

Abstract
Acetylcholine (ACh) is an important modulator of learning, memory, and synaptic plasticity in the basolateral amygdala (BLA) and other brain regions. Activation of muscarinic acetylcholine receptors (mAChRs) suppresses a variety of potassium currents, including sIAHP, the calcium-activated potassium conductance primarily responsible for the slow afterhyperpolarization (AHP) that follows a train of action potentials. Muscarinic stimulation also produces inositol 1,4,5-trisphosphate (IP3), releasing calcium from intracellular stores. Here, we show using whole-cell patch-clamp recordings and high-speed fluorescence imaging that focal application of mAChR agonists evokes large rises in cytosolic calcium in the soma and proximal dendrites in rat BLA projection neurons that are often associated with activation of an outward current that hyperpolarizes the cell. This hyperpolarization results from activation of small conductance calcium-activated potassium (SK) channels, secondary to the release of calcium from intracellular stores. Unlike bath application of cholinergic agonists, which always suppressed the AHP, focal application of ACh often evoked a paradoxical enhancement of the AHP and spike-frequency adaptation. This enhancement was correlated with amplification of the action potential-evoked calcium response and resulted from the activation of SK channels. When SK channels were blocked, cholinergic stimulation always reduced the AHP and spike-frequency adaptation. Conversely, suppression of the sIAHP by the β-adrenoreceptor agonist, isoprenaline, potentiated the cholinergic enhancement of the AHP. These results suggest that competition between cholinergic suppression of the sIAHP and cholinergic activation of the SK channels shapes the AHP and spike-frequency adaptation.
Keywords: learning, memory, nucleus, IP3, acetylcholine, mGluR
Introduction
The amygdaloid complex is a collection of deep temporal lobe nuclei involved in emotion and emotional learning. Within this complex, neurons in the basolateral amygdala (BLA) are proposed to be involved in assigning affective value to sensory stimuli. This association results in a change in the output [action potentials (APs)] of BLA projection neurons to the sensory stimulus. Functionally, neuronal output is determined by interactions between synaptic inputs impinging on neurons and their intrinsic excitability. Intracellular recordings have shown that BLA projection neurons in vivo discharge high-frequency bursts both spontaneously (Pare and Gaudreau, 1996) and in response to depolarizing current injections (Pare et al., 1995). Activation of calcium-activated potassium channels has been implicated in the generation of this bursting behavior (Lang and Pare, 1997; Chen and Lang, 2003). Similarly, the strongly adapting firing pattern observed in vitro is attributable to, in large part, the pronounced slow afterhyperpolarization (AHP) that follows trains of APs (Washburn and Moises, 1992a; Rainnie et al., 1993).
As in many other neurons, the AHP is primarily mediated by two calcium-activated potassium currents, IAHP and the sIAHP (Sah and Faber, 2002), which are activated by calcium influx during action potentials. The IAHP current is mediated by small conductance calcium-activated potassium (SK) channels, and its time course primarily follows cytosolic calcium, rising rapidly after APs and decaying with a time constant of 50 to several hundred milliseconds (Sah and Faber, 2002). In contrast, the kinetics of the sIAHP are slower, exhibiting a distinct rising phase and decaying with a time constant of 1–2 s (Sah, 1996). A variety of neuromodulators, including acetylcholine (ACh), noradrenaline, and glutamate acting via G-protein-coupled receptors, suppress the sIAHP and thus reduce spike-frequency adaptation (Nicoll, 1988).
The BLA receives one of the densest cholinergic innervations in the CNS (Ben-Ari et al., 1977; Mash and Potter, 1986). Consistent with this, neurons in the BLA express a variety of muscarinic (Levey et al., 1991) and nicotinic (Zhu et al., 2005) receptors. Although the precise mechanism of action is unclear, activation of cholinergic receptors have been implicated in amygdala-dependent learning (McGaugh, 2004; Tinsley et al., 2004; Barros et al., 2005). They are also known to modulate long-term synaptic plasticity within the amygdala (Watanabe et al., 1995). Physiologically, activation of muscarinic receptors on BLA neurons suppresses several potassium currents, including the sIAHP, as well as the voltage-sensitive M-current (Washburn and Moises, 1992b; Womble and Moises, 1993). These receptors also activate a nonspecific cation conductance (Yajeya et al., 1999). All these actions enhance neuronal excitability and are thought to underlie the cellular actions of acetylcholine. However, muscarinic stimulation also generates inositol 1,4,5-trisphosphate (IP3) that releases calcium from intracellular stores, a process that is amplified by AP-evoked calcium influx (Power and Sah, 2005, 2007). Store-released calcium activates SK calcium-activated potassium channels in midbrain dopamine neurons (Fiorillo and Williams, 1998, 2000; Morikawa et al., 2003) and cortical pyramidal neurons (Yamada et al., 2004; Gulledge and Stuart, 2005; Hagenston et al., 2007), consequently modulating neuronal excitability.
In this study, we examine the roles of calcium-activated potassium channels in modulating the firing properties of basal amygdala projection neurons. We show that, in these neurons, the AHP is mediated by a combination of apamin-sensitive SK channels and the apamin-insensitive sIAHP. Calcium release from intracellular stores, initiated by activation of muscarinic receptors, activates somatic SK channels and can enhance the AHP under some circumstances. These results show that muscarinic activation of SK channels competes with muscarinic suppression of the sIAHP to shape the AHP and spike-frequency adaptation.
Materials and Methods
Coronal brain slices (350–400 μm) were prepared using standard techniques (Power and Sah, 2002). Wistar rats (21–28 d of age) were anesthetized with halothane and decapitated. Slices were prepared using a vibrating microslicer (DTK-1000; Dosaka, Kyoto, Japan). These procedures were conducted in accordance with the guidelines of the University of Queensland Animal Ethics Committee. Slices were incubated at 33°C for 30 min and then maintained at room temperature in artificial CSF (ACSF) solution containing the following (in mm): 119 NaCl, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 1.0 Na2H2PO4, 26.2 NaHCO3, 11 glucose, equilibrated with 95% CO2, 5% O2. Slices were perfused with ACSF heated to 33°C, and whole-cell recordings were made from the soma of BLA neurons using infrared differential interference contrast video microscopy. Patch pipettes (2–5 MΩ) were filled with an internal solution containing the following (in mm): 135 KMeSO4, 8 NaCl, 10 HEPES, 2 Mg2-ATP, 0.3 Na3-GTP, 0.1 spermine (pH 7.3 with KOH, osmolarity 280–290 mOsm). Electrophysiological signals were amplified with either an Axopatch 1D or a Multiclamp 700A amplifier (Molecular Devices, Union City, CA), filtered at 2–5 kHz and digitized at 5–10 kHz with an ITC-16 board (Instrutech, Port Washington, NY), and controlled using Axograph 4.9 (AxoGraph Scientific, Sydney, Australia). Whole-cell recordings were obtained from projection neurons in the basal nucleus of the BLA. Only cells that had resting potentials more negative than −55 mV, AP amplitudes >100 mV, and membrane resistances (Rm) > 60 MΩ were included in the data set. Projection neurons were distinguished from local circuit interneurons based on the brevity of their action potentials (>0.7 ms), fast AHP (<15 mV), and their frequency-dependent spike broadening.
Whole-field fluorescence measurements were made using a monochromator-based imaging system, Polychrome II (T.I.L.L. Photonics, Munich, Germany) with 50 μm Oregon Green BAPTA-1 (Invitrogen, San Diego, CA) included in the internal solution. Neurons were visualized using a BX50 microscope (Olympus Optical, Tokyo, Japan) equipped with a 60× water immersion objective (numerical aperture, 0.9; Olympus) and illuminated with 488 nm light. Images were acquired with an interline transfer, cooled CCD camera (T.I.L.L. Photonics) in which the scan lines were binned by two in both horizontal and vertical directions, giving a spatial resolution of 0.33 μm per pixel. Frames were collected at 25–33 Hz with a 10 ms exposure time. Images were analyzed off-line using Vision (T.I.L.L Photonics). Calcium signals were calculated as the relative change in fluorescence over baseline fluorescence (ΔF/F). ΔF/F(t) = [F(t) − F0]/(F0 − B), where Ft is the fluorescence at time t, F0 is the average baseline fluorescence before the stimulus, and B is the background fluorescence measured in an adjacent extracellular region.
Two-photon fluorescence images were obtained using a Zeiss (Oberkochen, Germany) Axioskop 2FS with a 510 laser scanning head equipped with a Chameleon laser (Coherent, Santa Clara, CA) for two-photon excitation. For two-photon experiments, the green fluorescent calcium indicator Fluo 5F (300 μm; Kd, 2 μm; Invitrogen, Carlsbad, CA) was added to the internal solution along with the red fluorescent calcium-insensitive Alexa 594 (30 μm; Invitrogen). Fluo 5F and Alexa 594 were excited at 810 nm. The emitted light was split with a dichroic (DT560), bandpass filtered (green channel, 500–560 nm; red channel, 575–640 nm), and detected with separate nondescanned detectors. Fluorescence images were acquired in line scan-mode (100–500 Hz) at a resolution of 10–20 pixels μm−1. Small segments were selected over each subcellular region, and the fluorescence over this area was averaged. For two-photon sequences, calcium signals were calculated as the change in green fluorescence (Fluo 5F) normalized to the red fluorescence (Alexa 594), ΔG/R(t). Calcium signals were analyzed off-line using custom software written using LabVIEW (National Instruments, Austin, TX).
