

Abstract
Spike timing-dependent synaptic plasticity (STDP) plays an important role in neural development and information processing in the brain; however, the mechanism by which spike timing information is encoded into STDP remains unclear. Here, we show that a novel allosteric kinetics of NMDA receptors (NMDARs) is required for STDP. We developed a detailed biophysical model of STDP and found that the model required spike timing-dependent distinct suppression of NMDARs by Ca2+-calmodulin. This led us to predict an allosteric kinetics of NMDARs: a slow and rapid suppression of NMDARs by Ca2+-calmodulin with prespiking → postspiking and postspiking → prespiking, respectively. We found that the allosteric kinetics, but not the conventional kinetics, is consistent with specific features of amplitudes and peak time of NMDAR-mediated EPSPs in experiments. We found that the allosteric kinetics of NMDARs was also valid for synaptic plasticity induced by more complex spike trains in layer II/III of visual cortex. We extracted an essential synaptic learning rule by reduction of the allosteric STDP model and found that spike timing-dependent bidirectional role of postspiking in synaptic modification, which depends on the allosteric kinetics, is the essential principle in STDP. Thus, we propose a simple hypothesis of the allosteric kinetics of NMDARs that can coherently explain critical features of spike timing-dependent NMDAR-mediated EPSPs and synaptic plasticity.
Keywords: LTP, LTD, spike timing-dependent plasticity, NMDA receptor, kinetic simulation, systems neurobiology
Introduction
In the mammalian brain, activity-dependent bidirectional modification of synaptic strength, including long-term potentiation (LTP) and long-term depression (LTD), has been thought to be the molecular and cellular basis of learning and memory (Bi and Poo, 2001). There are various forms of LTP and LTD, but NMDA receptor (NMDAR)-dependent LTP and LTD at excitatory synapses are the most extensively studied and the prototypic form of long-term synaptic plasticity (Malenka and Bear, 2004). The induction of LTP and LTD has been shown recently to depend on the timing between presynaptic and postsynaptic spiking (postspiking), known as spike timing-dependent synaptic plasticity (STDP) (Bi and Poo, 2001; Dan and Poo, 2004). The hallmark of STDP is a spike sequence-dependent bidirectional modification of synaptic strength within a narrow time window between prespiking and postspiking on the order of milliseconds. STDP provides significant computational advantages as a synaptic learning rule, allowing neural circuits to learn temporal sequences of inputs, to shape the temporal and spatial dynamics of neural circuits, and to code causality of external events (Gerstner et al., 1996; Song et al., 2000; Senn, 2002). Additionally, STDP reportedly contributes to the effects of sensory stimuli in the refinement of the retinotectal system in vivo (Mu and Poo, 2006; Vislay-Meltzer et al., 2006).
In STDP, prespiking → postspiking is thought to trigger high Ca2+ influx via NMDARs and voltage-gated Ca2+ channels (VGCCs), which depends on the depolarization-induced removal of the Mg2+ block of NMDARs and supralinear summation of EPSPs and backpropagating action potentials (bpAPs) (Hoffman et al., 1997; Stuart and Hausser, 2001; Kampa et al., 2004). The high Ca2+ influx is thought to induce activation of Ca2+-calmodulin (CaM)-dependent kinase II (CaMKII), resulting in induction of LTP (Lisman, 1989, 2001; Wang et al., 2005). In contrast, postspiking → prespiking is thought to be mediated by depolarization-dependent suppression of NMDARs (Froemke et al., 2005), which may lead to low Ca2+ influx via NMDARs and VGCCs. The low Ca2+ influx is thought to induce calcineurin (CaN) activation, resulting in induction of LTD (Lisman, 1989, 2001; Wang et al., 2005). Some forms of LTD in STDP depend on cannabinoid receptor (CB1)-dependent suppression of transmitter release, in conjunction with activation of presynaptic NMDA autoreceptors (Sjostrom et al., 2003), whereas other forms of LTD do not require CB1 receptor signaling (Seol et al., 2007). A new study revealed that such controversial results are attributable to different types of neurons or developmental stages (Corlew et al., 2007). Furthermore, recent studies indicate that STDP induction depends not only on the time interval between prespiking and postspiking but also on nonlinear interactions among complex timing prespiking and postspiking (Boettiger and Doupe, 2001; Sjostrom et al., 2001; Froemke and Dan, 2002; Tzounopoulos et al., 2004; Wang et al., 2005; Froemke et al., 2006). Although some phenomenological models have been proposed to describe the interactions among complex timing prespiking and postspiking (Froemke and Dan, 2002; Froemke et al., 2006; Pfister and Gerstner, 2006), the coding mechanism by which spike times are translated into STDP remains unclear because of complexity in signaling mechanisms and properties of STDP.
To address the coding mechanism of STDP, here, we focused on STDP induced in layer II/III lateral connections of visual cortex (Froemke and Dan, 2002; Froemke et al., 2005, 2006) and developed an in-depth biophysical model. We found that spike timing-dependent LTP, but not LTD, could be reproduced. The failure of induction of LTD led us to predict a requirement of slow and rapid suppression of NMDARs by Ca2+ · CaM with prespiking → postspiking and postspiking → prespiking, respectively. We found that the predicted allosteric kinetics of NMDARs, but not the conventional kinetics (the binding of glutamate and Ca2+ · CaM do not affect their binding to NMDARs each other), appears to be valid for experimental features of NMDAR-mediated EPSPs and for synaptic plasticity induced by more complex spike trains (Froemke and Dan, 2002; Froemke et al., 2006). We extracted an essential synaptic learning rule and found bidirectional regulation of NMDARs by postspiking with allosteric kinetics: the positive regulation of NMDAR activation and Ca2+ influx with prespiking → postspiking and the negative regulation of NMDAR activation and Ca2+ influx with postspiking → prespiking. We propose that the simple allosteric kinetics of NMDARs is a fundamental coding mechanism for STDP.
Materials and Methods
Development of the STDP model.
The STDP model consists of the postsynaptic membrane potential model (see Fig. 1A), the postsynaptic signaling cascades model (see Fig. 1B), and the AMPA receptors (AMPARs) trafficking model (see Fig. 1C). The GENESIS simulator (version 2.2) with a Kinetikit interface was used to numerically simulate spatiotemporal dynamics of electrophysiological and biochemical reactions in the STDP model (Bhalla and Iyengar, 1999). Detailed descriptions of these models, including block diagrams, reactions, and parameters, are described in the supplemental information (http://www.kurodalab.org/info/STDP/Urakubo2008SI.pdf), and the constructed programs are available for download (http://www.kurodalab.org/info/STDP/index.html). Here, a brief overview of the STDP model is presented as follows.
Figure 1.

Development of the STDP model. A, Electrophysiological compartment model. B, Postsynaptic signaling cascades model, which includes Ca2+ influx and removal and biochemical interactions. C, AMPAR trafficking model. The arrows and lines with a black circle indicate stimulatory and inhibitory interactions, respectively. Molecules required for LTP and LTD are indicated by red and blue, respectively. The detailed description of the models is provided as supplemental information (available at http://www.kurodalab.org/info/STDP/Urakubo2008SI.pdf).
Modeling of postsynaptic neuron.
We constructed a multicompartment model of a postsynaptic neuron with Hodgkin–Huxley-like channel kinetics on the basis of the previous models (see Fig. 1A) (Migliore et al., 1999; Poirazi et al., 2003). The model has one somatic and 20 dendritic compartments (see Fig. 1A), which incorporate spike-generating Na+ channels, persistent Na+ channels, delayed rectifier K+ channels, A-type K+ channels, and L-type VGCCs with spatially inhomogeneous densities. The synaptic site is assumed to be 60 μm apart from the soma (third dendritic compartment from the soma). Presynaptic stimulation leads to probabilistic release of glutamate from presynaptic terminals, which activates postsynaptic AMPARs and NMDARs. Postsynaptic stimulation (current injection) elicits APs.
Modeling of postsynaptic signaling cascades.
We modeled Ca2+ influx to a spine via NMDARs and VGCCs, as well as Ca2+ uptake from the spine, and simulated postsynaptic Ca2+ concentration [Ca2+]PSD in response to presynaptic and postsynaptic stimulation on the basis of the previous observations (see Fig. 1B) (Sabatini et al., 2001). We also modeled intracellular signaling cascades within the spine on the basis of the previous models (Bhalla and Iyengar, 1999; Kuroda et al., 2001; Doi et al., 2005) and observations (Winder and Sweatt, 2001; Lisman et al., 2002). The spine is divided into cytosol and postsynaptic density (PSD) compartments. We incorporated interactions among Ca2+, CaM, CaMKII, CaN, protein kinase A (PKA), adenylnyl cylase 1/8 (AC1/8), cAMP, phosphorylated inhibitor-I (I-1), protein phosphatase 1 (PP1), and protein phosphatase 2A (PP2A). The molecules diffuse between the cytosol and PSD compartments, where CaMKII, PP1, and PP2A interact with scaffold (cytoskeletal scaffolds). We assumed that the molecules within each compartment are well mixed, and that they deterministically interact on the basis of mass assumption.
Modeling of AMPAR trafficking.
