

Abstract
Glaucoma is characterized by retinal ganglion cell (RGC) pathology and a progressive loss of vision. Previous studies suggest RGC death is responsible for vision loss in glaucoma, yet evidence from other neurodegenerative diseases suggests axonal degeneration, in the absence of neuronal loss, can significantly affect neuronal function. To characterize RGC degeneration in the DBA/2 mouse model of glaucoma, we quantified RGCs in mice of various ages using neuronal-specific nuclear protein (NeuN) immunolabeling, retrograde labeling, and optic nerve axon counts. Surprisingly, the number of NeuN-labeled RGCs did not decline significantly until 18 months of age, at which time a significant decrease in RGC somal size was also observed. Axon dysfunction and degeneration occurred before loss of NeuN-positive RGCs, because significant declines in RGC number assayed by retrograde tracers and axon counts were observed at 13 months. To examine whether axonal dysfunction/degeneration affected gene expression in RGC axons or somas, NeuN and neurofilament-heavy (NF-H) immunolabeling was performed along with quantitative reverse transcription-PCR for RGC-specific genes in retinas of aged DBA/2 mice. Although these mice had similar numbers of NeuN-positive RGCs, the expression of neurofilament light, Brn-3b, and Sncg mRNA varied; this variation in RGC-specific gene expression was correlated with the appearance of NF-H immunoreactive RGC axons. Together, these data support a progression of RGC degeneration in this model of glaucoma, beginning with loss of retrograde label, where axon dysfunction and degeneration precede neuronal loss. This progression of degeneration suggests a need to examine the RGC axon as a locus of pathology in glaucoma.
Keywords: RGC, death, axon, degeneration, optic nerve, axon
Introduction
Glaucoma, an optic neuropathy affecting nearly 60 million people (Quigley and Broman, 2006), is the second leading cause of blindness worldwide. Visual field changes in glaucoma are believed to be caused by the loss of retinal ganglion cells (RGCs), although the exact cause of RGC degeneration is still unknown (Marcic et al., 2003; Kuehn et al., 2005). A spontaneous model of glaucomatous neurodegeneration is available in DBA/2 mice that develop a progressive form of pigmentary angle-closure glaucoma (Chang et al., 1999; Anderson et al., 2002). Within these mice, intraocular pressure (IOP) becomes elevated by 6 months of age, and this elevation persists, resulting in progressive RGC degeneration over time. This is similar to what is observed in primary open angle glaucoma (John et al., 1998), making this an attractive model to the study of RGC neurodegeneration in the context of this disease.
Ganglion cell loss in glaucoma has been examined using techniques that rely on axon integrity and axonal transport, the expression of RGC-specific markers, and nuclear or cytoplasmic stains (Danias et al., 2003; Thanos and Naskar, 2004; Jakobs et al., 2005; Libby et al., 2005; Filippopoulos et al., 2006; Huang et al., 2006). Differences in the relative number of labeled RGCs before and after glaucomatous insult are interpreted as an indirect measure of RGC death. Significant RGC apoptosis has been observed in glaucoma models (Garcia-Valenzuela et al., 1995; Quigley, 1995; Mittag et al., 2000; Cordeiro et al., 2004; Libby et al., 2005), but evidence of RGC apoptosis in human glaucoma has been scarce (Kerrigan et al., 1997; Okisaka et al., 1997; Tatton et al., 2001). Interestingly, inhibiting apoptosis in the DBA/2 chronic glaucoma model prevented RGC loss, yet axonal degeneration still occurred (Libby et al., 2005).
Elevated IOP has been correlated to RGC axon degeneration (Mabuchi et al., 2003; Inman et al., 2006), although it is unknown whether axon pathology follows or initiates RGC loss. Evidence of axon degeneration occurring separate from and before somal degeneration has been observed in many neurodegenerative diseases (Coleman and Perry, 2002; Raff et al., 2002; Whitmore et al., 2005). Therefore, we set out to determine a timeline of RGC degeneration in the DBA/2 mouse using neuronal-specific nuclear protein (NeuN) immunohistochemistry, retrograde labeling, and optic nerve axon counts in DBA/2 mice of various ages. Ganglion cell number assayed by retrograde tracers and axon density decreased significantly by 13 months of age; however, RGC loss via NeuN immunolabeling, which was primarily RGC specific in DBA/2 retina, was not observed until 18 months. Similarly, RGC somal shrinkage was not significant until this time point. In a cohort of aged DBA/2 mice, decreased retinal expression of neurofilament light, Brn-3b, and Sncg (γ-synuclein) was observed, implying downregulation of a RGC-specific genetic program before RGC loss. Together, these data suggest RGC axonal changes precede RGC loss in this model, and that visual field changes in glaucoma may be caused by axonal dysfunction rather than by cell loss, therefore opening new therapeutic avenues in glaucoma research.
Materials and Methods
Subjects.
DBA/2 mice were originally obtained from The Jackson Laboratory (Bar Harbor, ME) and then bred and housed in a specific pathogen-free barrier facility at Harborview Medical Center (Seattle, WA). C57BL/6 mice at 5 months (The Jackson Laboratory) and 13 months (National Institute on Aging, Bethesda, MD) of age served as nonglaucoma controls. Mice were maintained in a 12 h light/dark cycle with standard rodent chow available ad libitum. The University of Washington Institutional Animal Care and Use Committee approved all experimental procedures.
Intraocular pressure measurement.
Intraocular pressure was measured in DBA/2 using the Tono-Pen XL (Medtronic Solan, Jacksonville, FL) as described previously (Inman et al., 2006). In brief, mice were anesthetized (Avertin, 1.3% tribromoethanol, 0.8% tert-amyl alcohol), and proparacaine (0.5% proparacaine hydrochloride ophthalmic solution; Bausch & Lomb, Tampa, FL) was applied to the eye before IOP measurement. Twenty Tono-Pen measurements were taken and recorded for each eye. IOP was recorded once per month between two and 10 months of age.
Labeling of RGCs.
For anterograde labeling of RGCs, mice were anesthetized with Avertin and placed supine in a plasticine support mold. A 32 ga Hamilton needle attached to a microinjection device was used to deliver 1 μl of 10 μg/ml Alexa Fluor 488 cholera toxin subunit B conjugate (CTB; Invitrogen, Carlsbad, CA) to the mouse eye posterior chamber. Injections took place over 2 min, and injections were made in both eyes. Retrograde RGC labeling was undertaken as described previously (Soto et al., 2008). In brief, anesthetized mice in a stereotaxic device had bilateral holes drilled over the superior colliculus, and 1 μl of 1% FluoroGold (FG) (hydroxystilbamidine, methanesulfonate; Invitrogen, Eugene, OR) in dH2O or 5% DiI (dialkylcarbocyanine dye; Invitrogen) was injected bilaterally via Hamilton syringe. Gelfoam pledgettes soaked in 5% FG or 5% DiI were also placed on the surface of the colliculus.
Immunohistochemistry.
Five days after CTB, FG, or DiI injection, mice were killed with 300 mg/kg sodium pentobarbital and then perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB), pH 7.4; eyes were dissected out, superior cut made, and the cornea and lens were removed before placing the eyecup in 4% PFA for 1 h. Eyecups were washed in PB, retinas dissected out, and vitreous removed using angled jeweler forceps and paint brushes. Retinas were cryoprotected by overnight incubation in 30% sucrose before three consecutive freeze-thaw cycles in 30% sucrose before immunolabeling. Retinas were rinsed three times (10 min each) in 0.1 m Tris-Base buffer (TB) (pH 7.4) and then blocked for 1 h in 5% donkey serum (Jackson ImmunoResearch, West Grove, PA) and 0.4% Triton X-100 (Sigma-Aldrich, St. Louis, MO) in TB (block solution). Tissue was incubated overnight in block solution and primary antibodies against NeuN (1:500; Chemicon, Temecula, CA); FG (1:500; Chemicon); phosphorylated (SMI-31) and/or nonphosphorylated (SMI-32) neurofilaments (1:500; Sternberger Monoclonals, Covance Research Products, Berkeley, CA); neurofilament-200 (1:500; Sigma-Aldrich); acetyl-cholinesterase (ChAT) (1:100; Chemicon); or neurofilament-70 (1:500; Chemicon). Retinas were then rinsed three times (10 min each) in TB, incubated for 30 min in block solution, and then incubated for 2 h in secondary antibodies diluted in blocking solution. Secondary antibodies were primary antibody species-specific IgG conjugated to either Cy2, Cy5, or RedX fluorophores (Jackson ImmunoResearch). Cell nuclei were labeled through incubation with 4′,6-diamidino-2-phenylindole (DAPI), whereas Nissl substance was labeled using NeuroTrace Nissl (Invitrogen) before final rinsing. After final rinsing (three times in TB), whole-mount retinas were mounted on SuperFrost slides (Fisher Scientific, Fairlawn, NJ); all slides were coverslipped in gelvitol.
