

Abstract
The flotillins/reggie proteins are associated with noncaveolar membrane microdomains and have been implicated in the regulation of a clathrin- and caveolin-independent endocytosis pathway. Endocytosis is required for the amyloidogenic processing of the amyloid precursor protein (APP) and thus to initiate the release of the neurotoxic β-amyloid peptide (Aβ), the major component of extracellular plaques found in the brains of Alzheimer's disease patients. Here, we report that small interference RNA-mediated downregulation of flotillin-2 impairs the endocytosis of APP, in both neuroblastoma cells and primary cultures of hippocampal neurons, and reduces the production of Aβ. Similar to tetanus neurotoxin endocytosis, but unlike the internalization of transferrin, clathrin-dependent endocytosis of APP requires cholesterol and adaptor protein-2 but is independent of epsin1 function. Moreover, on a nanoscale resolution using stimulated emission depletion microscopy and by Förster resonance energy transfer with fluorescence lifetime imaging microscopy, we provide evidence that flotillin-2 promotes the clustering of APP at the cell surface. We show that the interaction of flotillin-2 with APP is dependent on cholesterol and that clustering of APP enhances its endocytosis rate. Together, our data suggest that cholesterol/flotillin-dependent clustering of APP may stimulate the internalization into a specialized clathrin-dependent endocytosis pathway to promote amyloidogenic processing.
Keywords: neurons, endocytosis, amyloid β, Alzheimer's disease, flotillin, cholesterol
Introduction
Flotillin-1/reggie-2 and flotillin-2/reggie-1 are two highly conserved proteins associated with lipid rafts that have been implicated in a wide range of cellular processes such as axonal regeneration, insulin signaling, and recently also in endocytosis (Langhorst et al., 2005). Flotillins are palmitoylated and myristoylated proteins that are anchored to the cytoplasmic membrane bilayer, in which they form multimeric scaffolds (Morrow et al., 2002; Neumann-Giesen et al., 2004; Frick et al., 2007; Solis et al., 2007). Expression of flotillin is particular high in cells that lack caveolin, e.g., lymphocytes and neurons (Lang et al., 1998; Rajendran et al., 2003), and an accumulation of flotillin was found in the endosomal system of neurons from amyloid precursor protein × presenilin 1 (APP×PS1) transgenic mice and patients with Alzheimer's disease (AD) (Girardot et al., 2003; Langui et al., 2004). An additional link between flotillin and AD is the association of flotillin with cholesterol-rich detergent resistant membranes, which have been implicated in the generation of the β-amyloid peptide (Aβ) (Kojro et al., 2001; Riddell et al., 2001; Cordy et al., 2003; Ehehalt et al., 2003; Puglielli et al., 2003; Vetrivel et al., 2004; Kalvodova et al., 2005).
Aβ is liberated from the APP by the concerted action of two proteases, termed β- and γ-secretase (Kao et al., 2004; Small and Gandy, 2006; Haass and Selkoe, 2007). Endocytosis of APP is required to initiate cleavage by the β-secretase BACE (β-secretase activity of the β-site APP-cleaving enzyme) (Small and Gandy, 2006; Vetrivel and Thinakaran, 2006). The “YENPTY” motif located near the C terminus of APP mediates its internalization and also serves as docking site for several APP adaptor proteins with phosphotyrosine-binding domains, such as X11, Fe65, Shc, and JIP (King and Scott Turner, 2004). Interestingly, some of these adaptor proteins link APP to apolipoprotein E (apoE) receptors of the low-density lipoprotein receptor (LDLR) family (Zerbinatti and Bu, 2005). By this mechanism, the uptake of cholesterol-rich apoE particles via LDLR also modulates the endocytosis and the amyloidogenic processing of APP (Kounnas et al., 1995; Pietrzik et al., 2002; Ye et al., 2005), and several studies have demonstrated that membrane cholesterol levels influence the generation of Aβ (Simons et al., 1998; Frears et al., 1999; Fassbender et al., 2001; Abad-Rodriguez et al., 2004).
Here, we further tighten the relationship of cholesterol and APP by demonstrating a role of flotillin and cholesterol in endocytosis and β-cleavage of APP in neurons.
Materials and Methods
Transgenic animals, cell culture, transfection, and viral infection.
Heterozygous B6C3 swAPP/PS1dE9 mice harboring the Swedish mutation and human PS1 encoding the exon 9 deletion mutation were obtained from The Jackson Laboratory (Bar Harbor, ME).
Mouse neuroblastoma N2a cells were cultured as described previously (Rajendran et al., 2006). At 24 h after seeding of N2a cells on glass coverslips or into 3.5 cm dishes, cells were transfected with Fugene (Roche, Basel, Switzerland) according to the protocol of the manufacturer.
A stable N2a cell line expressing short hairpin RNA (shRNA) directed against flotillin-2 was created with the forward target sequence 5′GATCTCCGATCCTCAGCTTCACCATCAATTCAAGAGATTGATGGTGAAGCTGAGGATCTTTTTGGAAA3′ and the reverse target sequence 5′AGCTTTTCCAAAAAGATCCTCAGCTTCACCATCAATCTCTTGAATTGATGGTGAAGCTGAGGATCGGA3′. The annealed oligos were cloned into pSUPER, and retroviruses were produced in a Phoenix gag-pol retroviral packaging cell line and used to infect N2a cells. A control cell line was produced expressing the empty vector. N2a cells were selected for growth in hygromycin.
RNA oligonucleotides were delivered into N2a cells using Oligofectamine (Invitrogen, Karlsruhe, Germany) according to the instructions of the manufacturer. In experiments in which a combination of small interfering RNA (siRNA) and plasmid delivery was necessary, plasmids were transfected with Fugene 36 h after siRNA delivery. Endocytosis was assessed 16 h later. The following siRNAs were used: GUUCAUGGCAGACACCAAGTT against flotillin-1 and GAUCCUCAGCUUCACCAUCAATT against flotillin-2 (Ambion, Austin, USA), RNA duplex oligonucleotides with overhanging dTs directed to conserved regions in murine adaptor protein-2 (AP-2) μ2b GUGGAUGCCUUUCGCGUCAdTdT and UCAAGCGCAUGGCAGGCAUdTdT (Qiagen, Hilden, Germany) were used in combination. Control siRNA was obtained from Ambion (control siRNA #1) or from Qiagen (MBP siRNA; N2a cells do not express MBP).
Primary hippocampal neurons derived from mouse embryos were cultured on poly-lysine-coated glass coverslips or plastic dishes as described previously (Simons et al., 1998). Neurons were kept under serum-free conditions in MEM with B27 supplement for 5 d before a 1 h infection with recombinant Semliki Forest virus (SFV) encoding human APP695-myc with the double-mutation K670N and M671L (Swedish mutation) or with recombinant adenovirus encoding cyan fluorescent protein (CFP)–APP bearing the Swedish double mutation K670N plus M671L. After medium exchange, the cells were incubated for 8 h in the case of SFV and 16 h in the case of adenovirus infection before additional experimental processing.
Flotillin-2 siRNA was delivered into hippocampal neurons by nucleofection with Amaxa (Cologne, Germany) basic neuron kit according to the protocol of the manufacturer shortly before plating. After 4 d in vitro, neurons were infected with adenovirus, and endocytosis experiments were performed 12–16 h later.
Cholesterol depletion.
