

Abstract
Basolateral amygdala (BLA) neurons provide a major excitatory input to medial prefrontal cortex (mPFC)–layer V pyramidal neurons. Under stressful conditions, commonly associated with chronic cocaine abuse, altered BLA-to-mPFC synaptic transmission could lead to defective emotional information processing and decision making within the mPFC and result in misguided and inappropriate behaviors. We examined the effects of cocaine administered chronically in vivo on EPSCs recorded from a putative BLA–mPFC pathway in vitro and their modulation by dopamine (DA), corticotropin-releasing factor (CRF), and their combination (DA plus CRF). In saline-treated animals, activation of D1/5 receptors depressed BLA–mPFC EPSCs, whereas CRF1 receptor activation alone had no effect on EPSCs. Activating D1/5 and CRF1 receptors in combination, however, worked synergistically through presynaptic and postsynaptic mechanisms to depress EPSCs to levels greater than D1/5 receptor activation alone. After chronic cocaine administration, the function of DA1/5 and CRF receptors switched from inhibitory to excitatory. In slices from cocaine-treated animals, putative BLA–mPFC EPSCs were depressed through a presynaptic mechanism. Now, activation of either D1/5 or CRF2 receptors increased the cocaine-induced, depressed EPSCs. Additionally, simultaneous activation of presynaptic D1/5 and CRF2 receptors led to further enhancement of EPSCs. These data indicate that CRF acting synergistically with DA normally potentiates D1/5-induced synaptic depression. However, after chronic cocaine, the combined synergistic actions of DA and CRF switched polarity to enhance facilitation of BLA–mPFC glutamatergic transmission. Also unmasked after acute withdrawal from chronic cocaine are endogenous, tonic-inhibitory D2-like and tonic-facilitatory CRF2 receptor actions. These multiple functional and receptor changes may underlie the altered, possibly aberrant, decision-making process after chronic cocaine.
Keywords: addiction, CRF, dopamine, glutamate, SKF81297, NBI30775, Astressin2B, stress
Introduction
Cocaine addiction is a chronic medical disorder characterized by compulsive cocaine use despite adverse consequences (Leshner, 1996), and is associated with changes within prefrontal cortex (PFC) networks (Everitt and Robbins, 2005).
The amygdala is an important component of PFC networks by its involvement in decision-making processes as it encodes the emotional value/intensity of environmental stimuli and outcome–action associations (Winston et al., 2005; De Martino et al., 2006). Basolateral amygdala (BLA) neurons relay this information by excitatory efferents to pyramidal neurons in the medial PFC (mPFC) (Krettek and Price, 1977; Gabbott et al., 2006; Orozco-Cabal et al., 2006a). However, after chronic cocaine use, BLA–mPFC neurotransmission may become aberrant as cocaine-dependent individuals exhibit inappropriate utilization of emotional information to guide behavior (Schoenbaum et al., 2006).
Dopamine (DA) and corticotrophin-releasing factor (CRF) are essential contributors within the PFC (Koob, 1999; Kelley, 2004) during cocaine abuse. DA mediates incentive salience of reward-related stimuli (Berridge, 2007), and is a critical modulator of pyramidal neuron excitability and excitatory synaptic transmission in the PFC (Seamans and Yang, 2004; Floresco and Tse, 2007). Pyramidal neurons in the mPFC and their afferents express receptors for DA (D1- and D2-like) (Smiley et al., 1994; Krimer et al., 1997). After chronic cocaine administration, DA neurotransmission in the PFC undergoes significant changes (Williams and Steketee, 2005), time-dependent changes that may be caused by increases/decreases in binding to DA receptor subtypes (Ben-Shahar et al., 2007). In cocaine addicts, changes in DA actions have been associated with executive dysfunction (Dackis and Gold, 1985; Volkow et al., 1996).
CRF plays a crucial role in stress responses by integrating endocrine and neural systems to facilitate coping in mammals (Vale et al., 1981). CRF effects are mediated by two G-protein-coupled receptors, CRF1 and CRF2. Extrahypothalamic CRF receptors regulate cellular excitability and synaptic plasticity within frontolimbic circuits (Liu et al., 2004), and because CRF is released (Yan et al., 1998) within layer V of the PFC, activation of PFC-CRF receptors may affect local neurotransmission.
Chronic cocaine administration is associated with CRF hypersecretion (Richter et al., 1995), whereas CRF receptor binding was increased or decreased (Goeders et al., 1990; Ambrosio et al., 1997). Chronic cocaine induced a switch from an expected CRF2-mediated depression of glutamatergic transmission to one of facilitation (Liu et al., 2005).
The effects of chronic cocaine administration on BLA–mPFC EPSCs remain unclear. In controls, exogenously applied DA induced depression of EPSCs at putative BLA–mPFC synapses. Furthermore, this DA-induced EPSC depression was enhanced significantly when combining DA with CRF, whereas CRF itself had no action. After chronic cocaine administration in vivo, a switch in function of DA and CRF actions became apparent. Presynaptically, D1-like receptors facilitated EPSCs with emergence of a D2-like depressant tone. CRF again enhanced the action of DA, but now the combination facilitated rather than depressed EPSCs. In addition, CRF itself now enhanced EPSCs via an endogenous facilitatory CRF2 receptor tone.
These multiple changes in DA and CRF modulation of putative BLA–mPFC EPSCs after chronic cocaine may provide aberrant emotional information that contributes to compulsive drug-seeking and drug-taking behaviors in cocaine-dependent subjects.
Materials and Methods
Subjects and treatment groups.
After 5 d of acclimatization to the colony room, male rats [postnatal day 22 (P22)–P25, 75 g, Sprague Dawley, Harlan, Houston, TX] were divided into two groups. Control rats (saline-treated/control group) received 0.9% saline injections (1 ml/kg, i.p.) in their home cages according to the same schedule as chronic cocaine-treated (chronic cocaine-treated group; 15 mg/kg, i.p., twice a day for 14 d at 9:00 A.M. and 4:00 P.M.) rats. The rats (P36–P39, 175–225 g) were decapitated within 17–24 h after receiving their last injection of saline or cocaine, and their brains were rapidly removed for brain slice preparation. Rats of this age group are considered adolescent to young adult and would be most comparable in age to the most prevalent age group experiencing drug abuse, because over one-half (51%) of America's teenagers have tried an illicit drug by the time they finish high school (Rice, 1999).
Animal experiments were performed in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals.
PFC slice.
The brains were immersed in a modified cold (∼4°C) artificial CSF (ACSF) bubbled continuously with 95% O2/5% CO2 to maintain proper pH (7.3–7.4). The composition of the ACSF solution was as follows (in mm): 117 NaCl, 3.5 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.3 MgCl2, 25 NaHCO3, and 11.5 glucose. Subsequently, brains were cut at the level of the optic chiasm, and 400 μm coronal slices containing the PFC [approximately +3.20 mm from bregma (Paxinos and Watson, 1998)] were made from the remaining tissue block using a Vibroslice (World Precision Instruments, Sarasota, FL). After 1 h in ACSF at 33 ± 2°C for recovery, individual slices were transferred and submerged in a cylindrical Plexiglas recording chamber (volume = 2 cc) and continuously perfused (2.0–2.5 ml/min) with ACSF maintained at 33 ± 2°C. The recording chamber was transilluminated to facilitate identification of mPFC cortical layers for electrode placement.
Stimulating and recording electrode placements were made within the slice obtained at +3.20 mm from bregma. Briefly, the concentric stimulating electrode was positioned within layer V of the infralimbic cortex (IL) at 400–450 μm lateral to the interhemispheric fissure (Fig. 1A). Positioning of the stimulating electrode is based on our previous quantitative analyses, which had determined that the highest labeling intensity, after injection of DiI into the BLA (Orozco-Cabal et al., 2006a), appeared along layer V in the infralimbic cortex. The recording electrode was then positioned within layer V of the prelimbic cortex (PrL) separated by 600–800 μm dorsal to the stimulating electrode. The recording electrode was also ∼600–700 μm lateral to the interhemispheric fissure; this position was aligned as a continuum from the recording site and represented the highest density of DiI labeling (see Fig. 2) (Orozco-Cabal et al., 2006a). After recording, biocytin labeling (Fig. 1B) of the recorded layer V–VI pyramidal neurons provided another monitor of these neurons within prelimbic layers V–VI. Positioning of the stimulating electrode within the slice was based on our previous published work (Orozco-Cabal et al., 2006a).
Figure 1.
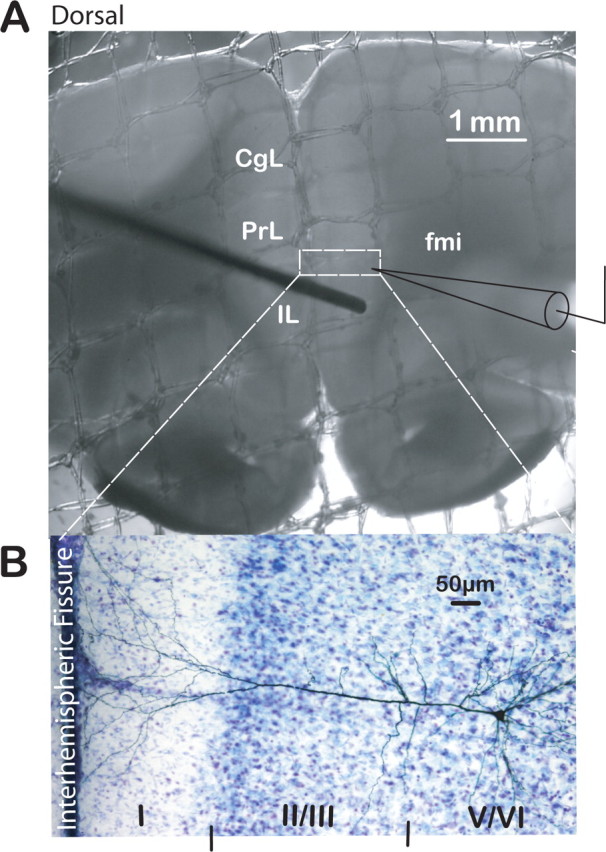
Brain slice preparation of the rat PFC used to study putative BLA–mPFC synapse. A, Photograph of a PFC slice submerged in our recording chamber depicting the position of stimulating (from the left) and recording (from the right) electrodes for EPSC recordings. CgL, Cingulate cortex; fmi, forceps minor. Scale bar, 1 mm. Dashed rectangle depicts area expanded below of typical biocytin-filled, layer V, PrL pyramidal neuron from which recordings were made. B, Photograph of DAB-stained layer V pyramidal neuron filled with biocytin in the cresyl violet-counterstained mPFC. Cortical layers are indicated by roman numerals at the bottom of the photograph. Scale bar, 50 μm.
Figure 2.

Depression of excitatory transmission at putative BLA–mPFC synapse mediated by the D1-like (D1/5) agonist SKF is enhanced through activation of CRF1 receptors in saline-treated control rats, whereas neither r/hCRF itself nor CRF antagonists affect evoked EPSCs. A, Representative traces of putative BLA–mPFC EPSCs at baseline and after increasing concentrations of SKF (top traces) and SKF plus 50 nm r/hCRF (bottom traces). Concentration–response curves for each treatment group are shown below. SKF (1 and 10 μm) significantly depressed EPSCs. Combining r/hCRF (50 nm) plus SKF (10 nm or higher concentrations) further depressed EPSCs, indicating that r/hCRF (50 nm) facilitated SKF effects at this putative BLA–mPFC synapse. B, Bar graph depicts changes in putative BLA–mPFC EPSC amplitudes at two different concentrations of SKF (1, 10 μm) alone and in combination with r/hCRF (50 nm) in the presence of selective antagonists for D1/5 or CRF1 receptors. Shown are SKF (1 μm, 10 μm) alone and in the presence of SCH, a selective D1/5 receptor antagonist, and SKF (1 μm, 10 μm) and r/hCRF (50 nm) together and with NBI, a selective CRF1 receptor antagonist. SCH (10 μm) prevented SKF-induced depression of EPSCs [1 μm SKF plus SCH, 101.4 ± 3.4%; 10 μm SKF plus SCH, 101.2 ± 3.3%; p > 0.05 compared with baseline; p < 0.05 compared with SKF (1, 10 μm) alone]. NBI (500 nm) blocked r/hCRF (50 nm)-mediated facilitation of SKF effects [1 μm SKF plus r/hCRF plus NBI, 87.4 ± 1.1%; 10 μm SKF plus r/hCRF plus NBI, 74.6 ± 4.2%; p < 0.05 compared with baseline; p < 0.05 compared with SKF (1, 10 μm) plus r/hCRF (50 nm)]. There were no significant differences (p > 0.05) between EPSC values after SKF (1, 10 μm) compared with SKF (1, 10 μm) plus r/hCRF (50 nm) in the presence of NBI. Ast2B, a specific CRF2 antagonist, did not affect EPSCs in saline-treated/control rats. C, Left, Bar graphs summarizing lack of effects of different r/hCRF concentrations on EPSCs. Neither NBI (500 nm; center) nor Ast2B (200 nm; right) had any significant effects on EPSCs before or after washout.
Whole-cell patch-clamp recordings.
