

Abstract
In the genesis of Alzheimer's disease (AD), converging lines of evidence suggest that amyloid-β peptide (Aβ) triggers a pathogenic cascade leading to neuronal loss. It was long assumed that Aβ had to be assembled into extracellular amyloid fibrils or aggregates to exert its cytotoxic effects. Over the past decade, characterization of soluble oligomeric Aβ species in the brains of AD patients and in transgenic models has raised the possibility that different conformations of Aβ may contribute to AD pathology via different mechanisms. The receptor for advanced glycation end products (RAGE), a member of the Ig superfamily, is a cellular binding site for Aβ. Here, we investigate the role of RAGE in apoptosis induced by distinct well characterized Aβ conformations: Aβ oligomers (AβOs), Aβ fibrils (AβFs), and Aβ aggregates (AβAs). In our in vitro system, treatment with polyclonal anti-RAGE antibodies significantly improves SHSY-5Y cell and neuronal survival exposed to either AβOs or AβAs but does not affect AβF toxicity. Interestingly, using site-specific antibodies, we demonstrate that targeting of the Vd domain of RAGE attenuates AβO-induced toxicity in both SHSY-5Y cells and rat cortical neurons, whereas inhibition of AβA-induced apoptosis requires the neutralization of the C1d domain of the receptor. Thus, our data indicate that distinct regions of RAGE are involved in Aβ-induced cellular and neuronal toxicity with respect to the Aβ aggregation state, and they suggest the blockage of particular sites of the receptor as a potential therapeutic strategy to attenuate neuronal death.
Keywords: Alzheimer's disease, amyloid-β, RAGE, Ig-like domains, cortical neurons, apoptosis
Introduction
The concept that cerebral accumulation of amyloid-β peptide (Aβ) induces Alzheimer's disease (AD) remains controversial, in large part because of the difficulty in providing direct mechanistic evidence that a particular Aβ species induces neuronal death. Early evidence suggested that Aβ-induced neurotoxicity in cell culture and in vivo was associated with insoluble fibrillar (AβF) and aggregated (AβA) forms of Aβ present in amyloid plaques of the AD brain (Pike et al., 1991; Lorenzo and Yankner, 1994; Estus et al., 1997; McLean et al., 1999; Naslund et al., 2000). In these studies, the Aβ neurotoxic effect persisted while aggregation was ongoing but diminished as the process of aggregation neared completion. Studies in human and transgenic mice revealed a weak correlation between amyloid plaque load, neuronal loss, and memory impairment (Terry et al., 1991; Dickson et al., 1995; Moechars et al., 1996; Irizarry et al., 1997a,b; Westerman et al., 2002). These observations are inconsistent with a mechanism for progressive dementia dependent on insoluble Aβ-induced neuronal death and indicate that other species may underlie neurodegeneration, particularly in the very early stages of AD. Recently, the amyloid cascade hypothesis was modified to include soluble oligomers (AβOs). Although they differ in structure, AβOs include dimers, trimers, dodecamers, and higher-molecular-weight complexes and possess a variety of biological activities, including the ability to disrupt cognitive function in vivo (Walsh et al., 2002; Cleary et al., 2005; Lesne et al., 2006; Lacor et al., 2007) and to induce neuronal apoptosis in vitro (Chong et al., 2006; Malaplate-Armand et al., 2006).
Several mechanisms could potentially target and concentrate Aβ on cellular elements. In this regard, the receptor for advanced glycation end products (RAGE) was identified as one of the cell-surface binding sites for Aβ (Yan et al., 1996). RAGE is a multiligand receptor composed of three extracellular Ig-like domains (Vd, C1d, C2d), a single transmembrane domain, and a short cytoplasmic tail. RAGE is overexpressed in the AD brain and acts as a binding site for Aβ at the plasma membrane of neurons, microglial cells, and endothelial cells of the vessel wall (Yan et al., 1996; Sasaki et al., 2001; Deane et al., 2003). Previous experiments indicate that RAGE mediates Aβ-induced oxidative stress and nuclear factor-κB activation (Yan et al., 1996) as well as neuronal expression of macrophage colony-stimulating factor (Du Yan et al., 1997), mitogen-activated protein (MAP) kinases signaling defects (Arancio et al., 2004), or cell death (Hadding et al., 2004).
The current study dissects the role of the distinct Ig-like domains of RAGE in Aβ-induced apoptosis. Therefore, we exposed RAGE-expressing SHSY-5Y cells and rat cortical neurons (RCNs) to AβO, AβF, or AβA conditioned media. In our in vitro system, simultaneous application of polyclonal anti-RAGE antibodies effectively prevented apoptosis induced by AβOs and AβAs. In contrast, this treatment did not affect AβF-induced SHSY-5Y cell death. Furthermore, using site-specific antibodies, we showed that attenuation of RAGE-mediated AβO- and AβA-induced toxicity required the blockage of specific and distinct Ig-like domains of the receptor, the Vd and C1d domains, respectively. Our data provide the first evidence that RAGE mediates Aβ-induced cellular and neuronal apoptotic events by mechanisms involving distinct sites of the receptor depending on the Aβ aggregation state. In addition, our data support the view that site-specific blockage of the Vd of RAGE may have cytoprotective effects especially with respect to preventing neuronal apoptosis early in the disease process.
Materials and Methods
Preparation and analysis of Aβ(1–40) conditioned media.
Synthetic Aβ(1–40) peptide (Bachem, Bubendorf, Switzerland) was dissolved in bidistilled water at 1 mm and adjusted to 10 μm with either RPMI-1640 (supplemented with 2 mm l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin) or Neurobasal (supplemented with B27, 2 mm l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin) medium. The conditioned media were incubated at 37°C and snap frozen in liquid N2. The relative proportions of soluble and fibrillar Aβ present in both media were determined by Congo red assay and transmission electron microscopy (TEM) at 0, 1, 3, 4, 6.5, 8, 10.5, 12, 15, and 24 h after peptide addition. According to Klunk et al. (1999), the absorbance of Congo red, known to specifically bind to amyloid fibers containing cross β-sheet structures, was recorded at 540 nm with an Anthos Labtec Instruments (Eugendorf, Austria) plate reader. Aβ structures were imaged using TEM. Briefly, samples were added (1 min) to 400-mesh copper grids, washed once with H2O, and negatively stained for 1 min with 2% uranyle acetate. Grids were air dried and examined on a Philips (Eindhoven, The Netherlands) CM12 electron microscope. Data analysis showed that both media predominantly contained spherical vesicles of Aβ with diameters of ∼5 nm, similar to previously described oligomers (Mastrangelo et al., 2006; Moore et al., 2007) at 0–8 h of incubation. Up to 12 h after peptide addition, Congo red assay revealed the presence of β-sheet-containing assemblies exhibiting typical fibril structures as imaged by TEM. Therefore, aliquots of Aβ-containing media were snap frozen in liquid N2 1 h after incubation at 37°C to generate AβOs. To generate AβF preparations, Aβ-containing medium was centrifuged (14,000 × g; 10 min) 12 h after incubation at 37°C, and the pellet containing the fibrils was resuspended in equal amounts of medium and snap frozen in liquid N2. The aliquots were kept at −80°C until use. AβAs were produced by dissolving the lyophilized peptide at 1 mm in PBS. After 2 h incubation at room temperature (RT), aggregates were collected by centrifugation at 6000 × g, resuspended in PBS, and adjusted to 10 μm in RPMI or Neurobasal medium. The presence and stability of the aggregated forms was confirmed as described above (Congo red binding assay and TEM), after 0, 12, and 24 h of incubation at 37°C. Because the formation of glycation end products during the experimental time course could influence Aβ aggregation and RAGE–Aβ interaction, we confirmed the absence of glycated Aβ in the conditioned media using MALDI (matrix-assisted laser desorption/ionization-time of flight). The spectra always showed a peak with a molecular mass of 4329 Da for Aβ, corresponding to the relative molecular mass of the peptide (data not shown).
Dot blot assay with A11 and 6E10.
