

Abstract
In the mature nervous system, changes in synaptic strength correlate with changes in neuronal structure. Members of the Nogo-66 receptor family have been implicated in regulating neuronal morphology. Nogo-66 receptor 1 (NgR1) supports binding of the myelin inhibitors Nogo-A, MAG (myelin-associated glycoprotein), and OMgp (oligodendrocyte myelin glycoprotein), and is important for growth cone collapse in response to acutely presented inhibitors in vitro. After injury to the corticospinal tract, NgR1 limits axon collateral sprouting but is not important for blocking long-distance regenerative growth in vivo. Here, we report on a novel interaction between NgR1 and select members of the fibroblast growth factor (FGF) family. FGF1 and FGF2 bind directly and with high affinity to NgR1 but not to NgR2 or NgR3. In primary cortical neurons, ectopic NgR1 inhibits FGF2-elicited axonal branching. Loss of NgR1 results in altered spine morphologies along apical dendrites of hippocampal CA1 neurons in vivo. Analysis of synaptosomal fractions revealed that NgR1 is enriched synaptically in the hippocampus. Physiological studies at Schaffer collateral–CA1 synapses uncovered a synaptic function for NgR1. Loss of NgR1 leads to FGF2-dependent enhancement of long-term potentiation (LTP) without altering basal synaptic transmission or short-term plasticity. NgR1 and FGF receptor 1 (FGFR1) are colocalized to synapses, and mechanistic studies revealed that FGFR kinase activity is necessary for FGF2-elicited enhancement of hippocampal LTP in NgR1 mutants. In addition, loss of NgR1 attenuates long-term depression of synaptic transmission at Schaffer collateral–CA1 synapses. Together, our findings establish that physiological NgR1 signaling regulates activity-dependent synaptic strength and uncover neuronal NgR1 as a regulator of synaptic plasticity.
Keywords: Nogo receptor, long-term potentiation, long-term depression, dendritic spine, synapse, FGF
Introduction
Long-term potentiation (LTP) and long-term depression (LTD) of synaptic transmission are opposing forms of synaptic plasticity that underlie aspects of learning and memory. LTP and LTD are regulated by multiple signaling pathways (Malenka and Bear, 2004), some of which regulate actin polymerization and changes in dendritic spine morphology (Lin et al., 2005). Dendritic spines are highly motile structures, and it is believed that their morphological plasticity reflects adaptive alterations in synaptic strength as a result of altered neural activity (Fischer et al., 2000; Yuste and Bonhoeffer, 2001). Neurotransmission regulates RhoA activity levels in dendritic spines, thereby causing changes in spine structure (Schubert and Dotti, 2007). In addition to neurotransmitter receptors, a growing number of nonglutamate receptors, including β-family integrins, trkB and ephrinB/EphB family members, have been shown to regulate spine actin dynamics by targeting Rho GTPases (Lamprecht and LeDoux, 2004).
There is evidence for extensive cross talk among actin regulatory pathways at multiple levels. For example, in the presence of brain-derived neurotrophic factor (BDNF), a positive modulator of LTP, subthreshold level theta burst stimuli are sufficient to induce LTP and increases in F-actin. However, BDNF alone has no effect on spine F-actin, demonstrating a synergistic interaction between different pathways that impinge on spine actin dynamics (Rex et al., 2007). The intricate relationship between spine actin structure and synaptic transmission suggests that many extracellular cues known to regulate the neuronal cytoskeleton may also influence spine structure and synaptic transmission.
The Nogo receptor family members Nogo-66 receptor 1 (NgR1) and NgR2 support high-affinity binding of myelin inhibitors and have been implicated in regulating neuronal structure (Xie and Zheng, 2008). NgR1 has been proposed to regulate RhoA activation through association with Lingo-1 and p75 or Taj/TROY (Yiu and He, 2006). In the mature CNS, NgR1 is abundantly expressed in projection neurons of the neocortex and hippocampus (Barrette et al., 2007) and is regulated in an activity-dependent manner (Josephson et al., 2003). Sensory deafferentation of somatosensory cortex leads to a downregulation of neuronal NgR1 expression in the cortex and correlates with increased cortical plasticity (Endo et al., 2007). Consistent with the idea that NgR1 limits neuronal plasticity, ocular dominance (OD) plasticity in NgR1 mutants is prolonged and continues into adulthood (McGee et al., 2005). The underlying molecular and cellular mechanisms of prolonged OD plasticity in NgR1 mutants, however, have not yet been defined, and the physiological role of NgR1 in the mature nervous system remains unknown.
Neurotrophic factors promote neuronal growth and plasticity, and may antagonize aspects of myelin inhibition by changing the intrinsic growth state of neurons. Fibroblast growth factors (FGFs) comprise a large family of polypeptides, members of which regulate a plethora of cellular processes during neural development and adulthood (Mason, 2007). The prototypic family member, FGF2, promotes axonal growth and sprouting after injury (Fagan et al., 1997) and has also been found to influence hippocampal synaptic plasticity (Terlau and Seifert, 1990).
Here, we report on a novel functional association between NgR1 and FGF2. In the hippocampus, physiological NgR1 signaling regulates dendritic spine morphology and activity-dependent synaptic plasticity.
Materials and Methods
cDNA constructs.
Human placental alkaline phosphatase (AP)-tagged fusion proteins were constructed by standard PCR cloning using the Tth-DNA polymerase. AP-Nogo-66-myc and AP-NiG have been described previously (Venkatesh et al., 2005). Additional constructs include AP-FGF2 (mouse), AP-FGF1 (human), AP-FGF4 (mouse), AP-FGF8 (mouse), AP-FGF9 (mouse), AP-FGF21 (mouse), and AP-vascular endothelial growth factor (VEGF)165 (a kind gift from M. Klagsbrun, Children's Hospital, Boston, MA).
Nogo receptor constructs for COS-7 cell binding studies shown in Figure 3 include rat NgR1 (construct I) and rat NgR3 (construct II) (Venkatesh et al., 2005); NgR1(C27–V311)/NgR3(S309–S424) fused by SpeI (construct III); NgR3(C25–P307)/NgR1(G314–G448) fused by SpeI (construct IV); NgR3(C25–V125)/NgR1(H131–G448) fused by HindIII (construct V); NgR1(C27–K277)/NgR2(V281–G399) fused by XbaI/NheI (construct VI); NgR2(C31–R278)/NgR1(G279–G448) fused by XbaI (construct VII). The receptor deletion construct NgR1Δstalk (lacking residues 373–448) was fused by XbaI to the NgR1 glycosylphosphatidylinositol (GPI) anchor consensus sequence.
Figure 3.
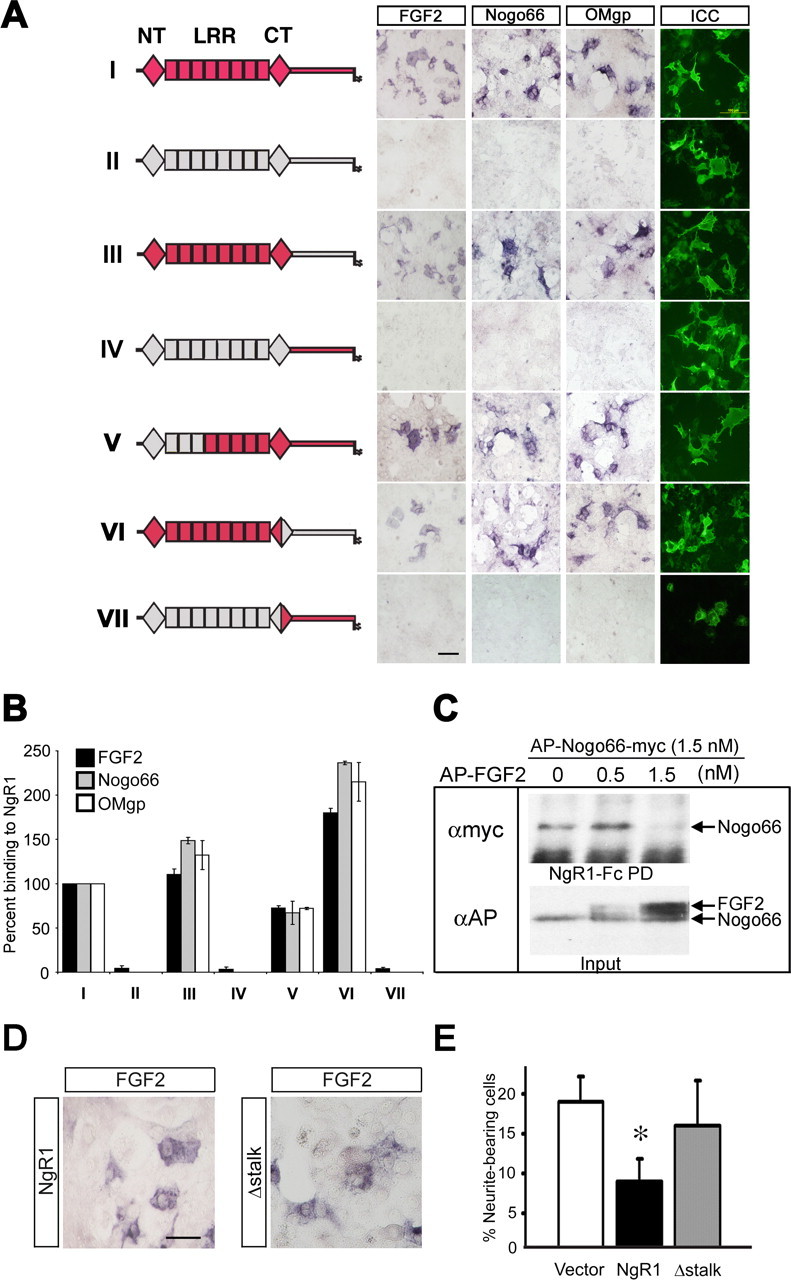
Structural basis of the NgR1–FGF2 association. A, Chimeric Nogo receptors were expressed on the surface of COS-7 cells and assayed for binding of AP-FGF2, AP-Nogo-66-myc, or AP-OMgp. NgR1 sequences are labeled in red and non-NgR1 sequences are labeled in gray. LRR, Leucine-rich repeats 1–8; NT, LRRNT cap domain; CT, LRRCT cap domain. Full-length NgR1 (construct I), but not NgR3 (construct II), supports binding of FGF2, Nogo-66, and OMgp. The NgR1 LRR cluster (composed of domains LRRNT-LRR-LRRCT) fused to the stalk region of NgR3 is sufficient to confer high-affinity ligand binding (see construct III). Construct IV, composed of the NgR3 LRR cluster and the NgR1 stalk region, does not support binding of FGF2, Nogo-66, or OMgp. Furthermore, the NgR1 LRRNT cap domain and the first three LRRs (construct V) and the NgR1 LRRCT distal portion and stalk region (construct VI) are dispensable for AP-FGF2, AP-Nogo-66-myc, or AP-OMgp binding. The NgR1 LRRCT distal region and stalk are not sufficient to support ligand binding (construct VII). Cell surface expression of chimeric Nogo receptor constructs was confirmed by ICC under nonpermeabilizing conditions. Scale bars: A, D, 30 μm. B, Quantification of ligand binding to chimeric Nogo receptors, normalized to wild-type NgR1 binding (=100%). Of note, the molecular basis for AP-FGF2-myc, AP-Nogo-66, and AP-OMgp is very similar. C, An NgR1-Fc pull-down (PD) assay was used for affinity precipitation of AP-Nogo-66-myc in the presence of increasing concentrations of AP-FGF2. AP-FGF2 competes with AP-Nogo-66-myc for NgR1 binding. D, Binding of AP-FGF2 to full-length NgR1 and NgR1Δstalk (Δstalk) transiently expressed on COS-7 cells. Binding of AP-FGF2 to wild-type NgR1 was normalized to 100%, and no significant change in AP-FGF2 binding was observed after deletion of residues T373–G448 of the NgR1 stalk (110 ± 9%). E, Experiments with PC12 cells stably expressing either NgR1 or NgR1Δstalk revealed that the NgR1 stalk region T373–G448 is important for the inhibition of FGF2-elicited PC12 cell differentiation. *p < 0.05, NgR1 versus vector. Error bars indicate SEM.