Whole-cell patch-clamp recordings were performed from mPFC–layer V pyramidal neurons using patch electrodes with tip resistances of 3–4 MΩ when filled with a solution consisting of the following (in mm): 122 K-gluconate; 0.3 CaCl2; 2 MgCl2; 1 EGTA; 10 HEPES; 5 Na2-ATP; 0.4 Na3-GTP; 5 N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium bromide (QX314), and 0.2% biocytin. The pH of the internal solution was adjusted to 7.2 with KOH, and osmolality was corrected to 280–290 mmol/kg with sucrose. In addition, a modified internal electrode solution containing 1 mm guanosine 5′-O-[β-thio] diphosphate (GDPβS) was used to inactivate postsynaptic G-protein-coupled receptors (GPCRs). The effectiveness of GDPβS at blocking postsynaptic GPCRs was assayed for the loss of a baclofen-induced (100 μm, drop applied) outward current in mPFC–layer V pyramidal neurons.
Recorded pyramidal neurons were identified based on the membrane electrical properties (Yang et al., 1996; Orozco-Cabal et al., 2006a) and were included for analyses if they maintained stability with a resting membrane potential more negative than −60 mV. Access resistance was monitored continuously throughout recordings; neurons were discarded if access resistance exceeded 30 MΩ.
Evoked EPSCs.
Putative excitatory afferents to the mPFC from the basolateral amygdala were stimulated at 0.05 Hz with a Grass S-88 stimulator (with isolation units) through a low-resistance concentric electrode (Frederick Haer, Bowdoinham, ME) placed in cortical layer V (400–450 μm from the interhemispheric fissure of the slice) and within the IL cortex (Fig. 1A) (Orozco-Cabal et al., 2006a). The recording electrode was situated within layer V of the PrL mPFC cortex (∼700 μm from the interhemispheric fissure) (Fig. 1A). Both stimulating and recording electrodes were positioned under visual control using a dissecting microscope (0.5×; Nikon Instruments, Melville, NY). We are aware that other glutamatergic afferents, arising from sources in addition to the BLA such as midline thalamic nuclei, the hippocampus, and septum, may course along our stimulation site. As a result, we have modified our description of the pathway we are stimulating and the synapse from which we are recording as the “putative” BLA–mPFC pathway and its synapse. Nonetheless, in support of our experimental results and description of the excitatory synapses from which we recorded as being putative BLA–mPFC synapses, Gabbott et al. (2006) concluded that excitatory BLA afferents to mPFC pyramidal neuron spines formed predominantly (>94%) asymmetric (Gray type I) monosynaptic synapses.
EPSCs were recorded at −70 mV holding membrane potential and in the presence of (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl] (phenylmethyl)phosphinic acid (CGP55845; 2 μm) to block GABAB receptors and d-2-amino-5-phosphonovaleric acid (d-APV; 10 μm) to partially block NMDA receptors while preventing signal contamination by recurrent activity within the slice preparation. The membrane holding potential (−70 mV) corresponds to the reversal potential for Cl− determined experimentally and is also based on the composition of the bath and pipette solutions; holding at this membrane potential controlled for GABAA receptor-mediated conductances (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). EPSCs obtained under these recording conditions consisted primarily of non-NMDA-mediated postsynaptic responses (Orozco-Cabal et al., 2006a). This recording solution would have prevented activation of possible disynaptic inhibitory currents arising from excitatory activation of inhibitory interneurons within our slice preparation. Data were acquired using an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA) with a switching frequency of 5–6 kHz (30% duty cycle; gain of 3 nA/mV, time constant = 20 ms). To test for drug effects, the stimulus intensity was set to elicit 50–70% of the maximal response. Current signals were low-pass filtered at 1 kHz with a 4-pole Bessel filter (Warner Instrument, Hamden, CT), digitized at 5.5 Hz (DigiData 3200 interface, Axon Instruments), and stored on a Dell computer for off-line analysis using pClamp (version 9.2; Axon Instruments) software.
Evoked fast IPSCs.
The stimulating electrode was placed in layer II–III of the PrL cortex adjacent to the recording electrode (100–200 μm interelectrode distance), which was located within layer V of the mPFC cortex (∼700 μm from the interhemispheric fissure). Stimulating and recording electrodes were positioned under visual control using a dissecting microscope (0.5×; Nikon Instruments). Evoked fast IPSCs (f-IPSCs) were acquired in the presence of CGP55845 (2 μm), 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX; 20 μm), and d-APV (50 μm), while the neuron was clamped at −55 mV membrane potential (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Pharmacological blockade of glutamate receptors did not have a depressant effect on f-IPSCs, suggesting that positioning of the stimulating electrode in layer II–III of the PrL cortex and in close proximity (100–200 μm) to the recording electrode yielded direct monosynaptic orthodromic stimulation of inhibitory interneurons in the slice preparation. Additionally, f-IPSCs could be completely blocked using a mixture of picrotoxin (PTX; 50 μm) and bicuculline methiodide (BMI; 10 μm), which confirmed the monosynaptic generation of these currents and their mediation by GABAA receptors (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). QX314 was not added to the internal pipette solution for f-IPSC recordings.
mEPSCs.
Action potential-independent miniature EPSCs (mEPSCs) were collected for 2 min after a 20 min period after the addition of tetrodotoxin (TTX; 1 μm) to the standard ACSF and analyzed off-line using Synaptosoft Mini Analysis software (Synaptosoft, Fort Lee, NJ). mEPSC events were defined as amplitudes above a preset baseline/noise level (5 pA). d-APV (50 μm), PTX (50 μm), and BMI (10 μm) were added to the perfusion solution during all mEPSC recordings. Holding membrane potential = −70 mV.
Drug application.
All drugs and drug combinations were dissolved in ACSF and superfused at known concentrations. A wash-in period of at least 12 min was allowed to establish equilibrium before drug effects were analyzed in all experiments. After reaching equilibrium concentrations, drug effects were analyzed over the next 15–20 min period. During washout, return of monitored parameters to predrug or drug combination baseline values was used as an indication of recovery and reversibility (usually 15–20 min).
The following drugs were used: rat/human corticotropin-releasing factor (r/hCRF), cyclo (31–34) [D-Phe11,His12,C(a)MeLeu13,39,Nle17,Glu31,Lys34] Ac-Svg(8–40) trifluoroacetate salt [Astressin2B (Ast2B)], d-APV, DNQX, BMI, PTX, dopamine hydrochloride (DA), S(−)-raclopride l-tartrate, SKF81297 (SKF), QX314, baclofen, TTX, EGTA, and GDPβS from Sigma-Aldrich (St. Louis, MO); CGP55845 and (R)-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride [SCH23390 (SCH)] from Tocris Cookson (Ellisville, MO); and 2,5-dimethyl-3-(6-dimethyl-4-methylpyridin-3-yl)-7-dipropylaminopyrazolo[1,5-a] pyrimidine [NBI30775 (NBI)/R121919] courtesy of D. E. Grigoriadis (Neurocrine Biosciences, San Diego, CA).
Data analyses.
To avoid cumulative effects of drugs, only one cell per slice was used for recording and data collection. The amplitudes of EPSCs and f-IPSCs were normalized to baseline responses and expressed as the mean percentage of baseline ± SEM. To test for the effects of concentration-dependent effects of drugs applied individually (r/hCRF, DA, SKF81297, NBI30775, Ast2B, and raclopride), we performed ANOVAs for repeated measures with Bonferroni correction using concentration as a repeated factor. To assess the influence of r/hCRF on the effects of SKF81297 or DA, we performed two-way mixed ANOVAs with Bonferroni correction using concentration as a repeated factor and treatment as between-subjects factor. To calculate IC50 and EC50 values for SKF81297, percentage change of EPSC amplitude after SKF81297 (10 μm) application was set as the maximal data point. For analyses of paired-pulse facilitation data, mixed ANOVAs with interstimulus interval and concentration as repeated-within-subjects factor, treatment as between-subjects factor, and post hoc tests were performed. The effects of chronic cocaine administration on r/hCRF and SKF or DA were tested using mixed ANOVAs with cocaine/saline-treatment groups as between-subjects factor and concentration as within-subjects factor with Bonferroni correction for multiple comparisons. Chronic cocaine effects on passive membrane electrical properties were analyzed using the Wald-Wolfowitz runs test. Significance level was defined as p ≤ 0.05, n indicates number of neurons, and data are presented as mean ± SEM.
Importantly, because brain slices from cocaine-treated rats were collected after only 17–24 h of withdrawal from cocaine administration in all chronic cocaine administration and acute withdrawal experiments, it is possible that cocaine withdrawal per se may have an effect on BLA–mPFC synaptic transmission and CRF and DA receptor-mediated modulation. However, our experimental design does not allow a clear distinction between the effects of chronic cocaine administration versus those of acute (17–24 h) cocaine withdrawal. Therefore, we are not making such a distinction and refer to our findings in this group of rats as chronic cocaine induced.
Results
Experiments in saline-treated/control rats
D1/5 receptor-mediated depression of excitatory transmission is enhanced on activating CRF1 receptors at mPFC–BLA synapse
We initiated our investigation of DA actions at the putative BLA–mPFC synapse with the selective D1/5 agonist SKF81297. Previous studies had confirmed that D1-like receptors are the most prevalent type of dopamine receptors within layer V of the mPFC. In addition, because DA and CRF systems are implicated in stress and cocaine abuse, we investigated a possible interaction between these two systems by examining the combined actions of SKF81297 and r/hCRF, a CRF agonist active at both CRF1 and CRF2 receptors.
SKF81297 depressed putative BLA–mPFC excitatory synaptic transmission in a concentration-dependent manner (Fig. 2A) (IC50 = 0.6 μm). SKF [1 μm (82.4 ± 1.3%; p < 0.05) and 10 μm (74.8 ± 1.6%; p < 0.05)] significantly depressed EPSCs (F(4,60) = 59.5; p < 0.01). Lower concentrations of SKF did not affect EPSCs (SKF at 10 nm, 96.7 ± 0.7%; p > 0.05; SKF at 100 nm, 97.0 ± 2.3%; p > 0.05). Consistent with a D1/5 receptor-mediated effect, the effects of SKF81297 (1 and 10 μm) on EPSCs were blocked by SCH23390 (10 μm) (Fig. 2B), a selective D1/5-antagonist. SCH (10 μm) prevented SKF-induced depression of EPSCs [SKF (1 μm) plus SCH, 101.4 ± 3.4%; SKF (10 μm) plus SCH, 101.2 ± 3.3%; p > 0.05 compared with baseline; p < 0.05 compared with SKF (1, 10 μm) alone]. In addition, SCH23390 did not affect membrane conductance or evoked EPSC amplitude from pyramidal neurons in our mPFC slice preparation.
Unlike SKF81297, r/hCRF (12.5–100 nm) did not affect putative BLA-to-mPFC EPSCs (Fig. 2C) (R/hCRF at 12.5 nm, 96.0 ± 1.6%; at 50 nm, 99 ± 1.3%; and at 100 nm, 95.8 ± 1.4%; F(3,21) = 1.52; p > 0.05). There was also no apparent CRF1- or CRF2-dependent endogenous tone after application of selective CRF receptor antagonists, NBI30775 (Heinrichs et al., 2002) and Astressin2B (Rivier et al., 2002), respectively (Fig. 2C). We observed no changes of EPSCs (95.9 ± 1.3%) after 20 min of continuous application of NBI30775 (500 nm) or after washout (98.7 ± 2.2%; F(2,8) = 1.89; p > 0.05). Similarly, Ast2B did not affect EPSCs (103.9 ± 1.8%) after 20 min of continuous application or after washout (98.7 ± 2.7%; F(2,8) = 1.89; p > 0.05). Together, these results demonstrated that neither exogenous r/hCRF (≤100 nm) or endogenous CRF modulates BLA–mPFC EPSCs in saline-treated control rats. The lack of exogenous and endogenous actions of CRF on evoked EPSCs at this synapse differed from CRF actions at the BLA–central amygdala nucleus synapse, where exogenously applied r/hCRF (IC50 = 0.32 nm) depressed evoked EPSCs by a postsynaptic CRF1-mediated action, and antagonists revealed tonic activation of CRF1 and CRF2 receptors (Liu et al., 2004).
Surprisingly, the combination of r/hCRF (50 nm; a concentration not affecting EPSCs) with SKF81297 (1 or 10 μm) synergistically depressed EPSCs (Fig. 2A,B), i.e., to a greater extent than SKF81297 alone. In these experiments, we compared changes in EPSCs induced by increasing concentrations of SKF81297 (10 nm to 10 μm) to those caused by the combination of SKF81297 (10 nm to 10 μm) with r/hCRF (50 nm). As shown in Figure 2A, r/hCRF shifted the concentration–response curve for SKF81297 downward and to the left (IC50 = 72 nm with r/hCRF vs 600 nm without r/hCRF; 44.9% EPSC reduction after 10 μm SKF81297 compared with 55.1% reduction after the combination [r/hCRF (50 nm) plus SKF81297 (10 μm)]), suggesting that r/hCRF (50 nm) acted to enhance D1/5 receptor-mediated depression of putative BLA–mPFC EPSCs in saline-treated control animals. In addition, the synergistic effects of r/hCRF (50 nm) on SKF81297-mediated effects could be blocked completely by NBI30775 (500 nm) (Fig. 2B), indicating that r/hCRF effects were mediated primarily by CRF1 receptors in saline-treated control animals.
r/hCRF1 facilitation of D1/5-mediated depression of putative BLA–mPFC EPSCs was independent of GABAA-mediated neurotransmission
Because CRF is known to colocalize with GABA in GABA terminals of PFC interneurons (Merchenthaler, 1984; Morin et al., 1999; Dautzenberg and Hauger, 2002; Gabbott et al., 2006), we tested whether the CRF1-induced enhancement of D1/5-mediated depression of EPSCs was related to r/hCRF-induced changes of GABAA neurotransmission. R/hCRF (50 nm) did not cause observable changes (100.9 ± 1.0% after 50 nm r/hCRF; 99.2 ± 1.6% after washout; r/hCRF effect, F(2,14) = 1.06; p > 0.05; n = 8) of fast GABAA-mediated IPSCs (f-IPSCs) recorded from mPFC–layer V pyramidal neurons (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), suggesting that enhancement of GABAA neurotransmission could not account for r/hCRF facilitation of D1/5 receptor-mediated EPSC depression. However, an effect of r/hCRF on GABAB neurotransmission could not be discounted, because all EPSC and f-IPSC recordings were performed in the presence of 2 μm CGP55845, a selective GABAB receptor antagonist that blocks both presynaptic and postsynaptic GABAB receptors.
