

Abstract
Huntington's disease (HD) is a hereditary neurological disease caused by expended CAG repeats in the HD gene, which codes for a protein called Huntingtin (Htt). The resultant mutant Huntingtin (mHtt) forms aggregates in neurons and causes neuronal dysfunction. In astrocytes, the largest population of brain cells, mHtt also exists. We report herein that astrocyte-conditioned medium (ACM) collected from astrocytes of R6/2 mice (a mouse model of HD) caused primary cortical neurons to grow less-mature neurites, migrate more slowly, and exhibit lower calcium influx after depolarization than those maintained in wild-type (WT) ACM. Using a cytokine antibody array and ELISA assays, we demonstrated that the amount of a chemokine [chemokine (C-C motif) ligand 5 (CCL5)/regulated on activation normal T cell expressed and secreted (RANTES)] released by R6/2 astrocytes was much less than that by WT astrocytes. When cortical neurons were treated with the indicated ACM, supplementation with recombinant CCL5/RANTES ameliorated the neuronal deficiency caused by HD-ACM, whereas removing CCL5/RANTES from WT-ACM using an anti-CCL5/RANTES antibody mimicked the effects evoked by HD-ACM. Quantitative PCR and promoter analyses demonstrated that mHtt hindered the activation of the CCL5/RANTES promoter by reducing the availability of nuclear factor κB-p65 and, hence, reduced the transcript level of CCL5/RANTES. Moreover, ELISA assays and immunocytochemical staining revealed that mHtt retained the residual CCL5/RANTES inside R6/2 astrocytes. In line with the above findings, elevated cytosolic CCL5/RANTES levels were also observed in the brains of two mouse models of HD [R6/2 and Hdh(CAG)150] and human HD patients. These findings suggest that mHtt hinders one major trophic function of astrocytes which might contribute to the neuronal dysfunction of HD.
Keywords: astrocyte, trophic, release, Huntington, neuron, transcription
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease characterized by chorea, dementia, and psychiatric symptoms. The causative mutation is a CAG trinucleotide expansion in exon 1 of the Huntington (HD) gene, which is translated into polyglutamine residues (polyQ) in the Huntingtin (Htt) protein. When the number of CAG repeats exceeds 36, Htt forms aggregates in the nuclei of neurons and might hijack a wide variety of proteins, including key transcriptional factors, which lead to transcriptional dysfunction, and eventually causes neuronal degeneration (Sugars and Rubinsztein, 2003; Li and Li, 2004). Previous studies also demonstrated that normal Htt protein functions in intracellular vesicle transport, whereas polyQ-expanded mutant Htt (mHtt) suppresses vesicle transport and disturbs the secretion of trophic factors [e.g., brain-derived neurotrophic factor (BDNF) (Gauthier et al., 2004)]. As a result, HD neurons exhibit shorter neurites, fewer numbers of dendritic spines, degenerated axons, abnormal neuronal activities, and eventual cell death in several specific brains areas, including the striatum and cortex (Klapstein et al., 2001; Li et al., 2001; Spires et al., 2004).
In addition to neurons, mHtt has also been found in many different types of cells, including glial cells in the brain, muscle cells, and liver hepatocytes (Orth et al., 2003; Shin et al., 2005; Chiang et al., 2007). In the CNS, astrocytes are the largest cell population and play multiple roles in the brain. The functions of astrocytes include promoting neuronal survival and plasticity, removing toxic materials (e.g., glutamate and free radicals), and providing gliotransmitters to neurons through neuronal-glial interactions (Volterra and Meldolesi, 2005). With these important characteristics, astrocytes have been implicated in several major neuronal degenerative diseases such as Parkinson's disease, Alzheimer's disease, and HD (Maccioni et al., 2001; Vila et al., 2001; Shin et al., 2005). The mHtt was previously found in reactive astrocytes of HD patients and mouse models and is believed to play a pathogenic role in HD (Singhrao et al., 1998; Hebb et al., 1999; Shin et al., 2005). In HD astrocytes, the glutamate transporter is impaired and thus disables their ability to protect neurons against glutamate-mediated excitotoxicity (Shin et al., 2005).
One major class of mediators for astrocytes comprises the chemokine family. In the present study, we found that primary astrocytes prepared from a transgenic mouse model of HD (R6/2) produced and secreted less chemokine (C-C motif) ligand 5 (CCL5)/regulated on activation normal T cell expressed and secreted (RANTES) (an important chemokine) than wild-type (WT) astrocytes. Through activating at least three different receptors (CCR1, CCR3, and CCR5), CCL5/RANTES exhibits diverse functions, including modulation of neuronal migration, regulation of proliferation and/or differentiation of astrocytes, and mediation of inflammation in neuronal diseases (Bolin et al., 1998; Appay and Rowland-Jones, 2001; Bakhiet et al., 2001). We demonstrate herein that mHtt reduced the expression of CCL5/RANTES at the transcriptional level in astrocytes as well as retained the residual CCL5/RANTES inside astrocytes. Such dysregulation of CCL5/RANTES in astrocytes greatly reduced the availability of CCL5/RANTES to neurons and is thought to possibly play an important role in the neuronal dysfunction of HD.
Materials and Methods
Primary astrocyte and neuron cultures.
Primary astrocytes were purified from neonatal (∼1–2 d of age) brains of R6/2 HD mice (Mangiarini et al., 1996) and their littermates as the WT control. In brief, the indicated brain areas (e.g., cortices, striatum, and cerebellum) were carefully removed, digested with 0.05% trypsin-EDTA (Invitrogen, Grand Island, NY) at 37°C for 10 min, and then mechanically dissociated by gentle pipetting and passed through a 70-μm-pore nylon mesh. Cells were plated onto poly-l-lysine-coated dishes and grown in DMEM (Invitrogen) supplemented with 10% v/v heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C in a humidified 5% CO2-containing atmosphere. Offspring were identified by a PCR-based genotyping technique of genomic DNA extracted from mice tails using the primers (5′-CCGCTCAGGTTCTGCTTTTA-3′ and 5′-GGCTGAGGAAGCTGAGGAG-3′) suggested by The Jackson Laboratory (Bar Harbor, ME). Mutant Htt expression was measured by a mouse monoclonal anti-human Htt antibody (EM48) (1:200 dilution; Millipore Bioscience Research Reagents, Temecula, CA). The purity of the primary astrocyte cultures was determined by immunocytochemical staining using an antibody against an astrocyte-specific marker (GFAP, dilution 1:1000; Sigma, St. Louis, MO) or a microglia-specific marker (anti-CD11b, dilution 1:200; Serotec, Oxford, UK). At 30 d in vitro (DIV), 99% of the primary cultured cells were GFAP positive. No detectable CD11b-positive cells (i.e., microglia) were found (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Primary neuronal cultures were prepared from brains of Sprague Dawley rat fetuses on embryonic day 18 (E18) to E19 as described previously (Brewer et al., 1993). Briefly, embryo cortices were digested with 0.25% trypsin-EDTA for 10 min at 37°C and mechanically dissociated by gentle pipetting in modified Eagle's medium supplemented with 5% v/v FBS, 5% v/v horse serum, 0.6% v/v glucose, 0.5 mm glutamine, 1% penicillin/streptomycin, and 1% insulin-transferrin-sodium selenite media supplement (ITS mixture; Sigma). Cells were plated on poly-l-lysine-coated culture dishes. After a 3 h incubation, the cultured medium was replaced with a Neurobasal medium supplemented with 0.5 mm glutamine, 12.5 μm glutamate, 2% B27, and 1% penicillin/streptomycin. The purity of neuronal cultures was determined by immunocytochemical staining using an antibody against a neuron-specific marker, class III β-tubulin (TUJ-1) (dilution, 1:1000; Promega, Madison, WI).
Astrocyte-conditioned medium and cytokine antibody arrays.
To prepare astrocyte-conditioned medium (ACM), primary astrocytes prepared from WT or R6/2 mice were cultured at the same density (30 DIV; 1.5 × 106 in 100 mm plates) in DMEM supplemented with 10% FBS for 2 d, washed twice with HBSS, and then cultured in serum-free DMEM for an additional 3 d. The ACM was then collected, centrifuged at 500 × g for 5 min to remove cell debris, and stored at −80°C until further analysis.
Cytokine antibody array.
Levels of cytokines/chemokines in the ACM were assessed using the mouse cytokine Ab array (RayBio; RayBiotech, Norcross, GA) following the protocol of the manufacturer. Signal intensities of each cytokine were quantified using the MetaMorph software and were normalized with the positive controls on the same membrane.
Immunochemical staining.
Cells or brain sections were fixed with 4% paraformaldehyde plus 4% sucrose in PBS, pH 7.4, at room temperature (RT) for 30 min and then permeabilized with 0.1% Triton X-100 at RT for an additional 30 min. Nonspecific antibody binding was blocked by incubating cells with 2% normal goat serum plus 2% bovine serum albumin (BSA) in PBS for 1 h at RT and incubated with the desired primary antibody at 4°C for 18 h, followed by incubation with the corresponding secondary antibody for 2 h at RT. The anti-mouse and anti-human antibodies of CCL5/RANTES and CCL2/monocyte chemoattractant protein-1 (MCP-1) were obtained from R&D Systems (Minneapolis, MN) and were used in immunochemical analyses following the protocols of the manufacture. Nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI). Fluorescence-immunostained samples were mounted with 50% glycerol. Patterns of immunostaining were analyzed with the aid of MetaMorph software (Universal Imaging Corporation, West Chester, PA) and a CCD microscope (Zeiss, Göttingen, Germany) or a confocal microscope (Radiance 2100 Confocal; Bio-Rad, Henel Hempstead, Hertfordshire, UK).
Neurite branching, sprouting, and outgrowth.
The neuronal fiber length was quantified based on an equation described in the MetaMorph software as follows: (1/4) × [P + √(P2 − 16A)], where P is the perimeter, and A is the area. The neurite branching was quantified by dividing the number of neurite endfeet by the number of neuronal sprouts.
Migration assay.
Primary cortical neurons, at a density of 2 × 106 cells/well, were plated onto transwell membrane inserts (8 μm pore size, 35 mm; Corning, New York, NY) and cultured in neurobasal medium plus B27 (NB) in both the upper and lower chambers for 4 d. Half of the medium in the lower chamber was then replaced with the indicated ACM containing the anti-CCL5/RANTES (0.2 μg/ml, R&D Systems), normal goat IgG (0.2 μg/ml) (Sigma), or recombinant mouse CCL5/RANTES (450 pg/ml; R&D Systems) for an additional 18 h. Cells migrating through the pores adhered to the underside of the transwell membrane and were fixed with 4% paraformaldehyde plus 4% sucrose in PBS for 30 min and counterstained with 0.1% crystal violet (Sigma) for 30 min. Cells on the upper side of the chamber were wiped out, and cells that had migrated to the underside were observed and photographed. Cells from five fields of each transwell were calculated. Three independent experiments were conducted.
Measurement of calcium influx.
