

Abstract
Endocannabinoid signaling is a key regulator of synaptic neurotransmission throughout the brain. Compelling evidence shows that its perturbation leads to development of epileptic seizures, thus indicating that endocannabinoids play an intrinsic protective role in suppressing pathologic neuronal excitability. To elucidate whether long-term reorganization of endocannabinoid signaling occurs in epileptic patients, we performed comparative expression profiling along with quantitative electron microscopic analysis in control (postmortem samples from subjects with no signs of neurological disorders) and epileptic (surgically removed from patients with intractable temporal lobe epilepsy) hippocampal tissue. Quantitative PCR measurements revealed that CB1 cannabinoid receptor mRNA was downregulated to one-third of its control value in epileptic hippocampus. Likewise, the cannabinoid receptor-interacting protein-1a mRNA was decreased, whereas 1b isoform levels were unaltered. Expression of diacylglycerol lipase-α, an enzyme responsible for 2-arachidonoylglycerol synthesis, was also reduced by ∼60%, whereas its related β isoform levels were unchanged. Expression level of N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D and fatty acid amide hydrolase, metabolic enzymes of anandamide, and 2-arachidonoylglycerol's degrading enzyme monoacylglycerol lipase did not change. The density of CB1 immunolabeling was also decreased in epileptic hippocampus, predominantly in the dentate gyrus, where quantitative electron microscopic analysis did not reveal changes in the ratio of CB1-positive GABAergic boutons, but uncovered robust reduction in the fraction of CB1-positive glutamatergic axon terminals. These findings show that a neuroprotective machinery involving endocannabinoids is impaired in epileptic human hippocampus and imply that downregulation of CB1 receptors and related molecular components of the endocannabinoid system may facilitate the deleterious effects of increased network excitability.
Keywords: DGL, CRIP, cannabinoid, CB1, 2-AG, temporal lobe epilepsy
Introduction
The endocannabinoid system consists of a family of lipid signaling molecules, several enzymes involved in their metabolism, and the cannabinoid receptors (Piomelli, 2003). In the brain, the major physiological task revealed for endocannabinoids to date is the mediation of retrograde synaptic communication (Alger, 2002; Wilson and Nicoll, 2002; Hashimotodani et al., 2007). Endocannabinoids are released from postsynaptic neurons in an activity-dependent manner and engage presynaptic CB1 cannabinoid receptors, which attenuates neurotransmitter release from several types of axon terminals (Freund et al., 2003). Thus, on-demand retrograde endocannabinoid signaling may represent a key physiological mechanism, which controls excess presynaptic activity in situations when neuronal excitability becomes unbalanced (Lutz, 2004). Indeed, accumulating evidence suggests that endocannabinoid levels are strongly elevated after diverse neuronal insults (Hansen et al., 2001; Panikashvili et al., 2001; Franklin et al., 2003), including distinct experimental paradigms of epilepsy (Sugiura et al., 2000; Marsicano et al., 2003; Wallace et al., 2003; Wettschureck et al., 2006) (but see Chen et al., 2003). In these in vivo animal models, activation of CB1 receptors by their endogenous ligands, 2-arachidonoylglycerol (2-AG) or anandamide, as well as by selective CB1 agonists, has prominent neuroprotective effects and prevents epileptic seizures (Nagayama et al., 1999; Panikashvili et al., 2001; van der Stelt et al., 2001a,b; Wallace et al., 2001, 2002, 2003; Shafaroodi et al., 2004). Conversely, genetic ablation of CB1 receptors or their acute pharmacological blockade by specific antagonists reduces seizure threshold, facilitates epileptogenesis, and increases neuronal cell death (Wallace et al., 2002, 2003; Marsicano et al., 2003; Shafaroodi et al., 2004; Bernard et al., 2005; Monory et al., 2006; Wettschureck et al., 2006; Chen et al., 2007).
These striking neuroprotective and anticonvulsive effects of the endocannabinoid system raise two important therapeutic implications. Cannabis extracts have been used to treat epilepsy since antiquity (Mechoulam and Lichtman, 2003). Although chronic administration of CB1 agonists is not feasible because of psychotropic side effects, selective enhancement of endocannabinoid levels may still produce significant beneficial effects (Pacher et al., 2006). However, epileptogenesis and recurrent seizures may impair the operation of this intrinsic protective system, which might speed up the progression of epilepsy, and may render the therapeutic exploitation of endocannabinoids more difficult. Therefore, it is of crucial importance to elucidate whether severe chronic epilepsy leads to adverse long-term reorganization of endocannabinoid signaling, especially in drug-refractory patients to whom novel targets for pharmacotherapy are needed.
To determine whether endocannabinoid signaling is affected in the epileptic human hippocampus, we performed comparative expression profiling and high-resolution electron microscopic analysis of the molecular components of the endocannabinoid system. We show that CB1 receptor expression and the fraction of glutamatergic axon terminals equipped with CB1 are downregulated in the epileptic hippocampus. Furthermore, expression levels of an interacting protein of CB1 and the enzyme synthesizing 2-AG, CB1's endogenous ligand, are also decreased. These findings suggest that the protective endocannabinoid signaling pathway is disrupted in severe chronic epilepsy, and this impairment may account for reduced seizure threshold and increased neuronal damage in epileptic patients.
Materials and Methods
Human tissue samples.
Hippocampal samples were obtained from patients with therapy-resistant temporal lobe epilepsy. The seizure focus was identified by multimodal studies including video-EEG monitoring, magnetic resonance imaging, single photon emission computed tomography, and/or positron emission tomography. Only patients with no gross temporal lobe damage based on postoperative histological analysis were included in the study. Patients with intractable temporal lobe epilepsy underwent surgery in the National Institute of Neurosurgery in Budapest, Hungary within the framework of the Hungarian Epilepsy Surgery Program. A written informed consent for the study was obtained from every patient before surgery. Standard anterior temporal lobectomies were performed (Spencer and Spencer, 1985): the anterior third of the temporal lobe was removed together with the temporomedial structures. Epileptic hippocampal samples could be classified into several types in accordance with previous studies (Wittner et al., 2002, 2005; de Lanerolle et al., 2003; Toth et al., 2007), of which we selected samples from the two major types (supplemental Table 1, available at www.jneurosci.org as supplemental material) based on the principal cell loss and interneuronal changes present at the light microscopic level as follows. Nonsclerotic samples (n = 16) showed only minimal cell loss in the CA1 region, pyramidal cells were abundant, layers were visible, and their borders were clearly identified. In some samples, a patchy cell loss could be detected in the CA1 pyramidal cell layer, but these segments of the CA1 region were never atrophic. In contrast, in sclerotic samples (n = 14), the CA1 region was robustly shrunken and atrophic, >90% of principal cells were lost, only scattered pyramidal cells remained in the CA1 region, and separation of the layers became impossible. In addition, mossy fiber sprouting in the dentate gyrus and considerable changes in the distribution and morphology of interneurons throughout the hippocampal formation could be observed in the samples of this group (Maglóczky et al., 2000).
Control hippocampi (n = 11) were kindly provided by the Lenhossek Human Brain Program (Semmelweis Medical University, Budapest, Hungary). Control subjects died suddenly from causes unrelated to any brain disease (supplemental Table 1, available at www.jneurosci.org as supplemental material), and were processed for autopsies in the Department of Forensic Medicine of the Semmelweis University Medical School. Examination of medical records of control subjects at the autopsy confirmed the absence of any neurological disorders or alcohol or drug abuse during the preceding 10 years. Brains were removed 2–5 h after death. Harvesting of tissues was approved by the local ethics committee, and informed consent was obtained from the next of kin. The study was approved by the ethics committee at the Regional and Institutional Committee of Science and Research Ethics of Scientific Council of Health (TUKEB 5-1/1996, further extended in 2005) and performed in accordance with the Declaration of Helsinki.
Mouse tissue samples.
Adult female CD1 mice (12; all 100 d of age) were first deeply anesthetized with Equithesin (4.2% w/v chloral hydrate, 2.12% w/v MgSO4, 16.2% w/w Nembutal, 39.6% w/w propylene glycol, and 10% w/w ethanol in H2O; 0.3 ml/100 g, i.p.). The mice were then separated into three groups. Control mice (n = 3 for the study of the effect of postmortem period and n = 3 for the study of the effect of anesthesia) were killed by cervical dislocation as soon as they were deeply anesthetized, and their brains were immediately processed. To determine the potential effects of postmortem period, a second group of mice (n = 3) were also killed by cervical dislocation as soon as anesthesia was established, but their brains were processed after 4 h of postmortem period at room temperature. To determine the potential effects of anesthesia, mice (n = 3) were deeply anesthetized for 4 h before cervical dislocation. Afterward, the brains were immediately processed. The brains in all cases were removed from the skull, and the left hippocampi were isolated and immediately placed into RNAlater (Ambion, Austin, TX) to prevent RNA degradation. The entire process was performed within 4–5 min. These experiments were performed according to the guidelines of the institutional ethical code and the Hungarian Act of Animal Care and Experimentation (1998, XXVIII, Section 243/1998.).
Quantitative real-time PCR experiments.
Mouse or human nonsclerotic, sclerotic, and control hippocampal tissues containing the dentate gyrus, the CA3, the CA2, and the CA1 subfields as well as a small part of the subiculum at the CA1 border were homogenized by UH-50 ultrasonic homogenizer (SMT, Akita, Japan). Total RNA samples were isolated and purified from the cell lysates using the RNAqueous-4PCR kit (Ambion). Quantity and quality of the total RNA samples were determined by measuring their A260/A280 ratio as well as the integrity and density of 28S and 18S rRNA bands using spectrophotometry (BioPhotometer; Eppendorf, Hamburg, Germany) and denaturing gel electrophoresis, respectively. Only samples with a value between 1.7 and 2.0 and with no evidence of RNA degradation were included in further experiments.
For real-time PCR measurements, the cDNA samples were prepared by reverse transcribing 1 μg of total RNA using 3 μl of RevertAid H Minus M-MuLV reverse transcriptase (RT; Fermentas, Vilnius, Lithuania) in a mixture containing 9 μl of M-MuLV RT buffer, 2 μl oligo-dT (18) primer (10 pmol/μl), 1 μl of RNasin (Promega, Madison, WI), and 1.5 μl of dNTP mix (Fermentas), which was brought up to a final volume of 44.5 μl with 0.1% diethylpyrocarbonate-treated distilled water. The reverse transcription reaction was performed at 37°C for 1 h, stopped at 95°C for 5 min, followed by 5 min on ice, and finally stored at −70°C until the real-time PCR measurements. To rule out the presence of potential chromosomal DNA contamination in the cDNA samples, reverse transcription reactions were also run without the reverse transcriptase (RT-negative control), and were compared with samples treated with reverse transcriptase (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
Expression level of the target genes was determined from the cDNA samples using quantitative real-time PCR (Rotor-Gene 3000; Corbett Research, Sydney, Australia). Reactions were composed of a mixture of 1 μl of cDNA sample; 5 μl of SYBR Green qPCR master mix, including the hot start DNA polymerase and its corresponding buffer (DyNAmo HS SYBR Green qPCR Kit; Finnzymes, Espoo, Finland); 1 μl of forward primer (10 pmol/μl); 1 μl of reverse primer (10 pmol/μl); and 4 μl of distilled water. As negative controls, both the respective RT-negative control and distilled water were used. PCR cycling protocols were the following: initial denaturation, 95°C for 15 min; cycling, 95°C for 15 s, 60°C for 15 s, 72°C for 15 s [30 cycles for CB1, cannabinoid receptor-interacting protein-1a (CRIP1a), diacylglycerol lipase-α (DGL-α), monoacylglycerol lipase (MGL), N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D (NAPE-PLD), and fatty acid amide hydrolase (FAAH); 35 cycles for CRIP1b and DGLβ]; final extension, 72°C for 1 min; melting curve analysis, 72–99°C by 1°C steps for 1 s. For each sample and each gene, at least three parallel reactions were run.
PCR primers were designed with the Primer3 program (Rozen and Skaletsky, 2000) to generate an amplicon of 100–250 bp ideal for real-time PCRs. Primers for the corresponding human gene sequences were the following: CB 1, GenBank accession number, BC100968; forward primer, 5′-AAG GTG ACA TGG CAT CCA AAT; reverse primer, 5′-AGG ACG AGA GAG ACT TGT TGT AA; CRIP1a, GenBank accession number, BC011535; forward primer, 5′-TCG AGA CAG TGT GGC AAG TC; reverse primer, 5′-CAT CAG ACT GCG TGT CTC GT; CRIP1b, GenBank accession number, AY144596; forward primer, 5′-CCT GAT GGG GAC AGA GTT GT; reverse primer, 5′-GAG ATC TCT TGG GGT CGT TG; DGL-α, GenBank accession number, NM_006133; forward primer, 5′-TGC TCT TCG GCC TGG TCT AT; reverse primer, 5′-CGC ATG CTC AGC CAG ATG AT; DGL-β, GenBank accession number, BC027603; forward primer, 5′-GAG TGC TGT GGT GGA TTG GC; reverse primer, 5′-TCT CAT GCT GAC ACA CAT GA; MGL, GenBank accession number, BC000551; forward primer, 5′-CAA GGC CCT CAT CTT TGT GT; reverse primer, 5′-ACG TGG AAG TCA GAC ACT AC; NAPE-PLD, GenBank accession number, NM_198990; forward primer, 5′-GAA GCT GGC TTA AGA GTC AC; reverse primer, 5′-CCG CAT CTA TTG GAG GGA GT; FAAH, GenBank accession number, NM_001441; forward primer, 5′-GGC CAC ACC TTC CTA CAG AA; reverse primer, 5′-GTT TTG CGG TAC ACC TCG AT; GAPDH, GenBank accession number, NM_002046; forward primer, 5′-GAG TCA ACG GAT TTG GTC GT; reverse primer, 5′-GAC AAG CTT CCC GTT CTC AG; β-actin, GenBank accession number, NM_001101; forward primer, 5′-CGT CAC CAA CTG GGA CGA CA; reverse primer, 5′-GGG GTG TTG AAG GTC TCA AA. Primers for the corresponding mouse gene sequences were the following: CB 1, GenBank accession number, NM_007726; forward primer, 5′-CTG GTT CTG ATC CTG GTG GT; reverse primer, 5′-TGT CTC AGG TCC TTG CTC CT; β-actin, GenBank accession number, NM_007393; forward primer, 5′-TGT TAC CAA CTG GGA CGA CA; reverse primer, 5′-GGG GTG TTG AAG GTC TCA AA.
