

Abstract
Diacylglycerol (DAG) and phorbol esters strongly potentiate transmitter release at synapses by activating protein kinase C (PKC) and members of the Munc13 family of presynaptic vesicle priming proteins. This PKC/Munc13 pathway has emerged as a crucial regulator of release probability during various forms of activity-dependent enhancement of release. Here, we investigated the relative roles of PKC and Munc13-1 in the phorbol ester potentiation of evoked and spontaneous transmitter release at the calyx of Held synapse. The phorbol ester phorbol 12,13-dibutyrate (1 μm) potentiated the frequency of miniature EPSCs, and the amplitudes of evoked EPSCs with a similar time course. Preincubating slices with the PKC blocker Ro31-82200 reduced the potentiation, mainly by affecting a late phase of the phorbol ester potentiation. The Ro31-8220-insensitive potentiation was most likely mediated by Munc13-1, because in organotypic slices of Munc13-1H567K knock-in mice, in which DAG binding to Munc13-1 is abolished, the potentiation of spontaneous release by phorbol ester was strongly suppressed. Using direct presynaptic depolarizations in paired recordings, we show that the phorbol ester potentiation does not go along with an increase in the number of readily releasable vesicles, despite an increase in the cumulative EPSC amplitude during 100 Hz stimulation trains. Our data indicate that activation of Munc13 and PKC both contribute to an enhancement of the fusion probability of readily releasable vesicles. Thus, docked and readily releasable vesicles are a substrate for modulation via intracellular second-messenger pathways that act via Munc13 and PKC.
Keywords: synapse, readily releasable pool, release probability, second messenger, diacylglycerol, organotypic culture
Introduction
Synaptic transmission is highly regulated by processes of short- and long-term synaptic plasticity, as well as by intracellular second-messenger cascades. A particularly important signaling cascade that targets the presynaptic compartment of synapses is the protein kinase C (PKC)/Munc13 pathway. It is well known that phorbol esters potently enhance transmitter release at synapses (Malenka et al., 1986; Shapira et al., 1987; Hori et al., 1999; Yawo, 1999). Phorbol esters are functional analogues of the lipid-signaling molecule diacylglycerol (DAG), a phospholipid cleavage product that is generated by phospholipase C and that activates C1-domain-containing proteins, like PKCs (Newman, 1997). However, recent studies showed that phorbol esters also activate Munc13s, presynaptic vesicle priming proteins (Augustin et al., 1999; Richmond et al., 1999), which contain a C1 domain that binds DAG and phorbol esters (Betz et al., 1998; Lackner et al., 1999; Rhee et al., 2002).
Using a knock-in mouse model in which DAG/phorbol ester binding to the C1 domain of Munc13-1 is impaired (the Munc13-1H567K mutant) (Betz et al., 1998), it was shown that Munc13-1 plays a critical role for the phorbol ester-mediated potentiation of release in hippocampal neurons, and no evidence for an involvement of PKC was found (Rhee et al., 2002). However, it has been shown at various synapses that PKC inhibitors suppress at least part of the phorbol ester potentiation (Hori et al., 1999; Yawo, 1999; Oleskevich and Walmsley, 2000; Wu and Wu, 2001), implying a role for PKC as well. Activation of presynaptic PKC also plays a critical role for activity-dependent enhancement of transmitter release (Brager et al., 2003; Korogod et al., 2007), and a PKC-dependent phosphorylation of Munc18 is necessary for the phorbol ester potentiation of release in hippocampal synapses (Wierda et al., 2007). Thus, there is evidence that PKC-dependent mechanisms contribute to the enhancement of release during short-term plasticity and phorbol ester treatment, but the exact role(s) of activation of PKC versus Munc13-1 during DAG/phorbol ester stimulation of release have not been settled.
The potentiation of evoked release by phorbol esters also goes along with an increased frequency of miniature EPSCs (mEPSCs) (Shapira et al., 1987; Parfitt and Madison, 1993; Hori et al., 1999; Lou et al., 2005). A recent explanation for both effects assumes that phorbol esters modulate the fusion “willingness” of readily releasable vesicles, a modulation that was shown to influence both spontaneous release, as well as Ca2+-evoked release (Lou et al., 2005). However, imaging vesicle pool recycling has suggested that vesicles released spontaneously could use a separate recycling pathway (Sara et al., 2005) (but see Groemer and Klingauf, 2007), and styryl dye imaging also suggested that potentiation of evoked and spontaneous release is mediated by separate mechanisms (Waters and Smith, 2000).
Here, we address the question whether potentiation of release by phorbol ester at the calyx of Held is mediated by PKC, or by activation of Munc13-1. To this end, we used pharmacological tools, and developed a novel organotypic slice culture of the mouse brainstem, which allowed us to measure spontaneous transmitter release and its potentiation by phorbol esters in perinatally lethal Munc13-1H567K knock-in mice. We investigated the potentiation of spontaneous and of evoked release by phorbol esters and found that both a PKC-dependent and a Munc13-dependent mechanism contribute to enhance the effective fusion probability of a given readily releasable vesicle.
Materials and Methods
Slice preparation.
Native brainstem slices of 200 μm thickness containing the region of the medial nucleus of trapezoid body (MNTB) were made using 8- to 11-d-old Wistar rats, with postnatal day 0 (P0) referring to the day of birth. The slices were kept in a chamber at initially 36°C, which was allowed to cool to room temperature after 1 h, containing the following solution (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 25 glucose, 0.4 ascorbic acid, 3 myo-inositol, and 2 Na-pyruvate, continuously bubbled with 95% O2 5% CO2, pH 7.4. Recordings were made at room temperature (21–24°C) under visual control with an upright microscope (Zeiss FS2) equipped with a 60× objective (Olympus) and gradient contrast illumination (Luigs and Neumann). For afferent fiber stimulation (see Figs. 1, 2, 6, 7), a bipolar stimulation electrode custom made from Teflon-coated platinum/iridium wire (0.125 mm diameter; World Precision Instruments) was placed between the midline and the MNTB, and pulses of 100 or 200 μs length and 5–30 V were applied.
Figure 1.

Phorbol esters enhance the amplitude of evoked EPSCs and the frequency of mEPSCs in parallel. A, Bath perfusion of 1 μm PDBu enhances the amplitudes of EPSCs evoked by fiber stimulation (left) and the mEPSC frequency (right). Traces obtained before and after application of PDBu are shown in black and gray, respectively. B, Amplitude of evoked EPSCs (closed symbols) and frequency of spontaneous mEPSCs (open symbols) both normalized to their average values before application of PDBu. Note the parallel increase of evoked EPSC amplitude and mEPSC frequency. Basal EPSC amplitudes and mEPSC frequency are indicated. Same cell as shown in A. C, Average relative potentiation curves for evoked EPSC amplitudes (closed symbols) and mEPSC frequency (open symbols; n = 4 cells). These and the experiments shown in Figure 2 were done at slightly lowered extracellular [Ca2+] (1.2 mm).
Figure 2.
