

Abstract
Ubiquitin-positive intraneuronal inclusions are a consistent feature of the major human neurodegenerative diseases, suggesting that dysfunction of the ubiquitin proteasome system is central to disease etiology. Research using inhibitors of the 20S proteasome to model Parkinson's disease is controversial. We report for the first time that specifically 26S proteasomal dysfunction is sufficient to trigger neurodegenerative disease. Here, we describe novel conditional genetic mouse models using the Cre/loxP system to spatially restrict inactivation of Psmc1 (Rpt2/S4) to neurons of either the substantia nigra or forebrain (e.g., cortex, hippocampus, and striatum). PSMC1 is an essential subunit of the 26S proteasome and Psmc1 conditional knock-out mice display 26S proteasome depletion in targeted neurons, in which the 20S proteasome is not affected. Impairment of specifically ubiquitin-mediated protein degradation caused intraneuronal Lewy-like inclusions and extensive neurodegeneration in the nigrostriatal pathway and forebrain regions. Ubiquitin and α-synuclein neuropathology was evident, similar to human Lewy bodies, but interestingly, inclusion bodies contained mitochondria. We support this observation by demonstrating mitochondria in an early form of Lewy body (pale body) from Parkinson's disease patients. The results directly confirm that 26S dysfunction in neurons is involved in the pathology of neurodegenerative disease. The model demonstrates that 26S proteasomes are necessary for normal neuronal homeostasis and that 20S proteasome activity is insufficient for neuronal survival. Finally, we are providing the first reproducible genetic platform for identifying new therapeutic targets to slow or prevent neurodegeneration.
Keywords: neuronal death, neuropathology, genetics, transgenic, Parkinson's disease, dementia, proteolysis
Introduction
The ubiquitin proteasome system (UPS) is the major regulated protein degradation mechanism of the cell, involving covalent tagging of unwanted proteins with polyubiquitin chains as a signal for their degradation by the multiprotein 26S proteasome complex. This ATP-dependent complex is composed of a 20S proteolytic core particle (CP), directly abutted by 19S regulatory particles (RPs) (Voges et al., 1999; Pickart and Cohen, 2004; Schmidt et al., 2005; Demartino and Gillette, 2007). The cylindrical 20S CP is four heptameric rings involving seven distinct α and β subunits stacked in a α1–7–β1–7–β1–7–α1–7 arrangement. The N-terminal tails of the outer α subunits restrict substrate entry into the 20S CP and the inner proteolytic β subunits (Groll et al., 1997, 2000; Groll and Huber, 2003). Two multimeric arrangements make up the 19S RP, the base and the lid, which play roles essential for 26S proteasome function. A recent study into the spatial organization of subunits in the base of the 19S RP revealed that Rpn1/S2 and Rpn2/S1, two large non-ATPase base subunits, as well as the characterized six homologous AAA-ATPases (Rpt1–6) are responsible for coassembly of the 19S RP with the outer α-rings of the 20S CP (Rosenzweig et al., 2008). Rpn1 and Rpn2 form a stacked central unit, in which Rpn2 binds directly to the surface of the 20S CP, which is encircled by the six nonredundant AAA-ATPases forming a hexameric ring (Rubin et al., 1998; Rosenzweig et al., 2008). Rpt2 (PSMC1/S4) has a unique role during 26S proteasome formation and activation, opening the entry pore gated by the α subunits of the 20S CP, enabling protein substrates to be translocated into the proteolytic channel for degradation (Köhler et al., 2001; Smith et al., 2005, 2007). Polyubiquitin chains are recognized by subunits associated with the base of the 19S RP (e.g., non-ATPases Rpn1/S2, Rpn10/S5a, and Rpn13/ARM1 and ATPase Rpt5/S6a), ensuring selective degradation by the 26S complex (Elsasser et al., 2002; Lam et al., 2002; Husnjak et al., 2008). The 19S RP also has components that deubiquitinate proteins before their degradation (e.g., Rpn11/S13) (Koulich et al., 2008).
The UPS is necessary for intracellular homeostasis, not only by controlling the levels of short-lived regulatory proteins, which are involved in the cell cycle, transcription, and DNA repair, but importantly by removing aberrant proteins (Glickman and Ciechanover, 2002; Welchman et al., 2005). UPS-regulated proteolysis is emerging as part of neurodevelopment, synaptic function, and plasticity and the survival of neurons (Yi and Ehlers, 2007). Despite the recent interest in this field, a role for the ubiquitin system in neuronal physiology was first highlighted in chronic neurodegenerative disease. We have known for two decades that neuropathological inclusions in the majority of neurodegenerative diseases consistently contain ubiquitinated proteins, suggesting impairment of the UPS (Lowe et al., 1988). Genetic evidence also links ubiquitin system dysfunction to these diseases. The parkin gene, which is mutated in autosomal recessive juvenile Parkinson's disease (PD), is a ubiquitin protein ligase (Giasson and Lee, 2001). A functional role for proteasome impairment in PD pathogenesis has been controversial because of conflicting experimental data using inhibitors of the 20S proteolytic core (McNaught et al., 2004; Kordower et al., 2006), but changes in the ubiquitin system are involved in Huntington's disease neuropathology (Bennett et al., 2007; Wang et al., 2008). Genetic disruption of cellular ubiquitin pools was recently shown to cause selective neurodegeneration within the hypothalamic arcuate nucleus (Ryu et al., 2008). Also, inactivation of macroautophagy, the less-selective bulk intracellular protein degradation mechanism, in mouse brain neurons leads to neurodegeneration with ubiquitin pathology, suggesting that proteolytic dysfunction contributes to the neuropathology of neurodegenerative disease (Hara et al., 2006; Komatsu et al., 2006).
Notwithstanding the consistent links between the UPS, neuronal inclusions, and neurodegeneration, direct evidence involving primary inhibition of ubiquitin-mediated protein degradation by the 26S proteasome complex in neurons has still not been demonstrated. The aim of this work was to genetically disrupt specifically 26S proteasome degradation in mouse neurons, leaving the 20S proteolytic core complex, which is involved in ubiquitin-independent degradation, unaffected. Therefore, we generated a reproducible conditional knock-out mouse using the Cre/loxP method for an essential subunit of the 19S RP, PSMC1 (Rpt2/S4). Two Cre deletor mouse strains were used to spatially restrict inactivation of Psmc1 to predominantly the forebrain or substantia nigra, expressing Cre recombinase either under the control of the calcium calmodulin-dependent protein kinase IIα (CaMKIIα) promoter or from the tyrosine hydroxylase (TH) locus (Lindeberg et al., 2002, 2004), respectively. We show here that loss of PSMC1 leads to 26S proteasome depletion, which causes neurodegeneration and the formation of intraneuronal Lewy-like inclusions resembling human pale bodies (PBs) in neurons of the nigrostriatal pathway and forebrain. Our findings are significant advances in the experimental support for a direct role of the UPS in the pathogenic mechanisms of neurodegenerative disease.