Pheochromocytoma cell cultures.
Rat pheochromocytoma (PC12) cells (a kind gift from David Ginty, The Johns Hopkins University, Baltimore, MD) were maintained in DMEM supplemented with 10% FBS and 5% horse serum. For transient expression of NgR1, cells were transfected using the Amaxa Biosystems (Köln, Germany) nucleofection technology using the U29 pulsing parameter according to the manufacturer's recommendations. Stable cells were obtained by transfection of PC12 cells with rat NgR1 or NgR1Δstalk cDNA followed by selection with G418 over several weeks. Cells expressing NgR1 were identified by anti-NgR1 immunocytochemistry (ICC), and expression levels were assayed by Western blot analysis (Venkatesh et al., 2005).
For differentiation experiments, PC12 cells were plated at low density (10,000 cells/well) in a 24-well plate coated with poly-lysine (50 μg/ml). The next morning, the medium was changed to low serum media (DMEM supplemented with 0.5% FBS and 0.25% horse serum), and after 24 h FGF2 (25 ng/ml; Peprotech, Rocky Hill, NJ) was added, and cells were kept in low serum medium for 4 d. Fresh growth factor was added after 2 d. Cells were fixed in 4% paraformaldehyde in PBS and immunostained with anti-NgR1 antibody under nonpermeabilizing conditions (Venkatesh et al., 2005) and then double-stained using TuJ1 (anti-β-tubulin III antibody; Promega, Madison, WI) in the presence of 0.1% Triton X-100.
Axonal branching of primary cortical neurons.
Embryonic day 18 (E18) rat cortex was dissociated in 0.05% trypsin in Neurobasal medium, 0.5 mm EDTA, and 0.01% DNase I, followed by gentle trituration in DMEM with 10% FBS. The cell suspension was centrifuged at 100 × g for 5 min, resuspended in fresh DMEM with 10% FBS, and transfected with NgR1 or eGFP plasmid DNA using the Amaxa nucleofector as described previously (Chivatakarn et al., 2007). Cells (1.5 × 105/well) were plated on six-well plates coated with poly-lysine (50 μg/ml) and laminin (10 μg/ml). After overnight culture, medium was changed to low B27 medium (Neurobasal medium containing 1 μl/ml B27 supplement, 25 mm glucose, 1 mm glutamine, 50 U/ml penicillin/50 μg/ml streptomycin). One day after transfection, FGF2 (15 ng/ml) was added, and cells were cultured for 2 d. The cultures were fixed in PBS containing 4% paraformaldehyde and 0.4 m sucrose in PBS for 30 min at room temperature (RT), blocked in PBS containing 2.5% horse serum for 30 min, and incubated with anti-NgR1 antibody and double-stained using TuJ1 as described above. For quantification of branches, pictures of dissociated cortical neurons were taken and the number of axon branches >20 μm in length was counted. Branch length was measured from digitized images taken by UTHSCSA Image Tool for Windows, version 3.0.
Ligand–receptor binding studies.
All AP-fusion proteins were expressed in transiently transfected HEK293T cells as described previously (Giger et al., 2000). COS-7 binding studies were performed as described previously (Venkatesh et al., 2005). Cell surface expression of recombinant Nogo receptors was confirmed by immunolabeling under nonpermeabilizing conditions or binding of myelin-associated glycoprotein (MAG)-Fc (R&D Systems, Minneapolis, MN). For pull down of FGF2 by NgR1, NgR1-Fc (0.5 μg; R&D Systems) and AP-tagged ligands (1.5 nm final concentration) were mixed in DMEM containing 0.1% BSA and protease inhibitor mixture (Sigma, St. Louis, MO) and then incubated at 4°C for 2 h. AP-ligands bound to NgR1-Fc were precipitated with protein A/G agarose beads after 2 h at 4°C. Samples were rinsed in washing buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% NP-40, 5 mm EDTA) and analyzed by Western blotting using anti-AP antibody (ARP, Belmont, MA). For cross-linking experiments, NgR1-Fc, FGF receptor (FGFR)1α(IIIb)/Fc, TROY-Fc, or ephrinB3-Fc (R&D Systems) (each at 13 nm), were incubated with 40 nm 125I-FGF2 (specific activity, ∼50 μCi/μg; MP Biochemicals, Irvine, CA) with or without the indicated concentrations of cold FGF2 or insulin for 2 h at RT in PBS. Cross-linking was initiated by adding BS3 (bis[sulfosuccinimidyl]suberate) (Pierce, Rockford, IL) to a final concentration of 2 mm and incubating for 30 min at RT. The reaction was stopped by adding 2 μl of 1 m Tris-HCl, pH 7.4, and protein complexes were analyzed by SDS-PAGE. Gels were subsequently fixed, dried, and subjected to autoradiography.
Histology and dendritic spine analysis.
Brain samples of wild-type and NgR1 mutant mice (Zheng et al., 2005) at postnatal day 71 (P71) to P104 were removed and submerged in Golgi-Cox solution and processed as previously described (Gibb and Kolb, 1998). In area CA1 of the dorsal hippocampus of each animal, apical dendrites of the stratum radiatum of 8–15 randomly selected pyramidal neurons were examined. These neurons were required to have no breaks in staining along the dendrites. Measurement occurred at least 50 μm away from the soma on secondary and tertiary branches. Approximately 30 dendritic branches (∼10 μm each) were analyzed from each brain. A single rater blinded with respect to the genotype of specimens did all analysis. Images were taken on an Olympus (Tokyo, Japan) BX60FS microscope at an optical magnification of 1000× and oil immersion. For spine morphology, spines were assigned the morphological category (Harris et al., 1992) that most resembled the shape of the spine. Spine density was calculated by dividing the number of spines on a segment by the length of the segment and was expressed as the number of spines per 10 μm of dendritic length. Means for the density of spines were analyzed using SigmaStat 3.5.
For electron microscopy, 6- to 8-week-old wild-type (n = 4) and NgR1 mutant mice (n = 4) were perfused in parafomaldehyde (2%) and glutaraldehyde (2%) solution at 37°C. Coronal slices were cut and brains were postfixed overnight at 4°C in the same fixative. Perfused brain coronal sections were bisected to separate the hemispheres, postfixed in 1.0% osmium peroxide buffered in 0.1 m sodium cacodylate for 1 h. Brain slices were rinsed in the same buffer (three times) and distilled water, placed into 50% ethanol, and stained in a solution of 0.5% uranyl acetate/50% ethanol overnight at 4°C. The sections were rinsed in 50% ethanol and dehydrated in a graded series of ethanol to 100%, transferred to propylene oxide, and embedded with the epoxy resin mixture of EPON/Araldite. The blocks were polymerized for 2 d at 70°C, sectioned with glass knives at 1 μm, and stained with toluidine blue to determine the area of hippocampus to be further processed by thin sectioning with a diamond knife onto 200 mesh grids. The grids were stained sequentially 10 min each in uranyl acetate and lead citrate.
A Hitachi (Tokyo, Japan) 7100 transmission electron microscope attached to a MegaView III (Olympus Soft Imaging Systems, Lakewood, CO) digital camera was used to generate images at 15,000× magnification. Approximately 25 electron micrographs were taken of the CA1 dendritic field and analyzed per animal. Digital images were converted to tif files and later processed in Adobe Photoshop 7.0.1. Clearly discernible synapses consisting of presynaptic terminal with synaptic vesicles and a spine with a defined postsynaptic density were counted. Perforated synaptic junctions and multiple synapses of one spine with more than one axon terminal were treated as one synapse. Mean synaptic densities were calculated for each animal and combined for each group. Student's t test was used to measure significance.
Western blot analysis.
To analyze the specificity of SU5402 (Mohammadi et al., 1997), PC12 cells plated on poly-lysine-coated 24-well plates were serum-starved in DMEM with 0.1% FBS for 24 h. Cells were treated with increasing concentrations of 3-[3-(2-carboxyethyl)-4-methylpyrrol-2-methylidenyl]-2-indolinone (SU5402) (0–25 μm) and epidermal growth factor (EGF) (2 ng/ml; Sigma) or FGF2 (10 ng/ml) for 5 min at RT, lysed in 2× Laemmli sample buffer, and subjected to Western blotting using anti-phospho-specific p44/42 extracellular signal-regulated kinase (ERK) antibody (Cell Signaling, Danvers, MA).
Isolation of synaptosomes.
