

Abstract
Phrenic long-term facilitation (pLTF) is a serotonin-dependent form of pattern-sensitive respiratory plasticity induced by intermittent hypoxia (IH), but not sustained hypoxia (SH). The mechanism(s) underlying pLTF pattern sensitivity are unknown. SH and IH may differentially regulate serine/threonine protein phosphatase activity, thereby inhibiting relevant protein phosphatases uniquely during IH and conferring pattern sensitivity to pLTF. We hypothesized that spinal protein phosphatase inhibition would relieve this braking action of protein phosphatases, thereby revealing pLTF after SH. Anesthetized rats received intrathecal (C4) okadaic acid (25 nm) before SH (25 min, 11% O2). Unlike (vehicle) control rats, SH induced a significant pLTF in okadaic acid-treated rats that was indistinguishable from rats exposed to IH (three 5 min episodes, 11% O2). IH and SH with okadaic acid may elicit pLTF by similar, serotonin-dependent mechanisms, because intravenous methysergide blocks pLTF in rats receiving IH or okadaic acid plus SH. Okadaic acid did not alter IH-induced pLTF. In summary, pattern sensitivity in pLTF may reflect differential regulation of okadaic acid-sensitive serine/threonine phosphatases; presumably, these phosphatases are less active during/after IH versus SH. The specific okadaic acid-sensitive phosphatase(s) constraining pLTF and their spatiotemporal dynamics during and/or after IH and SH remain to be determined.
Keywords: plasticity, pattern, phosphatase, control of breathing, motor neuron, hypoxia
Introduction
Stimulation pattern (e.g., intermittent vs sustained) has a powerful influence on the expression of learning and memory as well as synaptic plasticity in the nervous system. For example, intermediate-term memory formation and synaptic facilitation in Aplysia (Mauelshagen et al., 1998; Sutton et al., 2002; Sherff and Carew, 2004), habituation in the crab Chasmagnathus (Freudenthal et al., 1998), olfactory memory formation in Drosophila (Isabel et al., 2004), and hippocampal long-term potentiation in rodents (Kauer, 1999; Nguyen et al., 2000; Woo et al., 2000; Scharf et al., 2002) all exhibit distinct pattern sensitivity to the training protocol; intermittent (or spaced) stimuli are more robust than sustained (or massed) stimulation in each case. The mechanistic basis of pattern sensitivity in neuroplasticity is not understood (Brennan and Keverne, 1997; Baker et al., 2001; Scharf et al., 2002).
Phrenic long-term facilitation (pLTF) is a pattern-sensitive form of neural plasticity expressed as a progressive increase in respiratory-related motor output (e.g., phrenic, hypoglossal) (Mitchell et al., 2001). pLTF is initiated by serotonin receptor activation on or near phrenic motoneurons after intermittent, but not sustained, exposure to hypoxia (Baker and Mitchell, 2000; Baker-Herman et al., 2004; McKay et al., 2004). Intermittent (but not sustained) serotonin receptor activation is sufficient to induce pLTF in in vitro neonatal rat brainstem–spinal cord preparations (Lovett-Barr et al., 2006) and in anesthetized adult rats (P. M. MacFarlane and G. S. Mitchell, unpublished observations), similar to serotonin-induced hypoglossal LTF in in vitro neonatal brainstem slice preparations (Bocchiaro and Feldman, 2004).
Protein kinases and phosphatases play a critical role in neural plasticity by regulating the phosphorylation state, and hence activity, of key proteins in the signaling networks that underlie synaptic alterations (Thiels and Klann, 2001; Winder and Sweatt, 2001; Colbran, 2004). Shifts in the balance of protein kinase versus phosphatase activation potentially shift net phosphorylation of key proteins from restraint toward the induction of plasticity (Wu et al., 2001; Ajay and Bhalla, 2004). For example, protein phosphatase 2B (PP2B; also known as calcineurin) constrains intermediate- and long-term facilitation in Aplysia, likely via effects on mitogen-activated protein kinases (Sharma et al., 2003). Similarly, protein phosphatases constrain long-term potentiation and depression in rats (Woo et al., 2002; Launey et al., 2004; Belmeguenai and Hansel, 2005; Tomita et al., 2005).
The pattern sensitivity of pLTF to hypoxia and serotonin receptor activation suggests that differential protein kinase/phosphatase activation may enable pLTF after intermittent but not sustained hypoxia. Indeed, multiple protein kinases and phosphatases are hypoxia sensitive (Seta et al., 2001, 2002; Truttmann et al., 2004; Liu et al., 2005), although the effects of the hypoxia pattern on protein kinase/phosphatase activity are not known. One possibility is that intermittent and sustained hypoxia differentially regulate protein kinase/phosphatase activity by differences in the generation of reactive oxygen species (ROSs) (Yuan et al., 2004). ROSs regulate the activity of both protein kinases and phosphatases (Klann and Thiels, 1999; Prabhakar and Kumar, 2004) and are necessary for pLTF expression (MacFarlane and Mitchell, 2006).
We hypothesized that protein phosphatases restrain pLTF after sustained hypoxia and that this phosphatase constraint is diminished during or after intermittent hypoxia. Thus, we predicted that (1) serine/threonine protein phosphatases are present and active in the ventral spinal region containing the phrenic motor nucleus; (2) intrathecal application of okadaic acid, a cell-permeable serine/threonine protein phosphatase inhibitor, would reveal serotonin-dependent pLTF after sustained hypoxia; and (3) pLTF elicited by intermittent hypoxia is unaffected when spinal protein phosphatases are inhibited by intrathecal okadaic acid. Collectively, our data demonstrate that pattern sensitivity in the expression of pLTF is regulated, at least in part, by the activity of okadaic acid-sensitive serine/threonine protein phosphatases.
Materials and Methods
Experiments were performed using 3- to 5-month-old adult male Sprague Dawley rats (colony PO4; Charles River, Wilmington, MA). Animals were individually housed in a controlled environment (12 h light/dark cycle), with food and water ad libitum. The School of Veterinary Medicine Animal Care and Use Committee at the University of Wisconsin approved all protocols.
Surgical preparation and nerve isolation.
Anesthesia was induced with isoflurane in a closed chamber, followed by inhalation of isoflurane through a nose cone (3.0–3.5% in 50% O2, balance N2). The trachea was cannulated to enable pump ventilation [tidal volume, 2–2.5 ml; fraction of inspired oxygen (FIO2) = 0.50; Rodent Respirator model 682; Harvard Apparatus, South Natick, MA], and isoflurane anesthesia was continued through the ventilator. Midcervical, bilateral vagotomy was performed to prevent phrenic nerve entrainment with the ventilator. Catheters were placed into the tail vein for fluid administration (1:11 by volume NaHCO3/lactated Ringer's; 2.5 ml/h) and the femoral artery to enable blood pressure measurement and blood sampling for arterial blood gas analysis. Body temperature was maintained at 37.5 ± 1.0°C using a rectal probe and custom-designed heated table. The left phrenic nerve was isolated using a dorsal approach, cut distally, desheathed, and submerged in a mixture of mineral oil and petroleum jelly. The muscles overlying the second cervical segment were removed, followed by a C2 laminectomy. If necessary, Gelfoam and/or Surgicel were used to control bleeding around the spinal cord. A small durotomy was performed and a soft silicone catheter (2 French; Access Technologies, Skokie, IL) containing artificial CSF or okadaic acid (see below) was inserted until just over the fourth spinal segment (∼2 mm). The phrenic nerve was then placed on bipolar silver electrodes.
Before beginning a protocol, rats were slowly converted to urethane anesthesia (1.6 g/kg, i.v.) and then paralyzed with pancuronium bromide to prevent spontaneous breathing movements (2.5 mg/kg, i.v., supplemented as necessary). End-tidal CO2 was measured throughout the experiment using a flow-through capnograph (Capnogard, model 1265; Novametrix, Wallingford, CT) with sufficient response time to measure expiratory gases in rats.
Hypoxic protocols.
The CO2 apneic threshold was determined by decreasing CO2 levels and/or increasing the ventilator rate until phrenic activity ceased, then slowly decreasing ventilator rate or increasing inspired CO2 levels until phrenic activity resumed (the “recruitment threshold”) as described previously (Mahamed and Mitchell, 2006).