Dot blot assay was performed as described previously (Kayed et al., 2003). Briefly, 25 μl of 10 μm Aβ samples were dripped onto 0.2 μm nitrocellulose membrane (Bio-Rad, Hercules, CA) and allowed to dry for 5 min. The membrane was blocked in 10% milk in TBST (Tris-buffered saline with 0.01% Tween 20) for at least 1 h. The blots were washed three times in TBST before and after incubation with a 1:10,000 dilution of rabbit anti-oligomer antibody (A11; BioSource, Camarillo, CA) and goat anti-rabbit horseradish peroxidase (Sigma, St. Louis, MO) in 5% milk in TBST. The blots were developed using the SuperSignal West Dura System (Pierce, Rockford, IL). Blots were stripped and reprobed with mouse monoclonal anti-human Aβ 6E10 (1:5000; Signet, Dedham, MA).
SHSY-5Y cells and rat primary neuronal cultures.
Human neuroblastoma SHSY-5Y cells (American Type Culture Collection, Manassas, VA) were grown in RPMI supplemented with 10% fetal calf serum (FCS), 2 mm l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. The cells were maintained at 37°C in a humidified incubator containing 95% air and 5% CO2.
For the RCN cultures, the frontal cortices of three rat embryos (embryonic day 18) were dissected and washed with PBS containing 5.5 mm glucose. The cells were sedimented and dissociated in PBS supplemented with 0.5 mg/ml papain (Sigma), 10 mm glucose, 1 mg/ml BSA, and 10 μg/ml DNAaseI (Roche Diagnostics, Mannheim, Germany) for 15 min at 37°C. Cells were washed with DMEM and mechanically dissociated in DMEM supplemented with 10% FCS, 2 mm l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Two hours after cells were plated (in 5% CO2) onto poly-l-lysine (100 μg/ml)-coated multiwells or coverslips, medium was removed and replaced with Neurobasal medium supplemented with B27, 2 mm l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. The cortical neuron cultures contained a small percentage of glial cells (<10%) as assessed by immunofluorescence using anti-PGP 9.5 and anti-glial fibrillary acidic protein (data not shown).
Cell culture treatments.
SHSY-5Y cells were serum deprived for 24 h before treatment. Rat cortical neurons were plated at a density of 30,000 cells/cm2 and kept in serum-free medium for ≥8 d. Respective conditioned media were added for 24 h to SHSY-5Y cells or to rat neuronal cultures 8 d after plating [8 d in vitro (8 DIV)]. AβO conditioned media were exchanged every 8 h to avoid fibril formation and toxicity, and AβF conditioned media were exchanged every 12 h to avoid generation of contaminating aggregates. Control cultures underwent similar medium changes. To investigate the role of RAGE in Aβ-induced cell death, the soluble RAGE (sRAGE; 50 μg/ml), containing the three extracellular Ig-like domains of RAGE, the recombinant Vd domain of RAGE (recVd; 18.5 μg/ml), and the different polyclonal antibodies (25 μg/ml) were added to the different conditioned media, and apoptosis was assessed after 24 h. The human recombinant sRAGE and the human recombinant Vd (recVd) were expressed and purified as described previously (Ostendorp et al., 2006; Dattilo et al., 2007). The polyclonal goat anti-human sRAGE antibody (anti-RAGE) was obtained from R & D Systems (Minneapolis, MN). The RAGE-Vd-specific antibodies (anti-Vd), RAGE-C1d-specific antibodies (anti-C1), and RAGE-C2d-specific antibodies (anti-C2) were produced in rabbit as described previously (Ostendorp et al., 2006). Residues 54–70, 158–179, and 272–293 of human RAGE were selected for the generation of the anti-Vd, anti-C1, and anti-C2 antibodies, respectively. Antisera were affinity purified by using a HiTrap protein A column (GE Healthcare, Little Chalfont, Buckinghamshire, UK) according to the manufacturer's protocol. The IgG concentrations of the antisera were determined by the BCA method (Pierce). The RAGE-C1dC2d-specific antibodies (anti-C1C2) and RAGE-VdC1dC2d-specific antibodies (anti-VdC1C2) were generated by mixing equal amounts of anti-Vd, anti-C1, and anti-C2. In control experiments, we used nonspecific IgG (R & D Systems) with respect to the species used in the treatment. The nonspecific antibodies had no effect on cell survival either in the presence or absence of Aβ (data not shown).
Cell viability assays.
Cell death was determined by fluorescence-activated cell sorting (FACS) using the cycleTEST Plus DNA kit (Becton Dickinson, Mountain View, CA) and a FACSCalibur flow cytometer. A total of 104 cells was analyzed for each condition, and data from three separate experiments were pooled. Apoptosis was scored by terminal deoxynucleotidyltransferase-mediated dUTP biotin nick end labeling (TUNEL) assay according to the manufacturer's protocol (Roche Diagnostics). Cells were counterstained with 4′,6-diamidino-2-phenylindole. SHSY-5Y cells and RCNs were counted on coverslips, and at least 10 fields per culture in triplicate cultures were analyzed per individual experiment. In addition to the TUNEL assay, caspase 3 and 7 activities were quantified using the Caspase-Glo 3/7 kit (Promega, Madison, WI). Each experiment was repeated four times.
Immunofluorescence.
Cortical rat neurons were fixed in 4% paraformaldehyde for 1 h at RT, permeabilized with 0.2% Triton X-100 in PBS, and blocked for 1 h in 5% horse serum/PBS. Cultures were incubated with rabbit anti-Vd (1:1000), mouse anti-PGP 9.5 (1:500; Abcam, Cambridge, MA), mouse anti-synaptophysin (1:500; Calbiochem, La Jolla, CA), and mouse anti-Aβ 6E10 (1:1000; Signet) for 1 h at RT, followed by incubation with fluorescent-conjugated secondary antibodies (Alexa; Invitrogen, Eugene, OR). Omission of the primary antibody resulted in complete loss of specific labeling. The fluorescence signals were visualized using a Leica (Nussloch, Germany) SP2 confocal laser microscope.
Immunoblotting.
SHSY-5Y cells, rat neuronal cultures, mouse neuronal cultures, and RAGE−/− mouse brains were lysed in 50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 1% Triton X-100, 10 nm NaF, and 1 mm Na3VO4 supplemented with complete proteinase inhibitor cocktail (Roche Diagnostics) at the indicated time points. Protein concentration of the samples was measured using the BCA method (Pierce). Equal amounts of protein (50 μg) were separated by 10% PAGE, blotted onto nitrocellulose membrane, and probed with anti-VdC1C2 (1:1000), anti-PGP 9.5 (1:500; Abcam), anti-phosphorylated extracellular signal-regulated kinase 1/2 (ERK1/2), anti-ERK1/2, anti-phosphorylated c-Jun N-terminal kinase (JNK), and anti-JNK (1:1000; Cell Signaling Technology, Beverly, MA). The blots were incubated with a secondary antibody conjugated with peroxidase (1:10,000; GE Healthcare). The bands were visualized using ECL solution (GE Healthcare). Densitometric values from gels were obtained using a Bio-Rad densitometer GS 800 and analyzed with Bio-Rad Quantity One software. The amounts of phosphorylated ERK and JNK were normalized to the total amount of ERK and JNK, respectively.
Statistics.
Unless specified, data are presented as mean ± SEM and were analyzed using one-way ANOVA followed by Bonferroni's post hoc test with the level of significance set at p < 0.01.