R/hCRF enhanced the depressant effect of DA on putative BLA–mPFC EPSCs in the presence of a D2/3 receptor antagonist
Because SKF81297 is not the endogenous ligand for D1/5 receptors, we tested the effects of DA (10 μm) in the presence of raclopride (10 μm), a selective D2/3 receptor antagonist, alone and in combination with r/hCRF (Fig. 3). Blocking D2/3 receptors was necessary because previous studies demonstrated that D2/3 receptor activation decreases non-NMDA EPSCs (Cepeda et al., 1992; Hsu et al., 1995). Raclopride (10 μm) did not cause significant changes in the amplitude of EPSCs (98.9 ± 3.9% of baseline; paired t = 0.27; p > 0.05; n = 5), suggesting that D2/3 receptors are not tonically activated by endogenous DA in slices from saline-treated animals [but see raclopride effects after chronic cocaine (Fig. 7A)].
Figure 3.
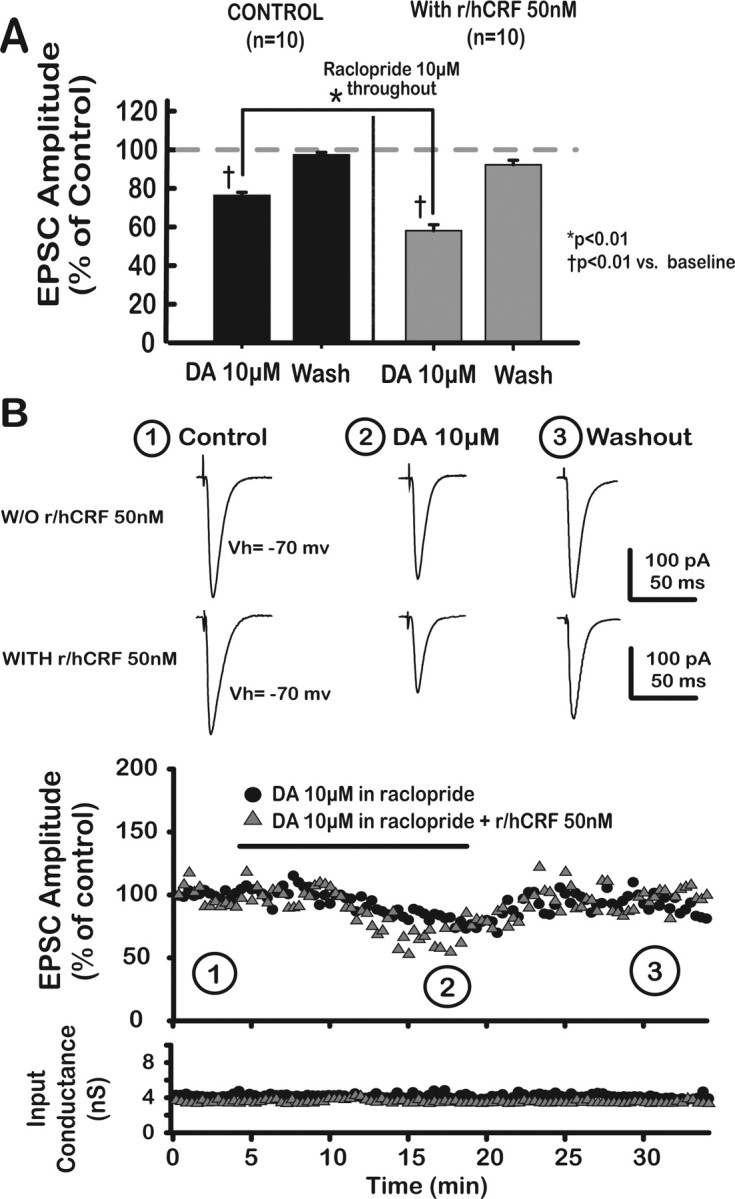
R/hCRF facilitated a DA-induced depression of putative BLA–mPFC EPSCs in the presence of raclopride, a D2-like antagonist, in saline-treated animals. A, Bar graph depicts reversible and significant DA-mediated depression of EPSCs in the presence of 10 μm raclopride. Raclopride, itself, did not have a significant effect on EPSC amplitude. DA (10 μm) combined with r/hCRF (50 nm) produced a greater depressive effect on EPSCs, indicating that r/hCRF (50 nm) facilitated DA-induced EPSC depression. B, Top, Representative averaged traces of EPSCs taken at different time points, indicated by numbers. Bottom, Input conductance plotted as a function of time.
Figure 7.
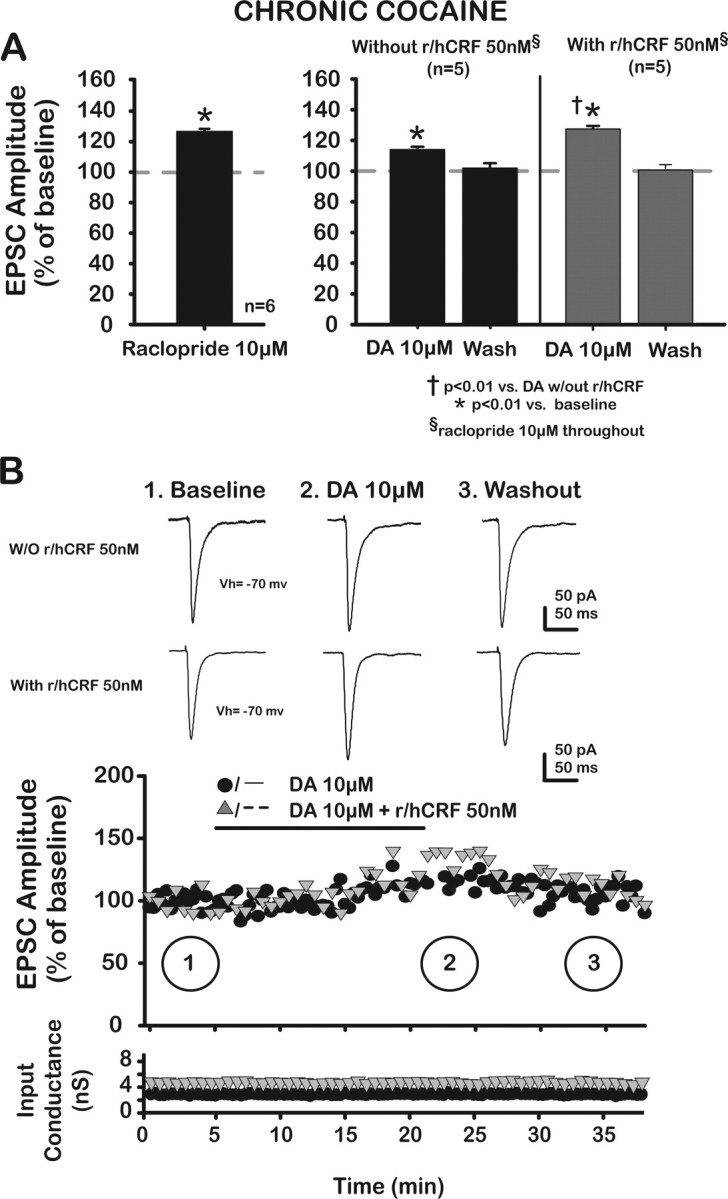
R/hCRF facilitated a DA-mediated increase of putative BLA–mPFC EPSCs in the presence of raclopride, a D2/3-like antagonist, in chronic cocaine-treated rats. A, Left, Application of raclopride (10 μm), a D2-like antagonist, unmasked tonic inhibitory action of D2/3 receptors at the synapse, thus increasing EPSCs (127.0 ± 1.4%; paired t = −19.17; p < 0.05). Right, DA (10 μm) caused a significant increase of EPSCs (112.2 ± 1.8%; p < 0.01), which returned to baseline (100.1 ± 3.1% after 20 min washout; p > 0.05 when compared with baseline with raclopride). R/hCRF (50 nm) enhanced DA (10 μm) effects on EPSCs. Note that raclopride (10 μm) is present throughout. B, Plots depict amplitude of EPSCs (percentage of baseline) as a function of time in each treatment group. Top, Representative averaged traces of EPSCs taken at different time points, indicated by numbers. Bottom, Input conductance is plotted as a function of time.
As shown in Figure 3, A and B, DA (10 μm) reversibly depressed EPSCs in the presence of raclopride (10 μm DA, 76.6 ± 1.5%; p < 0.01; after washout, 97.7 ± 1.3%; p > 0.05; DA effect, F(2,18) = 118.83; p < 0.01). Combining r/hCRF (50 nm) with DA resulted in an enhancement of DA effects on EPSCs [23.4% reduction after 10 μm DA compared with 40.7% reduction after the combination 10 μm DA plus 50 nm r/hCRF (r/hCRF × DA, F(2,24) = 19.07; p < 0.01; r/hCRF effect, F(1,18) = 53.93; p < 0.01; DA effect, F(2,24) = 281.75; p < 0.01)]. Because DA effects were tested in the presence of raclopride, our results confirmed that dopamine inhibitory effects were mediated through D1/5 receptors.
Postsynaptic CRF1 receptors mediated r/hCRF facilitation of presynaptic D1/5 receptor-induced depression of putative BLA–mPFC EPSCs
To determine the synaptic location (presynaptic vs postsynaptic) of D1/5 and CRF1 receptors, voltage-clamped pyramidal neurons were loaded with GDPβS (1 mm) dissolved in the patch electrode solution. GDPβS is a nonhydrolyzable GDP analog that locks GPCRs in their inactive state (Andrade et al., 1986). Because CRF1 and D1/5 receptors are both GPCRs (Missale et al., 1998; Grammatopoulos et al., 2001), these receptors would be inactivated by GDPβS if present postsynaptically. Consequently, putative presynaptic CRF1- and D1/5-mediated effects on EPSCs would remain unaltered by GDPβS loaded into the recorded neuron; GDPβS is not membrane permeable.
We assayed the effectiveness of GDPβS at blocking postsynaptic GPCRs by testing for the loss of an outward (hyperpolarizing) current elicited by GABAB receptor activation (Andrade et al., 1986; Yamada et al., 1999) with baclofen (100 μm). As shown in Figure 4A, GDPβS blocked effectively a GABAB-mediated outward current ∼20 min after whole-cell configuration was achieved. As a result, a 20 min waiting period was allowed before beginning to test for SKF81297 and r/hCRF effects in the presence of postsynaptic GDPβS.
Figure 4.