In situ hybridization. Digoxigenin (Dig) and FITC-labeled probes for Sncg (γ-synuclein) and GAD-67 were synthesized and hydrolyzed by standard procedures. In situ hybridization (ISH) proceeded as described previously (Soto et al., 2008); in brief, frozen sections were digested (Proteinase K, 20 μg/ml in PBS), postfixed (4% PFA, 10 min), and acetylated (0.25% Acetic Anhydride in 0.1 m Triethanolamine), followed by hybridization containing 1 μg/ml Dig- and FITC-labeled riboprobes. Bound probes were detected by nitroblue–tetrazolium–chloride/5-bromo-4-chlor-indolyl-phosphate reaction (Roche, Indianapolis, IN) or with peroxidase-conjugated anti-Dig and anti-FITC antibodies (Roche) followed by Cy-3 (PerkinElmer, Boston, MA) and Alexa488 tyramides (Invitrogen). Sections were stained with DAPI, coverslipped, and imaged using a Zeiss (Thornwood, NY) LSM inverted confocal microscope.
Quantitative reverse transcription-PCR.
Whole retinas were removed from DBA/2 mice, and RNA was isolated and amplified as described previously (Steele et al., 2005). Reverse transcriptase reactions were performed starting with 500 ng of total RNA for each sample, also using methods described previously (Soto et al., 2008). The following primer sets were used: tau forward 5′-CCT GGA GGA GGG AAT AAG AAG ATT G-3′, tau reverse 5′-AAG ACA CAT TGC TGA GGT GCC G-3′; neurofilament light (NF-L) forward 5′-AGA AGA AGA AGA AGG TGG TGA GGG-3′, NF-L reverse 5′-TGG AAT AGT TGG GAA TAG GGC TC-3′; Sncg forward 5′-GTC TCA ACC TGG CAC ACT GAA TG-3′, Sncg reverse 5′-AGA GGA CCA TAG GGT AAA AGG AGC-3′; Brn3b forward 5′-GCA GTC TCC ACT TGG TGC TTA CTC-3′, Brn3b reverse 5′-TTC CCC CTA CAA ACA AAC CTC C-3′. All quantitative reverse transcription-PCR (qRT-PCR) was performed in triplicate using six independent retinal samples with cycling conditions as described previously (Soto et al., 2008). SDS2.1 software (Applied Biosystems, Foster City, CA) was used to visualize the data. The standard curve method described in Applied Biosystems (Foster City, CA) User Bulletin #2 was used to determine relative changes in gene expression levels using β-actin as a reference, which did not change significantly in our samples.
Stereology.
Unbiased stereological analysis of cell number was undertaken in whole-mount retinas using StereoInvestigator software (MicroBrightField, Middlebury, VT). The optical fractionator module within StereoInvestigator was used to quantify density of NeuN-positive, FG-positive, and DiI-positive RGCs. NeuN-labeled DBA/2 retinas were separated into eight sectors consisting of four central and four peripheral quadrants that followed the retinal poles (superior, inferior, nasal, and temporal). A predefined tracing of the central quadrants (radius, 1500 μm) was centered on the optic nerve head, and the peripheral quadrants were traced along the retinal periphery. Total RGC density was determined by summing the total areas for each of the eight sectors and dividing by the summed value of the stereological estimate of RGC population for all sectors. FG- and DiI-labeled retinas were traced along the retinal periphery and did not receive sector-specific analysis.
A consistent proportion of ChAT-positive displaced amacrine cells expressed detectible levels of NeuN. Optical fractionator was used to quantify the total number of NeuN-positive cells that were also ChAT-positive in the ganglion cell layer (GCL) of 3, 12, and 18 month DBA/2 retinas. The relative percentage of ChAT-labeled NeuN-positive cells for each age group was used as a correction factor to exclude displaced amacrine cells from the NeuN counts. All NeuN-positive cell counts were corrected, with the exception of the 6–10 month age group, by subtracting the appropriate percentage for each age group. NeuN quantification data are presented in both corrected and uncorrected format (see Figs. 2 and 3 for comparison).
Figure 2.
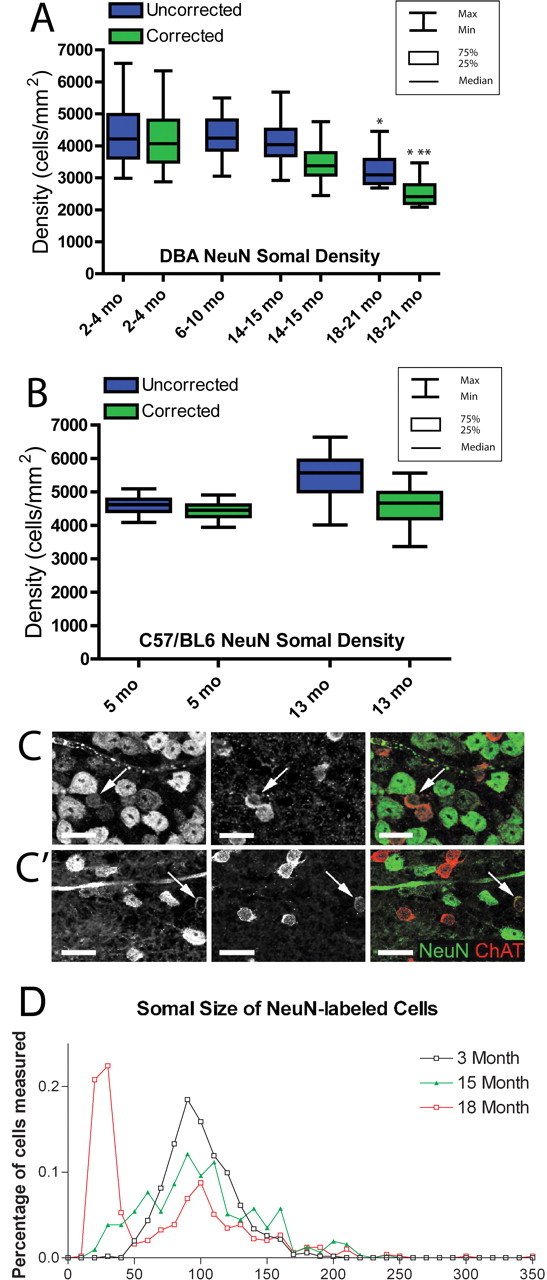
Unbiased stereological cell quantification and somal size measurements to assess RGCs across the disease spectrum of the DBA/2. A, Box plot graph showing the density of NeuN-positive cells in the GCL both before (blue) and after (green) application of the NeuN/ChAT-positive amacrine cell correction factor. NeuN-positive densities (green) include only NeuN-positive RGCs. Boxes extend from the 25th to the 75th percentile, with a line at the median cell density. Error bars show the highest and lowest values. The 2–4 month group has the largest range of NeuN-positive cell density, whereas median density is similar across all age groups until 18–21 months. The oldest group has significantly lower NeuN-positive cell density than all age groups (*p < 0.01). There was no significant difference in corrected NeuN-positive cell density between the 2–4 and 14–15 month groups, and the 18–21 month group had significantly lower corrected NeuN-positive cell density than both of the other corrected age groups. This difference was greater between the 2–4 month group (**p < 0.001) than the 14–15 month group (*p < 0.01). A correction factor was not created for 6–10 month retinas, so this group is represented by total NeuN-positive cells only. B, Box plot graph illustrating NeuN-positive cells in the GCL of C57BL/6 control retinas at 5 and 13 months of age. NeuN-positive cell densities are represented as in A. There is no significant difference in corrected NeuN-positive RGC density (p > 0.1). C, C′, Immunolabeling for NeuN and ChAT in DBA/2 whole-mount retina at 3 (C) and 18 (C′) months illustrating a visible reduction in both number and size of NeuN-positive cells by 18 months. Arrows indicate amacrine cells with both NeuN and ChAT immunofluorescence. Scale bar, 20 μm. D, Histogram of NeuN-positive somal area as measured from retinal whole mounts at 3, 15, and 18 months. The 3 month retinas have nearly 20% NeuN-positive cells with a somal area ∼100 μm2; this percentage decreased with age. From 15–18 months, there is a significant increase in the number of NeuN-positive cells with somal areas <50 μm2 (p < 0.05).
Figure 3.