Materials and Methods
Generation of floxed Psmc1 mice
Using homologous recombination, a loxP-flanked neomycin-thymidine kinase selection cassette and a loxP site were inserted into introns 1 and 3, respectively, of the Psmc1 gene (Gene ID 19179) in mouse 129/Sv embryonic stem (ES) cells (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). The selection cassette was excised by transient expression of Cre recombinase in vitro. PCR amplification identified correctly targeted ES cell clones. Male chimeric mice were generated using aggregation (Tanaka et al., 2001) with CD1-derived morulae and bred with CD1 females to establish homozygous floxed Psmc1 mice (Psmc1 fl/fl).
Region-specific inactivation of Psmc1
Forebrain-specific ablation of Psmc1 was achieved by crossing Psmc1 fl/fl mice with CaMKIIα-Cre mice (Lindeberg et al., 2002), generating Psmc1 fl/wt;CaMKIIα-Wt and Psmc1 fl/wt;CaMKIIα-Cre mice. Female Psmc1 fl/wt;CaMKIIα-Cre mice were mated with Psmc1 fl/fl males to produce mice in which Psmc1 was selectively inactivated in calcium calmodulin-dependent protein kinase IIα-expressing cells (Psmc1 fl/fl;CaMKIIα-Cre) and control mice (Psmc1 fl/fl;CaMKIIα-Wt or Psmc1 fl/wt;CaMKIIα-Wt). For catecholaminergic neurone-specific inactivation of Psmc1, Psmc1 fl/fl mice were crossed with TH Cre mice (Lindeberg et al., 2004) generating Psmc1 fl/wt;TH Cre and Psmc1 fl/wt;TH wt animals. Male Psmc1 fl/wt;TH Cre mice were bred with Psmc1 fl/fl females to produce mice deficient in Psmc1 specifically in tyrosine hydroxylase-expressing cells (Psmc1 fl/fl;TH Cre or Psmc1 fl/ko;TH Cre) and control mice (Psmc1 fl/fl;TH wt or Psmc1 fl/wt;TH Cre).
Genotyping
PCR amplification of ear biopsy DNA was used for genotyping.
Quantitative real-time RT-PCR
Total RNA was extracted using a QIAGEN RNeasy Micro kit. RNA was eluted with RNase-free water and quantified using a Nanodrop. A total of 500 ng of DNase 1 (Promega)-treated RNA provided the substrate for cDNA synthesis using oligo-dT18 and Invitrogen Superscript III Reverse Transcriptase according to manufacturers instructions. A total of 10 μl of real-time RT-PCRs was run using Stratagene Brilliant SYBR Green QPCR Master Mix and a Stratagene Mx3005P QPCR System. Primers (100 nm) for Psmc1 and glyceraldehyde phosphate dehydrogenase (GAPDH) were used. Analysis used Stratagene MxPro QPCR software, generating a standard curve from plasmid dilutions. Experimental samples were analyzed in triplicate, and gene expression was normalized to GAPDH and expressed as a fold change of the controls.
Sedimentation velocity analysis
This was performed as described previously (Tanahashi et al., 2000).
Western analysis
Microdissected brain regions were homogenized on ice in 50 mm Tris, pH 7.4, 175 mm NaCl, and 5 mm EDTA, and subjected to SDS-PAGE, and the separated proteins were transferred to nitrocellulose membrane. Incubation in the appropriate primary antibodies was for 1 h at room temperature [ubiquitin, 1:1000 (in-house), or synaptophysin, 1:1000 (Calbiochem)], and visualization used horseradish peroxidise-conjugated secondary antibodies (Sigma-Aldrich) and enhanced chemiluminescent substrate (Pierce). Protein analysis after adenoviral Cre recombinase treatment of mouse embryonic fibroblast cells was performed by recovering denatured protein from the cell lysate flowthrough during QIAGEN RNeasy Micro purification of total RNA as described in the handbook. Protein was subjected to SDS-PAGE as described above [PSMC1, 1:1000 (in-house), or actin, 1:1000 (Sigma-Aldrich)].
Histology
For light microscopy, mice were perfusion-fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, and brains were embedded in paraffin wax and sectioned (5 μm) according to standard protocols. General morphological examination used hematoxylin (Harris) and eosin staining. Immunostaining was performed as directed in Vector Laboratories M.O.M. immunodetection or Vectastain Elite rabbit IgG ABC kits. Antigen retrieval used microwave treatment in 0.01 m citrate buffer containing 0.05% Tween 20, pH 6, or 10 mm EDTA followed by formic acid treatment. Primary antibody incubation was for 1 h at room temperature: 1:2000 tyrosine hydroxylase (Sigma-Aldrich), 1:2000 ubiquitin (Dako), 1:4000 glial fibrillary acidic protein (GFAP) (Dako), 1:5 (Vector) or 1:10,000 (Dr. Diane Hanger, Institute of Psychiatry, King's College London, London, UK) α-synuclein, 1:1000 p62 (BIOMOL), 1:5 γ-tubulin, 1:100 activated caspase-9 (Cell Signaling), 1:600 cytochrome oxidase IV (cox IV) (Cell Signaling), 1:50 p53 (Calbiochem), and 1:1000 PSMC1 (BIOMOL). Analyses used an Olympus BX51 microscope and camera. For electron microscopic analyses, 3.2% paraformaldehyde, 0.2% glutaraldehyde, 1% sucrose, 3 mm CaCl2 in 0.1 m sodium cacodylate buffer was used. When cryosections were needed, brains were dissected and frozen in precooled isopentane over liquid nitrogen. For laser capture microdissection, cryostat sections (10 μm) were cut onto PEN membrane slides (Carl Zeiss) and fixed in precooled acetone for 5 min. The sections were immunostained using TH as described previously. Immunopositive TH cells were microdissected and laser pressure catapulted into adhesive caps using a PALM microbeam Microdissector (Carl Zeiss). For thioflavin S staining, sections were stained in hematoxylin for 5 min, followed by washing in water and then incubating in 1% thioflavin S for 10 min. Sections were washed in 50% ethanol and then water for 5 min each before mounting in Gel Mount (Sigma-Aldrich). Immunogold electron microscopy was performed by routine methods with 10 nm gold (Agar Scientific) as described previously (László et al., 1991).
Behavioral analyses
Open field.
Each mouse was monitored for 5 min in an open-field arena (350 × 300 × 300 mm; three walls transparent, one wall and floor dark gray). Activity in the open field was recorded and quantitated by a computer-operated animal activity system.
Morris water maze.
A hidden-platform test performed in an opaque pool at ∼25°C. Over 3 consecutive days, three blocks of six trials lasting 120 s each from two compass points located on the pool (18 trials total) were carried out. The time taken to reach the platform (latency to escape) was recorded for each trial. The 120 s probe trial in which the platform was removed was on day 4 from a start position opposite the original platform location.
Statistical analysis
Data analysis used two-tailed unpaired Student's t test with unequal variance. Statistical significance is indicated in the appropriate figure.