Primary cortical neurons were seeded onto 42 × 0.17 mm coverslides at a density of 3 × 106 cells per slide. At 4 DIV, half of the medium was removed and replaced with the desired ACM. Cells were then incubated for an additional 4 d before the calcium imaging experiments. To measure the intracellular free calcium levels, neurons were incubated with fura-2 (5 μm; Invitrogen) for 30 min at 37°C, and excess dye was removed. Basal-level Ca2+ imaging was first recorded for 50 s, and then cells were stimulated with a high external potassium concentration (60 mm) using a steady flow-through device. Fura-2 image ratios (with excitation at 340 and 380 nm and emission at 510 nm) were acquired using a CCD camera system (Cool SNAPHQ Digital monochrome charge-coupled device, ICX285AL; Roper Scientific, Duluth, GA). The intracellular Ca2+ concentration was calculated based on the 340 to 380 nm ratio (R340/380) using the Metafluor image analysis system (Universal Imaging Corporation, Philadelphia, PA). Net increases in the intracellular free calcium concentration ([Ca2+]i) were calculated by subtracting the basal level of [Ca2+]i from the peak level. Under each condition, at least 30 cells in each group were analyzed in every experiment. Three different experiments using different batches of ACM were performed. Student's t test was applied for the statistical analysis. Data are presented as the mean ± SEM.
ELISA.
Levels of CCL5/RANTES and CCL2/MCP-1 were determined using a mouse RANTES/CCL5 or a mouse CCL2/MCP-1 DuoSet ELISA Development System (R&D Systems) following the manufacturer protocol. Briefly, 96-well microplates were coated overnight with the Capture antibody, blocked with 1% BSA in PBS, and extensively washed. Samples or standards (100 μl) were added to the microplates, incubated for 2 h at RT, and washed extensively, followed by a 2 h incubation with the biotin-conjugated Detection antibody and a 30 min incubation with streptavidin-HRP plus substrate for signal development. The optical density of each well was detected using a microplate reader set to 450 nm, and readings were subtracted from those at 540 nm (ELISA Reader: Spectra MAX 340PC; Molecular Devices, Union City, CA). The amount of chemokines in each sample was calculated based on the standard curve prepared in the same experiment. To avoid potential interfering factors in cell lysates, the levels of chemokines in cell lysates were also neutralized with the corresponding neutralizing antibody following the manufacturer protocol.
RNA purification and quantitative-PCR.
RNA purification and cDNA synthesis were performed using TRIZOL and Superscript II (Invitrogen), respectively, following the manufacturer protocols. The quantitative real-time PCR (Q-PCR) was performed with the LightCycler FastStart DNA MasterPLUS SYBR Green I kit with a Roche Lightcycler (Roche Molecular Biochemicals, Mannheim, Germany) as described previously (Chou et al., 2005). The PCRs were processed under the following conditions: 95°C for 10 min, followed by 40 cycles of 95°C for10 s, 60°C for 10 s, and 72°C for 20 s. At least three different batches of cDNA and three independent PCRs were examined and normalized with the expression of the reference gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Data were analyzed using RelQuant software (Roche Molecular Biochemicals). Primer sets are listed in supplemental Table S1 (available at www.jneurosci.org as supplemental material).
Constructs.
The promoter region of the mouse CCL5/RANTES gene (GenBank accession number AB051897) was amplified using the PCR technique and subcloned into the pGL-3 basic vector harboring a luciferase reporter gene (Promega). The primers used in the PCRs are listed in supplemental Table S1 (available at www.jneurosci.org as supplemental material). The pcDNA3.1-Htt-(Q)25-hrGFP and pcDNA3.1-Htt-(Q)109-hrGFP constructs encoding an N-terminal fragment of Htt with the indicated number of polyQ residues fused to humanized Renilla green fluorescent protein (hrGFP) were created as described previously (Chiang et al., 2005). To create the DNA encoding DeRed:CCL5/RANTES, the coding region of mouse CCL5/RANTES was amplified from mouse brain cDNA using primers listed in Table S1 (available at www.jneurosci.org as supplemental material). The resultant DNA fragment was then subcloned into the C terminus of a red fluorescent protein, dsRed (Clontech, Palo Alto, CA) using standard molecular biological approaches. Expression constructs of nuclear factor κB (NFκB) and specificity protein 1 (Sp1) were generous gifts from Dr. Yi-Wen Liu (Graduate Institute of Biomedical and Biopharmaceutical Sciences, National Chiayi University, Chiayi, Taiwan) and Dr. Jan-Jong Hung (Center for Bioscience and Biotechnology, National Cheng Kung University, Tainan, Taiwan), respectively. The expression construct of high mobility group 1 protein (HMG-1) was purchased from Geneservice (Cambridge, UK). The CCL5/RANTES promoter mutants were created by a two-step PCR technique as described previously (Horton et al., 1989) with primers listed in supplemental Table S1 (available at www.jneurosci.org as supplemental material) and with pGL3-CCL5-P(-1064/-16) as the DNA template. The resultant DNA fragments contained double-point mutations at the NFκB-binding site. The PCR product was then subcloned into the pGL3-basic vector by standard molecular biology approaches. Mutations were confirmed by DNA sequencing.
Transfection and luciferase activity assay.
Two days before transfection, primary astrocytes were seeded onto a six-well plate with 5 × 105 cells per well. Cells were transiently transfected with the desired DNAs using Lipofectamine 2000 (Invitrogen) following the manufacturer protocol. Luciferase activities of 20 μl of lysate were determined following the manufacturer protocol using a TD-20/20 Luminometer (Promega) and normalized to the amount of proteins in the lysate. Four independent experiments using at least two different preparations of plasmids were performed for each condition.
SDS-PAGE and Western blotting.
Protein concentrations were determined using the Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad, Hercules, CA). Samples were loaded and separated on 10% SDS-polyacrylamide gels. After electrophoresis, the gels were transferred to nitrocellulose membranes and blocked with 5% skim milk in TBST (0.2 m Tris-base, 1.37 m NaCl, and 0.05% Tween 20) and probed with the indicated antibody at 4°C overnight. The membranes were then incubated with a peroxidase-conjugated secondary antibody for 1 h at RT and washed three times with PBS. The immunoreactive bands were stained using a light-emitting nonradioactive method (ECL; Fujifilm, Tokyo, Japan). For Western blot analyses, we used 1:1000 dilution for the anti-NFκB-p65 (Novus Biologicals, Littleton, CO).
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) assays were performed using a ChIP kit purchased from Upstate Biotechnology (Millipore Corporation, Billerica, MA). In brief, primary astrocytes were fixed with 1% formaldehyde for 10 min at RT for DNA cross linking. The reaction was terminated by adding glycine (125 mm). Cells were washed twice with ice-cold PBS, resuspended in L1 buffer (50 mm Tris, pH 8.0, 2 mm EDTA, 0.1% NP-40, 10% Glycerol, and 1× complete protease mixture (Roche, Nutley, NJ), and disrupted by passing them through G25 needles. Nuclear fractions were harvested by a 2 min centrifugation at 13,000 × g at 4°C and then resuspended in L2 buffer (50 mm Tris, pH 8.0, 5 mm EDTA, 1% SDS, and 1× complete protease mixture). DNA was fragmented into ∼500–1000 bp by a sonicator (Sonicator Ultrasonic processor; Misonix, Farmingdale, NY). The ChIP reactions were conducted following the manufacturer protocol using an anti-p65 antibody (Upstate Biotechnology) or an anti-histone H3 (Abcam, Cambridgeshire, UK). After immunoprecipitation, the DNA fragment harboring the p65 binding region of the CCL5/RANTES promoter was amplified by quantitative-PCR using the primers listed in supplemental Table S1 (available at www.jneurosci.org as supplemental material). Values for the relative intensities were determined by normalizing the DNA signal pulled down by the anti-p65 antibody with that obtained using the anti-histone H3 antibody in each group and are expressed as percentages of signals from WT astrocytes.
Human brain tissue.
Paraffin-embedded human brain sections were obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders (University of Maryland, Baltimore, MD) and the Mayo Clinic College of Medicine (Jacksonville, FL). The demographic data and clinical phenotypes of these subjects are summarized in Table 1.
Table 1.
Summary of the demographic data, neuropathology, and experimental results of human subjects
| Case | PMI | Brain area | Staining of CCL5 | Race | Age (years) | Gender | HD Vonsattel grade | Other pathology |
|---|---|---|---|---|---|---|---|---|
| HD-1b | 9 h | Frontal cortex | +++ | Caucasian | 80 | F | I | Cortical amyolid plaques |
| Caudate nucleus | +++ | |||||||
| HD-2b | <24 h | Frontal cortex | +++ | Caucasian | 70 | F | IV | Brainstem Lewy body |
| Caudate nucleus | +++ | |||||||
| HD-3a | 6 h | Frontal cortex | ++ | Caucasian | 82 | F | n.d. | Senile cerebral disease, Alzheimer type |
| Substantia nigra | ++ | |||||||
| HD-4a | 2 h | Frontal cortex | ++ | Caucasian | 78 | F | III | Senile cerebral disease, Alzheimer type |
| Substantia nigra | ++ | |||||||
| HD-5a | 17 h | Frontal cortex | +++ | Caucasian | 58 | M | n.d. | None |
| Substantia nigra | ++ | |||||||
| Non-HD-1b | 24 h | Frontal cortex | − | Caucasian | 88 | F | n.a. | Alzheimer disease Braak NFT III |
| Caudate nucleus | − | |||||||
| Non-HD-2a | 14 h | Frontal cortex | − | Caucasian | 79 | F | n.a. | None |
| Substantia nigra | − | |||||||
| Non-HD-3b | <24 h | Caudate nucleus | − | Caucasian | 81 | F | n.a. | None |
| Non-HD-4b | 12 h | Caudate nucleus | − | Caucasian | 80 | M | n.a. | Cortical amyolid plaques |
| Non-HD-5b | 4 h | Frontal cortex | − | Caucasian | 83 | M | n.a. | Alzheimer disease Braak NFT III |
| Caudate nucleus | − | |||||||
| Non-HD-6b | 6 h | Frontal cortex | − | Caucasian | 93 | M | n.a. | Alzheimer disease Braak NFT I |
| Caudate nucleus | − | |||||||
| Non-HD-7b | 24 h | Frontal cortex | − | Caucasian | 74 | M | n.a. | Alzheimer disease Braak NFT I-II |
| Caudate nucleus | − | |||||||
| Non-HD-8a | 8 h | Frontal cortex | + | Caucasian | 77 | F | n.a. | Lung disease, HBP, arthritis, and breast cancer |
| Substantia nigra | − | |||||||
| Non-HD-9a | 19 h | Frontal cortex | + | Caucasian | 72 | F | n.a. | Non-Hodgkin's lymphomia, heart disease, and diabetes |
| Substantia nigra | − | |||||||
| Non-HD-10a | 16 h | Frontal cortex | − | Caucasian | 75 | M | n.a. | None |
| Substantia nigra | − |
−, No detectable signal; +, weak staining; ++, moderate staining; +++, Intense staining; F, female; M, male; PMI, postmortem interval; NFT, neurofibrillary tangles; n.d., not determined; n.a., not applicable; HBP, high blood pressure.
aCases obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland.
bCases obtained from the Mayo Clinic College of Medicine.