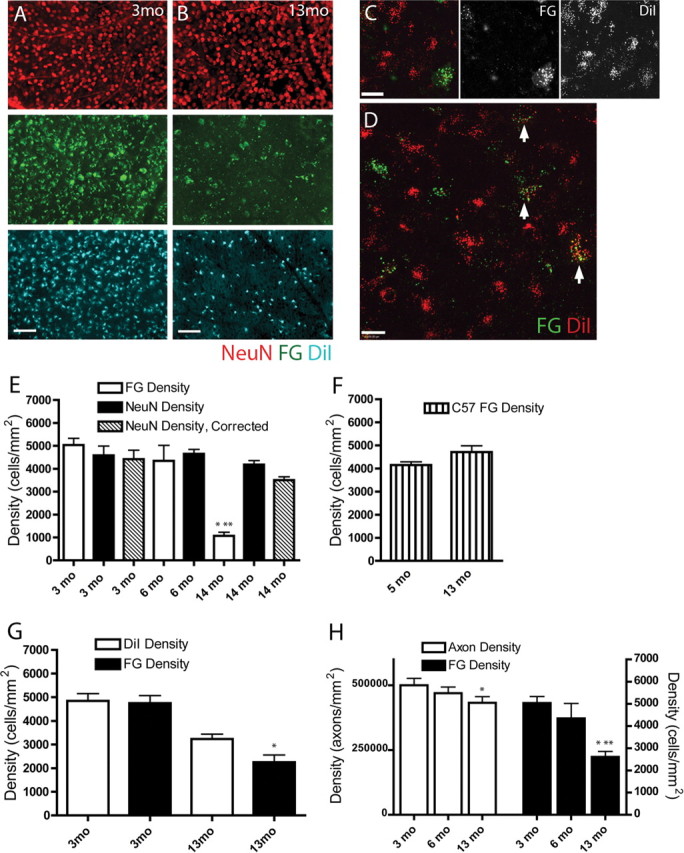
RGC axon pathology assessed by comparing two retrograde labeling methods. A, Separate fields from 3 month (3mo) whole-mount retina immunolabeled with NeuN (red) and retrogradely labeled with FG (green) and DiI (blue). NeuN, FG, and DiI showed similar numbers across all retinas at 3 months. Scale bar, 100 μm. B, By 13 months (13mo), FG labeling (middle panel, green) in all retinas was dramatically reduced, and DiI labeling (bottom panel, blue) was reduced to a lesser extent, whereas NeuN labeling was unaffected (top panel, red). Immunohistochemistry for FG was done to improve the tracer signal; puncta smaller than somal size in the FG panel are nonspecific secondary antibodies within the tissue. Scale bar, 100 μm. C, High-magnification image of typical whole-mount retinal field of RGCs retrogradely labeled with FG (green) and DiI (red) at 14 months showing more DiI-positive than FG-positive cells. The cell in the lower right is FG and DiI positive. Scale bar, 20 μm. D, High-magnification field of DiI-FG retrogradely labeled DBA/2 retina whole-mount. Arrows point to RGCs with varying levels of FG and DiI labeling. The majority of cells are DiI positive. Scale bar, 20 μm. E, Graph of RGC density in DBA/2 retinal whole mounts as quantified with NeuN immunolabel and FG tracer. Data from 3 month (3 mo) and 14 month (14 mo) retinas are presented with (hash marked) and without (black) the displaced amacrine cell correction factor. No correction factor was available for 6 month (6 mo) DBA/2 data. NeuN and FG labeled a similar number of RGCs at 3 and 6 months, but at 14 months, FG labeling was markedly reduced compared with earlier time points (*) and compared with NeuN immunolabel in 14 month retinas (**) (p < 0.01). F, Graph of FG-labeled RGC density in retinal whole mounts of 5 month (5 mo) and 13 month (13 mo) C57BL/6 normal controls. Unlike DBA/2 retinas, there was no reduction in FG-labeled RGCs in older C57BL/6 controls. G, Graph of RGC density in DBA/2 retinal whole mounts labeled with both FG and DiI. FG and DiI labeling was similar at 3 months (3mo). By 13 months (13mo), there was a visible but not statistically significant decrease in DiI-positive RGCs, whereas the FG-positive (*) RGCs were significantly reduced (p < 0.05). H, Counts of RGC axons in the optic nerve from FG-quantified retinas showed a significant decrease by 13 months (*) (p < 0.05). FG retrograde labeling of RGCs at 13 months was significantly decreased relative to FG labeling at both 3 (*) and 6 (**) months (p < 0.05). As a percentage of the total, FG loss in the DBA/2 retinas was larger than axon loss in the corresponding optic nerves.
Axon counts.
Ganglion cell axons were undertaken as described previously (Inman et al., 2006). In brief, 100× photomicrographs of each section were collected from 20–25 random, nonoverlapping positions using an Olympus Provis AX70 microscope (Olympus Optical, Tokyo, Japan). The entire surface area of the nerve was represented in each sample. Contrast and edge-enhancement using ImagePro (Media Cybernetics, Bethesda, MD) macro-routines allowed axon identification and quantification. The sampling area of the micrograph was also determined and used to calculate the axon density. For each nerve sample, axon density was calculated as the mean across the random micrographs. Measurements of the cross-sectional area of each nerve were used with axon density to estimate the total number of axons.
Somal size determination.
For measuring somal area from NeuN-labeled retinal whole-mount preparations, we collected photomicrographs using an Olympus AX70 Provis light microscope equipped with a motorized X-Y-Z stage, video camera, and a personal computer workstation interface mediated by ImagePro software (Media Cybernetics). For a particular location, an image containing the RGC layer was computed from a collapsed series of images collected within a 20 μm focus interval optimized to contain as many complete cells as possible. This stacking procedure ensured that irregularities in the plane of the RGC layer that arose during the whole-mount procedure did not induce an undersampling of labeled cells that lie outside the immediate focal plane. We optimized contrast and enhanced spatial edges for each collapsed series to minimize noise caused by optical blur. The perimeter of each labeled RGC was traced, and the area for each cell was calculated from the enclosed polygon.
Statistical analysis.
All data in this study and error bars in graphs are given as the mean ± SEM, except where otherwise indicated. Correlation coefficients and corresponding p values were calculated using GraphPad Prism 3.0 (GraphPad Software, San Diego, CA). Groups were compared using a Kruskal–Wallis test ANOVA assuming a nonparametric comparison. When the ANOVA detected significant variability in groups, a Dunn's Multiple Comparison's post hoc test was performed. Minimum significance was set at p < 0.05.
Results
NeuN can be used to monitor ganglion cells in the GCL
NeuN is a DNA-binding protein that identifies most mature neuronal populations, and recent studies have used NeuN as a marker for RGCs in the GCL of the retina (Canola et al., 2007; Dijk et al., 2007; Zhong et al., 2007). In addition to RGCs, the GCL is comprised of a large number of displaced amacrine cells. There was a lack of clear evidence that displaced amacrine cells that did not also express NeuN; thus we closely examined NeuN expression in both RGCs and displaced amacrine cells. Initially, FG-labeled retinas from young DBA/2 mice were sectioned and labeled with NeuN and the nuclear stain DAPI. Each marker was independently quantified to compare the relative number of labeled neurons. NeuN was found to label a consistent subpopulation of neurons residing primarily in the GCL (Fig. 1A). Importantly, NeuN and FG labeled a similar number of neurons, the number of DAPI-labeled neurons was larger than both NeuN and FG, and comparison of captured images showed NeuN label in all FG-positive cells (Fig. 1B). This provided evidence that NeuN labeled primarily RGCs and few, if any, displaced amacrine cells. To further analyze NeuN expression, sections of young FG-labeled DBA/2 retinas were labeled for NeuN by immunofluorescence, whereas in situ hybridization was used to visualize the amacrine marker GAD-67 and the RGC marker Sncg (Soto et al., 2008). Confocal imaging of quadruple-labeled retinas showed excellent correspondence between FG, Sncg, and NeuN (Fig. 1C), whereas NeuN was either absent or detected at very low levels in GAD-67-containing amacrine cells in the GCL (Fig. 1D). Whole-mount DBA/2 retinas from young and old age groups were then labeled for NeuN and ChAT immunofluorescence, and the number of colabeled neurons was counted to determine the number of NeuN-positive displaced amacrine cells likely included in NeuN counts. Analysis of colabeled cells showed that 3.6% of NeuN-positive cells were ChAT-positive displaced amacrine cells at 3 months, and 16.2% were ChAT-positive amacrines in 12 and 18 month retinas (quantitative data not shown) (see Fig. 2C for immunolabeling). These percentages were then used as a correction factor for all NeuN counts at relevant ages. Combined, these data demonstrate that NeuN immunofluorescence can be a useful tool for monitoring the RGC population in the GCL of the DBA/2 if appropriate corrections are made to exclude displaced amacrine cells.
Figure 1.