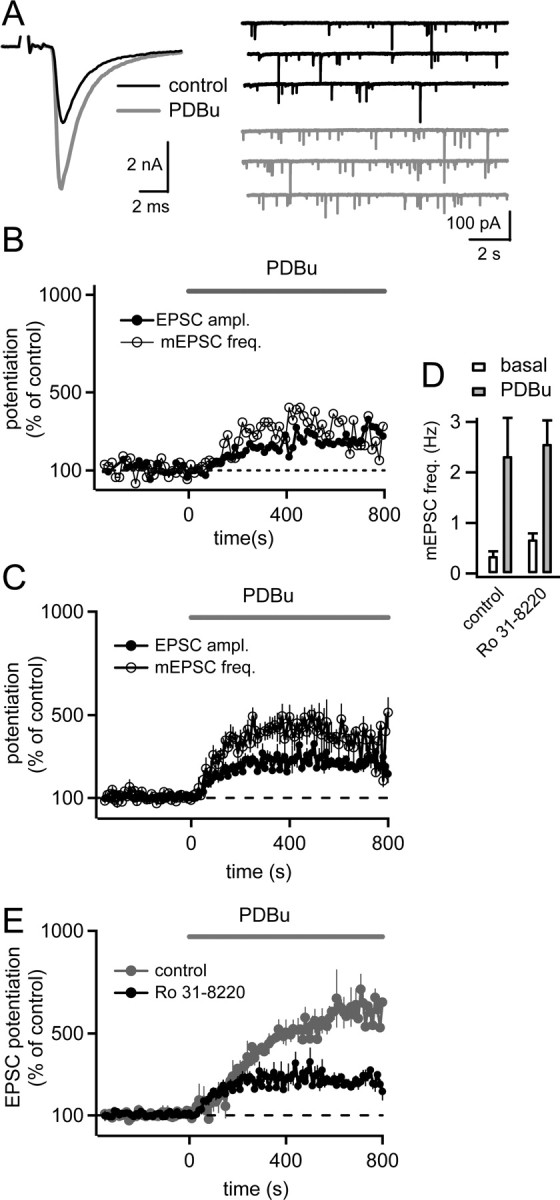
Suppression of the phorbol ester potentiation of evoked and spontaneous release by the PKC blocker Ro31-8220. A, Example traces of evoked EPSCs (left) and mEPSCs (right) from a slice preincubated with Ro31-8220 (3 μm), both before (black traces) and after bath application of 1 μm PDBu (gray traces). B, Time course of the relative potentiation of EPSC amplitudes (closed symbols) and mEPSC frequency (open symbols) for the same cell as shown in A. C, Average relative potentiation of evoked EPSC amplitudes and mEPSC frequency after preincubation with 3 μm Ro31-8220 (n = 11 cells). Note the smaller potentiation and the absence of a late phase of the potentiation compared with control conditions (Fig. 1). D, Average mEPSC frequency under basal conditions before PDBu application (open bars) and 400–800 s after application of 1 μm PDBu (gray bars), both under control conditions (Fig. 1) and after preincubation with 3 μm Ro31-8220. E, Average time course of the potentiation of evoked EPSC amplitudes by phorbol ester under control conditions (gray trace, replotted from Fig. 1C; n = 4 cells), as well as for slices preincubated with 3 μm Ro31-8220 (black trace; n = 11 cells). Note that Ro31-8220 preferentially blocked a late phase of the phorbol ester potentiation.
Figure 6.

PDBu increases the cumulative EPSC amplitude during 100 Hz stimulus trains. A, EPSC evoked by 25 stimuli at 100 Hz before (black trace) and after application of 1 μm PDBu (gray trace). Inset, The first five EPSCs on an expanded time scale. Note that in the presence of PDBu, the EPSC amplitudes late in the train were slightly increased (arrow), an observation made in several recordings. B, C, Plot of EPSC amplitudes (B) and of cumulative EPSC amplitudes (C), both before and after application of 1 μm PDBu (closed and open symbols, respectively). Each trace represents the average of n = 5 successive stimuli. The cumulative EPSC amplitude plot (C) was fitted in an intermediate range (0.1–0.15 s) with a line, which was back-extrapolated to time 0 (dashed line). D, Time course of the potentiation of the first EPSC amplitude in each 100 Hz train (filled symbols), and of the cumulative EPSC amplitude (open symbols). Note that a large part of the potentiation of the cumulative EPSC amplitude occurs at a time when also the first EPSC amplitude is potentiated. Data in A–D are from the same cell.
Figure 7.

Increasing the extracellular [Ca2+] also leads to an increase in the cumulative EPSC amplitude during 100 Hz trains. A–C, EPSC traces in response to 100 Hz trains (A), plot of EPSC amplitudes (B), and cumulative EPSC amplitude plot (C), both for the control condition (2 mm [Ca2+]) and after applying 4 mm [Ca2+]. A, Inset, The first five EPSCs at an expanded time scale. Note that the increase in cumulative EPSC amplitude mostly results from the increased first to fourth EPSC amplitudes. D, Time course of the increase of the first EPSC amplitude (filled symbols) and of the cumulative EPSC amplitude (open symbols). Data in A–D are from the same cell.
Electrophysiology and solutions.
Whole-cell patch-clamp recordings were made with a double EPC-10 amplifier (HEKA Elektronik). Series resistance (Rs) was 3–10 MΩ for postsynaptic recordings and was compensated by 50–85% through the Rs compensation circuit of the amplifier such that the remaining (uncompensated) Rs did not exceed 3 MΩ. The remaining Rs error in the postsynaptic current traces was compensated with an off-line routine (Wölfel et al., 2007). For postsynaptic recordings, including those in organotypic slices, the pipette solution contained (in mm) 130 Cs-gluconate, 20 tetraethylammonium (TEA)-Cl, 20 HEPES, 5 EGTA, 5 Na2-phosphocreatine, 4 MgATP, and 0.3 Na2GTP. The extracellular solution during recordings had the same composition as the slice keeping solution (see above), except for CaCl2, which was varied in some experiments (see Figs. 1, 2, 7) as indicated. The extracellular solution was complemented with 10 μm bicuculline and 2 μm strychnine for the experiments shown in Figures 1 and 2, or with 100 μm cyclothiazide (CTZ) and 1 mm kynurenic acid for the experiments shown in Figures 6 and 7. For simultaneous presynaptic and postsynaptic recordings (see Fig. 5), 100 μm CTZ, 1 mm kynurenic acid, 50 μm d-APV, 10 mm TEA, and 0.5 μm TTX were added to the extracellular solution. We aimed to obtain presynaptic Rs values not lower than 12 MΩ (range, 12–25 MΩ) by pulling patch pipettes with appropriate resistance (4–6 MΩ) and geometry. With presynaptic Rs values <10 MΩ, it was usually not possible to observe a potentiating effect of phorbol 12,13-dibutyrate (PDBu; from Sigma) on EPSCs, presumably because an intracellular factor important for the phorbol ester potentiation was diluted from the presynaptic nerve terminal.
Figure 5.

Phorbol esters do not increase the size of a readily releasable pool as defined by strong pool-depleting presynaptic depolarizations. A1, Presynaptic Ca2+ current (top) and the resulting EPSCs (bottom), both before (black traces) and after application of 1 μm PDBu (gray traces). Note the small increase (∼15%) of voltage-gated Ca2+ currents caused by PDBu (arrow), consistent with previous results (Korogod et al., 2007). The presynaptic voltage protocol is shown on top. A2, Transmitter release rate as obtained by EPSC deconvolution (top), and integrated release rate traces (bottom). B1, B2, Traces similar to those in A1 and A2, respectively, but in response to long (30 ms) presynaptic depolarizations, which were given between the short depolarizations. Note that EPSCs evoked by these pool-depleting stimuli were large (∼22 nA in this example) and not strongly potentiated by PDBu. The cumulative release rate traces in the bottom of B2 were fitted with double-exponential functions. The fast exponential functions are shown superimposed (dashed lines), as well as the difference trace that should indicate the slow release component (dotted traces). Note that neither of the two release components was strongly potentiated by PDBu. Scale bars in B1 and B2 are the same as those in A1, A2. C, Time course of the relative potentiation of the EPSCs evoked by short (round symbols) and by long depolarizations (square symbols) for the same cell as shown in A and B. D, Average relative potentiation by PDBu of EPSCs evoked by short (left bar) and by long depolarizations (right bar).
The PKC blocker 2-[1-(3-(amidinothio)propyl)-1H-indol-3-yl]-3-(1-methylindol-3-yl)maleimide methanesulfonate (Ro31-8220) was purchased from Calbiochem, and stock solutions were made in DMSO. For preincubation with Ro31-8220, slices were kept for at least 30 min in a small incubation chamber (∼25 ml volume, 36°C) that contained the standard Ringer's solution bubbled with 95% O2 and 5% CO2, to which 3 μm Ro31-8220 was added. For subsequent recordings from these slices, 3 μm Ro31-8220 was present in all perfusion solutions. After mounting on the microscope stage for recording, the slices were continuously perfused by a gravity-driven perfusion system at a rate of 1.3–2 ml/min. To guarantee the fastest possible exchange times of PDBu (1 μm), we minimized the volume of the recording chamber (∼700–900 μl).