For cholesterol depletion, cells were washed three times with HBSS, followed by a 2 h incubation with 1 mm methyl-β-cyclodextrin (mβCD) in HBSS at 37°C. For the subsequent experiments, we used medium that was supplemented with lipoprotein-free serum.
Immunoprecipitation and quantification.
For coimmunoprecipitation of APP and flotillin-2, N2a cells were grown in 3.5 cm dishes and cotransfected, and cell lysates were prepared in lysis buffer consisting of 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 150 mm NaCl, 5 mm EDTA, and 50 mm Tris, pH8, supplemented with a protease inhibitor mixture (Roche). Immunoprecipitates were recovered on protein A/G Plus agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). Samples were subjected to Western blot analysis; bands were quantified using the NIH ImageJ software. Metabolic labeling experiments, immunoprecipitation of medium and cell lysates, Tris-tricine or polyacrylamide electrophoresis, and quantification of the bands were done as described previously (Ehehalt et al., 2003).
Plasmids, antibodies, and reagents.
The following plasmids were used: mutant and wild-type (wt) cDNAs of green fluorescent protein (GFP)–dynaminII (Sandra Schmid, Scripps Research Institute, La Jolla, CA); p-cytomegalovirus (pCMV)–myc–AP180 C terminus and pCMV–myc–epsin (R63L+H73L) (Harvey McMahon, Medical Research Council, Laboratory of Molecular Biology, Cambridge, UK); pCMV–AP180–GFP; pCMV–epsin1–GFP (Peter McPherson, Montreal Neurological Institute, McGill University, Montreal, Quebec, Canada); pCMV–wtAPP–myc (Tobias Hartmann, Neurodegeneration and Neurobiology, University of Saarland, Saarland, Germany); pShuttleCMV–swAPP, pShuttleCMV–CFP–swAPP, pShuttleCMV–swAPP–yellow fluorescent protein (YFP), pShuttleCMV–swAPP, p–enhanced YFP (EYFP)–N1 (Clontech, Mountain View, CA), flotillin-1–GFP (Ben Nichols, Medical Research Council, Laboratory of Molecular Biology, Cambridge, UK), flotillin-2–GFP, flotillin-2–red fluorescent protein (RFP) (the GFP was removed using Age1 and BsrG1, and the mRFP generated by PCR amplification was cloned back into the original flotillin-2–GFP vector); pR4– proteolipid protein (PLP)–myc; and vesicular stomatitis virus protein (VSVG-A)–GFP–LTLM9 (Patrick Keller, Max-Planck-Institute for Molecular Cell Biology and Genetics, Dresden, Germany).
Antibodies were obtained from the following sources: mouse monoclonal antibodies against AP-2, flotillin-1, and flotillin-2, monoclonal rat anti-LAMP-1 (lysosome-associated membrane protein-1) (BD Biosciences, Heidelberg, Germany), mouse monoclonal antibody against γIII-tubulin (Promega, Mannheim, Germany), rabbit anti-calnexin (Stressgen Bioreagents, Victoria, British Columbia, Canada), mouse monoclonal anti-myc (Cell Signaling Technology, Denver, CO), rabbit anti-GFP (Abcam, Cambridge, UK), monoclonal antibody 6E10, directed against the 3–8 region of the Aβ peptide (Covance Research Products, Princeton, NJ), polyclonal antibody IP60 recognizes the C terminus of APP, rabbit antibody 70JE is raised against amino acids 1–11 of the Aβ peptide, polyclonal antibody ANJJ is directed against the C terminus of soluble swAPPβ, and B63-3 monoclonal antibody recognizes the C terminus of APP (kindly provided by Bart de Strooper, Department of Human Genetics, Katholieke Universiteit Leuven, Leuven, Belgium). O10 (monoclonal mouse IgM) recognizes PLP, and anti-VSVG antibody was provided by Patrick Keller.
The fluorophore-conjugated secondary antibodies Alexa Fluor 488, cyanine 3 (Cy3)–IgG, and Cy5 were purchased from Dianova (Hamburg, Germany), Alexa Fluor 488-f(ab)-fragment was from Invitrogen, and Cy3-f(ab)-fragment was from Jackson ImmunoResearch (Newmarket, UK).
The following reagents were used: rhodamine-conjugated transferrin, rhodamine–dextran (Mr of 10 kDa) (Invitrogen), DiI–LDL (generous gift from Jobst Landgrebe, Center for Biochemistry and Molecular Cell Biology, University of Göttingen, Göttingen, Germany), mβCD, and cycloheximide (Sigma, Schnellendorf, Germany).
Microscopy and analysis.
Fluorescence images were acquired on a Leica (Mannheim, Germany) DMRXA microscope or a Zeiss (Jena, Germany) LSM 510 confocal microscope with a 63× oil plan-apochromat objective (numerical aperture 1.4; Zeiss). Image processing and analysis was performed using Meta Imaging Series 6.1 software (Universal Imaging, Downingtown, PA). Quantification of fluorescence intensities was performed as reported previously (Trajkovic et al., 2006).
The stimulated emission depletion (STED) microscope has been described previously (Dyba et al., 2003). Fluorescence excitation was performed with a pulsed laser diode emitting 100 ps pulses at 470 nm (Picoquant, Berlin, Germany), whereas STED used 280 ps pulses with a repetition rate of 250 kHz at 603 nm, generated by a optic parametric amplifier being pumped through a regenerative amplified titanium:sapphire laser (Coherent, Santa Clara, CA). The conversion of the STED beam into a doughnut mode was accomplished by means of a spatial light modulator (Hamamatsu, Hamamatsu City, Japan) delivering a (0–2π) helical phase ramp. The excitation diode was electronically triggered by the RegA pulses; the appropriate delay between the pulses was adjusted with an electronic delay generator. The excitation and the STED beams were coupled into an oil immersion lens (HCX PL APO, 100×; Leica) with a 1.4 numerical aperture, by means of dichroic mirrors. The average power of the excitation beam and the STED beam in the sample was 50 nW and 240 μW, respectively. The fluorescence was collected by the same lens and directed into a counting avalanche photodiode (PerkinElmer, Vaudreuil-Dorian, Canada). The photodiode featured an opening diameter of ∼70% of the backprojected Airy disk at the detector plane. The image was obtained by scanning the sample with a piezo stage featuring a positioning accuracy <10 nm. For analysis of cluster sizes in STED images, the diameters of individual clusters were measured using MetaMorph software.
For fluorescence lifetime measurements, flotillin-2 or control siRNA-treated cells were transfected with swAPP cDNA. Anti-APP antibody 6E10 was incubated at a final concentration of 0.1 mg/ml with an eightfold molar excess of anti-mouse f(ab) in imaging buffer (PBS, 10 mm HEPES, pH 7.2, and 0.1% BSA) for 90 min at 4°C; f(ab) had been either coupled to Alexa 488 [digested from Alexa 488 f(ab)2 (Invitrogen) using immobilized papain (Pierce, Rockford, IL)] or to Cy3 (Jackson ImmunoResearch) or was unmodified (Jackson ImmunoResearch). After incubation, 6E10- f(ab)-Alexa 488 was mixed at a ratio of 1:3 with either 6E10–f(ab) or 6E10–f(ab)–Cy3; this corresponds to a ∼1:3 molar ratio of Alexa 488 to Cy3. At 24 h after swAPP transfection, cells were cooled to 4°C and incubated for 20 min with the freshly prepared antibody mixture at a final 6E10 concentration of 0.02 mg/ml, washed thoroughly, mounted in imaging buffer, and imaged at 20°C on a frequency-domain fluorescence lifetime imaging microscope (Esposito et al., 2005) using an argon laser (Innova 300; Coherent) at 488 nm and 250 mW. The filter cube contained a long-pass 495 dichroic and a band-pass 515/30 emission filter. The pixel lifetimes in all measured cells were globally fitted to correct for background fluorescence and absolute intensity effects (M. Gralle and F. S. Wouters, unpublished routines), and the distribution of true lifetimes was calculated. Förster resonance energy transfer (FRET) efficiencies in each FRET condition were calculated relative to the mean true lifetime of the corresponding donor only sample.