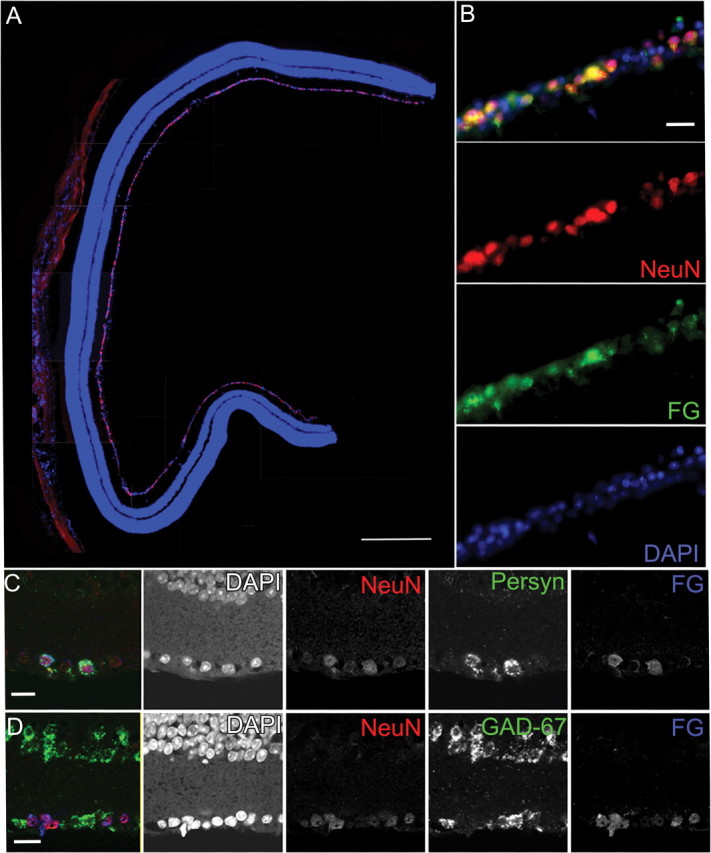
Immunohistochemistry and in situ hybridization of RGCs in the DBA/2 retina showing that NeuN antibody predominantly labels RGCs. A, A vertical section through a 3-month-old DBA/2 retina immunolabeled with NeuN (red) and the nuclear stain DAPI (blue). Although occasionally observed in the inner nuclear layer, the majority of NeuN-positive cells are observed in the GCL. Scale bar, 250 μm. B, Higher-magnification view of double immunolabeling in the GCL for NeuN (red) and FG (green) with DAPI (blue) shows that all retrogradely labeled FluoroGold-positive RGCs are also NeuN positive. The panels broken out by fluorescence color channel allow one to visualize the variations in labeling intensity for both NeuN and FG. Scale bar, 50 μm. C, In situ hybridization combined with immunohistochemistry permitted colocalization of NeuN immunolabel, Sncg mRNA, and FG immunolabel in the GCL of a 3-month-old DBA/2 retina. All FG-positive RGCs were also NeuN positive and had Sncg mRNA. Breakout panels show the individual fluorescence color channels. Scale bar, 50 μm. D, In situ hybridization for the amacrine marker GAD-67 combined with immunohistochemistry for NeuN and FG show that all FG-positive cells are NeuN positive; FG-positive/NeuN-positive cells are GAD-67 negative. The individual fluorescence channels allow one to discern light NeuN immunolabeling in a small number of GAD-67 displaced amacrine cells. Scale bar, 50 μm.
Ganglion cells persist in the glaucomatous DBA/2 retina
We initially performed a quantitative analysis of NeuN-labeled RGCs with the intention of assessing the relative timeline of RGC loss. Sixty-three DBA/2 retinas ranging in age from 2 to 21 months were processed as whole mounts and immunolabeled with NeuN. These retinas were separated into four groups: 2–4 months, 6–10 months, 14–15 months, and 18–21 months to represent young, middle-aged, old, and very old, respectively. A predefined template was centered on the optic disc of each retina, and the periphery was traced to allow for quantification within eight sectors corresponding to the retinal poles (superior, inferior, nasal, and temporal), with four central and four peripheral sectors. Neuron density quantification was performed across these sectors using unbiased stereology, and all counts were performed by a single observer (Fig. 2A). Where possible, NeuN-positive/ChAT-positive displaced amacrine cells (examples in Fig. 2C) have been excluded from the NeuN cell counts; both data sets, corrected (green) and not corrected (blue) for displaced amacrine cells, are shown (Fig. 2A,B). C57BL/6 retinas (5 and 13 months) were used as a nonglaucoma control group for changes in DBA/2 NeuN density (Fig. 2B). The NeuN-positive cell density was not different between the DBA/2 2–4 month age group and 5 month C57BL/6 retinas (p > 0.5), but by 14 months, DBA/2 NeuN cell density was significantly different than 13 month C57BL/6 NeuN cell density (p < 0.0001) (Fig. 2, compare A and B). NeuN-positive cell density in DBA/2 retinas up to 10 months was consistent with RGC density as assessed with FG retrograde labeling by others (Danias et al., 2003) (Fig. 2A), and it remained unchanged until 18–21 months of age, at which time a significant reduction in NeuN density (35.4%) was observed (p < 0.001) (Fig. 2A). Unless otherwise stated, all references to NeuN counts represent RGCs only. To test the reproducibility of the counting technique in whole-mount retinas, we conducted an intrarater reliability test by randomly recounting retinas. Variability in recounts was <1% (r = 0.999; p = <0.001). Although areas of very low NeuN-positive cell density were observed, we did not observe consistent loss of NeuN-positive cells within specific retinal poles.
Given the stability of NeuN densities in aging DBA/2 retinas, we measured somal sizes of NeuN-labeled DBA/2 cells in an attempt to establish a timeline of relative degeneration in glaucoma. It was not possible to specifically exclude the relatively small number of NeuN-positive displaced amacrine cells, and the somal size data therefore represent all NeuN-positive cells. There were no significant differences in somal size from 3 to 15 months, but somal size was significantly reduced by 18 months, which correlated in age with the first noticeable NeuN reduction (Fig. 2D). When NeuN-labeled cells were binned based on somal size (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), an ANOVA (Dunn's multiple comparison's test; p < 0.001) showed that the shift in somal size was represented by a net increase in abnormally small (10–50 μm2) somata at 18 months compared with either 3 or 15 months (Fig. 2D). These data suggest a general shrinkage of RGC somas before the loss of detectible NeuN immunolabeling in aged DBA/2 mice.
Disruption of FG retrograde label occurs before ganglion cell loss in DBA/2
We next explored evidence for glaucomatous axon pathology using retrograde tracers. FG, which requires active axon transport, was combined with the carbocyanine tracer DiI, which can move by passive diffusion and/or active transport within vesicles along the axon, and injected into the superior colliculi of two groups of mice, 3 and 13 months of age. Whole-mount retinas were immunolabeled with NeuN. In representative fields from 3-month-old DBA/2 retinas, the number of cells visualized with NeuN, FG, and DiI were similar within and across all retinas (Fig. 3A). In 13 month retinas, however, the number of FG-labeled somas was dramatically reduced (Fig. 3B), whereas a similar but less dramatic reduction of DiI-labeled somas was observed (Fig. 3B). The numbers of NeuN-labeled cells in 13 month retinas appeared comparable with densities observed at 3 months. Figure 3C shows a high-magnification view of a typical midcentral retinal field in a 13 month retina with notable differences in the number of RGCs positive for FG versus DiI, with punctate DiI-positive labeling predominant. When differential retrograde labeling was observed, the majority of cells were DiI-positive or DiI- and FG-positive (see colocalization in Fig. 3D).
To illustrate the disconcordance between retrograde labeling with FG and neuronal survival, we quantified FG- and NeuN-labeled RGGs in retinal whole mounts of DBA/2 mice at 3, 6, and 14 months of age (Fig. 3E). Although no significant difference in NeuN density was found in these mice, the number of FG-labeled RGCs was significantly reduced by 75% at 14 months of age. There was no reduction, however, in FG-positive RGCs from 5 to 13 months in C57BL/6 control retinas, suggesting the reduction in DBA/2 retina is not related to age. There is a significant difference in FG-positive RGC number in 14 month DBA/2 and 13 month C57BL/6 retinas (p < 0.0001) (Fig. 3, compare E and F). To distinguish between deficits in retrograde labeling by tracers that may have different properties of diffusion and thus different likelihood of uptake, stereological analysis of FG- and DiI-labeled RGCs in whole-mount DBA/2 retinas was performed. Although FG and DiI labeled similar numbers of RGCs at 3 months (4745 and 4846 cells/mm2, respectively), a significant decrease in FG (p < 0.05) and a visible, but not significant, decrease in DiI-labeled RGC somata was observed at 13 months (Fig. 3G). Notably, the decrease in DiI-labeled cells (4846–3235 cells/mm2) was less than that of FG (4745–2250 cells/mm2), yet this trend was not statistically significant. Finally, to establish whether the loss of FG labeling indicated retrograde labeling deficit or axonal loss, we compared optic nerve axon density and FG-labeled RGC numbers in a cohort of FG-injected DBA/2 mice at 3, 6, and 13 months of age. In 3 month DBA/2 optic nerves, axon density was 5.00E+05 axons/mm2; by 13 months, axon density was 4.33E+05 axons/mm2. Both axon density and the number of FG-labeled RGCs were significantly decreased at 13 months (p < 0.05), but the loss of FG-positive RGCs was more extensive than axonal loss (Fig. 3H). Although FG labeling experienced a more significant decrease than axon density in DBA/2 mice, the optic nerve at 13 compared with 3 months shows signs of severe degeneration with vacuolizations, axon swelling, glial hypertrophy, and abnormal myelin profiles (Fig. 4A–C). Anterograde labeling of DBA/2 RGCs with cholera toxin also demonstrated degenerative changes of RGC axons in the optic nerve; in a longitudinal section from 14 month DBA/2 optic nerve (Fig. 4D) retraction bulbs, vacuolizations, and intra-axonal accumulations are apparent. Accumulations of NF-70 in optic nerve at 6 months compared with 3 months gave way to decreases in NF-70 at 12 months (Fig. 4E). As a whole, these data demonstrate that axonal pathology occurs before RGC somal loss in the DBA/2 mouse.