Preparation of synaptosomes and subcellular fractionations were performed as described previously (Phillips et al., 2001). Briefly, rat hippocampi (adult or 2 weeks of age) were homogenized and synaptosomes were collected at the 1.25 m/1 m sucrose interface prepared in a sucrose gradient. From the isolated synaptosomes the extrasynaptic fraction was separated from the synaptic junction by extraction in 1% Triton X-100 at pH 6 and centrifugation at 40,000 × g for 30 min. The pellet comprising the synaptic junction fraction was further separated into presynaptic and postsynaptic fractions by extraction in 1% Triton X-100 at pH 8. Each fraction was adjusted to the same final protein concentration and separated by SDS-PAGE. Fractions were analyzed using antibodies specific for NgR1 and Lingo-1 (R&D Systems), NogoA (Millipore/Chemicon, Temecula, CA), FGFR1 and FGF receptor substrate 2α (FRS2α) (Santa Cruz, Santa Cruz, CA), Syntaxin 1A (StressGen, Ann Arbor, MI), postsynaptic density-95 (PSD-95) (Upstate, Lake Placid, NY), and synaptophysin (Sigma).
Electrophysiology.
All animal work was performed in compliance with the University of Rochester Committee on Animal Resources guidelines. NgR1 wild-type or mutant mice between 6 and 8 weeks of age were decapitated, and the brains were quickly removed and immediately placed in ice-cold artificial CSF (ACSF) (125 mm NaCl, 1.25 mm NaH2PO4, 25 mm glucose, 25 mm NaHCO3, 2.5 mm CaCl2, 1.3 mm MgCl2, 2.5 mm KCl saturated with 95% O2/5% CO2). Sagittal hippocampal slices (400 μm) were cut on a vibrating microtome and maintained in oxygenated ACSF at RT for at least 1 h. For recording, the slices were transferred to an immersion chamber, continuously perfused at 3 ml/min with oxygenated ACSF, and maintained at 32 ± 0.5°C.
Evoked field EPSPs (fEPSPs) were recorded from the CA1 stratum radiatum region. Briefly, a platinum/iridium concentric bipolar electrode (FHC, Bowdoinham, ME) was used to stimulate Schaffer collateral afferents. Recordings were taken with glass microelectrodes filled with ACSF (pipette resistance, ∼0.3–0.4 MΩ). Paired-pulse facilitation (PPF) was assessed at interpulse intervals of 25, 50, 100, 200, 300, 400, and 500 ms. Slices were monitored with stimuli consisting of constant current pulses of 0.1 ms duration at 0.067 Hz. After a baseline of ∼45 min (∼1 mV amplitude), LTP was induced at ≤50% of maximal amplitude by HFS (100 Hz; 1 s duration; two trains; interval, 10 s) as described previously (Meng et al., 2003). Slices that did not show a stable baseline for at least 30 min before stimulation were discarded.
For the induction of LTD, 3-week-old mice were used. MgCl2 and KCl concentrations in the ACSF solution were raised to 2.0 and 5.0 mm, respectively (Choi et al., 2005). A low-frequency stimulation of 900 pulses at 1 Hz was delivered with the stimulus intensity used during baseline recordings.
Recorded potentials were filtered at 3 kHz, digitized at 12.5 kHz, and stored for later analysis. fEPSPs were analyzed by fitting third-order polynomials to the sweeps, first to measure the peak and then to measure the slope at the 50% amplitude point. All fits were monitored visually on the oscilloscope screen. Data were normalized to the baseline average.
For local application, FGF2 and FGF8 (Peprotech) were diluted in ACSF to a final concentration of 10 μg/ml, loaded in the recording pipette, and delivered to CA1 directly through the recording pipette (Castro-Alamancos et al., 1995; Feldman, 2000; Pesavento et al., 2000). To visualize the diffusion and tissue distribution of focally applied molecules, we used Texas Red-conjugated dextran (molecular weight, 10 kDa) (Invitrogen, Carlsbad, CA) and pictures of the CA1 region were taken after 45 min using a Nikon (Tokyo, Japan) Diaphot (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). In addition, the NMDA receptor antagonist AP5 (d,l-2-amino-5-phospho-novalerate) (Tocris, Ellisville, MO) locally applied through the recording electrode (100 μm) significantly suppressed LTP, demonstrating successful drug delivery to the CA1 region in acute hippocampal slices (supplemental Fig. S3B,C, available at www.jneurosci.org as supplemental material). Data were analyzed statistically using Student's t test.
Results
Ectopic NgR1 attenuates FGF2-elicited differentiation of PC12 cells
NgR1 has been proposed to be part of a multicomponent receptor complex that also includes Lingo-1 and select members of the TNF (tumor necrosis factor) receptor superfamily, including p75 and Taj/TROY (Yiu and He, 2006). In vitro, ectopic expression of NgR1 confers Nogo-66 responsiveness on embryonic DRG neurons that are normally not inhibited by CNS myelin (Fournier et al., 2001). Conversely, loss of NgR1 in 2- to 4-week-old DRG neurons renders them more resistant toward Nogo-66-, MAG-, or oligodendrocyte myelin glycoprotein (OMgp)-mediated growth cone collapse (Kim et al., 2004; Chivatakarn et al., 2007). Interestingly, when myelin inhibitors are presented in substrate-bound form, NgR1 is not important for Nogo-66 (Zheng et al., 2005)-, OMgp (Chivatakarn et al., 2007)-, or MAG (Chivatakarn et al., 2007; Venkatesh et al., 2007)-mediated inhibition of neurite outgrowth. Moreover, recent evidence suggests that MAG, and perhaps other myelin inhibitors, employ cell type specific mechanisms for neurite outgrowth inhibition (Mehta et al., 2007; Venkatesh et al., 2007). To what extent and in which neuronal cell types p75 or TROY function as obligatory components in an NgR1 receptor complex to bring about growth cone collapse has not yet been tested. The restricted neuronal expression of p75 and virtual absence of neuronal TROY in the adult mammalian CNS (Barrette et al., 2007) argue for the existence of additional, as yet unidentified, coreceptors for NgR1.
Initially, to explore novel mechanisms for NgR1-mediated signal transduction, we embarked on a PC12 cell-based approach to examine whether ectopic expression of NgR1 leads to augmented responsiveness toward myelin inhibitors. Endogenous levels of NgR1 are very low in PC12 cells, and robust NgR1 expression is observed in transfected cells (data not shown). Differentiation and process outgrowth in PC12 cells was induced by FGF2 treatment. Consistent with previous reports, PC12 cells extend long neurite-like processes in the presence of FGF2 (Rydel and Greene, 1987). The number of green fluorescent protein (GFP)-transfected PC12 cells bearing neurites longer than two cell body diameters increased from 0% in the absence of FGF2, to 14 ± 2.6% in the presence of FGF2 (Fig. 1 A). To assay CNS myelin responsiveness, NgR1-transfected PC12 cells were plated on control substrate or CNS myelin. Remarkably, ectopic NgR1 significantly attenuated FGF2-elicited PC12 cell differentiation compared with the GFP-transfected cells (p < 0.05) even in the absence of exogenously applied myelin inhibitor (Fig. 1 A). The number of neurite-bearing PC12 cells transfected with NgR1 increased from 0% in the absence of FGF2, to 7.6 ± 1.4% in the presence of FGF2. This suggests that ectopic NgR1, by some unknown mechanism, interferes with FGF2-elicited neurite outgrowth in PC12 cells.
Figure 1.
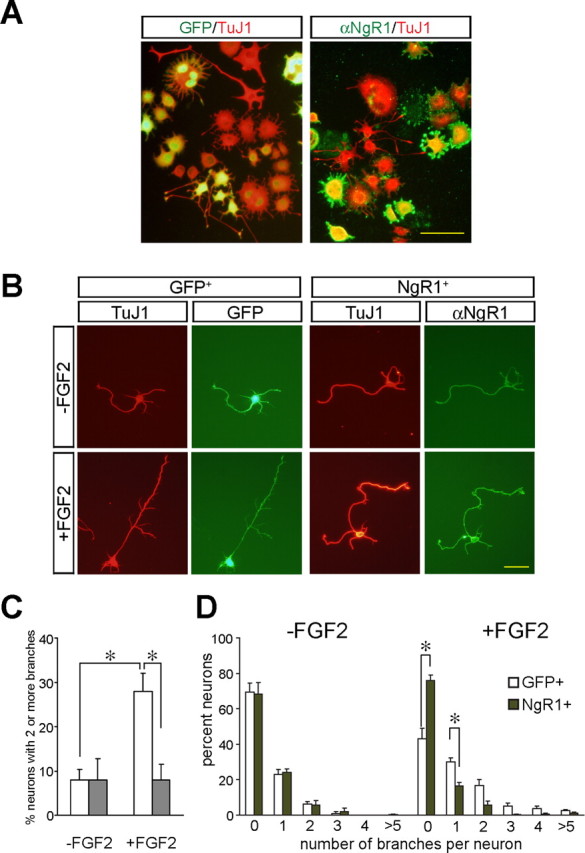
Ectopic expression of NgR1 blocks FGF2-elicited differentiation of PC12 cells and axonal branching in primary cortical neurons. A, FGF2-elicited differentiation is suppressed in PC12 cells. PC12 cells were transfected with NgR1 or eGFP plasmid DNA and then cultured in the presence of FGF2. Double immunofluorescence with anti-NgR1 and TuJ1 or anti-GFP and TuJ1 antibodies identified NgR1+ and GFP+ cells. Quantification of PC12 cell differentiation in the presence of FGF2 revealed significantly fewer processes in NgR1+ cells compared with GFP+ cells, whereas in the absence of FGF2 no cells with processes longer than two cell bodies in diameter were found. B, FGF2-mediated axon branching of rat E18 cortical neurons is suppressed in neurons transfected with NgR1. Neurons transfected with NgR1 or eGFP were cultured in the presence (+FGF2) or absence (−FGF2) of FGF2 and immunostained as described above. C, Quantification of neurons with two or more axonal branches observed in GFP+ (white) and NgR1+ (gray) neurons. D, Frequency histogram of axonal branches. In the presence of FGF2, GFP+ but not NgR1+ neurons show significantly enhanced axonal branching. Data plotted in C and D were from the same experiments. Error bars indicate SEM. *p < 0.05. Scale bars: A, 100 μm; B, 50 μm.