Endocytosis assays and quantification.
Cells expressing the appropriate CFP–APP or APP–YFP constructs (delivered by either transfection or viral infection) were washed with cold HBSS, cooled down, and labeled for 20–30 min at 4°C with whole serum rabbit anti-GFP antibody (1:100) in HBSS. Unbound antibody was removed by three washes with HBSS before placing the cells in prewarmed (37°C) normal growth medium (supplemented with lipoprotein-free serum in the case of previous cholesterol depletion). After internalization of bound label for various time intervals, medium was washed off with HBSS, and surface retained anti-GFP antibody was labeled at 4°C for 30–40 min with Cy5 anti-rabbit antibody (7.5 ng/μl) in HBSS, followed by extensive washing with cold HBSS and fixation. Cells were then permeabilized, and anti-GFP antibody was immunostained with Cy3 anti-rabbit antibody. AlexaFluor 555–transferrin endocytosis was performed as a continuous uptake.
For quantification, confocal images were taken with identical acquisition parameters. To quantify APP endocytosis, fluorescence intensities of cell-surface retained APP (Cy5 signal originating from the cell surface) and internalized APP (Cy3 signal from the cell interior) were measured with MetaMorph software. The region, corresponding to the cell surface, was identified by the Cy5 signal, and the cell interior was defined as the region within. The ratio of cell interior-derived Cy3/surface-derived Cy5 fluorescence intensity was calculated as a measure for endocytosis.
For clustering experiments, swAPP-expressing cells were labeled with monoclonal antibody 6E10 for 30 min at 4°C, washed three times, and incubated with bivalent IgG Cy3–IgG anti-mouse antibody, with monovalent Cy3–f(ab) anti-mouse fragment, or without secondary antibody for an additional 30 min at 4°C. After extensive washing, cells were shifted for various time intervals to 37°C to allow endocytosis to occur. Cells were then fixed and stained with wheat germ agglutinin (WGA), and confocal images were taken. The cell surface outline was identified by the WGA staining. The internalization of APP was quantified with MetaMorph software as the ratio of fluorescence intensity originating from the cell interior versus intensity of the total cell. Clustering experiments for VSVG or PLP transfected cells were performed according to the same protocol with anti-VSVG or anti-PLP antibodies.
Biotinylation and internalization of cell surface APP.
N2a cells were grown on 5 cm dishes and transfected with myc–APP 12 h before surface biotinylation. Cholesterol depletion or siRNA-mediated knockdown of flotillin-2 was done as described above. Cells were cooled to 4°C, washed with PBS, and labeled with 0.125 mg/ml reducible sulfo-NHS-SS-Biotin (Pierce) in PBS for 20 min at 4°C. After washing with PBS and quenching of unreacted biotin with 50 mm glycine, cells were either subjected to 15 min endocytosis by a 37°C incubation in prewarmed medium (supplemented with delipidated medium in cholesterol depletion experiments) or left at 4°C to determine the amount of biotinylated surface APP. In the case of endocytosis, remaining cell-surface biotin was cleaved with 100 mm DTT, and free DTT was quenched with 5 mg/ml Iodacetamide in PBS. Cells were lysed in PBS, 1% NP-40, 0.1% SDS, and a protease inhibitor mixture. Five percent of total lysates were used to determine transfection levels by Western blotting. Biotinylated surface or biotinylated endocytosed proteins were precipitated with neutravidin beads, and biotinylated APP was detected by Western blotting and normalized against the amount of APP in the respective total cell lysates. Blots were scanned, and the ratios of endocytosed versus surface APP were determined.
Results
Flotillin influences APP processing
To study the role of flotillin in APP processing (Fig. 1A), we used the neuronal cell line mouse neuroblastoma N2a, because these cells lack caveolae but abundantly express flotillin. By siRNA-mediated knockdown, the expression of flotillin-1 and -2 was reduced by ∼70% (Fig. 1B). Flotillin knockdown cells were subsequently infected with adenovirus to express the Swedish mutant of APP (swAPP), which is dominantly β-cleaved, resulting in several fold higher production of Aβ compared with wild-type APP (Citron et al., 1992). After 40 min of metabolic labeling and a 2 h chase, cell lysates and medium were subjected to immunoprecipitation with antibodies against the various APP fragments (Fig. 1A) (Rajendran et al., 2006). Immunoprecipitation from conditioned medium revealed that the generation of both Aβ and the soluble β-cleaved ectodomain (sAPPβ) were reduced by flotillin-2 knockdown (37 and 59% reduction, respectively) (Fig. 1C,D). Analysis of the remaining COOH-terminal fragments of APP (CTFs) in the cell lysate shows that both α- and β-CTFs were diminished (40 and 29% reduction) (Fig. 1C,D). In contrast, knockdown of flotillin-1 levels did not interfere with the processing of APP.
Figure 1.

Flotillin-2 regulates amyloidogenic processing of APP. A, Schematic representation of human APP, membrane topology, its cleavage products, and epitopes of the antibodies used. B, Western blot analysis of N2a cell lysates transfected with flotillin-1 (left) or flotillin-2 (right) specific siRNA. Transferrin-receptor (Tf-Rec) levels (top) are used as a loading control; the bottom shows the same lysates, probed with either a flotillin-1 (left) or flotillin-2 (right) specific antibody. C, Metabolic labeling of mock, flotillin-1, or flotillin-2 siRNA-transfected N2a cells. After adenovirus-mediated delivery of swAPP, cells were metabolically labeled and chased for 2 h. Immunoprecipitates from cell lysates and medium were performed with the antibodies IP60, 70JE, and ANJJ to detect swAPP, CTFs, Aβ, and soluble swAPPβ (s-swAPPβ), respectively. D, Full-length APP and cleavage products were quantified; values of mock transfected controls were normalized to 1, and all values are given as the mean + SEM from four experiments.
Whereas RNAi-mediated knockdown of flotillin-1 only affected the expression of flotillin-1, knockdown of flotillin-2 decreased the protein levels of both flotillin-1 and flotillin-2 (supplemental Fig. 1A–C, available at www.jneurosci.org as supplemental material). This is consistent with a recent report that showed proteasomal degradation of flotillin-1 after silencing of flotillin-2 (Solis et al., 2007). We therefore used flotillin-2 RNA interference (RNAi) for all subsequent experiments to obtain a knockdown of both flotillin isoforms.