Figure 4.
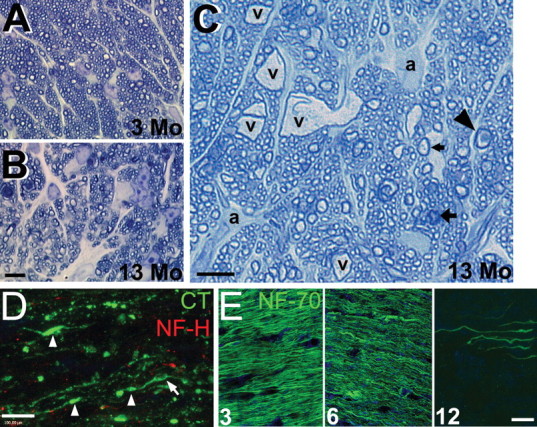
Optic nerves of DBA/2 mice show degenerative changes. A, Cross section of DBA/2 optic nerve at 3 months (3 Mo) processed for high-magnification light microscopy shows normal fascicle packing and regular myelin wraps on axons. B, Optic nerve cross section at 13 months (13 Mo) shows disruption of axon fascicles, increases in connective tissue septae, and lower axon numbers. C, Additional cross section of 13 month optic nerve showing vacuolization (v), large astrocytic processes (a), and arrows pointing to abnormal axon profiles. Arrowhead, Degenerating axon; wide arrow, myelin and axon pathology; narrow arrow, normal axon. Scale bars: (in B) A, B, 30 μm; C, 30 μm. D, Cholera toxin (CT) endocytosed by RGCs and transported through ganglion cell axons into the optic nerve showed numerous varicosities or swellings (arrowheads) and vacuolization (arrow) in addition to notable loss of NF-H immunoreactivity. Scale bar, 8 μm. E, Three, 6, and 12 month optic nerve immunolabeled with NF-70 showing accumulation of small neurofilaments from 3 to 6 months. By 12 months, NF-70 is nearly absent from many nerves. Scale bar, 10 μm.
Brn-3b, Sncg, and NF-L expression decrease with age in the glaucomatous retina
To examine whether decreases in retrograde labeling of RGCs was associated with protein changes in the axon or gene expression changes in the soma, we coanalyzed NeuN-positive RGC number with neurofilament heavy (NF-H) in whole-mount DBA/2 retinas in combination with quantitative PCR. We observed dramatically altered NF-H-immunolabeled axon numbers in DBA/2 retinas from 3 to 14 months of age, although there was no significant change in NeuN-positive RGC density. Figure 5A-D′ illustrates the comparison of NeuN-positive cells and NF-H axon immunoreactivity in the whole-mount retina of a typical 3 month DBA/2 (Fig. 5A–B′) versus a 14 month DBA/2 retina with visible reduction in NF-H labeling without concurrent NeuN loss (Fig. 5C–D′). To further understand the axon changes in the context of persistent NeuN-positive RGCs, retinas from six 14 month DBA/2 mice were divided in two pieces, one processed for immunohistochemistry and the other for qRT-PCR. Mice were matched for elevated IOP. Quantification of NeuN (corrected to exclude displaced amacrine cells) demonstrated no reduction in RGC density (4289 ± 548 cells/mm2). Despite similar density of NeuN-positive RGCs, individual retinas had significant variation in the number of NF-H-positive RGC axons (Fig. 5E–H). NF-H labeling was dramatically reduced in retinas in Figure 5F–H, whereas NF-H-positive axons were maintained in others (Fig. 5E), although all retinas had normal NeuN-positive RGC density.
Figure 5.
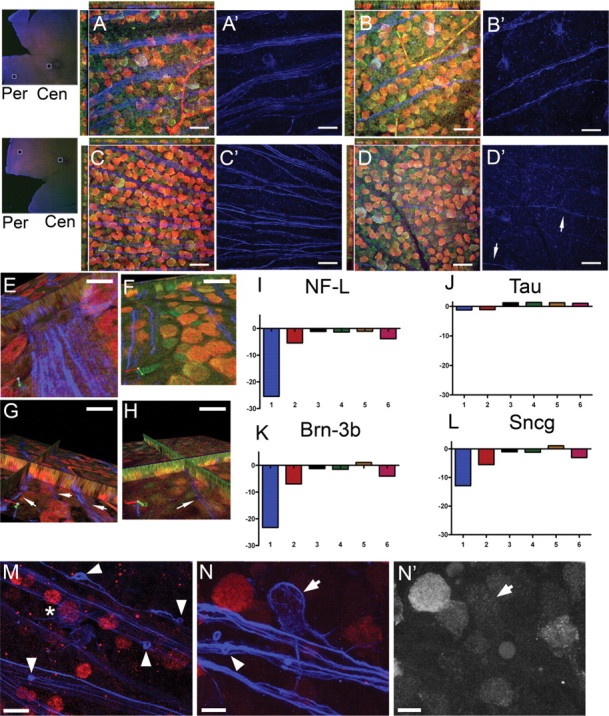
Comparison of RGC-specific gene expression with NF-H immunolabel across 14 month DBA/2 retinas. A–D′, Photomicrographs taken from peripheral (Per) (3 month retina in A, A′; 14 month retina in C, C′) and central (Cen) (3 month retina in B, B′; 14 month retina in D, D′) whole-mount retina immunolabeled with NeuN (red) and NF-H (blue) and stained with NeuroTrace Nissl (green). A, B, This 3 month retina appeared to have a normal GCL, including healthy, well-labeled axon fascicles and strong NeuN immunolabeling. C, D, This 14 month retina did not exhibit loss of NeuN immunolabeling in the GCL, but there appeared to be a significant decrease in NF-H-positive axon fascicles. The axons present were thin and difficult to visualize (D′, arrows). E–H, A panel of photomicrographs from 14 month DBA/2 retinas immunolabeled with NeuN (red), NF-H (blue), and NeuroTrace Nissl (green). One half of the retina was removed for quantitative RT-PCR analysis of RGC-specific genes (I–L). E, F, The axon fascicles in this retina appeared robust, particularly near the central retina. The corresponding qPCR data for this retina are represented in lane 3 of I–L. G, H, This retina demonstrated a dramatic reduction in the size and number of axon fascicles (G, H, arrows) compared with the age- and IOP-matched retina in E and F. The corresponding qPCR data for this retina are represented in lane 1 of I–L. I–L, Quantitative PCR results for NF-L (I), tau (J), Brn3b (K), and Sncg (L) from six separate half retinas (lanes 1–6). There was no change in mRNA for tau, a neuronal protein, across these 14 month retinas, but there was significant downregulation of RGC-specific mRNAs for NF-L, Brn3b, and Sncg in some retinas, particularly the retina in lane 1. Photomicrographs of the retina in lane 3 (E, F) allow one to observe how retinas with normal RGCs and RGC axons had no changes in NF-L, tau, Brn3b, and Sncg mRNA (lane 3), whereas retinas with diminished axon fascicles in the nerve fiber layer (retina in G and H) also had RGCs with significantly downregulated RGC-specific genes (lane 1 in I–L). The magnitude of changes in NF-L, Brn3b, and Sncg are similar for individual retinas, suggesting a shared upstream regulation of these mRNAs. The variability of qPCR results illustrate the inconsistency in the glaucoma phenotype across 14 month DBA/2 mice. M–N′, Whole-mount retina immunolabeled with NeuN (red) and NF-H (blue) shows end bulbs of degenerating RGC axons (M, N, arrowheads) and downregulation of NeuN in RGCs. Prominent NF-H-positive RGCs were observed with moderate NeuN immunolabel (right of asterisk in M) or almost no NeuN immunolabel (arrow in N and N′). Scale bars: A–D′, 20 μm; E–H, M, 8 μm; N, N′, 5 μm.