Organotypic slices cultures.
Organotypic slices at the level of the superior olivary complex (Lohmann et al., 1998) of newborn mice (P0) were made using a standard slice culture technique (Stoppini et al., 1991). After decapitation of a P0 mouse and careful dissection of the brain, 350-μm-thick coronal hindbrain slices were made with a tissue chopper. Two to three slices of the region of the auditory nerve were cultured from each mouse pup on a membrane (Millicell Organotypic; Millipore) in Neurobasal medium (75%) supplemented with horse serum (25%) in the presence of 0.2% penicillin–streptomycin and 25 mm KCl. The elevated [K+] might enhance activity and lead to a better preservation of the cytoarchitecture of the auditory nuclei (Lohmann et al., 1998). On the day of recording, the location of the MNTB nuclei in the organotypic slices was assessed by morphological criteria. Whole-cell recordings of neurons located in the MNTB were made under visual control in an upright microscope, similar as for recordings in native slices (see above). The extracellular solution was a bicarbonate-buffered standard solution (see above) to which 0.5 μm TTX, 50 μm d-AP5, 10 μm bicuculline, and 2 μm strychnine were added.
We bred heterozygous Munc13-1H576K/wild-type mice in a Munc13-2 knock-out background (Varoqueaux et al., 2002) to eliminate a possible contribution of the Munc13-2 isoform to DAG signaling. The offspring of such breeding pairs were used to make organotypic slices, with the aim to obtain slices from both Munc13-1 wild-type mice as a control group, and from homozygous Munc13-1H567K/H567K mice (these homozygous mutant mice are designated Munc13-1H567K in what follows). Because homozygous Munc13-1H567K mice die early after birth (Rhee et al., 2002), the preparation was made soon (1–4 h) after the birth of the litter, using two or three of the weakest pups (which often turned out to be homozygous Munc13-1H567K after subsequent genotyping), and another two or three healthy pups of the same litter. In all experiments, tail biopsies were kept for genotyping. After 5–10 d in vitro, recordings were made either from MNTB neurons in organotypic slices obtained from homozygous Munc13-1H567K mice, or, as a control group, from homozygous Munc13-1wild-type mice of the same litter.
Analysis of release rate and mEPSC detection.
Analysis was done with routines written in IgorPro (WaveMetrics). Transmitter release rates (see Fig. 5A2,B2, top) were calculated by deconvolving the measured and off-line-corrected EPSC traces with an idealized mEPSC waveform as described previously (Neher and Sakaba, 2001; Wölfel et al., 2007). Spontaneous (miniature) EPSCs were detected with a template matching routine, using current recordings of ∼9.7 s that were acquired between single-fiber stimulations (see Figs. 1, 2). Although these spontaneous EPSCs needed to be recorded in the absence of TTX, they likely represented quantal mEPSCs originating from calyces of Held for the following reasons. First, application of TTX does not lead to a change in the amplitude distribution of spontaneous EPSCs at the calyx of Held (Ishikawa et al., 2002). Second, the frequency of mEPSCs in MNTB principal cells can be enhanced by depolarizing changes in the holding potential of the calyx (Sahara and Takahashi, 2001) or by infusing the calyx with Ca2+-buffered solutions (Lou et al., 2005). This indicates that spontaneous EPSCs mostly originate from the calyx of Held. However, a small contribution of spontaneous EPSCs arising from noncalyceal inputs onto MNTB principal cells (Hamann et al., 2003) cannot be excluded, but should not greatly affect our conclusions. For the recordings in organotypic slices (see Figs. 3, 4), 0.5 μm TTX was present in the extracellular solution at all times.
Figure 3.

Morphological properties and spontaneous excitatory synaptic transmission in organotypic slice cultures of the MNTB. Aa–Ac, An organotypic slice of the superior olivary complex, made from a P0 mouse and kept for 8 d in vitro. Staining with an anti-parvalbumin antibody (see Materials and Methods) shows the MNTB as groups of parvalbumin-positive neurons on both sides of the midline, as well as parvalbumin-positive fibers that cross the midline. Images taken at a higher resolution from within the MNTB (Ab, Ac) show parvalbumin-positive cell bodies (asterisks), fibers (arrows), and parvalbumin-positive nerve endings close to the principal cells (arrowheads). Scale bars, 20 μm. B, Gradient-contrast infrared image of an organotypic slice with a whole-cell patch-pipette attached to an MNTB principal cell. C, mEPSCs in a MNTB neurons from an organotypic slice culture before (top) and after (bottom) application of 2 μm NBQX. D, Superimposed mEPSCs (n = 40) together with the average trace (black trace), which was fitted with an exponential function with time constant of 1 ms (white trace). E, Amplitude distribution of mEPSCs, with a mean amplitude of 42 pA in this cell. Data in C–E are from the same cell.
Figure 4.
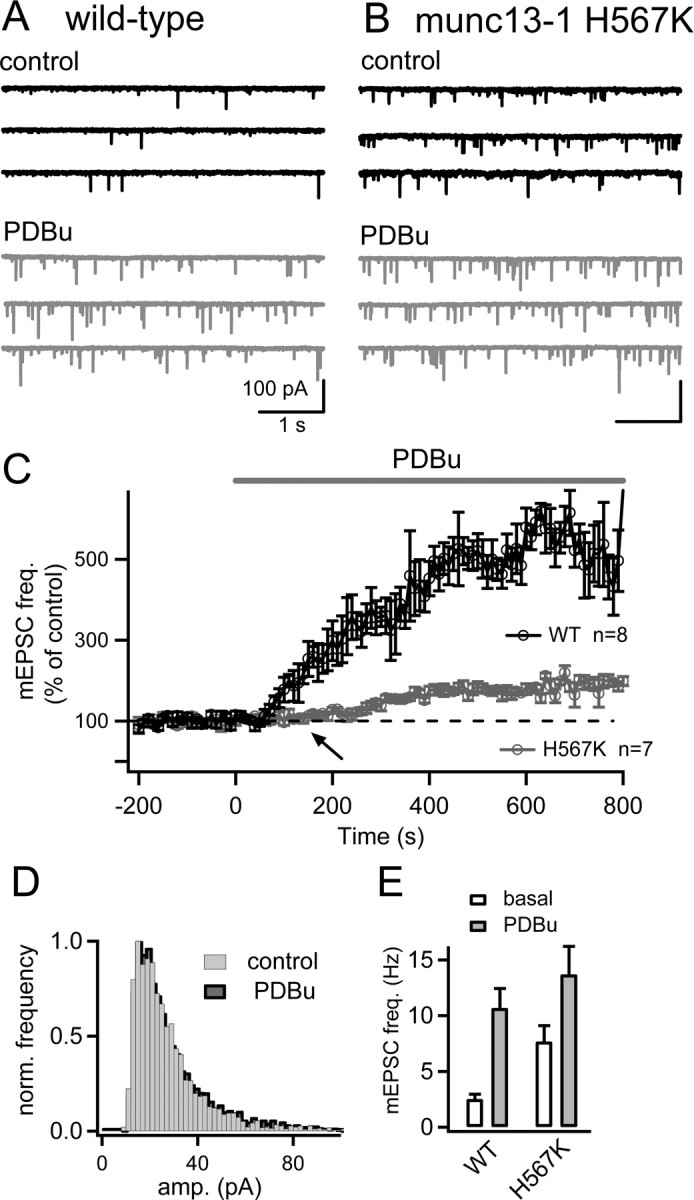
The potentiation of mEPSC frequency by phorbol ester is strongly suppressed in organotypic slices from Munc13-1H567K knock-in mice. A, Postsynaptic current traces before (black) and after application of 1 μm PDBu (gray) in an organotypic slice of a wild-type mouse. B, Similar experiment as in A, but using an organotypic slice of a homozygous Munc13-1H567K knock-in mouse. Note the higher basal mEPSC frequency and the smaller increase of mEPSC frequency after PDBu application. C, Average time course of the relative mEPSC frequency in control mice (black symbols) and in homozygous Munc13-1H567K knock-in mice (gray symbols). D, The mEPSC amplitude distribution for a Munc13-1H567K cell before (gray bars) and after (black bars) application of 1 μm PDBu, demonstrating that PDBu does not lead to change in the average mEPSC amplitude. E, Average mEPSC frequency under basal conditions (open bars) and after application of 1 μm PDBu (gray bars) both for wild-type (left) and homozygous Munc13-1H567K knock-in neurons. Note the significantly higher basal mEPSC frequency in Munc13-1H567K neurons (p = 0.004).