For focal application of cholinergic agonists, ACh or muscarine were loaded into a patch pipette and applied using either a picospritzer for pressure application (10–30 psi, 50–500 ms; Parker Hannifin Fairfield, NJ) or the 700A amplifier for iontophoretic application. For pressure application, drugs were dissolved in ACSF at a concentration of 10–20 μm. For iontophoretic application, acetylcholine (10–25 mm; pH 4 with HCl) was applied with a 2–50 nA ejection current and retained with a −5 nA retention current. The metabotropic glutamate receptor agonist, (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (t-ACPD) (2 mm; pH 8.5), was applied using a −50 nA ejection current and a 10 nA retention current. All other drugs were bath applied. Cyclopiazonic acid (CPA), 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl] (phenylmethyl) phosphinic acid (CGP 55845) were prepared as stock solutions in DMSO and diluted in ACSF when required. NBQX, CGP 55845, and 2-amino-5-phosphonovaleric acid (APV) were purchased from Tocris Bioscience (Bristol, UK). All other drugs were obtained from Sigma-Aldrich (St. Louis, MO). Drugs were prepared as 1000× stock solutions and stored frozen (−20°C) until required.
Depletion of intracellular calcium stores was prevented by maintaining the neuron at a slightly depolarized membrane potential (Vm, −50 mV) or evoking AP between ACh applications (Power and Sah, 2005). The AHP was evoked by a 100 Hz train of 5–10 ms depolarizing current injections, where each current step evoked a single AP. Occasionally two trains (200 ms intertrain interval) of depolarizing current injections were given to evoke both a robust AHP and calcium response. Measurements of the AHP included the peak amplitude, integrated area, mean afterpotential (0–5 s after the last current step), and the amplitude of the AHP 100 ms, 1 s, 2 s, and 5 s after the last current step. The peak and integrated area refer to negative (hyperpolarized) values only. Calcium response measures included the peak amplitude, integrated area, and the mean response 0–5 s after the last current step. For experiments using linopiridine the AHP and spike-frequency adaptation were measured alternatively at 1 min intervals before and during application of channel blockers. Inhibition was considered at steady state levels 7–10 min after the onset of drug application. AHP measurements were made from the average of two to three responses immediately before drug application and after blockers reached steady-state levels. Spike-frequency adaptation was measured from each current injection, and the responses were averaged. Statistical comparisons were made using a paired Student's t test, unless otherwise indicated. All data are presented as mean ± SEM unless otherwise indicated.
Results
Whole-cell patch-clamp recordings were made from projection neurons within the basal nucleus of the BLA. These neurons had resting potentials of −65.0 ± 4.3 mV (mean ± SD) and input resistances of 120 ± 66 MΩ (mean ± SD; n = 154). Projection neurons in the basal nucleus are traditionally separated into two classes based on their firing patterns in response to depolarizing current injections (Washburn and Moises, 1992a), although it is unclear whether these two classes represent distinct neuronal populations or are on opposite ends of a firing property continuum (Kroner et al., 2005). The majority of neurons (80%) had a pyramidal-like firing pattern with strong spike-frequency adaptation and a prominent AHP (Fig. 1A,B). Less frequently (20%), we encountered neurons that showed a late-firing phenotype where the onset of spike firing was delayed (>300 ms) near threshold (Fig. 1A). As described previously (Washburn and Moises, 1992a), late-firing neurons tended to show less spike-frequency adaptation than pyramidal-like neurons, and their AHP was generally smaller in amplitude (Fig. 1A,B, middle). However, nearly half of the late-firing neurons we encountered exhibited robust spike-frequency adaptation and pronounced AHPs (Fig. 1A,B, right). The AHP amplitude was 2.0 ± 0.5 mV for late-firing neurons (n = 17) compared with 2.7 ± 0.2 mV for pyramidal-like neurons (n = 48; p = 0.18). It has been reported that late-firing neurons are more hyperpolarized than pyramidal-like neurons, and the kinetics of their AHP differs from that in pyramidal-like neurons (Washburn and Moises, 1992a; but see Kroner et al., 2005). We did not observe any differences in either their resting membrane potential (late-firing neurons, −65.6 ± 0.8 mV; pyramidal-like neurons, −64.8 ± 0.4 mV; p = 0.35) or the membrane resistance (late-firing neurons, 137 ± 9 MΩ; pyramidal-like neurons, 115 ± 7 MΩ; p = 0.11) between classes. Although the AHP that followed a brief AP train was generally smaller in amplitude for late-firing neurons that did not accommodate, we found that both the calcium responses (data not shown) and the AHP time courses (Fig. 1B) were indistinguishable between these two types of cells, and the data have been combined. The AHP showed slow kinetics in both cell types with 32 ± 7 and 37 ± 2% of the AHP amplitude remaining 2 s after the pulse train for late-firing (n = 17) and pyramidal-like (n = 48) neurons, respectively (p = 0.38). To limit variability, however, spike-frequency adaptation was examined solely in pyramidal-like neurons.
Figure 1.
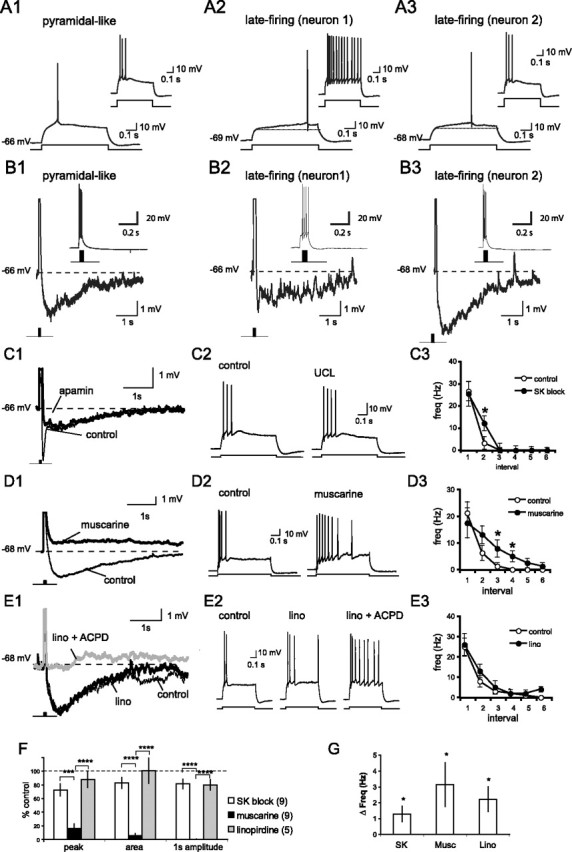
The slow AHP and spike-frequency adaptation are mediated by the muscarine-sensitive sIAHP. A, Projection neurons in the basal nucleus were classified as either pyramidal like or late firing based on the AP latency near threshold. Typical responses to 600 ms current steps just above the action potential threshold for a pyramidal-like (A1) and two late-firing (A2, A3) neurons are shown. Insets show the response to a 400 pA current injection. B1–B3, Typical AHP that follows a brief 4 AP (100 Hz) train for the pyramidal and late-firing neurons shown in A1–A3. Top and bottom traces show the same four-spike AHP on a different scale. Note the slow kinetics shown by both classes of neurons, reminiscent of the sIAHP. C, Application of apamin (100 nm) or UCL 1848 (UCL; 1 μm) has a modest effect on the AHP and reduces spike-frequency adaptation. A typical AHP evoked by a burst of four APs before and after bath application of apamin is shown in C1. Note SK channel blockade suppresses the early but not the late component of the AHP. APs have been truncated. A typical response to a depolarizing current injection (600 ms, 400 pA) before (thin traces) and after (thick traces) bath application of UCL 1848 is shown in C2. The mean spike frequency for each 100 ms interval before and after SK block is shown in C3. D, Application of muscarine blocks the AHP and reduces spike-frequency adaptation. A typical AHP evoked by a burst of four APs before (thin traces) and after (thick traces) bath application of 5 μm muscarine is shown in D1. A typical response to a depolarizing current injection (600 ms, 400 pA) before and after bath application of muscarine is shown in D2. The mean spike frequency for each 100 ms interval before and after muscarine is shown in D3. E, Application of the M-current blocker linopirdine (lino; 10 μm) reduces spike-frequency adaptation but has little effect on the AHP after a brief AP train. A typical AHP evoked by a burst of four APs before (thin traces) and after (thick traces) sequential application of linopirdine (lino) and t-ACPD (10 μm; gray traces), which blocks the sIAHP, is shown in E1. A typical response to a depolarizing current injection (600 ms, 400 pA) before and after bath application of linopirdine (lino) and ACPD (10 μm) is shown in E2. The mean spike frequency for each 100 ms interval before and after linopirdine (lino) is shown in E3. F, Columns show the fraction (mean ± SEM) of the peak amplitude of the AHP (peak), the integrated area of the AHP (area), and the amplitude of the AHP 1 s after the AP train (1 s amplitude), present after application of SK blockers (UCL 1848 or apamin), muscarine, or linopirdine. The number of neurons is shown in parentheses. Summary data showing the increase in spike frequency in response to a 600 ms, 400 pA current step after bath application of SK blockers, muscarine, or linopirdine (Lino) are given in G. Note that the APs have been truncated in C1, D1, and E1. *p < 0.05, paired t test; ***p < 0.001 and p < 0.0001 for post hoc comparisons. Depolarizing potentials were scored as 0 mV.