To establish baseline conditions, end-tidal CO2 levels were set 1–2 torr above the recruitment threshold. After phrenic nerve activity had stabilized (∼1 h), an initial blood sample was taken to characterize baseline levels of PaO2, PaCO2, pH, and base excess (0.3 ml in 0.5 ml heparinized glass syringe; ABL-500; Radiometer, Copenhagen, Denmark; unused blood was returned to the animal). Rats were given either of the following: (1) intermittent hypoxia, consisting of three 5 min hypoxic episodes (FIO2 = 0.11 ± 0.1, PaO2 = 38 ± 1 mmHg), separated by 5 min intervals of baseline conditions (FIO2 = 0.50); or (2) a single, 25 min hypoxic episode (i.e., sustained hypoxia, FIO2 = 0.11 ± 0.1, PaO2 = 38 ± 1 mmHg). Previous studies demonstrate that a sustained hypoxia exposure that is equivalent to or greater than the cumulative hypoxia presented during an intermittent hypoxia exposure fails to elicit phrenic LTF (Baker and Mitchell, 2000). Phrenic activity was monitored for 60 min after hypoxia. Arterial blood samples were drawn and analyzed during the final minute of the first hypoxic episode and 30 and 60 min after the final hypoxic episode. Additional rats that did not receive intermittent or sustained hypoxia (time controls) were used to verify the stability of the preparation over a similar time period under equivalent conditions and to enable measurements of baseline tissue protein levels. Following nerve recording protocols, the rats were killed by exsanguination during continued anesthesia, and C4–C5 spinal segments were harvested, frozen ventral side down on dry ice, and stored at −80°C for protein analysis (see below). Throughout the protocol, isocapnic conditions (±1 mmHg from baseline PaCO2) were maintained by adjusting ventilator frequency and/or inspired CO2.
Pharmacological treatments.
A stock solution of okadaic acid (10 μg/ml distilled deionized water) was prepared, aliquoted, and stored at −20°C. The stock okadaic acid solution was mixed with freshly made artificial CSF (aCSF; 120 mm NaCl, 3 mm KCl, 2 mm CaCl, 2 mm MgCl, 23 mm NaHCO3, 10 mm glucose bubbled with 95% O2–5% CO2) to a final concentration of 25 nm just before placement of the intrathecal catheter. Ten microliters of aCSF or okadaic acid was injected over 2 min into the intrathecal space ∼40 min before the beginning of hypoxia or time control protocols. A subset of animals received the broad-spectrum serotonin receptor antagonist methysergide (4 mg/kg, i.v.) ∼20 min before the onset of hypoxia, along with intrathecal application of okadaic acid.
Electrophysiological data analysis.
Phrenic nerve activity was amplified (10,000×), bandpass filtered (100 Hz to 10 kHz; model 1700; A-M Systems, Carlsborg, WA), and integrated (time constant = 50 ms; model MA-821RSP; CWE, Ardmore, PA). Integrated signals were digitized and processed with commercially available software (WINDAQ software; DATAQ Instruments, Akron, OH). Peak integrated phrenic burst amplitude, burst frequency, and mean arterial blood pressure were calculated over a 60 s period just before the first hypoxic episode (baseline), at the end of the first hypoxic episode or the equivalent time point during sustained hypoxia (short-term hypoxic response), and 30 and 60 min after hypoxia. Data were included in the analysis only if isocapnic conditions were maintained. Amplitude data are expressed as the change in phrenic burst amplitude as a percentage change from baseline values. Frequency data are reported as the change from baseline in bursts per minute. Data were compared using a one-way ANOVA or two-way ANOVA with repeated-measures design if applicable (Fisher's least significant difference post hoc test if necessary; SigmaStat 2.03; SPSS, Chicago, IL).
Immunohistochemistry.
A subset of rats were anesthetized in a closed chamber containing isoflurane followed by a beuthanasia injection (0.3 ml, i.p.), then perfused with heparinized PBS (0.01 m) followed by ice-cold fixative (4% paraformaldehyde, 0.05% gluteraldehyde in PBS). The spinal cord was removed and placed in fixative overnight at 4°C, then transferred to a refrigerated cryoprotectant solution (30% sucrose in PBS) until saturated. Serial coronal sections from cervical regions containing phrenic motoneurons (C3–C5, 40 μm) were prepared using a freezing microtome and processed for the detection of serine/threonine protein phosphatases. Briefly, tissues were washed with Tris-buffered saline (TBS), treated with 1% H2O2 in TBS for 30 min, and then washed several times with 0.1% Triton X-100 in TBS (TBS-T). After incubation in 5% normal goat serum, serial sections were incubated overnight at 4°C in TBS-T containing primary antibodies against protein phosphatase 1 (rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA), protein phosphatase 2A (rabbit polyclonal; Millipore, Billerica, MA), or protein phosphatase 5 (mouse monoclonal; BD Biosciences, San Jose, CA). The primary antibody concentration for all protein phosphatases was 1:500. Sections were then washed several times with TBS-T and incubated with biotinylated secondary antibody (PP1 and PP2A, 1:1000 goat anti-rabbit; PP5, 1:1000 goat anti-mouse) for 1 h at room temperature (RT). After several more washes in TBS-T, tissue sections were incubated with ABC solution (Vectastain ABC kit; Vector Laboratories, Burlingame, CA) for 1 h. The presence of protein phosphatases was visualized using a DAB solution kit (Vector Laboratories) according to the manufacturer's instructions. Sections were then washed in TBS, placed on gelatin-coated slides, dried at RT, dehydrated in a graded alcohol series, and then cleared with xylenes and mounted with Eukitt medium. All images were captured (100× and 400× magnifications) and analyzed with a digital camera (SPOT II; Diagnostic Instruments, Sterling Heights, MI). The final photomicrographs were created with Adobe Photoshop software (Adobe Systems, San Jose, CA). All images received equivalent adjustments to tone scale, gamma, and sharpness. Putative phrenic motor neurons were determined based on size (>30 μm) and location within the ventral horn (Goshgarian and Rafols, 1981).
Cervical segment preparation for phosphatase activity assay.
To determine protein phosphatase activity, cervical spinal segments were collected from a subset of rats ∼2 h after surgery. Animals were naive to hypoxia or drug treatment before tissue collection. Rats were killed via exsanguination while still anesthetized, and C4–C5 spinal segments were harvested, frozen ventral side down on dry ice, and then stored at −80°C. On the day of analysis, frozen C4–C5 spinal segments were placed dorsal side up on a freezing microtome, and 50–100 μm sections were removed until the central canal was visible. Ventral cervical spinal segments were thawed, quickly weighed, and then homogenized (Tissue Tearor; Biospec Products, Bartlesville, OK) in 1000 μl of ice-cold phosphatase homogenization buffer (50 mm Tris-HCl, 1 mm EDTA, 0.1% β-mercaptoethanol in distilled, deionized water with Roche Complete Protease Inhibitor Cocktail; Roche, Indianapolis, IN). Cell extracts were spun at 100,000 × g at 4°C for 60 min (Beckham Ultracentrifuge; Beckman Coulter, Fullerton, CA), and the supernatant (cytosolic fraction) was removed and immediately analyzed for protein phosphatase activity (see below) or aliquoted and stored at −80°C for total protein analysis. The membrane (S100) pellet was immediately frozen on dry ice and stored at −80°C for later analysis. Before measuring phosphatase activity in the membrane fraction, frozen S100 pellets were thawed, resuspended in 600 μl of ice-cold phosphate homogenization buffer, and quickly spun to 14,000 × g, and then the supernatant was removed and immediately analyzed for protein phosphatase activity or stored at −80°C in aliquots for total protein analysis.
Total protein concentration in each sample was measured using the bicinchoninic acid method with modifications for the presence of a reducing agent (Pierce Biotechnologies, Rockford, IL) according to manufacturer's instructions.
Protein phosphatase activity assay.