Results
AβO, AβF, and AβA conditioned media generation and toxicity
The mechanism by which Aβ aggregates is not fully understood, although it has been shown that peptide concentration, ions, pH, and temperature influence the Aβ oligomerization process and fibril conversion (Isaacs et al., 2006; Ha et al., 2007). In the present study, we generated two conditioned media (RPMI and Neurobasal) containing 10 μm analogs of soluble AβOs, AβFs, or amorphous AβAs. Under our experimental conditions, the formation of fibrils containing β-sheet structures started 9 h after the addition of soluble Aβ(1–40) peptide in both media, as revealed by enhanced absorbance in Congo red binding assay (Fig. 1A). Maximal absorbance was obtained after 12 h and thereafter slowly decreased during an additional 12 h (Fig. 1A). Electron microscopy confirmed the presence of typical 100–200 nm AβF structures in preparations at 12–24 h (Fig. 1B, AβFs). Electron microscopy analysis performed at 0–8 h of incubation showed small spherical Aβ assemblies that resembled previously described oligomers (Fig. 1B, AβOs) (Losic et al., 2006; Mastrangelo et al., 2006; Moore et al., 2007). These observations conform to previous studies indicating that mature amyloid fibrils occur through a number of intermediate structural forms referred to as oligomers (Arimon et al., 2005; Shahi et al., 2007). Although the precise oligomer stoichiometry remains unclear, the preparations were predominantly free of protofibrils and entirely free of fibrils as indicated by the absence of significant variation in Congo red binding during this period of time (Fig. 1A). AβAs were prepared as described previously (Lorenzo and Yankner, 1994) by dissolving Aβ(1–40) (1 mm) directly into PBS before adjusting the concentration to 10 μm in RPMI or Neurobasal medium. The prevalence and stability of the aggregates were confirmed by TEM (Fig. 1B, AβAs). The absence of variation in Congo red binding confirmed that AβAs did not coexist with β-sheet-containing structures (Fig. 1A). Based on these observations, we produced cell culture media enriched in AβOs, AβFs, or AβAs (see Materials and Methods). The A11 antibody, which reacts well with the soluble oligomers but not with soluble monomers or mature amyloid fibers (Kayed et al., 2003), was used to further characterize the distinct Aβ preparations. In accord with our previous observations, the oligomer-specific antibody detected Aβ in AβO conditioned media but did not react with Aβ in AβF and AβA preparations (Fig. 1C). The mouse monoclonal antibody 6E10, which recognizes Aβ independently of its conformational state, confirmed the presence of Aβ peptide in the different preparations (Fig. 1C).
Figure 1.
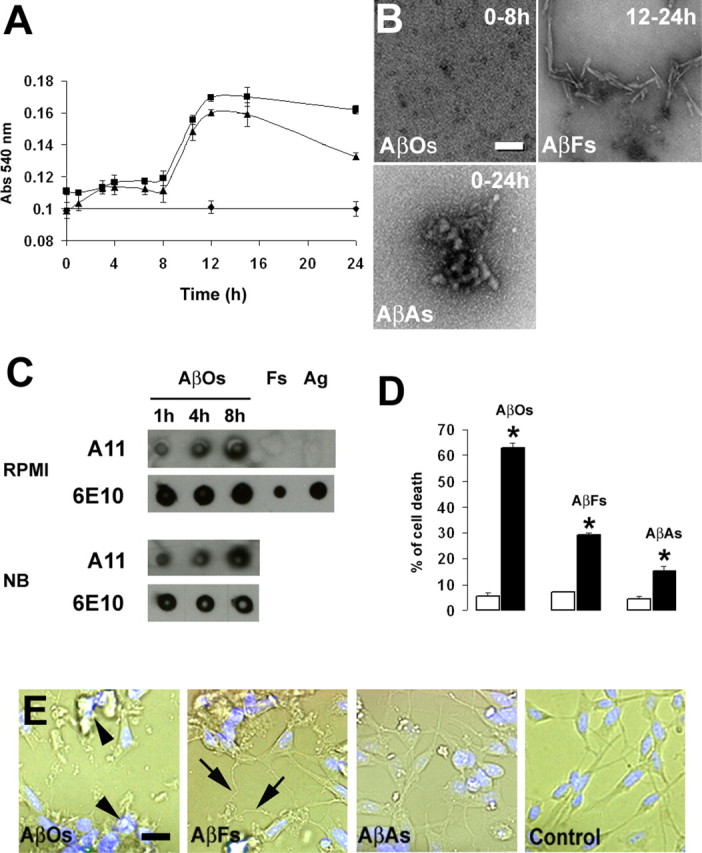
Analysis of Aβ aggregation and toxicity. A, Congo red binding analysis of soluble Aβ(1–40), adjusted to 10 μm in RPMI (▴) or Neurobasal (■) medium, or preaggregated Aβ (1 mm in PBS), brought to 10 μm in RPMI or Neurobasal medium (♦). Error bars indicate ±SD (n = 9). B, Electron microscopy analysis of aliquots of media. Typical soluble oligomers (AβOs) are present at 0–8 h of incubation. Up to 12 h, the vast majority of Aβ is fibrillar (AβFs). Preaggregated Aβ (AβAs) forms stable structures exhibiting typical aggregate morphology in RPMI. Scale bar, 100 nm. C, Oligomer-specific immunoreactivity of AβO, AβF, and AβA conditioned RPMI and Neurobasal (NB) media. AβOs were incubated at 37°C for the indicated time periods. AβOs, AβFs (Fs), and AβAs (Ag) were applied to a nitrocellulose membrane and first probed with the A11 antibody and reprobed after stripping with 6E10. A11 antibody is specific for AβOs, whereas 6E10 recognizes all Aβ species. D, FACS analysis of SHSY-5Y cell death exposed to the distinct Aβ-containing media. AβO-, AβF- and AβA-containing media (filled bars) induced a significant increase in cell death compared with their respective control (open bars). Error bars indicate ±SD. Statistical significance was determined by mean values (n = 3) of the ANOVA variance, followed by Student's t test. Significance was accepted for *p < 0.01. E, Phase-contrast microscopy of SH-SY5Y cells exposed to AβOs, AβFs, or AβAs for 24 h. AβOs (arrowheads) induced apoptotic features such as disintegration of processes, swelling of cell bodies, and nuclear condensation. AβFs (arrows) produced dystrophic effects on processes. Scale bar, 10 μm.
We subsequently investigated the toxicity of the distinct Aβ preparations on RAGE-expressing SHSY-5Y cells (Sajithlal et al., 2002). For this purpose, neuroblastoma cells were exposed to RPMI containing 10 μm AβOs, AβFs, or AβAs for 24 h, and the cell death was measured by FACS. To avoid AβO conversion into fibrils during the time course of the experiment, AβO conditioned medium was exchanged twice (every 8 h). Similarly, AβF conditioned medium was changed after 12 h (the same protocols were used in the following cell death experiments). FACS analysis revealed that the distinct Aβ conformations significantly increased cell death in our in vitro system (Fig. 1D). Chronic exposure to AβOs caused massive cell death, and after 24 h, >60% of the cells were dead (Fig. 1D, AβOs). In contrast, addition of AβF and AβA conditioned media resulted in moderate effects with ∼30 and 15% cell death, respectively (Fig. 1D, AβFs and AβAs). When AβF and AβA preparations were centrifuged (5 min at 16,000 × g), only the pellet containing the insoluble Aβ fraction was toxic, whereas the supernatant did not elicit any toxicity, indicating that small amounts of contaminating soluble AβOs were not responsible for AβF and AβA toxicity (data not shown). Moreover, AβOs induced apoptotic features including disintegration of processes, swelling of cell bodies, and nuclear condensation (Fig. 1E, AβOs), whereas AβFs produced dystrophic effects on neuroblastoma cell processes (Fig. 1E, AβFs). These results corroborate work done previously (Grace and Busciglio, 2003; Deshpande et al., 2006). In contrast, AβAs (Fig. 1E, AβAs) did not induce degenerative morphology when compared with control cells (Fig. 1E, control). These data indicate that AβO, AβF, and AβA conditioned media consistently trigger SHSY-5Y cell death with a difference in toxicity correlating with the Aβ aggregation state.
RAGE is implicated in AβO- and AβA-induced apoptosis in SHSY-5Y cells
Extracellular Aβ may induce neurotoxicity by interacting with putative candidate receptors, resulting in the activation of a number of cell-death signaling pathways leading to apoptosis (Estus et al., 1997; Yaar et al., 1997; Yao et al., 2005; St. John, 2007). Previous work provided strong evidence that RAGE interacts with Aβ at the membrane of various cell types promoting its adhesion and toxic effects (Yan et al., 1996; Deane et al., 2003). However, a direct link between RAGE and Aβ-induced cell death has not yet been demonstrated. We therefore investigated whether RAGE directly contributes to Aβ-induced cell death in our cellular system. For this purpose, SHSY-5Y cells were exposed for 24 h to AβO, AβF, or AβA conditioned media in the presence or absence of polyclonal anti-RAGE antibodies (anti-RAGE), which neutralize the three extracellular Ig-like domains (Vd, C1d, C2d) of the receptor. Treated cultures and control cells were processed for two parameters associated with apoptosis: DNA fragmentation and the activation of caspase 3/7 pathways. In accordance with our FACS studies, AβOs induced massive cell death with a fivefold increase in the mean percentage of TUNEL-positive cells (Fig. 2A) and a 380% increase in caspase activity (Fig. 2B). Anti-RAGE treatment resulted in a significant attenuation of cell death in SH-SY5Y cells exposed to either AβO or AβA preparations as indicated by a decrease in cells undergoing DNA fragmentation (Fig. 2A) and a significant reduction in AβO- or AβA-induced caspase activation (Fig. 2B). These effects were specific because treatment of cells with a control isotype IgG did not affect Aβ-induced cell death (data not shown). In contrast, treatment with anti-RAGE neither affected DNA fragmentation (Fig. 2A) nor the activation of the caspase pathways (Fig. 2B) induced by AβFs. Thus, our data indicate that RAGE is implicated, at least in part, in AβO- and AβA-induced apoptosis.