In most neurons, the AHP is primarily mediated by either the apamin-sensitive IAHP or the apamin-insensitive sIAHP and, in many cases, a combination of these two currents (Sah and Faber, 2002). It was initially reported that the apamin-sensitive IAHP is not present in BLA projection neurons (Womble and Moises, 1993). However, subsequent reports have indicated that SK calcium-activated potassium channels, which mediate the IAHP, are present in projection neurons in the lateral nucleus of the BLA (Faber and Sah, 2002; Chen and Lang, 2003; Faber et al., 2006). To determine whether the SK channels (IAHP) contribute to the AHP in projection neurons in the basal nucleus, we examined the AHP evoked by four APs (100 Hz) before and after SK channel blockade by bath application of either apamin (100 nm) or 8,14-diaza-1,7(1, 4)-diquinolinacyclotetradecaphane (UCL 1848; 1 μm) (Chen et al., 2000).
Application of specific SK channel blockers had a modest but significant effect on the AHP, reducing its peak by 28 ± 9% (p = 0.020; n = 7) and its integrated area by 17 ± 8% (p = 0.068; n = 7) (Fig. 1C). Consistent with the relatively rapid kinetics of the IAHP (Sah and Faber, 2002), the effect of SK channel blockade was greatest shortly after the spike train but produced only a modest reduction (19 ± 7%; p = 0.033; n = 7) in the AHP amplitude measured 1 s after the spike train (Fig. 1C). In response to a depolarizing current injection, pyramidal-like neurons fire a burst of APs, which rapidly accommodates. Blockade of SK channels produced a modest attenuation of this spike-frequency adaptation (Fig. 1C3), increasing the mean AP frequency evoked by a 600 ms, 400 pA current injection from 4.8 ± 0.8 to 6.1 ± 1.0 Hz (p = 0.023; n = 9).
Consistent with previous reports (Womble and Moises, 1993), suppression of the sIAHP by bath application of muscarine (5–20 μm) resulted in a reduction in both the AHP and spike-frequency adaptation (Fig. 1D). Muscarine reduced the peak amplitude of the AHP by 84 ± 7% (p = 0.00052), the integrated area by 94 ± 3% (p = 0.00546), and completely abolished the AHP measured 1 s after the AP train (100 ± 0% block; p = 0.00036). In many instances, the latter portion of the AHP was replaced by a slow afterdepolarization (Fig. 1D). The result was an increase in the mean spike frequency during a 600 ms, 400 pA current injection from 4.4 ± 0.8 to 7.5 ± 1.6 Hz (p = 0.047; n = 8).
In addition to the sIAHP, muscarinic stimulation also suppresses the voltage-sensitive M-current, which has been shown to modulate the AHP that contributes to spike-frequency adaptation in other neuronal types (Gu et al., 2005). To determine whether the M-current contributes to the AHP and spike-frequency adaptation, we applied the specific M-current blocker linopiridine (Schnee and Brown, 1998). Linopirdine (10 μm) had no effect on the AHP to a brief four-AP train (Fig. 1E). The peak amplitude and area of the AHP in the presence of linopirdine were 87 ± 11% (p = 0.43) and 101 ± 18% (p = 0.64) of control, respectively (n = 5). In BLA neurons, the sIAHP and the M-current are also suppressed by metabotropic glutamate receptor (mGluR) activation (Womble and Moises, 1994). In the presence of linopirdine, application of the mGluR agonist t-ACPD suppressed the AHP (Fig. 1E) (n = 2), indicating that muscarinic suppression of the AHP results from blockade of the sIAHP rather than the suppression of the M-current. This current does, however, play a role in spike frequency adaptation as linopirdine increased the average AP frequency during a 400 pA, 600 ms current step from 6.0 ± 1.1 to 8.2 ± 1.2 Hz (p = 0.034; n = 5). Thus, in projection neurons in the basal nucleus, both IAHP and sIAHP contribute to the AHP with the apamin-insensitive sIAHP making the greatest contribution (Fig. 1F). The M-current, although present, is not active during the AHP but does affect spike-frequency adaptation.
Release of calcium from intracellular stores activates SK channels
Brief focal application of ACh or muscarine evokes large (>1 μm) wave-like rises in intracellular calcium in the soma and proximal dendrites that result from release of calcium from IP3-sensitive intracellular calcium stores (Power and Sah, 2007). Interestingly, although release of calcium from IP3-sensitive intracellular calcium stores can also be observed in response to bath application of muscarinic agonists, focal stimulation is much more effective in evoking robust calcium rises (Power and Sah, 2002, 2007). This difference may result from desensitization of muscarinic receptors as receptor occupancy rises slowly during bath application. Focally evoked calcium waves were frequently associated with an outward current in voltage-clamp mode (Fig. 2A) or with membrane hyperpolarization with the cell in current clamp. Both the evoked calcium rise and outward current were only seen in response to agonist application onto the soma or proximal dendrites and were not observed (0 of 4) when agonists were applied to dendrites further than 60 μm from the soma (Power and Sah, 2007).
Figure 2.

Focal application of ACh activates SK channels. A, Picospritzer application of muscarine onto the soma and proximal dendrites (10 μm; 20 psi 600 ms) evokes an outward current. The whole-cell current (Vm, −50 mV) is shown above the calcium response (Oregon Green BAPTA-1), plotted as ΔF/F for the soma and proximal dendrite. B, Iontophoretic application of ACh evokes a robust somatic calcium response and outward current (Vm, −50 mV) that was blocked by bath application of atropine (1 μm). The whole-cell current together with the somatic calcium rise (Fluo 5F), plotted as ΔG/R, evoked by iontophoresis of ACh (2 s 40 nA) are shown before and after application of atropine. Mecalmylamine (10 μm), picrotoxin (50 μm), and kynurenic acid (2 mm) were included in the ACSF. CTL, Control. C, CPA blocks the somatic calcium rise and outward current evoked by iontophoretic application of ACh. The whole-cell current along with the somatic calcium rise (Fluo 5F), plotted as ΔG/R, evoked by iontophoresis of ACh (2 s 15 nA) are shown before and 15 min after bath application of CPA (15 μm), and 1 h after washout of CPA. D, Representative current and calcium responses to iontophoretic application of ACh before and after priming of intracellular calcium stores. ACh was applied at 2 min intervals. Calcium stores were primed by a 1 min depolarization to −50 mV between the third and fourth application of ACh. E, A projection of confocal image stacks of a BLA neuron filled with Alexa 594 and Fluo 5F is shown in E1. The scan line, somatic (soma) and dendritic (dend) regions of interest, and the location of the iontophoretic pipette are indicated on the projection image. The current and calcium response to ACh iontophoresis is shown in E2. The onset of the whole-cell current, indicated by the dashed line, is correlated with the onset of calcium wave propagation into the soma. F, Application of apamin (100 nm) eliminated the ACh-evoked outward current without affecting the calcium response. The whole-cell current together with the somatic calcium rise (Fluo 5F), plotted as ΔG/R, evoked by iontophoresis of ACh (2 s, 15 nA) at 150 s intervals is shown before and during bath application of apamin in F1. The whole-cell current and calcium response before apamin (control; thin traces) is overlaid with the response 5 min after application of apamin (apamin; thick traces) in F2. G, To test whether the activation of SK channels by iontophoretic application of ACh (inset) was caused by cholinergic modulation of SK channels, we compared the AHP current (Vm, −50 mV) evoked by a depolarizing voltage step (500 ms, 70 mV) with and without iontophoretic application of ACh (gray traces). The AHP current (top traces) and somatic calcium response (bottom traces) are shown to sequential application of CPA (15 μm) and apamin (100 nm). The apamin-sensitive current (“apamin + CPA” minus “CPA”) are shown on the right. Application of ACh augmented the somatic calcium rise and the early portion of AHP current but suppressed the current at later time points. Bath application of CPA abolished the cholinergic augmentation of the calcium response and the AHP current. Apamin produced a comparable reduction in the AHP current in the presence and absence of ACh, indicating that ACh did not modulate the apamin-sensitive (SK) current. H, Summary data (mean ± SEM) showing the effect of atropine, CPA, and apamin on the cholinergic-evoked outward current (filled bars) and the somatic calcium rise (empty bars).
