. Finally, after the probe trial, a visible platform was placed in the training quadrant 1 cm above the surface of the water. As in the place-learning task, the escape latencies were measured during at least four trials per session except that the test was conducted in a single day.
Statistical analysis.
We performed statistical analyses using StatView software (SAS Institute) by applying an unpaired two-tailed Student's t test and ANOVA followed by Fisher's exact test for single and multiple comparisons, respectively. To calculate survival curves, the Kaplan–Meier method was used and the survival periods were compared with the log rank test (Mantel–Cox) for univariate analysis. We performed experiments for quantification in a blinded manner.
Results
Expression of ALDH2*2 in the brain and vulnerability of ALDH2*2-expressing neurons to HNE
The mouse version of the dominant-negative form of ALDH2 (ALDH2*2) was introduced under an EF1α promoter (Fig. 1A), which constitutively expresses genes in most tissues (Wakabayashi-Ito and Nagata, 1994). Because the expression extent depends on the location inserted, we paid attention to the expression of ALDH2*2 in the brain and selected three independent transgenic founders (DAL101, DAL102, and DAL103) and confirmed expression of ALDH2*2 by RT-PCR in the brain (Fig. 1B). One founder (DAL101), which expressed the highest amount of ALDH2*2, was maintained as homozygotes and mainly used for additional experiments. The amount of exogenous ALDH2*2 transcripts relative to endogenous ALDH2 transcripts was higher in the olfactory bulb, cortex, hippocampus, and midbrain in DAL101 (Fig. 1C).
To evaluate the effect of ALDH2*2 in neurons, we examined the ability of HNE detoxification and found that exposure to HNE resulted in a more rapid decrease of viable cells in the ALDH2*2-expressing neurons. At first, hippocampal neurons were prepared from embryonic 17 d DAL101. Moderate neuritogenesis was seen after 1 d in culture. Then, cells were treated with HNE and incubated for an additional 1 d. Immunostaining with anti-MAP2 antibody showed that neurite length of neurons prepared from DAL101 was shorter than that from non-Tg control (CTL) mice (C57BL/6) (Fig. 2A,B). After 9 d in culture, when a mature synaptic network was established, the hippocampal neurons were treated with HNE and further incubated for 1 d. Immunostaining with anti-MAP2 antibody revealed that the MAP2-positive neurites derived from DAL101 mice were decreased by the treatment with 3 μm HNE (Fig. 2C,D). Additionally, apoptosis was more induced by the treatment with HNE in neurons derived from DAL101 mice (Fig. 2E,F). Furthermore, cortical neurons were prepared from embryonic 17 d DAL101 and cultured for 3 d. Twenty-four hours after treatment with 1 μm HNE, 77.1% of ALDH2*2-expressing neurons were injured and detached from the dish, whereas control neurons prepared from CTL mice were attached and showed normal morphology with neuron-specific TUJ-1-positive neurites (Fig. 2G). The sensitivity of ALDH2*2-expressing neurons to HNE was dose-dependent (Fig. 2H). At that time, a higher amount of HNE was accumulated in ALDH2*2-expressing neurons (Fig. 2I), indicating that the metabolism ability of exogenous HNE decreased. These findings clearly show that the decline of ALDH2 activity makes neurons less resistant to HNE.
Figure 2.

Neuronal vulnerability of DAL mice against HNE and its accumulation in the brain. Primary-cultured cells isolated from the brain of DAL101 (red bar) and C57BL/6 (blue bar) were treated with HNE and further incubated for 1 d. A, B, After 1 d in culture, hippocampal cells were treated with 1 μm (A) or the indicated amount (B) of HNE and immunostained with the dendrite-specific antibody to MAP2. Scale bar, 100 μm. Neurite length of each cell was measured under a microscope in at least 10 cells per well (mean ± SD; n = 4). *p < 0.05. C, D, Nine days after the culture, hippocampal cells were treated with the indicated amount of HNE and immunostained with anti-MAP2 antibody. Scale bar, 100 μm. Dead cells were washed out in the staining procedure and living cells were enumerated under a fluorescent microscope in four fields of view (FOVs) per well (mean ± SD; n = 4). *p < 0.05. E, F, Nine days after the culture, hippocampal cells were treated with 1 μm (E) or the indicated amount (F) of HNE and stained with TUNEL (green) and propidium iodide (PI) (red). Scale bar, 100 μm. Quantification of TUNEL-positive cells was performed under a fluorescent microscope in four FOVs per well and compared with the total number of cells (analyzed by PI staining) (mean ± SD; n = 4). *p < 0.05. G, H, Three days after the culture, cortical cells were treated with the indicated amount of HNE and surviving neurons were fixed and immunostained with the neuron-specific antibody to TUJ-1. Scale bar, 200 μm. Dead cells were washed out in the staining procedure, and living cells were enumerated under a fluorescent microscope in four FOVs per well (mean ± SD; n = 4). *p < 0.05; ***p < 0.001. I, HNE–protein conjugates were calculated from pixel intensity in cells stained with anti-HNE antibody. Intensity was measured with NIH Image (mean ± SD; n = 4). *p < 0.05; ***p < 0.001.
Accumulation of HNE in the brain is accelerated and lifespan is shortened
To clarify the relationship of ALDH2*2-expressing neurons with HNE, we measured HNE in the brain and found that a higher amount of HNE was accumulated in the brain of old DAL mice (Fig. 3A). These results indicated that ALDH2*2 inhibited the degradation of HNE and enhanced oxidative stress in the brain.
Figure 3.