Quantitative analysis of the expression of NF-L, tau, and the RGC-specific genes Brn-3b and Sncg (Soto et al., 2008) demonstrated a range of expression in these individual retinas that correlated with relative NF-H-positive axon number. For example, observe Retina 3 in Figure 5E and lane 3 in Figure 5I. This retina had near normal NF-H axon labeling and no significant change in expression of NF-L, tau, Brn-3b, or Sncg. In contrast, Retina 1 (Fig. 5G,H,I, lane 1) had few NF-H labeled axonal processes, and NF-L, Brn-3b, and Sncg levels were significantly decreased; tau remained unchanged. At 14 months, RGCs identified by the accumulation of NF-H in their somata were observed with variable levels of NeuN expression (Fig. 5M,N). In a region with visible degenerative end bulbs (Fig. 5M), one NF-H-positive RGC with NeuN expression (right of asterisk) is neighbor to another NF-H-positive RGC (below) that is NeuN negative. Another NeuN-negative RGC is in Figure 5N, illustrating NeuN protein downregulation in an identifiable RGC. NeuN-negative/NF-H-positive RGCs were not observed in appreciable numbers earlier than 13–14 months. In summary, these data demonstrate that preservation of the RGC somas may persist for a significant period even in the presence of profound axonal dysfunction/loss and the downregulation of axon maintenance genes within the RGC. Combined, these data suggest that glaucomatous axon pathology in the DBA/2 involves axon damage and varying degrees of RGC degeneration in the absence of somal death.
Figure 6 illustrates a proposed timeline of changes within the DBA/2 retina in the course of glaucoma development, primarily based on data presented here but also data from recent studies about the DBA/2 (Jakobs et al., 2005). The timeline begins at 0 months, with the observed IOP increases commencing at 6 months (Inman et al., 2006). The first observed decreases in FG labeling occur between 6 and 8 months in the DBA/2 and are significant at 13 months (Fig. 3G). Decreases in FG retrograde labeling may indicate a number of problems, including axon transport problems for RGCs or dying back of RGC axons from targets in the superior colliculus, conditions that precede degeneration or elimination of axons. Visible changes in retrograde DiI (Fig. 3G) and significant decreases in axon number in optic nerve are evident by 13 months (Fig. 3H), suggesting that RGC axons may be dying back from target areas or degenerating at points between the superior colliculus and the retina 3–6 months after retrograde labeling deficits are identified. Immunohistochemistry for NF-H shows axon thinning at 12–14 months and beyond (Fig. 5A–D′). Concomitantly, mRNA for RGC-specific genes such as Brn3b, Sncg, and NF-L decrease to a degree proportional to disease severity (Fig. 5I–L). Cell shrinkage has been observed at 15 months (Filippopoulos et al., 2006) and documented in this study at 18 months (Fig. 2D). The first significant decreases in NeuN-positive RGCs in the DBA/2 occur beginning at 18 months (Fig. 2A).
Figure 6.

Timeline of RGC changes in DBA/2 chronic secondary glaucoma. This timeline represents our conception of the pathogenesis of glaucoma from the point of view of the RGC. Gradients represent general onset and intensity of specified pathology over time. The timeline begins at 0 months; IOP increases commence at 6 months (Inman et al., 2006). The first observed decreases in FluoroGold labeling occur between 6 and 8 months in the DBA/2 and are significant at 13 months. Visible changes in DiI and significant decreases in axon number are evident by 13 months, suggesting that RGC axons may be dying back from target areas or degenerating at points between the superior colliculus and the retina 3–6 months after deficits in retrograde labeling are identified. Immunohistochemistry for NF-H shows axon thinning and degenerative end bulbs in retina and optic nerve at 12–14 months and beyond, whereas mRNA for RGC-specific genes such as Brn3b, Sncg, and NF-L decreases to a degree proportional to disease severity in this late period as well. Cell shrinkage has been observed at 15 months (Filippopoulos et al., 2006) and documented in this study at 18 months. The first significant decreases in NeuN-positive RGCs in this study occurred beginning at 18 months; this could represent downregulation of the NeuN protein or cell death.
Discussion
The data presented here suggest an alternative explanation to the prevailing opinion in glaucoma that RGC death begins early and leads to blindness. Mechanisms of glaucoma-related RGC loss have been shown to include apoptosis (Kerrigan et al., 1997; McKinnon, 1997; Quigley, 1999; Schuettauf et al., 2004), ischemia-induced apoptosis (Tezel and Wax, 2000; Chen et al., 2003), and even excitotoxicity (Vorwerk et al., 1996), but this evidence does not exclude the possibility that vision loss occurs well in advance of cell death. Our data suggest that RGCs give notice of growing dysfunction via somal shrinkage, retrograde labeling deficits, and downregulation of RGC-specific genetic programs before death.
Many RGC studies have relied on retrograde labeling (Schuettauf et al., 2002; Vecino et al., 2002; Danias et al., 2003) to study and quantify this heterogeneous, specialized population of retinal neurons. FluoroGold has been the tracer of choice for such studies, and this method requires active transport mechanisms to move endocytic vesicles packed with FG back to the RGC soma (Koebbert et al., 2000); hence, FG and similar tracers do not accumulate in ganglion cells with compromised axon transport. Our combined methods of retrograde tracing and direct RGC labeling yielded data that suggest retrograde labeling deficit precedes both axon and somal loss of RGCs in the DBA/2.
NeuN-positive RGCs persist through axon degeneration
NeuN is a DNA-binding (Mullen et al., 1992) neuronal phosphoprotein with isoforms that exist in the nucleus and cytoplasm (Lind et al., 2005). NeuN has been used to identify neurons in the retina (Wang et al., 2000), with some groups observing NeuN expression exclusive to RGCs in the GCL (Canola et al., 2007; Dijk et al., 2007; Zhong et al., 2007). The GCL is comprised of approximately equal numbers of RGCs and displaced amacrine cells (Jeon et al., 1998), and, to date, there has not been a study that presents evidence for the unequivocal absence of NeuN in displaced amacrine cells. In Figure 1, we show that NeuN strongly labels all RGCs and lightly labels a small but consistent population of displaced amacrine cells. The relative number of NeuN-positive displaced amacrine cells increased with age from 3.6% in young to 16% in old DBA/2. Even after correcting the cell counts for NeuN-positive displaced amacrine cells, there was no significant difference in NeuN-positive RGCs between the 2–4 and 14–15 month age groups. These data validated the use of NeuN as a method to accurately monitor the RGC population in the DBA/2. We found that NeuN persisted in retinas with presumptive degeneration, as indicated by marked reductions in FG-labeled RGCs. This suggests that RGCs are a CNS neuronal population, similar to rubrospinal neurons (McPhail et al., 2004), that preserve NeuN antigenicity despite degenerative changes. Relatedly, some groups have relied on histological stains of Nissl substance to assess RGC number, despite destruction then disappearance of Nissl granules in neurons undergoing degeneration (Sunderland, 1991).
By 18 months, there is a significant decrease in NeuN+ cell density in the DBA/2 retina. The cells were quantified in central versus peripheral retina and along the poles (e.g., superior-nasal, superior-temporal) to determine any topographic changes in cell loss; such changes were not observed. Jakobs et al. (2005) showed RGC loss in sectors radiating from the optic nerve head, but unlike the arcuate scotoma observed in human retina, the same sectors did not experience cell loss in every retina studied. Recent data suggest that, although it is the case that sectors of axon loss in the retina correspond to groups of fascicle loss in the optic nerve (Howell et al., 2007), it is interesting to note that each retina from a glaucomatous DBA/2 mouse has a unique pattern of loss. Loss or compromise of large optic nerve regions would be sufficient to eliminate nerve fibers or functional transport from large sectors of the retina proper.
Decreases in FG label in DBA/2 retina preceded changes in somal size and RGC loss, implying that retrograde labeling deficit is an early event on the path to vision loss in glaucoma secondary to increased IOP. There was no reduction in FG label in 13 month C57BL/6 control retinas indicating FG reduction in the DBA/2 was not age related. Previous studies in monkey and rat eyes demonstrated retrograde transport block to a degree that was proportional to the magnitude and duration of IOP (Minckler et al., 1977; Johansson, 1988). Rats with acute IOP elevation experienced a 50–74% decrease in transport of radiolabeled BDNF, coincident with RGC axons abnormally dilated with accumulating vesicles (Quigley et al., 2000). Studies with the Wlds mouse have demonstrated that failure to deliver microtubules and neurofilaments to the axon is not sufficient to cause axon degeneration, but the interruption likely triggers a proactive axon death program (Coleman, 2005). Axon degeneration without cell body death was demonstrated in a study of the effect of Bax gene dosage on RGC survival in the DBA/2 (Libby et al., 2005). DBA/2-Bax−/− mice underwent age and IOP-related optic nerve degeneration while retaining their RGCs, even after optic nerve crush, suggesting that the optic nerve in DBA/2 glaucoma is a locus of injury and the degeneration may have a mechanical component (Libby et al., 2005). An axon-cell death dichotomy lends credence to the concept of the axon compartment as a specific site of injury in glaucoma.