To ensure reaction specificity and accurate quantification, melting curve analysis was performed after each reaction, which confirmed the lack of primer–dimer artifacts or contamination in all cases (supplemental Figs. 1B, 2A, available at www.jneurosci.org as supplemental material). In addition, the length of the amplified fragments was further verified by gel electrophoresis (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material), and their identity was finally established by sequencing.
Analysis of real-time PCR measurements.
Each reaction was repeated three times in parallel, and the values included in statistical analysis were the means of these measurements. Coefficients of variation for both intra-assay and interassay variability were always very low (CV < 1%), demonstrating high precision and reproducibility, respectively.
Expression level of the target genes was normalized to the expression level of two reference genes. The target gene and the first reference gene were measured together within the same experiment, then the target gene was measured once more together with a second reference gene, but under the same conditions as before. As reference genes, the housekeeping genes β-actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used, because these genes did not show significant changes in their expression levels in human temporal lobe epilepsy patients (Becker et al., 2002; Jamali et al., 2006). These two housekeeping genes fulfill different cell biological functions, and their expression levels may be regulated differentially by unknown factors in the epileptic hippocampus. Yet, the GAPDH/β-actin ratio did not vary significantly between the nonsclerotic or sclerotic populations and the control samples, supporting the validity of these reference genes in our patient samples as well (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
To compare the expression level of target genes between the different experimental groups, the efficiency calibrated model of Pfaffl was applied (Pfaffl, 2001). Relative expression ratio of target genes normalized to housekeeping genes was determined as E target meanCt(control,target)−meanCt(epileptic,target)/ E housekeeping meanCt(control,housekeeping)− meanCt(epileptic,housekeeping), where E is the efficiency of the real-time PCR. Efficiency values in each experiment for a given gene were averaged, and the mean value was established as E, whereas cycle threshold (C t) values were determined at the same threshold (at normalized fluorescence intensity of 0.0316) for every experiment. Both values were calculated by the Rotor Gene 5 software (Corbett Research, Sydney, Australia). Mean ± SEM C t values for each experimental group and genes were obtained as averages of the C t values. Epileptic in the equation indicates values either from the nonsclerotic or from the sclerotic experimental group.
Statistical analysis of data were performed by the Relative Expression Software Tool (for details, see Pfaffl et al., 2002) developed specifically for statistical evaluation of relative gene expression measurements. Differences in gene expression level between experimental groups were considered significant when the p level was <0.05. Data are presented as mean expression ratio ± SEM. Because the presented expression ratio is the exponential of the original ΔC t values, SEM of these ΔC t values were also transformed exponentially to obtain the presented SEM. When the expression ratio of certain genes is given in the text, we also show the mean − SEM and mean + SEM values belonging to the given ratio in parentheses.
Immunocytochemistry.
After surgical or postmortem removal, the hippocampal tissue was immediately dissected into 3- to 4-mm-thick blocks, and immersed in a fixative containing 4% paraformaldehyde, 0.1% glutaraldehyde, and 0.2% picric acid in 0.1 m phosphate buffer (PB; pH 7.4). The blocks were first rinsed for 6 h at room temperature in the fixative, which was replaced every hour with a fresh solution. The blocks were then postfixed in the same fixative solution, but without glutaraldehyde overnight. In the case of one control brain, both the internal carotid and vertebral arteries were cannulated 4 h after death, and the brain was perfused with physiological saline (2 L in 30 min) followed by a fixative solution containing 4% paraformaldehyde and 0.2% picric acid in 0.1 m PB (5 L in 3.5 h). The hippocampus was removed after perfusion and cut into 3- to 4-mm-thick blocks, and was postfixed in the same fixative solution overnight.
From the blocks, 60-μm-thick slices were cut on a Vibratome (Vibratome, St. Louis, MO), and sections were processed for immunostaining. After washing in 0.1 m PB (six times for 20 min each), the sections were immersed in 30% sucrose in 0.1 m PB for 1–2 d, then freeze thawed over liquid nitrogen four times. Subsequently, all washing steps and dilutions of the antibodies were done in 0.05 m Tris-buffered saline (TBS; pH 7.4). After blocking endogenous peroxidase activity by 1% H2O2 for 10 min and extensive washing in TBS (five times for 10 min each), the sections were first blocked with 5% normal goat serum for 45 min and then incubated in affinity-purified guinea pig anti-CB1 antibody (1 μg/ml) for 48 h at 4°C. The specificity of antibody was confirmed by the lack of immunostaining in hippocampal sections derived from CB1 knock-out mice both in the laboratory of origin (Fukudome et al., 2004) and in our laboratory (Katona et al., 2006). After primary antibody incubation, the sections were washed extensively in TBS (3 times for 10 min each) and then treated first with biotinylated goat anti-guinea pig IgG (1:300; Vector Laboratories, Burlingame, CA) for 2 h, washed again three times in TBS, and then incubated with avidin biotinylated–horseradish peroxidase complex (1:500; Elite-ABC; Vector Laboratories) for 1.5 h. The immunoperoxidase reaction was developed using 3,3′-diaminobenzidine (DAB) as the chromogen and 0.01% H2O2 dissolved in Tris buffer (TB, pH 7.6). Mossy cell somata were visualized by immunostaining with a pan-glutamate receptor 2 and 3 subunit antibody (GluR2/3; 1:100; Millipore, Billerica, MA). The staining procedure was performed in a similar way, except that sections were blocked in a mixture of 5% milk powder and 2% bovine serum albumin (Sigma, St. Louis, MO) in TBS, and as secondary antibody, biotinylated goat anti-rabbit antibody (1:250; Vector Laboratories) was used. After development of the immunostaining, the sections were treated with 1% OsO4 in 0.1 m PB for 20 min, dehydrated in an ascending series of ethanol and propylene oxide, and embedded in Durcupan (ACM; Fluka, Buchs, Switzerland). During dehydration, the sections were treated with 1% uranyl acetate in 70% ethanol for 20 min.
To ensure identical antibody penetration, development time, and poststaining processing, each section was photographed before starting the experiment. Then sections from the three experimental groups were processed together within the same well, each well containing one section from one experimental group. After the reaction, each section could be unequivocally identified from the photographs.
Light microscopic analysis of CB1-positive interneuron density and GluR2/3-immunoreactive mossy cell density.
To determine whether the density of CB1-positive GABAergic interneurons or hilar mossy cells is affected in the epileptic hippocampus, quantitative light microscopic analysis was performed. CB1- or GluR2/3-positive cell bodies were drawn by camera lucida at 63–200× magnification from the entire dentate gyrus or from the hilus, respectively (n = 11–31 sections from n = 12 samples altogether, four human tissue samples from the control, nonsclerotic, and sclerotic groups). To measure the respective area, the drawings were scanned, and the area of interest was determined by the NIH Image J program (National Institutes of Health, Bethesda, MD). In each analysis, a cell was included if its immunonegative nucleus was visible. Cell number was determined per unit area (mm2) in a 60-μm-thick section. Statistical analysis of data obtained from the light microscopic measurements was performed by Statistica 6 (StatSoft, Tulsa, OK). Significance of changes in the density of CB1-positive GABAergic interneurons or GluR2/3-positive mossy cells between experimental groups was tested using Kruskal–Wallis and post hoc Mann–Whitney U test (one-tailed in case of mossy cells). Differences were considered significant when p < 0.05. Data are presented as mean ± SEM.
Quantitative electron microscopic analysis of CB1-positive axon terminal density.
To determine whether the density of CB1-positive axon terminals forming glutamatergic or GABAergic synapses is affected in the epileptic hippocampus, we used the physical disector counting technique applied to electron microscopic profiles (Geinisman et al., 1996). Three randomly selected sampling fields containing the inner molecular layer of dorsal dentate gyrus were selected for analysis from hippocampal sections immunostained for CB1 receptor (n = 9 altogether, three human tissue samples from the control, nonsclerotic, and sclerotic groups). The selected areas were reembedded and resectioned, and a series of consecutive ultrathin sections (70 nm thick) were collected on Formvar-coated single-slot grids and counterstained with lead citrate for 2 min. Electron micrographs were taken at 20,000× magnification with a Hitachi (Yokohama, Japan) 7100 electron microscope.
From each block, three grids were selected for analysis, and from each grid, three counting frames with an area of 16.2 μm2 were analyzed through six consecutive ultrathin sections. Thus, a set of serial sections containing five disectors produced a defined volume of the structure of ∼5.67 μm3. This volume and the number of counted objects were used to establish the estimated numerical density (est N v) of CB1-positive and CB1-negative axon terminals. Because the availability of sections with preserved good-quality ultrastructure was limited from control subjects, it was impossible to establish an estimation of volume of the structure, est V(ref), with high precision. Thus, the estimated numerical density, est N v, was used for the statistical comparisons, instead of using estimated total synapse number (est N), and it is also presented as the representative value throughout this study. Note that the results obtained in this way assume a similar volume of hippocampal tissue between the different groups. Because a strongly reduced est N v was measured in our epileptic samples, an increase in hippocampal volume of epileptic patients would contradict our conclusions. However, hippocampal formation atrophy of a magnitude of ∼10–15% is one of the hallmarks of mesial temporal lobe epilepsy in imaging studies (for review, see Theodore and Gaillard, 2002), which suggests a minor masking effect of volume reduction on our numerical data, and supports the validity of conclusions based on estimated numerical density values alone.
As objects, CB1-positive and CB1-negative axon terminals forming either asymmetric or symmetric synapses were counted in the counting frames from the inner molecular layer close to the granule cells, which helped to follow the counting frames in serial sections as well as provided sampling consistency between the experimental groups. Asymmetric synapses were distinguished from symmetric synapses by the presence of a prominent postsynaptic density. An axon terminal was considered CB1 immunoreactive if electron-dense DAB precipitate was found consistently throughout the serial sections within the bouton. Note that false-negative staining in immunocytochemical procedures should always be expected, because of several factors (e.g., antibody penetration); therefore, the presented numerical ratio and estimated numerical density should always be considered a minimal value. In an effort to minimize false-negative staining as a result of penetration problems, the counting frames were positioned at the border of tissue and Durcupan in the ultrathin sections. Furthermore, the ratio of CB1-positive excitatory axon terminals was not altered significantly between the first and third grids in any experimental group (paired t test, p = 0.329), indicating that quantification was not modified by inconsistent penetration of the CB1 antibody in the depth range we used for analysis (the upper 2 μm from the surface).
Statistical analysis of data obtained from the electron microscopic measurements was performed by Statistica 6 (StatSoft). Significance of changes in the ratio of CB1-positive axon terminals among all boutons forming asymmetric or symmetric synapses were tested using χ2 test. Comparison of the estimated numerical density of CB1-positive and CB1-negative axon terminals between experimental groups was performed using ANOVA and Dunnett's post hoc test, because data in all three experimental groups were normally distributed according to Shapiro-Wilk's W test. Differences were considered significant, when the p value was <0.05. Data are presented as mean ± SEM.
Dependence of CB1 immunostaining and gene expression levels on age, fixation, and postmortem delay.
In a preliminary experiment, we examined 11 control brains with different postmortem delays and age in both genders. The general intensity and distribution of CB1 immunoreactivity was similar in each case and was similar to perfused mouse hippocampal tissue (Katona et al., 2006). On the other hand, longer postmortem delay influenced the quality of ultrastructure at the electron microscopic level. Weaker membrane preservation was found in immersion-fixed subjects with postmortem delays longer than 6 h; therefore, these control subjects were excluded from the quantitative analysis. Those tissues with 2–4 h of postmortem delay revealed acceptable ultrastructural preservation even in the immersion-fixed controls, although it was inferior to the perfused tissue. The preservation of the postmortem-perfused control (C11) was comparable with the immediately fixed epileptic samples and perfusion-fixed mouse tissues (Katona et al., 2006). Because the ratio of CB1-positive excitatory axon terminals was similar in the control subjects independent of the postmortem delay and the fixation protocol (χ2 test, p > 0.1 for control subjects), but differed significantly from the epileptic subjects (χ2 test, p < 0.001), we concluded that the differences found between control and epileptic tissues in the present study are likely to be associated with epilepsy.
