

Abstract
During the development and maturation of sensory neurons, afferent activity is required for normal maintenance. There exists a developmental window of time when auditory neurons, including neurons of the anteroventral cochlear nucleus (AVCN), depend on afferent input for survival. This period of time is often referred to as a critical period. The cellular and molecular mechanisms that underlie AVCN neuron susceptibility to deafferentation-induced death remain unknown. Here, we show that only during this critical period deafferentation of mouse AVCN neurons by in vivo cochlea removal results in rapid nuclear translocation and activation of the transcription factor NFATc4 (nuclear factor of activated T-cells isoform 4). NFAT activation is abolished by in vivo treatment with the calcineurin inhibitor FK506 and the specific NFAT-inhibitor 11R-VIVIT. Inhibition of NFAT significantly attenuates deafferentation-induced apoptosis of AVCN neurons and abolishes NFAT-mediated expression of FasL, an initiator of apoptotic pathways, in the cochlear nucleus. These data suggest that NFAT-mediated gene expression plays a role in deafferentation-induced apoptosis of cochlear nucleus neurons during a developmental critical period.
Keywords: auditory, activity-dependent, FasL, AVCN, calcineurin, mouse
Introduction
Activity-dependent survival of neurons during a critical period of development is a common theme shared by sensory neurons in the CNS (Simmons et al., 1981; Trune, 1982; Born and Rubel, 1985; Lipton, 1986; Hashisaki and Rubel, 1989; Catsicas et al., 1992; Mostafapour et al., 2000). In the auditory system, cochlea ablation (CX) eliminates the primary source of excitatory afferent input to the ipsilateral cochlear nucleus and therefore provides a useful model for studying the effects of deafferentation on the survival or death of cochlear nucleus neurons (Parks, 1999; Oertel and Young, 2004; Lu et al., 2007). Although recent studies suggest reinnervation of deafferented anteroventral cochlear nucleus (AVCN) neurons by excitatory synapses (Lu et al., 2007), this does not occur until 5–9 d after cochlea ablation, long after the cell death process is complete. Developmentally, there is a limited time during which removal of afferent activity results in the death of a significant subpopulation of auditory neurons in several animal models, including AVCN of the mouse and gerbil (Tierney et al., 1997; Mostafapour et al., 2000), as well as nucleus magnocellularis (NM) of the chick (Born and Rubel, 1985), the avian homolog of the mammalian AVCN. For example, 61% of AVCN neurons die in mice deafferented at postnatal day 5 (P5); this percentage declines with age and is absent by P14. During maturation, the neurons undergo a transformation from being susceptible to deafferentation-induced death to being resistant to the same manipulation. The biological switch that transforms the way in which these neurons respond to removal of afferent input is yet to be characterized, although many likely cellular and molecular candidates have been suggested in both the auditory (Rubel et al., 2004; Harris et al., 2005) and visual (Taha and Stryker, 2005) systems.
After deafferentation of chick NM neurons, a rapid rise in intracellular calcium concentration ([Ca2+]i) (Zirpel et al., 1995) activates the transcription factor cAMP response element-binding protein (CREB), leading to transcription of prosurvival proteins (Zirpel et al., 2000). All of the NM neurons show an increase in [Ca2+]i, but only a subpopulation activate CREB and survive deafferentation. The pathway culminating in NM neuron death has not been characterized but has been shown to be Ca2+ dependent (Zirpel et al., 1998). Neurons in the AVCN of mice also show an increase in [Ca2+]i after deafferentation, both during and after the critical period of activity-dependent survival (L. Zirpel, unpublished observations). It is therefore plausible to hypothesize that this increase in [Ca2+]i causes a cascade of calcium-mediated intracellular signaling events that elicit a survival or death response in AVCN neurons within the critical period but not beyond. Therefore, a cellular mechanism must exist that differentiates the response to increased [Ca2+]i during the critical period, but no longer functions in this context, is functionally offset by a different intracellular environment because of development (e.g., different gene products and proteins as suggested by Harris and Rubel, 2005), or no longer exists after the critical period.
Calcium/calmodulin activation of the phosphatase calcineurin (CaN) (protein phosphatase 2B) results in dephosphorylation of target proteins (Klee et al., 1979; Gupta et al., 1985). One such target is nuclear factor of activated T-cells (NFAT), specifically isoforms c1–c4 (Northrop et al., 1994; Ho et al., 1995; Ruff and Leach, 1995; Molkentin et al., 1998). Dephosphorylation of NFATc leads to exposure of a nuclear localization sequence, translocation from the cytoplasm to the nucleus (Jain et al., 1993; Rao et al., 1997), and transcription of specific target genes (Ruff and Leach, 1995; Luo et al., 1996; Graef et al., 1999).