Flotillin modulates the endocytosis of APP
Previous studies have shown that APP endocytosis is required for β-cleavage to occur. Although a significant fraction of the Swedish mutant of APP is already processed by β-secretase during transport through the biosynthetic pathway (Haass et al., 1995), inhibition of endocytosis in N2a cells markedly reduces β-cleavage of swAPP in N2a cells (Perez et al., 1996; Ehehalt et al., 2003; Rajendran et al., 2006). Given a recently proposed flotillin-dependent endocytosis pathway (Glebov et al., 2006) and the reduction of amyloidogenic APP processing after flotillin knockdown detected here, we investigated whether flotillin regulated the endocytosis of APP. We created stable N2a cell lines expressing shRNA directed against flotillin-2 and control cells expressing the backbone vector without the flotillin-2 shRNA. The successful depletion of flotillin mRNA (∼90%) and protein levels (∼70%) was confirmed by real-time reverse transcription-PCR and Western blotting (data not shown). To analyze whether the reduction of flotillin-2 affected the endocytosis of APP, we transfected a CFP–swAPP fusion protein (Ehehalt et al., 2003) into the stably expressing flotillin-2 shRNA and control cells and performed a quantitative immunofluorescence-based APP endocytosis assay. In this assay, cell surface APP was labeled by incubating cells on ice with an antibody against the N-terminal CFP. After washing, cells were incubated at 37°C for different time spans (5, 20, and 45 min) to allow internalization of antibody-bound APP. Cells were subsequently incubated on ice with a Cy5-conjugated secondary antibody to visualize cell-surface retained APP. After fixing and permeabilization, internalized APP was visualized using a secondary Cy3-conjugated antibody directed against the rabbit anti-CFP primary antibody. During the time course of internalization, APP gradually disappeared from the cell surface and accumulated within the cell (Fig. 2A). No antibody uptake was observed when untransfected cells were subjected to this endocytosis assay. In flotillin-2 knockdown cells, a significant reduction of swAPP internalization was observed in this assay compared with control cells (44% reduction) (Fig. 2B), whereas overexpression of flotillin-2 stimulated the endocytosis of APP (Fig. 2D). Importantly and consistent with a previous report, flotillin-2 knockdown did not interfere with the uptake of rhodamine–transferrin, ruling out unspecific off-target effects on clathrin-dependent endocytosis (Fig. 2B) (Glebov et al., 2006). We also observed a reduction of the endocytosis of wild-type APP after depleting flotillin-2 with siRNA oligonucleotides (Fig. 2B). In contrast to the effect observed for flotillin-2 knockdown on APP internalization, APP endocytosis was not impaired by knockdown of flotillin-1 (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material), which is consistent with our finding, that only flotillin-2 affects the processing of APP.
Figure 2.

Flotillin-2 knockdown inhibits APP endocytosis in N2a cells and primary hippocampal neurons. A, CFP–swAPP-transfected N2a cells were labeled at 4°C with anti-GFP antibody, washed, and incubated at 37°C for 5, 20, and 45 min to allow internalization of antibodies bound to CFP–swAPP. To visualize cell-surface retained APP, cells were labeled with Cy5-conjugated secondary antibody at 4°C (pseudocolored in blue). After fixing, cells were permeabilized and stained with a Cy3-conjugated secondary antibody to resolve internalized APP (red). The cell surface was identified on confocal sections by the Cy5 label, and APP endocytosis was measured by calculating the intensity ratio of (cell interior Cy3)/(surface Cy5) labeled CFP–swAPP. The graph shows the increase in internalized APP over the time course of 45 min, the corresponding confocal images display the increasing amount of internalized APP (Cy3), whereas Cy5-labeled surface APP decreases over time (n > 95 cells, values represent means + SEM; *p < 0.05; ***p < 0.0005, t test). Scale bar, 10 μm. B, Antibody-uptake experiment of transiently transfected CFP–swAPP in flotillin-2 knockdown (gray bars) and control cells (white bars). Endocytosis of APP (40 min, 37°C) was determined as ratio of Cy3- to Cy5-labeled CFP–swAPP. swAPP endocytosis is reduced by 44% after flotillin-2 downregulation, whereas uptake of rhodamine–transferrin is not significantly altered. Similar results were obtained for wtAPP, in which the uptake was reduced by 60% after flotillin-2 knockdown. Values are shown as means + SEM [n = 990 cells (swAPP endocytosis), n = 1251 cells (transferrin uptake), n = 154 cells (wtAPP endocytosis); **p < 0.005; ***p < 0.0005, t test]. The confocal images depict the reduced amount of internalized swAPP after flotillin-2 knockdown (left) compared with control cells (right). Scale bar, 10 μm. C, Primary hippocampal neurons were transfected with flotillin-2-specific siRNA or a control siRNA before plating. After 4 d, cells were infected with adenovirus to express CFP–swAPP, and APP uptake was measured. Representative confocal images are shown. Scale bar, 10 μm. D, N2a cells were transiently transfected with pEYFP–N1 vector or increasing amounts of flotillin-2–GFP DNA (80 and 180 ng DNA/cm2), and endocytosis of cotransfected CFP–swAPP was measured as described in A–C. Values of internalized/surface retained APP were normalized to YFP-expressing controls and given as means + SEM, n = 208 cells, ***p < 0.0005, t test.
Similar results were obtained in primary cultures of hippocampal neurons, which, like N2a cells, do not express detectable levels of caveolin but produce large amounts of flotillin (Cameron et al., 1997). Flotillin-2-specific siRNA was delivered into hippocampal neurons by nucleofection, which resulted in an ∼70% reduction of both flotillin-1 and flotillin-2 protein levels (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material). Neurons were infected with recombinant adenovirus to express CFP–swAPP 4–5 d after nucleofection. Antibody-uptake experiments revealed that flotillin-2 knockdown reduces the internalization of APP by ∼68% compared with control siRNA nucleofected neurons (n = 154 cells, p < 0.0005, t test) (Fig. 2C). Together, our results show that flotillin-2 modulates β-secretase-mediated processing of APP by regulating its endocytosis.