One inherent problem with human studies is the lack of age-matched controls, which may have modified the conclusions derived from the real-time PCR and the quantitative electron microscopic analysis. Indeed, control patients were significantly older than patients with temporal lobe epilepsy in the real-time PCR, light microscopic, and electron microscopic experiments. Mean ± SEM of control, nonsclerotic, and sclerotic experimental groups were 60 ± 5, 32 ± 6, and 38 ± 3 years, respectively (for real-time PCR experiments); 59 ± 3, 37 ± 4, and 38 ± 2 years, respectively (for CB1 immunostaining at the light microscopic level); 62 ± 3, 36 ± 8, and 31 ± 2 years, respectively (for electron microscopic analysis of CB1-positive axon terminals) (ANOVA, p < 0.05 in all three cases).
To determine whether this age difference accounted for some of the findings of the present study, we performed a detailed correlation analysis between the expression level of all genes and the age of the human subjects. Neither linear regression nor Spearman's rank correlation coefficient revealed any significant correlation between gene expression level and the age of the subjects (p > 0.05 in all cases). Importantly, there was a nonsignificant decreasing trend of CB1 expression level with age, similar to previous reports (Westlake et al., 1994; Mato and Pazos, 2004). Because the major finding reported in this study is the robust downregulation of CB1 expression level and density of CB1-positive axon terminals in epileptic patients (who should have higher CB1 level because of their younger age), this decreasing trend suggests a minor masking effect of age difference on the measured expression level and axon terminal ratio, but it does not interfere with our major conclusions.
Results
CB1 cannabinoid receptor mRNA level is downregulated in the hippocampus of temporal lobe epilepsy patients
Previous studies provided ample evidence that the CB1 cannabinoid receptor plays an important protective role against epileptic seizures in several animal models (Wallace et al., 2001, 2002, 2003; Marsicano et al., 2003; Monory et al., 2006). To test the hypothesis that the endogenous signaling pathway involving CB1 receptors may be impaired in human epileptic patients, we compared the mRNA level of CB1 receptors in surgically removed hippocampal samples of patients with drug-refractory temporal lobe epilepsy and in postmortem hippocampal samples from subjects who had no signs of neurological disorders. Samples from patients with temporal lobe epilepsy were further divided into two groups identified as nonsclerotic and sclerotic based on established morphological criteria [for details, see Materials and Methods and Wittner et al. (2002, 2005)]. Hippocampal mRNA level was measured by real-time PCR; gene expression level was normalized in independent experiments to the expression level of two functionally different housekeeping genes, β-actin and GAPDH. Only those expression changes that were replicated with both reference genes were accepted for analysis.
Quantitative real-time PCR measurements revealed that CB1 receptor mRNA level was robustly downregulated both in the nonsclerotic (n = 7, throughout the real-time PCR experiments) and in the sclerotic (n = 6, throughout the real-time PCR experiments) epileptic human hippocampus compared with the expression level in control subjects (n = 7, throughout the real-time PCR experiments) in the case of both housekeeping genes (Fig. 1 A,B). In nonsclerotic epileptic hippocampus, mRNA level of CB1 was decreased to 37.1% (26.5–51.8%) or 46.5% (33.2–65.2%) of the control values established as 100% (87.9–113.7%) or 100% (86.7–115.3%), normalized to β-actin (p < 0.001) or GAPDH (p = 0.032) levels, respectively (Fig. 1 B). Likewise, in the sclerotic epileptic hippocampus, expression level of CB1 was reduced to 28.9% (19.8–42.1%) or 30.1% (18.6–48.6%), normalized to β-actin (p < 0.001) or GAPDH (p < 0.01) levels, respectively (Fig. 1 B).
Figure 1.
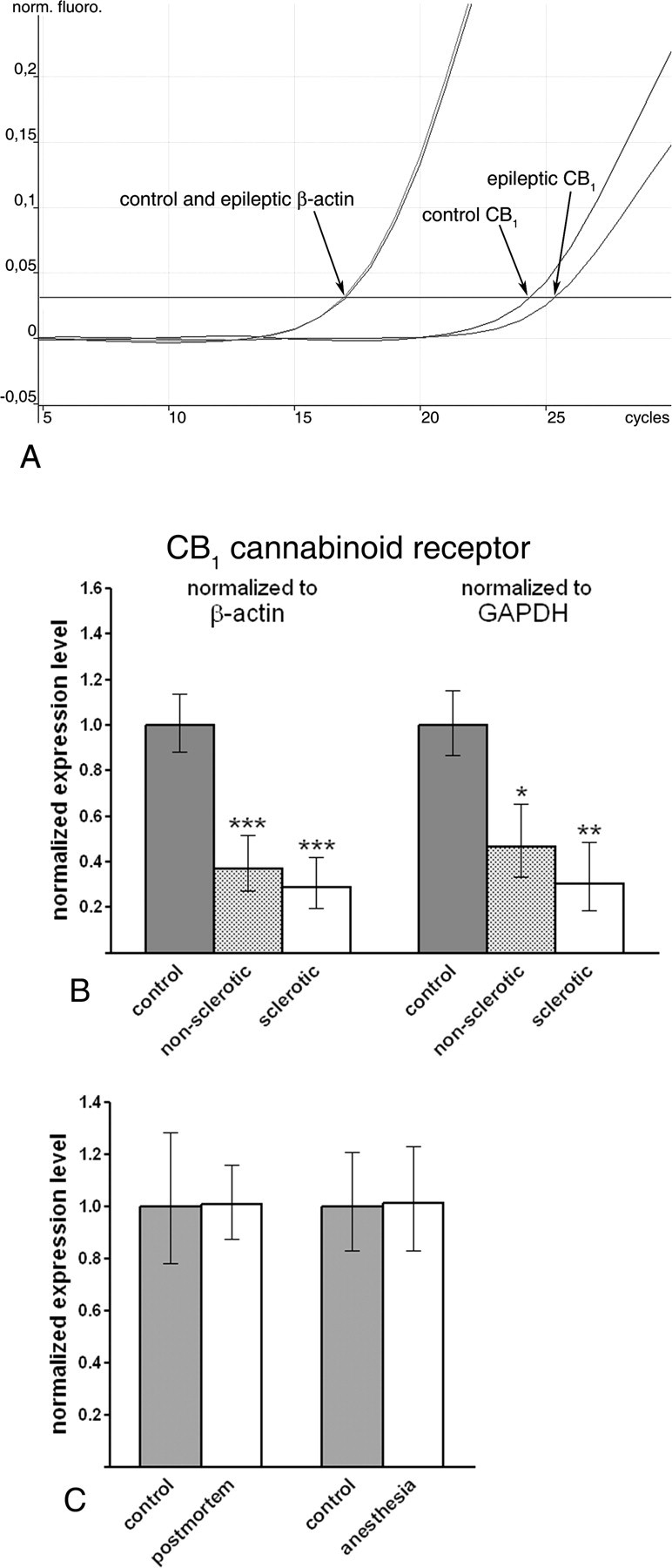
CB1 receptor mRNA level is downregulated in the epileptic human hippocampus. A, Representative real-time PCR measurement of CB1 cannabinoid receptor mRNA level in control and epileptic human hippocampus. Note that the housekeeping gene β-actin reaches threshold of normalized fluorescence intensity at identical values in both the control and epileptic hippocampus. In contrast, when CB1 cannabinoid receptor mRNA is measured, the exponential phase begins later and reaches threshold approximately one cycle later in a representative sample from the epileptic hippocampus. One cycle difference in the cycle threshold value indicates ∼50% difference in the original mRNA level because of the exponential nature of the PCR. B, Gene expression level of the CB1 receptor is robustly downregulated in both nonsclerotic (n = 7) and sclerotic (n = 6) epileptic hippocampi compared with control tissue (n = 7). Note that the direction and magnitude of expression level changes were identical in parallel experiments using two distinct housekeeping genes, β-actin and GAPDH. C, Bar graphs demonstrate that neither postmortem delay nor sustained anesthesia influence CB1 mRNA level in the mouse hippocampus. Data are presented as mean expression ratio ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
This striking downregulation of CB1 receptors in the epileptic human hippocampus raises two important methodological issues, which may influence the interpretation of the above measurements. First, epileptic tissue samples were frozen in liquid nitrogen immediately after surgical removal, whereas control tissues were processed after a postmortem period of 2–5 h. Second, epileptic samples were derived from patients undergoing sustained anesthesia for several hours during operation. Thus, epilepsy may not be the salient cause of reduced CB1 mRNA level, but other factors may also account for the observed changes in gene expression. To investigate the possibility that either the postmortem delay or the sustained anesthesia caused the observed downregulation in the CB1 mRNA level, we repeated real-time PCR measurements in two animal models. Mice were kept either for a 4-h-long postmortem period at room temperature or under sustained anesthesia for 4 h, and were compared with hippocampal tissue from control mice removed immediately after establishing deep anesthesia.
A postmortem period of 4 h did not affect the mRNA level of CB1 receptor normalized to β-actin level [100.6% (87.6–115.6%) for postmortem samples; 100% (78.0–128.1%) for control samples; n = 3 animals; p = 0.920] (Fig. 1 C). Similarly, sustained anesthesia for 4 h did not change CB1 receptor expression level normalized to β-actin [101.1% (83.2–122.8%) for samples from anesthetized animals; 100% (82.8–120.8%) for control samples; n = 3 animals; p = 0.700] (Fig. 1 C). These experiments indicate that postmortem delay or anesthesia is unlikely to account for the observed downregulation of CB1 receptors in the human epileptic hippocampus.
Expression level of the cannabinoid receptor-interacting protein CRIP1a is decreased in the sclerotic epileptic hippocampus
In the hippocampus, CB1 cannabinoid receptors are located presynaptically on both glutamatergic and GABAergic axon terminals (Katona et al., 2006; Kawamura et al., 2006; Monory et al., 2006). Recently, a binding partner for CB1 called CRIP has been identified and suggested to regulate its activity in the axon terminals (Niehaus et al., 2007). In the primate and human brain, two splice variants of CRIP have been described, which are called 1a and 1b (Niehaus et al., 2007).
Because CB1 receptors present on glutamatergic axon terminals were shown to be responsible for the anticonvulsant effects of endocannabinoids (Marsicano et al., 2003; Monory et al., 2006), we sought to determine whether CRIP expression level is also altered in the epileptic human hippocampus. Quantitative real-time PCR measurements uncovered that indeed, in the sclerotic epileptic hippocampus, CRIP1a mRNA level was significantly decreased to 69.6% (60.3–80.3%) or 68.2% (61.2–75.9%) of the control values established as 100% (92.8–107.7%) or 100% (91.4–109.5%), normalized to β-actin (p = 0.026) or GAPDH (p = 0.016) levels, respectively (Fig. 2 A). In the nonsclerotic hippocampus, mRNA level of CRIP1a showed a decreasing trend, which was close to, but did not reach, significance threshold, 83.7% (77.8–89.9%) or 81.4% (77.0–86.1%), normalized to β-actin (p = 0.108) or GAPDH (p = 0.069) levels, respectively (Fig. 2 A).
Figure 2.
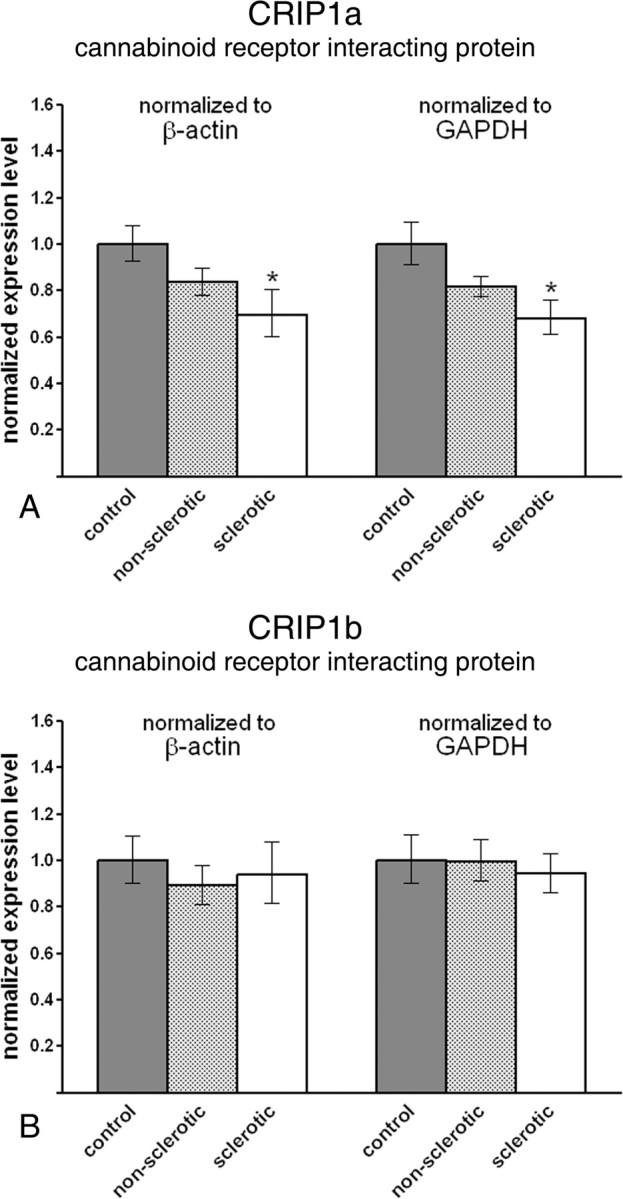
Gene expression level of cannabinoid receptor-interacting protein CRIP1a, but not CRIP1b, is reduced in the sclerotic epileptic hippocampus. A, CRIP1a mRNA level is significantly decreased in the sclerotic epileptic hippocampus (n = 6) compared with control values (n = 7). In contrast, the decrease in the nonsclerotic hippocampus (n = 7) did not reach significance. The direction and magnitude of expression level changes in the sclerotic hippocampus were identical in parallel experiments using two distinct housekeeping genes, β-actin and GAPDH. B, In contrast, CRIP1b mRNA levels did not differ significantly between nonsclerotic and sclerotic epileptic hippocampi compared with control values. Data are presented as mean expression ratio ± SEM. *p < 0.05.