Expression of the membrane-bound death receptor ligand FasL is mediated by NFAT (Latinis et al., 1997; Holtz-Heppelmann et al., 1998). When FasL binds to its receptor Fas, the intracellular machinery associated with the death receptor Fas is activated and eventually leads to apoptosis by caspase activation and subsequent DNA cleavage (Itoh et al., 1991; Jayanthi et al., 2005). Through this pathway, NFAT-dependent gene expression can actively regulate the induction of apoptosis (Kondo et al., 2003; Jayanthi et al., 2005).
The goals of this study were to determine whether or not NFAT plays a role in deafferentation-induced death of cochlear nucleus neurons during the developmental critical period and to describe a possible pathway through which NFAT mediates neuron death after deafferentation. We show that NFAT activation and FasL expression are increased in the cochlear nucleus after deafferentation during the critical period. Inhibition of NFAT significantly attenuates the increase of FasL expression and neuronal death after deafferentation. These results introduce a novel pathway through which deafferentation-induced death of sensory neurons might be mediated.
Materials and Methods
Animals and surgery.
P7, P14, and P21 C57BL mice (Harlan, Indianapolis, IN) were used for these studies, and procedures were approved by the Institutional Animal Care and Use Committee of the University of Minnesota. For all experiments, mice at these three ages were unilaterally deafferented by cochlea ablation (Mostafapour et al., 2000; Zirpel et al., 2000) while anesthetized with isoflurane. Ages stated for data are the age at which the surgery was performed. CX was performed by opening the tympanic membrane and removing the middle ear bones with fine forceps. The cochlear chamber was then penetrated with fine forceps, and the cochlear epithelium and modiolus were completely aspirated from the chamber through a pulled glass pipette with an approximate tip diameter of 0.7 mm. For P7 mice, a small incision was made inferior to the pinna to expose the ear canal. This incision was closed with cyanoacrylate glue after surgery. Mice were then returned to their cages with their mother until they were killed. After the animals were killed, a thick coronal section containing the brainstem was made with a razor blade and immediately immersed in oxygenated artificial CSF, whereas cochlear nuclei or whole brainstems were dissected and collected. Complete removal of the cochlea and modiolus was verified by examination of the cochlear bulla. For collections of cochlear nuclei, the nuclei on the side of the brain ipsilateral to CX were collected as deafferented tissue, and the nuclei on the side of the brain contralateral to CX were collected as control, or nondeafferented tissue. Tissue margins were determined using Figures 73–78 in The Mouse Brain Atlas (Paxinos and Franklin, 2001) as a guide to track the AVCN as well as the superficial glial zone and granular layer of the cochlear nucleus that mark the termination of the AVCN and the start of the posterior cochlear nucleus and the dorsal cochlear nucleus.
Drugs and administration.
11-R VIVIT (Calbiochem, Darmstadt, Germany), a cell-permeable peptide NFAT inhibitor, was dissolved in sterile saline to 8.40 mm and injected at concentrations of either 20 or 30 mg/kg. A higher dose of 35 mg/kg was injected but resulted in death of the animals shortly after injection. FK506 (Sigma, St. Louis, MO), a CaN inhibitor, was dissolved in DMSO to 3 mg/ml and then further diluted in sterile saline to 1.0 ng/μl and injected at a dose of 1 mg/kg. Injections were made with a 10 μl microsyringe and 30.5 gauge needle. Mice were anesthetized, and the area posterior to the bulla on the right side of the head was sterilized with 70% ethanol before receiving injection into the CSF. The area posterior to the bulla and just beneath the skull is in close contact with the right cochlear nucleus, allowing for efficient drug delivery to the cochlear nucleus neurons. This was verified by injection of fast green dye and visual verification of dye diffusion to the CSF surrounding the cochlear nucleus and staining of the cochlear nucleus neurons with the fast green dye. Saline injections of 2 μl were administered for vehicle control (2 μl was the maximum volume injected into a drug-treated animal). Drug injections were performed 90 min before surgery.
Fractionation of cellular lysates.