We constructed a simplified model of phosphorylation-dependent AMPAR [glutamate receptor 1 (GluR1)] trafficking on the basis of the previous experiments (see Fig. 1C) (Malinow and Malenka, 2002; Collingridge et al., 2004; Seol et al., 2007). An AMPAR subunit, GluR1, is phosphorylated by PKA at Ser845, and PDZ (PSD-95/Discs large/zona occludens-1)-containing proteins, which associate AMPARs, are phosphorylated by CaMKII. AMPARs are involved in the following recycling processes: (1) AMPARs with phosphorylated PDZs are trapped by scaffold at PSD fraction, (2) AMPARs are diffused laterally in the membrane, and (3) dephosphorylated AMPARs (GluR1 s at Ser845) are internalized by endocytosis, and phosphorylated AMPARs are reinserted into the membrane via exocytosis. We assumed that AMPAR number trapping at scaffolds of PSD corresponds to synaptic conductance.
Electrophysiology.
Acute visual cortical slices were prepared from 2- to 4-week-old Sprague Dawley rats. Somatic whole-cell recordings were made from layer II/III pyramidal cells in current-clamp mode. EPSPs were evoked by focal extracellular stimulation, and postsynaptic APs were elicited with current injection through the recording electrode. Synaptic strength was measured as the initial 2 ms slope of the EPSP at 11–20 min after the end of induction. The detailed experimental procedures are described previously (Froemke and Dan, 2002; Froemke et al., 2005, 2006).
Results
Development of the STDP model
To understand the mechanism of spike-timing detection in STDP, we developed a realistic biophysical model of synaptic plasticity within a single postsynaptic spine, on the basis of the literature and recent experimental findings on ion channels, intracellular signaling cascades, and receptor trafficking (Fig. 1 A–C) (supplemental information, http://www.kurodalab.org/info/STDP/Urakubo2008SI.pdf). In this model, prespiking and postspiking are inputs, and the change of synaptic conductance is the output. Prespiking represents glutamate release from the presynaptic terminal, which involves short-term depression of transmitter release probability during high-frequency bursts (>2 Hz) (Tsodyks and Markram, 1997; Matveev and Wang, 2000; Froemke et al., 2006). The postspiking signal is a large increase of membrane potential via bpAPs (Hoffman et al., 1997; Stuart and Hausser, 2001), which involves frequency-dependent attenuation (Froemke et al., 2006). We used 1 Hz prespiking for 100 s over a range of constant membrane potentials, which corresponds to the postspiking signal (Artola et al., 1990; Feldman, 2000; Lisman, 2001; Stiefel et al., 2005), as test stimuli to constrain the electrophysiological and biochemical dynamics of the STDP model (Fig. 2).
Figure 2.

Voltage-dependent changes of synaptic strength. A–E, Time courses of Ca2+ concentration (A), CaMKII activity (B), PP1 activity (C), PKA activity (D), and CaN activity (E) at PSD in response to 100 prespikes at 1 Hz with the indicated voltage clamp of somatic membrane potential during the stimulation. Colors correspond to the voltage as indicated in E. Arrowheads indicate the onset of stimulation. The inset in A shows its time courses by a single prespiking. F, Voltage-dependent Ca2+ concentration at PSD. The black and gray lines indicate the mean and maximal amplitude of Ca2+ concentration, respectively. G, Voltage-dependent CaN, PKA, PP1, and CaMKII activities. Integrated PP1 activity (red; normalized with the maximal activity, 0.33), integrated PKA activity (dark blue; normalized by 3.68), integrated CaN activity (green; normalized by 1.75), and final CaMKII activity (light blue) against the indicated membrane potentials are plotted. H, Time courses of changes of synaptic conductance. I, Voltage-dependent changes of synaptic conductance. Data were taken at 2 and 60 min after the onset of stimulation (Obs 1 and Obs 2, respectively) as indicated in H.
At membrane potentials positive from −45 mV, the Mg2+ block of NMDARs was removed, and the high Ca2+ influx via NMDARs (Fig. 2A,F) produced sufficient Ca2+ · CaM. This then bound to each subunit of a holoenzyme of CaMKII and induced successive autophosphorylation of CaMKII subunits, which led to the persistent activation of CaMKII (Fig. 2B,G) (Lisman et al., 2002). The persistent CaMKII activation required the inhibition of PP1 activity (Fig. 2C,G), which dephosphorylates CaMKII. The high Ca2+ influx also activated PKA via AC1/8 (Fig. 2D,G) and CaN (Fig. 2E,G). PP1 was inactivated by phosphorylated I-1, the dephosphorylation and phosphorylation of which depend on CaN and PKA, respectively. The high Ca2+ influx inhibited PP1 via phosphorylated I-1, because its phosphorylation by PKA counteracts with dephosphorylation by CaN (Figs. 1B, 2D,E,G). Therefore, PKA activation is necessary to trigger the persistent CaMKII activation via inhibition of PP1, which is consistent with the observation that PKA acts as a gate signal for LTP induction (Blitzer et al., 1998).
In contrast, at hyperpolarized membrane potentials such as −85 mV, the low Ca2+ influx (Fig. 2A,F) also produced Ca2+ · CaM; however, this activated CaN without sufficient activation of PKA and CaMKII (Fig. 2G), and the activated CaN dephosphorylated I-1 and activated PP1 (Fig. 2C,E,G) (Winder and Sweatt, 2001). Therefore, the low Ca2+ influx specifically induced activation of protein phosphatases rather than of protein kinases, consistent with previous observations (Bear and Malenka, 1994). The sensitivities to Ca2+ influx between protein phosphatases and kinases were attributable to the distinct sensitivities of CaN and PKA to Ca2+ · CaM (Fig. 2G). However, in contrast to CaMKII activation, PP1 activation was transient and returned to basal levels after stimulation (Fig. 2C).
The specific activation of signaling molecules regulated trafficking of AMPARs and thus regulated AMPAR synaptic conductance (Figs. 1C, 2H) (Malinow and Malenka, 2002; Collingridge et al., 2004; Seol et al., 2007). Persistent activation of CaMKII phosphorylated PDZs, including synapse-associated protein 97 and stargazin. The phosphorylation facilitated AMPAR trapping by cytoskeletal scaffolds (scaffolds) at PSD (Fig. 1C) and resulted in a persistent increase of AMPARs at PSD and of conductance, resulting in expression of LTP (Fig. 2H). Note that the persistent CaMKII activation was the only stabilizing mechanism for LTP and that no other stabilizing mechanisms, such as protein synthesis and AMPAR clustering, were modeled in this study. In contrast, both PP1 and CaN dephosphorylated PDZs and GluR1 at Ser845 (a subunit of AMPARs). Dephosphorylation of PDZs and GluR1 at Ser845 resulted in the dissociation of AMPARs from PSD and facilitated the internalization of AMPARs into the endosome, respectively (Fig. 1C). The internalized AMPARs were maintained in the dephosphorylated state by the action of PP1 and CaN, which prevented the AMPARs from being reinserted, and resulted in a decrease of conductance through AMPAR channels (Fig. 2H). However, because the activation of CaN and PP1 was transient, the number of AMPARs within the PSD and, correspondingly, the total prespike-evoked synaptic conductance eventually returned to the basal level. This result indicates that the stabilization of LTD cannot be explained with the experimentally observed time constants of AMPAR trafficking and suggests the presence of an unidentified pathway or pathways that are responsible for the maintenance of LTD. The mechanisms of persistency for LTD remain unknown; however, the saturation-dependent trafficking of AMPARs (Hayer and Bhalla, 2005) or the clustering of interacting receptors (Shouval, 2005) may be possible mechanisms. Although depression was not persistent, short-term, voltage-dependent changes of synaptic strength in our model were highly correlated with depolarization-dependent LTP and hyperpolarization-dependent LTD observed experimentally (Fig. 2I) (Artola et al., 1990; Stiefel et al., 2005). Similarly, we experimentally found that, by use of pharmacological manipulation, high, moderate, and low NMDAR activities are correlated with potentiation, no change (neither potentiation nor depression), and depression, respectively (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Because the NMDAR activity depends on the membrane potential, this result is consistent with synaptic plasticity induced by direct depolarization (Malenka and Bear, 2004). Therefore, we confirmed that our STDP model is, in principle, consistent with membrane potential-dependent synaptic plasticity and with related molecular events.
Prediction of the allosteric kinetics of NMDARs
We next explored whether the experimentally determined dynamics of STDP can be reproduced in our biophysical model. We used a brief current injection to the somatic compartment to trigger postspiking and bpAPs within the spine. Neither prespiking nor postspiking with 1 Hz for 100 s alone induced synaptic plasticity (see Fig. 4E) (Johnston et al., 2003). Induction of LTP by prespiking → postspiking is thought to be triggered by accelerated Ca2+ influx via NMDARs and VGCCs, which depends on the depolarization-induced removal of the Mg2+ block of NMDARs and supralinear summation of EPSPs and bpAPs (Hoffman et al., 1997; Stuart and Hausser, 2001; Kampa et al., 2004). Induction of LTD by postspiking → prespiking has been shown recently to be mediated by depolarization-dependent suppression of NMDARs (Froemke et al., 2005), which may lead to reduced Ca2+ influx through NMDARs and VGCCs. Moreover, the direct binding of Ca2+ · CaM to NMDARs reduces mean open time and leads to an overall reduction of open probability, resulting in suppression of NMDARs (Ehlers et al., 1996; Rycroft and Gibb, 2002, 2004b). Therefore, we first examined whether accelerated or suppressed Ca2+ influx can be induced by prespiking → postspiking or postspiking → prespiking via NMDARs, respectively (Fig. 3). Prespiking → postspiking led to accelerated Ca2+ influx via bpAP-dependent removal of the Mg2+ block of NMDARs (Fig. 3A,B), which led to induction of LTP (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). However, postspiking → prespiking did not suppress Ca2+ influx (Fig. 3A,B), in contrast to the experimental evidence (Froemke et al., 2005), and did not induce LTD (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This result indicates the existence of an unknown mechanism for suppression of Ca2+ influx.