Figure 2.

Effect of anti-RAGE antibody on Aβ-induced apoptosis. A, Simultaneous application of anti-RAGE (25 μg/ml) significantly reduces the toxic effect of AβOs and AβAs as measured by TUNEL, but it did not influence AβF-induced DNA fragmentation (*p < 0.01). B, Similarly, anti-RAGE treatment significantly attenuates AβO- and AβA-dependent activation of the executioner caspase 3/7 but did not affect AβF-induced caspase activation (*p < 0.01). Error bars indicate mean ± SEM.
Attenuation of AβO- and AβA-induced apoptosis requires blockage of distinct RAGE Ig-like domains
RAGE is a multivalent receptor that binds several other ligands besides Aβ. These include advanced glycation end products (Schmidt et al., 1992), the chromatin-binding protein HMGB1 (Huttunen et al., 2000; Tian et al., 2007), as well as several members of the S100 family (Hofmann et al., 1999; Leclerc et al., 2007) leading to either a trophic or a toxic cellular effect. We recently showed that S100B and S100A6, two structurally closely related RAGE ligands, interact with distinct domains of the receptor and activate distinct signaling pathways suggesting that the cellular effects triggered by RAGE might be specific for each ligand (Leclerc et al., 2007). We therefore hypothesized that RAGE-mediated AβO- and AβA-induced apoptosis could involve distinct domains of the receptor. To investigate this hypothesis, we exposed SHSY-5Y cells to AβO or AβA conditioned media for 24 h in the presence or absence of site-specific antibodies targeting particular epitopes within the Vd (anti-Vd), the C1d (anti-C1), or the C1d and the C2d (anti-C1C2) domain of the receptor. Here again, AβO conditioned medium was exchanged every 8 h to avoid the formation of fibrils. In the presence of AβOs, we observed a significant decrease in cells undergoing DNA fragmentation and a reduction in caspase activity when the cultures were treated with anti-Vd (Fig. 3A), whereas anti-C1 or anti-C1C2 treatment did not affect AβO-induced apoptotic events (Fig. 3B,C). In contrast, AβA-induced cell death was unaffected by the anti-Vd treatment as scored by TUNEL (Fig. 3A), whereas anti-C1 or anti-C1C2 treatment significantly blocked DNA fragmentation as well as caspase activation induced by AβAs (Fig. 3B,C). Thus, our data indicate that blockage of the Vd of RAGE effectively protects SHSY-5Y cells from AβO-induced cell death, whereas attenuation of AβA toxicity requires antagonism at the C1d domain, suggesting that AβOs and AβAs interact with distinct sites of RAGE.
Figure 3.

Involvement of RAGE Ig-like domains in AβO- and AβA-induced cell death. Ten micromolar AβOs or AβAs were added to SHSY-5Y cell cultures for 24 h. A–C, Treatment with anti-Vd (A) significantly attenuates RAGE-mediated AβO-induced cell death as determined by the TUNEL method and caspase activity assays (*p < 0.01), whereas anti-C1 (B) and anti-C1C2 (C) treatment did not affect cell survival. In contrast, AβA-induced apoptosis was inhibited in the presence of anti-C1 (B), anti-C1C2 (C), or antibodies (*p < 0.01). D, Simultaneous application of sRAGE (50 μg/ml) significantly decreased AβO- and AβA-induced caspase activation, whereas recVd (rVd) treatment inhibited only AβO toxicity (*p < 0.01). Attenuation of RAGE-mediated AβO- and AβA-induced toxicity requires the blockage of distinct and specific domains of the receptor, Vd, and C1d, respectively. Error bars indicate mean ± SEM.
The truncated isoform of RAGE (sRAGE), corresponding to the extracellular domains only of the receptor, has been suggested to function as a decoy, abrogating RAGE-mediated cellular activation by interacting with circulating RAGE ligands. To confirm the involvement of RAGE in Aβ-induced apoptosis, we exposed SHSY-5Y cells to either AβOs or AβAs in the presence or absence of recombinant sRAGE or the recombinant form of the Vd (recVd). As expected, the treatment of cells with sRAGE significantly decreased AβO- and AβA-induced caspase activation (Fig. 3D). In addition, recVd treatment also attenuated AβO-induced caspase activation. In contrast, the addition of recVd did not affect the increase in caspase activity in cells exposed to AβAs (Fig. 3D). These results are in accordance with our previous observations suggesting that RAGE mediates AβO- and AβA-induced apoptosis via mechanisms involving distinct sites of the receptor.
Anti-Vd antibodies attenuate AβO-induced apoptosis in RCNs
We next asked whether the effects observed with AβOs and AβAs in human neuroblastoma cells could be reproduced in a more relevant model such as primary cultures of neurons. We therefore investigated the effect of the anti-Vd and anti-C1 antibodies on AβO- and AβA-induced apoptosis in RCNs. Initial tests were performed to validate the experimental model and the effectiveness of RAGE–Aβ interactions to induce cellular responses. Western blot analysis using anti-VdC1C2 revealed a band of ∼50 kDa in the homogenates of RCNs at 0, 8, and 14 DIV (Fig. 4A). In addition, closely migrating bands were detected that are attributable to differentially glycosylated RAGE. These observation had previously been described in AD brains (Sasaki et al., 2001). We also confirmed the specificity of the anti-Vd, anti-C1, and anti-C2 antibodies as indicated by the absence of immunoreactive bands in brain extracts of RAGE−/− mice (Fig. 4A). Interestingly, RAGE expression was more prominent at 8 DIV in RCNs (Fig. 4A), whereas mouse cortical neurons did not express the receptor at this stage (Fig. 4A). Thus, we decided to perform subsequent experiments using 8 DIV RCNs.
Figure 4.