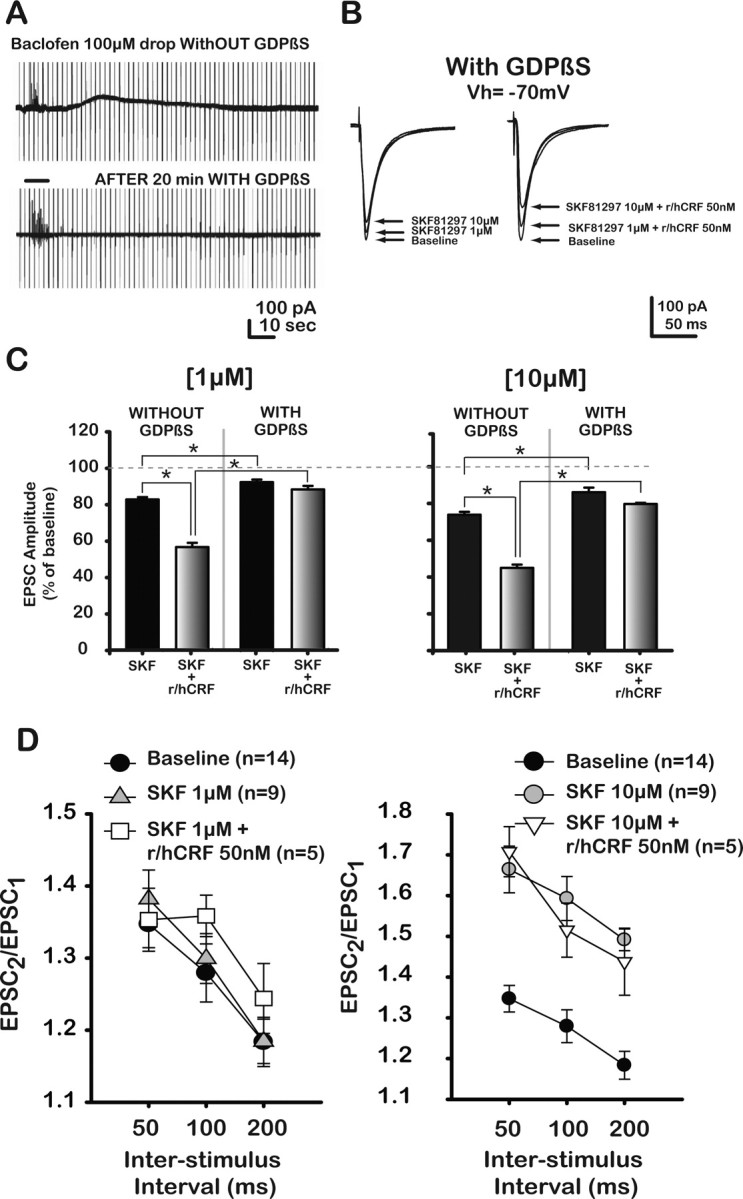
Postsynaptic CRF1 receptors enhanced presynaptic D1/5-mediated EPSC depression at the putative BLA–mPFC synapse in saline-treated control rats. A, Effects of GDPβS on GABAB receptor-mediated outward current. Current traces obtained under voltage clamp (−70 mV) from mPFC–layer V pyramidal neurons under control conditions and with GDPβS (1 mm) inside the recording electrode. Baclofen, a GABAB receptor agonist, was drop applied (horizontal bar between top and bottom traces) into the bath while changes in membrane conductance were monitored. Top trace shows slow outward, hyperpolarizing current in response to baclofen application. Loading pyramidal neurons with GDPβS (1 mm, ∼20 min) blocked GABAB receptor-mediated current (bottom trace). Stimulus artifacts have been partially cut. B, Representative traces of EPSCs from mPFC–layer V pyramidal neurons pyramidal neurons loaded with GDPβS at baseline and after two different concentrations of SKF alone (left) or with coapplication of r/hCRF (left). C, Bar graph depicts changes in EPSCs after SKF alone (1, 10 μm) or in combination with r/hCRF (50 nm) in the absence or presence of GDPβS. GDPβS blocked the effects of SKF at 1 μm, but did not block SKF at 10 μm. In addition, GDPβS blocked r/hCRF (50 nm)-induced facilitation of SKF (1 μm; 87.8 ± 2.0% vs 44.8%) and 10 μm (80.3 ± 0.7% vs 51.9%) effects on EPSCs. All treatments were significantly different from baseline. D, Paired-pulse ratios (PPR = EPSC2/EPSC1) plotted as a function of interstimulus interval. SKF (1 μm) did not change PPR values, but PPR values were significantly increased at all interstimulus intervals after 10 μm SKF. The combination SKF (1 μm) plus r/hCRF (50 nm) did not change PPR values from SKF alone or from baseline, whereas the combination SKF (10 μm) plus r/hCRF (50 nm) significantly increased PPR values at all interstimulus intervals compared with baseline; there were no differences (p > 0.05) between PPR values after 10 μm SKF in the presence or absence of CRF. *p < 0.05 versus baseline.
GDPβS, injected intracellularly into the recorded mPFC neuron, reduced the postsynaptic-GPCR-mediated actions of D1/5 and CRF1, which resulted in a reduced depression of EPSCs by DA and CRF ligands. Significantly, the actions of both concentrations of SKF81297 (1 or 10 μm) on EPSCs were diminished but not blocked by the presence of GDPβS (Fig. 4C). GDPβS reduced the depression by SKF at 1 μm (93.5 ± 1.4% vs 82.4 ± 1.3% without GDPβS; p < 0.01) and at 10 μm (87.1 ± 2.6% vs 74.9 ± 1.6%; p < 0.01). These results support presynaptic and postsynaptic locations for D1/5 receptors activated by SKF81297, and the presynaptic actions are consistent with previous reports of mPFC presynaptic D1/5 receptors on glutamatergic terminals in the mPFC, which when activated reduce the probability of glutamate release (Momiyama et al., 1996; Gao et al., 2001; Seamans et al., 2001a,b; Paspalas and Goldman-Rakic, 2004, 2005). Importantly, previous reports also indicated that activation of postsynaptic D1/5 receptors in layer V pyramidal neurons of the mPFC by DA receptor ligands at low micromolar concentrations enhanced NMDA-mediated excitatory responses, while having no or a small effect on non-NMDA EPSCs (Zheng et al., 1999; Zhou and Hablitz, 1999; Seamans et al., 2001a,b; Chen et al., 2004).
We also observed that GDPβS blocked r/hCRF (50 nm)-induced enhancement of the depression by SKF 1 μm (74.9 ± 1.6% vs 87.1 ± 2.6%) and 10 μm (45.0 ± 1.8% vs 80.3 ± 0.7%) effects on EPSCs. All treatments were significantly different from baseline. These data suggest that CRF1 receptors are located postsynaptically only (see Fig. 9, left) and add support for a D1/5 and CRF1 functional interaction postsynaptically. This result also confirms the effectiveness of the GDPβS postsynaptically.
Figure 9.

The type and function of CRF and DA receptors at the putative BLA–mPFC synapse were different in saline-treated controls compared with chronic cocaine-treated rats. Left, Activation of presynaptic and postsynaptic D1/5 receptors in saline-treated/control rats decreased EPSCs (inhibitory effect; red). Activation of postsynaptic CRF1 receptors (orange) by r/hCRF in the presence of glutamate and D1/5 agonists further depressed EPSCs. In addition, no tonic modulatory effects for D1/5, CRF1, or CRF2 receptors were measurable in saline-treated/controls. Right, After chronic cocaine administration, activation of presynaptic D1/5 receptors increased cocaine-induced depressed EPSCs. Moreover, activation of presynaptic CRF2 receptors, unmasked by chronic cocaine administration, led to EPSC enhancement. Furthermore, a synergistic interaction by coactivation of these receptors with low concentrations of r/hCRF and D1/5 agonists increased EPSC amplitude to values greater than occurred after activation of either receptor independently. On the other hand, CRF1 and D1/5 postsynaptic receptor functions were lost in chronic cocaine-treated rats. Our results also revealed a tonic facilitatory and depressant effect for CRF2 and D2-like receptors (shaded), respectively, that was unmasked after chronic cocaine administration. Thus, there was an apparent switch in the function of presynaptic D1/5 receptors along with a novel facilitatory effect for presynaptic CRF2 receptors. Tonic inhibitory activation of presynaptic D2/3 receptors may also contribute to depression of synaptic transmission within this putative BLA–mPFC synapse after chronic cocaine administration.
We further performed paired-pulse experiments (50, 100, and 200 ms interstimulus intervals) (Fig. 4D) to corroborate the findings with GDPβS. Paired-pulse ratios (PPR = EPSC2/EPSC1) were not significantly altered by SKF81297 (1 μm), a concentration that significantly reduced EPSCs, and suggested a postsynaptic location for D1/5 receptors (1.38 ± 0.04 at 50 ms; p > 0.05; 1.30 ± 0.03 at 100 ms; p > 0.05; 1.18 ± 0.03 at 200 ms; p > 0.05). In contrast, PPR values were increased significantly after SKF81297 (10 μm) at all interstimulus intervals (1.36 ± 0.02 to 1.66 ± 0.3 at 50 ms; p < 0.05; 1.28 ± 0.02 to 1.59 ± 0.02 at 100 ms; p < 0.05; 1.18 ± 0.2 to 1.49 ± 0.14; p < 0.05), suggesting a presynaptic decrease in transmitter release probability. These data suggest the presence of both presynaptic and postsynaptic D1/5 receptors at the putative BLA–mPFC synapse and validate results with GDPβS. Furthermore, coapplication of r/hCRF (50 nm) with SKF81297 (1, 10 μm) did not have a significant effect (p > 0.05) on PPR obtained with SKF alone (Fig. 4D), further corroborating GDPβS data, and supporting a postsynaptic location for CRF1 receptors.
Collectively, these results also indicate that activation of postsynaptic CRF1 receptors enhanced EPSC depression mediated by presynaptic and postsynaptic D1/5 receptors at the putative BLA–mPFC synapse in saline-treated control rats.
Experiments in chronic cocaine-treated rats
Chronic cocaine administration modified the passive membrane electrical properties of mPFC–layer V pyramidal neurons
Common membrane properties of 220 mPFC–layer V pyramidal neurons recorded from slices of 111 saline/control and 109 cocaine-treated rats are shown in Table 1. Compared with mPFC–layer V pyramidal neurons from saline-treated control rats, neurons recorded from chronic cocaine-treated rats exhibited more depolarized resting membrane potentials (p < 0.01), higher membrane resistances (Rm; p < 0.05), and slower membrane time constants (τ; p < 0.01). Mixed ANOVAs yielded no significant differences (F(1,38) = 0.96; p > 0.05; n = 40) in the current–voltage relationship between groups (data not shown).
Table 1.
Comparison of membrane electrical properties of layer V pyramidal neurons of the PL cortex recorded intracellularly from chronic cocaine- and saline-treated/control rats
| Saline/control group |
Cocaine-treated group |
||||
|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | pa | |
| RMP (mV) | −65.9 | 0.3 | −62.3 | 1.2 | <0.01 |
| Rm (MΩ) | 80.3 | 2.8 | 87.0 | 3.3 | <0.05 |
| τ (ms) | 6.7 | 0.2 | 7.4 | 0.2 | <0.01 |
| n | 111 | 109 | |||
RMP, Resting membrane potential; Rm, membrane resistance; τ, membrane time constant.
aWald-Wolfowltz test.
Evoked putative BLA–mPFC EPSCs were depressed and spontaneous mEPSC frequency but not amplitude was reduced in chronic cocaine-treated rats
We examined possible changes in EPSCs from brains of chronic cocaine-treated rats compared with saline-treated controls. The input–output relationship that measured the amplitude of EPSCs as a function of stimulus intensity was shifted downward and to the right in slices from chronic cocaine-treated rats (Fig. 5A), indicating that EPSCs were significantly depressed (from a maximum value at 12 V stimulus of 254 ± 17.0 pA in saline-treated/controls to 157 ± 11.1 pA in chronic cocaine-treated rats, a 55% reduction). EPSCs were reduced significantly (treatment × stimulus intensity, F(4,40) = 11.2; p < 0.01; chronic cocaine effect, F(1,8) = 41.0; p < 0.01; stimulus intensity effect, F(5,40) = 172.5; p < 0.01; n = 10) in chronic cocaine-treated rats (−7.8 ± 1.0 pA at 2 V; −26.2 ± 10.2 pA at 4 V; −85.2 ± 14 pA at 6 V; −100.4 ± 11.7 pA at 8 V; −134.2 ± 7.2 pA at 10 V; −157.4 ± 11.1 pA at 12 V) compared with saline-treated rats (−7.3 ± 3.6 pA at 2 V; −48.2 ± 5.5 pA at 4 V; −125.8 ± 6.1 pA at 6 V; −184.2 ± 9.1 pA at 8 V; −220.7 ± 9.0 pA at 10 V; −253.7 ± 17.0 pA at 12 V).
Figure 5.

Chronic cocaine administration affected presynaptic mechanisms to depress putative BLA–mPFC EPSCs and mEPSC frequency but not mEPSC amplitude. A, Left, Representative traces of EPSCs evoked at different stimulus intensities from saline-treated rats (top traces) and chronic cocaine-treated rats (bottom traces). Stimulation parameters noted above. Right, EPSC amplitude (in picoamperes) plotted as a function of stimulus intensity (in volts) for each treatment group. EPSCs were significantly reduced in chronic cocaine-treated rats compared with saline-treated rats. B, Representative traces of EPSCs evoked by pairs of stimuli at different interstimulus intervals in saline-treated (top traces) and chronic cocaine-treated rats (bottom traces). Right, Paired-pulse ratios (PPR = EPSC2/EPSC1) plotted as a function of interstimulus interval. PPR values were significantly increased in slices from chronic cocaine-treated rats compared with saline-treated rats. C, Representative records of mEPSC activity after TTX (1 μm) recorded from mPFC–layer V pyramidal neurons from saline-treated (left traces) and chronic cocaine-treated (right traces) rats. D, Cumulative fraction plots for mEPSC amplitude and interevent intervals calculated from a representative single recording with summary bar graphs. Note that mEPSCs were only included if they exceeded the preset baseline/noise level (5 pA). After chronic cocaine, mEPSP analyses showed a significant reduction in frequency, but not amplitude.
Additionally, PPR values in slices from chronic cocaine-treated rats were increased significantly compared with saline/controls (Fig. 5B), suggesting a decrease in glutamate release at the putative BLA–mPFC synapse after chronic cocaine administration. Paired-pulse ratios (PPR = EPSC2/EPSC1) were plotted as a function of interstimulus interval (Fig. 5B, right). PPR values were significantly increased (treatment × time interval, F(2,42) = 17.0; p < 0.01; chronic cocaine effect, F(1,25) = 11.6; p < 0.01; time interval effect, F(2,42) = 92.9; p < 0.05; n = 27) in slices from chronic cocaine-treated rats compared with saline-treated rats (1.3 ± 0.03 vs 1.6 ± 0.02 at 50 ms; p < 0.01; 1.2 ± 0.03 vs 1.3 ± 0.03 at 100 ms; p = 0.06; 1.1 ± 0.03 vs 1.1 ± 0.05 at 200 ms; p > 0.05).