Figure 4.

Prediction of the allosteric kinetics of NMDARs. A, Schematic (top) and detailed mechanism (bottom) of the allosteric kinetics of NMDARs. τ1 and τ2 were the time constants of Ca2+ · CaM binding to glutamate (Glu)-unbound and Glu-bound NMDARs, respectively, where Ca2+ · CaM concentration is 1 μm. Ca2+ · CaM bound and Ca2+ · CaM unbound NMDARs, which bind to Glu, show the low and high open probability, respectively (Ehlers et al., 1996; Rycroft and Gibb, 2002, 2004b). C0, C1, C2, C3, C0 · CaM, C1 · CaM, C2 · CaM, and C3 · CaM are the concentrations of closed-state NMDARs, and O and O · CaM are those of open-state NMDARs (bottom). CaM represents 2 Ca2+ · CaM or 3 Ca2+ · CaM, and the numerals on arrows are rate constants in μm−1s−1 or s−1, which were set on the basis of the experimental observation (bottom). The complete description of the allosteric kinetics of NMDARs is provided on-line (available at http://www.kurodalab.org/info/STDP/Urakubo2008SI.pdf). B, Time courses of spike timing-dependent [Ca2+]PSD by a single pairing of prespiking and postspiking in the allosteric model with the indicated spike timing in C. C, Spike timing-dependent [Ca2+]PSD in the allosteric model with the indicated τ1 and τ2. D, Time courses of spike timing-dependent synaptic conductance in the allosteric model. One hundred pairings of the prespiking and postspiking with 1 Hz were given. E, Spike timing-dependent synaptic conductance in the allosteric model at 2 min (Obs 1) and 60 min (Obs 2) after the onset of stimulation (left). The apparent time constants for LTP and LTD at 2 min were 36.1 and 53.0 ms, respectively. The synaptic conductance with 100 prespikes and postspikes alone at 2 min (black) and 60 min (gray) after the onset of stimulation is also plotted (right, bars). F, Integrated PKA activity (normalized by 3.06) and final CaMKII activity. G, Integrated PP1 activity (normalized by 0.34) and integrated CaN activity (normalized by 1.53).
Figure 3.
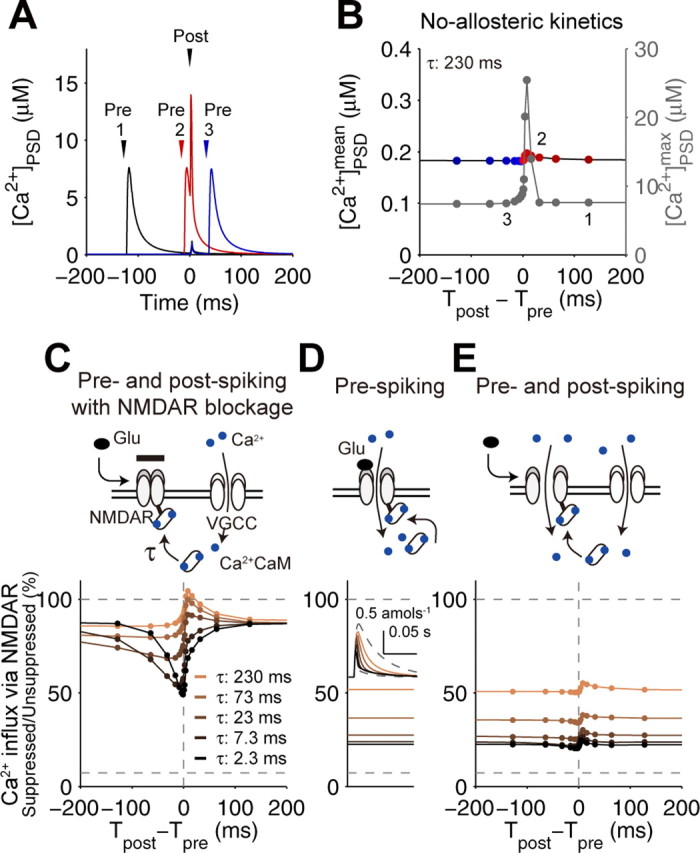
Requirement of the distinct suppression of NMDARs. A, Time courses of spike timing-dependent [Ca2+]PSD by a single pairing of prespiking and postspiking in the STDP model (the no-allosteric model) with the indicated spike timing in B; 1, prespiking → postspiking (Tpost − Tpre = +128 ms; black); 2, prespiking → postspiking (+16 ms; red); 3, postspiking → prespiking (−32 ms; blue). B, Spike timing-dependent Ca2+ increase in the no-allosteric model. The mean and maximum [Ca2+]PSD are indicated by black and gray, respectively. The mean denotes the integration of Ca2+ induced by a single pairing of prespiking and postspiking divided by 1 s. Tpost was defined as the time of 5 ms after the onset of current injection represented by an α-function (500 pA, 0.5 ms), and Tpre was defined as the time of the onset of glutamate release. The positive (red) and negative (blue) intervals correspond to the prespike → postspike and postspike → prespike timing, respectively. C, Spike timing-dependent suppression of NMDARs under the blockage of Ca2+ influx via NMDARs. Expected Ca2+ influx, if the NMDARs open, are plotted with the indicated time constant of Ca2+ · CaM binding to NMDARs, τ. Ca2+ influx via NMDARs was normalized by unsuppressed Ca2+ influx (C–E). D, Ca2+ influx via NMDARs induced by a prespiking alone with the indicated τ. The inset shows the time courses of the total NMDAR-mediated Ca2+ influx. E, Spike timing-dependent Ca2+ influx via NMDARs.
The failure to suppress Ca2+ influx by postspiking → prespiking led us to examine the mechanism of suppression of NMDARs by Ca2+ · CaM. We changed the time constant of Ca2+ · CaM binding to NMDARs under the blockage of NMDAR-dependent Ca2+ influx and examined the suppression of NMDARs solely by VGCC-dependent Ca2+ influx, which is triggered by postspiking (Fig. 3C). NMDARs were suppressed by VGCC-dependent Ca2+ influx with the rapid time constant (2.3 ms) but not with the slow time constant (230 ms). This indicates that suppression of NMDARs by VGCC-dependent Ca2+ influx, which may reflect the time window for LTD induction with postspike → prespike timing (Froemke et al., 2005), requires a rapid binding of Ca2+ · CaM to NMDARs. We also examined suppression of NMDARs by NMDAR-dependent Ca2+ influx induced by prespiking alone (Fig. 3D). Ca2+ influx was gradually reduced as the time constant of Ca2+ · CaM binding to NMDARs became rapid because of the self-suppression of NMDARs by Ca2+ influx through themselves. This indicates that the slow binding of Ca2+ · CaM to NMDARs is required to avoid self-suppression of NMDARs. We examined the suppression of NMDARs by both NMDAR- and VGCC-dependent Ca2+ influx and found that suppression of NMDARs did not change regardless of spike timing with any time constant and resulted in a failure of spike-timing detection (Fig. 3E). This result indicates that, with postspike → prespike timing, a rapid binding of Ca2+ · CaM to NMDARs is required to suppress NMDARs by VGCC-dependent Ca2+ influx, whereas with prespike → postspike timing, a slow binding of Ca2+ · CaM to NMDARs is required to avoid the rapid self-suppression of NMDARs.
This requirement of distinct suppression of NMDARs in a spike timing-dependent manner led us to predict an allosteric kinetics of NMDARs: rapid binding of Ca2+ · CaM to glutamate-unbound NMDARs and slow binding of Ca2+ · CaM to glutamate-bound NMDARs (Fig. 4A) (supplemental infor-mation, available at http://www.kurodalab.org/info/STDP/Urakubo2008SI.pdf). For simplicity, we changed the association and dissociation time constants without changing the affinity of Ca2+ · CaM to NMDARs. Hereafter, the STDP model (with or without the allosteric kinetics of NMDARs) denotes the allosteric or no-allosteric model, respectively. The allosteric model led to suppression of Ca2+ influx with postspike → prespike timing and accelerated Ca2+ influx with prespike → postspike timing (Fig. 4B,C), resulting in STDP (Fig. 4D,E), although any no-allosteric model with rapid or slow binding of Ca2+ · CaM to NMDARs did not produce the appropriate Ca2+ influx and subsequent STDP (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Note that the curve in LTP at 2 min was graded, but it became bistable at 60 min. Such slow increase is one of the unusual properties of spike timing-dependent LTP observed in experiments (Bi and Rubin, 2005).