Cytosolic and membrane fractions were analyzed for serine/threonine protein phosphatase activity according to the manufacturer's instructions (Promega Serine/Threonine Phosphatase Activity Assay; Promega, Madison, WI). Briefly, prepared samples were stripped of free phosphate groups using size exclusion columns. Samples were loaded in a 96-well plate with reaction buffer tailored toward measuring protein phosphatase 2A activity (250 mm imidazole, 1 mm EGTA, 0.1% β-mercaptoethanol, 0.5 mg/ml bovine serum albumin, and a phosphopeptide substrate [RRA(pT)VA]). We chose to focus on PP2A because of assay availability and because okadaic acid has a higher affinity for this phosphatase relative to other serine/threonine protein phosphatases (with the exception of PP5) (Honkanen and Golden, 2002). Multiple controls were assessed within each assay, including wells without sample, without phosphopeptide, with sample but terminated at time 0, and with sample and exogenous okadaic acid (final concentration = 0.05–5 nm okadaic acid). The plate was incubated for 30 min at 37°C, and then a molybdate solution was added to stop protein phosphatase activity and initiate a colorimetric reaction with free phosphates generated by cleavage of the phosphopeptide by the phosphatase. Plates were incubated at room temperature for 30 min and then read at 670 nm with a plate reader (Revelation 4.02; Dynex Technologies, West Sussex, UK). Protein phosphatase activity is presented as picomoles of phosphate generated per minute per microgram of total protein. Data were statistically compared with a one-way ANOVA (SigmaStat 2.03; SPSS).
Results
Serine/threonine protein phosphatases are present and active in ventral cervical spinal cord
Similar to previous observations (Strack et al., 1998; Gupta and Abou-Donia, 2001), immunohistochemical analysis demonstrated the presence of the serine/threonine protein phosphatases 1 (PP1), 2A (PP2A), and 5 (PP5) throughout cells in the cervical spinal cord, including putative phrenic motoneurons (Fig. 1). Whereas PP1 exhibited strong nuclear staining, PP2A and PP5 were more diffusely expressed.
Figure 1.
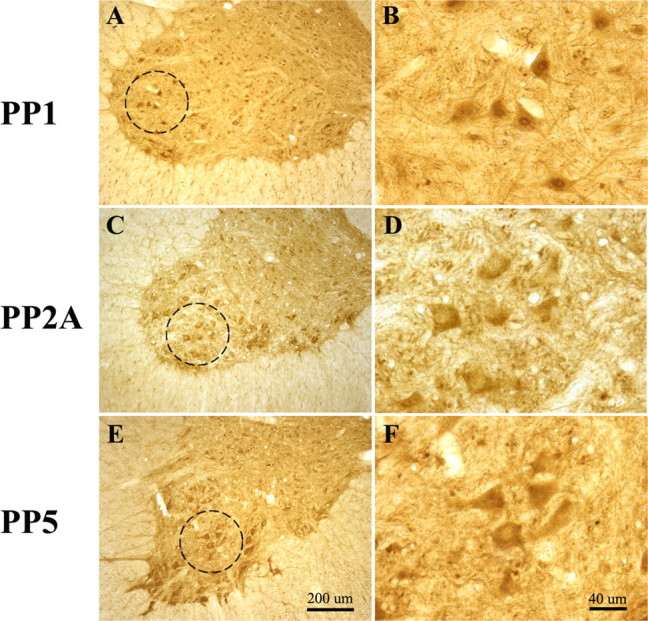
A–F, Representative photomicrographs of ventral C4–C5 spinal sections immunostained for protein phosphatases 1 (A, B), 2A (C, D), and 5 (E, F). Photomicrographs are presented at 100× (A, C, E) and 400× (B, D, F) magnification. Note that PP1 shows strong nuclear staining within putative phrenic motoneurons, whereas PP2A and PP5 are more diffusely expressed.
We used a serine/threonine protein phosphatase activity assay tailored toward measuring PP2A activity to demonstrate that at least one major okadaic acid-sensitive protein phosphatase is active within the ventral cervical spinal segments associated with the phrenic motor nucleus. PP2A activity was analyzed in ventral cervical spinal samples harvested ∼ 2 h after the end of surgery (n = 5). PP2A activity was present in the cytosolic fraction and was significantly greater in the S100 membrane fraction (11.2 ± 0.4 vs 18.2 ± 0.9 pmol of phosphate/min/μg protein, respectively) (Fig. 2A) (p < 0.05). To confirm that okadaic acid could inhibit PP2A activity in these samples, okadaic acid was added directly to cytosolic samples. Okadaic acid inhibited PP2A activity in a dose-dependent manner (Fig. 2B), with statistically significant decreases in basal PP2A activity at all concentrations used (0.05 nm okadaic acid, 15 ± 3% inhibition of basal PP2A activity; n = 11; 0.5 nm okadaic acid, 55 ± 4% inhibition of basal PP2A activity; n = 7; 5 nm okadaic acid, 69 ± 3% inhibition of basal PP2A activity; n = 13; p < 0.05 for all groups). Similar results were found when membrane fraction samples were studied (data not shown). These data confirm that at least one major okadaic acid-sensitive serine/threonine protein phosphatase is present and active in the cervical spinal region containing phrenic motoneurons.
Figure 2.
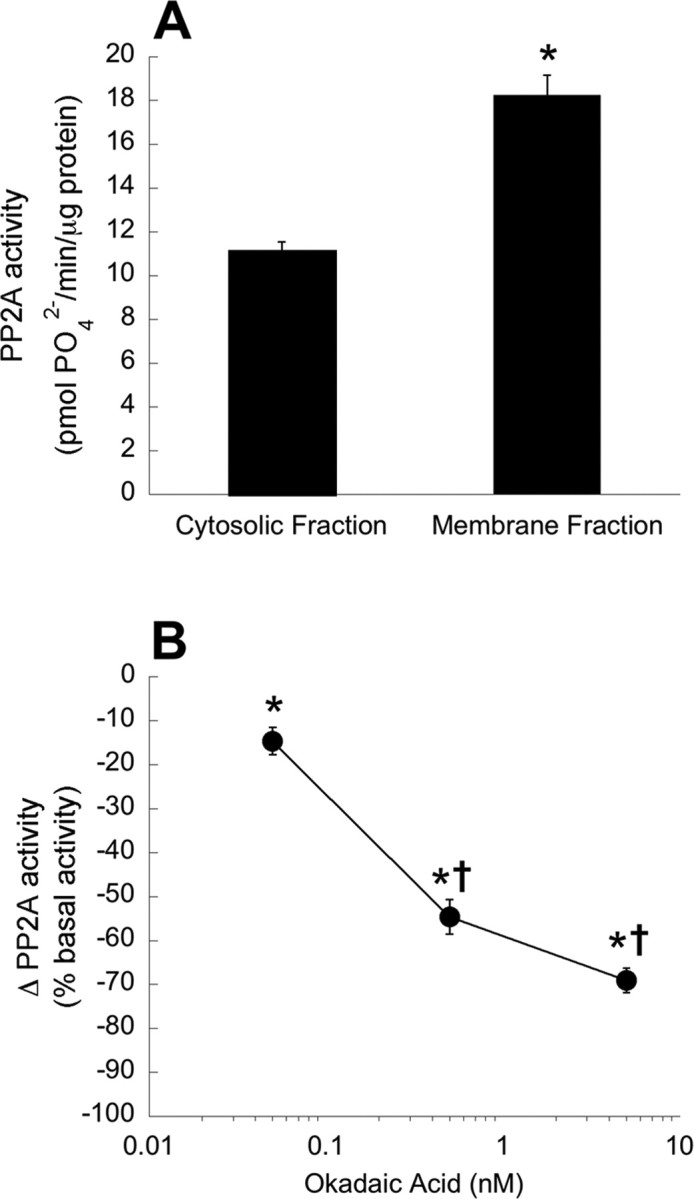
A, PP2A activity is present in cytosolic and membrane fractions of ventral cervical spinal cord homogenates (n = 5 per fraction). Data are presented as the mean ± SEM of picomoles of PO42− generated per minute per μg of protein. *Significantly increased compared with cytosolic fraction (p < 0.05). B, In vitro PP2A activity is dose-dependently decreased with okadaic acid. PP2A activity was measured in cytosolic fractions from ventral cervical spinal cords to which 0.05 nm (n = 11), 0.5 nm (n = 7), or 5 nm (n = 13) okadaic acid was added directly to the sample. Data are presented as the mean ± SEM of the change in PP2A activity as a percentage of basal PP2A activity (0 nm okadaic acid). *Significantly decreased from basal PP2A activity (p < 0.05). †Significantly decreased from 0.05 nm okadaic acid (p < 0.05).