Cytoplasmic and nuclear extracts were prepared using a nuclear and cytoplasmic extraction kit (NE-PER; Pierce, Rockford, IL). The protocol provided with the kit was followed with minor revisions. Cochlear nuclei were collected into 300 μl of cytoplasmic extraction reagent (CER)-I containing 1× protease inhibitor cocktail (Pierce) and 1× phosphatase inhibitor cocktail (Pierce). Samples were immediately homogenized with seven strokes (4× pestle A; 3× pestle B) in a 1 ml Dounce homogenizer, vortexed at highest setting for 15 s, and incubated on ice for 10 min. Then, 16.5 μl of ice-cold CER-II was added to each sample, vortexed for 5 s, and centrifuged at 14,000 × g for 5 min. The supernatant was collected as the cytoplasmic fraction. The nuclear pellet was suspended in 100 μl of nuclear extraction reagent containing 1× protease inhibitor (Pierce) and vortexed on high for 15 s every 10 min for 40 min. This was followed by centrifugation at 14,000 × g for 10 min and collection of the supernatant as nuclear lysate. All samples were kept on ice throughout the procedure. Centrifugation was performed at 4°C. Concentrations were determined using the BCA Protein Assay kit (Pierce). Lysates were stored at −80°C until used for Western blotting.
Western blot.
Cochlear nuclei were collected into ice-cold radioimmunoprecipitation assay buffer containing 50 mm Trizma, 1 mm EDTA, 1% Triton X-100 with 1× protease inhibitor cocktail (Pierce), and 1× phosphatase inhibitor cocktail (Pierce) and immediately homogenized with tissue tearor while on ice. Samples were then centrifuged at 14,000 × g for 15 min at 4°C. Supernatant was collected, and BCA assay was used to determine protein concentration. For a total sample volume of 37 μl, lysates containing 30 μg of protein were diluted with β-mercaptoethanol (1:10), 4× lithium dodecyl sulfate buffer (final concentration 1×), and dH2O, heated to 95°C for 10 min, and centrifuged for 5 min at 14,000 × g. Samples were separated into a 10% SDS polyacrylamide gel and blotted onto a nitrocellulose membrane. Membranes were then cut in half to assay the high-molecular-weight bands separately from the low-molecular-weight bands. Membranes were washed with 0.1 m PBS, pH 7.4, with 5% (w/v) powdered milk for 1 h and then incubated (overnight at 4°C) with one of the following primary antibodies diluted in PBS with 0.1% Triton X-100 (PBST) and 5% (w/v) powdered milk (antibody buffer): rabbit polyclonal anti-NFATc4 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal GAPDH (1:20,000; Millipore Bioscience Research Agents, Temecula, CA), or rabbit polyclonal anti-CREB (1:1000; Millipore, Billerica, MA). Membranes were washed in PBST five times for 5 min and incubated with one of the following secondary antibodies diluted in antibody buffer: goat anti-mouse Alexa-680 (1:20,000; Invitrogen, Carlsbad, CA) or goat anti-rabbit IRDye 800 (1:20,000; LI-COR Biosciences, Lincoln, NE). Membranes were washed seven times for 5 min and scanned with a LI-COR Odyssey infrared imaging system. The integrated intensity of fluorescence for each band (NFATc4, <120 kDa; FasL, 45 kDa; GAPDH, 38 kDa) was then determined. All levels were normalized to GAPDH levels. For fractionation analysis, the amount of nuclear translocation of NFATc4 was determined by first calculating the amount of nuclear NFATc4 in each sample set [control (CTL) or deafferented (CX)]: NFATc4nucleus/NFATc4total. These values were then used to calculate the percentage increase of nuclear NFATc4 after CX: [(nuclear NFATc4CX/nuclear NFATc4CTL) − 1) × 100]. CREB is localized to the nucleus in AVCN neurons (X.-Q. Tang, J. I. Luoma, and L. Zirpel, unpublished observations) and was used to verify complete separation of nuclear lysate from cytoplasmic lysate.
Immunohistochemistry.
After CX for the desired amount of time, brainstems were dissected from the cranium and immediately immersed and fixed in PBS with 4% paraformaldehyde at 4°C overnight and then 30% sucrose overnight at 4°C before embedding in OCT and freezing. Twenty micrometer cryosections were collected on gelatin-subbed slides and dried on a warming plate for 30 min. Immunohistochemistry for NFATc4 was performed by first rehydrating sections in PBS for 10 min, incubation in blocking buffer (0.1 m PBS, 1% bovine serum albumin, and 2% normal goat serum) 1 h at room temperature (∼70°F), and incubation with primary antibody [1:100, rabbit polyclonal anti-NFATc4 (Santa Cruz Biotechnology) or PBS for negative control] in blocking buffer with 0.3% Triton X-100 (antibody buffer) for 48 h at room temperature in a humidity chamber. Sections were then washed in PBS three times for 5 min and incubated with secondary antibody (1:1000, goat anti-rabbit Alexa-594; Invitrogen) in antibody buffer for 1 h at room temperature. Sections were washed five times for 5 min and mounted with FluorSave (Calbiochem). Apoptotic cells were identified using ApopTag Fluorescein In Situ Apoptosis Detection kit (Millipore Bioscience Research Agents). Product protocols were followed exactly. As a positive control, sections were pretreated with DNase buffer [30 mm Trizma, pH 7.2, 4 mm MgCl2, and 0.1 mm dithiothreitol (DTT)] for 5 min at room temperature, DNase I (1000 U/ml; Invitrogen) for 10 min at room temperature, and five times for 3 min in PBS washes before proceeding with the protocol. This resulted in ApopTag labeling of nearly all cells. As a negative control, terminal deoxynucleotidyl transferase was replaced with PBS, which resulted in no labeled cells.