Results
R6/2 astrocytes are defective in releasing CCL5/RANTES, a chemokine important for neuronal function
Shin et al. (2005) reported previously that aggregates of mHtt exist in astrocytes. To assess whether mHtt affects the trophic function of astrocytes, we cultured primary astrocytes from cortices of WT mice and R6/2 mice, a transgenic mouse model of HD (Mangiarini et al., 1996). Consistent with Shin et al. (2005), aggregates of mHtt were observed in HD astrocytes after 30 DIV but not in those prepared from WT mice (Fig. 1A,B) (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). Expression of mHtt was also detected in primary astrocytes from HD mice by both the reverse transcription-PCR method and Western blot analysis (data not shown). Likewise, aggregates of mHtt were detected in astrocytes of various brain areas of R6/2 mice but not in those of WT mice (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). Collectively, these data suggest that mHtt exists and forms aggregates in R6/2 astrocytes both in vivo and in vitro.
Figure 1.

ACM collected from R6/2 mice exhibited less activity in supporting neurite development of primary cortical neurons. A, B, Immunostaining of mHtt aggregates (green), GFAP (an astrocyte marker; red), and nucleus (DAPI; blue) in primary astrocytes (30 DIV) purified from the indicated mice. Representative merged images are shown. The corresponding images of mHtt, and GFAP and DAPI alone, are displayed in supplemental Figure S2A (available at www.jneurosci.org as supplemental material). The boxed region in B is enlarged to show the aggregates of mHtt. The arrow indicates mHtt aggregates in the nucleus. No aggregate was found in WT astrocytes. Scale bar, 20 μm. C–E, Immunocytochemical images of primary cortical neurons cultured with NB or the indicated ACM (50%, v/v) from 4 DIV. Cells were fixed at 8 DIV and stained with TUJ-1 (a neuron-specific marker; green) and DAPI (blue). F–H, The neurite length (F), the number of branchings (G), and the number of sproutings (H) of each neuron under the indicated condition were quantified using MetaMorph software. ap ≤ 0.001, compared with the NB group; bp ≤ 0.001, compared with the WT-ACM by Student's t test. Scale bar, 100 μm.
We then set out to determine whether the expression of mHtt alters the ability of astrocytes to release trophic factors and/or chemokines important for neuronal function and activities. Primary astrocytes (at 30 DIV) from WT or R6/2 mice seeded at the same density were cultured in serum-free DMEM for 3 d to produce ACM. Primary cortical neurons (at 4 DIV) were cultured in the presence of the indicated medium (50%, v/v) for 4 d. Neurites were visualized by immunostaining using a neuron-specific antibody (TUJ-1). As shown in Figure 1C–E, neurons cultured with R6/2-ACM exhibited shorter processes (Fig. 1F) and fewer branchings (Fig. 1G) than those with either WT-ACM or fresh NB. No difference in the number of sproutings was detected in neurons cultured with R6/2- and WT-ACM (Fig. 1H). These observations suggest that astrocytic factors present in ACM might be important for the development of neuronal processes and maturity. In addition, the expression of mHtt in astrocytes is likely to jeopardize the availability of astrocytic factor(s) in ACM.
We next examined the expression levels of 62 different cytokines and chemokines in WT- and R6/2-ACM using a cytokine antibody array. Because low levels of signals are prone to artifacts, spots with intensities of less than one-tenth of the positive controls on the same membrane were considered to be negative. By this standard, the levels of most chemokines/cytokines (including those of the interleukin family) were too low to be reliably detected. We verified this assumption by measuring the level of one chemokine (CCL11/eotaxin) with intensity that was approximately one-tenth of the positive control using ELISA. In 100 μl of WT- or R6/2-ACM, no trace of CCL11/eotaxin could be detected using a mouse CCL11/eotaxin ELISA kit (data not shown). In all, only 13 cytokines/chemokines were reproducibly detected in ACM (supplemental Table S2, available at www.jneurosci.org as supplemental material). Among these, only the level of CCL5/RANTES was consistently altered in R6/2-ACM compared with WT-ACM. As shown in Figure 2A, the level of CCL5/RANTES in R6/2-ACM was much lower than that in WT-ACM. ELISA analyses further demonstrated that the amount of CCL5/RANTES in WT-ACM was markedly higher than that in R6/2-ACM (320 ± 20 vs 80 ± 10 pg/ml, respectively; seven different batches of ACM) (Fig. 2B). In contrast, no significant difference in the amounts of another chemokine, CCL2/MCP-1, in WT-ACM and R6/2-ACM was detected. ELISA analyses revealed that the amounts of CCL2/MCP-1 in WT-ACM and R6/2-ACM were 414 ± 59 and 387 ± 81 pg/ml (three different batches of ACM), respectively. Expression of mHtt therefore appeared to selectively reduce the amount of CCL5/RANTES secreted by astrocytes. Most interestingly, defective release of CCL5/RANTES in cortical astrocytes prepared from R6/2 mice was also observed in primary astrocytes prepared from the striatum (but not the cerebellum) of R6/2 mice (Fig. 2C). This observation is of great interest, because the most notable damages of HD occur to the cortex and striatum, suggesting a potential functional relevance of the reduced availability of CCL5/RANTES in HD. In the following experiments, astrocytes prepared from cortices were used unless stated otherwise.
Figure 2.

Level of CCL5/RANTES was selectively reduced in ACM of R6/2 astrocytes. A, The cytokine antibody array revealed differential expression of CCL5/RANTES in ACM collected from WT or R6/2 cortical astrocytes as indicated. The intensities of the spots reflect the relative levels of the indicated proteins. Thick arrows, thin arrows, and the arrowhead mark the position of CCL5/RANTES, CCL2/MCP-1, and the positive control (IgG) on the same membrane, respectively. Quantization of CCL5/RANTES and CCL2/MCP-1 levels from cytokine array membranes is shown in the right panel. Data were normalized with an internal positive control on the same membrane and are presented as the mean ± SEM of four determinations from two independent batches of ACM and two sets of antibody arrays. *p < 0.05, compared with the WT-ACM, by Student's t test. B, Levels of CCL5/RANTES in the WT- and R6/2-ACM were determined by ELISA. To verify the specificity of the CCL5/RANTES signal, an anti-CCL5/RANTES neutralizing goat IgG (0.2 μg/ml) or a normal goat IgG (0.2 μg/ml) was added to the ACM as indicated. To reconstitute R6/2-ACM for further functional assays, recombinant mouse CCL5/RANTES (450 pg/ml) was added to R6/2-ACM and analyzed for the levels of CCL5/RANTES as shown. Values are presented as the mean ± SEM of 12 determinations from four different batches of ACM. Statistical analyses were conducted using Student's t test followed by the Mann–Whitney rank sum test. **p < 0.001, compared with WT-ACM containing the control IgG. C, Astrocytes from different brain areas of the indicated animals were prepared. Levels of CCL5/RANTES in the WT- and R6/2-ACM were determined by ELISA. Values are presented as the mean ± SEM from three different batches of ACM. Statistical analyses were conducted using Student's t test (*p < 0.05, compared with WT-ACM containing the control IgG).
To examine the function of CCL5/RANTES, the effect of a neutralizing anti-CCL5/RANTES antibody, which removed CCL5/RANTES from the ACM, was tested. As shown in Figure 2B, the addition of this neutralizing anti-CCL5/RANTES antibody effectively reduced CCL5/RANTES levels in WT-ACM. Supplementation of R6/2-ACM with recombinant CCL5/RANTES (450 pg/ml) consistently increased the levels of CCL5/RANTES detected (Fig. 2B).
To investigate the contribution of astrocytic CCL5/RANTES to neuronal functions, primary rat cortical neurons were prepared. At 4 DIV, the cortical neurons were cultured in the presence of the indicated ACM (50%, v/v) for an additional 4 d. At this stage (4 DIV), at least two receptors (CCR1 and CCR5) of CCL5/RANTES could be detected in cortical neurons using the reverse transcription-PCR technique (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). Note that the amino acid sequences of CCL5/RANTES and its major receptors are highly homologous between the mouse and rat (94% and 92% identities for CCL5/RANTES and CCR5, respectively). The function and regulation of CCL5/RANTES in the mouse and rat, therefore, are likely to be very similar, thus justifying the use of rat neurons in the following experiments.
We examined whether a deficiency of CCL5/RANTES in R6/2-ACM led to reduced neurite development as observed in Figure 1. Neurons cultured in WT-ACM, but not those in R6/2-ACM, developed long and well-branched neuronal processes (Fig. 3A,B). Adding the CCL5/RANTES neutralizing antibody to WT-ACM markedly suppressed the length and branching of cortical neurites (Fig. 3C). Supplementation of R6/2-ACM with recombinant CCL5/RANTES (450 ng/ml) to a level similar to that in WT-ACM significantly increased the length of the neuronal processes (Fig. 3D). The amount of CCL5/RANTES for the supplementation experiment was chosen to provide a sufficient amount of CCL5/RANTES found in WT ACM after a 4 DIV incubation. These data indicate that CCL5/RANTES secreted by astrocytes is critical for promoting neurite outgrowth in cortical neurons.
Figure 3.

CCL5/RANTES in ACM was critical for neurite development of primary cortical neurons. A–D, Immunocytochemical images of primary cortical neurons cultured with the indicated ACM (50%, v/v) containing the control, IgG, the anti-CCL5/RANTES IgG, or recombinant CCL5/RANTES (450 pg/ml) from 4 DIV. Note that the addition of an anti-CCL5/RANTES IgG completely removed CCL5/RANTES from WT-ACM as shown in Figure 2. Cells were fixed at 8 DIV and stained with TUJ-1. E, F, The neurite length (E) and the number of branchings (F) of each neuron under the indicated condition were quantified using MetaMorph software. Totals of ∼63–131 cells were scored in the examined conditions. Data points represent the mean ± SEM from four independent experiments. ap ≤ 0.001, compared with the WT-ACM containing control goat IgG; bp ≤ 0.001, compared with the R6/2-ACM containing control IgG, by Student's t test. Scale bar, 100 μm.
Because CCL5/RANTES has been shown to evoke neuronal migration (Bolin et al., 1998), we next examined the ability of WT- and R6/2-ACM to induce neuronal migration using a transwell approach. As shown in Figure 4, more neurons cultured with WT-ACM migrated to the underside of the transwell than those grown in R6/2-ACM. Adding the neutralizing anti-CCL5/RANTES antibody to WT-ACM reduced neuronal migration (Fig. 4C,E). It was interesting to find that, although supplementing R6/2-ACM with recombinant CCL5/RANTES increased neuronal migration, the effect of the added recombinant CCL5/RANTES was not as effective as WT-ACM (Fig. 4A,B,D,E), suggesting that CCL5/RANTES might not be the only molecule in ACM that plays important roles in the migration of cortical neurons. Alternatively, it is equally plausible that R6/2 astrocytes might secrete an undesirable factor at a low level that inhibits cortical neuron migration.
Figure 4.

CCL5/RANTES in ACM mediated neuronal migration. Transwell migration assays for neuron migration in response to the indicated ACM containing the control IgG (Con-IgG), anti-CCL5/RANTES IgG, or recombinant CCL5/RANTES (450 pg/ml) were performed. After 18 h, the undersides of the transwell membranes were fixed and stained with crystal violet. A–D, Representative photographs. Migrating neurons (indicated by red arrows) appear purple and can be observed near the membrane pores (indicated by blue arrows). E, Numbers of migrating cells from five different fields of each condition were quantified. Data points represent the mean ± SEM of four independent experiments. Statistical analyses were conducted using Student's t test followed by the Mann–Whitney rank sum test. ap ≤ 0.01, compared with WT-ACM containing the control IgG; bp ≤ 0.05, compared with R6/2-ACM containing the control IgG.