To eliminate movement artifacts and minimize problems related to calcium indicator saturation, neurons were focally stimulated by iontophoretic application of ACh and imaged using multiphoton fluorescence microscopy and the moderate affinity calcium indicator Fluo 5F (Kd, ∼2 μm). Because ACh also activates nicotinic receptors in these neurons (Klein and Yakel, 2006), experiments using ACh were performed in the presence of the nicotinic receptor antagonist mecamylamine (10 μm). The outward current evoked by ACh was blocked by atropine (1 μm, n = 3) (Fig. 2B) and was not affected by application of GABAergic (50 μm picrotoxin and 10 μm CGP 58845) and ionotropic glutamatergic blockers (2 mm kynurenic acid or 30–60 μm APV plus 20 μm NBQX), suggesting that it results from the direct actions of mAChR activation on BLA projection neurons rather than network effects. Moreover, the outward current was reversibly blocked by CPA, which blocks the endoplasmic reticulum (ER) Ca2+ ATPase and empties intracellular calcium stores (Fig. 2C) (n = 6). CPA (15–30 μm) abolished the cholinergic-evoked calcium rise and reduced the amplitude of the outward current by 82.5 ± 4.0% (p = 0.008) and the area of the outward current by 96.1 ± 1.8% (p = 0.022). On occasion, muscarinic stimulation evoked a small inward current with slow kinetics (Fig. 2C,D). This current was not blocked by CPA and, although we have not characterized it fully, its properties are consistent with activation of a nonspecific cationic conductance by muscarinic receptor stimulation (Yajeya et al., 1999). Release of calcium from intracellular stores is dependent on store content, with repetitive activation decreasing the store content and reducing release from intracellular calcium stores (Power and Sah, 2005). Conversely, calcium entry through voltage-gated calcium channels, during APs or depolarizations near threshold, primes the calcium stores, augmenting the calcium response to cholinergic stimulation (Power and Sah, 2005). Thus, when neurons were held at −65 mV, repeated application of ACh reduced both calcium store release and the outward current (Fig. 2D). Replenishing the calcium store with a 1 min membrane depolarization (−50 mV) augmented both store release and the outward current (Fig. 2D). We have shown previously that when cholinergic agonists are applied to the proximal dendrite, the calcium rise begins as a focal rise in the dendrite, which propagates into the soma (Power and Sah, 2007). Inspection of the timing of calcium rises in the soma and dendrite reveals that the onset of the outward current is best correlated with the somatic calcium rise (Fig. 2E), suggesting that the channels that underlie this current have a somatic location.
Calcium release from internal stores has been found to activate SK channels in other neurons (Fiorillo and Williams, 1998, 2000; Morikawa et al., 2003; Yamada et al., 2004; Gulledge and Stuart, 2005; Hagenston et al., 2007). Consistent with this mechanism, the outward current activated by muscarinic agonists in BLA neurons was suppressed by bath application of the SK channel blocker apamin (100 nm), with no effect on the calcium rise. Apamin reduced the amplitude of the outward current by 78.4 ± 3.2% (p = 0.006) and the area by 94.1 ± 0.3% (p = 0.025) (Fig. 2F) (n = 4), whereas the peak amplitude and the area of the calcium rise were unaffected (95.7 ± 3.5% of control; p = 0.33 and 97.0 ± 10.7% of control; p = 0.51, respectively). Because muscarinic receptors activate a second messenger cascade, we next examined whether SK channels could also be directly modulated by ACh. In the presence of CPA to block store release, we evoked an AHP current via a depolarizing voltage step (500 ms, 70 mV; Vm, −50 mV) with and without iontophoretic application of ACh (2 s, 50 nA). We subsequently evoked the AHP current with and without ACh in the presence of bath applied apamin. Subtracting the AHP currents evoked in apamin from their respective control currents, we were able to determine the apamin-sensitive (SK) current with and without application of ACh. When store release was blocked, iontophoretic application of ACh had no effect on the integrated area of the apamin-sensitive current (103 ± 3% of control; p = 0.47; n = 4) (Fig. 2G). Thus, muscarinic receptor activation does not directly modulate SK channels. These results, together with the effects of store depletion and priming, indicate that in BLA projection neurons, cholinergic stimulation releases calcium from intracellular stores, which in turn activates SK potassium channels.
In BLA projection neurons (Power and Sah, 2007) and elsewhere (Jaffe and Brown, 1994; Nakamura et al., 1999; Ross et al., 2005), calcium waves can also be evoked by exogenous application of group I mGluR agonists and synaptically through the cooperative action of mGluR and mAChR stimulation (Power and Sah, 2007). Thus, as with muscarinic stimulation, focal application of the mGluR agonist t-ACPD evoked a rise in somatic calcium and an outward current (Fig. 3A,C). As with muscarinic stimulation, apamin reduced the peak of the t-ACPD evoked current by 82 ± 4% (p = 0.010) and the area by 94 ± 4% (p = 0.030) without affecting the somatic calcium rise (peak, 89 ± 13%, p = 0.35; area, 98 ± 12%, p = 0.49; n = 4). Moreover, the outward current was not detected before the somatic calcium rise (Fig. 3B), regardless of whether the wave is initiated in the dendrite or the soma. Similarly, when calcium waves were evoked by synaptic stimulation (in the presence of GABAergic and iontotropic glutamatergic blockers) (Fig. 3D), somatic invasion of the calcium wave was usually (21 of 24 neurons) associated with an outward current. Muscarinic receptors are thought to play a critical role in somatic invasion of synaptically evoked calcium waves (Watanabe et al., 2006; Power and Sah, 2007). Application of atropine reduced the integrated area of the synaptically evoked somatic calcium rise by 82 ± 9% (p = 0.004) and the outward current by 83 ± 17% (p = 0.02; n = 4) (Fig. 3E,F), indicating that much of the somatic calcium rise and outward current are caused by mAChR activation. This outward current resulted from SK channel activation, because application of apamin reduced the amplitude and area of the synaptically activated outward current by 82 ± 8% (p = 0.006) and 96 ± 3% (p = 0.04), respectively (n = 5) (Fig. 3G–I). The peak amplitude and the area of the calcium rise were unaffected by apamin (118 ± 12% of control, p = 0.23, and 133 ± 20% of control, p = 0.26, respectively). Thus, regardless of how calcium waves were evoked (ACh, t-ACPD, synaptically), an apamin-sensitive outward current is activated with a time course that correlates well with somatic calcium rises. This is despite the fact that the calcium rise is largest in proximal dendrites (10–20 μm from the soma) typically exceeding 1 μm (Power and Sah, 2007). Given that the EC50 value for SK channels is 300–700 nm (Stocker, 2004), it seems unlikely that these SK channels are present on the proximal dendrite but are activated by calcium released from intracellular stores that are located on or near the soma.
Figure 3.

Synaptically evoked calcium waves activate SK channels. A, The somatic calcium rise (bottom traces) and membrane current (top traces; Vm, −50 mV) evoked by iontophoresis of ACPD (2 mm, −55 nA, 2 s) are shown before and during bath application of apamin. B, The membrane current (Vm, −50 mV) is shown together with the calcium rise in the soma and proximal dendrite in response to iontophoresis of ACPD (2 mm, −55 nA, 2 s) onto either the proximal dendrite (left) or the soma (right). Note the reversal of wave propagation as the location of agonist application changes. The amplitude and onset of the current were correlated with the somatic but not the dendritic calcium response. The onset of the somatic calcium rise is indicated by the dashed line. C, Columns show the fraction (mean ± SEM) of the peak amplitude (peak), integrated area (area) of the ACPD-evoked outward current, and the somatic calcium response remaining after bath application of apamin (100 nm). D, Calcium waves evoked by synaptic stimulation evoke outward currents. Calcium rises in the soma and dendrite together with the whole-cell current (w-c current) are shown in response to local synaptic stimulation (100 Hz, 1 s) in the presence of APV (60 μm), NBQX (20 μm), picrotoxin (100 μm), and CGP 58845 (10 μm). E, Application of atropine (1 μm) reduces somatic invasion of the calcium wave and the outward current evoked by local synaptic stimulation (100 Hz, 1 s). The outward currents and the somatic calcium rise are shown before (thin traces) and after (thick traces) application of atropine. F, Summary data (mean ± SEM; n = 4) showing the effect of atropine on the integrated area of the somatic calcium response (open circles) and outward current (filled circles) evoked at 3 min intervals before and during bath application of atropine. G, Synaptic stimulation activates SK type calcium-activated potassium channels. Calcium rises in the soma and dendrite, together with the whole-cell current (w-c current) are shown in response to local synaptic stimulation (100 Hz, 1 s) in the presence of APV (60 μm), NBQX (20 μm), picrotoxin (100 μm), and CGP 58845 (10 μm). Responses were evoked as 2 min intervals before (thin traces) and during (thick traces) application of apamin. The calcium rise and whole-cell current before (response 2) and after (response 5) apamin are shown overlaid in H. I, Columns show the fraction (mean ± SEM) of the peak amplitude (peak), integrated area (area) of the synaptically evoked outward current, and somatic calcium response remaining after bath application of apamin (100 nm).