Our results are surprising because flotillin-1 has been implicated previously in the regulation of a clathrin- and caveolin-independent pathway (Glebov et al., 2006), although it is well established that APP is internalized in a clathrin-dependent manner (Koo and Squazzo, 1994; Koo et al., 1996; Perez et al., 1999). Because the endocytosis of APP has mainly been studied in cells of non-neuronal origin, we speculated that APP may use an alternative pathway in neuronal cells. To test whether APP endocytosis is clathrin dependent in N2a cells, we performed internalization assays in cells, which were either depleted of AP-2 by siRNA or expressed dominant-negative mutants of AP180 or dynamin to interfere with the clathrin-dependent endocytosis machinery (Fig. 3). RNA interference with AP-2 resulted in an ∼60% downregulation of the protein (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material), which reduced the endocytosis of both APP and rhodamine–transferrin (Fig. 3). A similar inhibition of APP endocytosis and rhodamine–transferrin uptake was achieved by expression of dominant-negative mutants of AP180 and dynamin, indicating that APP requires clathrin function for its endocytosis in N2a cells (AP-2 RNAi: 99% reduction of APP endocytosis, 71% reduction of transferrin endocytosis; AP180 C terminus: 69% reduction of APP endocytosis, 79% reduction of transferrin uptake; dynaminK44a: 75% reduction of APP endocytosis, 62% reduction of transferrin uptake) (Fig. 3). However, when we expressed a dominant-negative form of the clathrin accessory protein epsin1, which is unable to bind to phosphatidylinositol-4,5-bisphosphate (Ford et al., 2002), we observed an interesting difference between APP and transferrin endocytosis. Although mutant epsin1 efficiently inhibited rhodamine–transferrin uptake as reported previously (Ford et al., 2002; Deinhardt et al., 2006), it did not show any effect on the endocytosis of APP (dominant-negative epsin1: 9% increase in APP endocytosis, 55% reduction of transferrin uptake) (Fig. 3). These results suggest that APP may be internalized by a specialized clathrin-dependent mechanism using a specific subset of accessory proteins. Our findings closely resemble an endocytosis pathway that has been described for the internalization of tetanus toxin into motor neurons (Deinhardt et al., 2006). The neurospecific tetanus toxin binds to polysialogangliosides within cholesterol-rich lipid microdomains before internalization by a clathrin-dependent pathway that requires AP-2 but is independent of epsin1 (Herreros et al., 2001; Deinhardt et al., 2006). APP can bind to gangliosides via its Aβ region (Yanagisawa et al., 1995). This and the fact that cholesterol depletion interferes with β-secretase processing of APP (Simons et al., 1998) led us to speculate whether cholesterol might play a role also in the endocytosis of APP. The conditions for cholesterol depletion were established so that endocytosis of transferrin was not affected. When N2a cells were pretreated for 2 h with 1 mm mβCD to extract plasma membrane cholesterol, we did not observe a reduction of rhodamine–transferrin or DiI–LDL uptake (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). However, the internalization of APP was significantly impaired (57% reduction, n = 1030 cells, p < 0.0005, t test), which was accompanied by reduced β-cleavage of APP (52% reduction, n = 228 cells, p < 0.0005, t test). Similar results were obtained in primary cultures of hippocampal neurons (90% reduction of APP endocytosis, 35% reduction of intracellular sAPPβ).
Figure 3.

APP endocytosis occurs in a distinct clathrin pathway independent of epsin1 and is regulated by cholesterol. N2a cells were cotransfected with CFP–swAPP and siRNA against AP-2 or control siRNA or plasmids encoding for the dominant-negative (dn) mutant myc–AP180–C terminus, the wild-type control AP180–GFP, dominant-negative dynaminK44A–GFP, wild-type dynamin–GFP, dominant-negative myc–epsin1, or wild-type epsin1–GFP. Expression time of all constructs was below 12 h, and cells did not display any signs of toxicity. APP endocytosis was determined by an antibody uptake assay with anti-GFP antibody; clathrin-dependent endocytosis was assessed by transferrin–rhodamine uptake. Internalization was quantified by analysis of confocal images (Cy3/Cy5 ratio for APP, rhodamine intensity for transferrin). Results are shown as means + SEM (n >100 cells for each condition, *p < 0.05; **p < 0.005; ***p < 0.0005, t test). Note that dominant-negative epsin1-expressing cells show a significant reduction of transferrin (Tf) uptake but no impairment of APP endocytosis.
When we compared the APP endocytosis rates of cholesterol-depleted or untreated cells using a biotinylation assay, we found a 62% reduction in APP endocytosis, confirming the results of the immunofluorescence-based assay (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). RNAi-mediated flotillin-2 knockdown led to a 87% reduction of APP in the biotin endocytosis assay compared with control siRNA-treated cells (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material).
Together, these results suggest that APP internalization occurs via a specialized clathrin-dependent pathway that is modulated by cholesterol and flotillin-2.
Flotillin modulates clustering of APP
Consistent with previous studies (Stuermer et al., 2001; Neumann-Giesen et al., 2007), we found flotillin in the endosomal system and at the plasma membrane of N2a cells and hippocampal neurons (data not shown), in which it colocalized partially with clusters of surface labeled APP (Fig. 4A). This observation together with the putative function of flotillin as a domain-organizing scaffold that entraps specific lipids and proteins in membrane microdomains prompted us to investigate whether flotillin could organize APP in larger clusters and whether such clustering could play a role in APP endocytosis.
Figure 4.
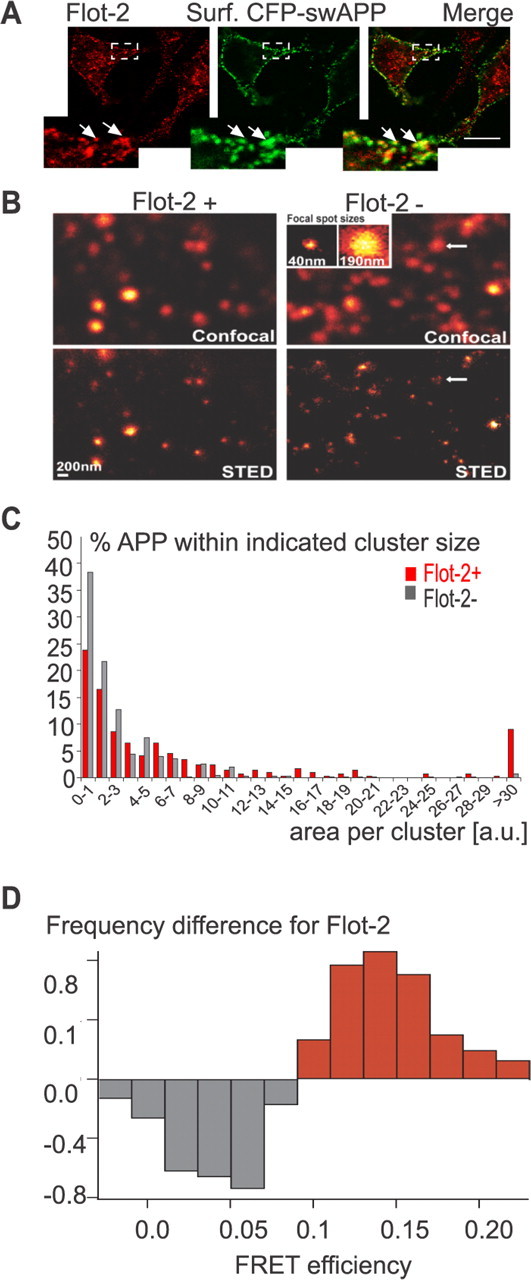
Flotillin-2 increases the size of cell-surface APP clusters on a nanoscale resolution. A, Partial colocalization (arrows) of surface APP and flotillin-2. N2a cells were transfected with CFP–swAPP, and surface APP was stained at 4°C with an antibody directed against CFP. After fixation, anti-CFP labeling and endogenous flotillin-2 were visualized by indirect immunofluorescence (surface APP green, endogenous flotillin red). Scale bar, 10 μm. B, N2a cells either stably expressing a control shRNA (Flot-2 +, left) or shRNA directed against flotillin-2 (Flot-2 −, right). Cells were transfected with CFP–swAPP, and surface APP was labeled with anti-GFP antibody for 30 min on ice before fixation. Cells were visualized by confocal microscopy (top) and by T-Rex STED microscopy for resolution of a nanometer scale (bottom). Inset, Smallest spots resolved for confocal (right) and STED microscopy (left) determining the minimum resolution of both imaging modes (4 individual clusters averaged). Note that, especially in case of smaller cluster sizes in the flotillin-2 knockdown cells, the confocal image cannot recall the individual, separate clusters as identified by the STED image (arrows). C, The histogram depicts the percentage of APP clusters within the respective indicated cluster size limit (gray bars, flotillin-2 knockdown cells; red bars, control cells). Note that, in flotillin-2-depleted cells, most clusters are found in the range of 0–700, whereas cluster sizes are shifted to higher values in control cells. Cluster sizes are given as numbers of pixels and represent mean values (n = 290 and 616 clusters for control and knockdown cells, respectively). D, Effect of flotillin-2 knockdown on APP homotypic interaction measured by FRET in live cells. N2a cells were treated with either flotillin-2 or control siRNA and transfected with swAPP. Surface APP was stained with 6E10 and dye-coupled secondary antibody f(ab) fragments, and energy transfer from donor dye Alexa 488 to acceptor dye Cy3 was measured by the reduction of the lifetime of Alexa 488. The difference in FRET efficiency distribution of control cells, compared with flotillin-2-depleted cells, is shown. Red bars, FRET efficiencies more populated in control cells. Gray bars, FRET efficiencies more populated in flotillin-2-depleted cells.