In contrast to CRIP1a, the expression level of CRIP1b, an alternative splice variant present only in primates (Niehaus et al., 2007), did not show any changes in expression in either the nonsclerotic epileptic hippocampus or the sclerotic epileptic hippocampus. In nonsclerotic epileptic patients, expression level was 89.2% (81.3–97.9%) or 99.6% (90.1–110.1%), normalized to β-actin (p = 0.387) or GAPDH (p = 0.978) levels, respectively (Fig. 2 B). In sclerotic epileptic hippocampus, expression level was 93.9% (81.8–107.8%) or 94.1% (86.0–103.0%), normalized to β-actin (p = 0.703) or GAPDH (p = 0.663) levels, respectively (Fig. 2 B). SEMs of control values in this experiment were 90.5–110.6% for β-actin or 90.0–111.2% for GAPDH.
Together, these experiments show that CRIP1a expression level is decreased in the epileptic hippocampus, whereas its splice variant CRIP1b remains unchanged.
Expression profiling of biosynthetic and inactivating enzymes of 2-arachidonoylglycerol, the major synaptic endocannabinoid in the epileptic hippocampus
The proposed endogenous ligand of CB1 cannabinoid receptors, as well as the most abundant endocannabinoid in the hippocampus, is 2-AG (Stella et al., 1997) (for review, see Sugiura et al., 2006). 2-AG is synthesized by two closely related diacylglycerol lipases, DGL-α and DGL-β (Bisogno et al., 2003), and degraded by MGL (Dinh et al., 2002). Recent evidence indicates that DGL-α is postsynaptically localized to glutamatergic synapses in the hippocampus (Katona et al., 2006; Yoshida et al., 2006), whereas MGL colocalizes with CB1 receptors in both excitatory and inhibitory axon terminals (Gulyas et al., 2004). Thus, 2-AG is an ideal candidate to be the mediator of neuroprotective and anticonvulsant endocannabinoid effect. To elucidate whether the metabolic machinery for 2-AG signaling is altered in the epileptic hippocampus, we determined the gene expression level of the three metabolic enzymes using quantitative real-time PCR measurements.
Remarkably, in the sclerotic epileptic hippocampus, DGL-α mRNA level was strongly diminished to 44.6% (34.5–57.8%) or 37.9% (24.9–57.7%) of the control values of 100% (80.3–124.5%) or 100% (74.2–134.8%), normalized to β-actin (p = 0.028) or GAPDH (p = 0.037) levels, respectively (Fig. 3 A). In contrast, we found no differences in the expression level of DGL-α between the control and nonsclerotic epileptic hippocampal tissues [76.2% (57.2–101.5%) for β-actin; p = 0.460; 97.1% (72.8–129.6%) for GAPDH; p = 0.932] (Fig. 3 A).
Figure 3.
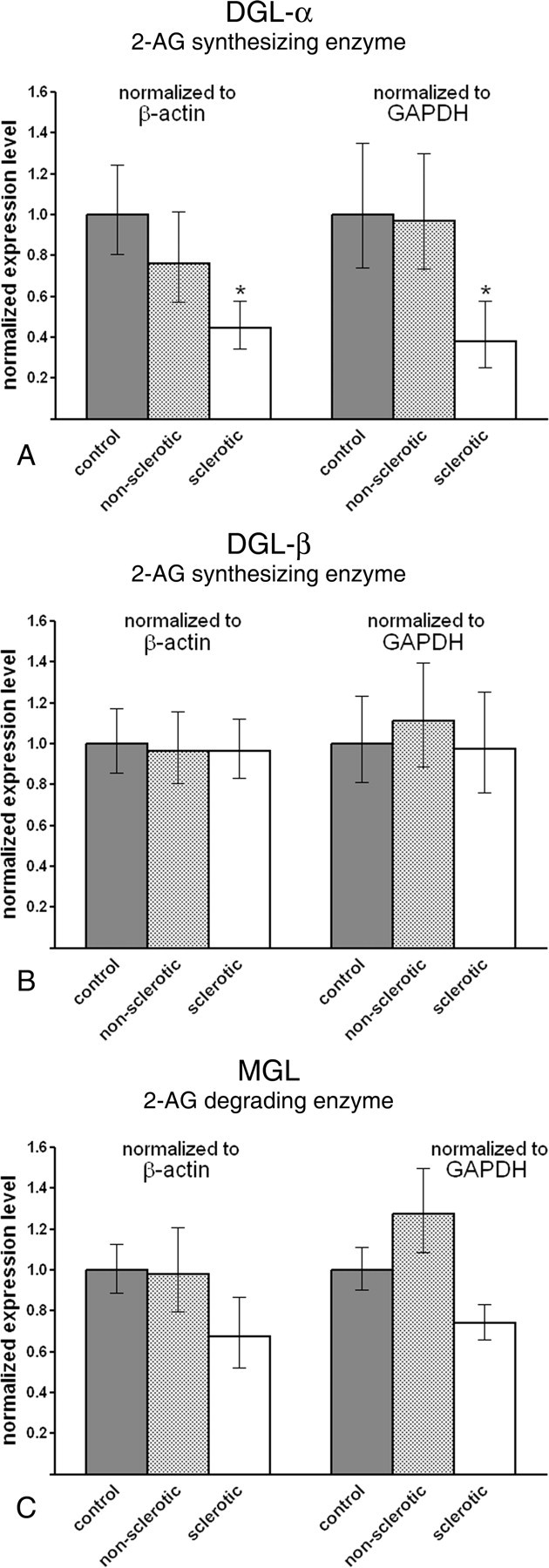
Gene expression level of DGL-α, the biosynthetic enzyme of 2-AG, is diminished in the sclerotic epileptic hippocampus. A, DGL-α mRNA level is decreased to one-half of its control level in the sclerotic epileptic hippocampus. In contrast, significant difference in mRNA level was not observed between the nonsclerotic hippocampal samples and control subjects. Importantly, the direction and magnitude of expression level changes were identical in parallel experiments using two distinct housekeeping genes, β-actin and GAPDH. B, The related isoenzyme DGL-β is unaffected in the epileptic hippocampus. Real-time PCR measurement did not reveal significant changes in mRNA level either in the nonsclerotic or in the sclerotic epileptic hippocampus as compared with control values. C, MGL, the enzyme responsible for elimination of 2-AG, showed a slight but insignificant decrease in mRNA level in the sclerotic hippocampus [normalized to β-actin (p = 0.171) or to GAPDH (p = 0.079)]. In the nonsclerotic hippocampal samples, there was no indication of any subtle changes in gene expression level. Data are presented as mean expression ratio ± SEM. *p < 0.05.
DGL-β is a closely related enzyme and may contribute to 2-AG synthesis (Bisogno et al., 2003); however, its subcellular localization and functional role have not yet been described in the hippocampus. Quantitative real-time PCR experiments revealed that DGL-β mRNA levels remained unchanged both in nonsclerotic and sclerotic epileptic hippocampus. Expression level of DGL-β in the nonsclerotic experimental group was 96.6% (80.8–115.5%) or 111.0% (88.2–139.7%), normalized to β-actin (p = 0.891) or GAPDH (p = 0.736) levels, respectively (Fig. 3 B), whereas in the sclerotic experimental group it was 96.6% (83.3–112.1%) or 97.3% (75.7–125.1%), normalized to β-actin (p = 0.868) or GAPDH (p = 0.936) levels, respectively (Fig. 3 B). SEMs of control values in this experiment were 85.5–117.0% for β-actin or 81.0–123.4% for GAPDH.
Gene expression level of MGL, the degrading enzyme of 2-AG was also statistically similar in all three experimental groups. Note, however that a decreasing trend was observed in the sclerotic epileptic hippocampus, which was close to but did not reach statistical significance. MGL mRNA level in these patients was 67.2% (52.1–86.7%) or 74.0% (65.8–83.2%) of the control values 100% (88.9–112.5%) or 100% (90.2–110.9%), normalized to β-actin (p = 0.171) or GAPDH (p = 0.079) levels, respectively (Fig. 3 C). In contrast, there was no hint of any change in MGL expression level in the nonsclerotic hippocampal tissue [97% (79.4–120.9%) or 127.3% (108.3–149.7%), normalized to β-actin (p = 0.931) or GAPDH (p = 0.231) levels, respectively] (Fig. 3 C).
These results suggest that biosynthesis of 2-AG at glutamatergic synapses may be impaired in the sclerotic epileptic hippocampus as a result of the robust reduction in DGL-α mRNA level (>50%). This value indicates that both reduced transcriptional activity and cell loss in the sclerotic CA1 region may account for the observed reduction, but only in patients with a more severely progressed form of epilepsy, because there was no significant change in the nonsclerotic samples.
Neither a biosynthetic nor a degrading enzyme of anandamide is altered in the hippocampus of epileptic patients
Beside 2-AG, anandamide is the other most well characterized endocannabinoid (Devane et al., 1992). One of its biosynthetic pathways involves the NAPE-PLD (Okamoto et al., 2004), and it is primarily degraded by FAAH (Cravatt et al., 1996). Importantly, several studies have reached conflicting conclusions on whether the effect of anandamide is proconvulsant or anticonvulsant (Ameri et al., 1999; Clement et al., 2003; Marsicano et al., 2003). To determine whether chronic temporal lobe epilepsy affects the biosynthetic or degrading enzymes of anandamide, we measured mRNA levels of NAPE-PLD and FAAH in the epileptic human hippocampus.
NAPE-PLD expression level was found to be comparable in both the nonsclerotic [94.8% (77.7–115.7%) or 103.1% (85.8–123.8%), normalized to β-actin (p = 0.798) or to GAPDH (p = 0.919), respectively] and the sclerotic [83.8% (67.0–105.0%) or 112.5% (102.0–124.0%), normalized to β-actin (p = 0.498) or to GAPDH (p = 0.678), respectively] epileptic hippocampus compared with the control human hippocampus [100% (92.2–108.4%) for β-actin, 100% (80.4–124.4%) for GAPDH] (Fig. 4 A).
Figure 4.
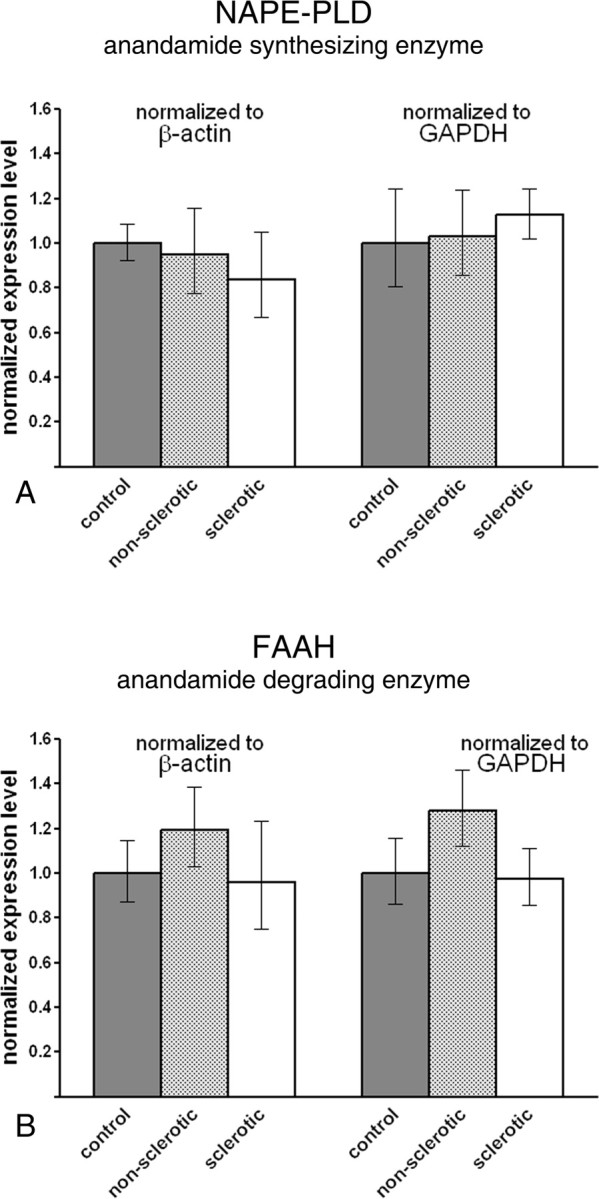
Metabolic enzymes of anandamide are not downregulated in the epileptic hippocampus. A, Real-time PCR measurement did not reveal alterations in the gene expression level of NAPE-PLD, a key synthetic enzyme of anandamide. The normalized expression level was similar in all three experimental groups and in the parallel experiments, which used β-actin or GAPDH as reference genes. B, FAAH, the degrading enzyme of anandamide, did not show significant expression changes in the epileptic hippocampal samples. Data are presented as mean expression ratio ± SEM.