Competing effects of ACh on the AHP
We have shown that muscarinic stimulation has two actions. First, it enhances neuronal excitability by reducing the AHP (Fig. 1D). However, activation of muscarinic receptors also causes release of calcium from intracellular calcium stores, which activates SK calcium-activated potassium channels (Fig. 2F). The resultant hyperpolarization would be expected to reduce neuronal excitability. We therefore examined how these competing actions of cholinergic stimulation on calcium-activated potassium currents shape the AHP. We first compared the AHP evoked by an AP train with and without iontophoretic application of ACh. Calcium is a coagonist at IP3 receptors (Bezprozvanny et al., 1991; Finch et al., 1991; Mak et al., 1999) and, in the presence of IP3, AP-evoked calcium influx can be amplified through IP3-assisted calcium-induced calcium release (IP3-CICR) (Taylor and Marshall, 1992; Nakamura et al., 1999; Power and Sah, 2002). An example of IP3-CICR is shown in Figure 4. In BLA projection neurons, AP trains evoke a rapid rise in calcium in the soma and dendrite that decays immediately after the last AP. The AP train also evokes an AHP, which shows a slower time course than the calcium response (Fig. 4A). In the same neurons, focal application of lower levels of ACh (obtained by reducing the duration and amplitude of the ejection current) produces little or no calcium response or corresponding membrane hyperpolarization. Pairing the same level of ACh with an AP train results in a calcium rise in the soma and proximal dendrites that is larger than the arithmetic sum of the separate ACh and AP presentations (Fig. 4B). In some instances, the amplified calcium response was accompanied by a clear enhancement of the AHP (Fig. 4A,B). IP3-CICR is also seen in response to bath application of mAChR agonists (Power and Sah, 2002, 2007), although the amplification is markedly smaller than that observed in response to focal stimulation (Fig. 4C). This amplification is largest in response to the first AP train and then declines, presumably because of desensitization of receptors in the continued presence of agonist (Fig. 4C).
Figure 4.

Synergistic action of ACh and APs on the calcium response in the soma and proximal dendrite. A, The calcium response in the soma and proximal dendrite (dend) are shown above the membrane potential in response to a four AP train (100 Hz; APs only), iontophoretic application of ACh (30 nA, 900 ms; ACh only), and paired presentation of ACh and APs (paired). B, The calcium and voltage response to paired presentation of ACh and APs is overlaid with the arithmetic sum of unpaired presentations of ACh and APs. C, IP3-CICR is also seen with bath application of muscarine, albeit to a lesser extent. The calcium and voltage response to a four AP train (100 Hz) is shown before (control) and during bath application of muscarine (5 μm). AP trains were evoked at 30 s intervals 5 min after the onset of muscarinic stimulation. The response to the first AP train (muscarine 1) is shown together with the averaged response to the fourth and fifth AP train (muscarine 4–5). The control response is an average of five responses.
The mean response to focal application of ACh was a slight (0.67 mV) enhancement of the AHP (n = 30), although this response was heterogeneous (Fig. 5). In some neurons focal application of ACh augmented the AHP, resulting in a more negative afterpotential (Figs. 4, 5A), whereas in others, it suppressed the AHP, resulting in a more positive afterpotential (Fig. 5B). A histogram of the distribution of the cholinergic modulation of the AHP peak amplitude is shown in Figure 5C. Because the AHP is a negative potential, enhancement of the AHP is plotted as a negative change in the AHP, whereas suppression of the AHP is plotted as a positive change in the AHP. We found that the variability of the response was attributable to, in part, variations in ACh-evoked store release. Here, the effect of ACh on the AHP was correlated with the extent to which ACh augmented the AP-evoked calcium response (Fig. 5D), with larger calcium responses being associated with an enhanced AHP. There was a clear correlation between the amplitude of the amplified calcium response (measured as the integrated area of the amplified portion of the response) and the change in the peak (r = 0.637; p < 0.0001), and the integrated area (r = 0.541; p = 0.0079), of the AHP as well as the amplitude of the AHP 100 ms (r = 0.710; p < 0.0001), 1 s (r = 0.675; p < 0.0001), and 2 s (r = 0.659; p < 0.0001) after the last AP.
Figure 5.

Heterogeneous effects of the focal application of ACh on the AHP. Examples where focal cholinergic stimulation enhanced and depressed the AHP are shown in A and B, respectively. A histogram showing the distribution of the response on the AHP peak is shown in C. The mean response (0.67 mV enhancement of the AHP peak) is indicated by the arrow. The normal distribution is shown overlaid on the histogram. D, Plot of the correlation between the cholinergic enhancement of the AP-evoked somatic calcium response and the change in the peak amplitude of the AHP (n = 30).
As shown above (Fig. 4), mAChR activation generates an outward current that augments the AHP. This augmentation was blocked by apamin or UCL 1848, showing that the AHP evoked during muscarinic stimulation is primarily caused by activation of SK-type calcium-activated potassium channels (Fig. 6A). Moreover, in neurons where cholinergic stimulation evoked IP3-CICR but produced a net reduction in the AHP, the cholinergic reduction of the AHP was often greater when SK channels were blocked (Fig. 6B). These results show that activation of SK channels via IP3-CICR masks the suppression of the AHP typically seen in response to bath application of mAChR agonists. Cholinergic suppression of the sIAHP occurs through activation of muscarinic receptors linked to phospholipase C (Dutar and Nicoll, 1989; Washburn and Moises, 1992b), possibly through activation of calcium calmodulin-dependent protein kinase II (Muller et al., 1992; Krause and Pedarzani, 2000). Thus, cholinergic phosphoinositol turnover both releases calcium from IP3-sensitive ER calcium stores and suppresses the sIAHP. To isolate the cholinergic action on the AHP and IP3-CICR from its modulation of the sIAHP, we applied the β-adrenergic receptor agonist isoprenaline. Stimulation of β-adrenergic receptors suppresses the sIAHP (Nicoll, 1988; Huang et al., 1994; Faber and Sah, 2005) through protein kinase A and is not linked to stimulation of phospholipase C (Pedarzani and Storm, 1993). When the sIAHP was suppressed by bath application of isoprenaline (10 μm), focal application of ACh always enhanced the AHP, even in neurons where ACh previously suppressed it (Fig. 6C,E). Thus, a competition exists between cholinergic suppression of the sIAHP and cholinergic activation of the SK channels resulting from calcium release from intracellular stores. Blockade of SK channels unmasked the cholinergic suppression of the sIAHP (Fig. 6D), whereas suppression of the sIAHP by isoprenaline unmasked cholinergic activation of SK channels (Fig. 6E). Neither the suppression of the sIAHP nor the blockade of SK channels altered the amplification of the calcium response by ACh. Thus, under conditions of SK channel blockade, there was no longer a relationship between cholinergic augmentation of the calcium response and the AHP (Fig. 6F).
Figure 6.

Competition between cholinergic suppression of the sIAHP and cholinergic SK channel activation determines the effect of ACh stimulation on the AHP. A, Augmentation of the AHP, but not the calcium response, is blocked by bath application of the SK channel blockers apamin and UCL 1848. A representative example showing the AP-evoked voltage and somatic calcium response with and without ACh before and after bath application of apamin (100 nm). B, An example in which application of apamin enhanced the cholinergic suppression of the AHP. Voltage and calcium traces are shown for APs alone and for APs paired with ACh before and after bath application of apamin (100 nm). C, Blockade of the sIAHP boosts the enhancement of the AHP by ACh. A representative example showing AP-evoked voltage and somatic calcium responses with and without ACh before and after suppression of the sIAHP by bath application of isoprenaline (ISO; 10 μm). D, Summary data showing the effect of apamin on the modulation of the AHP by focal application of ACh. The lines show the effect of focal application of ACh on the amplitude of the AHP before and after apamin (n = 9). Mean ± SEM values are shown outside the lines. E, Summary data showing the effect of isoprenaline on the modulation of the AHP by focal application of ACh. Lines show the effect of focal application ACh on the amplitude of the AHP before and after application of isoprenaline (n = 6). Mean ± SEM values are shown outside the lines. F, Plot of the cholinergic enhancement of the AP-evoked somatic calcium response versus the change in the peak amplitude of the AHP after SK channel blockade (filled circles) or suppression of the sIAHP with isoprenaline (open circles). The regression line under control conditions (Fig. 5D) is shown for clarity.