In the allosteric model, accelerated Ca2+ influx with prespike → postspike timing specifically induced CaMKII and PKA activation and PP1 suppression (Fig. 4F,G), which induced LTP (Fig. 4D,E). Persistent CaMKII activation requires a suprathreshold Ca2+ influx because of the nature of its autophosphorylation loop, so the high Ca2+ influx, rather than the low and sustained Ca2+ influx, preferentially triggers CaMKII activation and subsequent LTP induction (Fig. 4C,E,F) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In contrast, suppressed Ca2+ influx with postspike → prespike timing specifically induced PP1 activation without PKA activation (Fig. 4C,F,G), which induced LTD (Fig. 4D,E). Because PKA was not fully activated by the suppressed Ca2+ influx (Fig. 2G), PP1 activity was not suppressed and exhibited the highest activity at the low Ca2+ level (Fig. 2G). Also, because the PP1 activation has several steps from Ca2+, the PP1 activation requires enough duration of Ca2+. Therefore, the low and sustained Ca2+ influx, rather than the high and transient Ca2+ influx, preferentially induces LTD (Fig. 4C,E,G; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These results indicate that the allosteric kinetics of NMDARs can reliably detect prespike and postspike timing, which lead to STDP.
Features of the allosteric kinetics of NMDARs
To test the prediction of the allosteric kinetics of NMDARs, we measured the NMDAR-mediated EPSPs (NMDAR-EPSPs), which should directly show the effect of the allosteric kinetics, under the conditions where any component of AMPAR-EPSPs was not involved (Fig. 5A). We compared the amplitudes and time of NMDAR-EPSPs between the models and the experiments (Fig. 5B). In the allosteric model, the peak amplitudes of the NMDAR-EPSPs decreased with postspike → prespike timing (Fig. 5C, top). The peak amplitudes of the NMDAR-EPSPs in the no-allosteric model with rapid suppression (τ = 2.3 ms) also slightly decreased; however, those with slower suppression (τ = 230 ms) did not decrease with postspike → prespike timing (Fig. 5C, top). Experimentally, the peak amplitudes of the NMDAR-EPSPs also decreased (Fig. 5C, bottom). This experimental result is consistent with the results of both the allosteric model and the no-allosteric model with rapid suppression. We then compared the peak time of NMDAR-EPSPs (Fig. 5D). In the allosteric model and the no-allosteric model with slow suppression, the peak time of the NMDAR-EPSPs remained constant regardless of the spike timing, whereas the peak time of the NMDAR-EPSPs in the no-allosteric model with rapid suppression was delayed with postspike → prespike timing, because the decay of NMDAR-EPSPs is suppressed by Ca2+ influx via NMDARs themselves (Fig. 5D, top). In the experiments, the peak time of the NMDAR-EPSPs was not delayed and remained constant (Fig. 5D, bottom). This experimental result is consistent with the results in the allosteric model and in the no-allosteric model with slow suppression. We further examined the amplitude at the decay phase (decay amplitude), because the effect of the allosteric kinetics can appear more clearly during the decay phase. The decay amplitudes of the NMDAR-EPSPs in the allosteric model decreased; however, those in the no-allosteric models did not decrease with postspike → prespike timing (Fig. 5E, top). Experimentally, the decay amplitudes of the NMDAR-EPSPs also decreased (Fig. 5E, bottom). This experimental result is consistent with the result of the allosteric model but not with that of the no-allosteric models. We confirmed that the allosteric model gives a better fit to the experimental data than the no-allosteric models by measuring S(θ, Σ), a sum of variance and covariance of mean squared errors between experiment and simulation in peak amplitude, peak time, and decay amplitude, normalized by the best-fit simulation (Fig. 5F). As S(θ, Σ) becomes closer to 1, the model fit improves. S(θ, Σ) of the allosteric model was closer to 1 than that of the no-allosteric models, regardless of the time constant. This indicates that the allosteric model gives a much better fit to the experimental data than the no-allosteric models. Therefore, only the allosteric model appears to be consistent with the experimental properties of NMDAR-EPSPs.
Figure 5.

Features of the allosteric kinetics in NMDAR-EPSP. A, Derivation of NMDAR-EPSPs by postspiking → prespiking. NMDAR-EPSPs (right) was derived by the subtraction of AP alone from the NMDAR-EPSP plus AP in the absence of AMPAR activation (left, solid line) in presence of CNQX, a specific AMPAR antagonist (Froemke et al., 2005) (left, dashed line). B, Time courses of NMDAR-EPSPs by postspiking → prespiking in experiment (Tpost − Tpre = −10 ms, red; prespike-only as control, gray), in the no-allosteric model (−1, −2, −4,…, −256 ms) with rapid (τ1 = τ2 = 2.3 ms) and slow (τ1 = τ2 = 230 ms) suppression, and in the allosteric model (−1, −2, −4,…, −256 ms; τ1 = 2.3 ms and τ2 = 230 ms), as indicated. C–E, Peak amplitude (C), peak time (D), and decay amplitude (E) of NMDAR-EPSPs in the models (top) and the experiments (bottom). The responses were normalized by the control responses. The decay amplitude was taken at the time when three times longer than the largest peak time in each set. The responses of the no-allosteric model with rapid and slow suppression (blue and green, respectively) and the allosteric model (red) are plotted (top). The timing-dependent NMDAR-EPSPs in the experiments were fitted by a single exponential curve using the least square method (bottom). The apparent time constants of the responses of NMDAR-EPSPs versus the spike timing, τapp, are indicated. F, Evaluation of goodness of fit of the no-allosteric and allosteric models with the experimental data. The time constant in the no-allosteric model was changed as indicated, and the measure S(θ, Σ) was plotted. S(θ, Σ) is a sum of variance and covariance of errors between experiment and simulation in peak amplitude, peak time, and decay amplitude, normalized by variance and covariance of residuals in the best-fit model. S(θ, Σ) is given by
 , where f (xi, θ) is the fitting function, and θ is the parameter vector, Σ is the variance–covariance matrix of residuals of the best-fit model (τ1 = 2.3 ms and τ2 = 2.3 × 104 ms), Yi is a column vector of the peak amplitude, peak time, and decay amplitude of ith NMDAR-EPSP, and xi is Tpost − Tpre of the EPSP (Gallant, 1987). Note that the closer to 1 the S(θ, Σ) becomes, the better the model fits the experimental results. Arrowheads denote the time constants used in the no-allosteric and allosteric models. G, Goodness of fit against various time constant of Ca2+ · CaM binding to NMDARs, τ2, in the allosteric model. The τ2 was changed as indicated, and the S(θ, Σ) was measured. The arrowhead indicates τ2 (230 ms) used in the allosteric model.
, where f (xi, θ) is the fitting function, and θ is the parameter vector, Σ is the variance–covariance matrix of residuals of the best-fit model (τ1 = 2.3 ms and τ2 = 2.3 × 104 ms), Yi is a column vector of the peak amplitude, peak time, and decay amplitude of ith NMDAR-EPSP, and xi is Tpost − Tpre of the EPSP (Gallant, 1987). Note that the closer to 1 the S(θ, Σ) becomes, the better the model fits the experimental results. Arrowheads denote the time constants used in the no-allosteric and allosteric models. G, Goodness of fit against various time constant of Ca2+ · CaM binding to NMDARs, τ2, in the allosteric model. The τ2 was changed as indicated, and the S(θ, Σ) was measured. The arrowhead indicates τ2 (230 ms) used in the allosteric model.
We also examined the robustness of the allosteric kinetics of NMDARs by measuring of goodness of fit against various time constants of Ca2+ · CaM binding to NMDARs (Fig. 5G). We fixed τ1 as 2.3 ms from the experimental NMDAR suppression (Fig. 3C) (Froemke et al., 2005) and changed τ2 as indicated. We found that the allosteric kinetics can fit the NMDAR-EPSPs well in a wide range over 230 ms (100-fold difference between the time constants τ1 and τ2). In general, allosteric kinetics often accompanies a huge difference in time constants. For well-known allosteric kinetics of Ca2+ · CaM binding to nonphosphorylated and phosphorylated CaMKII, the difference in time con-stants is ∼1000-fold (supplemental information, available at http://www.kurodalab.org/info/STDP/Urakubo2008SI.pdf) (Meyer et al., 1992). Compared with such allosteric kinetics, a difference in time constants in the allosteric model of NMDARs (100-fold) is likely to be reasonable and physiological.