To further assess the role of CCL5/RANTES in regulating neuronal activity, we determined the depolarization (high K+, 60 mm)-induced Ca2+ elevations of neurons treated with ACM. As described above, cortical neurons were cultured with either WT- or R6/2-ACM from 4 to 8 DIV. In addition to the deficiency in neurite development, neurons maintained in the presence of R6/2-ACM also exhibited much lower Ca2+ elevation after depolarization when compared with those cultured in WT-ACM. Again, removing CCL5/RANTES from WT-ACM using the anti-CCL5/RANTES neutralizing antibody suppressed the calcium response, whereas supplementation of R6/2-ACM with recombinant CCL5/RANTES restored the calcium response (Fig. 5).
Figure 5.
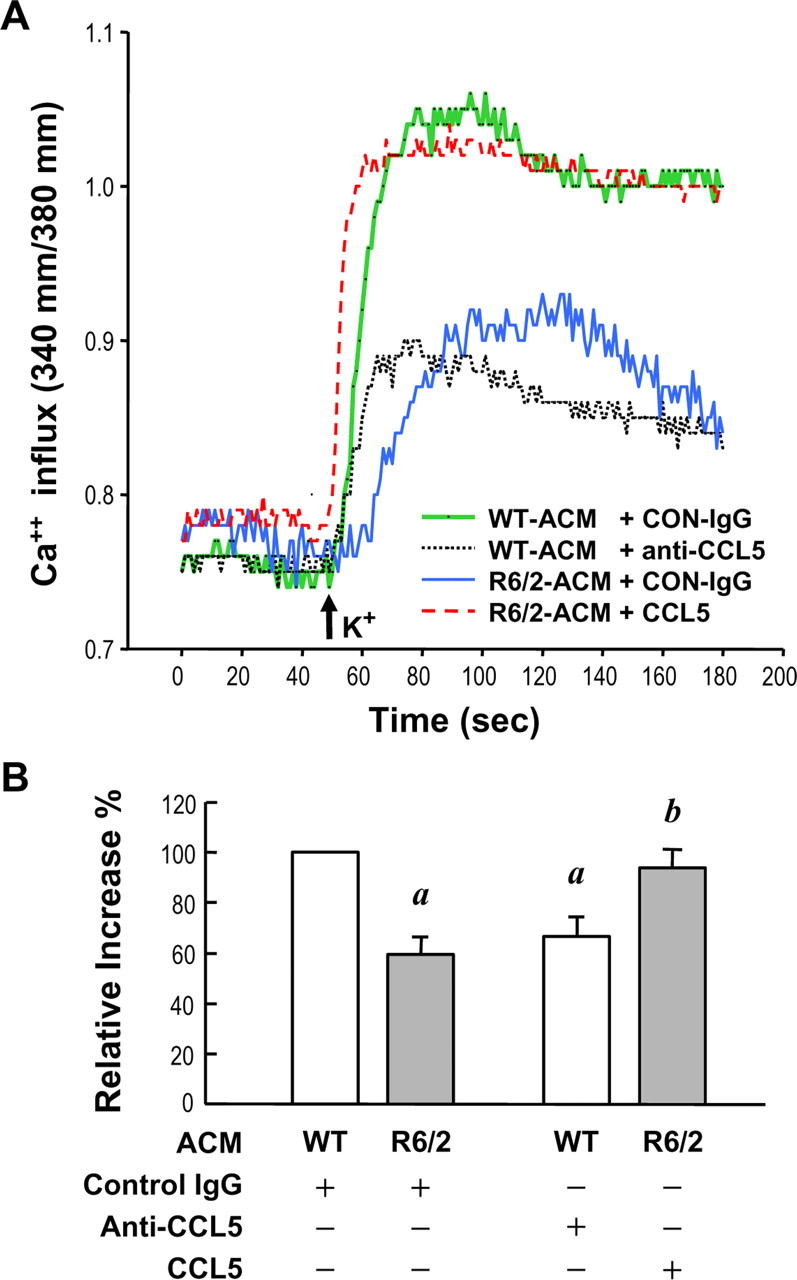
CCL5/RANTES in ACM maintained proper neuronal activity. Primary neurons (at 4 DIV) cultured in the indicated ACM containing the control IgG (0.2 μg/ml), the anti-CCL5/RANTES IgG (0.2 μg/ml), or the recombinant CCL5/RANTES (450 ng/ml) as indicated for 4 d. Cells were loaded with fura-2 (5 μm) and stimulated with 60 mm K+ (marked by the arrow). A, Representative traces depict the [Ca2+]i elevation evoked by the high external K+ as indicated. B, The maximal calcium response after depolarization was quantified using Metafluor software. At least 100 cells for each condition were analyzed. Values are expressed as percentages of the calcium in WT astrocytes and represent the mean ± SEM of four independent experiments. ap < 0.05, compared with WT-ACM containing the control IgG; bp < 0.05, compared with R6/2-ACM containing the control IgG, by Student's t test followed by the Mann–Whitney rank sum test.
Collectively, the above data demonstrate that CCL5/RANTES is an important astrocytic factor for neuronal maintenance of proper functions, and that expression of mHtt in astrocytes suppresses their ability to release CCL5/RANTES.
mHtt reduces the expression of CCL5/RANTES at the transcriptional level in astrocytes
mHtt has been shown to suppress the activities of many transcription factors (e.g., cAMP response element-binding protein, NFκB, and Sp1) and subsequently causes transcriptional dysfunction (Dunah et al., 2002; Chiang et al., 2005, 2007; Zhai et al., 2005). Because a conserved NFκB/Sp1 binding site (−151 to −126, +1 as the transcriptional start site) is present in the CCL5/RANTES promoter [GenBank accession number AB051897 (Miyamoto et al., 2000; Kim et al., 2004)], we thus investigated whether mHtt suppressed the amount of secreted astrocytic CCL5/RANTES by reducing the gene expression of CCL5/RANTES in astrocytes. Using a Q-PCR technique, we found that the level of CCL5/RANTES transcript was lower than that in WT astrocytes. Primary astrocytes (30 DIV) were harvested for total RNA collection and used for the quantitative RT-PCR analysis. Expression levels of CCL5/RANTES were normalized to that of GAPDH. When compared with those in WT astrocytes, the relative transcript levels of CCL5/RANTES in R6/2 astrocytes were 52 ± 19% (mean ± SEM of four independent experiments; p < 0.05, Student's t test). To determine whether mHtt suppresses the expression of CCL5/RANTES by inhibiting its promoter, a pGL3-CCL5/RANTES promoter construct, pGL3- CCL5- P(−1064/−16), was created and transfected into primary WT or R6/2 astrocytes for 48 h. The luciferase activity of each transfectant was measured and normalized to the protein content. Values are expressed as percentages of the promoter activity in WT astrocytes. When compared with that in WT astrocytes, the relative promoter activity of pGL3-CCL5-P(−1064/−16) in R6/2 astrocytes was also less (69.9 ± 5.2%; mean ± SEM of 12 determinants from four independent experiments; p < 0.001, Student's t test).
To verify this finding, we also cotransfected the CCL5/RANTES promoter construct with either Htt-(Q)25-hrGFP or Htt-(Q)109-hrGFP (Chiang et al., 2005) into primary WT astrocytes. Consistently, expression of polyQ-expanded Htt suppressed the CCL5/RANTES promoter by ∼35% (Fig. 6A). mHtt thus appeared to suppress the expression of CCL5/RANTES at the transcriptional level in astrocytes. To locate the DNA region responsible for the above suppressive effect, four additional 5′-deleted CCL5/RANTES promoter constructs were created and transfected into WT astrocytes along with mHtt harboring 25Q or 109Q. The suppressive effect of 109Q-Htt persisted when the CCL5/RANTES gene fragment was 5′-deleted to −159 bp. Promoter activity was not detectable when the promoter fragment was deleted to −76 bp (Fig. 6A). The core promoter of mouse CCL5/RANTES gene therefore appears to be located in the region of −159 to −16 bp. This finding is consistent with the reported core promoter (−194 to −1 bp) of the human CCL5/RANTES gene (Miyamoto et al., 2000). Moreover, mHtt inhibited astrocytic expression of CCL5/RANTES by suppressing its core promoter activity. Interestingly, a shared binding site for two transcription factors (NFκB and Sp1), the functions of which are affected by mHtt (Dunah et al., 2002; Takano and Gusella, 2002; Zhai et al., 2005), is located in the core promoter region (−118 to −107 bp). To determine the transcription factor involved in the suppression of the CCL5/RANTES promoter by mHtt, we transfected WT astrocytes with the CCL5/RANTES promoter construct pGL3-RANTES-P(−1064/−16), an expression construct of the desired transcription factor, and the construct of mHtt harboring 25Q or 109Q as indicated. In addition to NFκB and Sp1, two additional transcription factors (HMG-1 and p53), which do not bind to the core promoter region of CCL5/RANTES gene, were also tested. p53 is of interest, because its activity was shown to be altered in HD (Bae et al., 2005), and therefore it can serve as a useful control in this experiment. As shown in Figure 6B, elevation of NFκB/p65, but not the other three transcription factors including Sp1, effectively recovered the suppressive effect of mHtt on the CCL5/RANTES promoter. Although transfection of Sp1 elevated the promoter activity, mHtt remained effective in suppressing the CCL5/RANTES promoter. Sp1 thus appeared to be important for astrocytic expression of CCL5/RANTES but might not be involved in the mHtt-mediated inhibition of CCL5/RANTES observed in the present study. In contrast, dysregulation of NFκB/p65 might contribute to the reduced CCL5/RANTES expression by mHtt. To verify the importance of NFκB/p65 in suppressing CCL5/RANTES by mHtt, we mutated the NFκB- response element of the CCL5/RANTES promoter. Two mutants that contain mutations at the p65 binding site (−159 to approximately −142; M1) or the binding sites of p65 plus p50 (another NFκB member, −161 to approximately −139; M2) were created. The resultant CCL5/RANTES promoter mutants, M1 and M2, exhibited lower promoter activity and were not suppressed by mHtt (Fig. 6C). Overexpression of NFκB/p65 reversed only the inhibition of the CCL5/RANTES promoter (pGL3-CCL5-P(−1064/−16)) but not that of the mutated M1 or M2 promoter. These results support the hypothesis that a functional NFκB site is located in the promoter region of the CCL5/RANTES gene and that NFκB is involved in mHtt-evoked suppression of the CCL5/RANTES gene. Consistent with this hypothesis, Western blot analyses of nuclear extracts of WT and R6/2 astrocytes showed that the level of NFκB/p65 in R6/2 astrocytes was lower than that in WT astrocytes (Fig. 6D). ChIP assays further demonstrated that chromatin, containing the −245 to −16 bp region of the CCL5/RANTES gene, could be pulled down by an anti-NFκB/p65 antibody and detected by quantitative-PCR. Primary WT or R6/2 astrocytes (30 DIV) were cross linked and processed for the ChIP assay using an anti-NFκB-p65 or anti-histone H3 antibody as described in Materials and Methods. Values for the relative signal intensities were determined by normalizing the DNA signal pulled down by the anti-p65 antibody with that obtained using the anti-histone H3 antibody in each group and are expressed as a percentage of the signal from WT astrocytes. When compared with those in WT astrocytes, the relative ChIP signal obtained using an anti-p65 antibody in R6/2 astrocytes were 26.4 ± 9.8% (mean ± SEM of seven independent experiments; p < 0.001, Student's t test). Therefore, NFκB/p65 is important for the astrocytic expression of the CCL5/RANTES gene in vivo. Collectively, these findings suggest that mutant Htt reduces the availability of NFκB/p65 and subsequently hinders transcription of the CCL5/RANTES gene in astrocytes.