Results
Generation of neuron-specific 26S proteasome-depleted mice
To study the direct effect of impaired 26S-mediated degradation of ubiquitinated proteins in neurons, a key subunit of the 19S RP was genetically targeted, ATPase Psmc1 (Gene ID 19179) (Rubin et al., 1998; Köhler et al., 2001; Smith et al., 2007). As anticipated, Psmc1 knock-out mice (Psmc1 ko/ko) were embryonic lethal (data not shown). Therefore, Psmc1 conditional knock-out mice (Psmc1 fl/fl) were generated (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). To spatially restrict ablation of Psmc1, Psmc1 fl/fl mice were crossed with Cre deletor mouse strains, expressing Cre recombinase under the control of either the CaMKIIα promoter (Psmc1 fl/fl;CaMKIIα-Cre) or from the TH locus (Psmc1 fl/fl;TH Cre) (Lindeberg et al., 2002, 2004). The CaMKIIα promoter directs expression to neurons predominantly in forebrain regions (Burgin et al., 1990; Mayford et al., 1996; Tsien et al., 1996). Expression is negligible during prenatal and perinatal development, and then dramatically upregulated during postnatal weeks 2 and 3, permitting normal CNS development before Cre-mediated recombination of Psmc1 in postmitotic neurons. In Psmc1 fl/fl;TH Cre mice, TH is expressed from midgestation in midbrain dopaminergic neurons (Bayer et al., 1995).
Cre-mediated recombination of Psmc1 fl/fl in the mouse brain was confirmed by PCR analysis of Psmc1 fl/fl;CaMKIIα-Cre and Psmc1 fl/fl;TH Cre mice (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material) (data not shown). Because Cre deletor mice only target specific neurons, the mixed cell population of the brain proved unsuitable for analysis of Psmc1 mRNA expression. Therefore, we captured Psmc1-targeted neurons using laser capture microdissection (e.g., TH-expressing neurons from the substantia nigra or adrenal medulla of Psmc1 fl/fl;TH Cre mice). No cycle threshold value (Ct) was obtained for Psmc1 expression by real-time RT-PCR at 3 weeks of age. Immunostaining of cortex sections from 6-week-old Psmc1 fl/fl;CaMKIIα-Cre and control mice showed deletion of PSMC1 in cortical neurons of Psmc1 fl/fl;CaMKIIα-Cre mice. In addition, sequential sections of 1 week substantia nigra were stained with anti-TH and anti-PSMC1 antibodies, demonstrating the loss of PSMC1 in residual TH+ cells of Psmc1 fl/fl;TH Cre mice (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material).
To substantiate gene expression changes in a homogenous cell population, we established an in vitro model of Psmc1 ablation using mouse embryonic fibroblasts (MEFs) derived from mating Psmc1 ko/wt and Psmc1 fl/fl mice. Psmc1fl/wt (control) and Psmc1fl/ko (mutant) MEFs were transduced with adenoviral Cre recombinase green fluorescent protein (GFP) to inactivate the floxed Psmc1 allele. GFP-expressing cells were collected by fluorescence-activated cell sorting, and real-time RT-PCR analysis demonstrated loss of Psmc1 mRNA in mutant MEFs from 2 d after transduction (Table 1). Western analysis at 4 d after transduction confirmed that Cre-mediated recombination of floxed Psmc1 resulted in loss of PSMC1 protein (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material).
Table 1.
mRNA changes for selected 26S proteasomal, autophagic, and apoptotic genes after ablation of Psmc1
| Subunit | Increase (fold) |
|||
|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Day 4 | |
| Psmc1 | 0.1** | — | — | — |
| α7 | 1.4 | 1.1 | 3.6* | 1.5 |
| β5 | 0.8 | 1.1 | 2.4** | 1.5 |
| SUG1 | 0.8 | 1.4 | 2.6* | 0.9 |
| POH1 | 1 | 1.1 | 2.6** | 1.6** |
| Atg7 | ND | ND | 1.7 | ND |
| Atg8 | ND | ND | 0.8 | ND |
| Bcl2 | ND | ND | 0.2** | ND |
Psmc1fl/wt (control) and Psmc1fl/ko(mutant) MEFs were transduced on day 0 with adenoviral Cre recombinase (multiplicity of infection, ∼850; Vector Laboratories), and mRNA analysis was performed by real-time RT-PCR over 4 d. Representative proteasomal subunits are α7 and chymotryptic β5 of the 20S core complex, and SUG1/p45 ATPase and POH1 from the base and lid of the 19S regulatory particle, respectively. Atg7, Atg8, and Bcl2 represent changes in macroautophagy and apoptotic mechanisms, respectively. Gene expression was normalized to GAPDH and expressed as a fold change compared with the controls. —, No expression; ND, not done.
**p < 0.01,
*p < 0.05 by two-tailed Student's t test.
PSMC1 is necessary for assembly and activity of the 26S proteasome
Control and Psmc1 fl/fl;CaMKIIα-Cre (mutant) cortical tissue was subjected to glycerol density gradient centrifugation and fractions were assayed for chymotrypsin-like activity. The characteristic bimodal profile of 20S and 26S proteasome activity was evident in controls, with the 26S complex contributing the majority of proteasomal activity (Fig. 1 A). Western analyses confirmed proteasome sedimentation positions (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). At 2 weeks of age, the proteasomal distribution was comparable in control and mutant cortex (Fig. 1 A). In 3-week-old mice, less 26S complexes were observed in the mutant compared with the control, with an increase in lower molecular weight fractions corresponding to 20S (Fig. 1 A). The altered profile of proteasome activity in the Psmc1 fl/fl;CaMKIIα-Cre cortex is clear at 4 and 6 weeks (Fig. 1 A), at which 20S is the predominant complex. Because CaMKIIα-Cre activity is upregulated in mutant cortical neurons during postnatal weeks 2 and 3, the data indicate that ensuing progressive depletion of PMSC1 disrupts 26S formation. Residual 26S proteasome activity is present because of nontargeted neurons and gliosis (Fig. 2 D). This is the first report of mammalian 26S proteasome disruption in vivo (cf. Wójcik and DeMartino, 2002; Koulich et al., 2008); the gradual change in proteasomes between 2 and 4 weeks indicates a relatively long half-life associated with the 26S proteasome. As intended, 20S proteolytic core formation, and therefore ubiquitin-independent proteasomal degradation, was not affected in our model.
Figure 1.