Characterization of DAL mice. A, A higher amount of HNE was accumulated in the brain of DAL101 in an age-dependent manner. Filled bars, DAL101; open bars, C57BL/6. HNE levels were normalized against protein contents (mean ± SD; n = 6). *p < 0.05. B, No significant difference in body weight was found between 1-year-old DAL101 (filled bars) and C57BL/6 (open bars) (mean ± SD; n = 4). C, No significant difference in walking distance in the open-field test was found between 1-year-old DAL101 (filled bars) and C57BL/6 (open bars) (mean ± SD; n = 4). D, Time to exhaustion and fall in hang test. Significant muscle weakness between 1-year-old DAL101 (filled bars) and C57BL/6 (open bars) was found in male (mean ± SD; n = 4). **p < 0.01. E, F, Hair decolorization in 1-year-old male DAL101. Scale bar, 50 μm. G, Kaplan–Meier curve representing the survival of DAL101 and C57BL/6. Significant shortening of lifespan was found in DAL101 mice (p < 0.0001). Bold lines, Total male and female mice; thin line, male mice; thin dotted line, female mice.
One year after birth, female DAL101 showed no differences in weight (Fig. 3B), general health, sensory responses, or locomotor activity (Fig. 3C); however, male DAL mice showed muscle weakness (Fig. 3D) and some hair decolorization (n = 8 of 8) (Fig. 3E,F). It is noteworthy that the median lifespan of DAL101 was significantly shorter than that of CTL mice (96 vs 126 weeks; p < 0.0001) (Fig. 3G). The maximal lifespan of DAL101 was 129 weeks compared with 153 weeks for the CTL mice. The causes of death were not determined in many cases. In all the cases, no inflammation, other indicators of infectious disease, or tumors above levels seen in DAL mice were found.
ALDH2 deficiency by itself induces age-dependent brain degeneration
To examine whether ALDH2 deficiency induces neurodegeneration, we looked for abnormal neuronal loss in aged DAL mice by hematoxylin and eosin (H&E) staining and found the degeneration (Fig. 4A–D) and decrease in pyramidal neurons in the CA1 region of the hippocampus (Table 1). At 1 year of age, 20% of DAL mice showed neurodegeneration, whereas CTL mice did not (1 of 5 DAL vs 0 of 5 CTL; p = 0.2918). At 1.5 year of age, 77.8% of DAL mice showed neurodegeneration, whereas CTL mice did not (7 of 9 DAL vs 0 of 8 CTL; p = 0.0053). The obvious neurodegeneration in a 1 year DAL mouse was similar to that in 1.5 year DAL mice. In the hilus of the dentate gyrus of aged DAL mice, the neurons were also decreased (Table 1). Neuronal loss in the CA1 area of DAL mice was confirmed by immunostaining with anti-NeuN antibody (Tang et al., 2005) (Fig. 4E–H). Furthermore, marked increase in immunostaining with anti-GFAP antibody, a marker of activation of astrocytes, was found in the hippocampus of old DAL mice. At 1 year of age, 60% of DAL mice showed activation of astrocytes, whereas CTL mice did not (3 of 5 DAL vs 0 of 5 CTL; p = 0.1667). At 1.5 year of age, 55.6% of DAL mice showed activation of astrocytes, whereas CTL mice did not (5 of 9 DAL vs 0 of 8 CTL; p = 0.0447) (Fig. 4I,J). Because the increase of reactive astrocytes is a sensitive marker of brain injury (Isacson et al., 1987), these results indicate obvious impairment in neuronal function in these mice. As expected, we found neither the neuronal degeneration nor increase in reactive astrocytes in the brain of DAL mice at 6 months of age (Fig. 5, Table 1).
Figure 4.
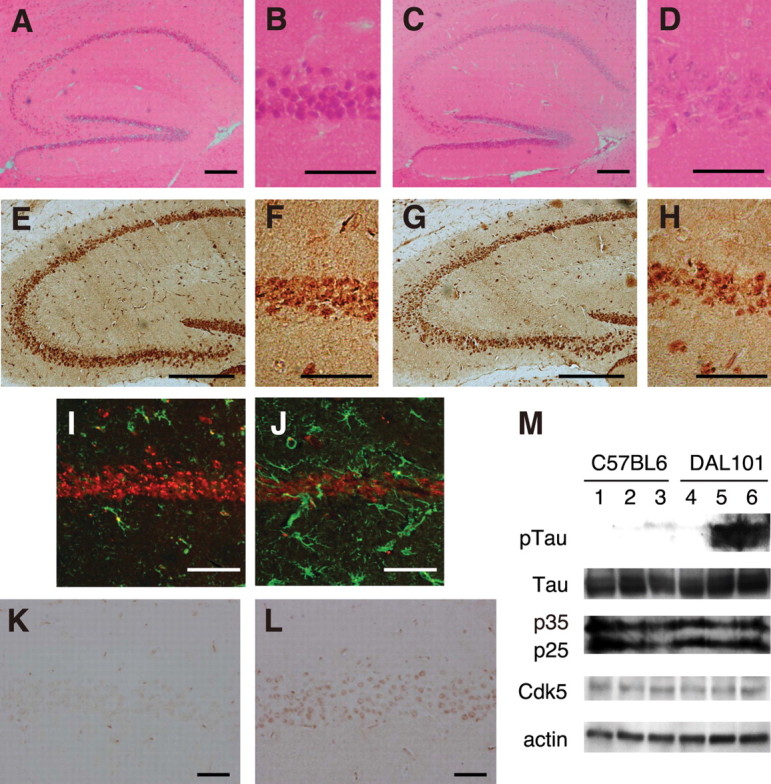
Hippocampal degeneration in DAL mice. A, B, K, Twelve-month-old C57BL/6. E, F, I, Eighteen-month-old C57BL/6. C, D, L, Twelve-month-old DAL101. G, H, J, Eighteen-month-old DAL101. A, C, Hippocampus was stained with H&E. Scale bar, 200 μm. B, D, The CA1 region stained with H&E was expanded. Scale bar, 50 μm. E, G, Hippocampus was stained with the antibody to NeuN. Scale bar, 250 μm. F, H, The CA1 region stained with the antibody to NeuN was expanded. Scale bar, 50 μm. I, J, The CA1 region was stained with antibodies to GFAP (green) and NeuN (red). Scale bar, 50 μm. K, L, The CA1 region was stained with anti-phosphorylated tau antibody (AT8). Scale bar, 50 μm. M, Representative immunoblots of hippocampal lysates from 18-month-old C57BL/6 (lanes 1–3) and DAL101 (lanes 4–6) mice probed with antibodies to phosphorylated tau (pTau), pan-tau (Tau), Cdk5 activator proteins (p25 and p35), Cdk5, and actin.