Results
PKC activation contributes to phorbol ester potentiation of evoked EPSC and mEPSC frequency
We started by investigating the contribution of a PKC-dependent mechanism to the phorbol ester potentiation of transmitter release using the PKC inhibitor Ro31-8220. In these experiments, we wished to quantify the potentiation of evoked release, as well as the potentiation of spontaneous release by phorbol ester. To do so, we stimulated afferent fibers at a low stimulation frequency (every 10 s) to assess evoked release. In the intervals between stimuli, we recorded spontaneous EPSCs, which mostly represent mEPSCs originating from the calyx of Held (Ishikawa et al., 2002) (see Materials and Methods). The experiments were performed at a lowered extracellular [Ca2+] (1.2 mm) with the aim to maximize the potentiation of evoked release by phorbol ester. Under control conditions (in the absence of Ro31-8220), bath application of the phorbol ester PDBu (1 μm) led to a strong potentiation of EPSC amplitudes (Fig. 1A, left). The time course of the EPSC potentiation was plotted as the EPSC amplitudes relative to their control value before the application of PDBu (Fig. 1B, filled symbols). This plot shows that the potentiation developed over several minutes, reaching 576 ± 73% (n = 4 cells) of the control EPSC amplitude as quantified during an interval of 400–800 s after the start of the PDBu application. After prolonged recording with 1 μm PDBu, the extracellular [Ca2+] was increased to 2 mm. This resulted in a strong additional increase of the evoked EPSC amplitude (to 22.5 nA in the example of Fig. 1A) (data not shown), demonstrating that EPSC amplitudes and presynaptic release probability were not maximal in the presence of PDBu at 1.2 mm [Ca2+].
Application of PDBu also caused an increase of the mEPSC frequency (Hori et al., 1999; Oleskevich and Walmsley, 2000; Lou et al., 2005). To analyze the time course of this effect, the mEPSCs were counted in each 10 s recording sweep, converted to frequency, and then normalized to the control frequency before PDBu application (Fig. 1B, open symbols). In the cell illustrated in Figure 1, the time course of the potentiation of mEPSC frequency and EPSC amplitudes were indistinguishable (Fig. 1B, compare open and, closed symbols). When the relative potentiation curves for evoked release and mEPSC frequency were averaged across all cells investigated under control conditions (n = 4 cells), the potentiation of evoked EPSC amplitudes and of mEPSC frequency was also very similar (Fig. 1C). On average, the increase in mEPSC frequency measured in a time window of 400–800 s after the start of the PDBu application was 667 ± 72% of the control mEPSC frequency. The similar time course and amount of the potentiation of mEPSC frequency and evoked EPSC amplitude is consistent with the idea that the potentiation of spontaneous as well as of Ca2+-evoked release reflects the same modulatory process of the release machinery (but see Waters and Smith, 2000; Lou et al., 2005; Basu et al., 2007).
To investigate the contribution of PKC-dependent mechanisms to the phorbol ester potentiation of transmitter release, we next preincubated slices with 3 μm the PKC inhibitor Ro31-8220 for at least 30 min, and then measured the potentiation of EPSC amplitude and mEPSC frequency in the presence of Ro31-8220 in all recording solutions (Fig. 2). Under these conditions, a potentiation of evoked EPSC amplitudes and mEPSC frequency was still observed (Fig. 2A) (279 ± 22% and 391 ± 30%, respectively; n = 11 cells), but the potentiation was significantly smaller than that observed under control conditions (see above) (p = 0.02 and 0.019 for EPSC amplitudes and mEPSC frequency, respectively). Figure 2B shows the time course of the phorbol ester potentiation for the cell illustrated in Figure 2A. In this cell, the relative potentiation of mEPSC frequency was slightly larger than the potentiation of evoked EPSC amplitudes (Fig. 2B, open and closed data points, respectively). This observation was confirmed when the relative potentiation curves were averaged for all cells (n = 11) (Fig. 2C). In this plot, it becomes apparent that the potentiation that remained in the presence of 3 μm Ro31-8220 was slightly larger for mEPSC frequency (391 ± 30% of control) than for EPSC amplitudes (279 ± 22%). This indicates that a PKC-dependent mechanism is somewhat more important for the potentiation of Ca2+-evoked release, than for the potentiation of spontaneous release (see Discussion).
In the slices preincubated with the PKC blocker Ro31-8220, the control EPSC amplitudes before application of phorbol ester were not significantly different from those recorded under control conditions (control, 1.74 ± 0.7 nA, n = 4 cells; Ro31-8220, 2.4 ± 0.4 nA, n = 11 cells; p = 0.45; 1.2 mm [Ca2+]). Thus, the baseline synaptic strength does not depend on a constitutive activation of PKC, in agreement with previous results (Korogod et al., 2007). We found, however, that the mEPSC frequency under baseline conditions (i.e., before PDBu application) was slightly higher after preincubation with Ro31-8220 (0.67 ± 0.12 Hz; n = 10 cells) than under control conditions (0.34 ± 0.1 Hz; n = 4 cells) (Fig. 2D, open bars). However, because of the cell-to-cell variability of basal mEPSC frequency, this effect did not reach statistical significance (p = 0.054).
In the presence of Ro31-8220, the phorbol ester potentiation of EPSC amplitudes and of mEPSC frequency reached a steady-state quite early (Fig. 2B,C), whereas under control conditions, the potentiation of both forms of release continued for a longer time and reached higher final values (Fig. 1B,C). Thus, it seems that Ro31-8220 decreased the amount of the phorbol ester potentiation mainly by inhibiting a late phase of the potentiation. This can be seen more directly by overlaying the average relative potentiation curves of EPSC amplitudes under control conditions, and in the presence of Ro31-8220 (Fig. 2E, gray and black data points, respectively). Based on the overlay of the SEM values, both data sets are not significantly different within the first ∼200 s after application of phorbol ester; at this time, a potentiation of ∼250% of control was attained (Fig. 2E). However, between 200 and 800 s after the onset of the application of PDBu, the EPSC amplitude potentiation continued under control conditions, whereas the EPSC amplitudes remained nearly constant in the presence of the PKC inhibitor Ro31-8220 (Fig. 2E, black and gray traces, respectively). This suggests that a PKC-dependent mechanism is responsible for a late phase of the phorbol ester potentiation, whereas an early phase of the potentiation is mostly independent of PKC.
Phorbol ester binding to the C1 domain of Munc13 is essential for the potentiation of spontaneous release
It is possible that the PKC-independent component of phorbol ester potentiation is mediated by phorbol ester binding to Munc13s, presynaptic vesicle-priming proteins with a phorbol ester-binding C1 domain (Betz et al., 1998; Augustin et al., 1999; Lackner et al., 1999). To test the role of Munc13-1, we wished to investigate Munc13-1H567K knock-in mice (Rhee et al., 2002), which carry a point mutation in the C1 domain that abolishes phorbol ester and DAG binding (Betz et al., 1998). However, these knock-in mice die at birth (Rhee et al., 2002), and patch-clamp recordings in native brain slices are therefore not feasible. To circumvent this problem, we adapted a method of organotypic slice cultures of the superior olivary complex (Lohmann et al., 1998) using mouse brains obtained shortly after birth (P0) (see Materials and Methods). After maintaining the slices for 5–10 d in vitro, we recorded spontaneous excitatory transmission in MNTB neurons.