Ectopic NgR1 blocks FGF2-elicited axon branching in primary cortical neurons
Next, to independently assess the role of NgR1 in blocking FGF2 signaling, we assayed the ability of NgR1 to modulate axonal branching of primary cortical neurons (Szebenyi et al., 2001). E18 rat cortical neurons express low levels of endogenous NgR1. Bath-applied FGF2 elicits an axonal branching response within 2–3 d. After NgR1 transfection, cortical neurons produce high levels of NgR1, as assessed by anti-NgR1 immunofluorescence (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Quantification of primary axonal branches in enhanced GFP (eGFP)-transfected cultures showed an FGF2-mediated increase in branching of GFP+ neurons. The number of neurons with two or more branches increased from 7 to 28%. In marked contrast, FGF2 treatment failed to induce axonal branching in NgR1-transfected cortical neurons. With or without FGF2 treatment, only 8% of NgR1+ neurons showed two or more branches (Fig. 1 B,C). Quantification of GFP+ and NgR1+ neurons bearing 0, 1, 2, 3, 4, or >5 branches in the presence of FGF2 revealed a significant decrease in unbranched GFP+ neurons compared with NgR1+ neurons, and conversely, a significant increase in GFP+ neurons bearing one or more axonal branches compared with NgR1+ neurons (Fig. 1 D). Collectively, these results show that, in primary cortical neurons, ectopic NgR1 leads to a significant reduction of FGF2-elicited axonal branching.
NgR1 supports binding of select members of the FGF family
The newly identified functional link between NgR1 and FGF2 prompted us to further examine how NgR1 influences FGF2 signaling. To ask whether NgR1 associates with FGFRs, immunoprecipitation experiments were performed using PC12 cells and cortical neurons ectopically expressing NgR1. However, we found no evidence for an interaction between NgR1 and FGFR1 suggesting either a weak or more indirect link between the two receptors (data not shown). Next, to examine whether NgR1 modulates FGF2 binding to FGFRs, we generated AP-tagged FGF2. These studies revealed that NgR1 expressed in transiently transfected COS cells is sufficient to confer robust binding of AP-FGF2. Binding appears to be specific, because AP-FGF2 does not bind to NgR2 or NgR3 (Figs. 2 A, 3 A). Initial binding experiments were performed with the 18 kDa (154 aa) isoform of mouse FGF2. To explore the possibility that other members of the FGF family also bind to Nogo receptors, we generated AP-FGF1, AP-FGF4, AP-FGF8, AP-FGF9, and AP-FGF21 fusion proteins. Similar to FGF2, FGF1 binds robustly to NgR1 but not to NgR2. A much weaker association was found between NgR1 and FGF4. Under similar conditions, no association of FGF8, FGF9, or FGF21 with NgR1 was detected (Fig. 2 A). Scatchard plot analysis of AP-FGF1 and AP-FGF2 binding to NgR1 expressed on the surface of COS-7 cells revealed dissociation constants (K D values) of 8.3 ± 0.3 nm (FGF1) and 16.9 ± 0.9 nm (FGF2) (Fig. 2 B,C).
Figure 2.

NgR1 supports binding of select members of the FGF family. A, NgR1 expressed on the surface of COS-7 cells supports binding of select members of the FGF family. None of the FGF family members tested binds to NgR2. Binding of MAG-Fc to NgR1 and NgR2 is shown as a positive control. B, C, Scatchard plot analysis of AP-FGF1 (B) and AP-FGF2 (C) binding to NgR1 expressed in COS-7 cells. The insets show saturation binding curves. D, Pull-down experiments using NgR1-Fc and AP-fusion proteins revealed a direct interaction of NgR1 with FGF2 and Nogo-66, but not NiG, or VEGF. Excess IgG competes with NgR1-Fc for binding to protein A/G beads and blocks the pull down of AP-FGF2. E, Cross-linking of 125I-FGF2 to NgR1-Fc and FGFR1-Fc, in the presence or absence of excess “cold” FGF2 or insulin. Troy-Fc and ephrinB3-Fc were used as negative controls. Complexes of 125I-FGF2:NgR1-Fc (∼220 kDa) and 125I-FGF2:FGFR1-Fc (∼250 kDa) were resolved by 7% SDS-PAGE, and the dried gel was exposed to x-ray film. The arrowhead denotes radiolabeled complexes. Scale bar, 30 μm.
To address whether NgR1 interacts with FGF2 directly, we performed affinity precipitation experiments. NgR1-Fc selectively and specifically forms a complex with AP-FGF2 and AP-Nogo-66 but not with AP-VEGF165 or AP-NiG, an inhibitory fragment of amino-Nogo as shown by pull-down experiments (Fig. 2 D). In a parallel approach, we used 125I-FGF2 to independently confirm the specificity of the NgR1–FGF2 association. 125I-FGF2 can be cross-linked selectively to NgR1-Fc and FGFR1-Fc but not to TROY-Fc or ephrinB3-Fc. Moreover, formation of the 125I-FGF2 complex with NgR1-Fc or FGFR1-Fc is specific and efficiently competed by excess unlabeled FGF2 but not by insulin. The complexes of 125I-FGF2:NgR1-Fc and 125I-FGF2:FGFR1-Fc run at apparent molecular weights of 220 and 250 kDa, respectively. Higher molecular weight complexes containing 125I-FGF2 are detected as well (Fig. 2 E). Together, we identified novel and direct interactions between NgR1 and select members of the FGF family.
Molecular basis of the NgR1–FGF2 association
To study the molecular basis of the FGF2–NgR1 association, chimeric Nogo receptor mutants were constructed and expressed on the surface of COS-7 cells. The leucine-rich repeat (LRR) cluster of NgR1 (comprised of domains LRRNT-LRR-LRRCT) adopts a superhelical quaternary structure (Barton et al., 2003; He et al., 2003). We reasoned that chimeric receptor variants are more likely to maintain this structure and, thus, may be advantageous over receptor deletion constructs for the mapping of ligand binding epitopes. In parallel to AP-FGF2, we compared binding of AP-Nogo-66 and AP-OMgp to chimeric Nogo receptors. As shown in Figure 3, A and B, full-length NgR1 (construct I) but not NgR3 (construct II) supports binding of FGF2, Nogo-66, and OMgp. Experiments with chimeric Nogo receptor variants revealed that the NgR1 LRR cluster (residues 27–310) fused to the stalk region of NgR3 is sufficient to confer high-affinity AP-FGF2 binding (construct III). Conversely, a chimeric receptor composed of the NgR3 LRR cluster and the NgR1 stalk region (construct IV) does not support binding of FGF2, Nogo-66, or OMgp. Furthermore, the NgR1 LRRNT cap domain and the first three LRRs (residues C25–G130) are dispensable for AP-FGF2, AP-Nogo-66, or AP-OMgp binding (construct V). Also, the distal portion of the NgR1 LRRCT cap domain and the stalk region (F278–G448) are not necessary for ligand binding, indicating that residues H131–K277 of NgR1 harbor the binding sites for FGF2, Nogo-66, and OMgp (Fig. 3 A,B).
The interaction between AP-Nogo-66-myc and NgR1-Fc was inhibited in the presence of AP-FGF2 as shown by pull-down experiments (Fig. 3 C). The competition of the two ligands for NgR1 association suggests that either some binding epitopes are shared or located in close proximity. Previous studies have mapped Nogo-66 (Schimmele and Pluckthun, 2005) and OMgp (Lauren et al., 2007) binding to LRRs 4–7. Binding epitopes are located on the concave face of the curved NgR1 LRR clusters. Based on these previous studies, we propose a similar location for the FGF2 binding epitope(s) on NgR1.
Because NgR1 binds FGF2 directly and with high affinity, we wanted to exclude the possibility that ectopic NgR1 functions as a decoy FGF2 receptor that binds ligand nonproductively, thereby sequestering it away from FGFRs. To address this possibility, we generated PC12 cells stably expressing either full-length NgR1 or an NgR1 deletion mutant that lacks residues T373–G448 of the stalk (NgR1Δstalk). Consistent with our chimeric receptor binding studies, deletion of stalk sequences (Δstalk) does not alter FGF2 binding to NgR1 (Fig. 3 D). Compared with wild-type NgR1 (100%), binding of AP-FGF2 to NgR1Δstalk is increased to 110 ± 9%. Similar to transiently transfected PC12 cells, stable expression of full-length NgR1 suppressed extension of neurite-like processes in the presence of FGF2. This is in marked contrast to PC12 cells expressing NgR1Δstalk, which were found to extend neurite-like processes in the presence of FGF2 (Fig. 3 E). Based on these observations, we conclude that ectopic NgR1 does not simply function as a decoy receptor that competes with FGFRs for FGF2 binding. Furthermore, the NgR1 stalk region T373–G448 is important for attenuation of FGF2 signaling.
NgR1 mutant mice exhibit a dendritic spine phenotype
Because gain of NgR1 function in embryonic neurons inhibits FGF2-elicited axonal branching, we wondered whether loss of NgR1 function in mutant mice results in altered neuronal morphology in vivo. Consistent with a previous study (Kim et al., 2004), NgR1 mutant brains show no obvious defects at the gross anatomical level, as assessed by Nissl staining (Fig. 4 A). During embryonic development, neural expression levels of NgR1 are low but rapidly increase postnatally. In the hippocampus, for example, NgR1 protein levels increase rapidly in the first postnatal week, peak during the second week, and remain high throughout adulthood (Fig. 4 B). In the postnatal brain, FGF2 is robustly expressed in the hippocampus (Williams et al., 1996; Monfils et al., 2006). To examine neuronal structure in NgR1 mutants, we used Golgi staining. No apparent alterations in dendritic orientation or gross neuronal architecture were observed in adult NgR1 mutant hippocampus or neocortex (Fig. 4 C) (data not shown). NgR1 has been detected at presynaptic and postsynaptic sites (Wang et al., 2002); however, our analyses at the electron microscopic level revealed no significant change (p = 0.939) in synaptic density in the CA1 dendritic field between adult NgR1 wild-type (0.68 synapses/μm2 ± 0.05; 1730 synapses; n = 4 animals) and age-matched mutant mice (0.69 synapses/μm2 ± 0.06; 1864 synapses; n = 4 animals) (Fig. 4 G).
Figure 4.