Physiological variables
No significant differences were observed in the CO2 recruitment thresholds of any experimental group (intermittent hypoxia, 40 ± 1 mmHg; n = 13; intermittent hypoxia with okadaic acid, 39 ± 1 mmHg; n = 7; sustained hypoxia, 40 ± 1 mmHg; n = 11; sustained hypoxia with okadaic acid, 38 ± 1 mmHg; n = 10; sustained hypoxia with okadaic acid and methysergide, 40 ± 1 mmHg; n = 6; artificial CSF, 42 ± 1 mmHg; n = 12; 25 nm okadaic acid time controls, 40 ± 1 mmHg; n = 8; p > 0.05).
Relative to baseline, phrenic burst amplitude and burst frequency significantly increased during hypoxia (Table 1) (all p < 0.05). Rats treated with sustained hypoxia, okadaic acid, and methysergide showed a significant decrease in phrenic burst amplitude (percentage change from baseline) during hypoxia compared with rats treated with sustained hypoxia and okadaic acid only (p < 0.05); neither group was significantly different from rats treated with sustained hypoxia and artificial CSF (Table 1) (p > 0.05). Rats treated with sustained hypoxia and okadaic acid showed a significant increase in burst frequency relative to rats treated with sustained hypoxia and vehicle; neither group was significantly different from rats treated with intermittent hypoxia (Table 1). Time control groups, which did not receive hypoxia, showed no significant changes in phrenic burst amplitude or burst frequency at equivalent time points (Table 1) (all p > 0.05).
Table 1.
Phrenic short-term hypoxic response in burst amplitude and frequency in rats intrathecally injected with aCSF or 25 nm okadaic acid (25 nm OK)
| Treatment | n | Δ Burst amplitude (% baseline) | Δ Burst frequency from baseline (bursts/min) |
|---|---|---|---|
| TC (aCSF) | 12 | −1 ± 5 | −3 ± 2 |
| TC (25 nm OK) | 8 | 2 ± 4 | 0 ± 2 |
| IH (aCSF) | 13 | 108 ± 15*† | 11 ± 3*† |
| IH (25 nm OK) | 7 | 81 ± 13*† | 14 ± 3*† |
| SH (aCSF) | 11 | 90 ± 13*† | 9 ± 2*†§ |
| SH (25 nm OK) | 10 | 126 ± 32*† | 15 ± 3*† |
| SH (25 nm OK, methysergide) | 6 | 65 ± 10*†§ | 9 ± 3*†§ |
TC, Time control; OK, okadaic acid.
*Significantly different from baseline within treatment group.
†Significantly different from TC (aCSF and 25 nm OK).
§Significantly different from SH (25 nm OK).
Arterial blood pressure did not differ among experimental groups, nor were there differences in the blood pressure decrease during hypoxia (Table 2). There were no significant changes in blood pressure at equivalent time points in groups that were not exposed to hypoxia (p > 0.05). There was a general tendency for blood pressure to decrease with time after hypoxia, or in the groups not exposed to hypoxia, but these time-dependent changes did not differ among groups.
Table 2.
Temporal changes in mean arterial blood pressure and phrenic burst frequency in rats intrathecally injected with aCSF or 25 nm okadaic acid (25 nm OK)
| Treatment (Trt) | Mean arterial blood pressure |
Burst frequency |
||||
|---|---|---|---|---|---|---|
| Baseline | Trt | 60 min post-Trt | Baseline | Trt | 60 min post-Trt | |
| TC (aCSF) | 113 ± 6 | 114 ± 5 | 108 ± 5 | 44 ± 2 | 42 ± 2 | 46 ± 2 |
| TC (25 nm OK) | 117 ± 6 | 116 ± 6 | 106 ± 5 | 41 ± 3 | 40 ± 3 | 45 ± 2 |
| IH (aCSF) | 113 ± 5 | 69 ± 6* | 103 ± 5* | 38 ± 3 | 49 ± 2* | 42 ± 2 |
| IH (25 nm OK) | 106 ± 9 | 73 ± 13* | 94 ± 10 | 41 ± 1 | 56 ± 3* | 41 ± 3 |
| SH (aCSF) | 114 ± 5 | 68 ± 8* | 103 ± 4* | 43 ± 2 | 51 ± 2* | 46 ± 3 |
| SH (25 nm OK) | 112 ± 6 | 82 ± 8* | 99 ± 4* | 38 ± 3 | 53 ± 2* | 44 ± 4* |
| SH (25 nm OK) (methysergide) | 100 ± 9 | 69 ± 12* | 99 ± 8 | 40 ± 3 | 49 ± 4* | 43 ± 5 |
TC, Time control; OK, okadaic acid.
*Significantly different from baseline within treatment group.
To ensure that phrenic burst amplitude and frequency changes were not caused by alterations in arterial CO2, PaCO2 was strictly maintained within 1 mmHg of the baseline value throughout every experimental protocol.
Sustained hypoxia elicits pLTF after intrathecal okadaic acid
Representative tracings of phrenic nerve activity before, during, and after intermittent hypoxia, sustained hypoxia, or sustained hypoxia with intrathecal okadaic acid are shown in Figure 3. Mean responses after hypoxia (or time controls) are shown in Figure 4. Phrenic burst amplitude was significantly increased from baseline at 30 min (21 ± 11% baseline; p < 0.05) and 60 min (50 ± 9% baseline; p < 0.01) after intermittent hypoxia, confirming the development of pLTF (Fig. 4). In contrast, phrenic burst amplitude after sustained hypoxia was not significantly changed at either 30 min (1 ± 9% baseline) or 60 min (1 ± 8% baseline; both p > 0.5) (Fig. 4).
Figure 3.

Representative integrated phrenic neurograms before, during, and 60 min after intermittent (top) or sustained (middle, bottom) hypoxia in rats intrathecally injected with aCSF (top, middle) or 25 nm okadaic acid (OK; bottom). Relative to baseline (indicated by dotted line), phrenic burst amplitude was increased in aCSF- and OK-treated rats 60 min after intermittent and sustained hypoxia, respectively, but not in aCSF-treated rats exposed to sustained hypoxia.
Figure 4.

Serine/threonine protein phosphatase inhibition reveals pLTF after sustained hypoxia. Rats treated with intrathecal aCSF and IH (•) or 25 nm okadaic acid and sustained hypoxia (SH+OK; ■) showed increased phrenic burst amplitude after hypoxia, indicating pLTF. pLTF was not observed in rats treated with intrathecal aCSF and SH (♦) or rats treated with intrathecal aCSF (TC; ○) or 25 nm okadaic acid (TC+OK; □), but not hypoxia (time controls). Data are presented as mean ± SEM of the change in phrenic burst amplitude as a percentage of baseline. *Significantly increased from baseline (p < 0.05). †Significantly increased compared with aCSF- or okadaic acid-treated time controls and aCSF-treated rats with sustained hypoxia (p < 0.05).
In rats that received intrathecal okadaic acid (25 nm) followed by sustained hypoxia, phrenic burst amplitude was significantly increased above baseline at 30 min (34 ± 22% baseline; p < 0.05) and 60 min (52 ± 21% baseline; p < 0.01) after hypoxia, demonstrating that pLTF was revealed after sustained hypoxia in animals treated with okadaic acid (Fig. 4). At 60 min after hypoxia, pLTF in the intermittent and sustained hypoxia plus okadaic acid groups was significantly increased relative to sustained hypoxia alone (p ≤ 0.01), although these values were not significantly different 30 min after hypoxia (Fig. 4). pLTF in intermittent versus sustained hypoxia plus okadaic acid groups was not significantly different at any time point (p > 0.9).
Time control rats that received intrathecal aCSF (30 min, −1 ± 9% baseline; 60 min, 11 ± 6% baseline; p > 0.20) or 25 nm okadaic acid (30 min, 8 ± 11% baseline; 60 min, 5 ± 7% baseline; p > 0.60) without hypoxia did not show increased phrenic burst amplitude over a time course similar to hypoxia-treated rats (Fig. 4).
pLTF after sustained hypoxia with intrathecal okadaic acid requires serotonin receptor activation
Inhibition of okadaic acid-sensitive protein phosphatases alone is insufficient to elicit pLTF; sustained hypoxia is also required (Fig. 4). Thus, an additional factor is necessary, which we hypothesized to be serotonin receptor activation. In rats treated with intravenous methysergide (a broad spectrum serotonin receptor antagonist) and intrathecal okadaic acid (n = 6), sustained hypoxia was no longer able to elicit pLTF at 30 min (−4 ± 15% baseline; p > 0.05) or 60 min (−3 ± 13% baseline; p > 0.05) (Fig. 5A) after hypoxia, demonstrating that this form of pLTF requires serotonin receptor activation similar to intermittent hypoxia-induced pLTF (Baker-Herman and Mitchell, 2002).