Complex spike train coding by the allosteric kinetics
We further tested the allosteric model with more complex spike patterns, shown previously to also induce STDP (Froemke and Dan, 2002; Wang et al., 2005; Froemke et al., 2006). We found that the allosteric model correctly predicted the direction of synaptic plasticity in spike triplet experiments (Fig. 6A,B) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) (Froemke and Dan, 2002), which indicated that the allosteric model can be extrapolated over STDP induced with more complex spike trains. We also validated the allosteric model with spike quadruplet experiments (Fig. 6C) (Froemke and Dan, 2002). We found a high correlation of the direction of synaptic plasticity between the allosteric model and the experiments (Fig. 6C) (Froemke and Dan, 2002). Finally, we validated the allosteric model using paired spike bursts instead of paired prespikes and postspikes (Fig. 7) (Froemke et al., 2006). The presynaptic burst after a postspike induced LTD at a similar extent, regardless of numbers of prespikes both in simulation and experiment (Fig. 7A). This is because of decreased probability of glutamate release during high-frequency stimulation (Tsodyks and Markram, 1997; Matveev and Wang, 2000) and of saturation of glutamate binding to NMDARs by a single prespike. The presynaptic burst before a postspike induced LTP, but the amplitude of LTP decreased as the number of prespikes increased (Fig. 7B). This is because a burst of five presynaptic spikes induced glutamate release mostly during the first or second spiking in the burst resulting from short-term depression; thereby the effective interval between prespiking and postspiking was prolonged, resulting in a reduction of the amplitude of LTP. The amplitudes of LTP with one or two prespikes in experiment are higher than those in the allosteric model because of the slower increase of LTP in the allosteric model; however, the amplitudes of LTP in experiment and the allosteric model became similar at longer periods (>60 min) (data not shown), which is in principle not inconsistent with the experimental data (Froemke et al., 2006). The postsynaptic burst with a single prespike induced a gradual shift from LTD to LTP as the number of postspikes increased both in experiment and in the allosteric model (Fig. 7C). The increase of the number of postspikes induced repetitive removal of the Mg2+ block from NMDARs, and this action overcame the suppression of NMDARs by Ca2+ · CaM, resulting in a gradual shift from LTD to LTP (Fig. 7C). The burst frequency-dependent synaptic plasticity was also similar between the experiments and the allosteric model (Fig. 7D) (Froemke et al., 2006). This result is consistent with the frequency-dependent synaptic plasticity at other synapses (Sjostrom et al., 2001). Therefore, the allosteric model appeared to be consistent with a wide range of complex spike train-dependent synaptic plasticity in layers II/III of visual cortex.
Figure 6.
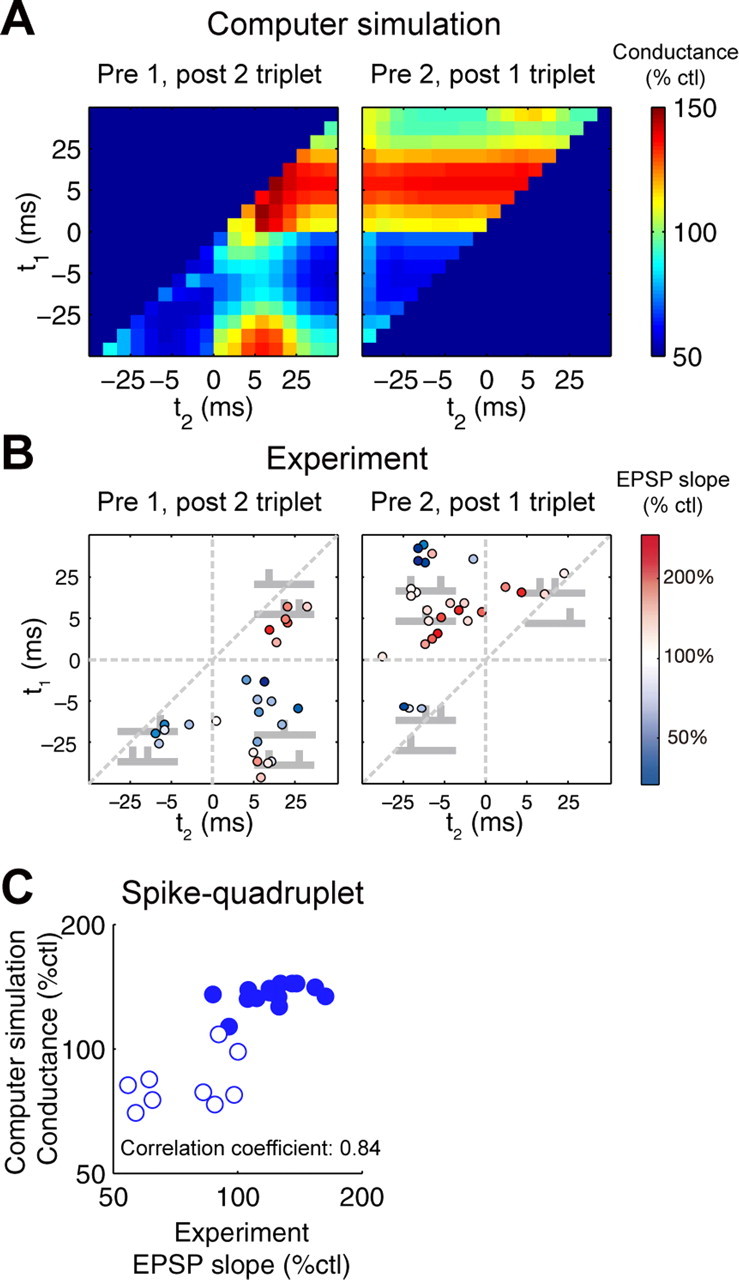
Complex spike timing-dependent synaptic plasticity. A, B, Changes of synaptic strength by spike triplets in the allosteric model (A) and in the experiments (B) (Froemke and Dan, 2002) (reproduced with permission from Nature). One prespiking and two postspiking (left), where t1 indicates the relative timing of the first postspiking to prespiking (Tpost 1 − Tpre), and t2 indicates the relative timing of the second postspiking to prespiking (Tpost 2 − Tpre). Two prespiking and one postspiking (right), where t1 = Tpost − Tpre 1 (Tpre 1, first prespiking) and t2 = Tpost − Tpre 2 (Tpre 2, second prespiking). Data in A were taken at 2 min after onsets of stimulation and averaged across three trials with different profiles of glutamate release based on presynaptic short-term depression (SEM <0.26%). C, Changes of synaptic strength by spike quadruplets. Synaptic conductance in the allosteric model versus correspondence in the experiments was plotted (Froemke et al., 2005). Open symbols, Prespiking → postspiking → postspiking → prespiking; filled symbols, postspiking → prespiking → prespiking → postspiking. Simulation data were averaged across three trials (SEM <0.38%).
Figure 7.

Burst-dependent synaptic plasticity. A, B, Changes of synaptic strength by presynaptic bursts with a single postsynaptic spike. The presynaptic spikes (with the indicated numbers) with 100 Hz after (A) or before (B) a postsynaptic spike was given at 1 Hz for 100 times. The interval between the postspiking and the nearest prespiking was 6 ms. Closed circles indicate synaptic conductance in the allosteric model (average of three trials; SEM <0.21%), and open circles indicate the correspondence in experiment (mean ± SEM) (Froemke et al., 2006) (A–D). C, Changes of synaptic strength by postsynaptic bursts with a single presynaptic spike. The postspikes (with the indicated numbers) with 100 Hz together with a prespike were given at 1 Hz for 100 times, and synaptic conductance was plotted. The interval between the prespiking and the first postspiking was 6 ms. D, Burst frequency-dependent synaptic plasticity. Five pairings of prespiking → postspiking (circles) and postspiking → prespiking (squares) with the indicated frequencies were given at 1 Hz for 100 times, and synaptic conductance was plotted (average of three trials; SEM <0.26%).
Requirement of postsynaptic NMDARs for the induction of LTD
We further compared the role of NMDARs in the induction of LTD between the allosteric model and the experimental results. Based on experiments, two NMDAR-dependent scenarios have been proposed for the induction of LTD by spike pairing. These two scenarios differ on the presumed synaptic location of NMDARs: one depends on activation of presynaptic NMDA autoreceptors (Sjostrom et al., 2003; Bender et al., 2006), and the other depends on postsynaptic NMDARs (Froemke et al., 2005; Seol et al., 2007). Therefore, we experimentally examined the requirement of postsynaptic NMDARs for the induction of LTD.
Through the recording electrodes, we loaded postsynaptic neurons with (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) (1 mm), an NMDAR antagonist, and examined whether LTD could be induced (Fig. 8). Postspike → prespike pairing did not induce LTD in cells filled with MK-801 (change in EPSP slope, 5.6 ± 9.5%; n = 3; p < 0.03) but induced LTD in nearby cells (<30 μm; change in EPSP slope; −35.9 ± 9.0%), which were recorded at the same time, indicating that injection of MK-801 fully blocked the induction of LTD. We confirmed that intracellular MK-801 (1 mm) blocked NMDARs to the same degree as bath-applied AP-5 (50 μm); intracellular MK-801 reduced NMDAR-EPSP amplitude by −76.9 ± 5.5% (n = 5), whereas extracellular APV reduced NMDAR-EPSP amplitude by −80.2 ± 4.8% (n = 4; p > 0.6, compared with intracellular MK-801) in the presence of CNQX (20 μm) to block AMPARs and picrotoxin (10 μm) to block GABAA receptors. These results indicate that postsynaptic NMDARs are required for the induction of LTD and support our hypothesis that postsynaptic NMDARs are responsible for the induction of LTD at this synapse.
Figure 8.

Requirement of postsynaptic NMDARs for the induction of LTD in STDP. Intracellular MK-801 (1 mm) prevented pairing-induced LTD. Examples of data from two simultaneously recorded neurons in the same slice are shown. Top, Recording with MK-801 in the internal solution. Bottom, Recording from a nearby neuron (∼25 μm away). At the time indicated by the arrow, postspike → prespike pairing [Tpost − Tpre = −9.6 ms (top), −10.3 ms (bottom)] was repeated. Red and blue lines indicate increase (11.8%; p > 0.2) and decrease (−44.8%; p < 10−10) in synaptic strength. EPSP slope is shown in the top panel, and input resistance (Ri) is shown in the bottom panel.