Table 1.
Number of neurons in the hippocampus of C57BL/6 and DAL101 mice
| C57BL/6 | DAL101 | p | |
|---|---|---|---|
| Old | n = 6 | n = 8 | |
| CA1 (no./mm2) | 2172.01 ± 259.76 | 1593.00 ± 356.38 | 0.0057 |
| Hilus (no./mm2) | 591.67 ± 88.86 | 462.50 ± 84.98 | 0.0172 |
| Young | n = 5 | n = 4 | |
| CA1 (no./mm2) | 2023.20 ± 218.47 | 2083.50 ± 197.39 | 0.6810 |
| Hilus (no./mm2) | 434.00 ± 44.50 | 427.50 ± 67.02 | 0.8658 |
The brain sections of old (1–1.5 years of age) and young (0.5 year of age) mice were stained with H&E, and the total numbers of neurons in area CA1 and hilus of the dentate gyrus were counted. Data are presented as mean ± SD.
Figure 5.
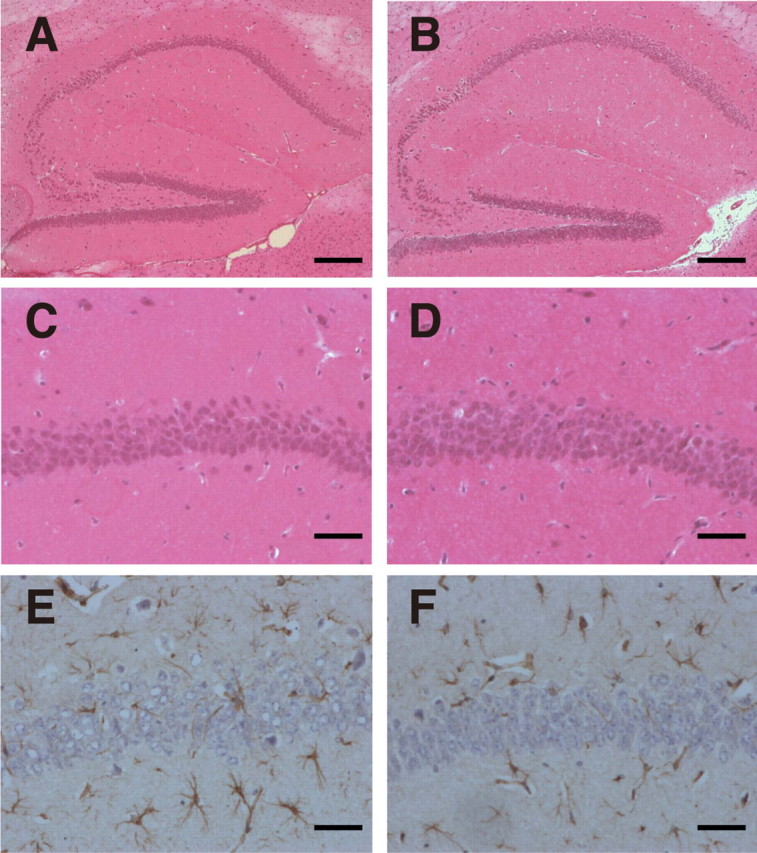
No obvious hippocampal degeneration was observed in the hippocampus in young DAL mice. A, B, Hippocampus was stained with H&E. Scale bar, 200 μm. C, D, The CA1 region stained with H&E was expanded. Scale bar, 50 μm. E, F, The CA1 region was stained with anti-GFAP antibody. Scale bar, 50 μm. A, C, E, Six-month-old C57BL/6. B, D, F, Six-month-old DAL101.
Next, we focused on the hyperphosphorylation of the tau protein. Tau-related pathologies are hallmarks of AD and other tauopathies (Takashima, 2006), and have been characterized in mice (Liou et al., 2003). The antibody, AT8, requires serine 202 to be phosphorylated to generate its epitope and is used frequently to assess the hyperphosphorylation of tau (Biernat et al., 1992). Staining brain sections of DAL mice at 1 and 1.5 year of age with AT8 yielded labeling of pyramidal neurons in the hippocampus of 44.4% of DAL mice, whereas those of wild-type littermates did not (4 of 9 DAL vs 0 of 13 CTL; p = 0.0361) (Fig. 4K,L). No obvious alternation was observed in the other brain regions. Immunoblot analysis also confirmed an increase in hyperphosphorylation of tau at the AT-8 site in the hippocampus of DAL mice (Fig. 4M). We examined the relationship between the DAL mice-specific increase in hyperphosphorylation of tau and expression levels of Cdk5 activator p25, p35, and Cdk5 (Cruz and Tsai, 2004); however, no obvious change in p25, p35, and Cdk5 levels was observed (Fig. 4M), suggesting that the other putative factors contribute to the abnormal hyperphosphorylation of tau in DAL mice.
Onset of cognitive impairment correlates with brain degeneration
To examine whether ALDH2 deficiency affects cognitive impairments, three independent genetic lines (DAL101, DAL102, and DAL103) were applied to the simple vORT. Because of relatively low physical demands in the test, male mice were used (Fig. 3D). Memory formation depends on the hippocampus (Miyamoto et al., 2005). During 10 min of training, two objects (global, a; triangle, b) were presented to a mouse. C57BL/6 mice, which were used as a control, and all DAL mice approached or sniffed each object approximately an equal number of times during the training period (Fig. 6A), indicating comparable attention, motivation, and visual perception. One day after training, one of the conditioned objects was replaced with a novel object (cubic, c). During 5 min of testing, young C57BL/6 (6 months of age) and DAL101 (6–8 months of age) mice and aged C57BL/6 (12 months of age) mice, but not aged DAL101 (11–14 months of age), DAL102 (12 months of age), and DAL103 (12 months of age) mice, approached or sniffed the novel object more times than the original object (Fig. 6B). These results indicated that aged DAL101 showed a deficit in visual recognition memory, whereas young DAL101 and aged CTL mice showed intact ability to detect a novel object. This age-dependent deficit in recognition memory agrees with the histological neuronal degeneration described above. Similar disability was observed in other lines of aged DAL mice (DAL102 and DAL103), indicating that the memory deficit was not specific to aged DAL101.