Fluorescently labeled cells were visualized with a Nikon (Tokyo, Japan) TE300 Eclipse microscope using either a 100× or 40× Fluor oil objective, filter cube sets 31004 (Alexa-594), 31000v2 [4′,6′-diamidino-2-phenylindole (DAPI)], and 41017 (ApopTag) from Chroma Technology (Rockingham, VT), and a xenon light source (Sutter Instruments, Novato, CA). Digital images were acquired with a cooled CCD camera (Photometrics CoolSNAP Hq or Cascade 512f; Roper Scientific, Tucson, AZ) and MetaMorph Software (Universal Imaging, West Chester, PA). Images were processed/analyzed using Adobe Photoshop (Adobe Systems, San Jose, CA). Neurons in the AVCN were discerned from glia by morphology. For each animal, three images of both the left and right AVCN were taken from each of four separate sections (12 images of each AVCN) and used for cell counting. Values were averaged to obtain a single value for the left (control) and right (CX) AVCN per animal. Counts made were of neurons positively labeled for ApopTag, NFATc4 (cytoplasmic and/or nuclear), or only nuclear NFATc4. Nuclear staining was determined by overlap of NFATc4 staining with DAPI nuclear staining. To calculate the relative amount of nuclear NFATc4, the number of neurons positive for nuclear NFATc4 was divided by the total number of neurons positive for NFATc4 (cytoplasmic and/or nuclear). The effect of CX on the level of nuclear NFATc4 was determined by calculating the percentage increase of nuclear NFATc4: [(the ratio of nuclear NFATc4 on CX side/CTL side − 1) × 100]. For immunohistochemical analysis, the change in nuclear NFATc4 was also verified by comparing the total area of NFATc4 and DAPI colocalization with the total area per image and was consistent with the reported cell count data.
Cell counts.
At least four animals were used for each experimental group. Frozen sections, 20 μm thick, were collected on microscope slides and stained using the following cresyl violet Nissl staining procedure: 100% EtOH for 2 min, xylenes for 2 min, 100% EtOH for 2 min, 70% EtOH for 2 min, 20% EtOH for 2 min, dH2O for 5 min, cresyl violet solution [0.1% (w/v) cresyl violet and 3% acetic acid] for 5 min, two dips in dH2O, two dips in differentiation solution 1 (70% EtOH and 10% acetic acid), two dips in differentiation solution 2 (100% EtOH and 10% acetic acid), 100% EtOH for 1 min, xylenes for 2 min, and then mounted with Permount (Sigma).
Four sections of each AVCN were chosen for counting, making sure to include anterior, middle, and posterior portions of the AVCN along the rostral/caudal axis. For each section, four 40× images of the AVCN were acquired to include ventral, middle, and dorsal portions along the medial/lateral axis of the nucleus in the cell counts. The total number of neurons in each 40× image was obtained, excluding glial cells based on morphology. Counting was performed by an unbiased observer with no knowledge of treatment group being counted. The average number of neurons per 40× image was then calculated for each AVCN nucleus analyzed. The averages were used to compare the number of AVCN neurons ipsillateral to CX (CX) versus contralateral to CX (CTL) for each animal. The percentage of neuron loss after deafferentation was calculated for each animal using the following equation: [1 − (CX # of neurons/CTL # of neurons)] × 100.
PCR.