Figure 6.
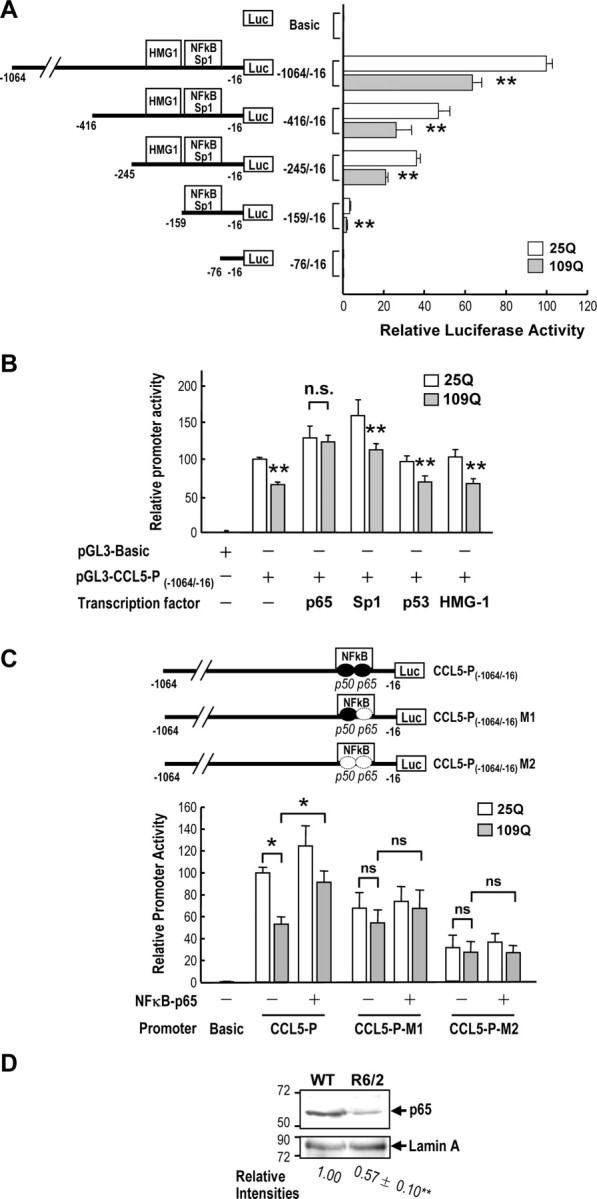
mHtt reduced the gene expression of CCL5/RANTES in astrocytes via reducing the nuclear level of NFκB p65. A, The pcDNA3.1-(Q)25-Htt-hrGFP or pcDNA3.1-(Q)109-Htt-hrGFP construct was cotransfected with the indicated pGL3-CCL5 promoter construct at a ratio of 7:2 into primary WT astrocytes (30 DIV) for 48 h. The luciferase activity was measured and normalized to the protein content. Values are expressed as percentages of the promoter activity of pGL3-CCL5-P(−1064/−16) in the presence of pcDNA3.1-(Q)25-Htt-hrGFP and represent the mean ± SEM of four independent experiments. **p < 0.001, compared with that of the 25Q transfectant of each group, by paired t test. B, The pcDNA3.1-(Q)25-Htt-hrGFP or pcDNA3.1-(Q)109-Htt-hrGFP construct was cotransfected with pGL3-CCL5-P(−1064/−16) and an expression construct of the indicated transcription factor (NFκB-p65, Sp1, p53, or HMG-1) at a ratio of 2:1:2 into primary WT astrocytes for 48 h. The luciferase activity was measured and normalized to the protein content. Values are expressed as percentages of the promoter activity of pGL3-CCL5-P(−1064/−16) in the presence of pcDNA3.1-(Q)25-Htt-hrGFP, but no overexpression of any transcription factor, and represent the mean ± SEM of four independent experiments. **p < 0.001, compared with that in the 25Q transfectant of each group; paired t test. C, The indicated Htt construct with 109 copies of polyQ was cotransfected with the WT or the indicated NFκB-null CCL5/RANTES promoter construct [pGL3-CCL5-P(−1064/−16)-M1 or pGL3-CCL5-P(−1064/−16)-M2] and an expression construct of NFκB-p65 as described above. The luciferase activity was measured and normalized to the protein content. Values are expressed as percentages of the promoter activity of pGL3-CCL5-P(−1064/−16) in the presence of 25Q without any transcription factor and are presented as the mean ± SEM of three independent experiments. *p < 0.01, Student's t test. D, Nuclear fractions (22 μg per lane) collected from the indicated astrocytes were subjected to Western blot analysis. The level of nuclear NFκB-p65 in R6/2 astrocyte was normalized with an internal control (lamin A) and compared with that of WT astrocytes. Data are presented as the mean ± SEM from four independent experiments. **p < 0.001 compared with that in WT astrocytes, t test.
Besides CCL5/RANTES, the transcript level of another NFκB-driven gene (CCR5, a CCL5/RANTES receptor) was markedly lower in HD astrocytes (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). This observation is consistent with previous studies where parallel regulation of CCL5/RANTES and CCR5 was reported (Kim et al., 2006; Li et al., 2006). However, expressions of three other genes [manganese superoxide dismutase 1 (SOD1), manganese superoxide dismutase 2 (SOD2), and adenosine A2A receptors (A2ARs) (Das et al., 1995; Xu et al., 1999; Morello et al., 2006)], which also contain one or more NFκB binding sites in their promoter regions were not altered in HD astrocytes. When compared with those in WT astrocytes, the relative transcript levels of SOD1, SOD2, and A2ARs in R6/2 astrocytes were 93.9 ± 1.6, 108.0 ± 1.2, and 94.0 ± 9.3%, respectively (mean ± SEM of three independent experiments). This discrepancy in the responses to a reduction in NFκB is not surprising, because NFκB-response elements from different genes can have different affinities toward NFκB. In addition, other transcription factors might manipulate and contribute to regulating of NFκB-mediated genes (Henriquet et al., 2007).
mHtt hinders the secretion of residual CCL5/RANTES from astrocytes
Note that although the level of CCL5/RANTES in R6/2-ACM was reduced by ∼75% when compared with that of WT-ACM (Fig. 2), mHtt suppressed the expression of CCL5/RANTES gene only by ∼50%. mHtt therefore might inhibit the production of astrocytic CCL5/RANTES by at least two pathways. Besides transcription disruption, mHtt has also been shown to abnormally bind with intracellular proteins and alter protein secretion (Fusco et al., 2003; Gunawardena and Goldstein, 2005; Desai et al., 2006). In contrast to the lower extracellular level of CCL5/RANTES in R6/2 astrocytes, immunocytochemical analyses also detected much stronger intracellular CCL5/RANTES signals in R6/2 astrocytes than in WT astrocytes (Fig. 7A). The CCL5/RANTES fluorescence intensities in 3 d cultures of WT and R6/2 astrocytes were quantified and are expressed as percentages of fluorescence levels in R6/2 astrocytes. When compared with those in R6/2 astrocytes, the relative CCL5/RANTES intensity in WT astrocytes was 23.5 ± 4.6% (mean ± SEM of at least 200 cells from three independent experiments; p < 0.001, Student's t test followed by the Mann–Whitney rank sum test). The average intracellular intensity of CCL5/RANTES signals in R6/2 astrocytes was approximately fourfold that in WT astrocytes. In line with this finding, ELISA analyses revealed that the cytosolic contents of CCL5/RANTES in R6/2 astrocytes were much higher than those in WT astrocytes. The levels of cytosolic CCL5/RANTES in WT and R6/2 astrocytes were 292.5 ± 31.5 and 965.0 ± 248.9 pg/ng lysate (16 determinations from eight different batches; p < 0.05, Student's t test), respectively. The intracellularly accumulated CCL5/RANTES might reflect a defect in the release of CCL5/RANTES by astrocytes in the presence of mHtt as observed in Figure 2B. To test the above hypothesis, we created a construct encoding CCL5/RANTES fused with a red fluorescence protein (DsRed; Clontech) at its N terminus. Similar to the distribution of endogenous CCL5/RANTES, transfection of the resultant cDNA (DsRed:CCL5) and Htt-(Q)25-hrGFP to WT astrocytes led to even distribution of DsRed:CCL5 in the cytoplasmic organelles. On the contrary, expression of Htt-(Q)109-hrGFP caused a marked increase in the intracellular accumulation of DsRed:CCL5/RANTES (Fig. 7B). This finding supports our hypothesis that mHtt jeopardizes the secretion of CCL5/RANTES by astrocytes.
Figure 7.
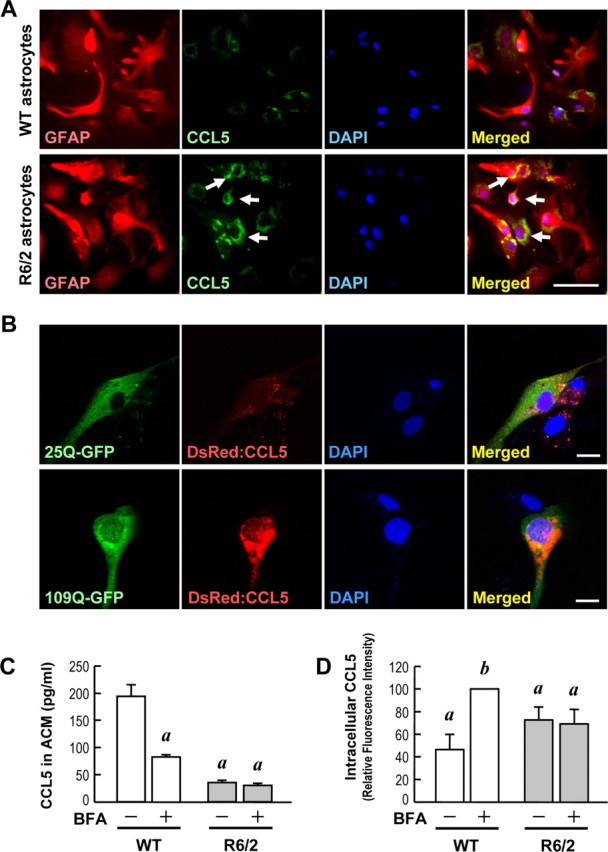
mHtt suppressed the release of CCL5/RANTES from primary astrocytes. A, Immunostaining of CCL5/RANTES (green) and GFAP (red) was performed in primary astrocytes. Nuclei were stained with Hoechst 33258 (blue). CCL5/RANTES was enriched inside of R6/2 astrocytes (bottom) but not in WT astrocytes (top). Scale bar, 100 μm. B, WT astrocytes were transfected with DsRed:CCL5 along with the Htt-(Q)25-hrGFP or Htt-(Q)109-hrGFP construct at a ratio of 1:1 for 48 h. Cells were fixed, and the fluorescent signals were detected using confocal microscopy. Scale bars, 20 μm. C, D, Both WT and R6/2 astrocytes were treated with either 0.1% DMSO as a control or BFA (10 μg/ml) for 4 h to block secretion of any newly synthesized CCL5/RANTES protein. Intracellular CCL5/RANTES levels were assessed by immunocytochemical staining using an anti-CCL5/RANTES. CCL5/RANTES accumulation was enhanced by BFA in WT astrocytes, but not in R6/2 astrocytes, as shown in supplemental Figure S4 (available at www.jneurosci.org as supplemental material). C, CCL5/RANTES released extracellularly from astrocytes was collected and detected using a CCL5/RANTES ELISA assay. D, The relative CCL5/RANTES fluorescence intensities of both vehicle- and BFA-treated WT and R6/2 astrocytes were quantified. Results are given as the mean ± SEM from three independent assays and normalized to the CCL5/RANTES intensity in BFA-treated WT astrocytes. Approximately 1000 cells were quantified in each group. ap < 0.001 compared with the DMSO-treated control in WT astrocytes; bp < 0.001 compared with the BFA-treated WT astrocytes by Student's t test.