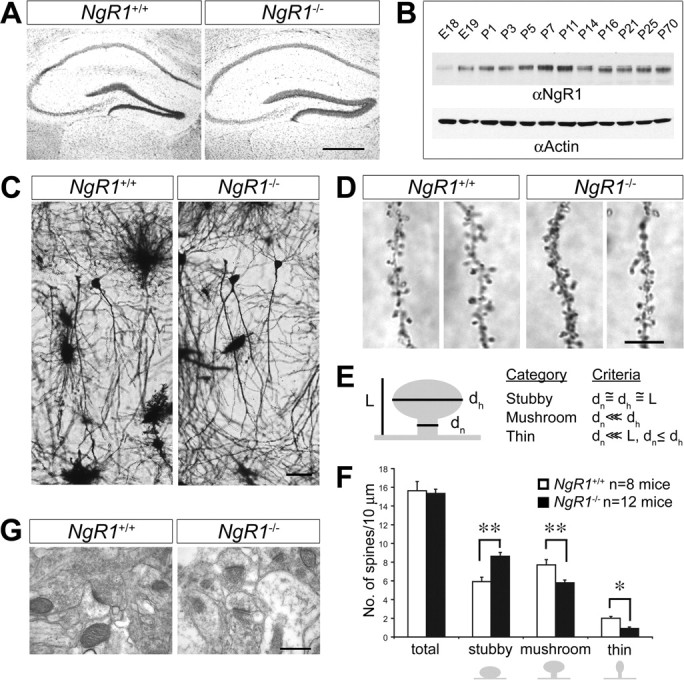
NgR1 mutant mice show altered distribution of CA1 dendritic spine morphologies in the adult hippocampus. A, Nissl staining of coronal sections of adult wild-type (NgR1 +/+) and mutant (NgR1 −/−) hippocampus. B, Time course of rat hippocampal NgR1 protein expression from E18 to P70 assessed by immunoblotting and normalized to actin. C, Golgi-Cox staining of dendrites of adult NgR1 +/+ and NgR1 −/− CA1 pyramidal neurons revealed no obvious morphological differences between the two genotypes. D, Representative images of dendritic spines of adult NgR1 +/+ and NgR1 −/− CA1 pyramidal neurons along apical dendrites. E, Morphological categories to which individual spines were assigned (Harris et al., 1992). F, Quantification of spine morphologies: assigning individual spines into different classes (stubby, mushroom, and thin) revealed a significantly altered spine distribution profile in NgR1 −/− (n = 12 mice) compared with NgR1 +/+ controls (n = 8 mice). **p < 0.001; *p < 0.05. G, Ultrastructural image of synapses in area CA1 of adult NgR1 +/+ (1730 synapses/n = 4 animals) and NgR1-null mutants (1864 synapses/n = 4 animals). Calculation of synapse density per square micrometer revealed no significant difference (p = 0.939) between NgR1 +/+ (0.68 ± 0.05) and NgR1 −/− (0.69 ± 0.06) mice. Scale bars: A, 200 μm; C, 25 μm; D, 5 μm; G, 0.2 μm. Error bars indicate SEM.
Next, we assessed dendritic spine morphology. Consistent with the ultrastructural analysis, Golgi impregnation revealed no changes in dendritic spine density along secondary or tertiary branches of CA1 apical dendrites of wild-type (15.5 spines/10 μm; n = 8 mice; 2356 spines) and NgR1 mutants (15.3 spines/10 μm; n = 12 mice; 5855 spines; p = 0.51) (Fig. 4 D,F). However, when the adult wild-type and NgR1 mutant dendritic spines were categorized into specific morphological spine categories (stubby, mushroom, or thin) (for details, see Fig. 4 E), analysis of spine morphologies revealed a shift in distribution toward more stubby (p < 0.001) and less mushroom-shaped (p < 0.001) and thin spines (p = 0.04) in NgR1 mutants compared with wild-type controls (Fig. 4 D,F; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The increase in stubby spines is primarily at the expense of mushroom-shaped and, to a lesser extent, of thin spines, implying that NgR1 function is necessary for the proper development or maintenance of mushroom-shaped spines. In summary, our anatomical studies show that NgR1 is an important regulator of dendritic spine structure in vivo.
NgR1 is enriched in synaptosomal membranes
The changes in spine morphology in NgR1 mutants suggest that NgR1 may play a role in spine maturation, maintenance, or stability in the hippocampus. Similar to NgR1, FGF2 expression in the hippocampus increases postnatally and persists throughout adulthood (Williams et al., 1996; Monfils et al., 2006). FGFR1 (but not FGFR2, FGFR3, or FGFR4) is expressed in CA1 pyramidal neurons in the adult hippocampus (Yazaki et al., 1994; Weickert et al., 2005). To examine whether NgR1 and FGFR1 signaling components are present at synapses, we isolated synaptosomal fractions of juvenile and adult rat hippocampi (Fig. 5). Synaptosomal fractions were further separated into extrasynaptic junction, synaptic junction, presynaptic and postsynaptic fractions. NgR1 is strongly enriched in synaptosomes compared with crude hippocampal tissue homogenate (Fig. 5). Furthermore, NgR1 is localized to presynaptic and postsynaptic sites, and in the adult hippocampus NgR1 protein levels are highest in the postsynaptic fraction (Fig. 5). Interestingly, FGFR1 and the FGFR docking protein FRS2α (Gotoh et al., 2004) are also enriched postsynaptically and expression levels are higher in the adult hippocampus. Nogo-A is also found presynaptically and postsynaptically but, unlike NgR1, appears not to be enriched at synapses. Notably, the previously identified NgR1 receptor component Lingo-1 shows little synaptic colocalization with NgR1 in the adult hippocampus. Lingo-1 is nearly exclusively found at presynaptic sites. Of note, p75 was not detectable in synaptosomal fractions of the adult hippocampus (data not shown). Together, these studies show that, in the hippocampus, NgR1 and FGFR1 are colocalized to postsynaptic sites.
Figure 5.

FGFR1 shows postsynaptic colocalization with NgR1. Hippocampal homogenate (homog) was used to isolate synaptosomes from 2-week-old and adult rats. Synaptosomes (synapto) were further separated into extrasynaptic junction (extra SJ), synaptic junction (SJ), presynaptic (pre), and postsynaptic (post) fractions and analyzed by immunoblotting. Fractions were probed with antibodies specific for NgR1, Lingo-1, Nogo-A, FGFR1, FRS2α, and the synaptic markers, Synaptophysin, Synthaxin 1A, and PSD-95. At both 2 weeks and in adulthood, NgR1 is enriched at synaptic junctions. In adult hippocampus, NgR1 is preferentially localized to postsynaptic sites and colocalizes with FGFR1 and FRS2α. Of note, Lingo-1 is almost exclusively found presynaptically. Nogo-A is found at synapses but is not enriched compared with crude hippocampal homogenate.
NgR1 regulates hippocampal LTP in an FGF2-dependent manner
Growing evidence suggests that altered dendritic spine structure impacts synaptic physiology, and reciprocally, changes in synaptic strength lead to altered spine morphology (Engert and Bonhoeffer, 1999). The previously reported role of FGF2 in modulating synaptic function in the hippocampus (Terlau and Seifert, 1990) coupled with the colocalization of NgR1, FGFR1, and FRS2α at postsynaptic sites prompted us to investigate whether physiological NgR1 signaling modulates synaptic transmission in the presence of exogenously applied FGF2.
Electrophysiological studies were conducted in acute hippocampal slices of 6- to 8-week-old NgR1 wild-type and mutant mice. Basal transmission at Schaffer collateral–CA1 synapses was unaltered between slices prepared from wild-type (n = 8 slices/5 animals) and NgR1-null (n = 10 slices/4 animals) mice as assessed by input/output (I/O) curves. I/O curves were constructed using three stimulus levels. No changes in single stimulus-evoked responses were observed between the two genotypes, suggesting that lack of NgR1 does not alter basal synaptic transmission (Fig. 6 A). To examine whether loss of NgR1 has an effect on long-term synaptic plasticity, we assessed LTP at the Schaffer collateral–CA1 synapses in acute hippocampal slices. To induce LTP, we applied two trains of high-frequency stimulation (HFS) (100 Hz; 1 s; separated by a 10 s interval) and measured the mean fEPSP slope as a percentage of baseline 40–45 min after stimulation. LTP of synaptic transmission in wild-type (mean fEPSP, 144 ± 4.5% of baseline; n = 11 slices/9 animals) and NgR1-deficient mouse slices (mean fEPSP, 145 ± 5.8%; n = 10 slices/9 animals) was robust and indistinguishable (Fig. 6 B,C). LTP saturation experiments induced by four trains of HFS (100 Hz; 1 s) separated by 10 min, revealed no significant difference between wild-type (fEPSP, 194 ± 10% of baseline; n = 3 slices/2 animals) and NgR1 mutants (fEPSP, 198 ± 15%; n = 3 slices/2 animals) (data not shown). Together, these experiments provide additional evidence that proper development and patterning of the hippocampal CA3–CA1 circuitry does not require NgR1 function, and importantly, loss of the NgR1 gene does not affect either induction or expression of LTP.
Figure 6.

FGF2 enhances hippocampal LTP in NgR1 mutants. Recording of fEPSPs at Schaffer collateral–CA1 synapses in acute hippocampal slices from NgR1 wild-type (+/+) and mutant (−/−) mice. A, Input–output curves for basal synaptic transmission revealed no differences between NgR1 +/+ and NgR1 −/− slices. B, Summary of LTP experiments in NgR1 +/+ and NgR1 −/− slices. fEPSPs were recorded at CA1 synapses and slopes were plotted against time before and after tetanic stimulation (2 trains of stimuli at 100 Hz for 1 s, separated by a 10 s interval; 144 ± 4.5 vs 145 ± 5.8%). C, Quantification of LTP at 40–45 min in NgR1 +/+ and NgR1 −/− slices revealed no difference in fEPSP slope ratio (p = 0.978). D, In the presence of FGF2 locally applied via the recording electrode, NgR1 −/− slices show significantly enhanced LTP compared with NgR1 +/+ (165 ± 4.6 vs 140 ± 9.4%; p < 0.05). E, Local application of FGF8, an FGF family member that does not bind to NgR1, does not result in enhanced LTP in NgR1 −/− slices (141 ± 5.7 vs 141 ± 5.9%). F, Quantification of LTP at 40–45 min in the presence of FGF2 and FGF8 in NgR1 +/+ and NgR1 −/− slices. Representative traces before and after LTP are shown as insets. Calibration: 0.5 mV, 5 ms. All values are mean ± SEM. *p < 0.05.