Figure 6.

Decreased object recognition memory in aged DAL mice. ALDH2 deficiency impaired memory for object recognition. A, Training for visible object recognition. During 10 min of training, two objects (a and b) were presented to a mouse. C57BL/6 mice, which were used as a control (CTL), and all DAL mice had equal preferences for two objects, a and b. Filled bars, Aged mice; open bars, young mice (see Materials and Methods). The numbers on the bars indicate the number of animals examined. B, Testing for novel object recognition. During 5 min of testing, one of the conditioned objects was replaced by a novel object (c). Young mice and aged CTL mice, but not aged DAL mice, showed recognition memory 1 d after a training session. #p < 0.05, ##p < 0.01 versus index in a training session; *p < 0.05, **p < 0.01.
To examine more carefully whether ALDH2 deficiency affects cognitive impairment, DAL101 was tested in the Morris water maze (MWM), a hippocampus-dependent paradigm used to assess the ability to acquire, process, and recall spatial information by using escape latency as an indicator of learning (D'Hooge and De Deyn, 2001). Because of the physical demands of this study, only female mice were used. At 6 months of age, no obvious pathology was apparent in any brain region of DAL mice (Fig. 5, Table 1). Nevertheless, analysis of cognitive function at this age is of great importance to evaluate whether mice are born with learning and memory impairment or whether they develop cognitive deficit in an age-dependent manner. We found that, at 6 months of age, DAL mice had intact spatial learning and memory, and no deficits in probe trials (Fig. 7A,B). They required 3 d of training to reach criterion (escape latency of <20 s) and spent significantly more time over the trained quadrant compared with other quadrants during probe trials. No obvious difference between DAL and CTL mice was observed. These results indicated that DAL mice were born normally and did not develop cognitive deficit during young adulthood.
Figure 7.

Age-dependent onset of cognitive impairment in DAL mice. Performance on the MWM test. A, At the age of 6 months, time to reach a hidden platform (latency) was similar in the DAL101 and C57BL/6 mice. However, the 12- and 18-month-old DAL101 mice showed profound impairment compared with C57BL/6 mice. B, Probe test performed 1 h after the hidden platform acquisition period. At the age of 6 months, time spent in the target quadrant was similar in the DAL101 (filled bars) and C57BL/6 (open bars) mice. However, the 12- and 18-month-old DAL101 mice spent a shorter time searching in the target quadrant than C57BL/6 mice (mean ± SD). *p < 0.05. C, Time to reach visible platform locations. Filled bars, DAL101; open bars, C57BL/6. D, Swimming speed during probe trial.
At 1 and 1.5 year of age, DAL mice were still able to learn the MWM task; however, they required 5 d of training to reach criterion (escape latency of <20 s) compared with the 3 d of age-matched CTL mice (Fig. 7A). Furthermore, time spent in the target quadrant of DAL mice during probe trials was significantly shorter than that of CTL mice and not significantly more compared with other nontarget quadrants (Fig. 7B). To assess whether the spatial learning and memory deficits observed in DAL mice were attributable to deficient escape motivation or impairment of visual and/or motor performance, the visible platform was used. Escape latencies after the training in this experiment were not different between DAL and CTL mice (Fig. 7C). Swimming speeds were also determined to exclude the possibility of different swimming abilities. No significant difference in speeds between DAL and CTL mice was observed, whereas speeds decreased age-dependently (Fig. 7D), suggesting that sensorimotor disturbances were not differentially affecting their performance.
Cognitive impairments in DAL mice are accelerated by APOE-knock-out
We reported previously that the ALDH2*2 allele is a synergistic risk for AD with ε4 allele of the apolipoprotein E gene (Kamino et al., 2000). We then sought to examine whether apoE affect phenotypes of DAL mice and examined DAL mice without apoE (DAL/ApoE−/−) at 6 months of age in the MWM. They were not able to learn the MWM task, whereas both DAL and ApoE−/− mice required only 3 d of training to reach criterion (escape latency of <20 s) (Fig. 8A). Furthermore, time spent in the target quadrant of DAL/ApoE−/− mice during probe trials was significantly shorter than that of both DAL and ApoE−/− mice (Fig. 8B). Escape latencies after training using the visible platform were not different among DAL/ApoE−/−, DAL, and ApoE−/− mice (Fig. 8C), suggesting that sensorimotor disturbances were not differentially affecting their performance. Thus, these results indicate that the ApoE depletion enhances the learning and memory deficits of DAL mice.
Figure 8.

Onset of cognitive impairment in DAL mice was enhanced by lacking in ApoE. Performance on the MWM test. A, At the age of 6 months, DAL101 mice in the absence of ApoE (DAL101/ApoE−/−) showed profound impairment compared with DAL101 and ApoE−/− mice. p < 0.05 against DAL101 and ApoE−/− mice, respectively. B, Probe test performed 1 h after the hidden platform acquisition period. DAL101/ApoE−/− spent a shorter time searching in the target quadrant than DAL101 and ApoE−/− mice (mean ± SD). *p < 0.05. C, Time to reach visible platform locations. D, Swimming speed during probe trial.