To verify whether the secretory machinery of CCL5/RANTES was impaired by mHtt in R6/2 astrocytes, we next treated astrocytes with Brefeldin A (BFA) (10 μg/ml) (Fig. 7C,D) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material) for 4 h to disrupt the function of Golgi apparatus and thus block the release of cytokines (Hunziker et al., 1992; Bandeira-Melo et al., 2002). ELISA and immunocytochemical analyses were used to detect the amount of CCL5/RANTES in ACM and the intracellular level of CCL5/RANTES, respectively. In agreement with the blocking of cytokine release by BFA, the level of CCL5/RANTES was markedly reduced in BFA-treated WT-ACM. No effect of BFA was found in the amount of CCL5/RANTES in R6/2-ACM (Fig. 7C). Quantitative analyses of the intracellular CCL5/RANTES level measured by immunocytochemical staining revealed that intracellular CCL5/RANTES was significantly enhanced by BFA treatment in WT astrocytes but not in R6/2 astrocytes (Fig. 7D) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). In this set of experiments for BFA treatment, the level of intracellular CCL5/RANTES was determined after 4 h incubation, which was much shorter than the standard 3 d incubation used in other experiments. Because of the shorter incubation in Figure 7D, the difference in the intracellular CCL5/RANTES levels (4 h accumulation) between WT and R6/2 astrocytes in control condition was less significant than that of 3 d accumulation. Nevertheless, these observations collectively indicate that CCL5/RANTES is constitutively secreted from WT astrocytes and that the secretory machinery is defective in R6/2 astrocytes. Note that BFA treatment effectively reduced the release of CCL2/MCP-1 from WT and R6/2 astrocytes to a comparable extent (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material). Therefore, the sensitivities of R6/2 and WT astrocytes to BFA were similar.
Because a reduced ability to secrete CCL5/RANTES by astrocytes is expected to cause abnormal accumulation of CCL5/RANTES, we next performed experiments to verify whether abnormal accumulation of CCL5/RANTES also occurred in vivo. Immunohistochemical staining revealed that the accumulation of CCL5/RANTES in astrocytes was found in the brains of two mouse models of HD [R6/2 (Fig. 8A) and Hdh(CAG)150 (Fig. 8B)]. R6/2 mice express exon 1 of the human Htt containing 144 repeats and exhibit pathological characters of HD at ∼9–11 weeks of age (Mangiarini et al., 1996). Hdh(CAG)150 mice harbor the Htt homolog gene (Hdh) containing 150 copies of polyQ and show neurological abnormalities at ∼15 months of age when only one copy of the poly-Q-expanded Hdh gene exists (Lin et al., 2001). Consistent with our hypothesis, CCL5/RANTES signals were readily found in the brains of both HD mouse models but only very rarely and at much lower levels in WT brains (Fig. 8C). Most importantly, marked accumulation of CCL5/RANTES in astrocytes was also found in the frontal cortex, substantia nigra, and caudate nucleus of human HD patients (Table.1). In the brains of age-matched non-HD controls, signal of CCL5/RANTES in astrocytes was not detected in 80% of the cases examined. Only two cases of non-HD controls exhibited weak signals in the frontal cortex. In sharp contrast, all brain areas of HD patients tested contained strong CCL5/RANTES signals, which had a punctuate/aggregate appearance similar to those in the primary R6/2 astrocytes (Fig. 9, Table 1). These CCL5/RANTES signals were tightly associated with those of GFAP, strongly arguing that CCL5/RANTES accumulated in the astrocytes of HD patients. Unlike CCL5/RANTES, only sparse signals of CCL2/MCP-1 were detected within astrocytes in the brains of both WT and R6/2 mice (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material). Most importantly, no difference in the cytosolic CCL2/MCP-1 was found in the astrocytes of HD patients and non-HD controls (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). The release machinery of CCL2/MCP-1 in astrocytes therefore remained intact in HD. Together, these findings suggest that abnormal accumulation of CCL5/RANTES in astrocytes indeed occurred in HD patients, and that the abnormal accumulation of CCL5/RANTES was caused by a defect that selectively affected the secretion of CCL5/RANTES.
Figure 8.

Abnormal accumulation of astrocytic CCL5/RANTES in the brains of HD mice. A–C, Immunofluorescence staining of CCL5/RANTES (green) and GFAP (red) in brains of R6/2 mice (12 weeks of age; A), Hdh(CAG)150 mice (16 months of age; B), and WT mice (12 weeks of age; C) was conducted. Nuclei were stained with DAPI (blue). Scale bar, 20 μm.
Figure 9.
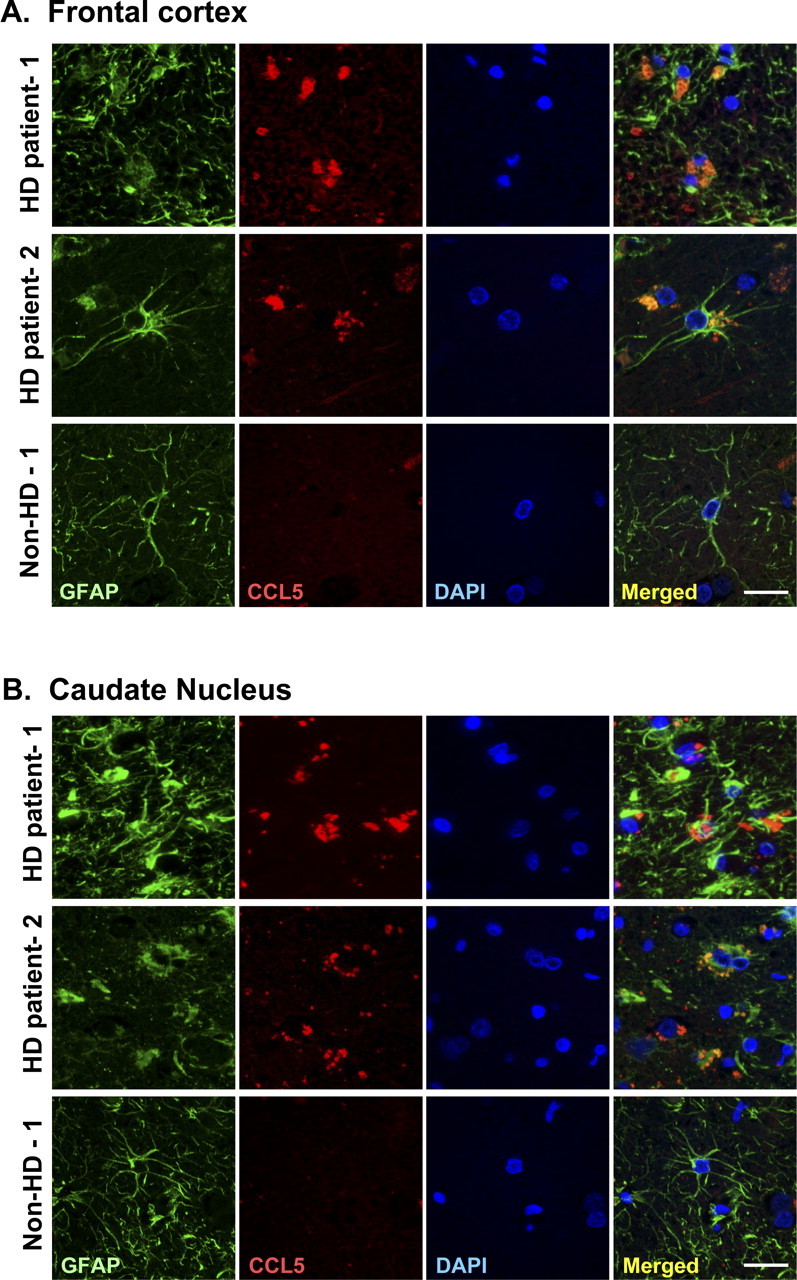
Abnormal accumulation of astrocytic CCL5/RANTES in brains of HD patients. A, B, The frontal cortex (A) and caudate nucleus (B) of HD patients and age-matched controls were analyzed. Immunofluorescence staining of CCL5/RANTES (red) and GFAP (green) was conducted. Nuclei were stained with DAPI (blue). Significant amounts of CCL5/RANTES were found in astrocytes of HD patients but not in astrocytes of the age-matched controls. Scale bars, 20 μm.
Discussion
The most important finding of the present study is the identification of an astrocytic defect that might contribute to neuronal dysfunction in HD. Our data reveal that mHtt suppresses the availability of astrocytic CCL5/RANTES to neurons through at least two different mechanisms: reduced transcription and defective release. This observation is consistent with previous studies demonstrating that many chemokines (including CCL5/RANTES) are simultaneously regulated at the gene transcription and protein secretion levels by a single stimulant/stress (Casola et al., 2002). It appears that defects in both transcription and release of CCL5/RANTES markedly affect the availability of astrocytic CCL5/RANTES in HD.
Several lines of evidence suggest that mHtt might cause transcriptional dysregulation of CCL5/RANTES in astrocytes. First, the gene expression and promoter activity of CCL5/RANTES in R6/2 astrocytes were markedly reduced. Second, elevation of NFκB (but not Sp1) ameliorated suppression of the RANTES/CCL5 gene by mHtt (Fig. 6B). The importance of NFκB was further verified by mutating the NFκB binding site on the CCL5/RANTES promoter (Fig. 6C) and by performing the ChIP assay using an anti-p65 antibody. NFκB is a very important transcription factor that has been implicated previously in the neurotoxicity of HD. Using an HD model induced by a mitochondrial toxin, 3-nitropropionic acid (3NP), genetic removal of p50 (a subunit of NFκB) suppressed NFκB activity and enhanced the damage caused by 3NP in striatal neurons (Yu et al., 2000). Aberrant regulation of NFκB was also observed in neurons of two genetic mouse models of HD (Khoshnan et al., 2004). Therefore, NFκB is believed to exert a neuronal protective effect in HD. In the present study, our data reveal a novel regulation of NFκB by mHtt in astrocytes, and that elevation of NFκB might exert a protective role by providing a sufficient level of this vital astrocytic trophic factor for neurons.