Figure 3 illustrates morphological properties of organotypic slice cultures and spontaneous excitatory transmission in MNTB brainstem neurons from these cultures. In Figure 3A, a slice culture established from a P0 mouse and maintained for 8 d in vitro was stained with an anti-parvalbumin antibody. Parvalbumin is present in MNTB principal cells and in nerve fibers of the trapezoid body, including calyces of Held, from ∼P6–P8 onwards (Lohmann and Friauf, 1996; Felmy and Schneggenburger, 2004). Indeed, parvalbumin-positive fibers crossing at the midline of the slice, and parvalbumin-positive groups of neurons were observed in organotypic slice cultures (Fig. 3Aa). Higher-magnification images show parvalbumin-positive neurons (Fig. 3Ab,Ac, stars), parvalbumin-positive fibers that likely represent axons (Fig. 3Ac, arrows), and thickenings of parvalbumin-positive fibers located opposite to cell bodies (Fig. 3Ab,Ac, arrowheads). Based on their location and parvalbumin-staining, we identify the groups of neurons present bilaterally from the midline as the MNTB (Fig. 3A).
Figure 3B shows a whole-cell patch-pipette attached to a MNTB neuron in a slice culture maintained for 7 d in vitro. We recorded spontaneous synaptic events in the presence of 0.5 μm TTX and also added bicuculline (10 μm) and strychnine (2 μm) to block GABAergic and glycinergic synaptic currents. The spontaneous currents shown in Figure 3C were glutamatergic, non-NMDA-receptor-mediated mEPSCs, based on their sensitivity to 2 μm NBQX (Fig. 3C, bottom) and on their fast decay time constant (∼1 ms) (Fig. 3D). The amplitude distribution of spontaneous mEPSC showed a mean of ∼40 pA in this cell, with an average value of 35 ± 3 pA across cells (n = 11 cells from wild-type cultures). This value is in good agreement with the average mEPSCs amplitudes of AMPA receptor (AMPA-R)-mediated EPSCs typically measured in MNTB neurons in native slices from P8–P10 mice (Fernández-Chacón et al., 2004; Joshi et al., 2004). Thus, AMPA-R-mediated mEPSC can be regularly measured from MNTB neurons maintained in organotypic slice cultures.
We next used the organotypic slice preparation to compare the potentiation of mEPSC frequency in Munc13-1H567K mice, and their Munc13-1 wild-type littermates. In slice cultures from wild-type mice, bath application of 1 μm PDBu strongly potentiated the mEPSC frequency (Fig. 4A), with an average potentiation of 453 ± 47% of the control value (n = 8 cells) (Fig. 4A,C). In Munc13-1H567K knock-in mice, the PDBu-induced potentiation of mEPSC frequency was significantly smaller, with a relative increase to only 181% ± 9.3% of the control frequency (Fig. 4B,C). In addition, in Munc13-1H567K mice, the potentiation of mEPSC frequency only started after a delay of ∼100–200 s (Fig. 4C, arrow), whereas in wild-type mice the potentiation was apparent already after ∼30 s of the application of PDBu. This indicates that abolishing phorbol ester binding to Munc13-1 affects an early component of the phorbol ester potentiation most strongly.
In the Munc13-1H567K mice, the basal mEPSC frequency was significantly higher (7.7 ± 1.4 Hz) than in Munc13-1 wild-type mice (2.5 ± 0.45 Hz) (Fig. 4E, open bars) (p = 0.004). The final value of mEPSC frequency after application of 1 μm PDBu was, however, not significantly different between the two genotypes (p = 0.39) (Fig. 4E, filled gray bars). Thus, in Munc13-1H567K mice, the higher baseline mEPSC frequency might reduce the potential for a dynamic regulation of mEPSC frequency. This finding is consistent with the recent proposal that the H567K mutation in Munc13-1 acts as a gain-of-function mutation that by itself increases the fusion willingness of readily releasable vesicles (Basu et al., 2007).
Phorbol ester does not increase the size of the readily releasable vesicle pool as tested by pool-depleting stimuli
An increase in mEPSCs frequency indicates that the fusion probability of docked and readily releasable vesicles is increased (Lou et al., 2005). An alternative interpretation would be that the increased mEPSC frequency is (in part) caused by an increase in the number of vesicles in the readily releasable pool. Indeed, phorbol esters increase the size of the readily releasable pool in chromaffin cells (Gillis et al., 1996), and a pool size increase was also reported at hippocampal synapses (Stevens and Sullivan, 1998) (but see Basu et al., 2007; Wierda et al., 2007). However, experiments at the calyx of Held using direct presynaptic recordings have found no evidence for an increase in the readily releasable pool as tested in membrane capacitance measurements (Wu and Wu, 2001) and Ca2+ uncaging experiments (Lou et al., 2005).
To verify the conclusion that phorbol esters do not primarily act via increasing the number of readily releasable vesicles, we investigated possible pool size changes in paired presynaptic and postsynaptic whole-cell recordings using prolonged presynaptic depolarizations to empty the readily releasable pool (Fig. 5). The experiments were performed in the presence of CTZ (100 μm) and kynurenic acid (1 mm) to prevent AMPA-R desensitization and saturation, respectively (Neher and Sakaba 2001). We applied both prolonged (30 ms) as well as brief (∼1–2 ms) presynaptic depolarizations to 0 mV with the aim to probe the pool of readily releasable vesicles with the long depolarizations and to verify the efficiency of PDBu with the short depolarizations. After establishing a baseline for EPSCs evoked by short and long depolarizations (Fig. 5C), the recording solution was switched to 1 μm PDBu (Fig. 5A1,B1, gray traces). Phorbol ester strongly potentiated the EPSCs evoked by the short depolarizations (Fig. 5A1) (378 ± 97% of control) but left the EPSCs evoked by pool-depleting stimuli mostly unchanged (Fig. 5B1, D) (112 ± 9% of control; p = 0.103; n = 8). This indicates that phorbol esters potentiate release independent of a pool size increase, at least during an early phase of the potentiation (∼200–300 s).
Prolonged presynaptic depolarizations have been shown to induce a fast and a slow phase of transmitter release at the calyx of Held (Sakaba and Neher, 2001; Wadel et al., 2007; Wölfel et al., 2007). The two distinct phases of release might arise from two subpools of releasable vesicles: “fast-releasing” and the “slowly releasing” subpools (Sakaba and Neher, 2001). To further distinguish whether phorbol ester could modulate the fast and slow subpools of the readily releasable vesicles differentially, we deconvolved the measured EPSCs to obtain transmitter release rates and traces of cumulative release. Transmitter release in response to short presynaptic depolarizations was brief (half-width, 0.6 ms) (Fig. 5A2, top), and the cumulative release rate traces showed that these stimuli released ∼240 vesicles in this example (Fig. 5A2, bottom, black traces). As expected from the robust potentiation of EPSCs in response to the brief depolarizations (Fig. 5A1), PDBu readily increased the transmitter release in response to short depolarizations, such that a total of ∼550 vesicles were released after PDBu application (Fig. 5A2, bottom, gray traces). In contrast, long presynaptic depolarizations (30 ms) evoked two release components, as apparent after fitting the cumulative release rate traces with double-exponential functions (Fig. 5B2, bottom) (Sakaba and Neher, 2001; Wölfel et al., 2007). Application of PDBu caused a small increase in the amplitude of the fast release component (from 1953 to 2145 vesicles in this example) (Fig. 5B2, bottom), that, on average, was not statistically significant (111 ± 8% of control; p = 0.23, n = 5). Also, phorbol ester did not significantly increase the number of vesicles released up to the end of a 30 ms depolarization, with 2715 vesicles under control conditions and 2945 vesicles after PDBu in the example of Figure 5. On average, PDBu increased the number of vesicles released after 30 ms of the prolonged depolarization to 110 ± 5% of control (p = 0.125; n = 5 pairs). Thus, activation of the presynaptic PKC/Munc-13 pathway by phorbol esters does not markedly increase the number of fast-releasable vesicles, nor does it significantly increase the total number of readily releasable vesicles.