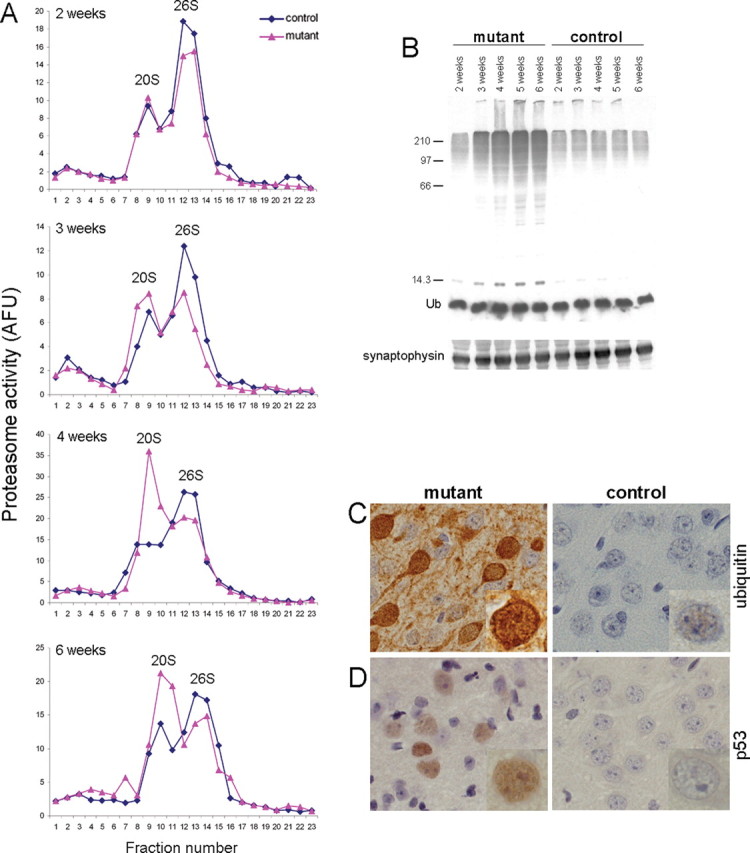
Ablation of Psmc1 causes 26S proteasome depletion. A, Representative proteasome profiles from cortex of 2-, 3-, 4-, and 6-week-old control and Psmc1 fl/fl;CaMKIIα-Cre (mutant) mice after glycerol density gradient centrifugation. Fractions 1 and 23 correspond to the top (10%) and bottom (40%) of the gradient, respectively. 20S and 26S proteasomal distribution was revealed by succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin hydrolysis activity of gradient fractions. Individual experiments at different ages are not directly comparable because of, for example, the amount of protein loaded and slight variability in fraction collection. AFU, Arbitrary fluorescent unit. B, Accumulation of polyubiquitinated proteins, but not free ubiquitin (Ub), in the Psmc1 fl/fl;CaMKIIα-Cre (mutant) cortex with increasing age, shown by Western analysis of cortex homogenates with an anti-ubiquitin antibody. Anti-synaptophysin was used as a loading control. C, D, Immunohistological staining of cortex sections from 6-week-old control and Psmc1 fl/fl;CaMKIIα-Cre (mutant) mice using anti-ubiquitin (C) and anti-p53 (D) antibodies (40×). Higher-magnification images are inset into the representative sections.
Figure 2.

26S proteasomal dysfunction causes neurodegeneration. A–D, Histological examination of coronal sections from control and Psmc1 fl/fl;CaMKIIα-Cre (mutant) mouse brains at 8 weeks of age. A, Hematoxylin and eosin (H&E)-stained whole-brain sections, shown at higher magnification in B (40×). The arrows point to pyknotic and fragmented nuclei in the mutant cortex. Immunohistochemistry with antibodies to cleaved caspase 9 (40×) (C) and GFAP (10×) (D) in cortex sections. Representative individual neurons are magnified and shown in the inset of appropriate figures. E, TH-stained horizontal sections of whole brain at the level of the nigrostriatal pathway from Psmc1 fl/fl;TH Cre (mutant) and control mice. F, TH and Nissl staining of sequential sections from the substantia nigra of control and Psmc1 fl/fl;TH Cre (mutant) mice (20×). G, Catecholamine and indolamine levels measured by HPLC with electrochemical detection in the striatum of Psmc1 fl/fl;TH Cre (mutant) and control mice. Graph represents mean ± SEM (controls, n = 5; mutants, n = 7). **p < 0.01 and ***p < 0.001 from control mice by unpaired Student's t test. Ctx, Cerebral cortex; cc, corpus callosum; Str, striatum; VZ, ventricular zone; Ob, olfactory bulb; SN, substantia nigra; LC, locus ceruleus.
26S proteasomal dysfunction was confirmed by accumulation of high molecular weight polyubiquitinated proteins with increasing age (Fig. 1 B) and significantly increased ubiquitin immunostaining in affected neurons (Fig. 1 C) in the Psmc1 fl/fl;CaMKIIα-Cre cortex (mutant) compared with control. The level of p53, which undergoes robust ubiquitin-dependent degradation, was also significantly elevated (Fig. 1 D).
Increased 20S proteasome activity in the Psmc1 fl/fl;CaMKIIα-Cre (mutant) cortex with age appears to be higher than would be expected for a direct shift of the 20S activity that would normally be associated with 26S complexes (Fig. 1 A). This may be explained by upregulation of proteasomal gene expression and biogenesis of active 20S complexes in response to 26S proteasome impairment (Wójcik and DeMartino, 2002; Meiners et al., 2003; Lundgren et al., 2005; Fuchs et al., 2008). In our MEF model, a significant increase in representative proteasome subunit mRNAs was observed 3 d after transduction (Table 1). This is most likely attributable to transcriptional activation (Meiners et al., 2003). Together with the observations in vivo, these results suggest that proteasome subunits are upregulated in an attempt to maintain proteasomal homeostasis in the absence of PSMC1.
Psmc1 fl/fl;CaMKIIα-Cre and Psmc1 fl/fl;TH Cre offspring are viable and indistinguishable from their littermates at birth. Subtle growth retardation was observed in Psmc1 fl/fl;CaMKIIα-Cre mice from 5 weeks of age (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material), and they die when aged 3–4 months, when they lack interest in locating food. These mice were significantly more anxious in open-field analysis and displayed obvious spatial learning deficits in the Morris water maze task at 6 and 8 weeks, respectively, suggesting 26S proteasomal function is essential for normal neurological function in mice (supplemental Fig. 3B,C, available at www.jneurosci.org as supplemental material). Psmc1 fl/fl;TH Cre mice became progressively runty from approximately day 14 and die before postnatal day 28 (see below).
26S proteasomal depletion causes neurodegeneration
Gross histological analysis of brains from control and Psmc1 fl/fl;CaMKIIα-Cre (mutant) mice revealed significant progressive atrophy of the forebrain, exemplified by reduced cortical thickness and expansion of the ventricular cavities (Fig. 2 A; supplemental Fig. 4, available at www.jneurosci.org as supplemental material). At 2 weeks of age, there were no degenerative changes evident in the Psmc1 fl/fl;CaMKIIα-Cre forebrain (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). However, we observed numerous pyknotic nuclei with hematoxylin and eosin staining at 4 weeks of age, which increased in 6-week-old mice, preceding the extensive neuronal loss by 8 weeks (Fig. 2 A,B; supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). Pyknotic nuclei suggested an apoptotic mechanism of neurodegeneration. This was confirmed by upregulation of the apoptosis signaling molecule, cleaved caspase-9 (Fig. 2 C). Decreased Bcl-2 expression was observed in the MEF model (Table 1). It remains possible that alternative death mechanisms contribute to degeneration of these neurons. Immunohistochemical staining with GFAP demonstrated extensive astrocytic gliosis accompanied the neuronal loss (Fig. 2 D). Neurodegeneration was evident in other CaMKIIα-expressing regions of the Psmc1 fl/fl;CaMKIIα-Cre brain, including the hippocampus, striatum, and amygdala (data not shown). We conclude that 26S proteasomal depletion directly causes neurodegeneration.
In Psmc1 fl/fl;TH Cre mice, 26S proteasomal dysfunction in dopaminergic neurons of the nigrostriatal pathway led to progressive degeneration and consequent loss of neuronal projections to the basal ganglia, as evidenced by TH immunostaining up to 3 weeks of age (Fig. 2 E) (data not shown). TH and Nissl staining of adjacent sections revealed this was neurodegeneration and not downregulation of TH expression (Fig. 2 F). In neurochemical support of the observations, significant catecholaminergic deficits were evident in the Psmc1 fl/fl;TH Cre brain, including decreases in dopamine and norepinephrine in the striatum (Fig. 2 G) and hypothalamus, and a decrease in norepinephrine in the hippocampus and brainstem (data not shown). Figure 3 emphasizes the ubiquitin accumulation in surviving neurons of the substantia nigra and extensive gliosis as a result of the neuronal damage. 26S proteasomal depletion in all catecholaminergic neurons of the Psmc1 fl/fl;TH Cre mice causes autonomic dysfunction (data not shown), explaining premature death.