Likewise, mRNA level of FAAH was not altered significantly in the nonsclerotic [119.3% (102.7–138.6%) or 127.8% (112.0–145.9%), normalized to β-actin (p = 0.390) or to GAPDH (p = 0.226), respectively] and in the sclerotic [96.1% (74.9–123.2%) or 97.5% (85.4–111.3%), normalized to β-actin (p = 0.905) or to GAPDH (p = 0.895), respectively] epileptic hippocampus compared with control values [100% (87.2–114.6%) for β-actin, 100% (86.3–115.9%) for GAPDH] (Fig. 4 B).
These data suggest that the expression levels of enzymes responsible for the metabolism of anandamide remain largely unaltered in the epileptic hippocampus.
The density of CB1 cannabinoid receptor immunoreactivity is reduced in the epileptic human hippocampus
In the experiments described above, we have measured mRNA levels of most of the genes identified to date to be components of the endocannabinoid system in the epileptic human hippocampus. Beside significant reduction in the expression level of CRIP1a and DGL-α in sclerotic hippocampi, the most robust change was observed for CB1, which was strongly downregulated at the mRNA level in both nonsclerotic and sclerotic epileptic cases. To extend this result to the protein level, and more importantly, to explore whether this downregulation varies with region, layer, or cell type in the hippocampal formation, we took advantage of a novel, highly sensitive antibody for CB1 receptors, which visualizes CB1-containing axons with an unprecedented sensitivity (Fukudome et al., 2004).
Immunostaining for CB1 in the control human hippocampus using this antibody revealed a very similar distribution of the receptor to that reported before in the perfused mouse hippocampus (Fig. 5 A,D) (Katona et al., 2006; Kawamura et al., 2006). A dense meshwork of CB1-immunoreactive fibers covered the entire hippocampal formation, yet the distribution of immunostaining followed the laminar structure of the hippocampus (Fig. 5 A,D). The most intense staining was seen in the inner molecular layer of the dentate gyrus (Fig. 5 D), followed by the outer molecular layer (Fig. 5 D) and the stratum radiatum of CA3, CA2, and CA1 subfields (Fig. 5 A). Strong CB1 immunostaining was also observed in strata pyramidale and oriens of these subfields (Fig. 5 A), the former containing several axon terminals in a basket-like array, which belong to GABAergic interneurons as described before in the human hippocampus (Katona et al., 2000). In contrast, the neuropil showed no labeling in the hilus and the stratum lucidum (Fig. 5 A,D), as expected from the lack of CB1 mRNA in granule cells of the human dentate gyrus (Westlake et al., 1994), but still contained large interneuron axons. Beside the widespread axonal immunostaining, scattered interneuron cell bodies immunoreactive for CB1 were also observed throughout the hippocampal formation. Staining in these neurons was limited to the perinuclear cytoplasm; no labeling was found in the somatic or dendritic membrane.
Figure 5.
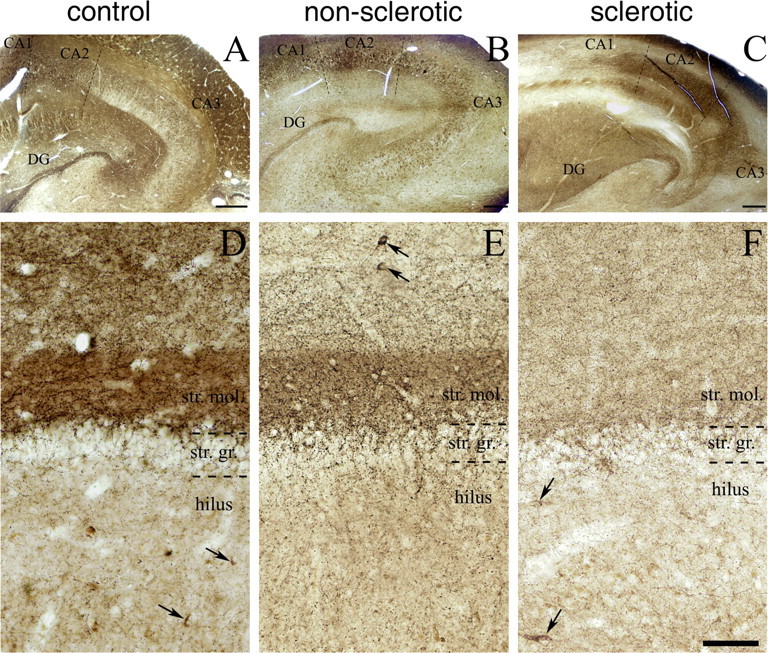
Immunostaining for CB1 cannabinoid receptor is reduced in the hippocampus of epileptic patients, particularly in the inner molecular layer of the dentate gyrus (DG). A, Light micrograph illustrating profound CB1 immunoreactivity throughout the human hippocampal formation of control subjects. By using a highly sensitive guinea pig antibody for CB1, the immunostaining highlights the different layers and subfields of the hippocampus according to the spatial arrangements of excitatory pathways. B, C, Although the general pattern of CB1 immunostaining is similar in the nonsclerotic and sclerotic epileptic hippocampi, the density of CB1 immunoreactivity is reduced in several layers. D–F, The most striking differences between the control and the epileptic hippocampi are visible in the dentate gyrus. The very dense neuropil-like labeling in the inner third of stratum moleculare (str. mol.) is evident in the control sample (D), but it is less so in the nonsclerotic epileptic sample (E), and it disappears almost completely in the sclerotic epileptic samples (F). In contrast, the stratum granulosum (str. gr.) and the hilus remained similar in all three experimental groups. Scattered cell bodies of GABAergic interneurons were also stained for CB1 (labeled by arrows), but conversely, there was no striking difference either in their distribution pattern or in their number between the control and epileptic human samples. Scale bars: A–C, 500 μm; D–F (in F), 100 μm.
Compared with the control hippocampus, general CB1 immunostaining was much weaker throughout the hippocampus of epileptic samples (Fig. 5). The most striking difference in CB1 immunoreactivity was the nearly complete disappearance of dense CB1-labeled neuropil from the inner molecular layer in sclerotic samples (Fig. 5 D,F). Labeling was also clearly diminished in this layer already in the nonsclerotic samples (Fig. 5 E). A robust downregulation of CB1 immunoreactivity was also visible in the stratum radiatum of CA3, CA2, and CA1 subfields in all epileptic subjects, as well as in the strata oriens and pyramidale of the sclerotic hippocampi (Fig. 5 C). In contrast, a modest increase in some sclerotic samples was observed in the stratum oriens of the CA3 and CA2 subfield as reported in a very recent work using an animal model of temporal lobe epilepsy (Falenski et al., 2007). Remarkably, the changes in CB1 immunoreactivity appeared to affect the two distinct types of CB1-positive axon terminals differentially. This was most striking in the dentate gyrus, where there was clearly visible reduction in the neuropil staining in the outer molecular layer, beside the powerful downregulation in the inner molecular layer (Fig. 5 D–F). This neuropil-like labeling pattern has been shown to belong to small excitatory axon terminals (Katona et al., 2006; Monory et al., 2006). In contrast, we did not experience conspicuous changes in the density of larger putative GABAergic axons, and CB1-positive scattered interneurons were also found in modest numbers in all three groups (Fig. 5 D–F).
Quantitative electron microscopic analysis of the density of CB1-positive excitatory and inhibitory axon terminals in the inner molecular layer of the dentate gyrus
To support the above qualitative observations, we performed a high-resolution quantitative analysis of CB1 receptor immunostaining in the control and epileptic human hippocampus. Because the most striking difference between control and epileptic hippocampi was observed in the inner molecular layer, we performed an unbiased stereological estimation of the numerical density of CB1-positive and CB1-negative axon terminals in this layer (Geinisman et al., 1996). First, we focused this analysis on those axon terminals that formed asymmetric, putative glutamatergic synapses with their postsynaptic target, for two reasons. Most importantly, two recent studies using animal models provided clear evidence that CB1 receptors located presynaptically on glutamatergic axon terminals, but not those that are positioned on GABAergic boutons, mediate the neuroprotective and anticonvulsant effects of cannabinoids (Marsicano et al., 2003; Monory et al., 2006). Furthermore, the inner molecular layer is the main termination zone of axonal arbors of hilar mossy cells (Amaral, 1978), a glutamatergic cell type implicated in epileptogenesis and the generation of network hyperexcitability (Santhakumar et al., 2000; Ratzliff et al., 2002).
CB1 immunoreactivity at the electron microscopic level was detected exclusively in axon terminals (Fig. 6). These boutons formed predominantly asymmetric, glutamatergic synapses, but axon terminals giving rise to symmetric, GABAergic synapses were also often found. CB1-positive axon terminals formed asymmetric synapses mainly on spine heads (Fig. 6 A,B) and rarely on dendritic shafts (Fig. 6 C). To establish and compare the estimated numerical density of CB1-positive and CB1-negative glutamatergic axon terminals in the control and epileptic human dentate gyrus, altogether 1092 disectors were analyzed in the three experimental groups (n = 3 subjects and n = 364 disectors from each group). In these samples, we identified 327 (235 positive, 92 negative for CB1), 224 (106 positive, 118 negative for CB1), and 197 (40 positive, 157 negative for CB1) excitatory axon terminals from control, nonsclerotic epileptic, and sclerotic epileptic sections, respectively (Fig. 7 A).
Figure 6.
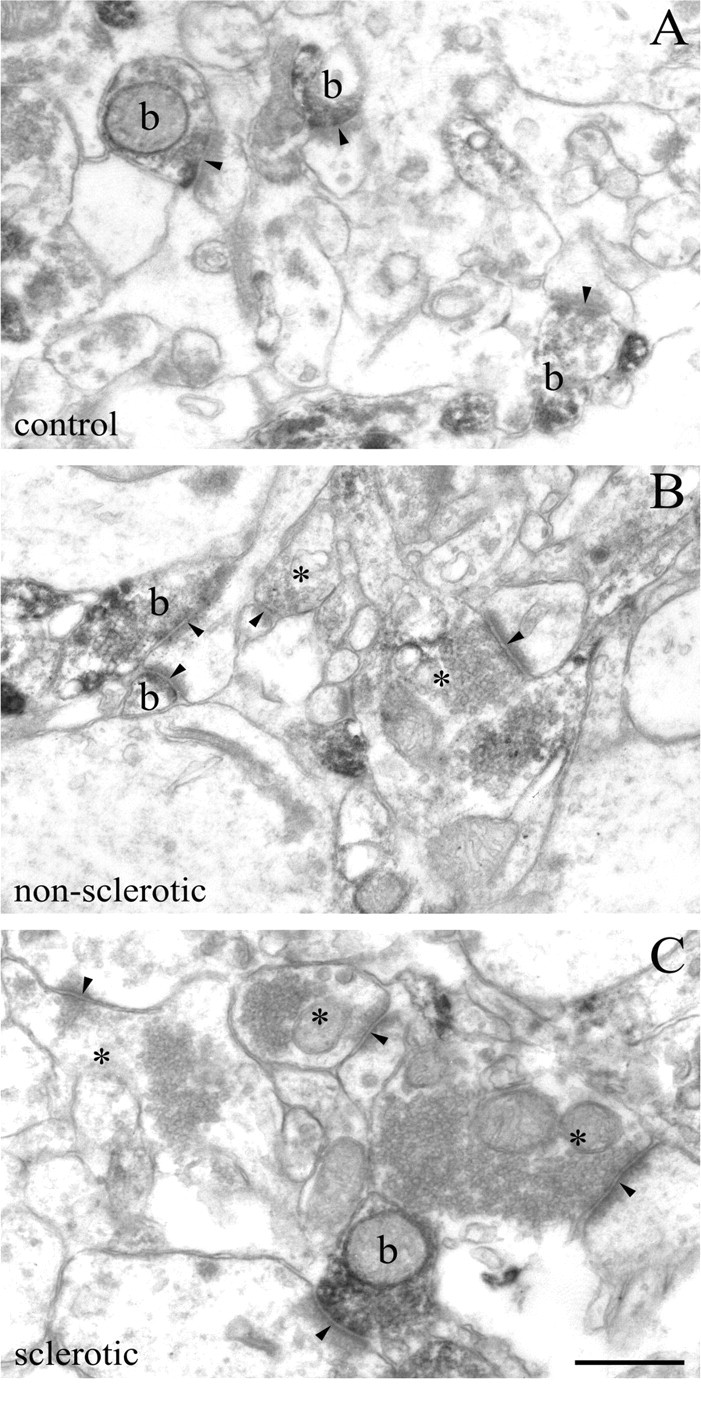
Density of glutamatergic axon terminals bearing presynaptic CB1 cannabinoid receptors is decreased in the epileptic human hippocampus. A–C, The electron micrograph demonstrates a robust accumulation of strong CB1 immunoreactivity within axon terminals in the inner third of the stratum moleculare of control subjects. These CB1-positive boutons (b) form the classic asymmetric synapses (arrowheads) with an extensive postsynaptic density on dendritic spine heads. In control samples, nearly all axon terminals with asymmetric synapses are positive for CB1, whereas in the nonsclerotic (B) and sclerotic (C) samples, the number of CB1-positive asymmetric synapses drops noticeably. Note that a lack of staining does not necessarily mean the complete absence of CB1 receptors, but it means that the antigen level fails to reach detection threshold in these CB1-negative boutons (depicted by asterisks). Scale bar, 0.5 μm.