Spike timing-dependent Ca2+ influx
Recent Ca2+ imaging experiments at a different synapse revealed that prespiking → postspiking triggered accelerated Ca2+ influx, whereas postspiking → prespiking did not show significant suppressed Ca2+ influx (Nevian and Sakmann, 2004). To investigate whether our model could reproduce this experimental result, we included in our simulation the presence of a Ca2+ indicator in the allosteric model (Fig. 9A). We found that suppression of Ca2+ influx (which was seen in the absence of the Ca2+ indicator) (Fig. 4B,C) was not triggered by postspiking → prespiking in the presence of the Ca2+ indicator. This is because rapid absorption of free Ca2+ by the Ca2+ indicator results in the reduction of Ca2+ binding to CaM, and the suppression of NMDARs by Ca2+ · CaM was not consequently triggered, resulting in disappearance of suppressed Ca2+ influx by postspiking → prespiking.
Figure 9.

Spike timing-dependent Ca2+ influx. A, Time courses of Ca2+-bound Ca2+ indicator in cytosol (Conc, 100 μm; Kd, 580 nm) by prespiking → postspiking (Tpost − Tpre = 4 ms; left) and postspiking → prespiking (−4 ms; center). Time courses of Ca2+-bound Ca2+ indicator by a prespike alone were overlaid. Maximum of Ca2+-bound Ca2+ indicator by prespiking, prespiking → postspiking and postspiking → prespiking are shown (right). B, Time courses of [Ca2+]PSD and [Ca2+]cytosol by prespiking → postspiking with the indicated timing.
In addition, Ca2+ influx via NMDARs in the allosteric model seems to be far higher than Ca2+ influx via VGCCs, although some Ca2+-imaging experiments have shown that Ca2+ influx via NMDARs is comparable with Ca2+ influx via VGCCs (Koester and Sakmann, 1998; Nevian and Sakmann, 2004) (Figs. 3A, 4B). This is simply because the observation site in the allosteric model was a PSD compartment, where NMDARs were highly concentrated, and the amplitudes of Ca2+ influx from NMDARs and VGCCs became comparable in the cytosol (Fig. 9B). Therefore, the ratio of Ca2+ influx via NMDARs and VGCCs is also consistent with the experimental results.
Extraction of an essential rule of STDP
We extracted a simple and essential learning rule by reduction of the allosteric model. The principle of Ca2+ influx in the allosteric model is that, with prespike → postspike timing, spiking prevents the suppression of NMDARs by Ca2+ · CaM via the allosteric kinetics of NMDARs, and postspiking triggers the removal of the Mg2+ block of NMDARs by depolarization of the postsynaptic membrane potential, resulting in accelerated Ca2+ influx and induction of LTP (Fig. 10A, left). In contrast, with postspike → prespike timing, postspiking triggers the suppression of NMDARs by Ca2+ · CaM via the allosteric kinetics of NMDARs, resulting in suppressed Ca2+ influx and induction of LTD (Fig. 10A, right). Note that increase of membrane potential by postspiking does not affect NMDARs with postspike → prespike timing, because membrane potential returns to the resting potential when prespiking arrives. Thus, postspiking positively and negatively regulates NMDAR activation via increase of membrane potential and suppression by Ca2+ · CaM, respectively. Based on this principle of the allosteric model, we write the simple STDP model as follows:
 |
 |
 |
 |
 |
where NMDAR(t), V(t), and Ca(t) represent NMDAR activity, membrane potential, and Ca2+ · CaM concentration, respectively; τNMDAR, τV, and τCa are the decay time constants of NMDAR(t), V(t), and Ca(t), respectively; Vrest is the resting membrane potential; AP is the amplitude of the backpropagating action potential; CaVGCC is the Ca2+ influx via VGCC; KCa is the effective Ca that gives the 50% inhibition of the maximal activity of NMDAR; tpre and tpost are the timings of the prespiking and postspiking, respectively; (Ca)max is the maximal value of Ca by a single pairing of prespiking and postspiking; θLTP and θLTD denote the thresholds of Ca for LTP and LTD, respectively; and ALTP, ALTD, and ALTP/LTD correspond to the amplitudes of LTP, LTD, and consequent synaptic strength, respectively. We set τNMDAR = 40 ms, τV = 6 ms, τCa = 20 ms, Vrest = −65 mV, AP = 40 mV, CaVGCC = 1.3, KCa = 0.3, θLTP = 6.2, θLTD = 4, ALTP = 40, and ALTD = 20.
Figure 10.

Simple STDP model. A, The principle of STDP. Postspiking positively regulates NMDARs (NMDAR) via increase of membrane potential (V) with prespiking → postspike timing (left, solid line), resulting in induction of LTP (left). With this timing, Ca2+ · CaM (Ca) does not affect NMDAR via the allosteric kinetics (left, dashed line). Postspiking negatively regulates NMDAR via Ca with postspike → prespike timing (right, solid line), resulting in induction of LTD (right). With this timing, V does not effectively affect NMDAR, because V returns to the basal level when a prespike arrives (right, dashed line). B, NMDAR(t), V(t), and Ca(t) by a single pairing of prespiking and postspiking with the indicated timing (Eqs. 1–3). The dashed line in NMDAR (top, right) indicates NMDAR with prespiking alone, and the dashed lines in Ca (bottom) indicate a linear sum of Ca induced by a prespike alone and a postspike alone with the indicated timing. C, Spike timing-dependent Ca (left) and synaptic strength (right). The high and low Ca induced by a single pairing of prespiking and postspiking with the indicated timing were converted into LTP (above the threshold, θLTP) and LTD (below the threshold, θLTD), respectively (Eqs. 4, 5). D, Synaptic plasticity induced with spike triplets. The relative spike timing of triplets, t1 and t2, is the same as in Figure 6.
Equations 1 and 2 represent the decays of NMDAR activity and membrane potential, respectively. Equation 3 represents that Ca2+ increase depends on the voltage-dependent activation of NMDARs with an exponential decay, where (0.0223[V(t) − Vrest] + 0.5) denotes the voltage dependence of NMDARs, which is obtained by the linearization of the voltage dependence of NMDARs in the allosteric model around the resting membrane potential (supplemental information, available at http://www.kurodalab.org/info/STDP/Urakubo2008SI.pdf). In Equation 4, at t = tpost, postspiking triggers the increase of voltage via bpAP and Ca2+ increase via VGCC, whereas at t = tpre, NMDAR is activated in a reciprocal manner of Ca. Equations 3 and 4 indicate that Ca acts on NMDAR only at t = tpre but not later, which means that the suppression of NMDARs reflects Ca2+ level at prespiking but not after prespiking. Therefore, Equations 3 and 4 represent the allosteric kinetics of NMDARs. The difference in Equation 4 used when t = tpre and t = tpost enables LTP induction with the prespike → postspike timing and LTD induction with the postspike → prespike timing. Equation 5 represents that the high Ca2+ influx results in LTP via CaMKII, and the low Ca2+ influx results in LTD via PP1.
The high and low Ca were induced with the prespike → postspike and the postspike → prespike timing, respectively (Fig. 10B). With the prespike → postspike timing, NMDAR is fully activated because of the absence of Ca at t = tpre, whereas with the postspike → prespike timing, NMDAR is weakly activated because of the presence of Ca at t = tpre (Fig. 10B) (Eq. 3). This dependence of NMDAR on Ca at t = tpre clearly indicates that the intracellular Ca2+ concentration, when NMDARs are activated, carries the previous prespike and postspike-timing information. The simple STDP model was capable of reproducing the spike timing-dependent Ca (Fig. 10C, left) and consequent STDP (Fig. 10C, right). Note that the high and low Ca, but not moderate one, triggered LTP and LTD, respectively. The time window for LTP depends on the decay time constant of NMDARs, and that for LTD on the decay time constant of Ca2+ increase by VGCC, which are consistent with the allosteric model. Synaptic plasticity induced by spike triplets was coherently reproduced (Fig. 10D). These results were similar to those in the allosteric model (Figs. 4B–E, 6A,B), indicating that the simple STDP model retains the essential properties of the allosteric model. Thus, the bidirectional role of postspiking via the allosteric kinetics of NMDARs in STDP, rather than the detailed parameters and complicated molecular interactions, is the essential principle of STDP.
Discussion
We show that the simple allosteric kinetics of NMDARs codes complex spike-timing information into STDP. The allosteric kinetics of NMDARs depends on the different time constants of Ca2+ · CaM to glutamate-bound and glutamate-unbound NMDARs. In general, the allosteric kinetics accompanies the changes of affinity, and it is possible that the allosteric kinetics of NMDARs also accompanies the changes of Ca2+ · CaM affinity for glutamate-bound and glutamate-unbound NMDARs. Similar results were obtained if the affinity of glutamate for Ca2+ · CaM-unbound NMDARs were over four times higher than that for Ca2+ · CaM-bound NMDARs (data not shown), although the affinity of glutamate to NMDARs is not significantly affected by binding Ca2+ · CaM to NMDARs (Zhang et al., 1998). The principle of our prediction is that any similar allosteric kinetics of NMDARs that depends on the timing between prespiking and postspiking can account for STDP, even if STDP is Ca2+ · CaM independent. Taking into consideration that STDP has been experimentally observed across a wide range of preparations, a single molecular property, such as an allosteric kinetics of NMDARs, seems more reasonable to account for STDP, rather than other complex molecular events, such as phosphorylation/dephosphorylation, because the former is more robust against variable expression levels of molecules.