Figure 3.
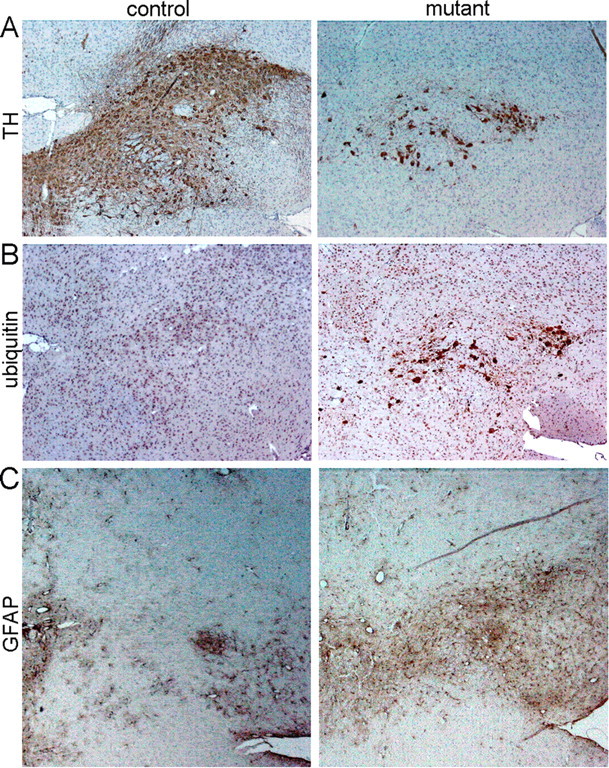
Neurodegeneration of the substantia nigra in Psmc1 fl/fl;TH Cre mice. A–C, Immunohistochemistry using antibodies to TH (A), ubiquitin (B), and GFAP (C) (10×). A, TH immunostaining identifies nigral neurons in control and Psmc1 fl/fl;TH Cre (mutant) horizontal brain sections at 3 weeks of age. B, C, Increased ubiquitin (B) and astrocytic gliosis (C) in the substantia nigra of the mutant brain.
Intraneuronal inclusions in 26S proteasome-depleted neurons
In parallel, we examined protein aggregation in the mouse brains using diagnostic antibodies for major human neurodegenerative diseases. Diffuse accumulation of ubiquitinated proteins was evident in a few forebrain neurons in Psmc1 fl/fl;CaMKIIα-Cre mice at 2 weeks, but no other differences were observed at this age (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). By 4 weeks of age, coincident with progressive 26S proteasomal impairment, there was significant ubiquitin pathology. However, we also observed numerous eosinophilic intraneuronal paranuclear inclusions, containing ubiquitin, α-synuclein, and p62, similar to Lewy bodies (LBs) seen in the brains of patients with dementia with Lewy bodies (DLB) (Fig. 4 A–D; supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Psmc1 fl/fl;TH Cre nigral neurons also demonstrated a similar phenomenon, resembling Lewy bodies of PD (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Although α-synuclein-immunoreactive neurites were not found in the mice, in contrast to human disease, some of these inclusions stained with γ-tubulin, PGP 9.5, αB crystallin, and neurofilament protein, supporting their similarity to Lewy bodies (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) (data not shown) (Shults, 2006). No Aβ (β-amyloid)-containing amyloid or tau protein deposits were evident on immunostaining (data not shown). Therefore, Lewy-like inclusion formation and neuronal loss are independent of amyloid formation in this model.
Figure 4.
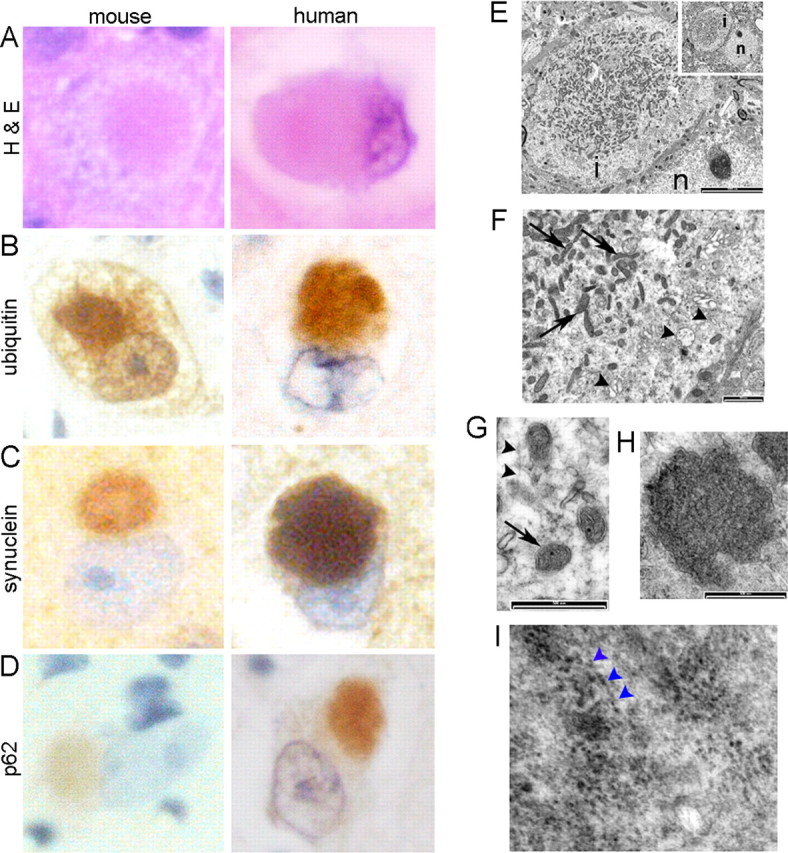
Lewy-like intraneuronal inclusions in 26S proteasome-depleted neurons. A–D, Representative Lewy-like inclusions in mouse cortical neurons at 6 weeks of age stained with H&E and ubiquitin, α-synuclein, and p62 antibodies, directly compared with Lewy bodies from the brains of patients with dementia with Lewy bodies (100×). No inclusions were observed in the control mouse brains (data not shown). E–I, Ultrastructure of mouse Lewy-like inclusions by EM. The paranuclear inclusion (E; inset; n, nucleus; i, inclusion) contains mitochondria (F, G, arrows), a background matrix of fine filaments (G, arrowheads) and granular material, with peripheral membrane-bound vesicles (F, arrowheads), including double-membraned autophagosome-like dense bodies (H). I, Filamentous α-synuclein is associated with 10 nm gold particles by immunoelectron microscopy. The arrowheads point to particles aligned along distinct filaments. Scale bars: E, 5000 nm; E, 1000 nm; G, H, 500 nm.