The major role of the slow AHP in neurons is thought be in spike-frequency adaptation (Sah, 1996). We next tested whether neurons that showed an enhanced AHP to focal ACh stimulation would also exhibit enhanced spike-frequency adaptation. AP trains (10 Hz, 10 s) were evoked using 50 ms current injections set 25 pA above threshold. Data were partitioned into 1 s bins, and spike frequency was calculated by counting the number of spikes within each 1 s bin. Under control conditions, the AP firing rate showed marked accommodation (Fig. 7A). In neurons where ACh iontophoresis enhanced the AHP, focal application of ACh resulted in an early transient enhancement of spike-frequency adaptation (reduced firing) that was followed by a prolonged period of reduced spike-frequency adaptation (enhanced firing). This enhanced spike-frequency adaptation was associated with an augmented somatic calcium rise. In contrast, neurons in which ACh suppressed the AHP showed little or no suppression of spike-frequency adaptation (Fig. 7B) and a less pronounced calcium rise (Fig. 7C). The prolonged period of enhanced firing did not differ between groups and presumably resulted from the cholinergic suppression of sIAHP and IM, together with activation of a mixed cation conductance. Blockade of SK channels eliminated the cholinergic enhancement of spike-frequency adaptation during the first two intervals (p values <0.05), without affecting the augmentation of the calcium rise (Fig. 8A–C) (n = 5). The prolonged reduction of spike-frequency adaptation was not affected by application of apamin. Consistent with the enhanced spike-frequency adaptation from SK channel activation being secondary to calcium store release, depletion of calcium stores with CPA blocked both the cholinergic enhancement of the AP evoked calcium response and the cholinergic enhancement of spike frequency adaptation (Fig. 8D–F) (n = 6). CPA significantly reduced the cholinergic suppression of spike-frequency adaptation during the first interval (p < 0.05), along with the cholinergic augmentation of the calcium response in the first five intervals (p values <0.05). Spike-frequency adaptation was reduced when the sIAHP was suppressed by bath application of isoprenaline (Fig. 8G) (n = 6; p values = 0.06–0.01). The early enhancement of spike-frequency adaptation by focal application of ACh was augmented after application of isoprenaline (Fig. 8G–I) (n = 6; interval 1, p < 0.05). Together, these results indicate that ACh modulates spike-frequency adaptation through the opposing actions of sIAHP suppression and SK channel activation.
Figure 7.

Biphasic effects of ACh on spike-frequency adaptation. A, APs were evoked by a 10 Hz, 10 s train of 50 ms current injections 25 pA above threshold before (control) and after (ACh) iontophoretic application of ACh onto the soma and proximal dendrites (dend). Calcium transients and graphs of the spike frequency (Freq.) (summary data mean ± SEM; n = 14) are shown below the voltage response. For clarity, the control response has been replotted with the response in ACh. B, Plot of the change in spike frequency (ACh − control) evoked by application of ACh over time (mean ± SEM) for neurons in which ACh enhanced the AHP (n = 10) or suppressed the AHP (n = 4). C, Plot of the augmentation (ACh − control) of the calcium response by application of ACh over time (mean ± SEM) for neurons in which ACh enhanced (n = 10) or suppressed (n = 4) the AHP. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 8.

Pharmacology of ACh modulation of spike-frequency adaptation. A, The initial enhancement of spike frequency (Freq) adaptation by ACh is blocked by apamin. APs evoked by a 10 Hz, 10 s train of 50 ms current injections are shown in the presence of apamin with (ACh in apamin) or without (apamin) iontophoretic application of ACh. Calcium transients and graphs of the spike frequency over time (population data mean ± SEM) are shown below the voltage response. The response of this neuron before application of apamin is shown in Figure 7A. For clarity, the response without ACh has been replotted with the response in ACh. Plots of the effect of ACh on the calcium response and spike-frequency adaptation over time (mean ± SEM; n = 5) before and after application of apamin are shown in B and C, respectively. D, The cholinergic enhancement of spike-frequency adaptation requires ER calcium stores. APs and somatic calcium responses are shown with (ACh iontophoresis) or without (control) iontophoretic application of ACh, before and after bath application of CPA. Plots of the spike frequency over time (population data mean ± SEM) are shown below the voltage response. Plots of the effect of ACh on the calcium response and spike frequency adaptation over time (n = 6) before and after application of CPA are shown in E and F, respectively. G, The cholinergic enhancement of spike-frequency adaptation is masked by the sIAHP. APs and somatic calcium responses are shown with (ACh iontophoresis) and without (control) iontophoretic application of ACh, before and after bath application of isoprenaline. Plots of the spike-frequency over time (population data mean ± SEM) are shown below the voltage response. Plots of the effect of ACh on the calcium response and spike-frequency adaptation over time (mean ± SEM; n = 6) before and after application of isoprenaline are shown in H and I, respectively. *p < 0.05; paired t test, control versus drug.
Discussion
Calcium entry via voltage-dependent calcium channels during the AP activates several types of calcium-activated potassium channels. These channels contribute to repolarization of the AP and play a dominant role in the AHP and spike-frequency adaptation during trains of APs. Two different calcium-activated potassium currents underlie the AHP. One current (IAHP) is mediated by apamin-sensitive SK type channels and has relatively rapid kinetics such that the macroscopic current tracks cytosolic calcium, decaying with a time constant of 50–100 ms. The other current (sIAHP) has much slower kinetics lasting for several seconds. In BLA projection neurons, the AHP that follows an AP train is mediated almost entirely by the apamin-insensitive sIAHP (Fig. 1) (Womble and Moises, 1993). In this regard, neurons in the basal nucleus are similar to projection neurons in the lateral amygdala (Faber and Sah, 2002) and pyramidal neurons of CA1 hippocampus (Oh et al., 2000), where blockade of SK channels has little effect on either the AHP or spike-frequency adaptation. Large rises in cytosolic calcium can also be generated by activation of metabotropic receptors coupled to phospholipase C, with subsequent release of calcium from IP3-sensitive ER calcium stores (Power and Sah, 2007). We have shown that mAChR-evoked store release preferentially activates SK calcium-activated potassium channels that are located close to the soma. This store release is also triggered by calcium-induced calcium release after calcium influx during APs and can act to enhance the AHP and reduce neuronal excitability.
In BLA projection neurons, SK channel activation was only observed with focal stimulation of the soma and proximal dendrites. The necessity for proximal stimulation is also seen in other brain regions (Fiorillo and Williams, 2000; Gulledge and Stuart, 2005; Hagenston et al., 2007) and may reflect a proximal distribution of IP3-sensitive calcium stores (Nakamura et al., 2002; Stutzmann et al., 2003; Power and Sah, 2007). Our data show (Figs. 2E, 3B,E) that the time course of activation of SK channels matches the somatic calcium signal but not the calcium signal of the proximal dendrites, suggesting that the outward current activated by calcium released from intracellular stores is primarily caused by activation of SK channels located on or near the soma. SK channels have also been shown recently to be present on dendritic spines in hippocampal and lateral amygdalar neurons (Faber et al., 2005; Ngo-Anh et al., 2005). However, given that IP3-evoked calcium release is primarily restricted to spine-sparse regions of the dendrite (Nakamura et al., 2002; Stutzmann et al., 2003; Power and Sah, 2007), it is unlikely that SK channels on spines would be activated by metabotropic receptor stimulation. Similar results are seen in pyramidal neurons within the prefrontal cortex, where SK channel activation by mGluR stimulation requires somatic invasion of the calcium wave (Hagenston et al., 2007). The relative apamin insensitivity of the AHP evoked by brief trains of APs may be caused by the apparent distribution of SK channels in the soma and spines but not on proximal dendrites. The amplitude of AP-evoked calcium rises is far greater in the dendrites than in the soma (Markram et al., 1995; Sah and Clements, 1999; Power and Sah, 2007), such that SK channels, with an EC50 value for calcium of 300–700 nm (Stocker, 2004), would show limited activation by the modest somatic calcium rise generated by AP trains. SK channels located on spines may be too electrotonically distant or decay too rapidly to have much impact on the somatic membrane potential.
Interestingly, the large rises in cytosolic calcium evoked by cholinergic stimulation do not appear to activate the calcium-activated potassium channels responsible for the sIAHP. There are a number of possible explanations for this result. First, in hippocampal pyramidal neurons, the channels that underlie the sIAHP have been proposed to be present on the dendritic tree (Sah and Bekkers, 1996; Bekkers, 2000). If a similar situation holds in BLA projection neurons, the lack of IP3-mediated calcium release in the distal dendritic tree (Power and Sah, 2007) may explain why they are not activated by an mAChR-evoked calcium response. Second, activation of the sIAHP has been proposed recently to occur through an interaction with a diffusible calcium sensor (Tzingounis et al., 2007). Thus, it is conceivable that this calcium sensor in BLA neurons does not have access to calcium released from internal stores. Finally, given that the sIAHP is suppressed by mAChR activation, it is possible that cholinergic suppression of the sIAHP occurs before store release.