Figure 5.
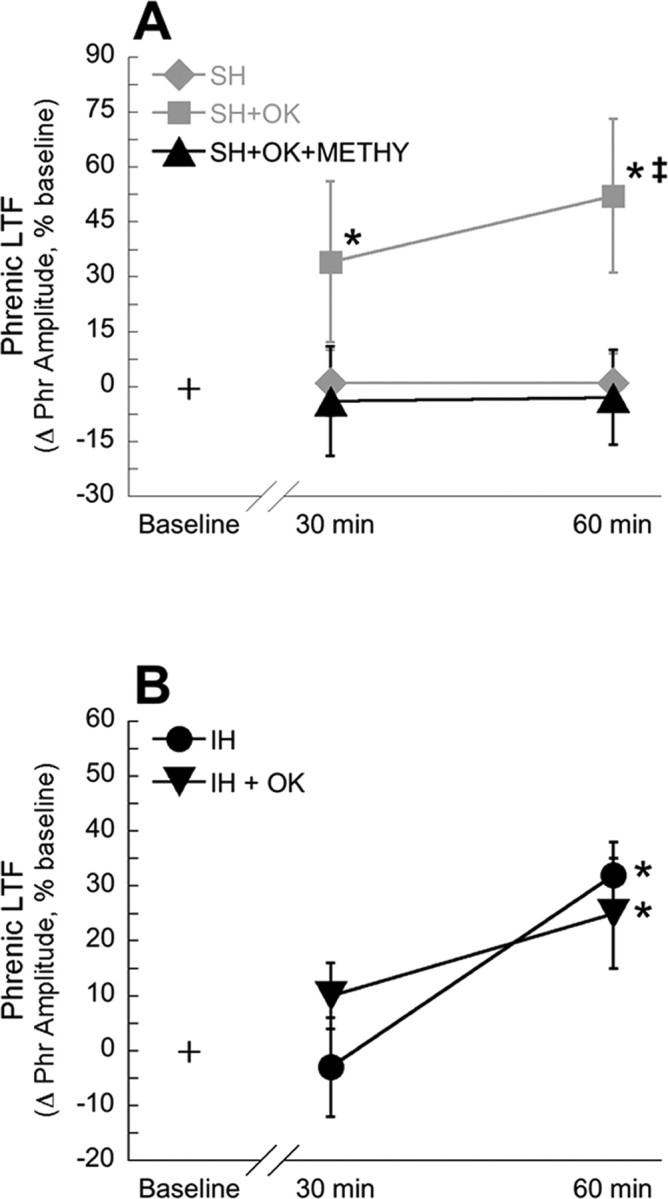
pLTF may be caused by similar mechanisms in rats treated with intermittent hypoxia or sustained hypoxia with concomitant protein phosphatase inhibition. A, Inhibition of serotonin receptors with methysergide abolishes pLTF in rats treated with intrathecal okadaic acid and sustained hypoxia (SH+OK+METHY; ▴). For comparison, data for rats treated with intrathecal aCSF and SH (♦) or 25 nm okadaic acid and sustained hypoxia (SH+OK; ■) are recapitulated from Figure 4. B, Okadaic acid does not alter pLTF in rats treated with intermittent hypoxia. Rats treated with intrathecal aCSF and IH (•) or 25 nm okadaic acid and intermittent hypoxia (IH + OK; ▾) showed increased phrenic burst amplitude after hypoxia, indicating pLTF. Data are presented as mean ± SEM of the change in phrenic burst amplitude as a percentage of baseline. *Significantly increased from baseline (p < 0.05). ‡Significantly increased relative to rats treated with okadaic acid, sustained hypoxia, and methysergide (p < 0.05).
Okadaic acid does not affect IH-induced pLTF
In a separate study, intrathecal okadaic acid did not affect pLTF in rats treated with intermittent hypoxia (n = 7). These animals developed significant pLTF 60 min after hypoxia (25 ± 10% baseline; p < 0.05) (Fig. 5B), but not 30 min after hypoxia (10 ± 6% baseline; p > 0.05) (Fig. 5B). Compared with rats that received intermittent hypoxia and artificial CSF (n = 4), the magnitude of pLTF was not significantly different (p > 0.05). These animals also developed pLTF 60 min after hypoxia (32 ± 6% baseline; p < 0.05), but not 30 min after hypoxia (−3 ± 9% baseline; p > 0.05) (Fig. 5B). Together, these data suggest that intermittent hypoxia inhibits spinal okadaic acid-sensitive protein phosphatases, thereby enabling pLTF. Thus, okadaic acid administration does not enhance IH-induced pLTF, because protein phosphatases are already inhibited. Overall, these data suggest that intermittent hypoxia and sustained hypoxia with concomitant protein phosphatase inhibition elicit similar mechanisms of pLTF.
Although okadaic acid did not increase the magnitude of pLTF relative to control animals studied at the same time, relatively low pLTF expression was observed in both control and okadaic acid-treated animals (Figs. 4, 5B, compare IH-induced pLTF). In previous studies of pLTF in our laboratory, we noted that multiple factors influence the magnitude of pLTF, including genetics (strain and substrain), age, sex, and prior experiences, such as preconditioning with intermittent hypoxia or neural injury (Baker et al., 2001; Mitchell et al., 2001; Behan et al., 2002; Mahamed and Mitchell, 2007). By having the same investigator perform concurrent experiments on control and okadaic acid-treated animals, any influence from these factors (which may explain variability in pLTF) does not affect the fundamental conclusions drawn. Thus, IH-induced pLTF is unaffected by intraspinal okadaic acid administration, most likely because spinal okadaic acid-sensitive protein phosphatases are already inhibited by intermittent hypoxia.
Frequency long-term facilitation
Whereas phrenic burst frequency significantly increased during hypoxia (Table 1), there was a small but significant increase in phrenic burst frequency observed 60 min after sustained hypoxia in rats treated with okadaic acid (7 ± 1 bursts/min) (Fig. 6) (p < 0.05). However, there was no significant frequency LTF in the other treatment groups (intermittent hypoxia, 4 ± 1 bursts/min; intermittent hypoxia with okadaic acid, 0 ± 3 bursts/min; sustained hypoxia, 3 ± 2 bursts/min; sustained hypoxia with okadaic acid and methysergide, 3 ± 3 bursts/min; no hypoxia with aCSF, 2 ± 1 bursts/min; no hypoxia with okadaic acid, 4 ± 2 bursts/min; p > 0.05 for all groups) (Fig. 6). No significant differences were observed between any treatment groups (p > 0.50), suggesting that frequency long-term facilitation is small and inconsistent, similar to other studies using the same experimental preparation (Baker-Herman et al., 2005).
Figure 6.

Serine/threonine phosphatase inhibition does not alter phrenic burst frequency long-term facilitation. Animals treated with okadaic acid and sustained hypoxia showed significant increases in delta burst frequency 60 min after hypoxia. Delta burst frequency did not significantly change in rats treated with intrathecal aCSF and IH (•) or SH (♦), sustained hypoxia and 25 nm okadaic acid (SH+OK; ■), sustained hypoxia and 25 nm okadaic acid with methysergide (SH+OK+METHY; ▴), or intermittent hypoxia and 25 nm okadaic acid (IH+OK; ▾). Non-hypoxia-treated rats that were intrathecally injected with aCSF (TC; ○) or 25 nm okadaic acid (TC+OK; □) did not have altered delta burst frequencies. Data are presented as mean ± SEM of the change in phrenic burst frequency from baseline. *Significantly increased from baseline (p < 0.05).