Total RNA was isolated from cochlear nuclei collected in RNAlater reagent using the RNeasy Micro kit and protocol (Qiagen, Valencia, CA). Tissue was homogenized with motorized tissue homogenizer followed by centrifugation through Qiashredder membrane (Qiagen). Total RNA was eluted with 13 μl of sterile distilled H2O and yielded ∼1 μg of RNA. cDNA was synthesized by adding the following to the total eluted RNA: 1 μl of oligo-DT20 (0.5 μg/ml) and 1 μl dNTP mix (10 mm). Mixture was heated to 65°C for 5 min, put on ice for 1 min and then 4 μl of 5× first-strand buffer, 1 μl of DTT (0.1 m), 1 μl of RNaseOUT (40 U/μl), and 1 μl of SuperScript III RT (200 U/μl) were added to each sample before incubation at 25°C for 5 min, 53°C for 50 min, and 70°C for 15 min. Finally, 1 μl of RNase-H was added to each cDNA sample and incubated at 37°C for 20 min. All RT reagents were purchase from Invitrogen. The cDNA was diluted to 40 ng/μl with sterile dH2O and 1 μl per reaction was used. Forward and reverse primers were diluted with sterile dH2O to 3.0 μm, and a mix was made containing both forward and reverse primers at 1.5 μm. Primer sequences used were ACCACAGTCCATGCCATCAC (forward GAPDH), TCCACCACCCTGTTGCTGTA (reverse GAPDH), AAGCTTCAGCTCTTCCACCTG (forward FasL), ATGAATTCCTGGTGCCCATG (reverse FasL), GAGAATTGCTGAAGACATGACAATCC (forward Fas), GTAGTTTTCACTCCAGACATTGTCC (reverse Fas), CCGAAGTGGAGCAGAAGAAG (forward S15), CTCCACTGGTTGAAGGTC (reverse S15), TCATTGACACCACCTCCAAA (forward RPL3), and GCACAAAGTGGTCCTGGAAT (reverse RPL3). Each 30 μl of PCR reaction contained 15 μl of DyNAmo HS SYBR green master mix (New England Biolabs, Ipswich, MA), 6 μl of primer mix, 6 μl of cDNA template, and 3 μl of dH2O. The thermocycling protocol was as follows: 95°C for 15 min, 94°C for 10 s, 60°C for 30 s, 72°C for 30 s, and fluorescence data acquisition for a total of 40 cycles. Melting curves were generated to verify the specificity of the generated products. Each reaction was run in triplicate. Controls for DNA contamination of reagents were run using dH2O in place of cDNA template, and all yielded no PCR product as expected. Fluorescence of the SYBR green was detected using the Opticon2 thermocycler (MJ Research, Waltham, MA). Ct (cycle at which threshold fluorescence is reached) values for each sample were then collected at a threshold level of fluorescence set within the linear phase of amplification. All samples were analyzed using the semiquantitative ΔΔCt method (Livak and Schmittgen, 2001) comparing expression between the deafferented and intact cochlear nuclei. Results are reported as mean fold changes in expression after CX (2−ΔΔCt). All values were normalized to GAPDH (or either S15 or RPL3 when noted) to control for loading. GAPDH was used again, because it has not been shown previously to be upregulated or degraded in the early stages of cell death. Upregulation of GAPDH in the nucleus has been shown in cultured neurons after the induction of oxidative stress by addition of AraC (Saunders et al., 1997; Chen et al., 1999).
Analysis and statistics.
At least three biological replications were performed for each experiment. For Western blots and semiquantitative reverse transcription (RT)-PCR, the cochlear nucleus tissue from three mice was pooled for each replicate. Control levels for each treatment group were not significantly different from each other, and the data were therefore normalized to their respective control, or nondeafferented values (CX/CTL). When ratios of CX/CTL were calculated, a paired t test was performed to determine whether the means were significantly different from the hypothetical value of 1.0 (indicated on graphs with a dotted line at 1.0), a value indicating no change after CX. Data are reported as the mean ± SEM. ANOVAs with Bonferroni's post hoc tests were used to determine the difference between group means. One value greater than 2 SDs from the mean was removed from the RT-PCR nondrug-treated CX group. All statistical analyses were calculated using Prism 4.0 software (GraphPad Software, San Diego, CA).