Phorbol esters and elevated extracellular Ca2+ lead to an increase of the cumulative EPSC amplitudes during high-frequency trains
The experiments illustrated in Figure 5 showed that phorbol esters potentiate transmitter release independent of a pool size increase. We wished to corroborate this finding with a different method for estimating the size of the readily releasable pool, by using 100 Hz train stimuli (Schneggenburger et al., 1999). In this method, depression observed during high-frequency stimulation is assumed to result from the depletion of a readily releasable vesicle pool, and the cumulative EPSC amplitudes, back-extrapolated to time 0, are used as a minimal pool size estimate.
We recorded EPSCs in response to brief 100 Hz trains of afferent fiber stimulations at the standard extracellular [Ca2+] concentration of 2 mm in the presence of 100 μm CTZ and 1 mm kynurenic acid. Trains of 100 Hz stimuli with 25 stimuli were applied every 45 s, and EPSC depression curves were constructed from five to eight such trains (Fig. 6A–C). Thereafter, the bath solution was switched to 1 μm PDBu, and 100 Hz trains were continued to be evoked every 45 s. PDBu increased the first EPSC amplitudes in response to 100 Hz trains to 217 ± 22% of the control EPSC amplitude, consistent with previous results (Lou et al., 2005). The EPSC potentiation was smaller than that observed in Figure 1 (562 ± 12% of control; see above), most probably because the lower extracellular Ca2+ concentration used in Figure 1 maximized the potentiation of evoked EPSCs by phorbol ester (see Discussion).
In addition to strongly increasing the first EPSC amplitude, PDBu also enhanced the second, third, and fourth EPSCs during the 100 Hz train (Fig. 6A). As a consequence of the strongly increased first few EPSC amplitudes, the plot of cumulative EPSC amplitudes versus time showed a much faster rise in the presence of PDBu, and this offset did not recover later during the train (Fig. 6C). Linear back-extrapolation of the cumulative EPSC amplitudes therefore gave significantly higher values in the presence of PDBu (26.5 ± 2 nA) than under the control conditions (17.6 ± 1.6 nA; p = 0.024; n = 5 cells). The relative increase of the back-extrapolated cumulative EPSC amplitude was, on average, 167 ± 22% (p = 0.034) of the corresponding control value.
The finding that phorbol ester causes an apparent pool size increase as tested with cumulative EPSC amplitude plots during 100 Hz stimulation is unexpected, because when release was stimulated by strong presynaptic depolarizations to probe the pool size, no significant increase of cumulative release was observed (Fig. 5) (Wu and Wu, 2001; Lou et al., 2005). To investigate at which time after PDBu application the increase in cumulative EPSC amplitude occurred, we plotted the time course of the potentiation of the first EPSC amplitude, and the cumulative EPSC amplitude (Fig. 6D). This time plot shows that most of the increase of the cumulative EPSC amplitude occurred during an early phase of the PDBu potentiation (<300 s), when also the first EPSC amplitude was most strongly potentiated. During this early phase of the PDBu potentiation, however, prolonged presynaptic depolarizations indicated no pool size increase (Fig. 5). Therefore, the increased cumulative EPSC amplitude during 100 Hz trains does probably not indicate a true increase in the number of readily releasable vesicles, but rather, a more complete release of the readily releasable pool after increasing the presynaptic release probability.
To verify this possibility, we tested whether increasing the extracellular [Ca2+] would also lead to an increase in the cumulative EPSC amplitude (Fig. 7). Increasing the extracellular [Ca2+] enhances the presynaptic Ca2+ influx during each AP (Schneggenburger et al., 1999), and this manipulation is generally accepted to enhance the effective release probability of any given readily releasable vesicle. When we increased the extracellular [Ca2+] from 2 to 4 mm (at a constant [Mg2+]), the first EPSC of a 100 Hz train was increased to 269 ± 26% of its control amplitude (Fig. 7A), in good agreement with previous results (Schneggenburger et al., 1999). Similarly to the action of PDBu, elevating [Ca2+] also significantly increased the back-extrapolated cumulative EPSC amplitudes to 182 ± 11% of control (p = 0.001), and in the corresponding time plot, the increase of the cumulative EPSC amplitude correlated well with the increase of the first EPSC amplitude (Fig. 7D). It seems, therefore, that a 100 Hz train under conditions of standard extracellular [Ca2+] of ∼2 mm does not fully deplete the pool of readily releasable vesicles. In this situation, increasing the release probability by enhancing the presynaptic Ca2+ influx (Fig. 7) or by activating the PKC/Munc13 pathway by phorbol ester (Fig. 6) can lead to an increase in the cumulative EPSC amplitude during a 100 Hz train that primarily reflects a more complete release of the readily releasable pool, rather than a true pool size increase. Together, these results indicate that activation of the presynaptic PKC/Munc13 pathway does not enhance the size of the readily releasable pool, but rather, that phorbol esters act by increasing the probability of transmitter release, in agreement with the results shown in Figure 5 (Wu and Wu, 2001; Lou et al., 2005).
Discussion
We studied the potentiation of evoked and spontaneous transmitter release by the DAG analog PDBu (a phorbol ester), using pharmacological and genetic tools to define the roles of protein kinase C and Munc13-1 in the phorbol ester modulation at the calyx of Held. We find that a PKC-dependent pathway and activation of Munc13-1 by phorbol ester converge to enhance both forms of release. Paired presynaptic and postsynaptic recordings showed that the activation of this PKC/Munc13 pathway does not go along with a measurable pool size increase. The results indicate that DAG can act via a PKC-dependent pathway and directly on Munc13-1 to increase the fusion probability of vesicles in the readily releasable pool.
PKC and Munc13-1 both regulate evoked and spontaneous release
We found that the time course, and the amount of the phorbol ester potentiation of evoked EPSCs and mEPSC frequency is similar, with an approximately sixfold potentiation (Fig. 1). A potentiation of spontaneous release of approximately sixfold agrees with previous studies (Lou et al., 2005; Wierda et al., 2007); however, a sixfold potentiation of the evoked EPSC amplitude is higher than what has typically been observed. It is likely that the use of low extracellular [Ca2+] in these experiments (1.2 mm), which decreased the initial release probability, was permissive for observing the full amount of the potentiation of evoked release because the potentiation of evoked release at normal extracellular [Ca2+] was much less (∼2.5-fold) (Fig. 6) (Lou et al., 2005). Thus, lowering the release probability maximizes the potentiation of evoked release by phorbol esters, a finding that is consistent with the idea that phorbol esters increase the release probability (Yawo, 1999; Lou et al., 2005).
In the presence of the PKC inhibitor Ro31-8220, the potentiation of evoked and spontaneous release was significantly smaller than under control conditions, indicating that a PKC-dependent mechanism contributes to the potentiation of both forms of release. However, the potentiation of evoked release was more strongly affected by the PKC inhibitor (a reduction of approximately one-half), whereas the potentiation of spontaneous release was reduced by only approximately one-third (Fig. 2C). The larger effect of the PKC blocker on the potentiation of evoked release is likely attributable to the fact that phorbol ester also slightly increases the presynaptic Ca2+ current (Fig. 5A1) (Lou et al., 2005; Korogod et al., 2007), a modulation that is expected to selectively affect evoked release. We found that in slice cultures of the Munc13-1H567K mice, in which phorbol ester and DAG binding to Munc13-1 is abolished (Betz et al., 1998; Rhee et al., 2002), the potentiation of mEPSC frequency was strongly suppressed, by approximately two-thirds (Fig. 4), corresponding to the (relatively) small effect of the PKC blocker on the potentiation of spontaneous release. This indicates that activation of Munc-13 by phorbol ester has a larger effect on mEPSC frequency than activation of PKC. In support of this view, we showed recently that Ro31-8220 greatly suppresses post-tetanic potentiation of evoked release (Korogod et al., 2007), suggesting that PKC might be activated physiologically during high-frequency stimulation (Wierda et al., 2007), but Ro31-8220 did not reduce the increased mEPSC frequency observed after high-frequency stimulation (Korogod et al., 2007). This again indicates that the PKC-dependent mechanism is less important for the potentiation of spontaneous release, but it is important to note that a PKC-dependent mechanism contributed to the increase in mEPSC frequency (Fig. 2).