Discussion
pLTF is pattern sensitive because it is elicited by intermittent but not sustained hypoxia of similar cumulative duration (Baker and Mitchell, 2000). We explored the role of serine/threonine phosphatases in pLTF pattern sensitivity by testing the hypothesis that okadaic acid-sensitive spinal protein phosphatases constrain the expression of pLTF uniquely during or after sustained (but not intermittent) hypoxia. We report that relevant okadaic-acid sensitive protein phosphatases are present (PP1, PP2A, and PP5) and active (PP2A) in or near phrenic motor neurons. Furthermore, protein phosphatase inhibition with intrathecal okadaic acid reveals pLTF after sustained hypoxia, but has no effect on intermittent hypoxia-induced pLTF. pLTF elicited by intermittent hypoxia and sustained hypoxia in rats treated with okadaic acid appears to rely on similar cellular mechanisms, because (1) spinal protein phosphatase inhibition alone was not sufficient to induce pLTF, (2) pLTF after sustained hypoxia with intrathecal okadaic acid requires serotonin receptor activation, and (3) intrathecal okadaic acid does not influence pLTF expression after intermittent hypoxia, suggesting that protein phosphatase activity did not pose a constraint in this group. Collectively, these data support the hypothesis that pattern sensitivity of pLTF arises (at least in part) from differential effects of intermittent and sustained hypoxia on the protein phosphatase constraint to pLTF. This mechanism may underlie pattern sensitivity in other forms of pattern-sensitive neuroplasticity.
Protein phosphatases and phrenic LTF
The overall phosphorylation state (and hence, activity) of key proteins in the cellular signaling networks that underlie neural plasticity is determined by protein phosphatases and protein kinases (Thiels and Klann, 2001; Winder and Sweatt, 2001; Colbran, 2004). For example, intermediate-term memory for tail shock-induced sensitization in Aplysia, a pattern-sensitive form of plasticity, is constrained by protein phosphatases, specifically PP2B (Sharma et al., 2003). Similarly, PP2B and PP1 constrain hippocampal LTP (Brown et al., 2000; Malleret et al., 2001). Although our data do not yet allow a determination of the specific phosphatase involved in constraining pLTF, multiple okadaic acid-sensitive serine/threonine phosphatases are found throughout the CNS (Hashikawa et al., 1995; Strack et al., 1998; Schmidt et al., 2002), including respiratory motoneurons (Back and Gorenstein, 1994; Bocchiaro et al., 2003). At the okadaic acid concentrations used in this study, inhibition of protein phosphatases 1, 2A, 4, and 5 is possible (Honkanen and Golden, 2002), but not protein phosphatases 2B (i.e., calcineurin) or 2C or protein tyrosine phosphatases (Bialojan and Takai, 1988; Suganuma et al., 1992). Thus, we focused attention on the presence and activity of these okadaic acid-sensitive phosphatases in the region of the phrenic motor nucleus (Figs. 1, 2).
Several possibilities exist as to how okadaic acid reveals pLTF after sustained hypoxia. For example, sustained hypoxia may increase protein phosphatase activity in the cervical spinal cord, thereby suppressing pLTF. However, available literature reports are inconsistent with respect to this possibility, because some studies report that sustained hypoxia increases protein phosphatase activity and expression (Seta et al., 2001; Arsham et al., 2003; Yung and Tolkovsky, 2003; Zhou et al., 2004), whereas others report the opposite (Ashraf et al., 2004; Mishra and Delivoria-Papadopoulos, 2004; Truttmann et al., 2004).
Another possibility is that intermittent hypoxia uniquely inhibits protein phosphatase activity, thereby enabling pLTF. In support of this hypothesis, intrathecal okadaic acid had no effect on the magnitude of pLTF after intermittent hypoxia (Fig. 5B), whereas it revealed pLTF after sustained hypoxia (Figs. 3, 4). Thus, the requirement of phosphatase inhibition is already satisfied by intermittent hypoxia. One candidate mechanism for unique suppression of phosphatase activity by intermittent, but not sustained, hypoxia is differential ROS formation. Intermittent hypoxia increases superoxide anion formation relative to sustained hypoxia in PC12 cells (Yuan et al., 2004), and ROSs are known to exert powerful influences on the activity of potentially relevant protein kinases and protein phosphatases (Klann and Thiels, 1999). Furthermore, ROSs are necessary for several forms of neural plasticity (Klann, 1998; Knapp and Klann, 2002), including pLTF (MacFarlane and Mitchell, 2006). For example, spinal ROS formation is necessary for pLTF expression after intermittent hypoxia (MacFarlane and Mitchell, 2006), most likely via NADPH oxidase activation (MacFarlane et al., 2007).
Another possible explanation for the unmasking of pLTF after sustained hypoxia in okadaic acid-treated rats is an indirect increase in the activity of protein kinases critical for pLTF resulting from protein phosphatase inhibition. However, spinal phosphatase inhibition alone was not sufficient to elicit pLTF and had no effect on the magnitude of pLTF induced by subsequent intermittent hypoxia (Figs. 4, 5B). Thus, the most likely explanation for our data is that intermittent, but not sustained, hypoxia removes a phosphatase constraint, most likely via differential ROS formation. As a consequence, pLTF is revealed after sustained hypoxia only with coincident, pharmacological inhibition of those same phosphatases.
Working model of phrenic LTF and pattern sensitivity
Our updated working model of cellular/synaptic mechanisms that underlie pLTF is shown in Figure 7[for review, see Mitchell et al. (2001), Feldman et al. (2003), and Mahamed and Mitchell, (2007)]. In brief, hypoxia is postulated to activate descending respiratory premotor neurons that activate phrenic motor neurons, giving rise to the short-term hypoxic phrenic response (Fig. 3). Simultaneously, serotonergic raphe neurons are activated, releasing serotonin near phrenic motor neurons and activating postsynaptic 5-HT2 receptors (Fuller et al., 2001), thereby leading to activation of protein kinases, such as protein kinase C (McGuire and Ling, 2004). We postulate that protein kinase activation initiates new protein synthesis, including synthesis of brain-derived neurotrophic factor (BDNF); subsequent BDNF release would activate its high-affinity receptor tyrosine kinase, TrkB (Baker-Herman et al., 2004). TrkB receptor activation in turn activates protein kinases, such as extracellular regulated kinases 1 and 2 (ERK 1/2) and protein kinase B (Akt) that are postulated to regulate glutamatergic receptor density at the postsynaptic membrane and, thus, motor neuron responses to descending respiratory drive (Huang and Reichardt, 2003).
Figure 7.

Sensitivity of pLTF to the pattern of hypoxia: postulated mechanisms. Top, SH induces serotonin release in the phrenic motor nucleus to activate 5-HT2 receptors, but a phosphatase constraint is present, thereby inhibiting the protein kinase cascade that initiates pLTF. Middle, IH induces serotonin release in the phrenic motor nucleus, activating 5-HT2 receptors and suppressing protein phosphatase activity, thus enabling the protein kinase cascade leading to pLTF (protein kinase to BDNF to TrkB to pLTF). Bottom, Sustained hypoxia initiates the serotonergic component of pLTF, and phosphatase activity is suppressed pharmacologically by okadaic acid (OK), thus enabling pLTF expression. Bold letters and symbols indicate activated pathways; faded gray symbols and letters indicate suppressed components of the cellular network. For further explanation, see Discussion.
Many physiological systems are subject to inhibitory constraints. Thus, proteins critical for pLTF expression appear to be restrained by serine/threonine protein phosphatases. This is most apparent by the lack of pLTF after sustained hypoxia unless this phosphatase constraint is removed pharmacologically. Serine/threonine phosphatases may regulate pLTF at multiple points in the proposed mechanism, including neurochemical receptors, translation factors, and, most likely, protein kinases necessary for pLTF expression (Ichinose et al., 1990; Millward et al., 1999; Bauman et al., 2000; Munton et al., 2004). Protein phosphatases may also act presynaptically to regulate neurotransmitter release (Sistiaga and Sanchez-Prieto, 2000; Silverman-Gavrila et al., 2005). The techniques used in this study do not allow us to address the specific actions of the relevant serine/threonine protein phosphatases. Regardless, our study demonstrates that okadaic acid-sensitive protein phosphatases are present and active in the cervical spinal cord and exert important effects on the expression of pLTF.
Significance of LTF
Although the physiological relevance of LTF remains unclear, an important role in preserving breathing stability has been suggested (Mahamed and Mitchell, 2007). For example, LTF may offset the tendency for upper airway collapse during sleep, thereby minimizing the incidence of apneas (Mahamed and Mitchell, 2007). LTF may also reflect more general mechanisms that allow the respiratory control system to adapt in the face of deteriorating mechanics or gas exchange associated with aging or lung disease and may enable compensatory respiratory plasticity after spinal cord injury or during the progression of neurodegenerative disease (Mitchell, 2007).