We also measured mEPSCs from mPFC–layer V pyramidal neurons in slices from chronic cocaine-treated rats and saline-treated rats in the presence of TTX (1 μm). Consistent with a decrease in evoked glutamate release, analyses of mEPSCs revealed a significant reduction of mEPSC frequency with no change in mEPSC amplitude in chronic cocaine-treated rats compared with saline-treated rats (Fig. 5C,D). After chronic cocaine, mEPSP analyses showed a significant reduction in frequency, but not amplitude (mEPSC amplitude = 15.9 ± 1.2 pA for saline-treated/controls; 14.8 ± 1.1 pA for cocaine-treated rats, t = 0.67; p > 0.05; n = 10; mEPSC interevent interval = 3571 ± 545 ms for saline-treated/controls; 6706 ± 576 ms for cocaine-treated rats; t = −3.94; p < 0.01; n = 10).
In summary, the decrease in PPR values and mEPSC analyses suggested a reduction of glutamate release probability at the putative BLA–mPFC synapse in chronic cocaine-treated rats compared with saline-treated rats.
Activation of D1/5 or CRF2 receptors increased putative BLA–mPFC EPSCs in chronic cocaine-treated rats
Subsequently, the D1/5 receptor-mediated modulation of EPSCs was analyzed in chronic cocaine-treated rats. As shown in Figure 6A, SKF81297 (10 μm) significantly increased EPSCs (114.1 ± 2.5%) (F(4,40) = 15.3; p < 0.01). Lower concentrations of SKF (10 nm, 100 nm, or 1 μm) did not affect EPSCs in chronic cocaine-treated rats, effects not significantly different from those observed in saline-treated/control rats (10 nm, 99.1 ± 1.3%; p > 0.05; 100 nm, 99.4 ± 1.7%; p > 0.05; 1 μm, 102.7 ± 2.3%; p > 0.05). This facilitatory effect was directly opposite to that observed in saline-treated/control animals, in which SKF81297 (10 μm) depressed EPSCs (compare Fig. 2) (p < 0.01). SKF81297 (1 μm) did not increase EPSCs significantly in chronic cocaine-treated rats compared with baseline. However, EPSC values were significantly (p < 0.01) different from those observed with an equivalent concentration of SKF81297 (1 μm) in saline-treated/control rats. Consistent with a D1/5 receptor-mediated effect, SCH23390 (10 μm), a selective D1/5 receptor antagonist, blocked completely the effects of SKF81297 (10 μm) (Fig. 6C) (SKF effect in the presence of SCH, F(3,12) = 1.01; p > 0.05). Also, SCH23390 (10 μm), itself, did not significantly affect EPSCs, suggesting that tonic activation of D1/5 receptors by endogenous DA could not account for changes in EPSCs in chronic cocaine-treated rats.
Figure 6.
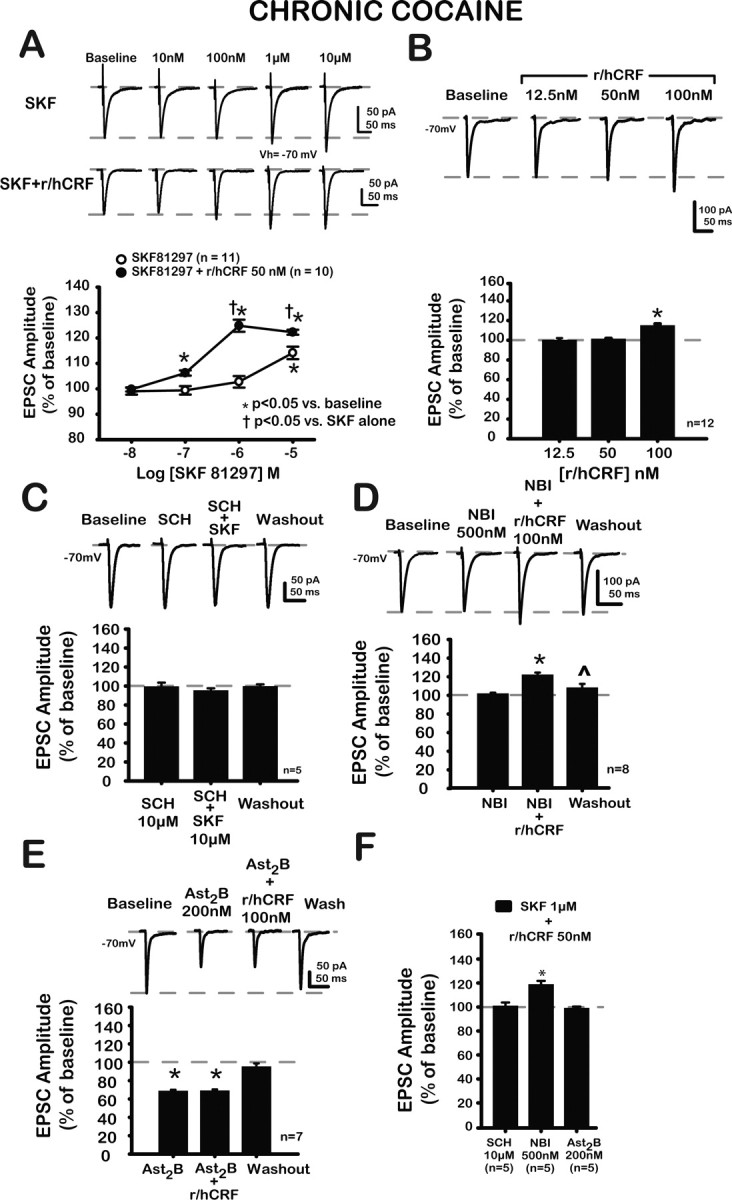
Actions of SKF alone, in combination with r/hCRF, with r/hCRF alone, and with antagonists were altered after chronic cocaine administration. A, Representative traces of EPSCs at baseline, after increasing concentrations of SKF (top traces), and SKF plus r/hCRF (50 nm; bottom traces). Bottom, Concentration–response curves for each treatment group. SKF (10 μm) significantly increased EPSCs (114.1 ± 2.5%). Lower concentrations of SKF did not affect EPSCs, indicating that r/hCRF (50 nm) synergistically enhanced SKF effects. B, Representative traces of EPSCs at baseline and after r/hCRF (12.5, 50, 100 nm). R/hCRF (100 nm) significantly increased EPSC amplitude (115.0 ± 1.2%; p < 0.01). Lower concentrations of r/hCRF (12.5, 50 nm) did not have a significant effect on EPSC amplitude (12.5 nm, 99.3 ± 1.3%; p > 0.05; 100.9 ± 1.1%; p > 0.05). C, Representative traces of EPSCs at baseline, after SCH (10 μm), after SCH (10 μm) plus SKF (10 μm), and after 20 min washout. Summary data are shown in bar graph below. SCH (10 μm) did not have a significant effect on EPSCs (99.4 ± 4.2%; p > 0.05). SCH (10 μm) blocked the effects of SKF (10 μm, 95.3 ± 2.4% after SCH plus SKF; p > 0.05; 99.6 ± 2.0% after washout; p > 0.05). D, Representative traces of EPSCs at baseline after NBI (500 nm), after r/hCRF (100 nm) plus NBI (500 nm), and after 20 min washout. Summary data are shown in bar graph below. NBI (500 nm) did not affect EPSCs (101.9 ± 0.9%; paired t = −2.02; p > 0.05), and NBI (500 nm) did not block the effects of r/hCRF (100 nm; r/hCRF effect, F(3,21) = 58.99; p < 0.01) on EPSCs (122.1 ± 1.7% after 500 nm NBI plus 100 nm r/hCRF; p < 0.01; 108.3 ± 2.3% after washout; p > 0.05). E, Representative traces of EPSCs at baseline, after Ast2B (200 nm), after r/hCRF (100 nm) plus Ast2B, and after 20 min washout. Summary data are illustrated in bar graph below. Ast2B itself depressed EPSCs (68.8 ± 1.2%; Ast2B effect, F(3,18) = 144.4; p < 0.01) and blocked r/hCRF (100 nm) effects on EPSCs (Ast2B plus r/hCRF, 69.1 ± 0.1%; p > 0.05 compared with Ast2B alone). F, Bar graph summarizes the effects of selective antagonists for D1/5 (SCH, 10 μm), CRF1 (NBI, 500 nm), and CRF2 (Ast2B, 200 nm) receptors on the effects of combined agonists. Ast2B and SCH blocked the effects of the combined agonists on EPSCs (SCH plus r/hCRF plus SKF, 100.8 ± 2.7%; paired t = 0.30; p > 0.05; Ast2B plus r/hCRF plus SKF, 98.9 ± 1.3%; paired t = 0.85; p > 0.05). NBI (500 nm) was unable to block the effects of the combined agonists (118.5 ± 2.9%; p < 0.05 compared with baseline; t = 1.6; p > 0.05 compared with SKF plus r/hCRF). *p < 0.05 versus baseline.
R/hCRF (100 nm) increased putative BLA–mPFC EPSCs (Fig. 6B), an action not present in saline-treated/controls. EPSC amplitudes after r/hCRF (12.5, 50 nm) were not significantly different (p > 0.05) from those obtained after r/hCRF (12.5, 50 nm) in saline-treated/controls. These data indicated that in chronic cocaine-treated rats, but not in saline-treated controls, r/hCRF itself increased EPSCs.
To characterize the type of CRF receptor mediating r/hCRF-induced increases in EPSCs, we tested r/hCRF (100 nm) with selective antagonists for CRF1 and CRF2 receptors, NBI30775 and Ast2B, respectively. As shown in Figure 6D, NBI30775 (500 nm) did not block the effects of r/hCRF (100 nm) on EPSCs. In addition, NBI30775 (500 nm) by itself did not affect EPSC amplitude. Together these data suggested that CRF1 receptors neither mediated r/hCRF (100 nm) effects on EPSCs, nor were activated by endogenous CRF in chronic cocaine-treated rats. Conversely, Ast2B itself significantly depressed EPSCs and blocked the effect of r/hCRF (100 nm) on EPSCs (Fig. 6E). Thus, our data demonstrated that activation of CRF2 receptors by r/hCRF applied exogenously or by endogenously released CRF-related peptides facilitated EPSCs in chronic cocaine-treated rats.
r/hCRF-facilitated D1/5 receptor-mediated increase of putative BLA–mPFC EPSCs was synergistically increased when combined with r/hCRF
Based on our results (Fig. 2A) showing that DA-mediated depression of mPFC EPSCs was synergistically enhanced when DA was combined with r/hCRF in saline-treated/controls, we also tested for modulatory effects of D1/5 receptor ligands combined with r/hCRF after chronic cocaine. R/hCRF (50 nm) plus SKF (100 nm or higher concentrations) further enhanced EPSCs (10 nm, 99.8 ± 0.1%; p > 0.05; 100 nm, 106.2 ± 1.1%; p < 0.05; 1 μm, 124.8 ± 2.3%; p < 0.05; 10 μm, 122.3 ± 0.1%; p < 0.05), indicating that r/hCRF (50 nm) synergistically enhanced SKF effects (r/hCRF × SKF concentration effect, F(4,76) = 18.7; p < 0.01; r/hCRF effect, F(1,19) = 35.0; p < 0.01; SKF concentration effect, F(4,76) = 70.2; p < 0.01; n = 21). As shown in Figure 6A, r/hCRF shifted the concentration–response for SKF81297 upward and to the left (EC50 = 183.6 nm compared with 6.6 μm without r/hCRF), resulting in a 22.3% facilitation after SKF81297 (10 μm) plus r/hCRF (50 nm) compared with 14.1% facilitation after SKF81297 (10 μm), suggesting that r/hCRF (50 nm) synergistically enhanced the SKF81297-induced increases of EPSCs in chronic cocaine-treated rats.
We examined the subtypes of DA and CRF receptors involved in the synergistic facilitatory effect of r/hCRF and SKF81297 by testing the combined effects of SKF81297 (1 μm) plus r/hCRF (50 nm) on EPSCs in the presence of selective antagonists for D1/5, CRF1, and CRF2 receptors (Fig. 6 C–F). The concentrations (1 μm SKF81297 plus 50 nm r/hCRF) used to probe for DA and CRF receptor interactions were chosen because each drug lacked significant effects on EPSCs at these concentrations either in saline-treated controls or chronic cocaine-treated rats when tested independently. Thus, if present, changes in EPSCs after SKF81297 (1 μm) plus r/hCRF (50 nm) would likely result from the synergistic action of CRF and DA receptors and their associated mechanisms, rather than from a summation of their individual effects.
As shown in Figure 6F, SCH23390 and Ast2B, but not NBI30775, blocked completely the effects of SKF81297 (1 μm) plus r/hCRF (50 nm) on EPSCs. These data suggested that after chronic cocaine, CRF2 and D1/5 receptors mediated the synergistic potentiation of EPSCs by SKF81297 (1 μm) plus r/hCRF (50 nm) in chronic cocaine-treated rats. In addition, CRF1 receptors do not contribute to the effects of SKF81297 (1 μm) plus r/hCRF (50 nm) in chronic cocaine-treated rats. Thus, based on our findings, there is an apparent loss of function for CRF1 receptors in the modulation of EPSCs after chronic cocaine administration. A similar result was obtained at the lateral septum mediolateral nucleus (LSMLN) synapse after chronic cocaine administration (Liu et al., 2005).
In chronic cocaine-treated rats, DA and r/hCRF synergistically enhanced putative BLA–mPFC EPSCs in the presence of a D2/3 receptor antagonist
To confirm our findings with SKF81297 and r/hCRF, we tested the endogenous ligand DA (10 μm) in combination with r/hCRF, in the presence of raclopride (10 μm), a selective D2/3 receptor antagonist (Fig. 7). In slices from cocaine-treated rats, exogenous application of raclopride (10 μm), itself, resulted in an increase of EPSCs, possibly because of the unmasking of a tonic depressant action of D2/3 receptors by an elevated endogenous DA (Fig. 7A).