Because NgR1 modulates FGF2 signaling in PC12 cells and primary cortical neurons, we next examined whether synaptic function in NgR1 mutants is altered in the presence of FGF2. LTP experiments in hippocampal slices were repeated in the presence of FGF2, locally applied to the CA1 dendritic field through the recording pipette (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). In wild-type slices, focal application of FGF2 (10 μg/ml in the recoding pipette) did not result in a significant alteration of HFS-induced LTP (mean fEPSP, 140 ± 9.4% of baseline; n = 6 slices/4 animals) (Fig. 6 D,F) compared with no-ligand controls (Fig. 6 B,C). In marked contrast, NgR1-null hippocampal slices showed an FGF2-dependent enhancement of LTP (mean fEPSP, 165 ± 4.6%; n = 8 slices/6 animals; p < 0.05) (Fig. 6 D,F). To ask whether the observed increase in LTP is specific for FGF2, we repeated the experiments with FGF8, a FGF family member that does not bind to NgR1 (Fig. 2 A). In the presence of locally applied FGF8 (10 μg/ml in the recording pipette), LTP is not enhanced in NgR1 mutants (fEPSP, 141 ± 5.7%; n = 4 slices/3 animals) or wild-type slices (fEPSP, 141 ± 5.9%; n = 4 slices/3 animals) and is comparable with no-ligand controls (Fig. 6 C,F). These results indicate that the observed ligand-dependent increase in LTP in NgR1 mutants is FGF2-specific. Based on these findings, we propose that, during LTP, NgR1 negatively regulates FGF2 signaling at the CA3–CA1 synapse.
Hippocampal LTD is attenuated in NgR1 mutants
LTP and LTD are opposing forms of long-lasting changes in synaptic strength. LTD of excitatory synaptic transmission is a persistent weakening of synaptic strength that is involved in learning and memory processes and neuronal development (Feldman et al., 1999). To examine whether loss of NgR1 influences hippocampal LTD at CA3–CA1 synapses, acute slices from 3-week-old mice were subjected to low-frequency stimulation (LFS) (900 pulses at 1 Hz) to induce LTD. As shown in Figure 7, A and B, in wild-type slices a long-lasting depression of the fEPSP was observed compared with baseline (84.9 ± 2.6%; n = 7 slices/4 animals). The depression continued for >1 h and was significantly lower than baseline at 55–60 min (p < 0.001). In NgR1 mutants, the same stimulation paradigm did not lead to a significant depression of the fEPSP (p > 0.05). The fEPSP depression stabilized at a higher level (96.9 ± 4.6%; n = 7 slices/4 animals) compared with age-matched wild-type controls (Fig. 7). This suggests that NgR1 plays an important role in long-lasting synaptic depression at the CA3–CA1 synapse. Importantly, the altered LTD observed in NgR1 mutants is independent of exogenously applied ligand, providing evidence for a physiological role of NgR1 as a regulator of long-lasting changes in synaptic strength.
Figure 7.

Loss of NgR1 attenuates hippocampal LTD. A, Summary of LTD experiments in NgR1 +/+ and NgR1 −/− hippocampal slices after LFS (900 pulses; 1 Hz). Evoked fEPSP slope ratios are shown as a function of time. Representative traces before and after LTD are shown as insets. Calibration: 0.5 mV, 5 ms. B, Quantification of LTD at 55–60 min after LFS in NgR1 +/+ and NgR1 −/− reveals significantly reduced depression in NgR1 −/− slices compared with NgR1 +/+ slices (96.9 ± 4.6 vs 84.9 ± 2.6%; *p < 0.05). Error bars indicate SEM. C, D, PPF, the increase in the second fEPSP slope over the first, was calculated in NgR1 +/+ (C) and NgR1 −/− (D) slices in the presence (+) or absence (−) of locally applied FGF2. Mean values were plotted against different interpulse intervals (25–500 ms).
Paired-pulse facilitation is unaltered in NgR1 mutants
Previous immunohistochemical studies (Wang et al., 2002) and our biochemical analyses show that NgR1 is present at both presynaptic and postsynaptic sites and NgR1 could therefore conceivably modulate synaptic strength at either locus. To determine the locus of NgR1 function, we examined the effects of FGF2 on a presynaptically driven form of short-term plasticity, PPF, at Schaffer collateral–CA1 excitatory synapses in acute hippocampal slices. PPF measures transient enhancement of neurotransmitter release induced by two closely spaced stimuli attributable to accumulation of intracellular calcium (Schulz et al., 1994). We measured PPF at interstimulus intervals of 25–500 ms and observed facilitation at all intervals tested. There was no difference detected in PPF between wild-type (n = 11 slices/7 animals) and NgR1 mutants (n = 10 slices/5 animals). Furthermore, no significant change in PPF was found in either animal group on exposure to FGF2 (wild type, n = 8 slices/3 animals; NgR1 mutant, n = 8 slices/4 animals) (Fig. 7 C,D). We also examined posttetanic potentiation (PTP), thought to be caused by enhanced presynaptic transmitter release attributable to calcium loading of the presynaptic terminal after high-frequency stimulation (Zucker and Regehr, 2002). PTP measured over a 0.25–2.5 min interval after tetanization was not altered between wild-type and NgR1 mutants (data not shown). Together, these findings provide additional confirmation that loss of NgR1 causes no apparent changes in net excitatory hippocampal synaptic activity. Furthermore, these electrophysiological assays argue against a presynaptic role of NgR1, either in the presence or absence of FGF2.
FGFR kinase activity is necessary for enhanced LTP in NgR1 mutants
Because NgR1 is GPI-linked to the neuronal cell membrane, it is not clear how NgR1 attenuates FGF2 signaling. In previous studies, Lingo-1 (Mi et al., 2004) and p75 or TROY were implicated as signal transducing components in the NgR1 receptor complex (Yiu and He, 2006). In the adult hippocampus, TROY is not expressed in neurons (Barrette et al., 2007) and p75 is not localized to synaptic membranes (data not shown). Lingo-1 is present at synapses but is almost exclusively located at presynaptic sites (Fig. 5). To test whether FGFR signaling participates in FGF2-elicited enhancement of LTP in NgR1 mutants, we locally applied the FGFR kinase inhibitor SU5402 (Mohammadi et al., 1997) to the CA1 dendritic field via the recording electrode. Exposure to SU5402 in the absence of FGF2 does not alter HFS-induced LTP in either wild-type or NgR1-null hippocampal slices compared with vehicle-treated slices (Fig. 8 A–C). However, when FGF2 was present, SU5402 blocked the enhancement of LTP in NgR1-null slices (n = 5 slices/3 animals), indicating that FGFR kinase activity is necessary for FGF2-elicited enhancement of hippocampal LTP in NgR1 mutants (Fig. 8 D,E). As a control for the specificity of SU5402, we show dose-dependent inhibition of FGF2- but not the EGF-mediated activation of the ERK1/2 pathway in PC12 cells (Fig. 8 F).
Figure 8.

FGFR kinase activity is necessary for FGF2-enhanced LTP in NgR1 mutants. A–C, LTP is not significantly altered in NgR1 +/+ (A) and NgR1 −/− (B) slices in the presence of SU5402. Induction of LTP in the presence of the FGFR kinase inhibitor, SU5402, locally applied via the recording electrode (1 mm; concentration in recording electrode), in NgR1 +/+ (−SU5402, n = 11 slices/9 animals; +SU5402, n = 6 slices/4 animals) and NgR1 −/− slices (−SU5402, n = 8 slices/8 animals; +SU5402, n = 6 slices/3 animals). The small insets show traces. Calibration: 0.5 mV, 5 ms. C, Quantification of LTP at 45 min shown in A and B. D, The FGF2-elicited enhancement of LTP in NgR1 −/− hippocampal slices (165.4 ± 4.6%) is not observed in the presence of the FGFR kinase inhibitor SU5402 (137.0 ± 3.7%). fEPSPs were recorded as described for Figure 6. Representative traces before and after LTP are shown as insets. Calibration: 0.5 mV, 5 ms. E, Quantification of LTP at 40–45 min in NgR1 −/− slices after local application of FGF2 in the presence of SU5402 revealed a significant reduction of the fEPSP slope ratio compared with application of FGF2 alone (*p < 0.05). Error bars indicate SEM. F, Dose-dependent inhibition of FGF2- but not EGF-elicited ERK1/2 activation in PC12 cells treated with SU5402. Cell lysates were analyzed by anti-phospho-ERK1/2 blotting and normalized to actin.
Together, electrophysiological and biochemical experiments suggest that NgR1 functions postsynaptically. Furthermore, the enhanced LTP observed in NgR1 mutants is FGFR kinase dependent, indicating that NgR1 attenuates postsynaptic FGFR1 signaling in hippocampal CA1 pyramidal neurons. When coupled with experiments in primary cortical neurons and PC12 cells, NgR1 appears to function as a negative regulator of FGF2–FGFR signaling.
Discussion
Here, we report on the identification of NgR1 as a novel regulator of synaptic plasticity in the juvenile and adult mammalian CNS. In the hippocampus, NgR1 is enriched at synapses and regulates dendritic spine morphology of CA1 neurons in vivo. In addition, NgR1 regulates activity-dependent synaptic strength at Schaffer collateral–CA1 synapses. Loss of NgR1 leads to a decrease in LTD in acute hippocampal slices and to enhanced LTP in the presence of FGF2. Biochemical studies revealed that NgR1 associates with select members of the FGF family. NgR1 and FGFR1 are colocalized to postsynaptic sites and NgR1 functions as a negative regulator of FGFR signaling at Schaffer collateral–CA1 synapses. The newly identified role of NgR1 in synaptic plasticity implicates NgR1 signaling in learning and memory. Furthermore, NgR1 antagonism after injury may promote synaptic plasticity and, thus, result in behavioral improvements in the absence of long-distance regenerative axonal growth.
NgR1 is a novel regulator of FGF2–FGFR signaling
NgR1 is a receptor for multiple myelin inhibitors, all of which signal neuronal growth inhibition through activation of the RhoA/Rho-kinase pathway (McGee and Strittmatter, 2003). Functional studies with Nogo-66, OMgp, and MAG revealed that NgR1 is important to bring about neuronal growth cone collapse after acute presentation of soluble inhibitors (Kim et al., 2004; Chivatakarn et al., 2007). The identification of two neurotrophic factors, FGF1 and FGF2, as high-affinity NgR1 ligands may thus have come as a surprise. On closer examination, however, it became clear that NgR1 does not promote FGF2 signaling, but rather functions as an antagonist of FGF2 signaling.