Results
Activation of NFATc4 occurs only within the critical period of dependence on afferent activity for survival
A critical period during development when AVCN neurons are susceptible to CX-induced death showed previously that deafferentation of AVCN neurons resulted in a 10-fold increase of apoptotic neurons at P7 and no increase at P21 as shown with terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay (Mostafapour et al., 2000). These results were reproduced in this study as described by the following experiment. Mice at ages P7, P14, and P21 underwent unilateral CX to remove afferent input from cochlear nucleus neurons but leave the contralateral side intact as a control to which the deafferented tissue could be compared. This control was chosen because no difference in neuron death is observed between the AVCN neurons of mice that have not undergone CX compared with the AVCN neurons contralateral to CX. The cochlear nucleus neurons were deafferented for 24 h and labeled with ApopTag, a fluorescent TUNEL stain that labels fragmented DNA, indicating induction of apoptosis. The 24 h time point was chosen because TUNEL of AVCN neurons was shown previously to be maximal after CX at this time (Mostafapour et al., 2000). Intact AVCN neurons contralateral to CX (CTL) at each age examined showed very few apoptotic neurons. However, in AVCN neurons ipsilateral to CX, apoptotic neurons at P7 were abundant. When CX was performed at either P14 or P21, there were very few apoptotic neurons in the CX AVCN (Fig. 1A). Quantification of these data showed that CX at P7 for 24 h leads to a significant increase in the number of apoptotic AVCN neurons compared with CTL. This is in contrast to deafferentation at P14 and P21 when CX did not result in an increase of apoptotic AVCN neurons (Table 1, Fig. 1B). To confirm that ApopTag-labeled AVCN neurons eventually lead to death and a decrease in neuron number in the AVCN, neuron counts were performed after 96 h of CX, a time at which loss of AVCN neurons after CX has reached a maximum (Mostafapour et al., 2000). In mice deafferented at P7, the percentage loss of AVCN neurons was significant, whereas at P14 and P21 the loss was not significant (Table 1, Fig. 1C). These data demonstrate that there is a developmental critical period during which the AVCN neurons are susceptible to CX-induced apoptosis that leads to neuron loss and that this critical period includes mice at age P7 but not ages P14 or P21, which are outside of this critical period. This is consistent with the results reported by Mostafapour et al. (2000).
Figure 1.
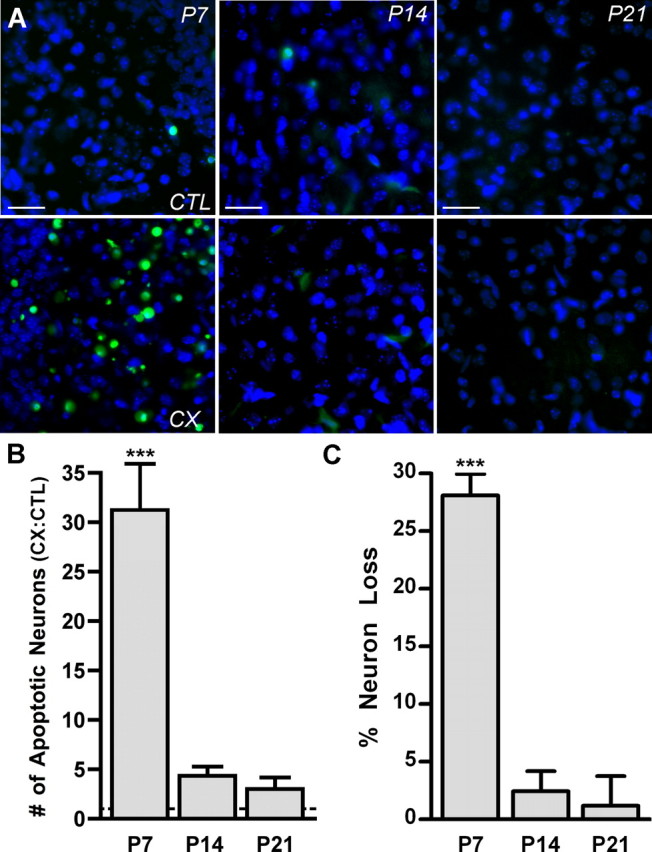
Apoptosis and neuron death result from deafferentation during a developmental critical period. A, Fluorescent photomicrographs of AVCN tissue labeled with ApopTag (green) and DAPI (blue) at P7, P14, and P21 after 24 h of CX. The AVCN contralateral to (CTL; top row) and ipsilateral to (bottom row) CX are shown at each age. ApopTag labeling is prevalent at P7 after CX but absent at P14 and P21. Scale bars, 75 μm. B, Comparison of the number of apoptotic AVCN neurons on the CX side versus the CTL side were made (CX:CTL) to demonstrate the effect of CX on neuron death in the AVCN. Cochlear nucleus neurons of P7 mice, but not P14 or P21 mice, showed significantly more apoptotic neurons on the CX versus the CTL side 24 h after surgery (31.26 ± 4.68; n = 6; t = 7.90; ***p < 0.001). Means were compared with 1.0. C, Percentage loss of AVCN neurons after 96 h of CX as assayed from neuron counts of cresyl violet-stained sections. At age P7, there is significant loss of AVCN neurons (28.08 ± 1.31; n = 4; t = 21.52; ***p < 0.001; means compared with 0.0) but no significant loss at P14 or P21.