This tonic endogenous depressant effect of D2/3 receptors on EPSCs unmasked by raclopride (raclopride, 131.3 ± 2.6; p < 0.05; after washout, 122.7 ± 8.3; p > 0.05; F(2,6) = 13.46; p < 0.05) in chronic cocaine-treated rats was not blocked by loading pyramidal neurons with GDPβS (data not shown), suggesting a presynaptic location for tonically active D2/3 receptors in chronic cocaine-treated animals. After raclopride (10 μm), exogenous application of DA 10 μm further increased EPSCs in a significant and reversible manner (DA effect, F(2,8) = 10.2; p < 0.01). In addition, coapplication of DA (10 μm) with r/hCRF (50 nm) enhanced DA effects on EPSCs (DA-control, 12.2% increase; DA plus r/hCRF, 25.4% increase) (Fig. 7A,B) (DA plus r/hCRF, 125.4 ± 1.2%; p < 0.01; washout, 99.1 ± 3.4%; p > 0.05; r/hCRF × DA concentration, F(2,16) = 7.36; p < 0.01; r/hCRF effect, F(1,8) = 5.33; p < 0.01; DA effect, F(2,16) = 55.5; p < 0.01; n = 10). The increases in EPSC amplitude by DA alone and the combination of DA plus CRF occurred independent of any significant postsynaptic change as monitored continuously as input conductance (Fig. 7B). These data with DA and raclopride confirmed our findings with SKF81297 and demonstrated that r/hCRF acted synergistically to enhance D1/5 receptor-mediated facilitating effects on EPSCs in chronic cocaine-treated rats.
CRF2 and D1/5 receptors mediating the effects of r/hCRF and DA ligands in chronic cocaine-treated rats are located presynaptically
To determine the location of CRF2 and D1/5 receptors at the putative BLA–mPFC synapse after treatment with chronic cocaine, voltage-clamped pyramidal neurons were loaded with GDPβS (1 mm) before testing the effects of r/hCRF, SKF81297, or the combination on EPSCs. The effectiveness of GDPβS was assayed as described previously (Fig. 3A). GDPβS (1 mm) did not block the effects of r/hCRF (100 nm; 120.1 ± 1.45%; paired t = −13.13; p < 0.01; n = 5), SKF (10 μm; 119.9 ± 1.5%; paired t = −13.11; p < 0.01; n = 5), or the combination (117.3 ± 2.8%; paired t = −6.2; p < 0.01; n = 5) on EPSCs. EPSCs from neurons loaded with GDPβS were not different from those obtained from neurons without GDPβS (100 nm r/hCRF, t = 2.40; p > 0.05; 10 μm SKF, t = −1.49; p > 0.05; the combination, t = −1.43; p > 0.05) (Fig. 8A).
Figure 8.
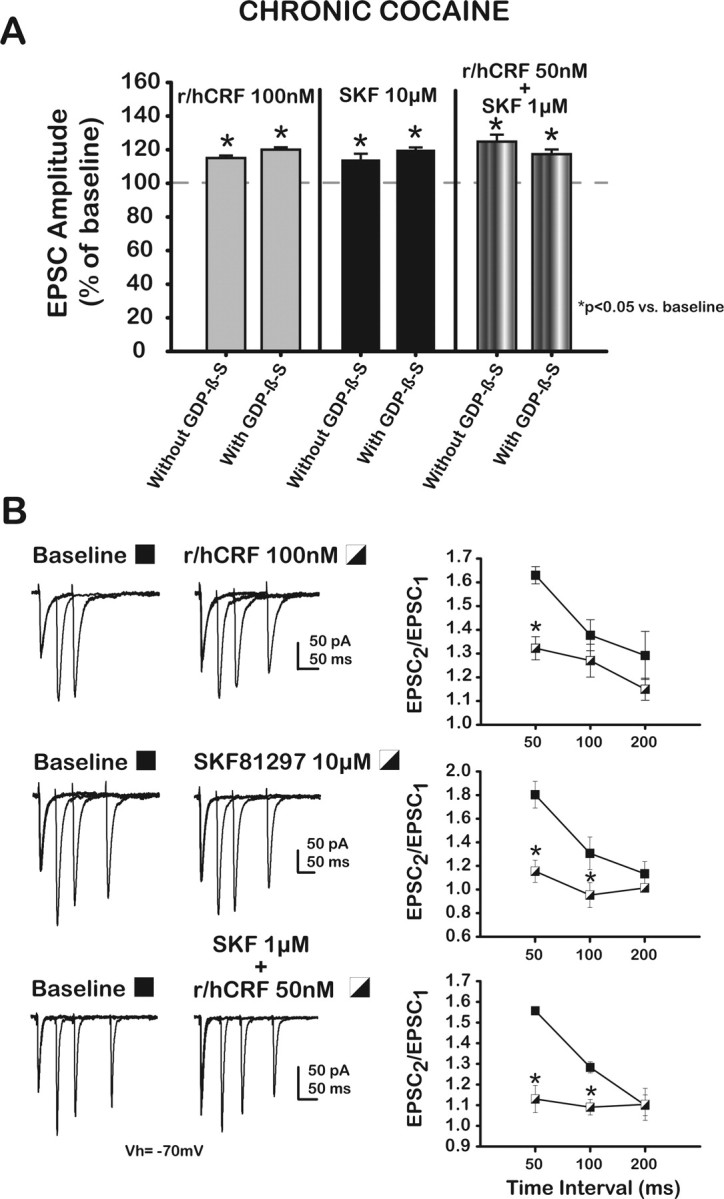
Presynaptic CRF2 and D1/5 receptors interact to increase putative BLA–mPFC EPSCs in chronic cocaine-treated rats. A, Bar graph summarizes the effects of r/hCRF (100 nm), SKF (10 μm), and a combination (50 nm r/hCRF plus 1 μm SKF) on EPSCs recorded from mPFC–layer V pyramidal neurons loaded with GDPβS (1 mm) from chronic cocaine-treated rats. Data from neurons without GDPβS are included for comparison. Vertical solid lines divide the data for each drug-treatment group. GDPβS (1 mm) did not block the effects of the individual ligands or their combination. B, Representative traces of EPSCs evoked by pairs of stimuli at different interstimulus intervals at baseline (left column) and after r/hCRF (100 nm; right column, top traces), SKF (10 μm; right column, middle traces), and the combination (50 nm r/hCRF plus 1 μm SKF). Right, Paired-pulse ratios (PPR = EPSC2/EPSC1) plotted as a function of interstimulus interval for each treatment group. PPR values were decreased significantly by r/hCRF (100 nm) compared with baseline only at the shortest interval (50 ms).
To validate these findings, we conducted paired-pulse experiments (50, 100, and 200 ms interstimulus intervals) (Fig. 8B). R/hCRF (100 nm), SKF81297 (10 μm), and the combination 50 nm r/hCRF plus 1 μm SKF81297 consistently decreased PPR values compared with baseline values at 50 ms (1.63 ± 0.0.04 vs 1.32 ± 0.05; p < 0.01). Less consistent were the effects of these compounds on PPR values measured at 100 and 200 ms interstimulus intervals (Fig. 8B) (100 ms, 1.38 ± 0.07 vs 1.27 ± 0.07; p > 0.05; 200 ms, 1.29 ± 0.1 vs 1.15 ± 0.05; p > 0.05; r/hCRF effect, F(1,4) = 64.44; p < 0.01; n = 5). PPR values were decreased significantly by SKF (10 μm) compared with baseline only at the two shorter intervals (50 ms, 1.80 ± 0.11 vs 1.15 ± 0.09; p < 0.05; 100 ms, 1.31 ± 0.14 vs 0.95 ± 0.11; p < 0.05; 200 ms, 1.13 ± 0.1 vs 1.01 ± 0.04; p > 0.05; SKF effect, F(1,4) = 215.82; p < 0.01; n = 5). PPR with the combination 50 nm r/hCRF plus 1 μm SKF was similarly reduced when compared with baseline (50 ms, 1.56 ± 0.02 vs 1.13 ± 0.07; p < 0.01; 100 ms, 1.28 ± 0.03 vs 1.09 ± 0.04; p < 0.05; 200 ms, 1.10 ± 0.05 vs 1.10 ± 0.08; p > 0.05; 50 nm r/hCRF plus 1 μm SKF effect, F(1,4) = 96.05; p < 0.01; n = 5). *p < 0.05 versus baseline.
These results suggested that CRF2 and D1/5 receptors are located presynaptically in chronic cocaine-treated rats and differed from those obtained in saline-treated controls (Fig. 1).
In addition, comparison of data, not shown but collected after chronic cocaine administration, demonstrated that EPSCs recorded in the presence of raclopride are still significantly depressed (149.4 ± 6.5 pA; p < 0.003) relative to EPSCs recorded from saline-treated neurons (205.8 ± 12.9 pA).
Similarly, EPSCs recorded in the presence of Astressin2B from neurons collected after chronic cocaine administration are depressed significantly (143.7 ± 14.9 pA; p < 0.04) relative to EPSCs recorded from saline-treated neurons (210.1 ± 24.7). These results support the concept of a chronic cocaine-induced tonic regulation of EPSCs by CRF2 and D2-like receptors, respectively, after chronic cocaine administration (Fig. 9).
Discussion
Experiments in saline-treated/control rats
We provide evidence for an interaction between DA and r/hCRF on glutamatergic synaptic transmission at a critical synapse carrying input from the basolateral amygdala to the prelimbic prefrontal cortex. The results demonstrated a novel process through which increased CRF, as might occur after a stressor, potentiates an ongoing modulation of excitatory transmission by DA. Specifically, we showed that a concentration-dependent D1/5 receptor activation depressed EPSCs (Fig. 1) and localized these receptors with GDPβS and PPR analyses (Figs. 3, 9) to both presynaptic and postsynaptic sites. Although demonstrating similar concentration sensitivity, these results differ from Onn et al. (2006), who report that D1/5 receptor activation facilitated corpus callosum afferent to type I mPFC (including the anterior cingulate, Cg3 area, and prelimbic cortex) pyramidal neuron EPSCs. Our findings that DA or SKF81297 depressed mPFC EPSCs are consistent with previous reports of presynaptic D1/5 receptors in the mPFC (Law-Tho et al., 1994; Momiyama et al., 1996; Gao et al., 2001; Urban et al., 2002; Paspalas and Goldman-Rakic, 2004; Mair and Kauer, 2006), and also support a postsynaptic locus for D1/5 receptors. D1/5 receptors enhance NMDA EPSCs (Zheng et al., 1999; Seamans et al., 2001a; Wang and O'Donnell, 2001; Tseng and O'Donnell, 2004), while having only a small or no effect on non-NMDA EPSCs. Because putative BLA–mPFC EPSCs lack a significant NMDA component (Orozco-Cabal et al., 2006a), our results indicated a significant D1/5 effect mediated through non-NMDA receptors. R/hCRF (≤100 nm) (Fig. 1C) alone did not have a significant effect on EPSCs, consistent with results in ventral tegmental DA neurons, in which r/hCRF did not affect non-NMDA EPSCs (Ungless et al., 2003). However, r/hCRF (50 nm) acted through postsynaptic CRF1 receptors (Figs. 2, 3, 9) to enhance EPSC depression induced by coapplied D1/5 receptor agonists. Synergistic EPSC depression by a combined activation of postsynaptic CRF1 and D1/5 receptors has not been reported previously but may be caused by activation and retrograde release of substances acting presynaptically and/or the modulation of an adjacent, third receptor type, e.g., metabotropic glutamate receptor (Ohno-Shosaku et al., 2002) or cannabinoid receptor (Melis et al., 2004).
Our data also demonstrated that r/hCRF alone or in combination with D1/5 receptor agonists does not affect GABAA receptor-mediated IPSCs in slices from either saline- or cocaine-treated animals (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
The concentrations of r/hCRF used in these studies are within physiological (endogenous) ranges. Based on our previous studies demonstrating significant effects of r/hCRF on EPSCs in limbic structures related to the PFC (Liu et al., 2004; Pollandt et al., 2006), concentrations of r/hCRF <100 nm were used here. Low nanomolar concentrations of r/hCRF closely mimic CRF concentrations (0.3 nm) reported for the amygdala after acute withdrawal from chronic cocaine administration (Richter et al., 1995; Merlo et al., 1995; Richter and Weiss, 1999). Also, comparable endogenous levels of CRF outside the hypothalamic–pituitary–adrenal axis have been reported in human plasma and CSF, respectively (Catalan et al., 1998; Baker et al., 1999).
Experiments in chronic cocaine-treated rats
Electrical and synaptic properties of mPFC–layer V pyramidal neurons
Electrical membrane properties at layer V prelimbic pyramidal neurons were different in chronic cocaine-treated rats compared with saline-treated controls (Table 1). The combination of depolarized membrane potential with increased input resistance would provide a more excitable state for these neurons. Our findings of increased input resistance values are consistent with previous reports for mPFC–layer V/VI pyramidal neurons after repeated cocaine administration despite differences in cocaine regimens, withdrawal periods, and recording techniques (Trantham et al., 2002; Nasif et al., 2005a,b).