CaN is required to suppress NMDARs with postspiking → prespiking (Froemke et al., 2005). Phosphatase activity of CaN is not likely to detect such short timing simply because Ca2+-dependent activation of phosphatase activity of CaN requires a longer time constant (Klee et al., 1998). In addition, dephosphorylation of NMDARs by CaN or other related phosphatases was required to associate Ca2+ · CaM to the NMDARs (Rycroft and Gibb, 2004a). Therefore, CaN activity may be a prerequisite condition for detecting the postspiking → prespiking, such as dephosphorylation of NMDARs, and this is implicitly incorporated into NMDAR kinetics in the allosteric model.
Although particular detailed parameters of our model may be unsupported by experimental observations, our main conclusion in this study is the bidirectional role of postspiking based on the allosteric kinetics of NMDARs in STDP as shown by the model reduction, and this does not critically depend on the exact details of most of the parameters. We emphasize that the signaling cascades in the allosteric model should be regarded as a tentative description, rather than a complete description. For example, some signaling cascades in the allosteric model, such as PP1, are based on the previous reports in other types of synaptic plasticity such as LTD induced by prolonged low-frequency presynaptic stimulation (LFS) (Mulkey et al., 1993), but they have not been shown to be directly involved in STDP. Also, in the allosteric model, LTP induced by high-frequency presynaptic stimulation, but not LTD induced by prolonged LFS (typically 900 times at 1 Hz), could be reproduced (data not shown) (Bear and Malenka, 1994). This implies the possibility that a missing pathway, requiring a long duration of the stimulus (>10 min), may be responsible for LTD induced by prolonged LFS. Note that 100 prespikes at 1 Hz alone did not induce LTD both in simulation and experiment (Fig. 4E) (Mizuno et al., 2001; Johnston et al., 2003). Additionally, another class of STDP has been shown to involve a distinct form of LTD that is independent of postsynaptic NMDARs and may be presynaptically expressed (Sjostrom et al., 2003; Bender et al., 2006). The STDP in layer II/III of visual cortex is not consistent with doublet-dependent STDP (Wittenberg and Wang, 2006) and triplet-dependent STDP (Wang et al., 2005) in hippocampal CA1 neurons. This suggests the possibility of requirement of an additional pathway(s) for the STDP in hippocampal CA1 neurons. The extension of the model by incorporating the future progress will shed light on the general principles of distinct classes of STDP.
A recent study reported that NMDAR subtypes are selective for the direction of synaptic modification in STDP (Gerkin et al., 2007). Our results indicate that the kinetics of NMDARs should detect the interaction between prespiking and postspiking. Together, the selectivity of NMDAR subtypes for the direction of synaptic modification may be attributable to the subtype-dependent different kinetics of NMDARs in Ca2+ · CaM binding or in the voltage dependence, both of which can be considered part of the postspiking signal.
Several biophysical models of STDP have been proposed in which postsynaptic Ca2+ influx also serves as a spike-timing detector (Karmarkar and Buonomano, 2002; Shouval et al., 2002; Rubin et al., 2005). These models assume that the basal Ca2+ influx induced by uncorrelated prespiking and postspiking (less than −200 or >100 ms) is far lower than that for LTD and LTP, and the postspiking → prespiking induces moderate Ca2+ influx for LTD, whereas the prespiking → postspiking induces high Ca2+ influx for LTP. However, with this assumption, it is difficult to avoid unnecessary LTD induction by the prespiking → postspiking with a long interval, which has not usually been observed in experiments (Sjostrom and Nelson, 2002), although some modifications have been proposed to avoid this (Rubin et al., 2005; Shouval and Kalantzis, 2005). Also, a biophysical model has been proposed to explain synaptic plasticity induced by spike triplets (Shah et al., 2006). However, this model uses the unnecessary LTD induction by the prespiking → postspiking to explain synaptic plasticity induced by spike triplets. In contrast, in the allosteric model, we set that the prespiking → postspiking, the uncorrelated spiking, and the postspiking → prespiking induced the high, moderate, and low Ca2+ influx, which led to potentiation, no change (neither potentiation nor depression), and depression, respectively (Koester and Sakmann, 1998; Cho et al., 2001; Lisman, 2001; Froemke et al., 2005) (Fig. 10C) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). This makes the allosteric model free from the unnecessary induction of LTD by the prespiking → postspiking, regardless of timing interval, and the allosteric model can coherently explain synaptic plasticity induced by a variety of complex spike trains within a single framework. Additionally, we experimentally found that the partial blockage of NMDAR activity induces depression, and the facilitation of NMDAR activity with removal of Mg2+ induces potentiation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The biphasic synaptic modification against NMDAR activity is consistent with the hypothesis of the three Ca2+ levels for synaptic plasticity, because Ca2+ influx dominantly depends on NMDARs. Together, the three Ca2+ levels for synaptic plasticity is substantially evident from both phenomenological experiments (synaptic plasticity induced by spike doublets, spike triplets, and more complex spike trains) and pharmacological experiments (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Our allosteric model belongs to the one-coincidence detector model. A model of two-coincidence detectors for STDP has also been proposed (Karmarkar and Buonomano, 2002). In the model, voltage-dependent Mg2+ removal from NMDARs detects prespiking → postspiking for LTP, whereas metabotropic GluRs (mGluRs) and Ca2+ influx via VGCCs detects postspiking → prespiking for LTD. Recently, some experiments have revealed that such secondary coincidence detectors are involved in LTD of STDP at other synapses: mGluRs at synapses between layer IV and layer II/III neurons in the somatosensory cortex (Bender et al., 2006) and CB1 receptors at synaptic connections between layer V pyramidal cells (Sjostrom et al., 2003). Taking our findings of requirement of rapid suppression by postspiking into consideration, these molecules should interact with a postspiking signal with the time constant on an order of a few ten milliseconds. Such rapid time constant may depend on the ion flux or changes of voltage, rather than phosphorylation/dephosphorylation processes or catalytic activities of downstream of mGluR1 and CB1 receptors.
We analyzed the sensitivity of the amplitude of STDP against the conductance of AMPARs, because the amplitude of LTP in STDP has been reported to be dependent on the size of the initial EPSPs (Bi and Poo, 1998; Sjostrom et al., 2001), which depends on the AMPAR activity, and this issue is theoretically related to the boundary condition on synaptic plasticity, whether the changes of synaptic strength depend on the current synaptic weight or not (Song et al., 2000; van Rossum et al., 2000; Cateau and Fukai, 2003). The relative amplitudes of LTP induced by prespiking → postspiking remained the same against the changes of conductance of AMPARs (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). This means that the amplitude of LTP is positively correlated with the size of the initial EPSPs, which is similar to the theoretical study of the multiplicative learning rule (van Rossum et al., 2000; Cateau and Fukai, 2003). However, additional experimental studies are necessary to conclude this issue.
In conclusion, the allosteric model appears to be consistent with specific features of STDP, such as spike timing-dependent NMDAR-EPSPs and spike doublet-, triplet-, quadruplet-, and burst-dependent synaptic plasticity within a single framework. This study provides a novel concept of the bidirectional role of postspiking in induction of synaptic plasticity by the allosteric kinetics of NMDARs.
Footnotes
This work was supported by a grant-in-aid for scientific research on priority areas “Systems Genomics” (17017005) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by the Grass Foundation. We thank K. Vance and S. Traynelis for valuable discussions, H. Bito for critically reading this manuscript, and Y. Dan for supporting the experimental portion of this study.