Table 1.
Number of AVCN neurons after 96 h of CX or number of apoptotic AVCN neurons after 24 h of CX
| Age at surgery | No. of neurons per sampled area (mean ± SEM) |
% Neuron loss (± SEM) |
No. of apoptotic neurons per sampled area (mean ± SEM) |
|||
|---|---|---|---|---|---|---|
| CX | Control | Control | n | CX:Control | n | |
| P7 | 36.46 ± 1.86 | 50.00 ± 1.61 | 28.08 ± 1.31 | 4 | 31.26 ± 4.68 | 6 |
| P7 (+20 mg/kg 11R-VIVIT) | n/a | 10.80 ± 2.98 | 7 | |||
| P7 (+30 mg/kg 11R-VIVIT) | 49.60 ± 2.84 | 57.82 ± 3.73 | 11.08 ± 0.91 | 4 | 6.45 ± 0.40 | 5 |
| P7 (+FK506) | 51.50 ± 1.32 | 55.63 ± 1.84 | 8.00 ± 1.41 | 4 | 4.31 ± 0.96 | 4 |
| P14 | 44.85 ± 2.12 | 45.00 ± 2.16 | 2.66 ± 0.85 | 4 | 2.43 ± 0.59 | 4 |
| P21 | 43.09 ± 1.25 | 43.92 ± 2.26 | 1.52 ± 1.44 | 5 | 3.00 ± 1.17 | 5 |
Because calcium-mediated events, such as rapid increase of intracellular calcium concentration or CREB phosphorylation, have been shown to result from removal of afferent input to auditory neurons (Zirpel et al., 1995, 2000) and the activation of the transcription factor NFATc4 is mediated by calcium (Jain et al., 1993; Crabtree and Olson, 2002), we wanted to determine whether NFATc4 was activated in AVCN neurons after CX. NFATc4 is the predominant NFATc isoform in nervous tissue and is expressed in mouse brainstem nuclei (Bradley et al., 2005). NFATc4 translocates from the cytoplasm to the nucleus on dephosphorylation and activation by CaN after a Ca2+ signal that activates CaM (Ruff and Leach, 1995; Woischwill et al., 2005). Therefore, NFAT activation was measured by the change in the amount of nuclear NFATc4 after CX by fractionation of the cytoplasmic and nuclear lysates of cochlear nucleus tissue followed by Western blot analysis. At P7, P14, and P21, NFATc4 was localized to the cytoplasm (C) of intact cochlear nucleus tissue (CTL). However, 15 min after CX, NFATc4 was detected in the nuclear lysate (N) at P7, but not P14 or P21 (Fig. 2A). Western blots (as shown in Fig. 2A) with cochlear nucleus tissue that underwent CX for 15, 30, or 45 min were analyzed and demonstrated a time course of NFATc4 nuclear translocation after CX. The amount of NFATc4 in nuclear lysate increased rapidly at age P7 by 15 min after CX and returned to control levels by 30 and 45 min (Fig. 2B). Together, these results demonstrate that cochlear nucleus neurons of P7 mice are within a critical period of susceptibility to CX-induced death and that CX for as little as 15 min results in rapid and transient activation of NFATc4 in AVCN tissue as demonstrated by an increase in nuclear NFATc4.
Figure 2.

NFATc4 is activated in the cochlear nucleus after deafferentation during a developmental critical period. A, Data of Western blots with fractionated AVCN cellular lysates. NFATc4 is localized to the nuclear lysate (N) 15 min after CX at P7, but not P14 or P21, when it is predominantly localized to the cytoplasm (C) on both the CX and the CTL AVCN. CREB is shown as a control for efficient separation of nuclear and cytoplasmic lysates, because it is localized to the nucleus in AVCN neurons. B, Quantification of Western blots of cytoplasmic and nuclear lysates. Percentage increase of nuclear NFATc4 15 min (as shown in A), 30 min, and 45 min after CX. Significant increase of nuclear NFATc4 is seen only at P7 15 min after CX (26.6 ± 1.28%; n = 3; t = 16.08; ***p < 0.001; means compared with 0.0 with paired t test).