EPSCs in the chronic cocaine-treated group were depressed compared with saline-treated controls (Fig. 5A). Paired-pulse experiments (Fig. 5B) and analyses of mEPSCs (Fig. 5C,D) suggested that EPSC depression was attributable to a reduction in glutamate release at BLA terminals. This reduction could result from cocaine-induced changes in modulatory systems regulating glutamate release at BLA terminals [e.g., autoreceptors (mGluRs) (Martin et al., 2007), heteroreceptors (DA) (Chen et al., 2007), associated signaling mechanisms (Bergson et al., 2003), or ligands (adenosine) (Steketee, 2005; Ladera et al., 2007)]. The finding (Fig. 7A) that presynaptic D2/3 receptors exert a tonic inhibitory effect on EPSCs suggests that this mechanism may contribute to depressed EPSCs in the chronic cocaine group.
In human cocaine addicts or animals exposed to cocaine, several studies using gambling tasks and reversal learning paradigms show that outcome-dependent decision processes are defective (Bechara and Damasio, 2002; Schoenbaum et al., 2006). Stalnaker et al. (2007) demonstrated that these deficits relate to persistent encoding of outdated associative information in the BLA. Our findings suggest that synaptic depression between the BLA and prelimbic pyramidal neurons might also contribute to abnormal processing of drug-related information leading to biased decision-making processes. Synaptic depression may reduce the dynamic range of BLA-to-mPFC synapse, thus limiting information processing. In fact, this hypothesis is in agreement with recent observations of congruent and abnormal metabolic activity after chronic cocaine administration in the PFC and temporal lobe structures such as the amygdala using a nonhuman primate cocaine-self administration model (Beveridge et al., 2006). The subsequent reversal in polarity of the actions of DA and CRF after chronic cocaine may be an adaptive process to compensate for the synaptic depression associated with chronic cocaine administration.
Modulatory effect of CRF and DA receptors on putative BLA–mPFC EPSCs
Exogenous r/hCRF (Fig. 6B,D) or endogenous CRF-related peptides (after Ast2B) (Fig. 6E) increased EPSCs in chronic cocaine-treated rats via CRF2 receptors (not detected in recordings from saline-treated/control slices). Unmasking of this presynaptic CRF2 receptor facilitatory function after chronic cocaine was similar to the previously reported occurrence of a CRF2 receptor-mediated facilitation at the LSMLN after chronic cocaine (Liu et al., 2005). Our findings do not support a role for postsynaptic CRF1 receptors in the modulation of EPSCs by r/hCRF alone or combined with DA receptor ligands in chronic cocaine-treated rats. This loss of function for postsynaptic CRF1 receptors in mPFC neurons after chronic cocaine is an illustration of the dynamic changes in CRF systems in response to chronic cocaine administration (cf. Liu et al., 2005). After chronic cocaine administration, a prominent PKA signaling process evident in saline-treated/control slices was reduced with a dominance of PKC signaling (Liu et al., 2005).
Importantly, stress levels have been shown to modulate the behavioral effects of CRF2 receptor activation (Henry et al., 2006), suggesting that “emotional states” may influence the function of CRF systems in the brain. We propose that as endogenous CRF concentrations change as a result of various stressors (e.g., mental disorders, including cocaine addiction), modulation of synaptic transmission by CRF receptors will be affected (Orozco-Cabal et al., 2006b; Gallagher et al., 2008). For example, unlike in saline-treated rats, postsynaptic CRF1 receptors did not play a role in the modulation of EPSCs by r/hCRF alone or combined with DA receptor ligands in chronic cocaine-treated rats.
The changes in CRF receptor modulation affecting putative BLA–mPFC EPSCs may be related to adaptations initiated during chronic cocaine administration, such as persistently elevated CRF levels. For example, CRF-like immunoreactivity is increased in the PFC during protracted withdrawal from cocaine-binge sessions in rats trained to self-administer cocaine, suggesting that delayed increases in CRF-like immunoreactivity “may reflect a subsequent failure to shut down compensatory CRF-mRNA synthesis” (Zorrilla et al., 2001). Their studies suggested that a massive overflow of CRF in the PFC could lead to CRF receptor changes, including upregulation of CRF2 and downregulation of CRF1 receptors.
In our studies, activation of presynaptic D1/5 receptors increased EPSCs in chronic cocaine-treated rats (Fig. 6), and this facilitatory effect was enhanced by activating presynaptic CRF2 receptors. Also, SKF81297, r/hCRF, and the combination decreased PPR values, suggesting that presynaptic D1/5 as well as CRF2 receptors increased glutamate release from BLA terminals. Interestingly, the effects of the combined agonists could be blocked completely by either SCH23390 or Ast2B, suggesting that coactivation of D1/5 and CRF2 receptors was required to increase EPSCs at these concentrations. These findings suggest that synergism between CRF2 and D1/5 receptors at BLA terminals may arise via one or more mechanisms, e.g., obligate cooperativity between two different receptors (Levenson, 2006), activation of one or the other receptors by a heterodimer of DA (Franco et al., 2007) and CRF (Milan-Lobo et al., 2006), involvement of common signaling pathways (Hillhouse and Grammatopoulos, 2006; Tseng and O'Donnell, 2004), etc. Importantly, these data also show that D1/5 receptor modulatory effects are changed in polarity from inhibitory (saline/controls) to facilitatory (chronic cocaine) at this synapse.
Postsynaptic D1/5 receptors did not appear to play a significant role in the modulation of putative BLA–mPFC EPSC in chronic cocaine-treated rats, whereas in saline-treated/control rats, an interaction between postsynaptic CRF1 and D1/5 receptors was evident. The lack of postsynaptic D1/5 receptor-mediated effects after chronic cocaine may be related to CRF1 and/or D1/5 receptor dysfunction. Alternatively, these observations may result from a decrease in overall function of D1/5 receptors after chronic cocaine administration. Stanwood and Levitt (2007) reported that prenatal exposure to cocaine resulted in substantially reduced D1-like receptor coupling to G(s)-protein, and D1-like receptor responsiveness was related to a reduced cell-surface localization of D1-like receptors. In addition, depressed D1/5 receptor function in the dorsomedial PFC has been suggested to explain the cocaine sensitization in rats after repeated administration of cocaine (Sorg et al., 1997), an effect that may contribute to the observed increase in behavioral responses (Ragozzino, 2002).
In summary, our results suggest that coactivation of CRF and DA receptors modulate putative BLA–mPFC EPSCs (Fig. 9). In saline-treated controls, CRF and DA receptor activation produced a net inhibitory effect on EPSCs. However, in chronic cocaine-treated rats, CRF and DA receptor activation caused the opposite effect (increased EPSCs). Interestingly, the enhancing effects of presynaptic D1/5 and CRF2 receptors coincide with a depression of EPSCs associated with chronic cocaine administration, per se. Perhaps, changes in CRF and DA function reflect compensatory processes at the synaptic level as an attempt to sustain normal communication during a stressful state.
Footnotes
This work was supported by Grants DA011991, DA017727, MH066996, MH058327, and the Center for Addiction Research at the University of Texas Medical Branch. We appreciate constructive discussion and review by Drs. G. Swanson and M. Joëls.
References
- Ambrosio E, Sharpe LG, Pilotte NS. Regional binding to corticotropin releasing factor receptors in brain of rats exposed to chronic cocaine and cocaine withdrawal. Synapse. 1997;25:272–276. doi: 10.1002/(SICI)1098-2396(199703)25:3<272::AID-SYN6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Andrade R, Malenka RC, Nicoll RA. A G protein couples serotonin and GABAB receptors to the same channels in hippocampus. Science. 1986;234:1261–1265. doi: 10.1126/science.2430334. [DOI] [PubMed] [Google Scholar]
- Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD., Jr Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry. 1999;156:585–588. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H. Decision-making and addiction (part 1): impaired activation of somatic states in substance dependent individuals when pondering decisions with negative future consequences. Neuropsychologica. 2002;40:1675–1689. doi: 10.1016/s0028-3932(02)00015-5. [DOI] [PubMed] [Google Scholar]
- Ben-Shahar O, Keeley P, Cook M, Brake W, Joyce M, Nyffeler M, Heston R, Ettenberg A. Changes in levels of D1, D2, or NMDA receptors during withdrawal from brief or extended daily access to IV cocaine. Brain Res. 2007;1131:220–228. doi: 10.1016/j.brainres.2006.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergson G, Levenson R, Goldman-Rakic PS, Lidow MS. Dopamine-receptor interacting proteins: the Ca(2+) connection in dopamine signaling. Trends Pharmacol Sci. 2003;24:486–492. doi: 10.1016/S0165-6147(03)00232-3. [DOI] [PubMed] [Google Scholar]
- Berridge KC. The debate over dopamine's role in reward: the case for incentive salience. Psychopharmacology (Berl) 2007;191:391–431. doi: 10.1007/s00213-006-0578-x. [DOI] [PubMed] [Google Scholar]
- Beveridge TJR, Smith HR, Daunais JB, Nader MA, Porrino LJ. Chronic cocaine self-administration is associated with altered functional activity in the temporal lobes of non-human primates. Eur J Neurosci. 2006;23:3109–3118. doi: 10.1111/j.1460-9568.2006.04788.x. [DOI] [PubMed] [Google Scholar]
- Catalan R, Gallart JM, Castellanos JM, Galard R. Plasma corticotropin-releasing factor in depressive disorders. Biol Psychiatry. 1998;44:15–20. doi: 10.1016/s0006-3223(97)00539-8. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Radisavljevic Z, Peacock W, Levine MS, Buchwald NA. Differential modulation by dopamine of responses evoked by excitatory amino acids in human cortex. Synapse. 1992;11:330–341. doi: 10.1002/syn.890110408. [DOI] [PubMed] [Google Scholar]
- Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci USA. 2004;101:2596–2600. doi: 10.1073/pnas.0308618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Bohanick JD, Nishihara M, Seamans JK, Yang CR. Dopamine D1/5 receptor-mediated long-term potentiation of intrinsic excitability in rat prefrontal cortical neurons: Ca2+-dependent intracellular signaling. J Neurophysiol. 2007;97:2448–2464. doi: 10.1152/jn.00317.2006. [DOI] [PubMed] [Google Scholar]
- Dackis CA, Gold MS. Bromocriptine as treatment of cocaine abuse. Lancet. 1985;1:1151–1152. doi: 10.1016/s0140-6736(85)92448-1. [DOI] [PubMed] [Google Scholar]
- Dautzenberg FM, Hauger RL. The CRF peptide family and their receptors: yet more partners discovered. Trends Pharmacol Sci. 2002;23:71–77. doi: 10.1016/s0165-6147(02)01946-6. [DOI] [PubMed] [Google Scholar]
- De Martino B, Kumaran D, Seymour B, Dolan R. Frames, biases, and rational decision-making in the human brain. Science. 2006;313:694–687. doi: 10.1126/science.1128356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Tse MT. Dopaminergic regulation of inhibitory and excitatory transmission in the basolateral amygdala-prefrontal cortical pathway. J Neurosci. 2007;27:2045–2057. doi: 10.1523/JNEUROSCI.5474-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Lluis C, Canela EI, Mallol J, Agnatoi L, Casadó V, Ciruela F, Ferré S, Fuxe K. Receptor-receptor interactions involving adenosine A1 or dopamine D1 receptors and accessory proteins. J Neural Transm. 2007;114:93–104. doi: 10.1007/s00702-006-0566-7. [DOI] [PubMed] [Google Scholar]
- Gabbott PLA, Warner TA, Busby SJ. Amygdala input monosynaptically innervates parvalbumin immunoreactive local circuit neurons in rat medial prefrontal cortex. Neuroscience. 2006;139:1039–1048. doi: 10.1016/j.neuroscience.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Gallagher JP, Orozco-Cabal LF, Liu J, Shinnick-Gallagher P. Synaptic physiology of central CRH system. Eur J Pharmacol. 2008 doi: 10.1016/j.ejphar.2007.11.075. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Krimer LS, Goldman-Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci USA. 2001;98:295–300. doi: 10.1073/pnas.011524298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeders NE, Bienvenu OJ, De Sousa EB. Chronic cocaine administration alters corticotropin-releasing factor receptors in the rat brain. Brain Res. 1990;531:322–328. doi: 10.1016/0006-8993(90)90794-c. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos DK, Randeva H, Levine M, Kanellopoulou K, Hillhouse E. Rat cerebral cortex corticotropin-releasing hormone receptors: evidence for receptor coupling to multiple G-proteins. J Neurochem. 2001;76:509–519. doi: 10.1046/j.1471-4159.2001.00067.x. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, De Souza EB, Schulteis G, Lapsansky JL, Grigoriadis DE. Brain penetrance, receptor occupancy and antistress in vivo efficacy of a small molecule corticotropin releasing factor type I receptor selective antagonist. Neuropsychopharmacology. 2002;27:194–202. doi: 10.1016/S0893-133X(02)00299-3. [DOI] [PubMed] [Google Scholar]