Several intracellular and extracellular regulators of the FGFR–Ras–mitogen-activated protein kinase (MAPK) pathway have been identified. Members of the Sef, Sprouty, Spred, and MAPK phosphatase families are negative regulators of FGF signaling. Conversely, the LRR protein FLRT3 and the HSPG binding protein anosmin-1 are positive regulators of FGFR signaling (Mason, 2007). More recently, activation of EphA4 by FGFRs has been found to potentiate FGF2 signaling (Yokote et al., 2005). Here, we report on yet another regulatory mechanism for FGF2–FGFR1 signaling, NgR1 negatively regulates neuronal FGF2 signaling and, thus, may sensitize neuronal responses toward growth inhibitory cues.
NgR1 regulates dendritic spine morphology in vivo
Dendritic spine shape and dynamics are regulated by the actin cytoskeleton (Ethell and Pasquale, 2005). Rho-GTPases have been implicated in regulating spine actin dynamics, and abnormalities in spine morphology have been associated with brain disorders such as mental retardation, schizophrenia, and Alzheimer's disease (Fiala et al., 2002; Schubert et al., 2006). NgR1 mutant mice show a substantial increase in stubby spines and fewer mushroom-shaped and thin spines compared with wild-type controls. The profile of spines along CA1 apical dendrites in adult NgR1 mutants resembles the morphology profile found in more immature rodents as described previously (Harris et al., 1992), suggesting that NgR1 may play a role in dendritic spine maturation or stabilization.
The growing number of NgR1 ligands, coupled with the neuronal expression of Nogo-A and OMgp, suggests that ligands other than FGF family members may influence dendritic spine morphology in an NgR1-dependent manner. Because NgR1 signaling has been shown to regulate the actin cytoskeleton in 3- to 4-week-old neurons in vitro (Chivatakarn et al., 2007), we speculate that NgR1 located at postsynaptic sites regulates RhoA activity levels and, thereby, influences spine morphogenesis or dynamic changes (Sfakianos et al., 2007). Alternatively, the spine phenotype observed in NgR1 mutant mice may be primarily a reflection of altered synaptic strengths, which leads to altered spine structure (Engert and Bonhoeffer, 1999).
NgR1 regulates activity-dependent synaptic strength
Basal synaptic parameters are unchanged in NgR1 mutants, as assessed by I/O curves and PPF or PTP, two forms of short-term plasticity. LTP at Schaffer collateral–CA1 synapses, induced by HFS, is indistinguishable between NgR1 wild-type and mutant mice. In the presence of FGF2, however, LTP is significantly enhanced compared with wild-type controls. The enhancement of LTP in NgR1 mutants is FGF2 specific, because it is not observed in the presence of FGF8 or in no-ligand controls. Based on these observations, we propose that NgR1 functions as a ligand-dependent regulator of long-lasting activity-induced changes in synaptic strength. Although previous studies have reported FGF2-elicited enhancement of LTP in CA1 neurons after tetanic stimulation in rat (Terlau and Seifert, 1990), we did not observe FGF2-dependent changes in HFS-induced LTP in wild-type mice. So, does FGF2 function at the synapse? FGF2 is a secreted protein (Zehe et al., 2006) and its neural expression is upregulated by learning and physical activity (Gomez-Pinilla et al., 1997). Interestingly, neural activity (kainic acid) and exercise (wheel running) have been reported to cause a rapid and significant downregulation of NgR1 mRNA in the hippocampus and neocortex (Josephson et al., 2003). Thus, the inverse regulation of NgR1 and FGF2 expression in the hippocampus may facilitate FGF2-elicited long-lasting increases in synaptic strength.
In addition to its role in regulating ligand-dependent LTP, we found that NgR1 is important for the induction and maintenance of LTD in the juvenile hippocampus. Loss of NgR1 attenuates LTD and leads to a significantly smaller depression of CA1 neurons compared with wild-type controls. Importantly, the changes in CA1 synaptic depression in NgR1 mutants are independent of exogenously applied reagents. Thus, we provide evidence that physiological NgR1 signaling regulates hippocampal synaptic plasticity. Additional studies are needed to elucidate which ligand–NgR1 interactions regulate LTD.
A recent study showed that NgR1 in the visual system is important to consolidate the neural circuitry established during experience-dependent plasticity (McGee et al., 2005). It has been proposed that NgR1 inhibits structural rearrangements in the mature cortex and thereby limits OD plasticity. Several studies have reported that long-lasting changes in synaptic strength influence OD plasticity and that LTP and LTD, perhaps regulated in a cortical layer-specific manner, are important to establish proper neural connectivity (Daw et al., 2004). The newly identified role for NgR1 in regulating LTP and LTD suggests that failure to consolidate neural connectivity in the mature visual cortex may be a reflection of altered synaptic function.
NgR1 negatively regulates FGFR signaling during LTP
Evidence for a role of NgR1 in regulating FGFR signaling in the adult CNS stems from electrophysiological studies. The FGF2-elicited enhancement of LTP in NgR1 mutants is blocked in the presence of FGFR kinase inhibitor. Among the four FGFRs, only FGFR1 shows robust expression in the adult hippocampus (Yazaki et al., 1994). Consistent with a role of NgR1 in regulating FGFR1 signaling at the synapse, NgR1, FGFR1, and FRS2α, are enriched at postsynaptic sites in adult hippocampus. Despite their functional interaction and colocalization to postsynaptic sites, we were not able to demonstrate a direct link between neural NgR1 and FGFR1 by immunoprecipitation. FGFRs and NgR1 may associate weakly or indirectly. Commensurate with this idea, we recently found that, similar to FGF2 and FGFR1, NgR1 also binds to heparan sulfate proteoglycans (HSPGs) (O. Chivatakarn and R. Giger, unpublished observations), and it will be interesting to examine whether NgR1 and FGFR1 associate in an HSPG-dependent manner. Alternatively, NgR1 may not be part of a neuronal FGFR1 complex but regulate FGFR kinase activity more indirectly. Similar mechanisms have been proposed for cell adhesion molecules of the IgCAM superfamily, members of which depend on FGFR kinase activity to exert their growth-promoting activity without direct FGFR association (Walsh and Doherty, 1997).
NgR1 and synaptic plasticity: implications for nervous system regeneration
It is well established that severed adult mammalian CNS axons show very little, if any, regenerative axonal growth. Nevertheless, substantial spontaneous sensory-motor recovery can be achieved by means of various forms of neuroplasticity (Bareyre et al., 2004). Recovery is use dependent, and exercise substantially improves sensory and motor recovery after incomplete transection of the spinal cord. After sensory deafferentation, as a result of midthoracic spinal cord injury, compensatory adaptations take place at multiple levels, and likely include plasticity at spinal and various supraspinal levels. Cortical representations, for example, undergo adaptive changes leading to an expansion and invasion of the spared forelimb area into adjacent sensory deprived hindlimb territory (Raineteau and Schwab, 2001). Importantly, a recent study found that cortical plasticity observed in injured animals is closely linked to downregulation of NgR1 transcriptional activity in the sensory-deprived and adjacent cortical areas (Endo et al., 2007). When coupled with our current observation that loss of the NgR1 gene facilitates activity-dependent synaptic strength, downregulation of NgR1 in injury may similarly enhance synaptic transmission and promote adaptive intracortical communication between nondeprived and deprived cortical neurons.
Recent studies reported positive behavioral effects in injured rodents after NgR1 ablation (Kim et al., 2004; Cafferty and Strittmatter, 2006), and the question arises of what the underlying cellular substrates are for the observed functional improvement in the absence of long-distance axonal regeneration. Enhanced structural plasticity observed in NgR1-null mice after unilateral pyramidotomy leads to the formation of collateral axonal sprouts that emanate from injured and uninjured upper motor neurons. Whether, and to what extent, collateral axonal sprouting contributes to the improved behavior observed in these animals, however, is not known. Based on our current findings, we propose an alternate, but not mutually exclusive mechanism for behavior improvements observed in injured NgR1 mutants. Loss of NgR1 facilitates existing intrinsic mechanisms of synaptic plasticity and thereby promotes spontaneous repair mechanisms in the adult CNS. Enhanced neuroplasticity leads to improved functional recovery in the absence of long-distance axonal regeneration.
In summary, these results suggest a new role for NgR1 at the synapse and increase our understanding of how NgR1 and its ligands mediate their diverse effects in steady state, nervous system injury, and disease.
Footnotes
This work was supported by Ruth L. Kirschstein Fellowship F31-NS049870 (K.V.); National Institutes of Health Training Grant T32 NS07489 (K.V.); the Dr. Miriam and Sheldon G. Adelson Research Medical Foundation's Adelson Program in Neural Repair and Rehabilitation (R.J.G.); the New York State Spinal Cord Injury Research Program (P.S., R.J.G.); and National Institute of Neurological Disorders and Stroke Grants NS048603 (H.H.Y.), NS17965 (P.S.), and NS047333 (R.J.G.). We thank C.-H. Chan for help with Golgi staining; K. Bentley (EM Core), J. Barbieri, D. Welch, C. McArdle, Y. Zhang, D. Fowler, J. Lee-Osbourne, and M. Lefort for excellent technical assistance; C. Francis for the FGF1 and FGF2 cDNA clones; G. Martin for FGF4 and FGF8b cDNA clones; D. Ornitz for the FGF9 and FGF21 cDNA clones; M. Tessier-Lavigne for NgR1 −/− mice; and D. Ginty for PC12 cells.