Figure 7.

Quantitative analysis of the ratio and density of CB1-positive excitatory axon terminals in the inner molecular layer of the human dentate gyrus. A, The number of excitatory axon terminals either positive or negative for CB1 cannabinoid receptor was established using an unbiased stereological estimation method (Geinisman et al., 1996). Altogether, 1092 disector pairs were analyzed, which resulted in 327 terminals in control, 224 terminals in nonsclerotic epileptic, and 197 terminals in sclerotic epileptic patients. The number of CB1-positive terminals decreased from 235 terminals in control subjects to 106 or 40 terminals in the nonsclerotic or sclerotic epileptic samples, respectively. In contrast, the number of CB1-negative terminals increased from 92 terminals in control subjects to 118 or 157 terminals in nonsclerotic or sclerotic epileptic samples, respectively. In the quantitative analysis, tissue samples from three individuals from each experimental group were used. B, The ratio of CB1-positive excitatory axon terminals versus all excitatory axon terminals was 72.8 ± 2.1% in control, 50 ± 2.8% in nonsclerotic epileptic, and 21 ± 3.8% in sclerotic epileptic samples (mean ± SEM). The difference between control and epileptic samples was highly significant (χ2 test, ***p < 0.001 both for nonsclerotic and sclerotic epileptic samples). C, The estimated numerical density of CB1-positive axon terminals in the inner molecular layer of the dentate gyrus of control subjects (0.648 ± 0.075/μm3) was strongly decreased in nonsclerotic epileptic patients (0.3 ± 0.051/μm3) and in sclerotic epileptic patients (0.112 ± 0.021/μm3) as well (values are mean ± SEM). This sharp decline in the density of CB1-positive axon terminals was statistically significant (ANOVA, p < 0.001). Significance exists between control values and both nonsclerotic and sclerotic epileptic patients (Dunnett's post hoc test, ***p < 0.001). D, The estimated numerical density of CB1-negative axon terminals was elevated in epileptic samples [density in control samples, 0.248 ± 0.038/μm3; in nonsclerotic samples, 0.326 ± 0.062/μm3; and in sclerotic samples, 0.454 ± 0.07/μm3 (mean ± SEM)]. Increase was significant between analyzed groups (ANOVA, p = 0.04); density of CB1-negative axon terminals in sclerotic epileptic patients was significantly increased compared with control values (Dunnett's post hoc test, *p = 0.037), but not in nonsclerotic patients (Dunnett's post hoc test, p = 0.546).
These data revealed that the ratio of CB1-positive axon terminals compared with all axon terminals with asymmetric, glutamatergic synapses differed in control, nonsclerotic, and sclerotic epileptic samples. Indeed, although in control samples the majority of glutamatergic boutons were positive for CB1 (72.8 ± 2.1%), this high ratio of CB1-positive excitatory axon terminals was significantly reduced in nonsclerotic epileptic samples to 50.0 ± 2.8% and was further diminished in sclerotic epileptic samples to 21.0 ± 3.8% (Fig. 7 B). The difference in the ratio of CB1-positive axon terminals forming asymmetric synapses in the inner molecular layer was significant for all three experimental groups (p < 0.001; χ2 test), as well as pairwise between control and nonsclerotic or sclerotic subjects (p < 0.001 in both cases) (Fig. 7 B).
To determine whether the robust reduction in the ratio of CB1-positive excitatory axon terminals is attributable to the disappearance of CB1 from surviving axon terminals or could be explained by the loss of mossy cells or both, we also calculated changes in the number of CB1-positive and CB1-negative excitatory axon terminals. The estimated numerical density (est N v) of CB1-positive axon terminals was 0.648 ± 0.075/μm3 in control subjects, which decreased to 0.300 ± 0.051/μm3 in nonsclerotic epileptic subjects and to only 0.112 ± 0.021/μm3 in sclerotic epileptic patients (Fig. 7 C). This sharp decline proved to be significant between the experimental groups (ANOVA, p < 0.001 and Dunnett's post hoc test, p < 0.001 for both experimental groups). In contrast, the density of CB1-negative axon terminals showed an increasing trend from 0.248 ± 0.038/μm3 in the control samples to 0.326 ± 0.062/μm3 in nonsclerotic epileptic and to 0.454 ± 0.07/μm3 in sclerotic epileptic patients (Fig. 7 D). ANOVA did reveal significant difference between the experimental groups (p = 0.04), which was further confirmed by Dunnett's post hoc test between the control and the sclerotic experimental groups (p = 0.037), but not between the control and the nonsclerotic experimental group (p = 0.546).
Mossy cells are especially vulnerable to neuronal insults. Hence, to further test the potential contribution of mossy cell loss to the findings above, we performed GluR2/3 immunostaining, which selectively visualizes the cell body of mossy cells, but not of GABAergic interneurons in the hilus of the dentate gyrus (Leranth et al., 1996; Freund et al., 1997). The density of GluR2/3-immunoreactive mossy cells was 70.9 ± 3.2 cells/mm2 in control subjects and 67.4 ± 12.0 cells/mm2 in nonsclerotic epileptic subjects, which decreased to 34.6 ± 9.3 cells/mm2 in sclerotic epileptic subjects (supplemental Figs. 4, 5, available at www.jneurosci.org as supplemental material). Kruskal–Wallis test did reveal significant difference between the experimental groups (p = 0.04), which was further confirmed by one-tailed Mann–Whitney U test between the control and the sclerotic experimental groups (p = 0.029), but there was no significant decrease in the density of mossy cells in the nonsclerotic experimental group compared with the control subjects (p = 0.886). These data are in agreement with previous findings in human temporal lobe epileptic samples (Blumcke et al., 2000) and further confirm that mossy cell loss alone, especially in the nonsclerotic subjects, cannot explain the reduced ratio of CB1-positive excitatory axon terminals in the inner molecular layer of the dentate gyrus.
CB1-positive axon terminals formed symmetric synapses mainly on thick proximal dendrites of granule cells (Fig. 8). In contrast to glutamatergic axon terminals, however, the ratio of CB1-positive boutons forming symmetric synapses was unaltered in the inner stratum moleculare of the epileptic dentate gyrus (Fig. 9). In the same sample of 1092 disectors, the majority of GABAergic boutons were positive for CB1 (67.9 ± 2.7%, 71.5 ± 1.3%, and 72.9 ± 3.3% in the control, nonsclerotic, and sclerotic epileptic samples, respectively) in all three experimental groups (Fig. 9 B). The difference in the ratio of CB1-positive axon terminals forming symmetric synapses in the inner molecular layer was insignificant (p = 0.698; χ2 test). Likewise, significant differences in the numerical density were found in neither the CB1-positive nor the CB1-negative axon terminals (p = 0.114 and p = 0.458, respectively; ANOVA) (Fig. 9 C,D). In accordance with the lack of changes in the density of CB1-positive GABAergic axon terminals, the density of the somata of CB1-immunoreactive interneurons in the dentate gyrus was also similar in all three experimental groups (6.36 ± 1.10 cells/mm2, 6.78 ± 0.49 cells/mm2, and 7.64 ± 0.88 cells/mm2 for control, nonsclerotic, and epileptic subjects, respectively; p = 0.52; Kruskal–Wallis test).
Figure 8.
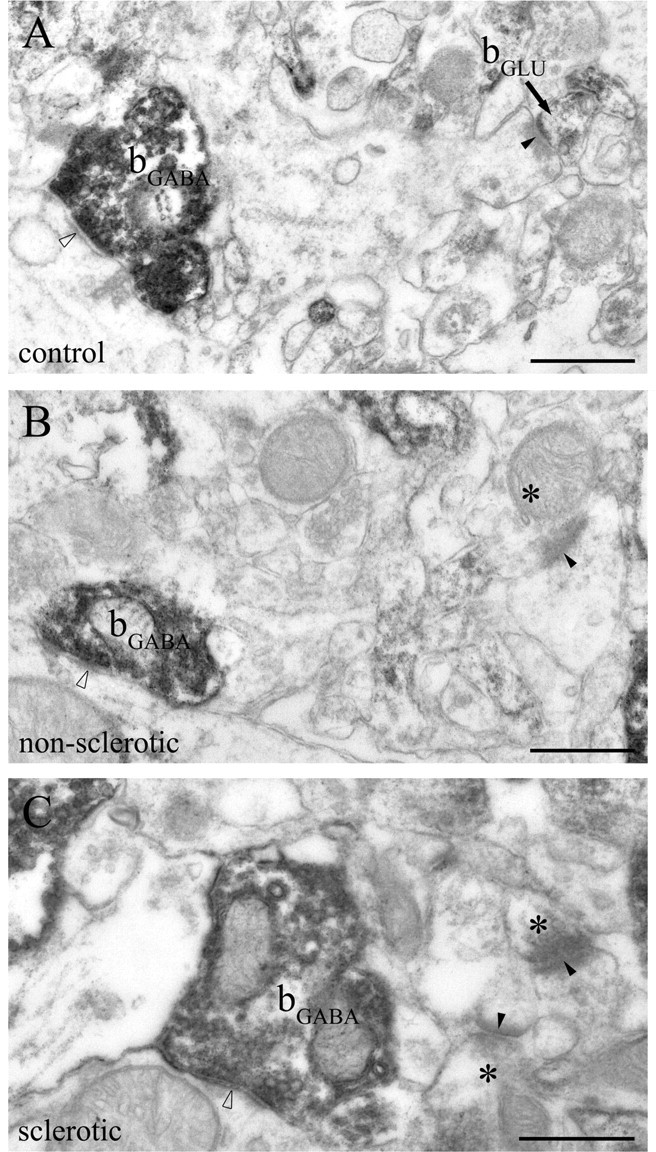
CB1-positive GABAergic axon terminals are intact in the epileptic human hippocampus. A–C, The electron micrographs show striking CB1 immunoreactivity within GABAergic axon terminals (bGABA) forming symmetric synapses (open arrowheads) in the inner third of stratum moleculare of the dentate gyrus. Dense accumulation of the end product of immunoperoxidase reaction (DAB) indicates that these GABAergic axon terminals are fully equipped with CB1 receptors in both the control and the epileptic hippocampi. Glutamatergic boutons terminate on dendritic spine heads with typical asymmetric synapses (closed arrowheads), which is characterized by broad postsynaptic density. In the control sample, the glutamatergic axon terminal (bGLU) is positive for CB1 (A), whereas CB1-negative boutons (asterisks) forming asymmetric synapses are shown in electron micrographs taken from the nonsclerotic (B) and sclerotic (C) samples. Note that CB1-positive axon terminals forming inhibitory synapses are larger than those giving excitatory synapses. Scale bars, 0.5 μm.
Figure 9.

Quantitative analysis of the ratio and density of CB1-positive inhibitory axon terminals in the inner molecular layer of the human dentate gyrus. A–D, The ratio and density of inhibitory axon terminals either positive or negative for CB1 cannabinoid receptor were established in a manner similar to that detailed in Figure 7 for excitatory terminals. A, Altogether, 1092 disector pairs were analyzed, which resulted in 102 inhibitory terminals in control, 85 terminals in nonsclerotic epileptic, and 107 terminals in sclerotic epileptic patients. The number of CB1-positive terminals forming symmetric synapses was 68 terminals in control subjects, and 60 or 77 terminals in the nonsclerotic or sclerotic epileptic samples, respectively. The number of CB1-negative terminals was 34 terminals in control subjects, and 25 or 30 terminals in nonsclerotic or sclerotic epileptic samples, respectively. B, The ratio of CB1-positive GABAergic axon terminals versus all GABAergic axon terminals was 67.8 ± 2.7% in control, 71.5 ± 1.3% in nonsclerotic epileptic, and 72.9 ± 3.3% in sclerotic epileptic samples (mean ± SEM). The difference between control and epileptic samples was not significant (χ2 test, p = 0.698). C, The estimated numerical density of CB1-positive inhibitory axon terminals in the inner molecular layer of the dentate gyrus of control subjects (0.16 ± 0.02/μm3) was similar in nonsclerotic epileptic patients (0.15 ± 0.01/μm3) and in sclerotic epileptic patients (0.19 ± 0.02/μm3) (ANOVA, p = 0.114). D, The estimated numerical density of CB1-negative inhibitory axon terminals was comparable in the three groups (density in control samples, 0.08 ± 0.01/μm3; in nonsclerotic samples, 0.06 ± 0.01/μm3; and in sclerotic samples, 0.07 ± 0.01/μm3; ANOVA, p = 0.458).
Together, these experiments provided evidence that CB1-positive excitatory axon terminals, but not inhibitory terminals, are severely downregulated in the inner molecular layer of the dentate gyrus in intractable epilepsy.