A number of observations suggest that axon transport deficits contribute to the progression of neurodegenerative diseases. Stokin et al. (2005) observed axon swellings with accumulated microtubule-associated and molecular motor proteins (among others) before Alzheimer's disease-related pathology in mouse model and human tissue. Furthermore, increased levels of amyloid-β, a product of amyloid β protein precursor proteolysis and component of amyloid plaques, have been observed in RGCs from rats with ocular hypertension (McKinnon, 2003). Patients with glaucoma have significant vitreal decreases in Aβ42 levels and significant increases in tau when compared with controls, conditions that echo the findings in Alzheimer's patient CSF (Yoneda et al., 2005).
NeuN-positive cells shrink in DBA/2
In addition to quantification, we measured RGC somal area of NeuN-positive cells, showing significant increases in RGCs smaller than 50 μm2 in 18 month DBA/2 with a corresponding decrease in RGCs larger than 50 μm2. This tremendous cell shrinkage, even from 15 to 18 months in the DBA/2, has been observed similarly by others (Jakobs et al., 2005) and in the DBA/2NNia mouse at 15 months (Filippopoulos et al., 2006). The cell shrinkage is progressive, as indicated by the absence of cells <50 μm2 at 3 months, but 5% at 15 months and >20% by 18 months (Fig. 2D). Cell shrinkage indicates severe dysfunction (Morgan, 2002); despite which, NeuN immunoreactivity was maintained in a majority of cells. In a monkey model of glaucoma, RGC morphological changes start with dendritic alterations followed by axon thinning and finally somal shrinkage (Weber et al., 1998). Evidence of retinal ganglion cell shrinkage in monkey and human (Morgan et al., 2000; Morgan, 2002) as well as analysis of patient functional data (Pearson et al., 2006) have combined to cast doubt on previous reports of selective large ganglion cell loss. Although dendritic alterations were not examined in this study, Jakobs et al. (2005) noted underbranched and misdirected RGC dendrites in 12-month-old DBA/2 retinas. These observations support a degenerative sequence of events, with somal loss occurring after dendritic changes, axon thinning, and somal shrinkage.
RGC-specific gene downregulation
In times of stress, specialized cells often stop expressing proteins specific to their function. We investigated this phenomenon in RGCs undergoing axon degeneration in DBA/2. Expression of NeuN and tau, both found in retinal neurons, did not change dramatically through the development of glaucoma in the DBA/2, whereas the ganglion cell-specific genes Sncg, NF-L, and Brn-3b showed varying degrees of mRNA downregulation. During development, the basic HLH transcription factor Math5 activates Brn-3b, thus directing the maturation of retinal progenitor cells to adopt a ganglion cell fate (Mu et al., 2005). Sncg expression decreases in the optic nerves of rats with experimental glaucoma (Surgucheva et al., 2002) and in RGCs of Alzheimer's patients (Surguchov et al., 2001). Retinas with normal NeuN-positive cell number showed downregulation of Sncg, NF-L, and Brn-3b in old DBA/2 retina, similar to the widespread downregulation of neuronal and RGC-specific genes concomitant with reduction in FG retrograde transport observed in another DBA/2 study (Soto et al., 2008). All of these data suggest that RGCs lose expression of these genes before death.
Our data suggest that retrograde labeling dysfunction or loss is an early step in the progression of glaucoma (Fig. 6). The number of FG-labeled ganglion cells decline in advance of decreases in DiI-labeled RGCs, axon number in optic nerve, and NeuN-positive RGCs, suggesting that retrograde axon transport becomes dysfunctional before axons and RGCs are lost. The significance of this observation, if confirmed in the human condition, supports assisting axon transport and/or preserving RGC-specific gene expression to slow the progression of glaucoma.
Footnotes
This work was supported by the Catalyst for a Cure initiative of the Glaucoma Research Foundation and by The Steven and Michele Kirsch Foundation (D.J.C., M.L.V., N.M.-A., and P.J.H.).
References
- Anderson MG, Smith RS, Hawes NL, Zabaleta A, Chang B, Wiggs JL, John SW. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat Genet. 2002;30:81–85. doi: 10.1038/ng794. [DOI] [PubMed] [Google Scholar]
- Canola K, Angenieux B, Tekaya M, Quiambao A, Naash MI, Munier FL, Schorderet DF, Arsenijevic Y. Retinal stem cells transplanted into models of late stages of retinitis pigmentosa preferentially adopt a glial or a retinal ganglion cell fate. Invest Ophthalmol Vis Sci. 2007;48:446–454. doi: 10.1167/iovs.06-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Smith RS, Hawes NL, Anderson MG, Zabaleta A, Savinova O, Roderick TH, Heckenlively JR, Davisson MT, John SW. Interacting loci cause severe iris atrophy and glaucoma in DBA/2J mice. Nat Genet. 1999;21:405–409. doi: 10.1038/7741. [DOI] [PubMed] [Google Scholar]
- Chen YG, Zhang C, Chiang SK, Wu T, Tso MO. Increased nuclear factor-kappa B p65 immunoreactivity following retinal ischemia and reperfusion injury in mice. J Neurosci Res. 2003;72:125–131. doi: 10.1002/jnr.10548. [DOI] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Perry VH. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 2002;25:532–537. doi: 10.1016/s0166-2236(02)02255-5. [DOI] [PubMed] [Google Scholar]
- Cordeiro MF, Guo L, Luong V, Harding G, Wang W, Jones HE, Moss SE, Sillito AM, Fitzke FW. Real-time imaging of single nerve cell apoptosis in retinal neurodegeneration. Proc Natl Acad Sci USA. 2004;101:13352–13356. doi: 10.1073/pnas.0405479101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danias J, Lee KC, Zamora MF, Chen B, Shen F, Filippopoulos T, Su Y, Goldblum D, Podos SM, Mittag T. Quantitative analysis of retinal ganglion cell (RGC) loss in aging DBA/2NNia glaucomatous mice: comparison with RGC loss in aging C57Bl/6 mice. Invest Ophthalmol Vis Sci. 2003;44:5151–5162. doi: 10.1167/iovs.02-1101. [DOI] [PubMed] [Google Scholar]
- Dijk F, Bergen AA, Kamphuis W. GAP-43 expression is upregulated in retinal ganglion cells after ischemia/reperfusion-induced damage. Exp Eye Res. 2007;84:858–867. doi: 10.1016/j.exer.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Filippopoulos T, Danias J, Chen B, Podos SM, Mittag TW. Topographic and morphologic analyses of retinal ganglion cell loss in old DBA/2NNia mice. Invest Ophthalmol Vis Sci. 2006;47:1968–1974. doi: 10.1167/iovs.05-0955. [DOI] [PubMed] [Google Scholar]
- Garcia-Valenzuela E, Shareef S, Walsh J, Sharma SC. Programmed cell death of retinal ganglion cells during experimental glaucoma. Exp Eye Res. 1995;61:33–44. doi: 10.1016/s0014-4835(95)80056-5. [DOI] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–1537. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Fileta J, Guo Y, Grosskreutz CL. Downregulation of Thy1 in retinal ganglion cells in experimental glaucoma. Curr Eye Res. 2006;31:265–271. doi: 10.1080/02713680500545671. [DOI] [PubMed] [Google Scholar]
- Inman DM, Sappington RM, Horner PJ, Calkins DJ. Quantitative correlation of optic nerve pathology with ocular pressure and corneal thickness in the DBA/2 mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2006;47:986–996. doi: 10.1167/iovs.05-0925. [DOI] [PubMed] [Google Scholar]
- Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005;171:313–325. doi: 10.1083/jcb.200506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson JO. Inhibition and recovery of retrograde axoplasmic transport in rat optic nerve during and after elevated IOP in vivo. Exp Eye Res. 1988;46:223–227. doi: 10.1016/s0014-4835(88)80079-4. [DOI] [PubMed] [Google Scholar]
- John SW, Smith RS, Savinova OV, Hawes NL, Chang B, Turnbull D, Davisson M, Roderick TH, Heckenlively JR. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Invest Ophthalmol Vis Sci. 1998;39:951–962. [PubMed] [Google Scholar]