Electron microscopy of Lewy-like inclusions revealed spherical structures with a lamellar appearance (Fig. 4 E; supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Numerous mitochondria, fine filaments, and granular material were evident in the central region, surrounded by a zone of clear vesicular and membranous material, as well as double-membraned autophagolysomes, suggesting the autophagic processes followed 26S proteasomal dysfunction (Fig. 4 E–H; supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Immunogold labeling with anti-α-synuclein revealed some filaments with 10 nm gold particles in the central region (Fig. 4 I). In support of this observation, a subset of inclusions stained with the amyloid-binding dye thioflavin S (supplemental Fig. 6, available at www.jneurosci.org as supplemental material).
Mitochondrial involvement in inclusion body formation
The pattern of α-synuclein aggregate in our model, containing mitochondria, has been recognized in human disease and is considered to be an early form of Lewy body (Gai et al., 2000). The paucity of literature on mitochondria in human disease led us to substantiate our findings by EM analysis of substantia nigra from PD patients. This revealed the presence of mitochondria in pale bodies, an α-synuclein-containing inclusion thought to be an early form of nigral Lewy body (Fig. 5 A). In the example shown, the larger mitochondria-containing PB is adjacent to a smaller LB, which is devoid of mitochondria. Mitochondria-rich inclusions were further investigated using cox IV immunostaining. cox IV was seen in a proportion of human cortical Lewy bodies as well as in nigral pale bodies, but was absent from classical dense core Lewy bodies (Fig. 5 B–D). Therefore, the mouse model probably represents filamentous α-synuclein aggregates containing mitochondria as “early-stage” intraneuronal inclusions. The inclusions do not progress to reveal Lewy bodies with a dense core and lamellar pattern as in human disease. The abundance of mitochondria in mouse Lewy-like bodies after 26S proteasomal impairment suggests that mitochondria might play a direct role in Lewy body formation and/or function.
Figure 5.
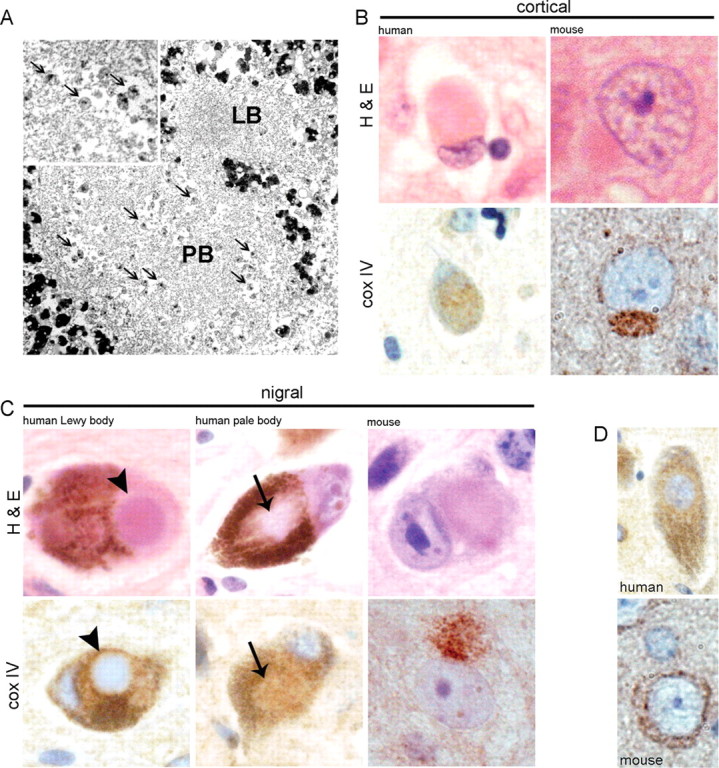
Mitochondria in human and mouse Lewy bodies. A, EM showing mitochondria (arrows) among filamentous material in a human nigral PB, which is adjacent to a classical LB that is devoid of mitochondria. Enlarged view of mitochondria within the pale body is shown in the inset. B, C, Shown is H&E and cox IV immunostaining of cortical Lewy bodies (B) and nigral Lewy and pale bodies (C) from human disease and Psmc1 fl/fl;CaMKIIα-Cre and Psmc1 fl/fl;TH Cre mice (100×). The arrows and arrowheads identify the pale and classical dense core Lewy bodies of human disease, respectively. D, The normal pattern of cox IV in human and mouse neurons (100×). The heavy brown pigment in H&E human nigral neurons is neuromelanin.
Discussion
We report a conditional knock-out mouse harboring 26S proteasomal dysfunction in neurons, enabling a definitive link between ubiquitinated protein degradation by the 26S proteasome and neuronal pathology to be established. Impaired 26S proteasomal function causes neurodegeneration and intraneuronal inclusion body formation, reflecting early forms of human Lewy bodies described as homogeneous Lewy bodies (Gai et al., 2000) or pale bodies (Dale et al., 1992). The intraneuronal inclusions in our mouse model are different from those seen in PD or DLB because the classical concentric Lewy body does not form. The reasons for this are currently unknown. It is possible that neurodegeneration proceeds before the time that would be necessary to form a late-stage concentric Lewy body. This is most likely a consequence of the genetic approach taken in this model, which rapidly ablates 26S proteasomal function. In contrast, progressive proteasomal inhibition with aging most likely occurs in human disease. Alternatively, there may be intrinsic differences in the pathophysiology between the mouse model and human disease.
Based on mutational studies of the ATPase subunits in the base of the 19S RP using yeast proteasomes as a model, we chose to inactivate Psmc1 (Rpt2/S4) to generate a novel mouse model of specifically 26S proteasomal dysfunction (Seeger et al., 1996; Rubin et al., 1998; Köhler et al., 2001). Because the six base ATPases are functionally nonredundant, deletion of PSMC1 would prevent hexameric ring biogenesis in the base of the 19S RP and coassembly with the 20S CP. PSMC1 also has a unique role in proteolysis activity of the 20S core. Using glycerol gradient analysis, we demonstrate here in higher eukaryotes that ablation of PSMC1 in mouse neurons disrupts 26S proteasome formation, but does not affect the 20S CP. Therefore, in contrast to the use of 20S proteasome inhibitors in models of PD, which do not differentiate between ubiquitin-dependent and ubiquitin-independent degradation or neurons and glia, the neuropathological changes evident in our mouse model are linked to neuronal 26S proteasomal dysfunction. This model also reveals the importance of the 26S proteasome versus the 20S proteolytic CP in neuronal homeostasis. The 20S core is unable to maintain neuronal survival in the absence of PSMC1. It is still possible that a decrease in 20S proteolytic activity of the proteasome occurs with aging in neurodegenerative disease. Importantly, this work demonstrates that the pathological consequences of a decrease in 20S CP activity are linked to the ubiquitin-mediated degradation pathway.