Because of the calcium dependence of the IP3 receptor, calcium store release is augmented when AP-evoked calcium influx occurs coincidently with mAChR stimulation (Fig. 4). In pyramidal neurons in visual cortex, where the AHP is predominantly mediated by the IAHP, IP3-CICR results in an enhancement of the AHP and spike-frequency adaptation (Yamada et al., 2004). In BLA projection neurons, the situation is more complex, because the sIAHP is the predominant current responsible for the AHP and spike-frequency adaptation. Results from the differential blockade of SK channels and the sIAHP demonstrate that a competition between these calcium-activated potassium channels determines the cholinergic action on the AHP and spike-frequency adaptation. In the absence of IP3-CICR, mAChR stimulation suppresses the AHP and reduces spike frequency adaptation. When IP3-CICR is functional, the resultant activation of SK channels counteracts the cholinergic suppression of the AHP and spike-frequency adaptation. This store-dependent calcium rise and SK activation lasts for several seconds, significantly longer than the IAHP in the absence of metabotropic stimulation. Both the IP3-CICR and the inhibitory effects of ACh are transient and are followed by a longer-lasting reduction in spike frequency adaptation, presumably because of the cholinergic suppression of the potassium channels and activation of mixed cation conductances (Washburn and Moises, 1992b; Womble and Moises, 1993; Yajeya et al., 1999). Because store content and its release potential are dependent on previous neuronal activity (Power and Sah, 2005), the calcium-store signal also conveys information about previous neuronal activity (Fig. 3D). Thus, the dependence of SK activation on calcium-store release, and the preferential location of these channels on the soma, close to the site of AP generation, provides a mechanism for coincident activity to modulate intrinsic excitability.
What is the functional role of the metabotropic-mediated hyperpolarization? It has been proposed that activation of calcium-activated potassium channels may be neuroprotective, delaying the onset of epileptogenesis during periods of enhanced neuronal activity (Rainnie et al., 1994). SK channel activation may also act to uncouple large postsynaptic calcium rises from neuronal output, ensuring that calcium-dependent changes necessary for synaptic plasticity are not passed on to downstream targets of BLA projection neurons. Interestingly, learning-associated reductions in the sIAHP are well correlated with learning and memory in the hippocampus and piriform cortex (Saar and Barkai, 2003; Disterhoft and Oh, 2006). Although it is unclear whether a similar reduction of the sIAHP occurs in BLA neurons, noradrenaline levels in the BLA have been shown to increase in response to emotionally arousing stimuli such as footshocks (Galvez et al., 1996). Our data show that a reduction of the sIAHP, through adrenergic stimulation or other mechanisms, enhances the transient inhibitory effects of cholinergic stimulation. This biphasic action of cholinergic stimulation on the AHP and spike-frequency adaptation may serve to tune neuronal output. Excitatory inputs that immediately follow cholinergic stimulation would be inhibited from producing APs, whereas inputs that are predictive of the cholinergic stimulation are unaffected. Excitatory inputs occurring after this brief window of inhibition are more likely to initiate neuronal output. Continuous excitation, which would normally result in a burst of APs that rapidly accommodate, may yield two bursts of APs, one before and one after the onset of cholinergic activity.
Footnotes
This work was supported by grants from the National Health and Medical Research Council of Australia and the Australian Research Council. J.M.P. is a recipient of a Smart State Fellowship from the Queensland State Government. We thank Matt Oh and Rowan Tweedale for comments on this manuscript.
References
- Barros et al., 2005.Barros DM, Ramirez MR, Izquierdo I. Modulation of working, short- and long-term memory by nicotinic receptors in the basolateral amygdala in rats. Neurobiol Learn Mem. 2005;83:113–118. doi: 10.1016/j.nlm.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Bekkers, 2000.Bekkers JM. Distribution of slow AHP channels on hippocampal CA1 pyramidal neurons. J Neurophysiol. 2000;83:1756–1759. doi: 10.1152/jn.2000.83.3.1756. [DOI] [PubMed] [Google Scholar]
- Ben-Ari et al., 1977.Ben-Ari Y, Zigmond RE, Shute CC, Lewis PR. Regional distribution of choline acetyltransferase and acetylcholinesterase within the amygdaloid complex and stria terminalis system. Brain Res. 1977;120:435–444. doi: 10.1016/0006-8993(77)90397-3. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny et al., 1991.Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Chen and Lang, 2003.Chen JC, Lang EJ. Inhibitory control of rat lateral amygdaloid projection cells. Neuroscience. 2003;121:155–166. doi: 10.1016/s0306-4522(03)00430-5. [DOI] [PubMed] [Google Scholar]
- Chen et al., 2000.Chen JQ, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH. bis-Quinolinium cyclophanes: 8,14-diaza-1,7(1, 4)-diquinolinacyclotetradecaphane (UCL 1848), a highly potent and selective, nonpeptidic blocker of the apamin-sensitive Ca(2+)-activated K(+) channel. J Med Chem. 2000;43:3478–3481. doi: 10.1021/jm000904v. [DOI] [PubMed] [Google Scholar]
- Disterhoft and Oh, 2006.Disterhoft JF, Oh MM. Learning, aging and intrinsic neuronal plasticity. Trends Neurosci. 2006;29:587–599. doi: 10.1016/j.tins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Dutar and Nicoll, 1989.Dutar P, Nicoll RA. Pharmacological characterization of muscarinic responses in rat hippocampal pyramidal cells. Exs. 1989;57:68–76. doi: 10.1007/978-3-0348-9138-7_7. [DOI] [PubMed] [Google Scholar]
- Faber and Sah, 2002.Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber and Sah, 2005.Faber ES, Sah P. Independent roles of calcium and voltage-dependent potassium currents in controlling spike frequency adaptation in lateral amygdala pyramidal neurons. Eur J Neurosci. 2005;22:1627–1635. doi: 10.1111/j.1460-9568.2005.04357.x. [DOI] [PubMed] [Google Scholar]
- Faber et al., 2005.Faber ES, Delaney AJ, Sah P. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci. 2005;8:635–641. doi: 10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- Faber et al., 2006.Faber ES, Sedlak P, Vidovic M, Sah P. Synaptic activation of transient receptor potential channels by metabotropic glutamate receptors in the lateral amygdala. Neuroscience. 2006;137:781–794. doi: 10.1016/j.neuroscience.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Finch et al., 1991.Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- Fiorillo and Williams, 1998.Fiorillo CD, Williams JT. Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature. 1998;394:78–82. doi: 10.1038/27919. [DOI] [PubMed] [Google Scholar]
- Fiorillo and Williams, 2000.Fiorillo CD, Williams JT. Cholinergic inhibition of ventral midbrain dopamine neurons. J Neurosci. 2000;20:7855–7860. doi: 10.1523/JNEUROSCI.20-20-07855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez et al., 1996.Galvez R, Mesches MH, McGaugh JL. Norepinephrine release in the amygdala in response to footshock stimulation. Neurobiol Learn Mem. 1996;66:253–257. doi: 10.1006/nlme.1996.0067. [DOI] [PubMed] [Google Scholar]
- Gu et al., 2005.Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol (Lond) 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge and Stuart, 2005.Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston et al., 2007.Hagenston AM, Fitzpatrick JS, Yeckel MF. MGluR-mediated calcium waves that invade the soma regulate firing in layer V medial prefrontal cortical pyramidal neurons. Cereb Cortex. 2007;18:407–423. doi: 10.1093/cercor/bhm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang et al., 1994.Huang CC, Tsai JJ, Gean PW. Actions of isoproterenol on amygdalar neurons in vitro. Chin J Physiol. 1994;37:73–78. [PubMed] [Google Scholar]
- Jaffe and Brown, 1994.Jaffe DB, Brown TH. Metabotropic glutamate receptor activation induces calcium waves within hippocampal dendrites. J Neurophysiol. 1994;72:471–474. doi: 10.1152/jn.1994.72.1.471. [DOI] [PubMed] [Google Scholar]
- Klein and Yakel, 2006.Klein RC, Yakel JL. Functional somato-dendritic alpha7-containing nicotinic acetylcholine receptors in the rat basolateral amygdala complex. J Physiol (Lond) 2006;576:865–872. doi: 10.1113/jphysiol.2006.118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause and Pedarzani, 2000.Krause M, Pedarzani P. A protein phosphatase is involved in the cholinergic suppression of the Ca(2+)-activated K(+) current sI(AHP) in hippocampal pyramidal neurons. Neuropharmacology. 2000;39:1274–1283. doi: 10.1016/s0028-3908(99)00227-0. [DOI] [PubMed] [Google Scholar]
- Kroner et al., 2005.Kroner S, Rosenkranz JA, Grace AA, Barrionuevo G. Dopamine modulates excitability of basolateral amygdala neurons in vitro. J Neurophysiol. 2005;93:1598–1610. doi: 10.1152/jn.00843.2004. [DOI] [PubMed] [Google Scholar]