Discussion
We reported here that transgenic mice expressing a mouse version of dominant-negative ALDH2 showed enhanced accumulation of ROS end products and age-dependent learning and memory deficits. ALDH enzymes efficiently oxidize HNE, a highly toxic product of lipid peroxidation (Ohta et al., 2004; Vasiliou et al., 2004). Interestingly, mitochondria ALDH2 metabolize nitroglycerin, which has been used for over a century in the therapy of cardiovascular disease (Chen et al., 2002). Nitroglycerin tolerance developed by chronic therapy is associated with a decrease in ALDH2 activity and an increase of ROS (Chen and Stamler, 2006), and, at that time, ALDH2 itself is inactivated by various oxidants (Wenzel et al., 2007). However, ALDH2 activity was found to increase in liver with age and in the brain of AD patients (Picklo et al., 2001; Yoon et al., 2006), suggesting that sufficient amount of active ADLH2 is needed to detoxify the increased HNE that occurs in aging and neurodegenerative diseases.
The construction of model animals by genetic manipulation is one of the best methods to analyze the involvement of particular genes in the defense against oxidative stress at the animal level. In mice defective in Mn-superoxide dismutase (SOD), oxidative stress accumulated, and mitochondrial dysfunction and subsequent cell death were observed (Melov et al., 1998). Because the model mice died during the first few postnatal weeks, their pathology was too severe to investigate age-dependent degeneration. Furthermore, mice defective in phospholipid hydroperoxide glutathione peroxidase (PHGPx) were embryonic lethal (Imai et al., 2003). PHGPx is the only known enzyme that directly reduces lipid hydroperoxide; however, knock-out mice deficient in Cu, Zn-SOD, cellular glutathione peroxidase, and catalase developed normally and showed no marked pathological changes under normal physiological conditions (Reaume et al., 1996; Esposito et al., 2000; Ho et al., 2004), indicating that their defects in major antioxidant defense mechanisms are strongly compensated by each other. In the case of DAL mice, no obvious pathological features, including cognitive function, were observed in young mice (6 months of age), indicating that no physiological and neurological changes would occur during their growth and development. After maturation, oxidative stress accumulated in the brain and cognitive deficits developed in aged DAL mice. The importance of this model mouse is the age-dependent onset of neurodegenerative disorders, which will be helpful for understanding human disease.
ALDH belongs to a large family consisting of at least 16 different genes in humans, and are involved in the metabolic systems of various alcohols and aldehydes according to their distribution and substrate specificity (Vasiliou and Nebert, 2005). It is likely that knocking out one such gene causes moderate change in aldehyde metabolism, because the knocked-out enzyme would be complemented with other members of the gene family. In fact, in ALDH2 knock-out mice, methoxyacetaldehyde metabolism is markedly reduced in liver, but no apparent abnormality was observed (Kitagawa et al., 2000). This discrepancy suggests that the phenotype observed in DAL mice is not simply attributable to the decrease in the ALDH2 activity. ALDH2*2 may inactivate not only ALDH2 but also the other member, such as ALDH5A, of the ALDH family presumably by forming heterotetramers. In humans, however, the suppression of ALDH2 activity by ALDH2*2 causes various disorders, presumably because of increased oxidative stress. For instance, ALDH2 deficiency was found to contribute to the risk of complication with diabetes mellitus, hypertension, myocardial infarction, osteoporosis, and cancer (Suzuki et al., 1996; Amamoto et al., 2002; Takagi et al., 2002; Yokoyama and Omori, 2003; Yamaguchi et al., 2006). Therefore, we expected the development of model animals closer to humans by introducing the ALDH2*2 gene. The resultant DAL mice showed accumulated oxidative stress and their lifespan decreased (Fig. 3). Aging is associated with changes in physiological characteristics, including muscle weakness and hair decolorization, and many physiological functions. The causes, however, are multifactorial and several studies have suggested that oxidative stress plays an important role. ALDH2 could reduce oxidative stress in multiple organs. Indeed, we have observed amytrophy developed in muscles of Tg-mice expressing ALDH2*2 (I. Ohsawa, K. Nishimaki, Y. Suzuki, T. Nukina, E. Kodaira, M. Sano, S. Yamaguchi, S. Yagihashi, and S. Ohta, unpublished observation). The molecular mechanisms that trigger dementia symptoms have been highly elusive; however, marked increases in oxidative stress, typically represented by HNE, were reported in the hippocampus and superior and middle temporal gyrus of patients with mild cognitive impairment and in those with early AD compared with age-matched healthy individuals (Williams et al., 2006). These results, which are in agreement with the results of LPO analysis in CSF (Praticó et al., 2002), suggest that oxidative stress, typically represented by HNE, is involved in the causes of the onset of dementia. As shown in Figure 3A, age-dependent accumulation of HNE in the brain of DAL mice was observed and the ALDH2-defective hippocampal and cortical neurons were highly susceptible to HNE (Fig. 2). These findings suggest that earlier accumulation of HNE in the ALDH2-deficient brain is responsible for a trigger of cognitive decline.
The accumulation of aldehydes, including HNE, not only induces neuronal death but also causes synaptic dysfunction because of mechanisms such as reducing Na+,K+-ATPase activity (Pedersen et al., 1999) and markedly inhibits microtubule formation and neurite outgrowth (Neely et al., 1999). Furthermore, there have been a number of reports on the relationship between neurofibrillary tangle (NFT) and oxidative stress (Zarkovic, 2003). Positive immunohistochemical staining of the DAL mouse brain with phosphorylation-specific monoclonal antibody against tau (Fig. 4L) suggested that the accumulation of HNE induced by ALDH2 deficiency causes age-dependent tau hyperphosphorylation, which is a pathological feature characteristic of AD. It has been further reported that HNE induces structural changes in phosphorylated tau by its modification of it and formation of NFT-like tau assemblies in vitro (Takeda et al., 2000; Liu et al., 2005). Cdk5 is thought to be responsible for hyperphosphorylation of tau in AD and activated by p35, which may be cleaved to more stable fragment p25 (Cruz and Tsai, 2004). However, although some experiments using animals overexpressing Cdk5, p35, or p25 have supported a causal relationship between Cdk5/p25 and NFT, there is still discrepancy among studies. As shown in Figure 4M, no obvious change of their expression level was observed, whereas tau was hyperphosphorylated in the hippocampus of old DAL mice. Thus, p35/p25 may be not essential for the tau hyperphosphorylation in DAL mice.