Regardless of its specific physiological relevance, a detailed understanding of the cellular mechanisms that underlie LTF may enable the development of new therapeutic approaches for severe ventilatory control disorders such as obstructive sleep apnea and respiratory insufficiency after cervical spinal injury or during the progression of motor neuron disease (Mahamed and Mitchell, 2007; Mitchell, 2007). For example, by manipulating key molecules that regulate LTF, it may be possible to harness the inherent capacity for plasticity in respiratory motor neurons, thereby improving the disorder (Mitchell, 2007). The results of this study suggest that strategies targeting relevant protein phosphatases may increase the capacity for LTF, thereby strengthening respiratory motor output in disorders where it may be compromised. Phosphatase inhibition with okadaic acid is of limited therapeutic potential because it is carcinogenic. However, by targeting the specific phosphatases that regulate pLTF with, for example, RNAi, it may be possible to strengthen respiratory motor output without risk of cancer (Janssens and Goris, 2001; Honkanen and Golden, 2002). Further investigations to define the relevant phosphatases are therefore of considerable interest.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants HL80209 and HL69064. J.E.R.W. was supported by NIH Training Grant HL07654. We thank Bradley Hodgeman for expert technical assistance.
References
- Ajay SM, Bhalla US. A role for ERKII in synaptic pattern selectivity on the time-scale of minutes. Eur J Neurosci. 2004;20:2671–2680. doi: 10.1111/j.1460-9568.2004.03725.x. [DOI] [PubMed] [Google Scholar]
- Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–29660. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- Ashraf QM, Haider SH, Katsetos CD, Delivoria-Papadopoulos M, Mishra O. Nitric oxide-mediated alterations of protein tyrosine phosphatase activity and expression during hypoxia in the cerebral cortex of newborn piglets. Neurosci Lett. 2004;362:108–112. doi: 10.1016/j.neulet.2004.02.069. [DOI] [PubMed] [Google Scholar]
- Back SA, Gorenstein C. Differential response of neutral endopeptidase 24.11 (“enkephalinase”), and cholinergic and opioidergic markers to hypoglossal axotomy. J Comp Neurol. 1994;340:149–160. doi: 10.1002/cne.903400202. [DOI] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol (Lond) 2000;529:215–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Fuller DD, Zabka AG, Mitchell GS. Respiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapnia. Respir Physiol. 2001;129:25–35. doi: 10.1016/s0034-5687(01)00280-8. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Wilkerson JER, Golder FJ, Mitchell GS. Long-term facilitation of respiratory burst frequency following intermittent hypoxia is dependent on prehypoxia burst frequency. Soc Neurosci Abstr. 2005;31:352–8. [Google Scholar]
- Bauman AL, Apparsundaram S, Ramamoorthy S, Wadzinski BE, Vaughan RA, Blakely RD. Cocaine and antidepressant-sensitive biogenic amine transporters exist in regulated complexes with protein phosphatase 2A. J Neurosci. 2000;20:7571–7578. doi: 10.1523/JNEUROSCI.20-20-07571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan M, Zabka AG, Mitchell GS. Age and gender effects on serotonin-dependent plasticity in respiratory motor control. Respir Physiol Neurobiol. 2002;131:65–77. doi: 10.1016/s1569-9048(02)00038-1. [DOI] [PubMed] [Google Scholar]
- Belmeguenai A, Hansel C. A role for protein phosphatases 1, 2A, and 2B in cerebellar long-term potentiation. J Neurosci. 2005;25:10768–10772. doi: 10.1523/JNEUROSCI.2876-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J. 1988;256:283–290. doi: 10.1042/bj2560283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchiaro CM, Feldman JL. Synaptic activity-independent persistent plasticity in endogenously active mammalian motoneurons. Proc Natl Acad Sci USA. 2004;101:4292–4295. doi: 10.1073/pnas.0305712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchiaro CM, Saywell SA, Feldman JL. Dynamic modulation of inspiratory drive currents by protein kinase A and protein phosphatases in functionally active motoneurons. J Neurosci. 2003;23:1099–1103. doi: 10.1523/JNEUROSCI.23-04-01099.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PA, Keverne EB. Neural mechanisms of mammalian olfactory learning. Prog Neurobiol. 1997;51:457–481. doi: 10.1016/s0301-0082(96)00069-x. [DOI] [PubMed] [Google Scholar]
- Brown GP, Blitzer RD, Connor JH, Wong T, Shenolikar S, Iyengar R, Landau EM. Long-term potentiation induced by theta frequency stimulation is regulated by a protein phosphatase-1-operated gate. J Neurosci. 2000;20:7880–7887. doi: 10.1523/JNEUROSCI.20-21-07880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran RJ. Protein phosphatases and calcium/calmodulin-dependent protein kinase II-dependent synaptic plasticity. J Neurosci. 2004;24:8404–8409. doi: 10.1523/JNEUROSCI.3602-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal R, Locatelli F, Hermitte G, Maldonado H, Lafourcade C, Delorenzi A, Romano A. Kappa-B like DNA-binding activity is enhanced after spaced training that induces long-term memory in the crab Chasmagnathus. Neurosci Lett. 1998;242:143–146. doi: 10.1016/s0304-3940(98)00059-7. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. discussion 2000. [DOI] [PubMed] [Google Scholar]
- Goshgarian HG, Rafols JA. The phrenic nucleus of th albino rat: a correlative HRP and Golgi study. J Comp Neurol. 1981;201:441–456. doi: 10.1002/cne.902010309. [DOI] [PubMed] [Google Scholar]
- Gupta RP, Abou-Donia MB. Enhanced activity and level of protein kinase A in the spinal cord supernatant of diisopropyl phosphorofluoridate (DFP)-treated hens. Distribution of protein kinases and phosphatases in spinal cord subcellular fractions. Mol Cell Biochem. 2001;220:15–23. doi: 10.1023/a:1011010824252. [DOI] [PubMed] [Google Scholar]
- Hashikawa T, Nakazawa K, Mikawa S, Shima H, Nagao M. Immunohistochemical localization of protein phosphatase isoforms in the rat cerebellum. Neurosci Res. 1995;22:133–136. doi: 10.1016/0168-0102(95)00886-x. [DOI] [PubMed] [Google Scholar]
- Honkanen RE, Golden T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr Med Chem. 2002;9:2055–2075. doi: 10.2174/0929867023368836. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Ichinose M, Endo S, Critz SD, Shenolikar S, Byrne JH. Microcystin-LR, a potent protein phosphatase inhibitor, prolongs the serotonin- and cAMP-induced currents in sensory neurons of Aplysia californica. Brain Res. 1990;533:137–140. doi: 10.1016/0006-8993(90)91806-r. [DOI] [PubMed] [Google Scholar]
- Isabel G, Pascual A, Preat T. Exclusive consolidated memory phases in Drosophila. Science. 2004;304:1024–1027. doi: 10.1126/science.1094932. [DOI] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA. Blockade of hippocampal long-term potentiation by sustained tetanic stimulation near the recording site. J Neurophysiol. 1999;81:940–944. doi: 10.1152/jn.1999.81.2.940. [DOI] [PubMed] [Google Scholar]
- Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- Klann E, Thiels E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: implications for hippocampal synaptic plasticity. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:359–376. doi: 10.1016/s0278-5846(99)00002-0. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory? J Neurosci Res. 2002;70:1–7. doi: 10.1002/jnr.10371. [DOI] [PubMed] [Google Scholar]
- Launey T, Endo S, Sakai R, Harano J, Ito M. Protein phosphatase 2A inhibition induces cerebellar long-term depression and declustering of synaptic AMPA receptor. Proc Natl Acad Sci USA. 2004;101:676–681. doi: 10.1073/pnas.0302914101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Pei JJ, Wang XC, Zhou XW, Tian Q, Winblad B, Wang JZ. Acute anoxia induces tau dephosphorylation in rat brain slices and its possible underlying mechanisms. J Neurochem. 2005;94:1225–1234. doi: 10.1111/j.1471-4159.2005.03270.x. [DOI] [PubMed] [Google Scholar]