Discussion
Despite the well known safeguarding role of endocannabinoid signaling against excess neuronal excitability, including epileptic seizures, vulnerability of this chemical messenger system has not yet been investigated in epileptic patients. Combining expression profiling and quantitative electron microscopic analysis, we show here a major downregulation of CB1 cannabinoid receptor, the predominant neuronal cannabinoid receptor responsible for neuroprotective and anticonvulsant effects of cannabinoids, in the hippocampal formation of patients with intractable temporal lobe epilepsy. We also found a parallel reduction in the expression level of CRIP1a, a regulator of CB1 activity, and of DGL-α, the main biosynthetic enzyme of 2-AG, the endogenous ligand of CB1. Conversely, gene expression level of the enzymes responsible for the synthesis or inactivation of anandamide, another major endocannabinoid, remained unaffected in the epileptic hippocampus.
Downregulation of CB1 receptor and other molecular components involved in endocannabinoid signaling in the epileptic human hippocampus
Temporal lobe epilepsy is a common neurological disorder with varied etiology. However, it is consistently manifested by a phenotype of seriously unbalanced network activity and recurrent seizures. Approximately one-third of epileptic patients have inadequate seizure control despite maximal medical therapy (Kwan and Brodie, 2000). First and foremost, the ineffectiveness of conventional pharmacological treatments in this patient population makes imperative the need to exploit novel therapeutically useful signaling pathways. The recently discovered endocannabinoid system is one such promising pathway. In addition, the lack of effective treatment for these patients may also derive from their extreme vulnerability to perturbations of network excitability; however, the underlying molecular and cellular mechanisms are poorly understood. Therefore, we believe that the massive decline of CB1 cannabinoid receptors, well established molecular “stout guards” in controlling excess network activity (Mechoulam and Lichtman, 2003), in the hippocampal formation of patients with drug-refractory epilepsy is the most significant finding of the present study.
Several results support the conclusion that CB1 receptors are downregulated in the epileptic human hippocampus. First of all, real-time PCR experiments with two distinct reference genes revealed that its mRNA level was decreased to less than one-half of its value in control individuals. Potential confounding factors could be the impact of anesthesia or postmortem delay. However, the lack of difference in CB1 expression level in the two animal models makes the first two factors unlikely. This robust reduction may also reflect immense cell loss in epileptic patients, which certainly contributes to reduced CB1 level to some extent. However, two observations suggest that gene-specific reduction of transcriptional activity is the main underlying reason. Importantly, the magnitude of decrease in CB1 expression level was comparable in both epileptic groups, although these are distinguished by the lack of cell loss in nonsclerotic hippocampal samples in contrast to extensive cell death, especially in the CA1 region, of sclerotic hippocampal samples (Wittner et al., 2002, 2005). Conversely, mRNA level of MGL, which is expressed by exactly the same cell populations as CB1 (Gulyas et al., 2004), did not change significantly. Similarly, expression level of FAAH and DGL-β remained unaltered, although these enzymes also show a largely similar expression pattern to CB1 (Tsou et al., 1998; Bisogno et al., 2003).
Interestingly, related molecular components of the endocannabinoid system also showed parallel downregulation in the epileptic hippocampus. Although the precise physiological role of CRIP1a has not yet been determined, it is a binding partner of CB1 (Niehaus et al., 2007); thus, its expression level may be regulated in tandem. DGL-α, the final enzyme in the biosynthetic pathway of 2-AG (Bisogno et al., 2003), was also downregulated in the epileptic hippocampus. DGL-α is expressed by principal neurons and colocalizes with mGluR5 through the scaffolding protein Homer in a perisynaptic annulus around the postsynaptic density of glutamatergic synapses opposite to presynaptic CB1 receptors (Katona et al., 2006; Yoshida et al., 2006; Jung et al., 2007). This striking colocalization suggests that mGluR5 and DGL-α is involved in the molecular cascade, which detects surplus excitatory activity and induces 2-AG release to attenuate further glutamate release and the escalation of excitotoxicity. Importantly, both mGluR5 and long Homer isoforms are robustly downregulated after status epilepticus (Kirschstein et al., 2007). Moreover, elimination of other upstream components in 2-AG's biosynthetic route such as Gq/G11 G-proteins or PLCβ1 and its target CB1 also led to increased seizure susceptibility (Kim et al., 1997; Marsicano et al., 2003; Wettschureck et al., 2006).
Decreased ratio of CB1-positive excitatory axon terminals in the epileptic human hippocampus
An important question is whether the reduction of CB1 transcription occurs in the glutamatergic or in the GABAergic cell population, because both express CB1 receptors (Westlake et al., 1994; Katona et al., 1999, 2000, 2006; Marsicano and Lutz, 1999; Marsicano et al., 2003). Several observations suggest that the majority of reduction, at least at the protein level, may occur in the glutamatergic cell population. Most importantly, CB1 immunostaining visualized a neuropil-like labeling pattern throughout the human hippocampal formation. This pattern resembles the distribution of glutamatergic axon terminals, as has been demonstrated in the rodent hippocampus (Katona et al., 2006; Kawamura et al., 2006; Monory et al., 2006). Here we provide direct anatomical evidence that CB1 receptors are also located presynaptically on glutamatergic axon terminals in the human hippocampus. Moreover, light microscopic analysis revealed that this neuropil-like labeling pattern was severely reduced in density throughout the hippocampal formation of epileptic patients. To estimate the magnitude of reduction, and to substantiate the above observations at the subcellular level, we have performed quantitative electron microscopic analysis of CB1-positive terminals forming glutamatergic asymmetric synapses. We selected the inner molecular layer of the dentate gyrus for detailed analysis, because it contained the highest density of CB1 immunostaining in the neuropil. CB1 receptors in this layer in mice are present mainly on axon terminals of the glutamatergic mossy cells (Monory et al., 2006). These cells are integrated into the dentate network in a central position receiving excitation from dentate granule cells, then propelling it back to their dendrites. This disynaptic feedback excitation is a key determinant of hyperexcitability (Santhakumar et al., 2000). Remarkably, the ratio of excitatory synapses formed by CB1-positive axon terminals was dropped by 32% in nonsclerotic hippocampus and by ∼70% in the sclerotic hippocampus. In addition, the density of CB1-positive axon terminals was also decreased in both epileptic groups. Three phenomena are likely to contribute to this decrease at the terminal level. Because the mossy cell is a particularly vulnerable cell type, their loss may contribute to the reduction in the density of CB1-positive glutamatergic axon terminals in this layer. However, significant numbers of mossy cells survive in epilepsy (Blumcke et al., 2000; Ratzliff et al., 2002; present study). Thus, the magnitude of reduction in the ratio of CB1-positive boutons, especially in the nonsclerotic samples (in which mossy cells are not decreased in density), indicates that a fraction of CB1 disappears from the axon terminals of surviving mossy cells. This loss of brake in feedback excitation will definitely have a proconvulsive effect. Finally, because granule cells do not express CB1 receptors, the increase in the density of CB1-negative boutons in the sclerotic population may also involve sprouting recurrent axon collaterals of granule cells (Sutula et al., 1989). Irrespectively of the different contribution of certain cell types, the electron microscopic analysis clearly demonstrate a sharp decline in the proportion of excitatory axon terminals controlled by CB1 receptors; in other words, the net effect is that most excitatory synapses innervating granule cell dendrites will remain without an important negative feedback control in the epileptic dentate gyrus. Importantly, in future experiments it will be necessary to determine whether the reduced control of glutamate release by CB1 receptors occurs in other key structures of epileptogenesis, such as the subiculum (Cohen et al., 2002; Fabó et al., 2008), and thus diminished endocannabinoid signaling at glutamatergic synapses may generally contribute to increased network excitability throughout the hippocampal formation.
Conversely, quantitative analysis did not reveal changes either in the number of CB1-positive GABAergic interneurons or in the ratio and number of CB1-positive GABAergic axon terminals in the inner molecular layer. This is an interesting observation in light of the crucial contribution of depolarizing GABA currents to spontaneous epileptic activity (Cohen et al., 2002), and further supports the striking dichotomy of distinct cortical interneuron types in the regulation of epileptic network activity, i.e., that parvalbumin-containing but not CB1-positive GABAergic interneurons are critically involved (Cossart et al., 2005; Maglóczky and Freund, 2005; Monory et al., 2006; Freund and Katona, 2007; Ogiwara et al., 2007).
Functional link between CB1 receptors, endocannabinoid signaling, and tight control of network excitability has been clearly demonstrated during the last decade (Lutz, 2004). The emerging view is that the beneficial effect of this endogenous signaling system derives from a defense mechanism in which endocannabinoids alleviate excitotoxicity by activating CB1 cannabinoid receptors on glutamatergic neurons (Marsicano et al., 2003; Monory et al., 2006). Hence, downregulation of CB1 receptors, especially on glutamatergic axon terminals, implies that protective endocannabinoid signaling is diminished in the hippocampal formation of patients with intractable temporal lobe epilepsy, and impairment of this synaptic circuit breaker will inevitably lead to increased network excitability and neuronal damage in epileptic patients.
Footnotes
This work was supported by Országos Tudományos Kutatási Alapprogramok (OTKA) Grants T046820 (T.F.F.) and F046407 (I.K.), Egészségügyi Tudományos Tanács Grant 561/2006 (I.K.), Nemzeti Kutatási és Fejlesztési Program Grant 1A/002/2004 (T.F.F.), by the Szentágothai János Knowledge Center (RET 5/2004), and by National Institutes of Health Grant MH54671 (T.F.F.). I.K. is a grantee of the János Bolyai scholarship. We are grateful to Sándor Koncz and Izinta Kft for their help with real-time PCR experiments and to Prof. Péter Sótonyi and Dr. Zsolt Borostyánkői (Semmelweis University, Budapest, Hungary) for providing control human tissue. We also thank for Katalin Lengyel, Katalin Iványi, Emőke Simon, Gabriella Urbán, and Győző Goda for excellent technical assistance; Ken Mackie, Barna Dudok, and Rita Nyilas for comments on this manuscript; Kinga Tóth for her help in cell counting; and Dr. Gábor Nyíri for help with the statistical analysis.