To examine the effects of ALDH2 deficiency on memory, we used two tests, vORT and MWM. In both tests, the form of memory depends on the hippocampus. Aged DAL mice showed a deficit in both visual recognition memory and spatial learning and memory, whereas young DAL mice showed intact ability to form these memories (Figs. 6, 7). This age-related deficit in memory agrees with histological neuronal degeneration in the hippocampus of aged DAL mice (Fig. 4). We observed a similar disability of recognition memory in three independent lines of aged DAL mice (DAL101, DAL102, and DAL103), indicating that the expression of ALDH2*2 in the brain is sufficient to induce deficit in hippocampal-dependent learning and memory.
We found that the deletion of ApoE enhances learning and memory deficits of DAL mice (Fig. 8). The strength of binding between human APOE and HNE was ε2 > ε3 > ε4, which was in agreement with the preventive activity of APOE against cell death caused by HNE (Pedersen et al., 2000). Thus, APOE eliminates free HNE and the possession of APOE-ε4, the APOE with the weakest HNE elimination ability, leading to the accumulation of HNE in neurons and increasing oxidative stress. These results agree with our previous epidemiological investigation (Kamino et al., 2000), in which ALDH2*2 and APOE-ε4 alleles synergistically increased the risk of AD. Together, the accumulation of HNE in ALDH2-deficient mice is further intensified by the loss of ApoE. However, no obvious neurodegeneration, activation of astrocyte, nor hyperphosphorylation of the tau protein was apparent in any brain region of DAL/ApoE−/− mice at 6 months of age. Thus, additional investigations will be required to clarify cellular and molecular mechanisms of cognitive impairments in these mice.
Because highly toxic aldehydes are generated spontaneously by lipid peroxidation, there are a variety of mechanisms for their detoxification including binding with glutathione and reduction by aldose reductase, ADH and ABAD. Also, multiple ALDHs are considered to oxidize aldehydes. It is quite interesting that mitochondrial ALDH5A as well as ALDH2 plays an important role in the detoxification of HNE in the CNS (Murphy et al., 2003). In this study, we established transgenic mice with reduced ALDH2 activity and found that age-dependent neurodegeneration accompanying memory loss was caused by the introduction of a single mutant gene. This study emphasizes that the contribution of toxic analdehyde(s) derived from oxidative stress is sufficient to develop age-dependent neurodegeneration with memory loss.
Footnotes
This work was supported by grants from the Ministry of Health, Labour and Welfare (Longevity Science, H17-Chouju-009; Nervous and Mental Disorders, 17A-10) (S.O.).
References
- Amamoto K, Okamura T, Tamaki S, Kita Y, Tsujita Y, Kadowaki T, Nakamura Y, Ueshima H. Epidemiologic study of the association of low-Km mitochondrial acetaldehyde dehydrogenase genotypes with blood pressure level and the prevalence of hypertension in a general population. Hypertens Res. 2002;25:857–864. doi: 10.1291/hypres.25.857. [DOI] [PubMed] [Google Scholar]
- Biernat J, Mandelkow EM, Schröter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, Mandelkow E. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosron WF, Li TK. Genetic polymorphism of human liver alcohol and aldehyde dehydrogenases, and their relationship to alcohol metabolism and alcoholism. Hepatology. 1986;6:502–510. doi: 10.1002/hep.1840060330. [DOI] [PubMed] [Google Scholar]
- Chen Z, Stamler JS. Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc Med. 2006;16:259–265. doi: 10.1016/j.tcm.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tsai LH. A Jekyll and Hyde kinase: roles for Cdk5 in brain development and disease. Curr Opin Neurobiol. 2004;14:390–394. doi: 10.1016/j.conb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- D'Hooge R, De Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev. 2001;36:60–90. doi: 10.1016/s0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- Esposito LA, Kokoszka JE, Waymire KG, Cottrell B, MacGregor GR, Wallace DC. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic Biol Med. 2000;28:754–766. doi: 10.1016/s0891-5849(00)00161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadoni E, Olivero A, Miglietta A, Bocca C, Gabriel L. Cytoskeletal modifications induced by 4-hydroxynonenal. Cytotechnology. 1993;11(Suppl 1):S62–S64. doi: 10.1007/BF00746057. [DOI] [PubMed] [Google Scholar]
- Hamann M, Meisler MH, Richter A. Motor disturbances in mice with deficiency of the sodium channel gene Scn8a show features of human dystonia. Exp Neurol. 2003;184:830–838. doi: 10.1016/S0014-4886(03)00290-5. [DOI] [PubMed] [Google Scholar]
- Higuchi S, Matsushita S, Masaki T, Yokoyama A, Kimura M, Suzuki G, Mochizuki H. Influence of genetic variations of ethanol-metabolizing enzymes on phenotypes of alcohol-related disorders. Ann NY Acad Sci. 2004;1025:472–480. doi: 10.1196/annals.1316.058. [DOI] [PubMed] [Google Scholar]
- Ho YS, Xiong Y, Ma W, Spector A, Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem. 2004;279:32804–32812. doi: 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, Kitamoto T, Hayasaka M, Hanaoka K, Nakagawa Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun. 2003;305:278–286. doi: 10.1016/s0006-291x(03)00734-4. [DOI] [PubMed] [Google Scholar]
- Isacson O, Fischer W, Wictorin K, Dawbarn D, Björklund A. Astroglial response in the excitotoxically lesioned neostriatum and its projection areas in the rat. Neuroscience. 1987;20:1043–1056. doi: 10.1016/0306-4522(87)90262-4. [DOI] [PubMed] [Google Scholar]