This model differs significantly from current models of neurodegenerative disease, in which neuronal loss is minimal and generally not seen in regions linked to disease (McGowan et al., 2006; Melrose et al., 2006) (e.g., the substantia nigra in α-synuclein-expressing transgenic mice). Neurodegeneration may be an expected consequence of PSMC1 and thence 26S proteasomal depletion in mouse neurons. The early Schizosaccharomyces pombe and Saccharomyces cerevisiae models of proteasomal dysfunction, with mutation of the homologous ATPase mts2 or rpt2, respectively, reflect the observations in our mouse model. Lack of the functional ATPase caused accumulation of ubiquitinated proteins and growth defects (Gordon et al., 1993; Seeger et al., 1996; Rubin et al., 1998). More recently, as anticipated, proteasome-dependent activities are being revealed as fundamental in the nervous system (Yi and Ehlers, 2007). Although a buildup of polyubiquitinated proteins and degeneration may be obvious effects of disrupted neuronal proteasomal homeostasis in this model, previous work did not suggest that intracellular paranuclear inclusion body formation would occur, resembling early-stage Lewy bodies seen in human disease. This could be explained by inherent differences in mechanisms between higher and lower eukaryotes. The formation of inclusion bodies with some of the currently accepted features of Lewy bodies (Shults, 2006) suggests 26S proteasome dysfunction is associated with a cascade of specific neuropathological changes that occur in disease progression.
Much research into the synucleinopathies, particularly Parkinson's disease, focuses on mechanisms involving α-synuclein, but more recently mitochondrial involvement in the biology of Lewy body disorders has been recognized. We demonstrate both α-synuclein and mitochondria are linked to neuropathological changes after proteasomal dysfunction, which itself is consistently associated with disease (Gandhi and Wood, 2005). Although proteasomal dysfunction may prevent the degradation of α-synuclein and other proteins, which then accumulate passively in paranuclear aggresomes (Lewy-like bodies), another interpretation is that α-synuclein may be an essential protein involved in the biogenesis of Lewy bodies and its accumulation is an active event. This is supported by the disproportionate and early accumulation of α-synuclein in inclusion bodies after 26S proteasomal dysfunction in neurons; α-synuclein is not recognized as a protein that has a high turnover mediated by the UPS, and has been reported to undergo ubiquitin-independent degradation (Tofaris et al., 2001).
Mitochondria are a striking component of the inclusion bodies in mouse neurons and we show current evidence for mitochondria in some human cortical and nigral early-stage Lewy bodies. Mitochondria have also been noted as a component of Lewy-like aggresomes induced by proteasome inhibition (Abou-Sleiman et al., 2006). The precise role of mitochondria in neurodegenerative disease remains uncertain. Evidence for mitochondrial dysfunction is provided by mutations in genes that are associated with mitochondria in familial PD [e.g., mitochondrial kinase PINK (PTEN-induced kinase 1) and Parkin (Valente et al., 2004; Poole et al., 2008)], and features of PD are caused by mitochondrial toxins (Bové et al., 2005). The observations made in the mouse models and reexamination of human pathology convinces us to propose a hypothesis that suggests that mitochondria are a key component of an early phase in the biogenesis of the Lewy body.
Another mechanistic possibility for the findings described here that will need additional investigation is that the roles of the 26S proteasome, 19S RP, and 20S catalytic core in gene expression (Gonzalez et al., 2002; Sikder et al., 2006) may be affected by the loss of PSMC1. Progranulin mutations result in neuronal cytoplasmic inclusions containing TDP43, which is normally involved in transcription and exon skipping and may cause neurodegeneration by errors in gene expression (Eriksen and Mackenzie, 2008).
The known options for the isolation and/or degradation of unwanted proteins in the absence of the UPS are the formation of aggresomes (Johnston et al., 1998) and macroautophagy. The neuroprotective function of macroautophagy has become an interesting focus in neurodegenerative disease, exemplified in Huntington's disease (Menzies et al., 2006; Mizushima et al., 2008), as well as in animal models (Hara et al., 2006; Komatsu et al., 2006). Whether this alternative nonselective proteolytic pathway plays a role during dysfunction of the UPS is still unclear. Proteasome function was found to be normal in autophagy-deficient brain (Komatsu et al., 2006). Preliminary experiments in 26S proteasome-ablated MEFs did not show significant upregulation of autophagic genes (Table 1), but in vivo EM analysis revealed double-membraned autophagolysomes.
This is the first mouse model that permits investigation into 26S proteasomal dysfunction in neurons. We demonstrate that impairment of neuronal 26S proteasomes in the neocortex or substantia nigra causes florid neurodegeneration and Lewy-like inclusion body formation, resembling pathophysiology evident in the brains of patients with DLB and PD, respectively. The models described here provide unparalleled novel platforms in which to investigate and exploit therapeutic approaches to prevent neurodegeneration in the brain.
Footnotes
This work was supported by The Wellcome Trust, Biotechnology and Biological Sciences Research Council, Alzheimer's Research Trust, Neuroscience Support Group at the Queen's Medical Centre, and the Parkinson's Disease Society. T.E. and D.U. were supported by the Swedish Research Council. We thank Dr. R. Kuhn for the CaMKIIα-Cre mice. Denise Watson, Lianne Finnerty, Janet Palmer, Neil Hand, and Howard Coleman (School of Molecular Medical Sciences, University of Nottingham Medical School) provided valuable technical assistance. We thank Drs. C. Gordon and S. Thomson (Medical Research Council Human Genetics, Western General Hospital, Edinburgh, UK) for detailed discussions and assistance leading to the inactivation of Psmc1 by the Cre/loxP system. The studies were performed with British Home Office Project License 40/2688.
References
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Wills KV, Triarhou LC, Ghetti B. Time of neuron origin and gradients of neurogenesis in midbrain dopaminergic neurons in the mouse. Exp Brain Res. 1995;105:191–199. doi: 10.1007/BF00240955. [DOI] [PubMed] [Google Scholar]
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- Bové J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson's disease. NeuroRx. 2005;2:484–494. doi: 10.1602/neurorx.2.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgin KE, Waxham MN, Rickling S, Westgate SA, Mobley WC, Kelly PT. In situ hybridization histochemistry of Ca2+/calmodulin-dependent protein kinase in developing rat brain. J Neurosci. 1990;10:1788–1798. doi: 10.1523/JNEUROSCI.10-06-01788.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale GE, Probst A, Luthert P, Martin J, Anderton BH, Leigh PN. Relationships between Lewy bodies and pale bodies in Parkinson's disease. Acta Neuropathol. 1992;83:525–529. doi: 10.1007/BF00310030. [DOI] [PubMed] [Google Scholar]
- Demartino GN, Gillette TG. Proteasomes: machines for all reasons. Cell. 2007;129:659–662. doi: 10.1016/j.cell.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Elsasser S, Gali RR, Schwickart M, Larsen CN, Leggett DS, Müller B, Feng MT, Tübing F, Dittmar GA, Finley D. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat Cell Biol. 2002;4:725–730. doi: 10.1038/ncb845. [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Mackenzie IR. Progranulin: normal function and role in neurodegeneration. J Neurochem. 2008;104:287–297. doi: 10.1111/j.1471-4159.2007.04968.x. [DOI] [PubMed] [Google Scholar]