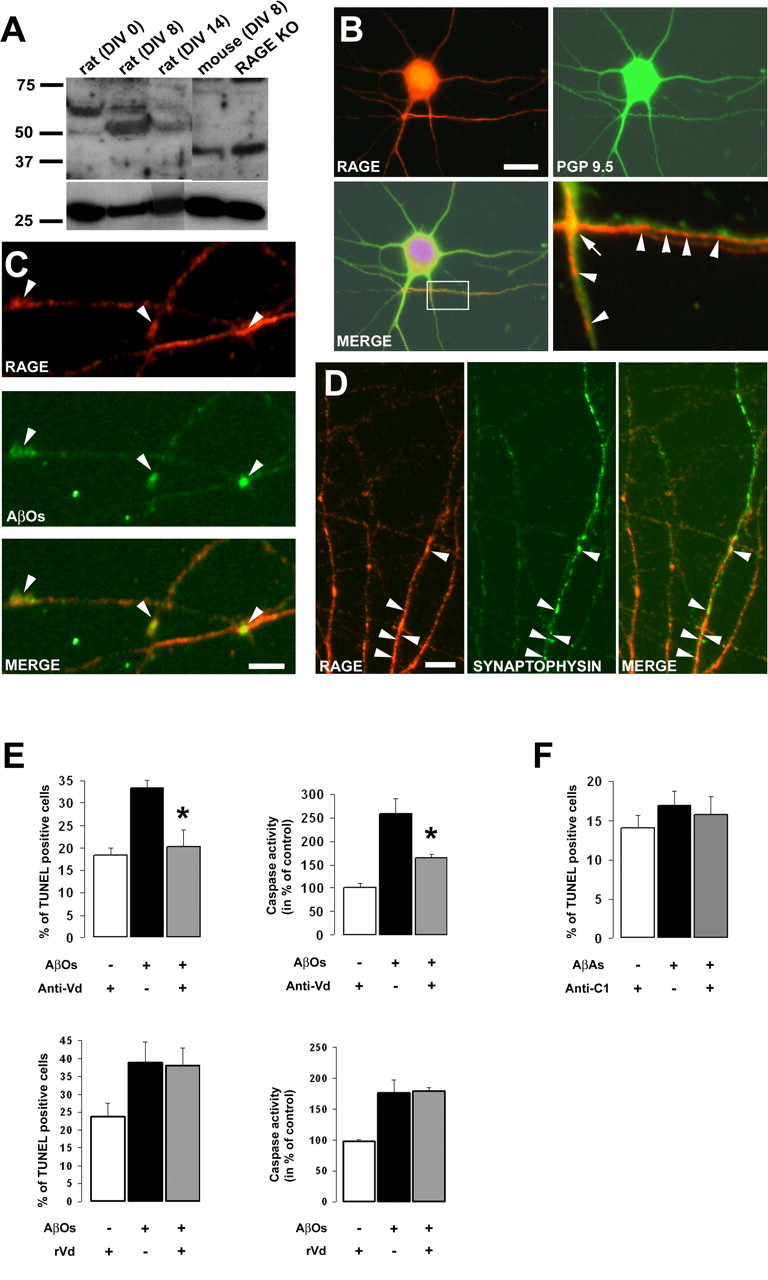
RAGE-Vd mediates AβO-induced neuronal death. A, Western blot analysis using anti-VdC1C2 antibodies (1:1000) of RAGE expression in rat cortical neuronal cultures at 0, 8, and 14 DIV. RAGE expression was more prominent at 8 DIV in RCNs. The bottom panels show representative loading control using anti-PGP 9.5 (1:1000). KO, Knock-out. B, Double labeling of 8 DIV RCNs with anti-Vd (red; 1:1000) and anti-PGP 9.5 (green; 1:500). The merged image shows expression of RAGE in neurons. Scale bar, 10 μm. Higher magnification of the boxed area in the merged image shows that RAGE is present at en passant synapses (bottom right, arrow) and at particular sites along neuronal processes (bottom right, arrowheads). C, Double immunofluorescence of 8 DIV RCNs with anti-Vd (red; 1:1000) and AβOs (6E10; green; 1:1000). Cultures were incubated with 10 μm AβOs for 2 h before fixation. The merged image shows the colocalization of RAGE and AβOs along neuronal processes (arrows). Scale bar, 2 μm. D, RCNs at 8 DIV were fixed and costained with anti-Vd (red; 1:1000) and anti-synaptophysin (green; 1:500). Partial colocalization is observed as light yellow spots (merge, arrowheads). Scale bar, 5 μm. E, F, RCNs were exposed to 10 μm AβOs or AβAs for 24 h. TUNEL and caspase assays were performed in the presence or absence of anti-Vd or anti-C1 treatment. AβOs (E), but not AβAs (F), induced a significant increase in caspase activity and DNA fragmentation events. In contrast to recVd (rVd) treatment, simultaneous application of anti-Vd improves neuronal survival exposed to AβOs (E) as measured by the same methods (*p < 0.01). Error bars indicate mean ± SEM.
The ability of RAGE to colocalize with AβOs in our model was evaluated by immunofluorescence. RAGE immunoreactivity (Fig. 4B, red) was detected in cell bodies and processes of RCNs as indicated by PGP 9.5 colabeling (Fig. 4B, merge). Higher-magnification images showed a prominent RAGE immunoreactivity at “en passant” synapses (Fig. 4B, arrow) and a clear punctuated staining defining submicrometer-sized subdomains along neuronal processes (Fig. 4B, arrowheads). Double immunolabeling of RCN cultures exposed to AβOs for 2 h revealed that a fraction of AβOs colocalizes with RAGE along neuronal processes (Fig. 4C, arrowheads) suggesting that RAGE might interact with AβOs. Previous work has shown a synaptic targeting of soluble Aβ species in rat hippocampal and human cortical neurons (Lacor et al., 2004; Deshpande et al., 2006). To determine whether RAGE is localized at synaptic sites, we performed multiple fluorescence labeling, and synapses were defined by using the presynaptic marker synaptophysin. We found a clear but restricted colocalization of RAGE with the synaptic marker along sections of neuronal processes (Fig. 4D, arrowheads). Interestingly, RAGE was also present along processes exhibiting poor synaptophysin immunoreactivity, whereas some other sections, rich in synaptic sites, did not contain the receptor (Fig. 4D).
Finally, RCN cultures were treated with AβO conditioned medium for 24 h in the presence or absence of either anti-Vd or recVd. Control and treated cells were processed for TUNEL and caspase activity. AβOs induced a significant increase in neuronal cell death as indicated by an increase in both TUNEL-positive cells and caspase activity (Fig. 4E). As observed with the human neuroblastoma cells, neuronal apoptosis induced by AβOs could be attenuated significantly by anti-Vd treatment as indicated by a reduction in DNA fragmentation and caspase activation (Fig. 4E). In contrast, recVd treatment did not affect DNA fragmentation nor caspase activity in RCNs exposed to AβOs (Fig. 4E). RCN cultures were also exposed to AβAs in the presence or absence of anti-C1. However, under our experimental conditions, the Aβ aggregates failed to significantly induce neuronal death (Fig. 4F). Thus, these data support the hypothesis that RAGE might participate in AβO-induced neuronal apoptosis and confirm that specific neutralization of the Vd is sufficient to significantly promote RCN survival.
RAGE mediates AβO-induced perturbation of ERK signaling pathway
We also explored the possible molecular mechanisms underlying RAGE-mediated AβO-induced cell death. Previous studies have indicated that RAGE–ligand interactions modulate MAP kinase pathways (Arancio et al., 2004; Monteiro et al., 2006). Furthermore, it has been suggested that defects in both ERK and JNK signaling underlie neuronal dysfunction such as caspase activation evoked by Aβ oligomers in various AD paradigms (Bell et al., 2004; Chong et al., 2006; Ma et al., 2007; Townsend et al., 2007; Yan and Wang, 2007). Therefore, using our models, we investigated whether RAGE could be involved in AβO-induced ERK and/or JNK signaling defects. In our experimental conditions, Western blot analysis of neuroblastoma cell extracts exposed to AβOs for 8 h revealed an increase in ERK activation (Fig. 5A). Densitometric analysis of gels from separate experiments demonstrated the reproducibility of these observations and revealed a 130% increase in phosphorylated ERK in SHSY-5Y cells exposed to AβOs as compared with control cells (Fig. 5A). Interestingly, simultaneous application of either anti-Vd or recVd consistently suppressed AβO-induced activation of ERK as indicated by the absence of significant variation in control and treated SHSY-5Y cells (Fig. 5A). In contrast, Western blot analysis of RCN extracts exposed to AβOs for 8 h revealed a downregulation of the phosphorylated form of ERK (Fig. 5C). Densitometric analysis of gels confirmed this observation and revealed that the phosphorylated form of ERK, normalized to the total amount of ERK, decreased to 70% of the control value in the presence of AβOs (Fig. 5C). A comparable reduction in ERK phosphorylation was observed at 24 h, whereas the addition of AβOs for 1, 2, and 4 h had no significant effect on the basal activity of ERK (data not shown). However, anti-Vd treatment consistently suppressed AβO-induced hypophosphorylation of ERK in RCNs as indicated by the absence of significant variation in control and treated cultures (Fig. 5C). In contrast, similar experiments revealed no change in phosphorylated JNK immunoreactivity in control and treated samples from SHSY-5Y cells and RCNs (Fig. 5B,D). These results suggest a role for RAGE in AβO-induced ERK signaling pathway defects in human SHSY-5Y cells and RCNs.
Figure 5.