- Lang and Pare, 1997.Lang EJ, Pare D. Synaptic and synaptically activated intrinsic conductances underlie inhibitory potentials in cat lateral amygdaloid projection neurons in vivo. J Neurophysiol. 1997;77:353–363. doi: 10.1152/jn.1997.77.1.353. [DOI] [PubMed] [Google Scholar]
- Levey et al., 1991.Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak et al., 1999.Mak DO, McBride S, Foskett JK. ATP regulation of type 1 inositol 1,4,5-trisphosphate receptor channel gating by allosteric tuning of Ca(2+) activation. J Biol Chem. 1999;274:22231–22237. doi: 10.1074/jbc.274.32.22231. [DOI] [PubMed] [Google Scholar]
- Markram et al., 1995.Markram H, Helm PJ, Sakmann B. Dendritic calcium transients evoked by single back-propagating action potentials in rat neocortical pyramidal neurons. J Physiol (Lond) 1995;485:1–20. doi: 10.1113/jphysiol.1995.sp020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mash and Potter, 1986.Mash DC, Potter LT. Autoradiographic localization of M1 and M2 muscarine receptors in the rat brain. Neuroscience. 1986;19:551–564. doi: 10.1016/0306-4522(86)90280-0. [DOI] [PubMed] [Google Scholar]
- McGaugh, 2004.McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- Morikawa et al., 2003.Morikawa H, Khodakhah K, Williams JT. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller et al., 1992.Muller W, Petrozzino JJ, Griffith LC, Danho W, Connor JA. Specific involvement of Ca(2+)-calmodulin kinase II in cholinergic modulation of neuronal responsiveness. J Neurophysiol. 1992;68:2264–2269. doi: 10.1152/jn.1992.68.6.2264. [DOI] [PubMed] [Google Scholar]
- Nakamura et al., 1999.Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura et al., 2002.Nakamura T, Lasser-Ross N, Nakamura K, Ross WN. Spatial segregation and interaction of calcium signalling mechanisms in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 2002;543:465–480. doi: 10.1113/jphysiol.2002.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo-Anh et al., 2005.Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Nicoll, 1988.Nicoll RA. The coupling of neurotransmitter receptors to ion channels in the brain. Science. 1988;241:545–551. doi: 10.1126/science.2456612. [DOI] [PubMed] [Google Scholar]
- Oh et al., 2000.Oh MM, Power JM, Thompson LT, Disterhoft JF. Apamin increases excitability of CA1 hippocampal pyramidal neurons. Neurosci Res Comm. 2000;27:135–142. [Google Scholar]
- Pare and Gaudreau, 1996.Pare D, Gaudreau H. Projection cells and interneurons of the lateral and basolateral amygdala: distinct firing patterns and differential relation to theta and delta rhythms in conscious cats. J Neurosci. 1996;16:3334–3350. doi: 10.1523/JNEUROSCI.16-10-03334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare et al., 1995.Pare D, Pape HC, Dong J. Bursting and oscillating neurons of the cat basolateral amygdaloid complex in vivo: electrophysiological properties and morphological features. J Neurophysiol. 1995;74:1179–1191. doi: 10.1152/jn.1995.74.3.1179. [DOI] [PubMed] [Google Scholar]
- Pedarzani and Storm, 1993.Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- Power and Sah, 2002.Power JM, Sah P. Nuclear calcium signaling evoked by cholinergic stimulation in hippocampal CA1 pyramidal neurons. J Neurosci. 2002;22:3454–3462. doi: 10.1523/JNEUROSCI.22-09-03454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power and Sah, 2005.Power JM, Sah P. Intracellular calcium store filling by an L-type calcium current in the basolateral amygdala at subthreshold membrane potentials. J Physiol (Lond) 2005;562:439–453. doi: 10.1113/jphysiol.2004.076711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power and Sah, 2007.Power JM, Sah P. Distribution of IP3-mediated calcium responses in basolateral amygdala neurons and their role in nuclear signalling. J Physiol (Lond) 2007;580:835–857. doi: 10.1113/jphysiol.2006.125062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie et al., 1993.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69:1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- Rainnie et al., 1994.Rainnie DG, Holmes KH, Shinnick-Gallagher P. Activation of postsynaptic metabotropic glutamate receptors by trans-ACPD hyperpolarizes neurons of the basolateral amygdala. J Neurosci. 1994;14:7208–7220. doi: 10.1523/JNEUROSCI.14-11-07208.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross et al., 2005.Ross WN, Nakamura T, Watanabe S, Larkum M, Lasser-Ross N. Synaptically activated Ca2+ release from internal stores in CNS neurons. Cell Mol Neurobiol. 2005;25:283–295. doi: 10.1007/s10571-005-3060-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saar and Barkai, 2003.Saar D, Barkai E. Long-term modifications in intrinsic neuronal properties and rule learning in rats. Eur J Neurosci. 2003;17:2727–2734. doi: 10.1046/j.1460-9568.2003.02699.x. [DOI] [PubMed] [Google Scholar]
- Sah, 1996.Sah P. Ca(2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Sah and Bekkers, 1996.Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah and Clements, 1999.Sah P, Clements JD. Photolytic manipulation of [Ca2+]i reveals slow kinetics of potassium channels underlying the afterhyperpolarization in hippocampal pyramidal neurons. J Neurosci. 1999;19:3657–3664. doi: 10.1523/JNEUROSCI.19-10-03657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah and Faber, 2002.Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Schnee and Brown, 1998.Schnee ME, Brown BS. Selectivity of linopirdine (DuP 996), a neurotransmitter release enhancer, in blocking voltage-dependent and calcium-activated potassium currents in hippocampal neurons. J Pharmacol Exp Ther. 1998;286:709–717. [PubMed] [Google Scholar]
- Stocker, 2004.Stocker M. Ca(2+)-activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci. 2004;5:758–770. doi: 10.1038/nrn1516. [DOI] [PubMed] [Google Scholar]
- Stutzmann et al., 2003.Stutzmann GE, LaFerla FM, Parker I. Ca2+ signaling in mouse cortical neurons studied by two-photon imaging and photoreleased inositol triphosphate. J Neurosci. 2003;23:758–765. doi: 10.1523/JNEUROSCI.23-03-00758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor and Marshall, 1992.Taylor CW, Marshall IC. Calcium and inositol 1,4,5-trisphosphate receptors: a complex relationship. Trends Biochem Sci. 1992;17:403–407. doi: 10.1016/0968-0004(92)90009-x. [DOI] [PubMed] [Google Scholar]
- Tinsley et al., 2004.Tinsley MR, Quinn JJ, Fanselow MS. The role of muscarinic and nicotinic cholinergic neurotransmission in aversive conditioning: comparing pavlovian fear conditioning and inhibitory avoidance. Learn Mem. 2004;11:35–42. doi: 10.1101/lm.70204. [DOI] [PubMed] [Google Scholar]
- Tzingounis et al., 2007.Tzingounis AV, Kobayashi M, Takamatsu K, Nicoll RA. Hippocalcin gates the calcium activation of the slow afterhyperpolarization in hippocampal pyramidal cells. Neuron. 2007;53:487–493. doi: 10.1016/j.neuron.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn and Moises, 1992a.Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci. 1992a;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn and Moises, 1992b.Washburn MS, Moises HC. Muscarinic responses of rat basolateral amygdaloid neurons recorded in vitro. J Physiol (Lond) 1992b;449:121–154. doi: 10.1113/jphysiol.1992.sp019078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe et al., 2006.Watanabe S, Hong M, Lasser-Ross N, Ross WN. Modulation of calcium wave propagation in the dendrites and to the soma of rat hippocampal pyramidal neurons. J Physiol (Lond) 2006;575:455–468. doi: 10.1113/jphysiol.2006.114231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe et al., 1995.Watanabe Y, Ikegaya Y, Saito H, Abe K. Roles of GABAA, NMDA and muscarinic receptors in induction of long-term potentiation in the medial and lateral amygdala in vitro. Neurosci Res. 1995;21:317–322. doi: 10.1016/0168-0102(94)00867-f. [DOI] [PubMed] [Google Scholar]
- Womble and Moises, 1993.Womble MD, Moises HC. Muscarinic modulation of conductances underlying the afterhyperpolarization in neurons of the rat basolateral amygdala. Brain Res. 1993;621:87–96. doi: 10.1016/0006-8993(93)90301-3. [DOI] [PubMed] [Google Scholar]
- Womble and Moises, 1994.Womble MD, Moises HC. Metabotropic glutamate receptor agonist ACPD inhibits some, but not all, muscarinic-sensitive K+ conductances in basolateral amygdaloid neurons. Synapse. 1994;17:69–75. doi: 10.1002/syn.890170202. [DOI] [PubMed] [Google Scholar]
- Yajeya et al., 1999.Yajeya J, de la Fuente Juan A, Bajo VM, Riolobos AS, Heredia M, Criado JM. Muscarinic activation of a non-selective cationic conductance in pyramidal neurons in rat basolateral amygdala. Neuroscience. 1999;88:159–167. doi: 10.1016/s0306-4522(98)00210-3. [DOI] [PubMed] [Google Scholar]
- Yamada et al., 2004.Yamada S, Takechi H, Kanchiku I, Kita T, Kato N. Small-conductance Ca2+-dependent K+ channels are the target of spike-induced Ca2+ release in a feedback regulation of pyramidal cell excitability. J Neurophysiol. 2004;91:2322–2329. doi: 10.1152/jn.01049.2003. [DOI] [PubMed] [Google Scholar]
- Zhu et al., 2005.Zhu PJ, Stewart RR, McIntosh JM, Weight FF. Activation of nicotinic acetylcholine receptors increases the frequency of spontaneous GABAergic IPSCs in rat basolateral amygdala neurons. J Neurophysiol. 2005;94:3081–3091. doi: 10.1152/jn.00974.2004. [DOI] [PubMed] [Google Scholar]