To visualize the clustering behavior of APP, we used STED microscopy (Donnert et al., 2006) that provides a nanoscale resolution (∼40 nm laterally, corresponding to the applied STED power in this study) (Fitzner et al., 2006; Willig et al., 2006). Using STED microscopy, we compared the size of APP clusters in the plasma membrane of control cells and cells that were depleted of flotillin-2 by RNAi (Fig. 4B). Remarkably, we found that the flotillin-2 knockdown resulted in a significant reduction of mean APP cluster sizes (77% reduction, 900 clusters counted, p < 0.0001, t test). The size distribution of APP clusters was shifted toward lower cluster sizes in flotillin-2-depleted cells (Fig. 4C), suggesting that flotillin-2 may be involved in the clustering of APP at the plasma membrane. In contrast, knockdown of flotillin-1 did not affect cluster size and size distribution of surface APP (data not shown).
The effect of flotillin-2 knockdown on APP clustering was also investigated by the technique of FRET, which is particularly suitable for observing phenomena occurring at a scale of <10 nm (Bunt and Wouters, 2004). Donor and acceptor dyes were exclusively coupled to surface swAPP molecules by incubation of swAPP-transfected N2a cells with antibody-coupled dyes at 4°C. Despite the low signal attributable to the low concentration of surface APP, it was possible to measure resonance energy transfer between donor and acceptor dye molecules.
In flotillin-2-depleted cells, the absolute intensity of the bound dyes was higher, apparently attributable to lower endocytosis rates; mean FRET efficiency was 3.2%. In control cells only, an additional subpopulation of swAPP underwent energy transfer with a higher FRET efficiency centered around 12%. This redistribution to a new subpopulation, with a loss at lower and gain at higher FRET efficiencies, induced by flotillin-2 is visible in the difference of the distributions (Fig. 4D) and introduces a significant difference (p < 0.01) between the mean FRET efficiencies of control and flotillin-2-depleted cells. APP is therefore more likely to homotypically interact in the presence of flotillin-2. This result is compatible with the reduction of APP cluster size during flotillin-2 knockdown as seen by STED microscopy. The low mean values of the average FRET efficiencies probably reflect the large distance between the dye molecules coupled to secondary f(ab) fragments on separate primary antibody molecules bound to separate swAPP molecules.
A previous study has provided evidence that flotillin interacts with APP via residues 189–282 of its proposed oligomerization domain (Chen et al., 2006), suggesting that direct protein–protein interaction between APP and flotillin oligomers might be the driving force in the formation of APP clusters. Indeed, by coimmunoprecipitation, we found an interaction of APP and flotillin-2 not only in N2a cell culture but also in brain lysates from heterozygous swAPP/PS1dE9 transgenic mice (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Because flotillin partitions into lipid rafts, we speculated that the interaction between APP and flotillin would be sensitive to cholesterol depletion. Indeed, the amount of flotillin-2 that was coimmunoprecipitated with APP was significantly reduced by cholesterol depletion (51% reduction) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), raising the possibility that, in addition to the direct protein interaction, protein–lipid interactions may contribute to the complex formation.
We hypothesized that the flotillin-driven clustering of APP might be a mechanism to influence its endocytosis. If this were to be the case, then artificial clustering of APP by antibody cross-linking should overcome the inhibitory effect of flotillin-2 knockdown on the endocytosis of APP. To test this possibility, we transfected swAPP–YFP into N2a cells and labeled APP at the cell surface with primary antibody at 4°C, followed by the subsequent incubation with monovalent f(ab) fragment or with bivalent IgG secondary antibody to induce clustering of APP. Cells were then shifted to 37°C to allow endocytosis to occur. Cross-linking with bivalent secondary antibody dramatically stimulated the endocytosis of APP (64% increase, n = 170 cells, p < 0.0005, t test) but not of two other transmembrane proteins [VSVG and PLP (supplemental Fig. 5, available at www.jneurosci.org as supplemental material)]. In line with the stimulation of endocytosis, we observed an increase in intracellular β-cleaved APP ectodomain during antibody induced clustering (52% increase, n = 406 cells, p < 0.005, t test) as reported previously (Ehehalt et al., 2003). Having shown that antibody-induced clustering promotes endocytosis of APP, we tested the possible role of flotillin in this process. APP was labeled at the cell surface with primary antibody and cross-linked with bivalent IgG secondary antibody or not in both control and flotillin-2-depleted cells. The rate of endocytosis was determined over a time course of 15 and 30 min. Consistent with our results above, we found that depletion of flotillin-2 reduced the internalization of APP in the absence of IgG secondary antibody (Fig. 5A). However, when endocytosis was promoted by incubating cells with bivalent secondary antibody, we observed that the inhibition by flotillin-2 knockdown was completely overcome (Fig. 5A). Antibody-induced clustering of APP might, thus, mimic the function of flotillin in modulating APP endocytosis.
Figure 5.

Antibody-induced clustering of APP increases endocytosis. A, CFP–swAPP was labeled with anti-GFP antibody on the cell surface in flotillin-2 knockdown (flot-2 −) or control N2a (flot-2 +) cells. Cells were subsequently either clustered with a bivalent Cy3-coupled secondary antibody or not and subjected to endocytosis for 15 min (left) or 30 min (right). To measure the internalization of APP, the ratios of Cy3 intensity originating from the cell interior and the total cell were determined. Ratios are shown as means + SEM (n > 70 cells for each condition). B, N2a cells were treated with 1 mm mβCD for 2 h or not before labeling CFP–swAPP on the cell surface with 6E10, followed by Cy3-conjugated secondary antibody clustering or not. Antibody uptake was performed in medium supplemented with lipoprotein-free serum for 15 and 30 min. Ratios of internalized to total labeled APP are shown as means + SEM. The inhibitory effect of cholesterol depletion on APP endocytosis can be reversed by clustering with bivalent secondary antibody. Ratios are shown as means + SEM (n > 75 cells for each condition).
Similar results were obtained in cells depleted of cholesterol, in which the inhibitory effect of cholesterol depletion could be reversed by secondary IgG antibody-mediated clustering of APP (Fig. 5B).