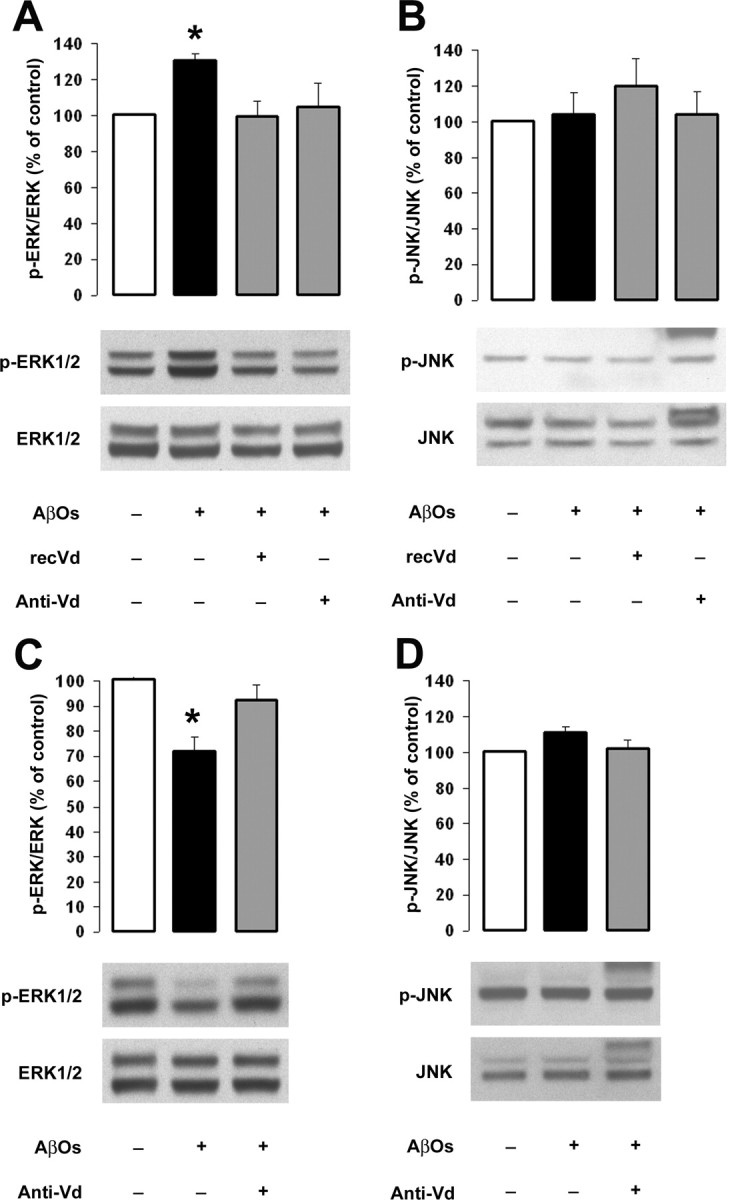
RAGE modulates the phosphorylation of ERK in response to AβOs in SHSY-5Y cells and RCNs. Cell cultures were incubated with AβOs for 8 h in the presence or absence of the indicated treatment. A, B, Equal amounts (50 μg) of total SHSY-5Y cell lysates were immunoblotted for phosphorylation of ERK (A) and JNK (B) using antibodies specific for the phosphorylated forms (1:1000). C, D, Equal amounts (50 μg) of total RCN lysates were immunoblotted for phosphorylation of ERK (C) and JNK (D). The top panels correspond to the quantification of phosphorylated ERK (p-ERK) and JNK (p-JNK) normalized to the total amount of ERK and JNK (*p < 0.01), and the bottom panels show representative immunoblots. Error bars indicate mean ± SEM.
Discussion
Aβ is thought to be the instigator of the neuronal death driving AD. Different conformations of Aβ peptide including oligomers, fibrils, and amorphous aggregates have been found in AD brains (Lorenzo and Yankner, 1994; McLean et al., 1999; Naslund et al., 2000). The aim of the present study was to rigorously investigate the contribution of RAGE in apoptosis induced by distinct well characterized Aβ conformations. First of all, we developed experimental conditions allowing the reproducible generation of particular Aβ peptide assemblies. For this purpose, we used synthetic Aβ(1–40) because Aβ(1–42) is much more prone to aggregation (Yan and Wang, 2006). In accordance with previous publications that routinely required micromolar concentration of Aβ to induce toxicity, we produced cell culture media containing distinct Aβ(1–40) conformations at a concentration of 10 μm based on the initial peptide mass. The presence of AβOs, AβFs, and AβAs was controlled by both Congo red binding assays and TEM. Under our conditions using multiple lots of synthetic peptides, we obtained consistent and reproducible results for the distinctly generated Aβ(1–40) conformations.
To investigate AβO, AβF, and AβA toxicity, we used an established human neuroblastoma cell line (SHSY-5Y) expressing RAGE endogenously (Sajithlal et al., 2002) and a primary culture of neurons (RCNs) as experimental paradigms. In our in vitro system, oligomeric and fibrillar preparations of Aβ produced distinct and reproducible patterns of toxicity that differed from aggregated preparations of the peptide as determined by FACS analysis (Fig. 1). Consistently, the different preparations promoted distinct morphological alterations in SHSY-5Y cells as revealed by light microscopy analysis (Fig. 1). AβOs were found to be the most toxic conformation promoting SHSY-5Y cell death approximately twofold more than AβFs and approximately fourfold more than AβAs. Similarly, we showed that AβO preparation promoted RCN apoptosis, whereas the presence of AβAs did not significantly affect neuronal survival (Fig. 4). Consistent with previous studies (Estus et al., 1997; Yao et al., 2005; Florent et al., 2006; Malaplate-Armand et al., 2006), we found that AβO-, AβF-, and AβA-induced SHSY-5Y cell death involved the activation of apoptotic pathways as revealed by TUNEL and caspase activity assays (Figs. 2–4). With respect to our FACS data, AβOs consistently induced the more dramatic alterations in both events in SHSY-5Y cells and RCNs compared with AβF or AβA effects. In accordance with recent studies using natural and synthetic Aβ(1–42) oligomers (Chong et al., 2006; Townsend et al., 2006, 2007), our distinct AβO(1–40) preparations were found to affect the pattern of ERK activation, indicating that cellular homeostasis is challenged (Fig. 5). In our experimental paradigm, AβOs induced a sustained activation of ERK in SHSY-5Y cells (Fig. 5A). In contrast, ERK phosphorylation was suppressed by AβO treatment in RCNs (Fig. 5C). Conflicting results with the stimulatory or inhibitory effects of Aβ on ERK in culture systems as well as in vivo have previously been reported (Chong et al., 2006; Ma et al., 2007; Townsend et al., 2007). Furthermore, soluble oligomers have been shown to initially stimulate, but later downregulate, ERK in hippocampal slice cultures (Bell et al., 2004), and studies in AD brain and AD mouse models suggest stage-dependent ERK activation followed by loss of active ERK (Dineley et al., 2001; Webster et al., 2006). However, studies investigating the effects of soluble oligomers on either SHSY-5Y neuroblastoma cells (Frasca et al., 2004, 2008) or RCNs (Tong et al., 2004; Florent et al., 2006) observed the same alterations of the ERK signaling pathway as reported in our work. Importantly, both the sustained activation and downregulation of the ERK survival-promoting pathway are associated with susceptibility to cell death (Dineley et al., 2001; Bell et al., 2004; Chong et al., 2006; Florent et al., 2006; Webster et al., 2006; Ma et al., 2007; Townsend et al., 2007). In contrast, AβAs did not affect the ERK phosphorylation state in SHSY-5Y cells at 8 and 24 h (data not shown). Consistently, MAP kinase pathway recruitment has been shown to be dependent on the Aβ conformational state (Bell et al., 2004; Echeverria et al., 2005). Our data are thus in good agreement with these and other reports (Deshpande et al., 2006; St. John, 2007) suggesting that Aβ exhibits specific and distinct toxic effects depending on a particular Aβ aggregation.
We next rigorously characterized the role of RAGE in mediating apoptosis induced by the different conformations of Aβ. Our study revealed that RAGE is involved in AβO- and AβA-induced apoptosis because simultaneous application of a polyclonal anti-RAGE antibody prevented both caspase activation and DNA fragmentation in SHSY-5Y cells (Fig. 2). Similar results were obtained with RAGE site-specific antibodies or sRAGE (Fig. 3). However, anti-RAGE antibody treatments still resulted in significant (∼50–60%) but not absolute prevention of AβO-induced neuronal and cell death. These findings are consistent with previous reports showing that other receptors/mechanisms may also participate in Aβ toxicity (Wogulis et al., 2005; Wright et al., 2007). In addition, anti-Vd-specific antibodies prevented AβO toxicity in RCNs (Fig. 4), supporting the specificity of RAGE contribution in Aβ signaling. Interestingly, in contrast to AβOs and AβAs, we showed that apoptosis induced by mature AβFs was not RAGE dependent. In this regard, previous reports indicated that Aβ toxicity occurs through distinct pathways depending on the peptide conformation (Sponne et al., 2004; Deshpande et al., 2006). Thus, our results suggest that AβO and AβA but not AβF signal, at least in part, through RAGE to induce apoptosis.