- Fuchs D, Berges C, Opelz G, Daniel V, Naujokat C. Increased expression and altered subunit composition of proteasomes induced by continuous proteasome inhibition establish apoptosis resistance and hyperproliferation of Burkitt lymphoma cells. J Cell Biochem. 2008;103:270–283. doi: 10.1002/jcb.21405. [DOI] [PubMed] [Google Scholar]
- Gai WP, Yuan HX, Li XQ, Power JT, Blumbergs PC, Jensen PH. In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp Neurol. 2000;166:324–333. doi: 10.1006/exnr.2000.7527. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Wood NW. Molecular pathogenesis of Parkinson's disease. Hum Mol Genet. 2005;14:2749–2755. doi: 10.1093/hmg/ddi308. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Lee VM. Parkin and the molecular pathways of parkinson's disease. Neuron. 2001;31:885–888. doi: 10.1016/s0896-6273(01)00439-1. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Gonzalez F, Delahodde A, Kodadek T, Johnston SA. Recruitment of a 19S proteasome subcomplex to an activated promoter. Science. 2002;296:548–550. doi: 10.1126/science.1069490. [DOI] [PubMed] [Google Scholar]
- Gordon C, McGurk G, Dillon P, Rosen C, Hastie ND. Defective mitosis due to a mutation in the gene for a fission yeast 26S protease subunit. Nature. 1993;366:355–357. doi: 10.1038/366355a0. [DOI] [PubMed] [Google Scholar]
- Groll M, Huber R. Substrate access and processing by the 20S proteasome core particle. Int J Biochem Cell Biol. 2003;35:606–616. doi: 10.1016/s1357-2725(02)00390-4. [DOI] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Groll M, Bajorek M, Köhler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D. A gated channel into the proteasome core particle. Nat Struct Biol. 2000;7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegeneration in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature. 2008;453:481–488. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler A, Cascio P, Leggett DS, Woo KM, Goldberg AL, Finley D. The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol Cell. 2001;7:1143–1152. doi: 10.1016/s1097-2765(01)00274-x. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Kanaan NM, Chu Y, Suresh Babu R, Stansell J, 3rd, Terpstra BT, Sortwell CE, Steece-Collier K, Collier TJ. Failure of proteasome inhibitor administration to provide a model of Parkinson's disease in rats and monkeys. Ann Neurol. 2006;60:264–268. doi: 10.1002/ana.20935. [DOI] [PubMed] [Google Scholar]
- Koulich E, Li X, Demartino GN. Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol Biol Cell. 2008;19:1072–1082. doi: 10.1091/mbc.E07-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YA, Lawson TG, Velayutham M, Zweier JL, Pickart CM. A proteasomal ATPase subunit recognises the polyubiquitin degradation signal. Nature. 2002;416:763–767. doi: 10.1038/416763a. [DOI] [PubMed] [Google Scholar]
- László L, Doherty FJ, Watson A, Self T, Landon M, Lowe J, Mayer RJ. Immunogold localisation of ubiquitin-protein conjugates in primary (azurophilic) granules of polymorphonuclear neutrophils. FEBS Lett. 1991;279:175–178. doi: 10.1016/0014-5793(91)80142-p. [DOI] [PubMed] [Google Scholar]
- Lindeberg J, Mattsson R, Ebendal T. Timing the doxycycline yields different patterns of genomic recombination in brain neurons with a new inducible Cre transgene. J Neurosci Res. 2002;68:248–253. doi: 10.1002/jnr.10213. [DOI] [PubMed] [Google Scholar]
- Lindeberg J, Usoskin D, Bengtsson H, Gustafsson A, Kylberg A, Söderström S, Ebendal T. Transgenic expression of Cre-recombinase from the tyrosine hydroxylase locus. Genesis. 2004;40:67–73. doi: 10.1002/gene.20065. [DOI] [PubMed] [Google Scholar]
- Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson's disease, Pick's disease, and Alzheimer's disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol. 1988;155:9–15. doi: 10.1002/path.1711550105. [DOI] [PubMed] [Google Scholar]
- Lundgren J, Masson P, Mirzaei Z, Young P. Identification and characterization of a Drosophila proteasome regulatory network. Mol Cell Biol. 2005;25:4662–4675. doi: 10.1128/MCB.25.11.4662-4675.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neurol. 2004;56:149–162. doi: 10.1002/ana.20186. [DOI] [PubMed] [Google Scholar]
- Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM, Krüger E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of mammalian proteasomes. J Biol Chem. 2003;278:21517–21525. doi: 10.1074/jbc.M301032200. [DOI] [PubMed] [Google Scholar]
- Melrose HL, Lincoln SJ, Tyndall GM, Farrer MJ. Parkinson's disease: a rethink of rodent models. Exp Brain Res. 2006;173:196–204. doi: 10.1007/s00221-006-0461-3. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Ravikumar B, Rubinsztein DC. Protective roles for induction of autophagy in multiple proteinopathies. Autophagy. 2006;2:224–225. doi: 10.4161/auto.2696. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5:177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Osmulski PA, Gaczynska M, Glickman MH. The central unit within the 19S regulatory particle of the proteasome. Nat Struct Mol Biol. 2008;15:573–580. doi: 10.1038/nsmb.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin DM, Glickman MH, Larsen CN, Dhruvakumar S, Finley D. Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J. 1998;17:4909–4919. doi: 10.1093/emboj/17.17.4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KY, Garza JC, Lu XY, Barsh GS, Kopito RR. Hypothalamic neurodegeneration and adult-onset obesity in mice lacking the Ubb polyubiquitin gene. Proc Natl Acad Sci U S A. 2008;105:4016–4021. doi: 10.1073/pnas.0800096105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Hanna J, Elsasser S, Finley D. Proteasome-associated proteins: regulation of a proteolytic machine. Biol Chem. 2005;386:725–737. doi: 10.1515/BC.2005.085. [DOI] [PubMed] [Google Scholar]
- Seeger M, Gordon C, Ferrell K, Dubiel W. Characteristics of 26S proteases from fission yeast mutants, which arrest in mitosis. J Mol Biol. 1996;263:423–431. doi: 10.1006/jmbi.1996.0586. [DOI] [PubMed] [Google Scholar]
- Shults CW. Lewy bodies. Proc Natl Acad Sci U S A. 2006;103:1661–1668. doi: 10.1073/pnas.0509567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikder D, Johnston SA, Kodadek T. Widespread, but non-identical, association of proteasomal 19 and 20 S proteins with yeast chromatin. J Biol Chem. 2006;281:27346–27355. doi: 10.1074/jbc.M604706200. [DOI] [PubMed] [Google Scholar]
- Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL. ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol Cell. 2005;20:687–698. doi: 10.1016/j.molcel.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell. 2007;27:731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanahashi N, Murakami Y, Minami Y, Shimbara N, Hendil KB, Tanaka K. Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J Biol Chem. 2000;275:14336–14345. doi: 10.1074/jbc.275.19.14336. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Hadjantonakis AK, Nagy A. Aggregation chimeras. Combining ES cells, diploid and tetraploid embryos. Methods Mol Biol. 2001;158:135–154. doi: 10.1385/1-59259-220-1:135. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, Spillantini MG. alpha-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang CE, Orr A, Tydlacka S, Li SH, Li XJ. Impaired ubiquitin-proteasome system activity in the synapses of Huntington's disease mice. J Cell Biol. 2008;180:1177–1189. doi: 10.1083/jcb.200709080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welchman RL, Gordon C, Mayer RJ. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol. 2005;6:599–609. doi: 10.1038/nrm1700. [DOI] [PubMed] [Google Scholar]
- Wójcik C, DeMartino GN. Analysis of Drosophila 26S proteasome using RNA interference. J Biol Chem. 2002;277:6188–6197. doi: 10.1074/jbc.M109996200. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]