Discussion
Endocytosis is required for the amyloidogenic processing of APP and thus to initiate the release of the neurotoxic Aβ peptide. Here, we demonstrate that flotillin-2 promotes the clustering of APP, and this may stimulate the sequential endocytosis of APP into a specialized clathrin-dependent endocytosis pathway. Clathrin-dependent and cholesterol-dependent endocytosis pathways have been primarily regarded as mutually exclusive. However, there are a number of examples suggesting an overlap between cholesterol- and clathrin-dependent pathways. An interesting example is the anthrax toxin that clusters its receptor into lipid rafts, thereby triggering clathrin-dependent endocytosis (Abrami et al., 2003). For efficient endocytosis, the anthrax toxin requires rafts to promote receptor clustering, and a recent report suggests that clustering involves a conformational change of the clustered receptor, which is a prerequisite for its subsequent ubiquitination and incorporation into clathrin-coated pits (Abrami et al., 2006). Other examples are the tetanus neurotoxin and the prion protein, which reside in lipid rafts from which they seem to dissociate after lateral sorting and association with the clathrin coat (Sunyach et al., 2003; Deinhardt et al., 2006). One might envision a similar mechanism for the endocytosis of APP. It is possible that APP transiently interacts with flotillin-2 in a cholesterol-dependent manner before being internalized. Additional experiments will be required to clarify the question whether clusters of APP and flotillin are internalized together or dissociate before entering clathrin-coated pits.
It will also be interesting to study how other APP-interacting proteins that are known to regulate its endocytosis are connected to this process. It was demonstrated recently that the low-density lipoprotein receptor-related protein (LRP) promotes the association of APP with detergent-resistant membranes (Yoon et al., 2007). LRP may, thus, function as an additional stabilizing factor for the interaction of APP with cholesterol-rich membrane domains, and this mechanism may also contribute to the regulation of APP endocytosis.
It is tempting to speculate that plasma membrane cholesterol may promote the assembly of an APP-containing protein complex involved in the uptake of cholesterol. Interestingly, the internalization of cholesterol-rich lipoprotein particles by apoE receptors also triggers the endocytosis and processing of APP (Ye et al., 2005; He et al., 2007). The physiological relevance of this link between cholesterol and APP internalization is not clear, but recent evidence points to a role of APP processing in the regulation of lipid homeostasis (Grimm et al., 2005). Future studies are necessary to elucidate whether flotillin might also be involved in the regulation of neuronal cholesterol uptake.
In summary, we propose a model in which flotillin acts as a scaffolding protein, forming a platform for the clustering of APP and subsequent endocytosis into a specialized clathrin-dependent pathway. It will be interesting to investigate whether flotillin-dependent clustering of APP also facilitates the dimerization of APP, which was shown recently to result in increased Aβ production by a direct modulation of the gamma-secretase complex (Munter et al., 2007).
Clearly more work is required to elucidate the molecular details of the regulation of APP endocytosis. Such studies are worthwhile in particular in light of the genetic association of late-onset AD with the neuronal sortilin-related receptor, Sorl1, a protein involved in the regulation of the endosomal trafficking of APP (Spoelgen et al., 2006; Rogaeva et al., 2007) and the known endocytic pathway abnormalities in Alzheimer's disease (Cataldo et al., 2003; Nixon, 2005; Laifenfeld et al., 2007).
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 523) and Deutsche Hirnliga. We are grateful to Dominik Wildanger and Lars Meyer for their expert assistance with the STED experiments.
References
- Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, Dingwall C, De Strooper B, Dotti CG. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol. 2004;167:953–960. doi: 10.1083/jcb.200404149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160:321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Leppla SH, van der Goot FG. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J Cell Biol. 2006;172:309–320. doi: 10.1083/jcb.200507067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunt G, Wouters FS. Visualization of molecular activities inside living cells with fluorescent labels. Int Rev Cytol. 2004;237:205–277. doi: 10.1016/S0074-7696(04)37005-1. [DOI] [PubMed] [Google Scholar]
- Cameron PL, Ruffin JW, Bollag R, Rasmussen H, Cameron RS. Identification of caveolin and caveolin-related proteins in the brain. J Neurosci. 1997;17:9520–9535. doi: 10.1523/JNEUROSCI.17-24-09520.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Peterhoff CM, Terio NB, Epstein CJ, Villar A, Carlson EJ, Staufenbiel M, Nixon RA. App gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of down syndrome. J Neurosci. 2003;23:6788–6792. doi: 10.1523/JNEUROSCI.23-17-06788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TY, Liu PH, Ruan CT, Chiu L, Kung FL. The intracellular domain of amyloid precursor protein interacts with flotillin-1, a lipid raft protein. Biochem Biophys Res Commun. 2006;342:266–272. doi: 10.1016/j.bbrc.2006.01.156. [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci USA. 2003;100:11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, Berninghausen O, Willison HJ, Hopkins CR, Schiavo G. Tetanus toxin is internalized by a sequential clathrin-dependent mechanism initiated within lipid microdomains and independent of epsin1. J Cell Biol. 2006;174:459–471. doi: 10.1083/jcb.200508170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnert G, Keller J, Medda R, Andrei MA, Rizzoli SO, Luhrmann R, Jahn R, Eggeling C, Hell SW. Macromolecular-scale resolution in biological fluorescence microscopy. Proc Natl Acad Sci USA. 2006;103:11440–11445. doi: 10.1073/pnas.0604965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyba M, Jakobs S, Hell SW. Immunofluorescence stimulated emission depletion microscopy. Nat Biotechnol. 2003;21:1303–1304. doi: 10.1038/nbt897. [DOI] [PubMed] [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito A, Gerritsen HC, Wouters FS. Fluorescence lifetime heterogeneity resolution in the frequency domain by lifetime moments analysis. Biophys J. 2005;89:4286–4299. doi: 10.1529/biophysj.104.053397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer's disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzner D, Schneider A, Kippert A, Mobius W, Willig KI, Hell SW, Bunt G, Gaus K, Simons M. Myelin basic protein-dependent plasma membrane reorganization in the formation of myelin. EMBO J. 2006;25:5037–5048. doi: 10.1038/sj.emboj.7601376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, McMahon HT. Curvature of clathrin-coated pits driven by epsin. Nature. 2002;419:361–366. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM. The role of cholesterol in the biosynthesis of beta-amyloid. NeuroReport. 1999;10:1699–1705. doi: 10.1097/00001756-199906030-00014. [DOI] [PubMed] [Google Scholar]
- Frick M, Bright NA, Riento K, Bray A, Merrified C, Nichols BJ. Coassembly of flotillins induces formation of membrane microdomains, membrane curvature, and vesicle budding. Curr Biol. 2007;17:1151–1156. doi: 10.1016/j.cub.2007.05.078. [DOI] [PubMed] [Google Scholar]
- Girardot N, Allinquant B, Langui D, Laquerriere A, Dubois B, Hauw JJ, Duyckaerts C. Accumulation of flotillin-1 in tangle-bearing neurones of Alzheimer's disease. Neuropathol Appl Neurobiol. 2003;29:451–461. doi: 10.1046/j.1365-2990.2003.00479.x. [DOI] [PubMed] [Google Scholar]