References
- Artola A, Brocher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283:381–387. doi: 10.1126/science.283.5400.381. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modification by correlated activity: Hebb's postulate revisited. Annu Rev Neurosci. 2001;24:139–166. doi: 10.1146/annurev.neuro.24.1.139. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Rubin J. Timing in synaptic plasticity: from detection to integration. Trends Neurosci. 2005;28:222–228. doi: 10.1016/j.tins.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- Boettiger CA, Doupe AJ. Developmentally restricted synaptic plasticity in a songbird nucleus required for song learning. Neuron. 2001;31:809–818. doi: 10.1016/s0896-6273(01)00403-2. [DOI] [PubMed] [Google Scholar]
- Cateau H, Fukai T. A stochastic method to predict the consequence of arbitrary forms of spike-timing-dependent plasticity. Neural Comput. 2003;15:597–620. doi: 10.1162/089976603321192095. [DOI] [PubMed] [Google Scholar]
- Cho K, Aggleton JP, Brown MW, Bashir ZI. An experimental test of the role of postsynaptic calcium levels in determining synaptic strength using perirhinal cortex of rat. J Physiol (Lond) 2001;532:459–466. doi: 10.1111/j.1469-7793.2001.0459f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Corlew R, Wang Y, Ghermazien H, Erisir A, Philpot BD. Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J Neurosci. 2007;27:9835–9845. doi: 10.1523/JNEUROSCI.5494-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Doi T, Kuroda S, Michikawa T, Kawato M. Inositol 1,4,5-trisphosphate-dependent Ca2+ threshold dynamics detect spike timing in cerebellar Purkinje cells. J Neurosci. 2005;25:950–961. doi: 10.1523/JNEUROSCI.2727-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, Zhang S, Bernhadt JP, Huganir RL. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Dan Y. Spike-timing-dependent synaptic modification induced by natural spike trains. Nature. 2002;416:433–438. doi: 10.1038/416433a. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Poo MM, Dan Y. Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature. 2005;434:221–225. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Tsay IA, Raad M, Long JD, Dan Y. Contribution of individual spikes in burst-induced long-term synaptic modification. J Neurophysiol. 2006;95:1620–1629. doi: 10.1152/jn.00910.2005. [DOI] [PubMed] [Google Scholar]
- Gallant AR. Nonlinear statistical models. New York: Wiley; 1987. Multivariate nonlinear regression; pp. 267–404. [Google Scholar]
- Gerkin RC, Lau PM, Nauen DW, Wang YT, Bi GQ. Modular competition driven by NMDA receptor subtypes in spike-timing-dependent plasticity. J Neurophysiol. 2007;97:2851–2862. doi: 10.1152/jn.00860.2006. [DOI] [PubMed] [Google Scholar]
- Gerstner W, Kempter R, van Hemmen JL, Wagner H. A neuronal learning rule for sub-millisecond temporal coding. Nature. 1996;383:76–81. doi: 10.1038/383076a0. [DOI] [PubMed] [Google Scholar]
- Hayer A, Bhalla US. Molecular switches at the synapse emerge from receptor and kinase traffic. PLoS Comput Biol. 2005;1:137–154. doi: 10.1371/journal.pcbi.0010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Johnston D, Christie BR, Frick A, Gray R, Hoffman DA, Schexnayder LK, Watanabe S, Yuan LL. Active dendrites, potassium channels and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci. 2003;358:667–674. doi: 10.1098/rstb.2002.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa BM, Clements J, Jonas P, Stuart GJ. Kinetics of Mg2+ unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J Physiol (Lond) 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmarkar UR, Buonomano DV. A model of spike-timing dependent plasticity: one or two coincidence detectors? J Neurophysiol. 2002;88:507–513. doi: 10.1152/jn.2002.88.1.507. [DOI] [PubMed] [Google Scholar]
- Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc Natl Acad Sci USA. 1998;95:9596–9601. doi: 10.1073/pnas.95.16.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda S, Schweighofer N, Kawato M. Exploration of signal transduction pathways in cerebellar long-term depression by kinetic simulation. J Neurosci. 2001;21:5693–5702. doi: 10.1523/JNEUROSCI.21-15-05693.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Three Ca2+ levels affect plasticity differently: the LTP zone, the LTD zone and no man's land. J Physiol (Lond) 2001;532:285. doi: 10.1111/j.1469-7793.2001.0285f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Matveev V, Wang XJ. Implications of all-or-none synaptic transmission and short-term depression beyond vesicle depletion: a computational study. J Neurosci. 2000;20:1575–1588. doi: 10.1523/JNEUROSCI.20-04-01575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- Migliore M, Hoffman DA, Magee JC, Johnston D. Role of an A-type K+ conductance in the back-propagation of action potentials in the dendrites of hippocampal pyramidal neurons. J Comput Neurosci. 1999;7:5–15. doi: 10.1023/a:1008906225285. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kanazawa I, Sakurai M. Differential induction of LTP and LTD is not determined solely by instantaneous calcium concentration: an essential involvement of a temporal factor. Eur J Neurosci. 2001;14:701–708. doi: 10.1046/j.0953-816x.2001.01679.x. [DOI] [PubMed] [Google Scholar]
- Mu Y, Poo MM. Spike timing-dependent LTP/LTD mediates visual experience-dependent plasticity in a developing retinotectal system. Neuron. 2006;50:115–125. doi: 10.1016/j.neuron.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Single spine Ca2+ signals evoked by coincident EPSPs and backpropagating action potentials in spiny stellate cells of layer 4 in the juvenile rat somatosensory barrel cortex. J Neurosci. 2004;24:1689–1699. doi: 10.1523/JNEUROSCI.3332-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister JP, Gerstner W. Triplets of spikes in a model of spike timing-dependent plasticity. J Neurosci. 2006;26:9673–9682. doi: 10.1523/JNEUROSCI.1425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirazi P, Brannon T, Mel BW. Arithmetic of subthreshold synaptic summation in a model CA1 pyramidal cell. Neuron. 2003;37:977–987. doi: 10.1016/s0896-6273(03)00148-x. [DOI] [PubMed] [Google Scholar]
- Rubin JE, Gerkin RC, Bi GQ, Chow CC. Calcium time course as a signal for spike-timing-dependent plasticity. J Neurophysiol. 2005;93:2600–2613. doi: 10.1152/jn.00803.2004. [DOI] [PubMed] [Google Scholar]
- Rycroft BK, Gibb AJ. Direct effects of calmodulin on NMDA receptor single-channel gating in rat hippocampal granule cells. J Neurosci. 2002;22:8860–8868. doi: 10.1523/JNEUROSCI.22-20-08860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycroft BK, Gibb AJ. Inhibitory interactions of calcineurin (phosphatase 2B) and calmodulin on rat hippocampal NMDA receptors. Neuropharmacology. 2004a;47:505–514. doi: 10.1016/j.neuropharm.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Rycroft BK, Gibb AJ. Regulation of single NMDA receptor channel activity by alpha-actinin and calmodulin in rat hippocampal granule cells. J Physiol (Lond) 2004b;557:795–808. doi: 10.1113/jphysiol.2003.059212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Maravall M, Svoboda K. Ca2+ signaling in dendritic spines. Curr Opin Neurobiol. 2001;11:349–356. doi: 10.1016/s0959-4388(00)00218-x. [DOI] [PubMed] [Google Scholar]
- Senn W. Beyond spike timing: the role of nonlinear plasticity and unreliable synapses. Biol Cybern. 2002;87:344–355. doi: 10.1007/s00422-002-0350-1. [DOI] [PubMed] [Google Scholar]
- Seol GH, Ziburkus J, Huang S, Song L, Kim IT, Takamiya K, Huganir RL, Lee HK, Kirkwood A. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron. 2007;55:919–929. doi: 10.1016/j.neuron.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NT, Yeung LC, Cooper LN, Cai Y, Shouval HZ. A biophysical basis for the inter-spike interaction of spike-timing-dependent plasticity. Biol Cybern. 2006;95:113–121. doi: 10.1007/s00422-006-0071-y. [DOI] [PubMed] [Google Scholar]
- Shouval HZ. Clusters of interacting receptors can stabilize synaptic efficacies. Proc Natl Acad Sci USA. 2005;102:14440–14445. doi: 10.1073/pnas.0506934102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval HZ, Kalantzis G. Stochastic properties of synaptic transmission affect the shape of spike time-dependent plasticity curves. J Neurophysiol. 2005;93:1069–1073. doi: 10.1152/jn.00504.2004. [DOI] [PubMed] [Google Scholar]
- Shouval HZ, Bear MF, Cooper LN. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc Natl Acad Sci USA. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Nelson SB. Spike timing, calcium signals and synaptic plasticity. Curr Opin Neurobiol. 2002;12:305–314. doi: 10.1016/s0959-4388(02)00325-2. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Song S, Miller KD, Abbott LF. Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat Neurosci. 2000;3:919–926. doi: 10.1038/78829. [DOI] [PubMed] [Google Scholar]
- Stiefel KM, Tennigkeit F, Singer W. Synaptic plasticity in the absence of backpropagating spikes of layer II inputs to layer V pyramidal cells in rat visual cortex. Eur J Neurosci. 2005;21:2605–2610. doi: 10.1111/j.1460-9568.2005.04094.x. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Hausser M. Dendritic coincidence detection of EPSPs and action potentials. Nat Neurosci. 2001;4:63–71. doi: 10.1038/82910. [DOI] [PubMed] [Google Scholar]
- Tsodyks MV, Markram H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci USA. 1997;94:719–723. doi: 10.1073/pnas.94.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- van Rossum MC, Bi GQ, Turrigiano GG. Stable Hebbian learning from spike timing-dependent plasticity. J Neurosci. 2000;20:8812–8821. doi: 10.1523/JNEUROSCI.20-23-08812.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vislay-Meltzer RL, Kampff AR, Engert F. Spatiotemporal specificity of neuronal activity directs the modification of receptive fields in the developing retinotectal system. Neuron. 2006;50:101–114. doi: 10.1016/j.neuron.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Wang HX, Gerkin RC, Nauen DW, Bi GQ. Coactivation and timing-dependent integration of synaptic potentiation and depression. Nat Neurosci. 2005;8:187–193. doi: 10.1038/nn1387. [DOI] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- Wittenberg GM, Wang SS. Malleability of spike-timing-dependent plasticity at the CA3-CA1 synapse. J Neurosci. 2006;26:6610–6617. doi: 10.1523/JNEUROSCI.5388-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL. Calmodulin mediates calcium-dependent inactivation of N-methyl-d-aspartate receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]