In an additional step, we aimed to map more precisely the domain(s) of RAGE involved in Aβ-induced apoptosis. For this purpose, we used site-specific antibodies directed against distinct epitopes within the Vd, the C1d, or the C2d domain of the receptor as well as the recombinant form of the Vd domain (recVd). We found that attenuation of RAGE-mediated AβA-induced apoptosis required the specific antagonism of the C1d of the receptor (Fig. 3). In contrast, the targeting of the Vd with specific antibodies (anti-Vd) or the recVd itself was necessary and sufficient to prevent AβO-induced SHSY-5Y cell death (Fig. 3A,D). Importantly, anti-Vd antibodies also prevented the toxic effects of AβOs in RCNs, providing evidence of the specificity and the relevance of the treatment (Fig. 4E). In accordance with these data, previous reports (Chaney et al., 2005; Mruthinti et al., 2007) demonstrated that soluble Aβ interacts with residues of RAGE, included in the Vd domain. Unexpectedly, recVd treatment did not affect AβO-induced neuronal apoptosis (Fig. 4E). Consistently, Mruthinti et al. (2007) reported that soluble Aβ(1–42) and RAGE(23–54) form a toxic complex for neuronal cells. In accordance with our previous observations, anti-Vd and recVd treatments, which inhibited caspase activation and DNA fragmentation (Fig. 3), were also found to block AβO-induced ERK signaling perturbations in neuroblastoma cells and RCNs (Fig. 5), highlighting the involvement of RAGE as a signal transduction receptor mediating the effects of AβOs. Chronic ERK perturbation might be an early and sustained signaling amplifier of AβO-induced cytotoxicity ultimately leading to the activation of caspase (Chong et al., 2006; Florent et al., 2006). Previous studies have revealed that RAGE-dependent activation of MAP kinases proceeds via an oxidant-sensitive mechanism involving p21ras (Lander et al., 1997), and more recently, the RAGE intracellular domain has been shown to interact directly with ERK (Ishihara et al., 2003). However, detailed mechanisms linking occupancy of RAGE to ERK modulation remain to be elucidated.
Our results suggest for the first time a model in which distinct sites of RAGE are involved in Aβ toxicity with respect to a particular Aβ conformational state. In this regard, previous work indicated that the calcium-binding proteins S100B (Dattilo et al., 2007; Xie et al., 2007), S100A12 (Dattilo et al., 2007; Xie et al., 2007), and S100A6 (Leclerc et al., 2007), which possess high structural homology, also interact with different Ig-like domains of RAGE, the Vd, C1d, and C2d, respectively. Although the in vitro system used in this study does not allow a distinction between the effects of Aβ monomers, dimers, trimers, and higher-order oligomers, it allows us to determine the involvement of RAGE in apoptosis induced by distinct well defined Aβ species. At the current stage of research, we cannot conclude that large insoluble aggregates, fibrils, or soluble oligomers represent the sole molecular pathogen in AD; indeed, various Aβ species may play relevant roles in neurotoxicity (Haass and Selkoe, 2007). Our findings provide a new insight into how multiple Aβ species may contribute to neurodegeneration. Furthermore, in AD pathophysiology, AβOs are present at very early stages and may coexist with AβAs at later stages of the disease. In addition, Yan et al. (1996) provided evidence that levels of RAGE are increased in the AD brain, particularly in neurons associated with aggregated deposits. Because RAGE expression increases and remains elevated as long as ligands are present, RAGE may be important in initiating and perpetuating Aβ neuronal toxicity “amplification loops.” These observations provide an interesting parallel with the Aβ-induced changes in RAGE expression observed recently in rat hippocampus (Minogue et al., 2007).
In summary, the current experiments demonstrate that RAGE can act as a receptor exacerbating critical effects of Aβ on several signaling molecules involved in the apoptotic pathway. Furthermore, these studies establish that RAGE mediates AβO- and AβA-induced apoptosis and suggest a novel mechanism in which the engagement of distinct nonoverlapping regions of the receptor by multiple Aβ species might contribute to neurodegeneration. Although RAGE–ligand interactions support normal cellular functions and homeostasis, our results suggest that the blockage of specific sites of the receptor using antibodies provide strategy to attenuate chronic activation of RAGE.
Footnotes
This work was supported by the National Centre for Competence in Research on Neural Plasticity and Repair and the Transregio-Sonderforschungsbereich Konstanz-Zurich. We thank Prof. R. Nitsch for valuable discussion, Dr. A. Bierhaus and Prof. P. Nawroth for providing RAGE−/− mice, L. Allen for proofreading, T. Ballard for help with electron microscopy, C. Siedler for providing rat cortex, and P. Kleinert for the MALDI analyses.
References
- Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, et al. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004;23:4096–4105. doi: 10.1038/sj.emboj.7600415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimon M, Diez-Perez I, Kogan MJ, Durany N, Giralt E, Sanz F, Fernandez-Busquets X. Fine structure study of Abeta1–42 fibrillogenesis with atomic force microscopy. FASEB J. 2005;19:1344–1346. doi: 10.1096/fj.04-3137fje. [DOI] [PubMed] [Google Scholar]
- Bell KA, O'Riordan KJ, Sweatt JD, Dineley KT. MAPK recruitment by beta-amyloid in organotypic hippocampal slice cultures depends on physical state and exposure time. J Neurochem. 2004;91:349–361. doi: 10.1111/j.1471-4159.2004.02722.x. [DOI] [PubMed] [Google Scholar]
- Chaney MO, Stine WB, Kokjohn TA, Kuo YM, Esh C, Rahman A, Luehrs DC, Schmidt AM, Stern D, Yan SD, Roher AE. RAGE and amyloid beta interactions: atomic force microscopy and molecular modeling. Biochim Biophys Acta. 2005;1741:199–205. doi: 10.1016/j.bbadis.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ. ERK1/2 activation mediates Abeta oligomer-induced neurotoxicity via caspase-3 activation and tau cleavage in rat organotypic hippocampal slice cultures. J Biol Chem. 2006;281:20315–20325. doi: 10.1074/jbc.M601016200. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Dattilo BM, Fritz G, Leclerc E, Kooi CW, Heizmann CW, Chazin WJ. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry. 2007;46:6957–6970. doi: 10.1021/bi7003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26:6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285–298. doi: 10.1016/0197-4580(95)00013-5. discussion 298–304. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Godman GC, Stern D, Schmidt AM. Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:5296–5301. doi: 10.1073/pnas.94.10.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverria V, Ducatenzeiler A, Chen CH, Cuello AC. Endogenous beta-amyloid peptide synthesis modulates cAMP response element-regulated gene expression in PC12 cells. Neuroscience. 2005;135:1193–1202. doi: 10.1016/j.neuroscience.2005.06.057. [DOI] [PubMed] [Google Scholar]
- Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE. Aggregated amyloid-β protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J Neurosci. 1997;17:7736–7745. doi: 10.1523/JNEUROSCI.17-20-07736.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florent S, Malaplate-Armand C, Youssef I, Kriem B, Koziel V, Escanye MC, Fifre A, Sponne I, Leininger-Muller B, Olivier JL, Pillot T, Oster T. Docosahexaenoic acid prevents neuronal apoptosis induced by soluble amyloid-beta oligomers. J Neurochem. 2006;96:385–395. doi: 10.1111/j.1471-4159.2005.03541.x. [DOI] [PubMed] [Google Scholar]
- Frasca G, Chiechio S, Vancheri C, Nicoletti F, Copani A, Angela Sortino M. Beta-amyloid-activated cell cycle in SH-SY5Y neuroblastoma cells: correlation with the MAP kinase pathway. J Mol Neurosci. 2004;22:231–236. doi: 10.1385/jmn:22:3:231. [DOI] [PubMed] [Google Scholar]
- Frasca G, Carbonaro V, Merlo S, Copani A, Sortino MA. Integrins mediate beta-amyloid-induced cell-cycle activation and neuronal death. J Neurosci Res. 2008;86:350–355. doi: 10.1002/jnr.21487. [DOI] [PubMed] [Google Scholar]