- Glebov OO, Bright NA, Nichols BJ. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat Cell Biol. 2006;8:46–54. doi: 10.1038/ncb1342. [DOI] [PubMed] [Google Scholar]
- Grimm MO, Grimm HS, Patzold AJ, Zinser EG, Halonen R, Duering M, Tschape JA, De Strooper B, Muller U, Shen J, Hartmann T. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol. 2005;7:1118–1123. doi: 10.1038/ncb1313. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ. The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- He G, Qing H, Tong Y, Cai F, Ishiura S, Song W. Degradation of nicastrin involves both proteasome and lysosome. J Neurochem. 2007;101:982–992. doi: 10.1111/j.1471-4159.2007.04449.x. [DOI] [PubMed] [Google Scholar]
- Herreros J, Ng T, Schiavo G. Lipid rafts act as specialized domains for tetanus toxin binding and internalization into neurons. Mol Biol Cell. 2001;12:2947–2960. doi: 10.1091/mbc.12.10.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, Simons K. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem. 2005;280:36815–36823. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- Kao SC, Krichevsky AM, Kosik KS, Tsai LH. BACE1 suppression by RNA interference in primary cortical neurons. J Biol Chem. 2004;279:1942–1949. doi: 10.1074/jbc.M309219200. [DOI] [PubMed] [Google Scholar]
- King GD, Scott Turner R. Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp Neurol. 2004;185:208–219. doi: 10.1016/j.expneurol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci USA. 2001;98:5815–5820. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Koo EH, Squazzo SL, Selkoe DJ, Koo CH. Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J Cell Sci. 1996;109:991–998. doi: 10.1242/jcs.109.5.991. [DOI] [PubMed] [Google Scholar]
- Kounnas MZ, Moir RD, Rebeck GW, Bush AI, Argraves WS, Tanzi RE, Hyman BT, Strickland DK. LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted beta-amyloid precursor protein and mediates its degradation. Cell. 1995;82:331–340. doi: 10.1016/0092-8674(95)90320-8. [DOI] [PubMed] [Google Scholar]
- Laifenfeld D, Patzek LJ, McPhie DL, Chen Y, Levites Y, Cataldo AM, Neve RL. Rab5 mediates an amyloid precursor protein signaling pathway that leads to apoptosis. J Neurosci. 2007;27:7141–7153. doi: 10.1523/JNEUROSCI.4599-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang DM, Lommel S, Jung M, Ankerhold R, Petrausch B, Laessing U, Wiechers MF, Plattner H, Stuermer CA. Identification of reggie-1 and reggie-2 as plasmamembrane-associated proteins which cocluster with activated GPI-anchored cell adhesion molecules in non-caveolar micropatches in neurons. J Neurobiol. 1998;37:502–523. doi: 10.1002/(sici)1097-4695(199812)37:4<502::aid-neu2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Langhorst MF, Reuter A, Stuermer CA. Scaffolding microdomains and beyond: the function of reggie/flotillin proteins. Cell Mol Life Sci. 2005;62:2228–2240. doi: 10.1007/s00018-005-5166-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langui D, Girardot N, El Hachimi KH, Allinquant B, Blanchard V, Pradier L, Duyckaerts C. Subcellular topography of neuronal Abeta peptide in APPxPS1 transgenic mice. Am J Pathol. 2004;165:1465–1477. doi: 10.1016/s0002-9440(10)63405-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow IC, Rea S, Martin S, Prior IA, Prohaska R, Hancock JF, James DE, Parton RG. Flotillin-1/reggie-2 traffics to surface raft domains via a novel golgi-independent pathway. Identification of a novel membrane targeting domain and a role for palmitoylation. J Biol Chem. 2002;277:48834–48841. doi: 10.1074/jbc.M209082200. [DOI] [PubMed] [Google Scholar]
- Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann-Giesen C, Falkenbach B, Beicht P, Claasen S, Luers G, Stuermer CA, Herzog V, Tikkanen R. Membrane and raft association of reggie-1/flotillin-2: role of myristoylation, palmitoylation and oligomerization and induction of filopodia by overexpression. Biochem J. 2004;378:509–518. doi: 10.1042/BJ20031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann-Giesen C, Fernow I, Amaddii M, Tikkanen R. Role of EGF-induced tyrosine phosphorylation of reggie-1/flotillin-2 in cell spreading and signaling to the actin cytoskeleton. J Cell Sci. 2007;120:395–406. doi: 10.1242/jcs.03336. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Perez RG, Squazzo SL, Koo EH. Enhanced release of amyloid beta-protein from codon 670/671 “Swedish” mutant beta-amyloid precursor protein occurs in both secretory and endocytic pathways. J Biol Chem. 1996;271:9100–9107. doi: 10.1074/jbc.271.15.9100. [DOI] [PubMed] [Google Scholar]
- Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- Pietrzik CU, Busse T, Merriam DE, Weggen S, Koo EH. The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. EMBO J. 2002;21:5691–5700. doi: 10.1093/emboj/cdf568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Masilamani M, Solomon S, Tikkanen R, Stuermer CA, Plattner H, Illges H. Asymmetric localization of flotillins/reggies in preassembled platforms confers inherent polarity to hematopoietic cells. Proc Natl Acad Sci USA. 2003;100:8241–8246. doi: 10.1073/pnas.1331629100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell DR, Christie G, Hussain I, Dingwall C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr Biol. 2001;11:1288–1293. doi: 10.1016/s0960-9822(01)00394-3. [DOI] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis GP, Hoegg M, Munderloh C, Schrock Y, Malaga-Trillo E, Rivera-Milla E, Stuermer CA. Reggie/flotillin proteins are organized into stable tetramers in membrane-microdomains. Biochem J. 2007;403:313–322. doi: 10.1042/BJ20061686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoelgen R, von Arnim CA, Thomas AV, Peltan ID, Koker M, Deng A, Irizarry MC, Andersen OM, Willnow TE, Hyman BT. Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and β-secretase β-site APP-cleaving enzyme. J Neurosci. 2006;26:418–428. doi: 10.1523/JNEUROSCI.3882-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuermer CA, Lang DM, Kirsch F, Wiechers M, Deininger SO, Plattner H. Glycosylphosphatidyl inositol-anchored proteins and fyn kinase assemble in noncaveolar plasma membrane microdomains defined by reggie-1 and -2. Mol Biol Cell. 2001;12:3031–3045. doi: 10.1091/mbc.12.10.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003;22:3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic K, Dhaunchak AS, Goncalves JT, Wenzel D, Schneider A, Bunt G, Nave KA, Simons M. Neuron to glia signaling triggers myelin membrane exocytosis from endosomal storage sites. J Cell Biol. 2006;172:937–948. doi: 10.1083/jcb.200509022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Thinakaran G. Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology. 2006;66:S69–S73. doi: 10.1212/01.wnl.0000192107.17175.39. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem. 2004;279:44945–44954. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature. 2006;440:935–939. doi: 10.1038/nature04592. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer's disease. Nat Med. 1995;1:1062–1066. doi: 10.1038/nm1095-1062. [DOI] [PubMed] [Google Scholar]
- Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci USA. 2005;102:18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon IS, Chen E, Busse T, Repetto E, Lakshmana MK, Koo EH, Kang DE. Low-density lipoprotein receptor-related protein promotes amyloid precursor protein trafficking to lipid rafts in the endocytic pathway. FASEB J. 2007;21:2742–2752. doi: 10.1096/fj.07-8114com. [DOI] [PubMed] [Google Scholar]
- Zerbinatti CV, Bu G. LRP and Alzheimer's disease. Rev Neurosci. 2005;16:123–135. doi: 10.1515/revneuro.2005.16.2.123. [DOI] [PubMed] [Google Scholar]