Recently, Wierda et al. (2007) identified Munc18 as an important target for PKC phosphorylation in the phorbol ester potentiation of release in hippocampal neurons (Fujita et al., 1996; Barclay et al., 2003). They also showed that a first brief application of phorbol ester rendered the PKC blockers ineffective during a second round of phorbol ester stimulation, suggesting a sequential action in which a PKC-dependent mechanism is a prerequisite for the activation of Munc13-1 by DAG. Although we have not been able to test conditioning applications of PDBu (because the effect of phorbol esters are nonreversible in slices) (Hori et al., 1999), our data are more compatible with a simple additive effect of both molecular mechanisms. Thus, the phorbol ester potentiation was blocked only partially by Ro31-8220 at a high concentration (3 μm), and a partial block was also observed with 2 μm bisindolymaleimide (data not shown). The Ro31-8220-resistant component of the potentiation likely represents activation of Munc13-1 by phorbol ester (Fig. 4); thus, Munc13-1 activation took place even without previous stimulation of PKC. However, we cannot exclude a certain degree of interdependence between both molecular mechanisms (Wierda et al., 2007).
Taken together, the following picture of the action of DAG and its analog phorbol ester emerges (Fig. 8). Because we showed that the potentiation is mostly independent of a pool-size increase (Figs. 5–7) (see below), the fusion probability of a given readily releasable vesicle must be modulated. This can occur by at least three different routes. First, activation of Munc13-1 by DAG directly modulates the fusion probability of readily releasable vesicles. This leads to an increase in both the spontaneous release rate (Fig. 4), and also in the Ca2+-evoked release because, in Ca2+ uncaging experiments, the phorbol ester potentiation of transmitter release was only partially blocked by Ro31-8220 (Korogod et al., 2007). The finding that Munc13-1 activation contributes to an increase in the fusion probability independent of a pool-size increase indicates a postpriming role for Munc-13 in modulating neurotransmitter release (Basu et al., 2007). Thus, Munc13 probably remains bound to the SNARE [soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor] complex and forms a DAG receptor. Second, a PKC-dependent pathway, acting via phosphorylation of SNAP-25 (Nagy et al., 2002; Shu et al., 2008) (but see Finley et al., 2003) and/or Munc18 (Wierda et al., 2007) also enhances the spontaneous release rate (Fig. 2) and leads to a Ro31-8220-sensitive increase in the Ca2+ sensitivity of vesicle fusion, as probed by presynaptic Ca2+ uncaging (Korogod et al., 2007). Thus, a PKC-dependent mechanism, as well as activation of Munc13-1 by DAG, converge to increase the fusion probability of a given readily releasable vesicle, via a mechanisms that increases the vesicle fusion willingness with a concomitant increase in the Ca2+ sensitivity of vesicle fusion (Lou et al., 2005; Basu et al., 2007). Finally, in a third route, PKC can enhance the release probability by increasing the presynaptic Ca2+ current (Fig. 8) (Lou et al., 2005; Korogod et al., 2007). However, this route probably plays only a minor role because the modulation of Ca2+ current is small (∼15%), and because a large part of the phorbol ester modulation is also observed when transmitter release is evoked by Ca2+ uncaging, completely independent of the opening of voltage-gated Ca2+ channels (Lou et al., 2005).
Figure 8.

Diagram of how a PKC-dependent pathway and the activation of Munc13 converge to increase the release probability of a given docked and readily releasable vesicle. There are at least three different mechanisms of actions of the phospholipase C product, DAG. First, DAG can bind to Munc13-1, causing an increased fusion probability of readily releasable vesicles that manifests itself as an increased mEPSC frequency, and probably also as an increased Ca2+ sensitivity of vesicle fusion. Second, DAG can recruit and activate PKC, which leads to phosphorylation of a protein in the vesicle fusion machinery, like Munc18 (Wierda et al., 2007) or other proteins. This again leads to a potentiation of the spontaneous transmitter release (Fig. 2), as well as to an increased Ca2+ sensitivity of vesicle fusion as probed by Ca2+ uncaging (Korogod et al., 2007). A third action is via a PKC-dependent phosphorylation of presynaptic voltage-gated Ca2+ channels, because presynaptic Ca2+ currents are slightly enhanced in a Ro31-8220-sensitive manner by phorbol esters (Korogod et al., 2007).
Phorbol esters act independent of a pool size increase
We found that transmitter release in response to pool-depleting long presynaptic depolarizations was not increased significantly by PDBu (Fig. 5), indicating that phorbol esters do not act via increasing the size of the readily releasable vesicle pool (Lou et al., 2005; Basu et al., 2007; Wierda et al., 2007). In additional control experiments, we showed that strong elevations of the extracellular [Ca2+] do not increase the number of vesicles released in response to a prolonged presynaptic depolarization (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), confirming that such stimuli release the pool of readily releasable vesicles, as shown previously (Schneggenburger and Neher, 2000; Sakaba and Neher, 2001; Sun and Wu, 2001). Phorbol ester application and elevation of the extracellular [Ca2+] did, however, increase the cumulative EPSC amplitude during 100 Hz trains. It seems, therefore, that 100 Hz trains do not fully deplete the pool of readily releasable vesicles at 2 mm [Ca2+], and that enhancing the effective release probability of any given readily releasable vesicle will lead to an increase in the cumulative EPSC amplitude. This noncomplete release of the readily releasable pool during 100 Hz trains agrees with previous findings of an increase in the apparent pool size after elevating the extracellular [Ca2+] in hippocampal neurons (Moulder and Mennerick, 2005). Also, presynaptic Ca2+ uncaging released only a fraction of the pool when low-intermediate [Ca2+]i stimuli were given, a phenomenon that has been termed “submaximal release” of fast-releasable vesicles (Wölfel et al., 2007). Submaximal release is also evident from EPSC responses to hypertonic solution application, when lower concentrations of sucrose are used (∼250 mm or less) (Basu et al., 2007; Wierda et al., 2007). Thus, when studying functional pool sizes during neuromodulation, care must be taken to ensure that the entire readily releasable pool is released effectively under control conditions.
Although we think, therefore, that the increase in cumulative release during 100 Hz trains does not indicate an increased pool size, we cannot exclude that an increase in the readily releasable pool could contribute to a late phase of the phorbol ester potentiation (>300 s) (Fig. 6D). At these late times, the potentiation of PDBu cannot be followed in paired recordings (Fig. 5C) (Lou et al., 2005). It has also been shown that PKC activation speeds pool recovery after high-frequency trains (Wierda et al., 2007), and there might be a relation between enhanced pool recovery and the onset of post-tetanic potentiation, which is also PKC dependent (Korogod et al., 2007). Thus, activation of PKC could influence the recovery kinetics of the readily releasable pool by a mechanism that needs to be explored in more detail, but during steady-state conditions, an enhancement of the pool size by phorbol esters is negligible.
Together, the fusion probability of docked and primed vesicles in the readily releasable pool can be modulated by the second-messenger DAG, via activation of both Munc13-1 and PKCs. The consequence of these modulations is an increased effective release probability of readily releasable vesicles, via an enhanced Ca2+-sensitivity of vesicle fusion (Lou et al., 2005). In synapses, where specific vesicle docking sites at active zones are probably limited in number and are thought to be mostly occupied under resting conditions (Neher, 2006), an increase in the vesicle fusion probability is an effective way to increase the transmitter output. Also, because the released fraction of the readily releasable pool is small in hippocampal synapses (Reim et al., 2001) and at the calyx of Held (∼1/10 or less) (for review, Schneggenburger et al., 2002), there is ample room for an increase in the amount of transmitter release via an increase in the release probability. Thus, vesicles in the readily releasable pool emerge as a novel substrate for a direct modulation via intracellular second-messenger pathways.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Schn451/4-1, 4-2 and a Heisenberg Fellowship) (R.S.), the Swiss National Science Foundation (3100A0-114069), and the network of European Neuroscience Institutes from the European Union. We thank Erwin Neher and Takeshi Sakaba for helpful discussions and Holger Taschenberger for help with mEPSC analysis.