In addition to transcriptional dysregulation of CCL5/RANTES by mHtt, we demonstrated that an additional layer of mHtt-evoked regulation occurs at the secretory process of CCL5/RANTES (Fig. 7). Via assessing the abnormal accumulation of astrocytic CCL5/RANTES, we found that the secretary pathway of CCL5/RANTES in R6/2 astrocytes was impaired both in vitro (primary astrocytes) (Fig. 7) and in vivo (two HD mouse models and human HD patients) (Figs. 8, 9) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Moreover, overexpression of mHtt also caused an accumulation of DsRed:CCL5 driven by the cytomegalovirus promoter in primary astrocytes (Fig. 7B), further supporting our hypothesis that cytosolic CCL5/RANTES (or DsRed:CCL5) cannot be properly released by R6/2 astrocytes and therefore accumulates inside astrocytes. This defective release observed in R6/2 astrocytes appears to be specific to CCL5/RANTES, because release of another chemokine (CCL2/MCP-1) by R6/2 astrocytes was not affected. No abnormal accumulation of CCL2/MCP-1 inside astrocytes was found in either R6/2 mice or HD patients (supplemental Figs. S5A, S6, available at www.jneurosci.org as supplemental material). Such selectivity might be attributable to the fact that CCL5/RANTES and CCL2/MCP-1 are stored in different compartments (Oynebraten et al., 2004). To date, the molecular machinery responsible for the secretion of CCL5/RANTES in astrocytes is primarily unknown. In CD8+ T cells, CCL5/RANTES is stored in a unique secretory vesicle [designated the RANTES storage vesicle (RSV) (Catalfamo et al., 2004)]. This CCL5/RANTES-specific RSV structure might explain the selective reduction in the astrocytic release of CCL5/RANTES by mHtt (supplemental Table S2, available at www.jneurosci.org as supplemental material). It is also interesting to note that CCL5/RANTES is colocalized with vesicle-associated membrane protein 2 (VAMP-2) in eosinophils. VAMP-2 is an important component of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes, which regulate membrane vesicle fusion and exocytosis (Lacy et al., 2001). Because the formation of mHtt aggregates has been suggested to cause abnormalities in the SNARE machinery (Morton and Edwardson, 2001; Molinari and Sitia, 2005), a defect in the SNARE machinery might account for the impairment of CCL5/RANTES secretion in R6/2 astrocytes. Alternatively, mHtt might interfere with other cellular components important for the secretion of CCL5/RANTES. For example, aggregation of mHtt has been shown to wear out the protein degradation machinery and cause endoplasmic reticulum (ER) stress, which might subsequently lead to accumulation of misfolded proteins in ER and dysregulation of the secretory pathway (Kouroku et al., 2002; Molinari and Sitia, 2005). This hypothesis is of particular interest, because BFA is an ER stressor (Elyaman et al. 2002). BFA treatment of R6/2 astrocytes did not cause further retention of CCL5/RANTES inside R6/2 astrocytes, nor did it reduce the levels of CCL5/RANTES in R6/2-ACM (Fig. 7C,D), which suggests that the same pathway might be used to suppress the secretion of CCL5/RANTES by both mHtt and BFA. Dysfunction of the ER and protein degradation machinery therefore might also mediate the defective secretion of CCL5/RANTES. Besides suppressing the secretion and transcription of CCL5/RANTES as presented herein, mHtt also inhibits the release and transcription of another critical neurotrophic factor, BDNF (Zuccato et al., 2001, 2003; Gauthier et al., 2004; Humbert and Saudou, 2005; del Toro et al., 2006). It is very likely that negative regulation of the secretion of trophic factors (e.g., CCL5/RANTES and BDNF) by mHtt is a crucial common pathway that significantly contributes to HD pathology.
Another interesting observation is that aggregates of mHtt in astrocytes could be found in both the cytoplasm and the nucleus. This is in contrast to what was observed in neurons, where aggregates of mHtt were primarily detected in the nucleus. In addition, the size of the aggregates in astrocytes was much smaller than that in neurons. These differences may be related to the different natures of astrocytes and neurons. Specifically, neurons cannot proliferate. Accumulated mHtt in neurons therefore cannot be diluted by proliferation and therefore has to somehow be removed through multiple mechanisms. In contrast, astrocytes possess the ability to proliferate, which might slow down the accumulation rate of mHtt. Note that CCL5/RANTES has also been shown to play complex regulatory roles in the differentiation and proliferation of astrocytes. CCL5/RANTES has opposite effects on the proliferation/survival of 5- and 10-week-old astrocytes. It was suggested that CCL5/RANTES can exhibit diverse functions at different stages (Bakhiet et al., 2001). Thus, in addition to disrupting astrocyte-neuron homeostasis as suggested in the present study, reduced CCL5/RANTES expression in HD astrocytes might also cause dramatic impacts on their own survival, proliferation, and other cellular functions.
The functions of astrocytes in HD have been speculated on and documented. Previous focuses were on the roles of astrocytes in protecting neurons from excitotoxic insults. Specifically, several laboratories reported decreased expression of an astroglial glutamate transporter in HD. The subsequent lower astrocytic glutamate uptake might accelerate excitotoxicity and thus contribute to the neuronal damage caused by HD (Lievens et al., 2001, 2005; Shin et al., 2005). The present study, in contrast, points to a novel aspect of an astrocytic defect that might cause neuronal dysfunctions (e.g., shorter neurites and a fewer number of spines) in HD (Spires et al., 2004). Our data demonstrate that among molecules secreted by astrocytes, CCL5/RANTES plays a key role in supporting and maintaining proper neurite development and neuronal activity after stimulation (Figs. 3–5). This observation is consistent with a previous report that demonstrated that CCL5/RANTES functions as a neurotrophic factor and induces the migration of primary dorsal root ganglion sensory neurons (Bolin et al., 1998). Note that CCL5/RANTES by itself can also trigger elevation of intracellular Ca2+ in these cells (Bolin et al., 1998). In the CNS, a major CCL5/RANTES receptor (CCR5) was found in both neurons and astrocytes. Activation of CCR5 causes inhibition of cAMP production, induction of Ca2+ influx, and activation of phosphatidyl inositol 3-kinase (PI3K) and mitogen-activated protein kinases (MAPK) (Huang et al., 2005). Because MAPK and PI3K are important for neurite outgrowth (Cheng et al., 2002; Shibata et al., 2003; Goold and Gordon-Weeks, 2005), it is likely that the action of CCL5/RANTES in neurite development of primary cortical neurons is mediated by MAPK and PI3K. Moreover, CCL5/RANTES may also modulate neuronal migration through upregulation of a protein (CDK5R2, p39) enriched in the growth cone, because p39 expression is induced by CCL5/RANTES stimulation (Valerio et al., 2004). A lack of availability of CCL5/RANTES to neurons in HD therefore might result in less-developed neuronal processes, altered neuronal properties, and degenerated motor coordination as reported previously (Klapstein et al., 2001; Spires et al., 2004; Ariano et al., 2005). In summary, the observed defect in releasing CCL5/RANTES in our primary astrocyte culture strongly suggests a dysregulation of neuronal-glial interactions in HD.
Footnotes
This work was supported by grants from the National Science Council (NSC94–2321-B-001–026), the Academia Sinica (AS-94-TP-B17), and the Institute of Biomedical Science, Academia Sinica (Clinical Research Center grants), Taipei, Taiwan. We are grateful to Dr. Yun-Chia Chou (Institute of Neurosciences, Yang-Ming University, Taipei, Taiwan) for teaching us how to prepare primary astrocyte cultures and to Dr. Yi-Chao Lee (Institute of Pharmacology, National Cheng-Gung University, Tainan, Taiwan), Dr. Ming-Jen Lee (Department of Medical Genetics, National Taiwan University Hospital, Taipei, Taiwan), and Dr. Richard M. Ransohoff (Lerner Research Institute, Cleveland Clinic, Cleveland, OH) for helpful comments and suggestions. We thank Dan Chamberlin for reading and editing this manuscript, Hue-Mei Chen for maintaining the animal colony, Hao-Hung Chang and Han-Yun Hsiao for imaging analyses, and Jiao-Perng Chen for providing the original CCL5/RANTES promoter construct.