- Jo SA, Kim E, Han C, Park MH, Jo I. P-166A Glu487Lys polymorphism in the gene for mitochondrial aldehyde dehydrogenase 2 is associated with mild cognitive impairment in elderly Korean men. Alzheimers Dement. 2007;3:S151. doi: 10.1016/j.cca.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Kamino K, Nagasaka K, Imagawa M, Yamamoto H, Yoneda H, Ueki A, Kitamura S, Namekata K, Miki T, Ohta S. Deficiency in mitochondrial aldehyde dehydrogenase increases the risk for late-onset Alzheimer's disease in the Japanese population. Biochem Biophys Res Commun. 2000;273:192–196. doi: 10.1006/bbrc.2000.2923. [DOI] [PubMed] [Google Scholar]
- Kim KS, Lee KW, Lee KW, Im JY, Yoo JY, Kim SW, Lee JK, Nestler EJ, Han PL. Adenylyl cyclase type 5 (AC5) is an essential mediator of morphine action. Proc Natl Acad Sci USA. 2006;103:3908–3913. doi: 10.1073/pnas.0508812103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Kawamoto T, Kunugita N, Tsukiyama T, Okamoto K, Yoshida A, Nakayama K, Nakayama K. Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Lett. 2000;476:306–311. doi: 10.1016/s0014-5793(00)01710-5. [DOI] [PubMed] [Google Scholar]
- Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- Liu Q, Smith MA, Aváil J, DeBernardis J, Kansal M, Takeda A, Zhu X, Nunomura A, Honda K, Moreira PI, Oliveira CR, Santos MS, Shimohama S, Aliev G, de la Torre J, Ghanbari HA, Siedlak SL, Harris PL, Sayre LM, Perry G. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic Biol Med. 2005;38:746–754. doi: 10.1016/j.freeradbiomed.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid β-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci. 1997;17:1046–1054. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yared E, Pak JH, Huang FL, Huang KP, Crawley JN. Neurogranin null mutant mice display performance deficits on spatial learning tasks with anxiety related components. Hippocampus. 2001;11:763–775. doi: 10.1002/hipo.1092. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Chen L, Sato M, Sokabe M, Nabeshima T, Pawson T, Sakai R, Mori N. Hippocampal synaptic modulation by the phosphotyrosine adapter protein ShcC/N-Shc via interaction with the NMDA receptor. J Neurosci. 2005;25:1826–1835. doi: 10.1523/JNEUROSCI.3030-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine KS, Olson SJ, Amarnath V, Whetsell WO, Jr, Graham DG, Montine TJ. Immunohistochemical detection of 4-hydroxy-2-nonenal adducts in Alzheimer's disease is associated with inheritance of APOE4. Am J Pathol. 1997;150:437–443. [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Ohsawa I, Kasahara T, Ohta S. Cytoprotective role of mitochondrial amyloid beta peptide-binding alcohol dehydrogenase against a cytotoxic aldehyde. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.07.002. in press. [DOI] [PubMed] [Google Scholar]
- Murphy TC, Amarnath V, Gibson KM, Picklo MJ., Sr Oxidation of 4-hydroxy-2-nonenal by succinic semialdehyde dehydrogenase (ALDH5A) J Neurochem. 2003;86:298–305. doi: 10.1046/j.1471-4159.2003.01839.x. [DOI] [PubMed] [Google Scholar]
- Murray IV, Liu L, Komatsu H, Uryu K, Xiao G, Lawson JA, Axelsen PH. Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid beta proteins. J Biol Chem. 2007;282:9335–9345. doi: 10.1074/jbc.M608589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely MD, Sidell KR, Graham DG, Montine TJ. The lipid peroxidation product 4-hydroxynonenal inhibits neurite outgrowth, disrupts neuronal microtubules, and modifies cellular tubulin. J Neurochem. 1999;72:2323–2333. doi: 10.1046/j.1471-4159.1999.0722323.x. [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Nishimaki K, Yasuda C, Kamino K, Ohta S. Deficiency in a mitochondrial aldehyde dehydrogenase increases vulnerability to oxidative stress in PC12 cells. J Neurochem. 2003a;84:1110–1117. doi: 10.1046/j.1471-4159.2003.01619.x. [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Kamino K, Nagasaka K, Ando F, Niino N, Shimokata H, Ohta S. Genetic deficiency of a mitochondrial aldehyde dehydrogenase increases serum lipid peroxides in community-dwelling females. J Hum Genet. 2003b;48:404–409. doi: 10.1007/s10038-003-0046-y. [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S, Ohta S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007;13:688–694. doi: 10.1038/nm1577. [DOI] [PubMed] [Google Scholar]
- Ohta S, Ohsawa I, Kamino K, Ando F, Shimokata H. Mitochondrial ALDH2 deficiency as an oxidative stress. Ann NY Acad Sci. 2004;1011:36–44. doi: 10.1007/978-3-662-41088-2_4. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Cashman NR, Mattson MP. The lipid peroxidation product 4-hydroxynonenal impairs glutamate and glucose transport and choline acetyltransferase activity in NSC-19 motor neuron cells. Exp Neurol. 1999;155:1–10. doi: 10.1006/exnr.1998.6890. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Chan SL, Mattson MP. A mechanism for the neuroprotective effect of apolipoprotein E: isoform-specific modification by the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 2000;74:1426–1433. doi: 10.1046/j.1471-4159.2000.0741426.x. [DOI] [PubMed] [Google Scholar]
- Picklo MJ, Olson SJ, Markesbery WR, Montine TJ. Expression and activities of aldo-keto oxidoreductases in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:686–695. doi: 10.1093/jnen/60.7.686. [DOI] [PubMed] [Google Scholar]
- Praticò D, Clark CM, Liun F, Rokach J, Lee VY, Trojanowski JQ. Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of Alzheimer disease. Arch Neurol. 2002;59:972–976. doi: 10.1001/archneur.59.6.972. [DOI] [PubMed] [Google Scholar]