- Kerrigan LA, Zack DJ, Quigley HA, Smith SD, Pease ME. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol. 1997;115:1031–1035. doi: 10.1001/archopht.1997.01100160201010. [DOI] [PubMed] [Google Scholar]
- Koebbert C, Apps R, Bechmann I, Lanciego JL, Mey J, Thanos S. Current concepts in neuroanatomical tracing. Prog Neurobiol. 2000;62:327–351. doi: 10.1016/s0301-0082(00)00019-8. [DOI] [PubMed] [Google Scholar]
- Kuehn MH, Fingert JH, Kwon YH. Retinal ganglion cell death in glaucoma: mechanisms and neuroprotective strategies. Ophthalmol Clin North Am. 2007;18:383–395. vi. doi: 10.1016/j.ohc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind D, Franken S, Kappler J, Jankowski J, Schilling K. Characterization of the neuronal marker NeuN as a multiply phosphorylated antigen with discrete subcellular localization. J Neurosci Res. 2005;79:295–302. doi: 10.1002/jnr.20354. [DOI] [PubMed] [Google Scholar]
- Mabuchi F, Aihara M, Mackey MR, Lindsey JD, Weinreb RN. Optic nerve damage in experimental mouse ocular hypertension. Invest Ophthalmol Vis Sci. 2003;44:4321–4330. doi: 10.1167/iovs.03-0138. [DOI] [PubMed] [Google Scholar]
- Marcic TS, Belyea DA, Katz B. Neuroprotection in glaucoma: a model for neuroprotection in optic neuropathies. Curr Opin Ophthalmol. 2003;14:353–356. doi: 10.1097/00055735-200312000-00006. [DOI] [PubMed] [Google Scholar]
- McKinnon SJ. Glaucoma, apoptosis, and neuroprotection. Curr Opin Ophthalmol. 1997;8:28–37. doi: 10.1097/00055735-199704000-00006. [DOI] [PubMed] [Google Scholar]
- McKinnon SJ. Glaucoma: ocular Alzheimer's disease? Front Biosci. 2003;8:s1140–s1156. doi: 10.2741/1172. [DOI] [PubMed] [Google Scholar]
- McPhail LT, McBride CB, McGraw J, Steeves JD, Tetzlaff W. Axotomy abolishes NeuN expression in facial but not rubrospinal neurons. Exp Neurol. 2004;185:182–190. doi: 10.1016/j.expneurol.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Minckler DS, Bunt AH, Johanson GW. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest Ophthalmol Vis Sci. 1977;16:426–441. [PubMed] [Google Scholar]
- Mittag TW, Danias J, Pohorenec G, Yuan HM, Burakgazi E, Chalmers-Redman R, Podos SM, Tatton WG. Retinal damage after 3 to 4 months of elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2000;41:3451–3459. [PubMed] [Google Scholar]
- Morgan JE. Retinal ganglion cell shrinkage in glaucoma. J Glaucoma. 2002;11:365–370. doi: 10.1097/00061198-200208000-00015. [DOI] [PubMed] [Google Scholar]
- Morgan JE, Uchida H, Caprioli J. Retinal ganglion cell death in experimental glaucoma. Br J Ophthalmol. 2000;84:303–310. doi: 10.1136/bjo.84.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Fu X, Sun H, Beremand PD, Thomas TL, Klein WH. A gene network downstream of transcription factor Math5 regulates retinal progenitor cell competence and ganglion cell fate. Dev Biol. 2005;280:467–481. doi: 10.1016/j.ydbio.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Okisaka S, Murakami A, Mizukawa A, Ito J. Apoptosis in retinal ganglion cell decrease in human glaucomatous eyes. Jpn J Ophthalmol. 1997;41:84–88. doi: 10.1016/s0021-5155(97)00013-0. [DOI] [PubMed] [Google Scholar]
- Pearson PM, Schmidt LA, Ly-Schroeder E, Swanson WH. Ganglion cell loss and age-related visual loss: a cortical pooling analysis. Optom Vis Sci. 2006;83:444–454. doi: 10.1097/01.opx.0000218432.52508.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA. Ganglion cell death in glaucoma: pathology recapitulates ontogeny. Aust N Z J Ophthalmol. 1995;23:85–91. doi: 10.1111/j.1442-9071.1995.tb00135.x. [DOI] [PubMed] [Google Scholar]
- Quigley HA. Neuronal death in glaucoma. Prog Retin Eye Res. 1999;18:39–57. doi: 10.1016/s1350-9462(98)00014-7. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, McKinnon SJ, Zack DJ, Pease ME, Kerrigan-Baumrind LA, Kerrigan DF, Mitchell RS. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000;41:3460–3466. [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Schuettauf F, Quinto K, Naskar R, Zurakowski D. Effects of anti-glaucoma medications on ganglion cell survival: the DBA/2J mouse model. Vision Res. 2002;42:2333–2337. doi: 10.1016/s0042-6989(02)00188-8. [DOI] [PubMed] [Google Scholar]
- Schuettauf F, Rejdak R, Walski M, Frontczak-Baniewicz M, Voelker M, Blatsios G, Shinoda K, Zagorski Z, Zrenner E, Grieb P. Retinal neurodegeneration in the DBA/2J mouse-a model for ocular hypertension. Acta Neuropathol (Berl) 2004;107:352–358. doi: 10.1007/s00401-003-0816-9. [DOI] [PubMed] [Google Scholar]
- Soto I, Oglesby E, Buckingham BP, Son JL, Roberson EDO, Steele MR, Inman DM, Vetter ML, Horner PJ, Marsh-Armstrong N. Retinal ganglion cells downregulate gene expression and lose their axons with the optic nerve head in a mouse glaucoma model. J Neurosci. 2008;28:548–561. doi: 10.1523/JNEUROSCI.3714-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele M, Inman DM, Sappington RM, Golestaneh N, Marsh-Armstrong N, Calkins DJ, Horner PJ, Vetter ML. Whole retinal microarray analysis of DBA/2J mice: a model for glaucoma. Invest Ophthalmol Vis Sci. 2005;47:977–985. [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Sunderland S. Axon degeneration. In: Sunderland S, editor. Nerve injuries and their repair. New York: Churchill Livingstone; 1991. pp. 79–89. [Google Scholar]
- Surgucheva I, McMahan B, Ahmed F, Tomarev S, Wax MB, Surguchov A. Synucleins in glaucoma: implication of gamma-synuclein in glaucomatous alterations in the optic nerve. J Neurosci Res. 2002;68:97–106. doi: 10.1002/jnr.10198. [DOI] [PubMed] [Google Scholar]
- Surguchov A, McMahan B, Masliah E, Surgucheva I. Synucleins in ocular tissues. J Neurosci Res. 2001;65:68–77. doi: 10.1002/jnr.1129. [DOI] [PubMed] [Google Scholar]
- Tatton NA, Tezel G, Insolia SA, Nandor SA, Edward PD, Wax MB. In situ detection of apoptosis in normal pressure glaucoma. A preliminary examination. Surv Ophthalmol. 2001;45:S268–S272. doi: 10.1016/s0039-6257(01)00204-1. discussion S273–S266. [DOI] [PubMed] [Google Scholar]
- Tezel G, Wax MB. Increased production of tumor necrosis factor-α by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci. 2000;20:8693–8700. doi: 10.1523/JNEUROSCI.20-23-08693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos S, Naskar R. Correlation between retinal ganglion cell death and chronically developing inherited glaucoma in a new rat mutant. Exp Eye Res. 2004;79:119–129. doi: 10.1016/j.exer.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Vecino E, Garcia-Grespo D, Garcia M, Martinez-Millan L, Sharma SC, Carrascal E. Rat retinal ganglion cells co-express brain derived neurotrophic factor (BDNF) and its receptor TrkB. Vision Res. 2002;42:151–157. doi: 10.1016/s0042-6989(01)00251-6. [DOI] [PubMed] [Google Scholar]
- Vorwerk CK, Lipton SA, Zurakowski D, Hyman BT, Sabel BA, Dreyer EB. Chronic low-dose glutamate is toxic to retinal ganglion cells. Toxicity blocked by memantine. Invest Ophthalmol Vis Sci. 1996;37:1618–1624. [PubMed] [Google Scholar]
- Wang X, Tay SS, Ng YK. An immunohistochemical study of neuronal and glial cell reactions in retinae of rats with experimental glaucoma. Exp Brain Res. 2000;132:476–484. doi: 10.1007/s002210000360. [DOI] [PubMed] [Google Scholar]
- Weber AJ, Kaufman PL, Hubbard WC. Morphology of single ganglion cells in the glaucomatous primate retina. Invest Ophthalmol Vis Sci. 1998;39:2304–2320. [PubMed] [Google Scholar]
- Whitmore AV, Libby RT, John SW. Glaucoma: thinking in new ways-a role for autonomous axonal self-destruction and other compartmentalised processes? Prog Retin Eye Res. 2005;24:639–662. doi: 10.1016/j.preteyeres.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Yoneda S, Hara H, Hirata A, Fukushima M, Inomata Y, Tanihara H. Vitreous fluid levels of beta-amyloid((1–42)) and tau in patients with retinal diseases. Jpn J Ophthalmol. 2005;49:106–108. doi: 10.1007/s10384-004-0156-x. [DOI] [PubMed] [Google Scholar]
- Zhong L, Bradley J, Schubert W, Ahmed E, Adamis AP, Shima DT, Robinson GS, Ng YS. Erythropoietin promotes survival of retinal ganglion cells in DBA/2J glaucoma mice. Invest Ophthalmol Vis Sci. 2007;48:1212–1218. doi: 10.1167/iovs.06-0757. [DOI] [PubMed] [Google Scholar]