References
- Appay V, Rowland-Jones SL. RANTES: a versatile and controversial chemokine. Trends Immunol. 2001;22:83. doi: 10.1016/s1471-4906(00)01812-3. [DOI] [PubMed] [Google Scholar]
- Ariano MA, Cepeda C, Calvert CR, Flores-Hernandez J, Hernandez-Echeagaray E, Klapstein GJ, Chandler SH, Aronin N, DiFiglia M, Levine MS. Striatal potassium channel dysfunction in Huntington's disease transgenic mice. J Neurophysiol. 2005;93:2565–2574. doi: 10.1152/jn.00791.2004. [DOI] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Bakhiet M, Tjernlund A, Mousa A, Gad A, Stromblad S, Kuziel WA, Seiger A, Andersson J. RANTES promotes growth and survival of human first-trimester forebrain astrocytes. Nat Cell Biol. 2001;3:150–157. doi: 10.1038/35055057. [DOI] [PubMed] [Google Scholar]
- Bandeira-Melo C, Sugiyama K, Woods LJ, Phoofolo M, Center DM, Cruikshank WW, Weller PF. IL-16 promotes leukotriene C4 and IL-4 release from human eosinophils via CD4- and autocrine CCR3-chemokine-mediated signaling. J Immunol. 2002;168:4756–4763. doi: 10.4049/jimmunol.168.9.4756. [DOI] [PubMed] [Google Scholar]
- Bolin LM, Murray R, Lukacs NW, Strieter RM, Kunkel SL, Schall TJ, Bacon KB. Primary sensory neurons migrate in response to the chemokine RANTES. J Neuroimmunol. 1998;81:49–57. doi: 10.1016/s0165-5728(97)00158-6. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Casola A, Henderson A, Liu T, Garofalo RP, Brasier AR. Regulation of RANTES promoter activation in alveolar epithelial cells after cytokine stimulation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1280–L1290. doi: 10.1152/ajplung.00162.2002. [DOI] [PubMed] [Google Scholar]
- Catalfamo M, Karpova T, McNally J, Costes SV, Lockett SJ, Bos E, Peters PJ, Henkart PA. Human CD8+ T cells store RANTES in a unique secretory compartment and release it rapidly after TcR stimulation. Immunity. 2004;20:219–230. doi: 10.1016/s1074-7613(04)00027-5. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Shih HM, Chern Y. Essential role of cAMP-response element-binding protein activation by A2A adenosine receptors in rescuing the nerve growth factor-induced neurite outgrowth impaired by blockage of the MAPK cascade. J Biol Chem. 2002;277:33930–33942. doi: 10.1074/jbc.M201206200. [DOI] [PubMed] [Google Scholar]
- Chiang MC, Lee YC, Huang CL, Chern Y. cAMP-response element-binding protein contributes to suppression of the A2A adenosine receptor promoter by mutant Huntingtin with expanded polyglutamine residues. J Biol Chem. 2005;280:14331–14340. doi: 10.1074/jbc.M413279200. [DOI] [PubMed] [Google Scholar]
- Chiang MC, Chen HM, Lee YH, Chang HH, Wu YC, Soong BW, Chen CM, Wu YR, Liu CS, Niu DM, Wu JY, Chen YT, Chern Y. Dysregulation of C/EBPalpha by mutant Huntingtin causes the urea cycle deficiency in Huntington's disease. Hum Mol Genet. 2007;16:483–498. doi: 10.1093/hmg/ddl481. [DOI] [PubMed] [Google Scholar]
- Chou SY, Lee YC, Chen HM, Chiang MC, Lai HL, Chang HH, Wu YC, Sun CN, Chien CL, Lin YS, Wang SC, Tung YY, Chang C, Chern Y. CGS21680 attenuates symptoms of Huntington's disease in a transgenic mouse model. J Neurochem. 2005;93:310–320. doi: 10.1111/j.1471-4159.2005.03029.x. [DOI] [PubMed] [Google Scholar]
- Das KC, Lewis-Molock Y, White CW. Activation of NF-kappa B and elevation of MnSOD gene expression by thiol reducing agents in lung adenocarcinoma (A549) cells. Am J Physiol. 1995;265:L588–L602. doi: 10.1152/ajplung.1995.269.5.L588. [DOI] [PubMed] [Google Scholar]
- del Toro D, Canals JM, Gines S, Kojima M, Egea G, Alberch J. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J Neurosci. 2006;26:12748–12757. doi: 10.1523/JNEUROSCI.3873-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai UA, Pallos J, Stockwell BR, Thompson LM, Marsh JL, Diamond MI. Biologically active molecules that reduce polyglutamine aggregation and toxicity. Hum Mol Genet. 2006;15:2114–2124. doi: 10.1093/hmg/ddl135. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- Elyaman W, Terro F, Suen KC, Yardin C, Chang RC, Hugon J. BAD and Bcl-2 regulation are early events linking neuronal endoplasmic reticulum stress to mitochondria-mediated apoptosis. Brain Res Mol Brain Res. 2002;109:223–238. doi: 10.1016/s0169-328x(02)00582-x. [DOI] [PubMed] [Google Scholar]
- Fusco FR, Zuccato C, Tartari M, Martorana A, De March Z, Giampa C, Cattaneo E, Bernardi G. Co-localization of brain-derived neurotrophic factor (BDNF) and wild-type huntingtin in normal and quinolinic acid-lesioned rat brain. Eur J Neurosci. 2003;18:1093–1102. doi: 10.1046/j.1460-9568.2003.02844.x. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Goold RG, Gordon-Weeks PR. The MAP kinase pathway is upstream of the activation of GSK3beta that enables it to phosphorylate MAP1B and contributes to the stimulation of axon growth. Mol Cell Neurosci. 2005;28:524–534. doi: 10.1016/j.mcn.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LSB. Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch Neurol. 2005;62:46–51. doi: 10.1001/archneur.62.1.46. [DOI] [PubMed] [Google Scholar]
- Hebb MO, Denovan-Wright EM, Robertson HA. Expression of the Huntington's disease gene is regulated in astrocytes in the arcuate nucleus of the hypothalamus of postpartum rats. FASEB J. 1999;13:1099–1106. doi: 10.1096/fasebj.13.9.1099. [DOI] [PubMed] [Google Scholar]
- Henriquet C, Gougat C, Combes A, Lazennec G, Mathieu M. Differential regulation of RANTES and IL-8 expression in lung adenocarcinoma cells. Lung Cancer. 2007;56:167–174. doi: 10.1016/j.lungcan.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Huang CD, Ammit AJ, Tliba O, Kuo HP, Penn RB, Panettieri RA, Jr, Amrani Y. G-protein-coupled receptor agonists differentially regulate basal or tumor necrosis factor-alpha-stimulated activation of interleukin-6 and RANTES in human airway smooth muscle cells. J Biomed Sci. 2005;12:763–776. doi: 10.1007/s11373-005-9008-z. [DOI] [PubMed] [Google Scholar]
- Humbert S, Saudou F. Huntington's disease: intracellular signaling pathways and neuronal death. J Soc Biol. 2005;199:247–251. doi: 10.1051/jbio:2005026. [DOI] [PubMed] [Google Scholar]
- Hunziker W, Whitney JA, Mellman I. Brefeldin A and the endocytic pathway: possible implications for membrane traffic and sorting. FEBS Lett. 1992;307:93. doi: 10.1016/0014-5793(92)80908-y. [DOI] [PubMed] [Google Scholar]
- Khoshnan A, Ko J, Watkin EE, Paige LA, Reinhart PH, Patterson PH. Activation of the IκB kinase complex and nuclear factor-κB contributes to mutant huntingtin neurotoxicity. J Neurosci. 2004;24:7999–8008. doi: 10.1523/JNEUROSCI.2675-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Park HR, Sul KH, Chung HY, Chung J. Induction of RANTES and CCR5 through NF-kappaB activation via MAPK pathway in aged rat gingival tissues. Biotechnol Lett. 2006;28:17–23. doi: 10.1007/s10529-005-4681-6. [DOI] [PubMed] [Google Scholar]
- Kim MO, Suh HS, Brosnan CF, Lee SC. Regulation of RANTES/CCL5 expression in human astrocytes by interleukin-1 and interferon-beta. J Neurochem. 2004;90:297–308. doi: 10.1111/j.1471-4159.2004.02487.x. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Kouroku Y, Fujita E, Jimbo A, Kikuchi T, Yamagata T, Momoi MY, Kominami E, Kuida K, Sakamaki K, Yonehara S, Momoi T. Polyglutamine aggregates stimulate ER stress signals and caspase-12 activation. Hum Mol Genet. 2002;11:1505–1515. doi: 10.1093/hmg/11.13.1505. [DOI] [PubMed] [Google Scholar]
- Lacy P, Logan MR, Bablitz B, Moqbel R. Fusion protein vesicle-associated membrane protein 2 is implicated in IFN-gamma-induced piecemeal degranulation in human eosinophils from atopic individuals. J Allergy Clin Immunol. 2001;107:671–678. doi: 10.1067/mai.2001.113562. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wang D, Wang Y, Xu Q. Simvastatin down regulates mRNA expression of RANTES and CCR5 in posttransplant renal recipients with hyperlipidemia. Transplant Proc. 2006;38:2899–2904. doi: 10.1016/j.transproceed.2006.08.136. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Rival T, Iche M, Chneiweiss H, Birman S. Expanded polyglutamine peptides disrupt EGF receptor signaling and glutamate transporter expression in Drosophila. Hum Mol Genet. 2005;14:713–724. doi: 10.1093/hmg/ddi067. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Maccioni RB, Munoz JP, Barbeito L. The molecular bases of Alzheimer's disease and other neurodegenerative disorders. Arch Med Res. 2001;32:367–381. doi: 10.1016/s0188-4409(01)00316-2. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Miyamoto NG, Medberry PS, Hesselgesser J, Boehlk S, Nelson PJ, Krensky AM, Perez HD. Interleukin-1beta induction of the chemokine RANTES promoter in the human astrocytoma line CH235 requires both constitutive and inducible transcription factors. J Neuroimmunol. 2000;105:78–90. doi: 10.1016/s0165-5728(00)00195-8. [DOI] [PubMed] [Google Scholar]
- Molinari M, Sitia R. The secretory capacity of a cell depends on the efficiency of endoplasmic reticulum-associated degradation. Curr Top Microbiol Immunol. 2005;300:1–15. doi: 10.1007/3-540-28007-3_1. [DOI] [PubMed] [Google Scholar]
- Morello S, Ito K, Yamamura S, Lee KY, Jazrawi E, Desouza P, Barnes P, Cicala C, Adcock IM. IL-1 beta and TNF-alpha regulation of the adenosine receptor (A2A) expression: differential requirement for NF-kappa B binding to the proximal promoter. J Immunol. 2006;177:7173–7183. doi: 10.4049/jimmunol.177.10.7173. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Edwardson JM. Progressive depletion of complexin II in a transgenic mouse model of Huntington's disease. J Neurochem. 2001;76:166–172. doi: 10.1046/j.1471-4159.2001.00059.x. [DOI] [PubMed] [Google Scholar]
- Orth M, Cooper JM, Bates GP, Schapira AH. Inclusion formation in Huntington's disease R6/2 mouse muscle cultures. J Neurochem. 2003;87:1–6. doi: 10.1046/j.1471-4159.2003.02009.x. [DOI] [PubMed] [Google Scholar]
- Oynebraten I, Bakke O, Brandtzaeg P, Johansen FE, Haraldsen G. Rapid chemokine secretion from endothelial cells originates from 2 distinct compartments. Blood. 2004;104:314–320. doi: 10.1182/blood-2003-08-2891. [DOI] [PubMed] [Google Scholar]
- Shibata A, Laurent CE, Smithgall TE. The c-Fes protein-tyrosine kinase accelerates NGF-induced differentiation of PC12 cells through a PI3K-dependent mechanism. Cell Signal. 2003;15:279–288. doi: 10.1016/s0898-6568(02)00089-x. [DOI] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhrao SK, Thomas P, Wood JD, MacMillan JC, Neal JW, Harper PS, Jones AL. Huntingtin protein colocalizes with lesions of neurodegenerative diseases: an investigation in Huntington's, Alzheimer's, and Pick's diseases. Exp Neurol. 1998;150:213–222. doi: 10.1006/exnr.1998.6778. [DOI] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Garry S, Cordery PM, Van Dellen A, Blakemore C, Hannan AJ. Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington's disease transgenic mice. Eur J Neurosci. 2004;19:2799–2807. doi: 10.1111/j.0953-816X.2004.03374.x. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Takano H, Gusella JF. The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neuroscience. 2002;3:15–27. doi: 10.1186/1471-2202-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio A, Ferrario M, Martinez FO, Locati M, Ghisi V, Bresciani LG, Mantovani A, Spano P. Gene expression profile activated by the chemokine CCL5/RANTES in human neuronal cells. J Neurosci Res. 2004;78:371–382. doi: 10.1002/jnr.20250. [DOI] [PubMed] [Google Scholar]
- Vila M, Jackson-Lewis V, Guegan C, Wu DC, Teismann P, Choi DK, Tieu K, Przedborski S. The role of glial cells in Parkinson's disease. Curr Opin Neurol. 2001;14:483–489. doi: 10.1097/00019052-200108000-00009. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Xu Y, Kiningham KK, Devalaraja MN, Yeh CC, Majima H, Kasarskis EJ, St Clair DK. An intronic NF-kappaB element is essential for induction of the human manganese superoxide dismutase gene by tumor necrosis factor-alpha and interleukin-1beta. DNA Cell Biol. 1999;18:709–722. doi: 10.1089/104454999314999. [DOI] [PubMed] [Google Scholar]
- Yu ZZD, Cheng G, Mattson MP. Neuroprotective role for the p50 subunit of NF-κB in an experimental model of Huntington's disease. J Mol Neurosci. 2000;15:31. doi: 10.1385/JMN:15:1:31. [DOI] [PubMed] [Google Scholar]
- Zhai W, Jeong H, Cui L, Krainc D, Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell. 2005;123:1241–1253. doi: 10.1016/j.cell.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]