References
- Alger, 2002.Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Amaral, 1978.Amaral DG. A Golgi study of cell types in the hilar region of the hippocampus in the rat. J Comp Neurol. 1978;182:851–914. doi: 10.1002/cne.901820508. [DOI] [PubMed] [Google Scholar]
- Ameri et al., 1999.Ameri A, Wilhelm A, Simmet T. Effects of the endogeneous cannabinoid, anandamide, on neuronal activity in rat hippocampal slices. Br J Pharmacol. 1999;126:1831–1839. doi: 10.1038/sj.bjp.0702478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker et al., 2002.Becker AJ, Chen J, Paus S, Normann S, Beck H, Elger CE, Wiestler OD, Blumcke I. Transcriptional profiling in human epilepsy: expression array and single cell real-time qRT-PCR analysis reveal distinct cellular gene regulation. NeuroReport. 2002;13:1327–1333. doi: 10.1097/00001756-200207190-00023. [DOI] [PubMed] [Google Scholar]
- Bernard et al., 2005.Bernard C, Milh M, Morozov YM, Ben-Ari Y, Freund TF, Gozlan H. Altering cannabinoid signaling during development disrupts neuronal activity. Proc Natl Acad Sci USA. 2005;102:9388–9393. doi: 10.1073/pnas.0409641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno et al., 2003.Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumcke et al., 2000.Blumcke I, Suter B, Behle K, Kuhn R, Schramm J, Elger CE, Wiestler OD. Loss of hilar mossy cells in Ammon's horn sclerosis. Epilepsia. 2000;41(Suppl 6):S174–S180. doi: 10.1111/j.1528-1157.2000.tb01577.x. [DOI] [PubMed] [Google Scholar]
- Chen et al., 2003.Chen K, Ratzliff A, Hilgenberg L, Gulyas A, Freund TF, Smith M, Dinh TP, Piomelli D, Mackie K, Soltesz I. Long-term plasticity of endocannabinoid signaling induced by developmental febrile seizures. Neuron. 2003;39:599–611. doi: 10.1016/s0896-6273(03)00499-9. [DOI] [PubMed] [Google Scholar]
- Chen et al., 2007.Chen K, Neu A, Howard AL, Foldy C, Echegoyen J, Hilgenberg L, Smith M, Mackie K, Soltesz I. Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures. J Neurosci. 2007;27:46–58. doi: 10.1523/JNEUROSCI.3966-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement et al., 2003.Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. Increased seizure susceptibility and proconvulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J Neurosci. 2003;23:3916–3923. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen et al., 2002.Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Cossart et al., 2005.Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Cravatt et al., 1996.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- de Lanerolle et al., 2003.de Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, Spencer DD. A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia. 2003;44:677–687. doi: 10.1046/j.1528-1157.2003.32701.x. [DOI] [PubMed] [Google Scholar]
- Devane et al., 1992.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Dinh et al., 2002.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabó et al., 2008.Fabó D, Maglóczky Z, Wittner L, Pék A, Erőss L, Czirják S, Vajda J, Sólyom A, Rásonyi G, Szucs A, Kelemen A, Juhos V, Grand L, Dombovári B, Halász P, Freund TF, Halgren E, Karmos G, Ulbert I. Properties of in vivo interictal spike generation in the human subiculum. Brain. 2008;131:485–499. doi: 10.1093/brain/awm297. [DOI] [PubMed] [Google Scholar]
- Falenski et al., 2007.Falenski KW, Blair RE, Sim-Selley LJ, Martin BR, DeLorenzo RJ. Status epilepticus causes a long-lasting redistribution of hippocampal cannabinoid type 1 receptor expression and function in the rat pilocarpine model of acquired epilepsy. Neuroscience. 2007;146:1232–1244. doi: 10.1016/j.neuroscience.2007.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin et al., 2003.Franklin A, Parmentier-Batteur S, Walter L, Greenberg DA, Stella N. Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. J Neurosci. 2003;23:7767–7775. doi: 10.1523/JNEUROSCI.23-21-07767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund and Katona, 2007.Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Freund et al., 1997.Freund TF, Hajos N, Acsady L, Gorcs TJ, Katona I. Mossy cells of the rat dentate gyrus are immunoreactive for calcitonin gene-related peptide (CGRP) Eur J Neurosci. 1997;9:1815–1830. doi: 10.1111/j.1460-9568.1997.tb00748.x. [DOI] [PubMed] [Google Scholar]
- Freund et al., 2003.Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Fukudome et al., 2004.Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, Watanabe M, Manabe T, Kano M. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signalling. Eur J Neurosci. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- Geinisman et al., 1996.Geinisman Y, Gundersen HJ, van der Zee E, West MJ. Unbiased stereological estimation of the total number of synapses in a brain region. J Neurocytol. 1996;25:805–819. doi: 10.1007/BF02284843. [DOI] [PubMed] [Google Scholar]
- Gulyas et al., 2004.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- Hansen et al., 2001.Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F, Manzanares J, Ikonomidou C, Schmid HH, Fernandez-Ruiz JJ, Hansen HS. Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem. 2001;78:1415–1427. doi: 10.1046/j.1471-4159.2001.00542.x. [DOI] [PubMed] [Google Scholar]
- Hashimotodani et al., 2007.Hashimotodani Y, Ohno-Shosaku T, Kano M. Endocannabinoids and synaptic function in the CNS. Neuroscientist. 2007;13:127–137. doi: 10.1177/1073858406296716. [DOI] [PubMed] [Google Scholar]
- Jamali et al., 2006.Jamali S, Bartolomei F, Robaglia-Schlupp A, Massacrier A, Peragut JC, Regis J, Dufour H, Ravid R, Roll P, Pereira S, Royer B, Roeckel-Trevisiol N, Fontaine M, Guye M, Boucraut J, Chauvel P, Cau P, Szepetowski P. Large-scale expression study of human mesial temporal lobe epilepsy: evidence for dysregulation of the neurotransmission and complement systems in the entorhinal cortex. Brain. 2006;129:625–641. doi: 10.1093/brain/awl001. [DOI] [PubMed] [Google Scholar]
- Jung et al., 2007.Jung KM, Astarita G, Zhu C, Wallace M, Mackie K, Piomelli D. A key role for diacylglycerol lipase-alpha in metabotropic glutamate receptor-dependent endocannabinoid mobilization. Mol Pharmacol. 2007;72:612–621. doi: 10.1124/mol.107.037796. [DOI] [PubMed] [Google Scholar]
- Katona et al., 1999.Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona et al., 2000.Katona I, Sperlagh B, Maglóczky Z, Santha E, Kofalvi A, Czirják S, Mackie K, Vizi ES, Freund TF. GABAergic interneurons are the targets of cannabinoid actions in the human hippocampus. Neuroscience. 2000;100:797–804. doi: 10.1016/s0306-4522(00)00286-4. [DOI] [PubMed] [Google Scholar]
- Katona et al., 2006.Katona I, Urban GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura et al., 2006.Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M. The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci. 2006;26:2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim et al., 1997.Kim D, Jun KS, Lee SB, Kang NG, Min DS, Kim YH, Ryu SH, Suh PG, Shin HS. Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature. 1997;389:290–293. doi: 10.1038/38508. [DOI] [PubMed] [Google Scholar]
- Kirschstein et al., 2007.Kirschstein T, Bauer M, Muller L, Ruschenschmidt C, Reitze M, Becker AJ, Schoch S, Beck H. Loss of metabotropic glutamate receptor-dependent long-term depression via downregulation of mGluR5 after status epilepticus. J Neurosci. 2007;27:7696–7704. doi: 10.1523/JNEUROSCI.4572-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan and Brodie, 2000.Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–319. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- Leranth et al., 1996.Leranth C, Szeidemann Z, Hsu M, Buzsaki G. AMPA receptors in the rat and primate hippocampus: a possible absence of GluR2/3 subunits in most interneurons. Neuroscience. 1996;70:631–652. doi: 10.1016/s0306-4522(96)83003-x. [DOI] [PubMed] [Google Scholar]
- Lutz, 2004.Lutz B. On-demand activation of the endocannabinoid system in the control of neuronal excitability and epileptiform seizures. Biochem Pharmacol. 2004;68:1691–1698. doi: 10.1016/j.bcp.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Maglóczky and Freund, 2005.Maglóczky Z, Freund TF. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005;28:334–340. doi: 10.1016/j.tins.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Maglóczky et al., 2000.Maglóczky Z, Wittner L, Borhegyi Z, Halász P, Vajda J, Czirják S, Freund TF. Changes in the distribution and connectivity of interneurons in the epileptic human dentate gyrus. Neuroscience. 2000;96:7–25. doi: 10.1016/s0306-4522(99)00474-1. [DOI] [PubMed] [Google Scholar]
- Marsicano and Lutz, 1999.Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Marsicano et al., 2003.Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W, Di Marzo V, Behl C, Lutz B. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- Mato and Pazos, 2004.Mato S, Pazos A. Influence of age, postmortem delay and freezing storage period on cannabinoid receptor density and functionality in human brain. Neuropharmacology. 2004;46:716–726. doi: 10.1016/j.neuropharm.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Mechoulam and Lichtman, 2003.Mechoulam R, Lichtman AH. Neuroscience. Stout guards of the central nervous system. Science. 2003;302:65–67. doi: 10.1126/science.1091256. [DOI] [PubMed] [Google Scholar]
- Monory et al., 2006.Monory K, Massa F, Egertova M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wolfel B, Dodt HU, Zieglgansberger W, Wotjak CT, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama et al., 1999.Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci. 1999;19:2987–2995. doi: 10.1523/JNEUROSCI.19-08-02987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus et al., 2007.Niehaus JL, Liu Y, Wallis KT, Egertova M, Bhartur SG, Mukhopadhyay S, Shi S, He H, Selley DE, Howlett AC, Elphick MR, Lewis DL. CB1 cannabinoid receptor activity is modulated by the cannabinoid receptor interacting protein CRIP 1a. Mol Pharmacol. 2007;72:1557–1566. doi: 10.1124/mol.107.039263. [DOI] [PubMed] [Google Scholar]
- Ogiwara et al., 2007.Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto et al., 2004.Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem. 2004;279:5298–5305. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- Pacher et al., 2006.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panikashvili et al., 2001.Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Pfaffl, 2001.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl et al., 2002.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli, 2003.Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Ratzliff et al., 2002.Ratzliff AH, Santhakumar V, Howard A, Soltesz I. Mossy cells in epilepsy: rigor mortis or vigor mortis? Trends Neurosci. 2002;25:140–144. doi: 10.1016/s0166-2236(00)02122-6. [DOI] [PubMed] [Google Scholar]
- Rozen and Skaletsky, 2000.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Santhakumar et al., 2000.Santhakumar V, Bender R, Frotscher M, Ross ST, Hollrigel GS, Toth Z, Soltesz I. Granule cell hyperexcitability in the early post-traumatic rat dentate gyrus: the ‘irritable mossy cell’ hypothesis. J Physiol (Lond) 2000;524:117–134. doi: 10.1111/j.1469-7793.2000.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafaroodi et al., 2004.Shafaroodi H, Samini M, Moezi L, Homayoun H, Sadeghipour H, Tavakoli S, Hajrasouliha AR, Dehpour AR. The interaction of cannabinoids and opioids on pentylenetetrazole-induced seizure threshold in mice. Neuropharmacology. 2004;47:390–400. doi: 10.1016/j.neuropharm.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Spencer and Spencer, 1985.Spencer DD, Spencer SS. Surgery for epilepsy. Neurol Clin. 1985;3:313–330. [PubMed] [Google Scholar]
- Stella et al., 1997.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Sugiura et al., 2000.Sugiura T, Yoshinaga N, Kondo S, Waku K, Ishima Y. Generation of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand, in picrotoxinin-administered rat brain. Biochem Biophys Res Commun. 2000;271:654–658. doi: 10.1006/bbrc.2000.2686. [DOI] [PubMed] [Google Scholar]
- Sugiura et al., 2006.Sugiura T, Kishimoto S, Oka S, Gokoh M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res. 2006;45:405–446. doi: 10.1016/j.plipres.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Sutula et al., 1989.Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- Theodore and Gaillard, 2002.Theodore WH, Gaillard WD. Neuroimaging and the progression of epilepsy. Prog Brain Res. 2002;135:305–313. doi: 10.1016/S0079-6123(02)35028-3. [DOI] [PubMed] [Google Scholar]
- Toth et al., 2007.Toth K, Wittner L, Urban Z, Doyle WK, Buzsaki G, Shigemoto R, Freund TF, Maglóczky Z. Morphology and synaptic input of substance P receptor-immunoreactive interneurons in control and epileptic human hippocampus. Neuroscience. 2007;144:495–508. doi: 10.1016/j.neuroscience.2006.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou et al., 1998.Tsou K, Nogueron MI, Muthian S, Sanudo-Pena MC, Hillard CJ, Deutsch DG, Walker JM. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett. 1998;254:137–140. doi: 10.1016/s0304-3940(98)00700-9. [DOI] [PubMed] [Google Scholar]
- van der Stelt et al., 2001a.van der Stelt M, Veldhuis WB, Bar PR, Veldink GA, Vliegenthart JF, Nicolay K. Neuroprotection by Delta9-tetrahydrocannabinol, the main active compound in marijuana, against ouabain-induced in vivo excitotoxicity. J Neurosci. 2001a;21:6475–6479. doi: 10.1523/JNEUROSCI.21-17-06475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stelt et al., 2001b.van der Stelt M, Veldhuis WB, van Haaften GW, Fezza F, Bisogno T, Bar PR, Veldink GA, Vliegenthart JF, Di Marzo V, Nicolay K. Exogenous anandamide protects rat brain against acute neuronal injury in vivo . J Neurosci. 2001b;21:8765–8771. doi: 10.1523/JNEUROSCI.21-22-08765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace et al., 2001.Wallace MJ, Wiley JL, Martin BR, DeLorenzo RJ. Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. Eur J Pharmacol. 2001;428:51–57. doi: 10.1016/s0014-2999(01)01243-2. [DOI] [PubMed] [Google Scholar]
- Wallace et al., 2002.Wallace MJ, Martin BR, DeLorenzo RJ. Evidence for a physiological role of endocannabinoids in the modulation of seizure threshold and severity. Eur J Pharmacol. 2002;452:295–301. doi: 10.1016/s0014-2999(02)02331-2. [DOI] [PubMed] [Google Scholar]
- Wallace et al., 2003.Wallace MJ, Blair RE, Falenski KW, Martin BR, DeLorenzo RJ. The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J Pharmacol Exp Ther. 2003;307:129–137. doi: 10.1124/jpet.103.051920. [DOI] [PubMed] [Google Scholar]
- Westlake et al., 1994.Westlake TM, Howlett AC, Bonner TI, Matsuda LA, Herkenham M. Cannabinoid receptor binding and messenger RNA expression in human brain: an in vitro receptor autoradiography and in situ hybridization histochemistry study of normal aged and Alzheimer's brains. Neuroscience. 1994;63:637–652. doi: 10.1016/0306-4522(94)90511-8. [DOI] [PubMed] [Google Scholar]
- Wettschureck et al., 2006.Wettschureck N, van der Stelt M, Tsubokawa H, Krestel H, Moers A, Petrosino S, Schutz G, Di Marzo V, Offermanns S. Forebrain-specific inactivation of Gq/G11 family G proteins results in age-dependent epilepsy and impaired endocannabinoid formation. Mol Cell Biol. 2006;26:5888–5894. doi: 10.1128/MCB.00397-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson and Nicoll, 2002.Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- Wittner et al., 2002.Wittner L, Erőss L, Szabo Z, Toth S, Czirják S, Halász P, Freund TF, Maglóczky ZS. Synaptic reorganization of calbindin-positive neurons in the human hippocampal CA1 region in temporal lobe epilepsy. Neuroscience. 2002;115:961–978. doi: 10.1016/s0306-4522(02)00264-6. [DOI] [PubMed] [Google Scholar]
- Wittner et al., 2005.Wittner L, Erőss L, Czirják S, Halász P, Freund TF, Maglóczky Z. Surviving CA1 pyramidal cells receive intact perisomatic inhibitory input in the human epileptic hippocampus. Brain. 2005;128:138–152. doi: 10.1093/brain/awh339. [DOI] [PubMed] [Google Scholar]
- Yoshida et al., 2006.Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, Watanabe M. Localization of diacylglycerol lipase-alpha around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci. 2006;26:4740–4751. doi: 10.1523/JNEUROSCI.0054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]