- Reagan LP, Magariños AM, Yee DK, Swzeda LI, Van Bueren A, McCall AL, McEwen BS. Oxidative stress and HNE conjugation of GLUT3 are increased in the hippocampus of diabetic rats subjected to stress. Brain Res. 2000;862:292–300. doi: 10.1016/s0006-8993(00)02212-5. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- Schneider C, Tallman KA, Porter NA, Brash AR. Two distinct pathways of formation of 4-hydroxynonenal. Mechanisms of nonenzymatic transformation of the 9- and 13-hydroperoxides of linoleic acid to 4-hydroxyalkenals. J Biol Chem. 2001;276:20831–20838. doi: 10.1074/jbc.M101821200. [DOI] [PubMed] [Google Scholar]
- Siems W, Grune T. Intracellular metabolism of 4-hydroxynonenal. Mol Aspects Med. 2003;24:167–175. doi: 10.1016/s0098-2997(03)00011-6. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Chandra A, Ansari NH, Srivastava SK, Bhatnagar A. Identification of cardiac oxidoreductase(s) involved in the metabolism of the lipid peroxidation-derived aldehyde-4-hydroxynonenal. Biochem J. 1998;329:469–475. doi: 10.1042/bj3290469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Muramatsu T, Taniyama M, Atsumi Y, Suematsu M, Kawaguchi R, Higuchi S, Asahina T, Murata C, Handa M, Matsuoka K. Mitochondrial aldehyde dehydrogenase in diabetes associated with mitochondrial tRNA(Leu(UUR)) mutation at position 3243. Diabetes Care. 1996;19:1423–1425. doi: 10.2337/diacare.19.12.1423. [DOI] [PubMed] [Google Scholar]
- Takagi S, Iwai N, Yamauchi R, Kojima S, Yasuno S, Baba T, Terashima M, Tsutsumi Y, Suzuki S, Morii I, Hanai S, Ono K, Baba S, Tomoike H, Kawamura A, Miyazaki S, Nonogi H, Goto Y. Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens Res. 2002;25:677–681. doi: 10.1291/hypres.25.677. [DOI] [PubMed] [Google Scholar]
- Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer's disease. J Alzheimers Dis. 2006;9:309–317. doi: 10.3233/jad-2006-9s335. [DOI] [PubMed] [Google Scholar]
- Takeda A, Smith MA, Avilá J, Nunomura A, Siedlak SL, Zhu X, Perry G, Sayre LM. In Alzheimer's disease, heme oxygenase is coincident with Alz50, an epitope of tau induced by 4-hydroxy-2-nonenal modification. J Neurochem. 2000;75:1234–1241. doi: 10.1046/j.1471-4159.2000.0751234.x. [DOI] [PubMed] [Google Scholar]
- Tang FR, Chia SC, Zhang S, Chen PM, Gao H, Liu CP, Khanna S, Lee WL. Glutamate receptor 1-immunopositive neurons in the gliotic CA1 area of the mouse hippocampus after pilocarpine-induced status epilepticus. Eur J Neurosci. 2005;21:2361–2374. doi: 10.1111/j.1460-9568.2005.04071.x. [DOI] [PubMed] [Google Scholar]
- Tjalkens RB, Cook LW, Petersen DR. Formation and export of the glutathione conjugate of 4-hydroxy-2,3-E-nonenal (4-HNE) in hepatoma cells. Arch Biochem Biophys. 1999;361:113–119. doi: 10.1006/abbi.1998.0946. [DOI] [PubMed] [Google Scholar]
- Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- Vasiliou V, Nebert DW. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum Genomics. 2005;2:138–143. doi: 10.1186/1479-7364-2-2-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou V, Pappa A, Estey T. Role of human aldehyde dehydrogenases in endobiotic and xenobiotic metabolism. Drug Metab Rev. 2004;36:279–299. doi: 10.1081/dmr-120034001. [DOI] [PubMed] [Google Scholar]
- Wakabayashi-Ito N, Nagata S. Characterization of the regulatory elements in the promoter of the human elongation factor-1 alpha gene. J Biol Chem. 1994;269:29831–29837. [PubMed] [Google Scholar]
- Wang B, Wang J, Zhou S, Tan S, He X, Yang Z, Xie YC, Li S, Zheng C, Ma X. The association of mitochondrial aldehyde dehydrogenase gene (ALDH2) polymorphism with susceptibility to late-onset Alzheimer's disease in Chinese. J Neurol Sci. 2008;268:172–175. doi: 10.1016/j.jns.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci. 2004;7:635–642. doi: 10.1038/nn1248. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Hink U, Oelze M, Schuppan S, Schaeuble K, Schildknecht S, Ho KK, Weiner H, Bachschmid M, Munzel T, Daiber A. Role of reduced lipoic acid in the redox regulation of mitochondrial aldehyde dehydrogenase (ALDH-2) activity. Implications for mitochondrial oxidative stress and nitrate tolerance. J Biol Chem. 2007;282:792–799. doi: 10.1074/jbc.M606477200. [DOI] [PubMed] [Google Scholar]
- Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease. Neurobiol Aging. 2006;27:1094–1099. doi: 10.1016/j.neurobiolaging.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Xie C, Lovell MA, Markesbery WR. Glutathione transferase protects neuronal cultures against four hydroxynonenal toxicity. Free Radic Biol Med. 1998;25:979–988. doi: 10.1016/s0891-5849(98)00186-5. [DOI] [PubMed] [Google Scholar]
- Yamaguchi J, Hasegawa Y, Kawasaki M, Masui T, Kanoh T, Ishiguro N, Hamajima N. ALDH2 polymorphisms and bone mineral density in an elderly Japanese population. Osteoporos Int. 2006;17:908–913. doi: 10.1007/s00198-006-0077-2. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Omori T. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and risk for esophageal and head and neck cancers. Jpn J Clin Oncol. 2003;33:111–121. doi: 10.1093/jjco/hyg026. [DOI] [PubMed] [Google Scholar]
- Yoon M, Madden MC, Barton HA. Developmental expression of aldehyde dehydrogenase in rat: a comparison of liver and lung development. Toxicol Sci. 2006;89:386–398. doi: 10.1093/toxsci/kfj045. [DOI] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkovic K. 4-Hydroxynonenal and neurodegenerative diseases. Mol Aspects Med. 2003;24:293–303. doi: 10.1016/s0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]