References
- Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Barclay JW, Craig TJ, Fisher RJ, Ciufo LF, Evans GJ, Morgan A, Burgoyne RD. Phosphorylation of munc-18 by protein kinase C regulates the kinetics of exocytosis. J Biol Chem. 2003;278:10538–10545. doi: 10.1074/jbc.M211114200. [DOI] [PubMed] [Google Scholar]
- Basu J, Betz A, Brose N, Rosenmund C. Munc-13-1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J Neurosci. 2007;27:1200–1210. doi: 10.1523/JNEUROSCI.4908-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- Brager DH, Cai X, Thompson SM. Activity-dependent activation of presynaptic protein kinase C mediates post-tetanic potentiation. Nat Neurosci. 2003;6:551–552. doi: 10.1038/nn1067. [DOI] [PubMed] [Google Scholar]
- Felmy F, Schneggenburger R. Developmental expression of the Ca2+-binding proteins calretinin and parvalbumin at the calyx of Held of rats and mice. Eur J Neurosci. 2004;20:1473–1482. doi: 10.1111/j.1460-9568.2004.03604.x. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón R, Wölfel M, Nishimune H, Tabares L, Schmitz F, Castellano-Muñoz M, Rosenmund C, Montesinos ML, Sanes JR, Schneggenburger R, Südhof TC. The synaptic vesicle protein CSPα prevents presynaptic degeneration. Neuron. 2004;42:237–251. doi: 10.1016/s0896-6273(04)00190-4. [DOI] [PubMed] [Google Scholar]
- Finley MF, Scheller RH, Madison DV. SNAP-25 Ser187 does not mediate phorbol ester enhancement of hippocampal synaptic transmission. Neuropharmacology. 2003;45:857–862. doi: 10.1016/s0028-3908(03)00283-1. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Sasaki T, Fukui K, Kotani H, Kimura T, Hata Y, Südhof TC, Scheller RH, Takai Y. Phosphorylation of Munc-18/n-Sec1/rbSec1 by protein kinase C. J Biol Chem. 1996;271:7265–7268. doi: 10.1074/jbc.271.13.7265. [DOI] [PubMed] [Google Scholar]
- Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Groemer T, Klingauf J. Synaptic vesicles recycling spontaneously and during activity belong to the same pool. Nat Neurosci. 2007;10:145–147. doi: 10.1038/nn1831. [DOI] [PubMed] [Google Scholar]
- Hamann M, Billups B, Forsythe ID. Non-calyceal excitatory inputs mediate low fidelity synaptic transmission in rat auditory brainstem slices. Eur J Neurosci. 2003;18:2899–2902. doi: 10.1111/j.1460-9568.2003.03017.x. [DOI] [PubMed] [Google Scholar]
- Hori T, Takai Y, Takahashi T. Presynaptic mechanism for phorbol ester-induced synaptic potentiation. J Neurosci. 1999;19:7262–7267. doi: 10.1523/JNEUROSCI.19-17-07262.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Sahara Y, Takahashi T. A single packet of transmitter does not saturate postsynaptic glutamate receptors. Neuron. 2002;34:613–621. doi: 10.1016/s0896-6273(02)00692-x. [DOI] [PubMed] [Google Scholar]
- Joshi I, Shokralla S, Titis P, Wang LY. The role of AMPA receptor gating in the development of high-fidelity neurotransmission at the calyx of Held. J Neurosci. 2004;24:183–196. doi: 10.1523/JNEUROSCI.1074-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Posttetanic potentiation critically depends on an enhanced Ca2+ sensitivity of vesicle fusion mediated by presynaptic PKC. Proc Natl Acad Sci U S A. 2007:15923–15928. doi: 10.1073/pnas.0704603104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM. Facilitation of synaptic transmission by EGL-30 Gqα and EGL-8 PLCβ: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Friauf E. Distribution of the calcium-binding proteins parvalbumin and calretinin in the auditory brainstem of adult and developing rats. J Comp Neurol. 1996;367:90–109. doi: 10.1002/(SICI)1096-9861(19960325)367:1<90::AID-CNE7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Ilic V, Friauf E. Development of a topographically organized auditory network in slice culture is calcium dependent. J Neurobiol. 1998;34:97–112. doi: 10.1002/(sici)1097-4695(19980205)34:2<97::aid-neu1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Lou X, Scheuss V, Schneggenburger R. Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature. 2005;435:497–501. doi: 10.1038/nature03568. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol ester. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Mennerick S. Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J Neurosci. 2005;25:3842–3850. doi: 10.1523/JNEUROSCI.5231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Matti U, Nehring RB, Binz T, Rettig J, Neher E, Sørensen JB. Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitments. J Neurosci. 2002;22:9278–9286. doi: 10.1523/JNEUROSCI.22-21-09278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. A comparison between exocytotic control mechanisms in adrenal chromaffin cells and a glutamatergic synapse. Pflugers Arch. 2006;453:261–268. doi: 10.1007/s00424-006-0143-9. [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Combining deconvolution and noise analysis for the estimation of transmitter release rates at the calyx of Held. J Neurosci. 2001;21:444–461. doi: 10.1523/JNEUROSCI.21-02-00444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AC. Regulation of protein kinase C. Curr Op Cell Biol. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- Oleskevich S, Walmsley B. Phosphorylation regulates spontaneous and evoked transmitter release at a giant terminal in the rat auditory brainstem. J Physiol. 2000;526:349–357. doi: 10.1111/j.1469-7793.2000.t01-1-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt KD, Madison DV. Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. J Physiol. 1993;471:245–268. doi: 10.1113/jphysiol.1993.sp019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Südhof TC, Takahashi M, Rosenmund C, Brose N. β phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by munc13s and not by PKCs. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara Y, Takahashi T. Quantal components of the excitatory postsynaptic currents at a rat central auditory synapse. J Physiol. 2001;536:189–197. doi: 10.1111/j.1469-7793.2001.00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/s0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Sara Y, Virmani T, Deák F, Liu X, Kavalali ET. An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron. 2005;45:563–573. doi: 10.1016/j.neuron.2004.12.056. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E. Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 2002;25:206–212. doi: 10.1016/s0166-2236(02)02139-2. [DOI] [PubMed] [Google Scholar]
- Shapira R, Silberberg SD, Ginsburg S, Rahamimoff R. Activation of protein kinase C augments evoked transmitter release. Nature. 1987;325:58–60. doi: 10.1038/325058a0. [DOI] [PubMed] [Google Scholar]
- Shu Y, Liu X, Yang Y, Takahashi M, Gillis KD. Phosphorylation of SNAP-25 at Ser187 mediates enhancement of exocytosis by a phorbol ester in INS-1 cells. J Neurosci. 2008;28:21–30. doi: 10.1523/JNEUROSCI.2352-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Sun JY, Wu LG. Fast kinetics of exocytosis revealed by simultaneous measurements of presynaptic capacitance and postsynaptic currents at a central synapse. Neuron. 2001;30:171–182. doi: 10.1016/s0896-6273(01)00271-9. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of munc-13-mediated vesicle priming. Proc Natl Acad Sci U S A. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadel K, Neher E, Sakaba T. The coupling between synaptic vesicles and Ca2+ channels determines fast neurotransmitter release. Neuron. 2007;53:563–575. doi: 10.1016/j.neuron.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Waters J, Smith SJ. Phorbol esters potentiate evoked and spontaneous release by different presynaptic mechanisms. J Neurosci. 2000;20:7863–7870. doi: 10.1523/JNEUROSCI.20-21-07863.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RFG, De Wit H, Brussard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54:275–290. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Wölfel M, Lou X, Schneggenburger R. A mechanism intrinsic to the vesicle fusion machinery determines fast and slow transmitter release at a large CNS synapse. J Neurosci. 2007;27:3198–3210. doi: 10.1523/JNEUROSCI.4471-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XS, Wu LG. Protein kinase C increases the apparent affinity of the release machinery to Ca2+ by enhancing the release machinery downstream of the Ca2+ sensor. J Neurosci. 2001;21:7928–7936. doi: 10.1523/JNEUROSCI.21-20-07928.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H. Protein kinase C potentiates transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J Physiol. 1999;515:169–180. doi: 10.1111/j.1469-7793.1999.169ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]