- Grace EA, Busciglio J. Aberrant activation of focal adhesion proteins mediates fibrillar amyloid beta-induced neuronal dystrophy. J Neurosci. 2003;23:493–502. doi: 10.1523/JNEUROSCI.23-02-00493.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha C, Ryu J, Park CB. Metal ions differentially influence the aggregation and deposition of Alzheimer's beta-amyloid on a solid template. Biochemistry. 2007;46:6118–6125. doi: 10.1021/bi7000032. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hadding A, Kaltschmidt B, Kaltschmidt C. Overexpression of receptor of advanced glycation end products hypersensitizes cells for amyloid beta peptide-induced cell death. Biochim Biophys Acta. 2004;1691:67–72. doi: 10.1016/j.bbamcr.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275:40096–40105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997a;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997b;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs AM, Senn DB, Yuan M, Shine JP, Yankner BA. Acceleration of amyloid beta-peptide aggregation by physiological concentrations of calcium. J Biol Chem. 2006;281:27916–27923. doi: 10.1074/jbc.M602061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K, Tsutsumi K, Kawane S, Nakajima M, Kasaoka T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003;550:107–113. doi: 10.1016/s0014-5793(03)00846-9. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Jacob RF, Mason RP. Quantifying amyloid beta-peptide (Abeta) aggregation using the Congo red-Abeta (CR-abeta) spectrophotometric assay. Anal Biochem. 1999;266:66–76. doi: 10.1006/abio.1998.2933. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid β oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem. 1997;272:17810–17814. doi: 10.1074/jbc.272.28.17810. [DOI] [PubMed] [Google Scholar]
- Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A. S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem. 2007;282:31317–31331. doi: 10.1074/jbc.M703951200. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losic D, Martin LL, Mechler A, Aguilar MI, Small DH. High resolution scanning tunnelling microscopy of the beta-amyloid protein (Abeta1–40) of Alzheimer's disease suggests a novel mechanism of oligomer assembly. J Struct Biol. 2006;155:104–110. doi: 10.1016/j.jsb.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Ma QL, Harris-White ME, Ubeda OJ, Simmons M, Beech W, Lim GP, Teter B, Frautschy SA, Cole GM. Evidence of Abeta- and transgene-dependent defects in ERK-CREB signaling in Alzheimer's models. J Neurochem. 2007;103:1594–1607. doi: 10.1111/j.1471-4159.2007.04869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaplate-Armand C, Florent-Bechard S, Youssef I, Koziel V, Sponne I, Kriem B, Leininger-Muller B, Olivier JL, Oster T, Pillot T. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol Dis. 2006;23:178–189. doi: 10.1016/j.nbd.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Mastrangelo IA, Ahmed M, Sato T, Liu W, Wang C, Hough P, Smith SO. High-resolution atomic force microscopy of soluble Abeta42 oligomers. J Mol Biol. 2006;358:106–119. doi: 10.1016/j.jmb.2006.01.042. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Minogue AM, Lynch AM, Loane DJ, Herron CE, Lynch MA. Modulation of amyloid-beta-induced and age-associated changes in rat hippocampus by eicosapentaenoic acid. J Neurochem. 2007;103:914–926. doi: 10.1111/j.1471-4159.2007.04848.x. [DOI] [PubMed] [Google Scholar]
- Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. Expression in brain of amyloid precursor protein mutated in the alpha-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 1996;15:1265–1274. [PMC free article] [PubMed] [Google Scholar]
- Monteiro FA, Sousa MM, Cardoso I, do Amaral JB, Guimaraes A, Saraiva MJ. Activation of ERK1/2 MAP kinases in familial amyloidotic polyneuropathy. J Neurochem. 2006;97:151–161. doi: 10.1111/j.1471-4159.2006.03716.x. [DOI] [PubMed] [Google Scholar]
- Moore RA, Hayes SF, Fischer ER, Priola SA. Amyloid formation via supramolecular peptide assemblies. Biochemistry. 2007;46:7079–7087. doi: 10.1021/bi700247y. [DOI] [PubMed] [Google Scholar]
- Mruthinti S, Capito N, Sood A, Buccafusc JJ. Cytotoxicity of Abeta1–42, RAGE23–54, and an Abeta-RAGE complex in PC-12 cells. Curr Alzheimer Res. 2007;4:581–586. doi: 10.2174/156720507783018325. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Ostendorp T, Weibel M, Leclerc E, Kleinert P, Kroneck PM, Heizmann CW, Fritz G. Expression and purification of the soluble isoform of human receptor for advanced glycation end products (sRAGE) from Pichia pastoris. Biochem Biophys Res Commun. 2006;347:4–11. doi: 10.1016/j.bbrc.2006.04.077. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. Aggregation-related toxicity of synthetic beta-amyloid protein in hippocampal cultures. Eur J Pharmacol. 1991;207:367–368. doi: 10.1016/0922-4106(91)90014-9. [DOI] [PubMed] [Google Scholar]
- Sajithlal G, Huttunen H, Rauvala H, Munch G. Receptor for advanced glycation end products plays a more important role in cellular survival than in neurite outgrowth during retinoic acid-induced differentiation of neuroblastoma cells. J Biol Chem. 2002;277:6888–6897. doi: 10.1074/jbc.M107627200. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Toki S, Chowei H, Saito T, Nakano N, Hayashi Y, Takeuchi M, Makita Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer's disease. Brain Res. 2001;888:256–262. doi: 10.1016/s0006-8993(00)03075-4. [DOI] [PubMed] [Google Scholar]
- Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, Esposito C, Hegarty H, Hurley W, Clauss M, Wang F, Pan YC, Tsang TC, Stern D. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. 1992;267:14987–14997. [PubMed] [Google Scholar]
- Shahi P, Sharma R, Sanger S, Kumar I, Jolly RS. Formation of amyloid fibrils via longitudinal growth of oligomers. Biochemistry. 2007;46:7365–7373. doi: 10.1021/bi7001136. [DOI] [PubMed] [Google Scholar]
- Sponne I, Fifre A, Koziel V, Oster T, Olivier JL, Pillot T. Membrane cholesterol interferes with neuronal apoptosis induced by soluble oligomers but not fibrils of amyloid-beta peptide. FASEB J. 2004;18:836–838. doi: 10.1096/fj.03-0372fje. [DOI] [PubMed] [Google Scholar]
- St John PA. Differential binding and activation of caspase-3 in cultured hippocampal neurons by assembly forms of Abeta(1–42) J Neurosci Res. 2007;85:1205–1214. doi: 10.1002/jnr.21251. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- Tong L, Balazs R, Thornton PL, Cotman CW. β-Amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol (Lond) 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Mehta T, Selkoe DJ. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282:33305–33312. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Webster B, Hansen L, Adame A, Crews L, Torrance M, Thal L, Masliah E. Astroglial activation of extracellular-regulated kinase in early stages of Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:142–151. doi: 10.1097/01.jnen.0000199599.63204.6f. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wogulis M, Wright S, Cunningham D, Chilcote T, Powell K, Rydel RE. Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J Neurosci. 2005;25:1071–1080. doi: 10.1523/JNEUROSCI.2381-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S, Malinin NL, Powell KA, Yednock T, Rydel RE, Griswold-Prenner I. Alpha2beta1 and alphaVbeta1 integrin signaling pathways mediate amyloid-beta-induced neurotoxicity. Neurobiol Aging. 2007;28:226–237. doi: 10.1016/j.neurobiolaging.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Xie J, Burz DS, He W, Bronstein IB, Lednev I, Shekhtman A. Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J Biol Chem. 2007;282:4218–4231. doi: 10.1074/jbc.M608888200. [DOI] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA. Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J Clin Invest. 1997;100:2333–2340. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- Yan Y, Wang C. Abeta42 is more rigid than Abeta40 at the C terminus: implications for Abeta aggregation and toxicity. J Mol Biol. 2006;364:853–862. doi: 10.1016/j.jmb.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Yan Y, Wang C. Abeta40 protects non-toxic Abeta42 monomer from aggregation. J Mol Biol. 2007;369:909–916. doi: 10.1016/j.jmb.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Yao M, Nguyen TV, Pike CJ. β-Amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J Neurosci. 2005;25:1149–1158. doi: 10.1523/JNEUROSCI.4736-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]