References
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- Barrette B, Vallieres N, Dube M, Lacroix S. Expression profile of receptors for myelin-associated inhibitors of axonal regeneration in the intact and injured mouse central nervous system. Mol Cell Neurosci. 2007;34:519–538. doi: 10.1016/j.mcn.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Barton WA, Liu BP, Tzvetkova D, Jeffrey PD, Fournier AE, Sah D, Cate R, Strittmatter SM, Nikolov DB. Structure and axon outgrowth inhibitor binding of the Nogo-66 receptor and related proteins. EMBO J. 2003;22:3291–3302. doi: 10.1093/emboj/cdg325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Strittmatter SM. The Nogo-Nogo receptor pathway limits a spectrum of adult CNS axonal growth. J Neurosci. 2006;26:12242–12250. doi: 10.1523/JNEUROSCI.3827-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Donoghue JP, Connors BW. Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. J Neurosci. 1995;15:5324–5333. doi: 10.1523/JNEUROSCI.15-07-05324.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivatakarn O, Kaneko S, He Z, Tessier-Lavigne M, Giger RJ. The Nogo-66 receptor NgR1 is required only for the acute growth cone-collapsing but not the chronic growth-inhibitory actions of myelin inhibitors. J Neurosci. 2007;27:7117–7124. doi: 10.1523/JNEUROSCI.1541-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Chang J, Jiang B, Seol GH, Min SS, Han JS, Shin HS, Gallagher M, Kirkwood A. Multiple receptors coupled to phospholipase C gate long-term depression in visual cortex. J Neurosci. 2005;25:11433–11443. doi: 10.1523/JNEUROSCI.4084-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw N, Rao Y, Wang XF, Fischer Q, Yang Y. LTP and LTD vary with layer in rodent visual cortex. Vision Res. 2004;44:3377–3380. doi: 10.1016/j.visres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Endo T, Spenger C, Tominaga T, Brene S, Olson L. Cortical sensory map rearrangement after spinal cord injury: fMRI responses linked to Nogo signalling. Brain. 2007;130:2951–2961. doi: 10.1093/brain/awm237. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB. Molecular mechanisms of dendritic spine development and remodeling. Prog Neurobiol. 2005;75:161–205. doi: 10.1016/j.pneurobio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Suhr ST, Lucidi-Phillipi CA, Peterson DA, Holtzman DM, Gage FH. Endogenous FGF-2 is important for cholinergic sprouting in the denervated hippocampus. J Neurosci. 1997;17:2499–2511. doi: 10.1523/JNEUROSCI.17-07-02499.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Feldman DE, Nicoll RA, Malenka RC. Synaptic plasticity at thalamocortical synapses in developing rat somatosensory cortex: LTP, LTD, and silent synapses. J Neurobiol. 1999;41:92–101. [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A. Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat Neurosci. 2000;3:887–894. doi: 10.1038/78791. [DOI] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Gibb R, Kolb B. A method for vibratome sectioning of Golgi-Cox stained whole rat brain. J Neurosci Methods. 1998;79:1–4. doi: 10.1016/s0165-0270(97)00163-5. [DOI] [PubMed] [Google Scholar]
- Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, Pickering S, Simmons D, Rastan S, Walsh FS, Kolodkin AL, Ginty DD, Geppert M. Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron. 2000;25:29–41. doi: 10.1016/s0896-6273(00)80869-7. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Dao L, So V. Physical exercise induces FGF-2 and its mRNA in the hippocampus. Brain Res. 1997;764:1–8. doi: 10.1016/s0006-8993(97)00375-2. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Laks S, Nakashima M, Lax I, Schlessinger J. FRS2 family docking proteins with overlapping roles in activation of MAP kinase have distinct spatial-temporal patterns of expression of their transcripts. FEBS Lett. 2004;564:14–18. doi: 10.1016/S0014-5793(04)00287-X. [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XL, Bazan JF, McDermott G, Park JB, Wang K, Tessier-Lavigne M, He Z, Garcia KC. Structure of the Nogo receptor ectodomain: a recognition module implicated in myelin inhibition. Neuron. 2003;38:177–185. doi: 10.1016/s0896-6273(03)00232-0. [DOI] [PubMed] [Google Scholar]
- Josephson A, Trifunovski A, Scheele C, Widenfalk J, Wahlestedt C, Brene S, Olson L, Spenger C. Activity-induced and developmental downregulation of the Nogo receptor. Cell Tissue Res. 2003;311:333–342. doi: 10.1007/s00441-002-0695-8. [DOI] [PubMed] [Google Scholar]
- Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- Lauren J, Hu F, Chin J, Liao J, Airaksinen MS, Strittmatter SM. Characterization of myelin ligand complexes with neuronal Nogo-66 receptor family members. J Biol Chem. 2007;282:5715–5725. doi: 10.1074/jbc.M609797200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Kramar EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mason I. Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci. 2007;8:583–596. doi: 10.1038/nrn2189. [DOI] [PubMed] [Google Scholar]
- McGee AW, Strittmatter SM. The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci. 2003;26:193–198. doi: 10.1016/S0166-2236(03)00062-6. [DOI] [PubMed] [Google Scholar]
- McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309:2222–2226. doi: 10.1126/science.1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta NR, Lopez PH, Vyas AA, Schnaar RL. Gangliosides and Nogo receptors independently mediate myelin-associated glycoprotein inhibition of neurite outgrowth in different nerve cells. J Biol Chem. 2007;282:27875–27886. doi: 10.1074/jbc.M704055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Jia Z. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron. 2003;39:163–176. doi: 10.1016/s0896-6273(03)00368-4. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, McMahon G, Sun L, Tang C, Hirth P, Yeh BK, Hubbard SR, Schlessinger J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science. 1997;276:955–960. doi: 10.1126/science.276.5314.955. [DOI] [PubMed] [Google Scholar]
- Monfils MH, Driscoll I, Melvin NR, Kolb B. Differential expression of basic fibroblast growth factor-2 in the developing rat brain. Neuroscience. 2006;141:213–221. doi: 10.1016/j.neuroscience.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Pesavento E, Margotti E, Righi M, Cattaneo A, Domenici L. Blocking the NGF-TrkA interaction rescues the developmental loss of LTP in the rat visual cortex: role of the cholinergic system. Neuron. 2000;25:165–175. doi: 10.1016/s0896-6273(00)80880-6. [DOI] [PubMed] [Google Scholar]
- Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon RE, Gawinowicz MA, Zhao Y, Colman DR. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- Raineteau O, Schwab ME. Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci. 2001;2:263–273. doi: 10.1038/35067570. [DOI] [PubMed] [Google Scholar]
- Rex CS, Lin CY, Kramar EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–3029. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydel RE, Greene LA. Acidic and basic fibroblast growth factors promote stable neurite outgrowth and neuronal differentiation in cultures of PC12 cells. J Neurosci. 1987;7:3639–3653. doi: 10.1523/JNEUROSCI.07-11-03639.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmele B, Pluckthun A. Identification of a functional epitope of the Nogo receptor by a combinatorial approach using ribosome display. J Mol Biol. 2005;352:229–241. doi: 10.1016/j.jmb.2005.06.073. [DOI] [PubMed] [Google Scholar]
- Schubert V, Dotti CG. Transmitting on actin: synaptic control of dendritic architecture. J Cell Sci. 2007;120:205–212. doi: 10.1242/jcs.03337. [DOI] [PubMed] [Google Scholar]
- Schubert V, Da Silva JS, Dotti CG. Localized recruitment and activation of RhoA underlies dendritic spine morphology in a glutamate receptor-dependent manner. J Cell Biol. 2006;172:453–467. doi: 10.1083/jcb.200506136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci. 1994;14:5325–5337. doi: 10.1523/JNEUROSCI.14-09-05325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfakianos MK, Eisman A, Gourley SL, Bradley WD, Scheetz AJ, Settleman J, Taylor JR, Greer CA, Williamson A, Koleske AJ. Inhibition of Rho via Arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability, and behavior. J Neurosci. 2007;27:10982–10992. doi: 10.1523/JNEUROSCI.0793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi G, Dent EW, Callaway JL, Seys C, Lueth H, Kalil K. Fibroblast growth factor-2 promotes axon branching of cortical neurons by influencing morphology and behavior of the primary growth cone. J Neurosci. 2001;21:3932–3941. doi: 10.1523/JNEUROSCI.21-11-03932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlau H, Seifert W. Fibroblast growth factor enhances long-term potentiation in the hippocampal slice. Eur J Neurosci. 1990;2:973–977. doi: 10.1111/j.1460-9568.1990.tb00009.x. [DOI] [PubMed] [Google Scholar]
- Venkatesh K, Chivatakarn O, Lee H, Joshi PS, Kantor DB, Newman BA, Mage R, Rader C, Giger RJ. The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci. 2005;25:808–822. doi: 10.1523/JNEUROSCI.4464-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh K, Chivatakarn O, Sheu SS, Giger RJ. Molecular dissection of the myelin-associated glycoprotein receptor complex reveals cell type-specific mechanisms for neurite outgrowth inhibition. J Cell Biol. 2007;177:393–399. doi: 10.1083/jcb.200702102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh FS, Doherty P. Neural cell adhesion molecules of the immunoglobulin superfamily: role in axon growth and guidance. Annu Rev Cell Dev Biol. 1997;13:425–456. doi: 10.1146/annurev.cellbio.13.1.425. [DOI] [PubMed] [Google Scholar]
- Wang X, Chun S, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci. 2002;22:5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Kittell DA, Saunders RC, Herman MM, Horlick RA, Kleinman JE, Hyde TM. Basic fibroblast growth factor and fibroblast growth factor receptor-1 in the human hippocampal formation. Neuroscience. 2005;131:219–233. doi: 10.1016/j.neuroscience.2004.09.070. [DOI] [PubMed] [Google Scholar]
- Williams TE, Meshul CK, Cherry NJ, Tiffany NM, Eckenstein FP, Woodward WR. Characterization and distribution of basic fibroblast growth factor-containing cells in the rat hippocampus. J Comp Neurol. 1996;370:147–158. doi: 10.1002/(SICI)1096-9861(19960624)370:2<147::AID-CNE2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Xie F, Zheng B. White matter inhibitors in CNS axon regeneration failure. Exp Neurol. 2008;209:302–312. doi: 10.1016/j.expneurol.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki N, Hosoi Y, Kawabata K, Miyake A, Minami M, Satoh M, Ohta M, Kawasaki T, Itoh N. Differential expression patterns of mRNAs for members of the fibroblast growth factor receptor family, FGFR-1-FGFR-4, in rat brain. J Neurosci Res. 1994;37:445–452. doi: 10.1002/jnr.490370403. [DOI] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokote H, Fujita K, Jing X, Sawada T, Liang S, Yao L, Yan X, Zhang Y, Schlessinger J, Sakaguchi K. Trans-activation of EphA4 and FGF receptors mediated by direct interactions between their cytoplasmic domains. Proc Natl Acad Sci USA. 2005;102:18866–18871. doi: 10.1073/pnas.0509741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Zehe C, Engling A, Wegehingel S, Schafer T, Nickel W. Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc Natl Acad Sci USA. 2006;103:15479–15484. doi: 10.1073/pnas.0605997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Atwal J, Ho C, Case L, He XL, Garcia KC, Steward O, Tessier-Lavigne M. Genetic deletion of the Nogo receptor does not reduce neurite inhibition in vitro or promote corticospinal tract regeneration in vivo. Proc Natl Acad Sci USA. 2005;102:1205–1210. doi: 10.1073/pnas.0409026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]