- Lovett-Barr MR, Mitchell GS, Satriotomo I, Johnson SM. Serotonin-induced in vitro long-term facilitation exhibits differential pattern sensitivity in cervical and thoracic inspiratory motor output. Neuroscience. 2006;142:885–892. doi: 10.1016/j.neuroscience.2006.06.036. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Respiratory long term facilitation evoked by acute intermittent hypoxia is impaired following intravenous injection of a superoxide dismutase mimetic. FASEB J. 2006;20:A372. [Google Scholar]
- MacFarlane PM, Satriotomo I, Mitchell GS. Reactive oxygen species generated by NADPH oxidase are necessary for phrenic long-term facilitation following acute intermittent hypoxia. FASEB J. 2007;21:918–10. [Google Scholar]
- Mahamed S, Mitchell GS. Does simulated apnea elicit respiratory long-term facilitation? 2006 Experimental Biology meeting abstracts. 2006 [on CD-ROM] Abstract 231.3. [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell. 2001;104:675–686. doi: 10.1016/s0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Mauelshagen J, Sherff CM, Carew TJ. Differential induction of long-term synaptic facilitation by spaced and massed applications of serotonin at sensory neuron synapses of Aplysia californica. Learn Mem. 1998;5:246–256. [PMC free article] [PubMed] [Google Scholar]
- McGuire M, Ling L. Activation of protein kinase C near/in phrenic motoneurons is required for phrenic long-term facilitation in rats. Am J Respir Crit Care Med. 2004;169:A433. Abstr Suppl. [Google Scholar]
- McKay LC, Janczewski WA, Feldman JL. Episodic hypoxia evokes long-term facilitation of genioglossus muscle activity in neonatal rats. J Physiol (Lond) 2004;557:13–18. doi: 10.1113/jphysiol.2004.064006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- Mishra OP, Delivoria-Papadopoulos M. Effect of hypoxia on the expression and activity of mitogen-activated protein (MAP) kinase-phosphatase-1 (MKP-1) and MKP-3 in neuronal nuclei of newborn piglets: the role of nitric oxide. Neuroscience. 2004;129:665–673. doi: 10.1016/j.neuroscience.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Mitchell GS. Respiratory plasticity following intermittent hypoxia: a guide for novel therapeutic approaches to ventilatory control disorders. In: Gaultier C, editor. Genetic basis for respiratory control disorders. New York: Springer; 2007. [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Munton RP, Vizi S, Mansuy IM. The role of protein phosphatase-1 in the modulation of synaptic and structural plasticity. FEBS Lett. 2004;567:121–128. doi: 10.1016/j.febslet.2004.03.121. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Duffy SN, Young JZ. Differential maintenance and frequency-dependent tuning of LTP at hippocampal synapses of specific strains of inbred mice. J Neurophysiol. 2000;84:2484–2493. doi: 10.1152/jn.2000.84.5.2484. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK. Oxidative stress in the systemic and cellular responses to intermittent hypoxia. Biol Chem. 2004;385:217–221. doi: 10.1515/BC.2004.015. [DOI] [PubMed] [Google Scholar]
- Scharf MT, Woo NH, Lattal KM, Young JZ, Nguyen PV, Abel T. Protein synthesis is required for the enhancement of long-term potentiation and long-term memory by spaced training. J Neurophysiol. 2002;87:2770–2777. doi: 10.1152/jn.2002.87.6.2770. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Kins S, Schild A, Nitsch RM, Hemmings BA, Gotz J. Diversity, developmental regulation and distribution of murine PR55/B subunits of protein phosphatase 2A. Eur J Neurosci. 2002;16:2039–2048. doi: 10.1046/j.1460-9568.2002.02274.x. [DOI] [PubMed] [Google Scholar]
- Seta K, Kim HW, Ferguson T, Kim R, Pathrose P, Yuan Y, Lu G, Spicer Z, Millhorn DE. Genomic and physiological analysis of oxygen sensitivity and hypoxia tolerance in PC12 cells. Ann NY Acad Sci. 2002;971:379–388. doi: 10.1111/j.1749-6632.2002.tb04500.x. [DOI] [PubMed] [Google Scholar]
- Seta KA, Kim R, Kim HW, Millhorn DE, Beitner-Johnson D. Hypoxia-induced regulation of MAPK phosphatase-1 as identified by subtractive suppression hybridization and cDNA microarray analysis. J Biol Chem. 2001;276:44405–44412. doi: 10.1074/jbc.M103346200. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Bagnall MW, Sutton MA, Carew TJ. Inhibition of calcineurin facilitates the induction of memory for sensitization in Aplysia: requirement of mitogen-activated protein kinase. Proc Natl Acad Sci USA. 2003;100:4861–4866. doi: 10.1073/pnas.0830994100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherff CM, Carew TJ. Parallel somatic and synaptic processing in the induction of intermediate-term and long-term synaptic facilitation in Aplysia. Proc Natl Acad Sci USA. 2004;101:7463–7468. doi: 10.1073/pnas.0402163101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman-Gavrila LB, Orth PM, Charlton MP. Phosphorylation-dependent low-frequency depression at phasic synapses of a crayfish motoneuron. J Neurosci. 2005;25:3168–3180. doi: 10.1523/JNEUROSCI.4908-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistiaga A, Sanchez-Prieto J. Protein phosphatase 2B inhibitors mimic the action of arachidonic acid and prolong the facilitation of glutamate release by group I mGlu receptors. Neuropharmacology. 2000;39:1544–1553. doi: 10.1016/s0028-3908(00)00034-4. [DOI] [PubMed] [Google Scholar]
- Strack S, Zaucha JA, Ebner FF, Colbran RJ, Wadzinski BE. Brain protein phosphatase 2A: developmental regulation and distinct cellular and subcellular localization by B subunits. J Comp Neurol. 1998;392:515–527. [PubMed] [Google Scholar]
- Suganuma M, Fujiki H, Okabe S, Nishiwaki S, Brautigan D, Ingebritsen TS, Rosner MR. Structurally different members of the okadaic acid class selectively inhibit protein serine/threonine but not tyrosine phosphatase activity. Toxicon. 1992;30:873–878. doi: 10.1016/0041-0101(92)90385-i. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ide J, Masters SE, Carew TJ. Interaction between amount and pattern of training in the induction of intermediate- and long-term memory for sensitization in aplysia. Learn Mem. 2002;9:29–40. doi: 10.1101/lm.44802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiels E, Klann E. Extracellular signal-regulated kinase, synaptic plasticity, and memory. Rev Neurosci. 2001;12:327–345. doi: 10.1515/revneuro.2001.12.4.327. [DOI] [PubMed] [Google Scholar]
- Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 2005;45:269–277. doi: 10.1016/j.neuron.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Truttmann AC, Ashraf Q, Mishra OP, Delivoria-Papadopoulos M. Effect of hypoxia on protein phosphatase 2A activity, subcellular distribution and expression in cerebral cortex of newborn piglets. Neuroscience. 2004;127:355–363. doi: 10.1016/j.neuroscience.2004.05.033. [DOI] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- Woo NH, Duffy SN, Abel T, Nguyen PV. Genetic and pharmacological demonstration of differential recruitment of cAMP-dependent protein kinases by synaptic activity. J Neurophysiol. 2000;84:2739–2745. doi: 10.1152/jn.2000.84.6.2739. [DOI] [PubMed] [Google Scholar]
- Woo NH, Abel T, Nguyen PV. Genetic and pharmacological demonstration of a role for cyclic AMP-dependent protein kinase-mediated suppression of protein phosphatases in gating the expression of late LTP. Eur J Neurosci. 2002;16:1871–1876. doi: 10.1046/j.1460-9568.2002.02260.x. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- Yuan G, Adhikary G, McCormick AA, Holcroft JJ, Kumar GK, Prabhakar NR. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J Physiol (Lond) 2004;557:773–783. doi: 10.1113/jphysiol.2003.058503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung HW, Tolkovsky AM. Erasure of kinase phosphorylation in astrocytes during oxygen-glucose deprivation is controlled by ATP levels and activation of phosphatases. J Neurochem. 2003;86:1281–1288. doi: 10.1046/j.1471-4159.2003.01946.x. [DOI] [PubMed] [Google Scholar]
- Zhou G, Golden T, Aragon IV, Honkanen RE. Ser/Thr protein phosphatase 5 inactivates hypoxia-induced activation of an apoptosis signal-regulating kinase 1/MKK-4/JNK signaling cascade. J Biol Chem. 2004;279:46595–46605. doi: 10.1074/jbc.M408320200. [DOI] [PubMed] [Google Scholar]