- Henry B, Vale W, Markou A. The effect of lateral septum corticotropin-releasing factor receptor 2 activation on anxiety is modulated by stress. J Neurosci. 2006;26:9142–9152. doi: 10.1523/JNEUROSCI.1494-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillhouse EW, Grammatopoulos DK. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev. 2006;27:260–286. doi: 10.1210/er.2005-0034. [DOI] [PubMed] [Google Scholar]
- Hsu KS, Huang CC, Yang CH, Gean PW. Presynaptic D2 dopaminergic receptors mediate inhibition of excitatory synaptic transmission in rat neostriatum. Brain Res. 1995;690:264–268. doi: 10.1016/0006-8993(95)00734-8. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Koob GF. Stress, corticotropin-releasing factor, and drug addiction. Ann NY Acad Sci. 1999;897:445–460. doi: 10.1111/j.1749-6632.1999.tb07876.x. [DOI] [PubMed] [Google Scholar]
- Krettek JE, Price JL. Projections from the amygdaloid complex to the cerebral cortex and thalamus in the rat and cat. J Comp Neurol. 1977;172:687–721. doi: 10.1002/cne.901720408. [DOI] [PubMed] [Google Scholar]
- Krimer LS, Jakab RL, Goldman-Rakic PS. Quantitative three-dimensional analysis of the catecholaminergic innervation of identified neurons in the macaque prefrontal cortex. J Neurosci. 1997;17:7450–7461. doi: 10.1523/JNEUROSCI.17-19-07450.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladera C, Godino MD, Martin R, Luján R, Shigemoto R, Cirule F, Torres M, Sánchez-Prieto J. The coexistence of multiple receptors in a single nerve terminal provides evidence for pre-synaptic integration. J Neurochem. 2007;103:2314–2326. doi: 10.1111/j.1471-4159.2007.04964.x. [DOI] [PubMed] [Google Scholar]
- Law-Tho D, Hirsch JC, Crepel F. Dopamine modulation of synaptic transmission in rat prefrontal cortex: an in vitro electrophysiological study. Neurosci Res. 1994;21:151–160. doi: 10.1016/0168-0102(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Leshner AI. Understanding drug addiction: implications for treatment. Hosp Pract (Minneap) 1996;31:47–49. doi: 10.1080/21548331.1996.11443361. [DOI] [PubMed] [Google Scholar]
- Levenson CW. Regulation of the NMDA receptor: implications for neuropsychological development. Nutr Rev. 2006;64:428–432. doi: 10.1111/j.1753-4887.2006.tb00228.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Yu B, Neugebauer V, Grigoriadis DE, Rivier J, Vale WW, Shinnick-Gallagher P, Gallagher JP. Corticotropin-releasing factor and urocortin I modulate excitatory glutamatergic synaptic transmission. J Neurosci. 2004;24:4020–4029. doi: 10.1523/JNEUROSCI.5531-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yu B, Orozco-Cabal L, Grigoriadis DE, Rivier J, Vale WW, Shinnick-Gallagher P, Gallagher J. Chronic cocaine administration switches corticotropin-releasing factor 2 receptor-mediated depression to facilitation of glutamatergic transmission in the lateral septum. J Neurosci. 2005;25:577–583. doi: 10.1523/JNEUROSCI.4196-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair RD, Kauer JA. Amphetamine depresses excitatory synaptic transmission at prefrontal cortical layer V synapses. Neuropharmacology. 2006;52:193–199. doi: 10.1016/j.neuropharm.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Martin T, Torres M, Sanchez-Prieto J. mGluR7 inhibits glutamate release through a PKC-independent decrease in the activity of P/Q type Ca2+ channels and by diminishing cAMP in hippocampal nerve terminals. Eur J Neurosci. 2007;26:312–322. doi: 10.1111/j.1460-9568.2007.05660.x. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchenthaler I. Corticotropin releasing factor (CRF)-like immunoreactivity in the rat central nervous system. Extrahypothalamic distribution. Peptides. 1984;5(Suppl 1):53–69. doi: 10.1016/0196-9781(84)90265-1. [DOI] [PubMed] [Google Scholar]
- Merlo PE, Lorang M, Yeganeh M, Rodriguez de FF, Raber J, Koob GF, Weiss F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan-Lobo L, Ruentzler D, Bonci A, Freissmuth M, Sitte HH. Visualizing the membrane mobility of corticotrophin-releasing-factor-type 2 receptors (CRF2-R) by fluorescence microscopy. Soc Neurosci Abstr. 2006;32:726–9. [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Momiyama T, Sim JA, Brown DA. Dopamine D1-like receptor-mediated presynaptic inhibition of excitatory transmission onto rat magnocellular basal forebrain neurones. J Physiol (Lond) 1996;495:97–106. doi: 10.1113/jphysiol.1996.sp021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin SM, Ling N, Liu XJ, Kahl SD, Gehlert DR. Differential distribution of urocortin- and corticotropin-releasing factor-like immunoreactivities in the rat brain. Neuroscience. 1999;92:281–291. doi: 10.1016/s0306-4522(98)00732-5. [DOI] [PubMed] [Google Scholar]
- Nasif FJ, Hu XT, White FJ. Repeated cocaine administration increases voltage-sensitive calcium currents in response to membrane depolarization in medial prefrontal cortex pyramidal neurons. J Neurosci. 2005a;25:3674–3679. doi: 10.1523/JNEUROSCI.0010-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasif FJ, Sidiropoulou K, Hu XT, White FJ. Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J Pharmacol Exp Ther. 2005b;312:1305–1313. doi: 10.1124/jpet.104.075184. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M. Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci. 2002;15:953–961. doi: 10.1046/j.1460-9568.2002.01929.x. [DOI] [PubMed] [Google Scholar]
- Onn SP, Wang X-B, Lin M, Grace AA. Dopamine D1 and D4 receptor subtypes differentially modulate recurrent excitatory synapses in prefrontal cortical pyramidal neurons. Neuropsychopharmacology. 2006;31:318–338. doi: 10.1038/sj.npp.1300829. [DOI] [PubMed] [Google Scholar]
- Orozco-Cabal L, Pollandt S, Liu J, Vergara L, Shinnick-Gallagher P, Gallagher JP. A novel rat medial prefrontal cortical slice preparation to investigate synaptic transmission from amygdala to layer V prelimbic pyramidal neurons. J Neurosci Methods. 2006a;151:148–158. doi: 10.1016/j.jneumeth.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Orozco-Cabal L, Pollandt S, Liu J, Shinnick-Gallagher P, Gallagher JP. Regulation of synaptic transmission by CRF receptors. Rev Neurosci. 2006b;17:279–307. doi: 10.1515/revneuro.2006.17.3.279. [DOI] [PubMed] [Google Scholar]
- Paspalas CD, Goldman-Rakic PS. Microdomains for dopamine volume neurotransmission in primate prefrontal cortex. J Neurosci. 2004;24:5292–5300. doi: 10.1523/JNEUROSCI.0195-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paspalas CD, Goldman-Rakic PS. Presynaptic D1 dopamine receptors in primate prefrontal cortex: target-specific expression in the glutamatergic synapse. J Neurosci. 2005;25:1260–1267. doi: 10.1523/JNEUROSCI.3436-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain. Orlando, FL: Harcourt; 1998. [Google Scholar]
- Pollandt S, Liu J, Orozco-Cabal L, Grigoriadis DE, Vale WW, Gallagher JP, Shinnick-Gallagher P. Cocaine withdrawal enhances long-term potentiation induced by corticotropin-releasing factor at central amygdala glutamatergic synapses via CRF, NMDA receptors and PKA. Eur J Neurosci. 2006;24:1733–1743. doi: 10.1111/j.1460-9568.2006.05049.x. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME. The effects of dopamine D(1) receptor blockade in the prelimbic-infralimbic areas on behavioral flexibility. Learn Mem. 2002;9:18–28. doi: 10.1101/lm.45802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DP. Economic costs of substance abuse, 1995. Proc Assoc Am Phys. 1999;111:119–125. doi: 10.1046/j.1525-1381.1999.09254.x. [DOI] [PubMed] [Google Scholar]
- Richter RM, Pich EM, Koob GF, Weiss F. Sensitization of cocaine-stimulated increase in extracellular levels of corticotropin-releasing factor from the rat amygdala after repeated administration as determined by intracranial microdialysis. Neurosci Lett. 1995;187:169–172. doi: 10.1016/0304-3940(95)11365-4. [DOI] [PubMed] [Google Scholar]
- Richter RM, Weiss F. In vivo CRF release in rat amygdala is increased during cocaine withdrawal in self-administering rats. Synapse. 1999;32:254–261. doi: 10.1002/(SICI)1098-2396(19990615)32:4<254::AID-SYN2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Rivier J, Gulyas J, Kirby D, Low W, Perrin MH, Kunitake K, DiGruccio M, Vaughan J, Reubi J-C, Waser B, Koerber SC, Martinez V, Wang L, Tache Y, Vale W. Potent and long-acting corticotropin releasing factor (CRF) receptor 2 peptide competitive antagonists. J Med Chem. 2002;45:4737–4747. doi: 10.1021/jm0202122. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Roesch MR, Stalnaker TA. Orbitofrontal cortex, decision-making and drug addiction. Trends Neurosci. 2006;29:116–124. doi: 10.1016/j.tins.2005.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci USA. 2001a;98:301–306. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR. Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J Neurosci. 2001b;21:3628–3638. doi: 10.1523/JNEUROSCI.21-10-03628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS. D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci USA. 1994;91:5720–5724. doi: 10.1073/pnas.91.12.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg BA, Davidson DL, Kalivas PW, Prasad BM. Repeated daily cocaine alters subsequent cocaine-induced increase of extracellular dopamine in the medial prefrontal cortex. J Pharmacol Exp Ther. 1997;281:54–61. [PubMed] [Google Scholar]
- Stalnaker TA, Roesch MR, Franz TM, Calu DJ, Singh T, Schoenbaum G. Cocaine-induced decision-making deficits are mediated by miscoding in basolateral amygdala. Nat Neurosci. 2007;10:949–951. doi: 10.1038/nn1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanwood GD, Levitt P. Prenatal exposure to cocaine produces unique developmental and long-term adaptive changes in dopamine D1 receptor activity. J Neurosci. 2007;27:152–157. doi: 10.1523/JNEUROSCI.4591-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steketee JD. Cortical mechanisms of cocaine sensitization. Crit Rev Neurobiol. 2005;17:69–86. doi: 10.1615/critrevneurobiol.v17.i2.20. [DOI] [PubMed] [Google Scholar]
- Trantham H, Szumlinski KK, McFarland K, Kalivas PW, Lavin A. Repeated cocaine administration alters the electrophysiological properties of prefrontal cortical neurons. Neuroscience. 2002;113:749–753. doi: 10.1016/s0306-4522(02)00246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A. Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron. 2003;39:401–407. doi: 10.1016/s0896-6273(03)00461-6. [DOI] [PubMed] [Google Scholar]
- Urban NN, Gonzalez-Burgos G, Henze DA, Lewis DA, Barrionuevo G. Selective reduction by dopamine of excitatory synaptic inputs to pyramidal neurons in primate prefrontal cortex. J Physiol (Lond) 2002;539:707–712. doi: 10.1113/jphysiol.2001.015024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Hitzemann R, Gatley SJ, MacGregor RR, Wolf AP. Cocaine uptake is decreased in the brain of detoxified cocaine abusers. Neuropsychopharmacology. 1996;14:159–168. doi: 10.1016/0893-133X(95)00073-M. [DOI] [PubMed] [Google Scholar]
- Wang J, O'Donnell P. D(1) dopamine receptors potentiate NMDA-mediated excitability increase in layer V prefrontal cortical pyramidal neurons. Cereb Cortex. 2001;11:452–462. doi: 10.1093/cercor/11.5.452. [DOI] [PubMed] [Google Scholar]
- Williams JM, Steketee JD. Time-dependent effects of repeated cocaine administration on dopamine transmission in the medial frontal cortex. Neuropharmacology. 2005;48:51–61. doi: 10.1016/j.neuropharm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Winston JS, Gottfried JA, Kilner JM, Dolan RJ. Integrated neural representations of odor intensity and affective valence in human amygdala. J Neurosci. 2005;25:8903–8907. doi: 10.1523/JNEUROSCI.1569-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Yu B, Gallagher JP. Different subtypes of GABAB receptors are present at pre- and postsynaptic sites within the rat dorsolateral septal nucleus. J Neurophysiol. 1999;81:2875–2883. doi: 10.1152/jn.1999.81.6.2875. [DOI] [PubMed] [Google Scholar]
- Yan XX, Baram TZ, Gerth A, Schultz L, Ribak CE. Co-localization of corticotropin-releasing hormone with glutatamate decarboxylase and calcium-binding proteins in infant rat neocortical interneurons. Exp Brain Res. 1998;123:334–340. doi: 10.1007/s002210050576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Seamans JK, Gorelova N. Electrophysiological and morphological properties of layers V–VI principal pyramidal cells in rat prefrontal cortex in vitro. J Neurosci. 1996;16:1904–1921. doi: 10.1523/JNEUROSCI.16-05-01904.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Zhang XX, Bunney BS, Shi WX. Opposite modulation of cortical N-methyl-d-aspartate receptor-mediated responses by low and high concentrations of dopamine. Neuroscience. 1999;91:527–535. doi: 10.1016/s0306-4522(98)00604-6. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Hablitz JJ. Dopamine modulation of membrane and synaptic properties of interneurons in rat cerebral cortex. J Neurophysiol. 1999;81:967–976. doi: 10.1152/jn.1999.81.3.967. [DOI] [PubMed] [Google Scholar]
- Zorrilla EP, Valdez GR, Weiss F. Changes in levels of regional CRF-like-immunoreactivity and plasma corticosterone during protracted drug withdrawal in dependent rats. Psychopharmacology (Berl) 2001;158:374–381. doi: 10.1007/s002130100773. [DOI] [PubMed] [Google Scholar]