To confirm that NFATc4 is activated after CX and that it takes place specifically within the neurons of the AVCN, the subcellular localization of NFATc4 after CX was analyzed using immunohistochemistry (Fig. 3A–D). AVCN sections were labeled with NFATc4 (red) and DAPI for identification of nuclei (blue). For ages within (P7) and outside of (P14 and P21) the critical period, there was very little colocalization of DAPI and NFATc4 in the control AVCN neurons contralateral to CX, indicating that NFATc4 is mostly located in the cytoplasm in an inactive state under normal conditions (Fig. 3A–D, top). In AVCN ipsilateral to CX, NFATc4 was significantly increased in the nuclei 15 min after CX at P7 as shown by the large amount of colocalization of NFATc4 and DAPI (green) (Fig. 3A,B). Magnification of the boxed area in Figure 3A is shown so that the change in colocalization can easily be visualized (Fig. 3B). At ages outside of the critical period (P14 and P21), the amount of colocalization remained visually unchanged after CX, indicating that NFATc4 does not translocate to the nucleus after CX at ages outside of the critical period (Fig. 3C,D). Quantification of the increase in nuclear NFATc4 in AVCN neurons after CX for 15, 30, 45, or 60 min showed that the amount of NFATc4 in the nucleus at P7 (□) increased significantly after CX for 15 min. This level decreased with time but remained significant 30 min and 45 min after CX. The increase at P7 was absent by 60 min after CX. No increase of nuclear NFAT occurred for any of the time points at P14 (▲) or P21 (○) after CX (Fig. 3E; means compared with two-way ANOVA and Bonferroni's post hoc analysis). These results indicate that there is a dramatic shift in the localization of NFATc4 from the cytoplasm to the nucleus in AVCN neurons after CX during the critical period described here and that NFATc4 remains in the nucleus for up to 45 min after CX.
Figure 3.

NFATc4 is activated in AVCN neurons after CX during a critical period of development. A–D, Fluorescent photomicrographs of AVCN. CTL and 15 min CX, labeled with NFATc4 (red), DAPI nuclear counterstain (blue), and the colocalization of NFATc4 and DAPI (green). A, Marked colocalization of NFATc4 and DAPI is shown on the side ipsilateral to CX at P7. B, Magnification of the boxed area is shown in A. C (P14) and D (P21), Little to no colocalization in either the CTL or CX AVCN neurons after CX. E, Analysis of immunohistochemical images of mice deafferented for 15, 30, 45, or 60 min shows a time course for nuclear translocation of NFATc4. Nuclear NFATc4 significantly increases in the AVCN neurons at P7 (□) 15 min (23.62 ± 4.51%; n = 3; t = 16.14; ***p < 0.001), 30 min (6.71 ± 4.51%; n = 3; t = 4.58; **p < 0.01), and 45 min (4.78 ± 4.50%; n = 3; t = 3.27; *p < 0.05) after CX. P14 (▲) and P21 (○) AVCN show no significant increase of nuclear NFATc4 after CX for any of the time points. Scale bars: A, C, D, 75 μm; B, 25 μm. Data analyzed with two-way ANOVA.
NFAT-dependent apoptosis
The cell-permeable peptide 11R-VIVIT binds to the calcium-mediated phosphatase CaN and specifically blocks its ability to activate NFAT, whereas other substrates targeted by CaN remain unaffected (Aramburu et al., 1999; Noguchi et al., 2004). Additionally, FK506 is a less specific inhibitor of NFAT that works by inhibition of CaN (Liu et al., 1991; Steiner et al., 1992).
To determine whether treatment with 11R-VIVIT or FK506 blocks NFATc4 activation in the cochlear nuclei of P7 mice in vivo after 15 min of deafferentation, mice were pretreated with 30 mg/kg 11R-VIVIT (VIV), 1 mg/kg FK506, or saline as vehicle control (sal). NFATc4 activation was determined by calculating the percent increase of nuclear NFATc4 within the AVCN on the side ipsilateral to CX compared with the side contralateral to CX by Western blot analysis of fractionated lysates (Fig. 4A). Pretreatment with VIV or FK506 abolished the CX-induced increase of nuclear NFATc4. Pretreatment with saline did not differ from the untreated group, and both groups showed an increase of nuclear NFATc4 after CX (Fig. 4A). These data demonstrate the specificity of the deafferentation-induced activation of NFATc4 by showing that pretreatment with a specific inhibitor of NFAT (Fig. 4A, VIV) or an inhibitor of CaN (FK506) (Fig. 4A) abolishes deafferentation-induced activation of NFATc4.
Figure 4.

Activation of NFATc4 is a precursor to induction of apoptosis during the developmental critical period. A, Data of Western blot analysis of cellular fractions. Pretreatment with 30 mg/kg of the NFAT inhibitor peptide 11R-VIVIT (VIV) or CaN inhibitor FK506 attenuated the increase of nuclear NFATc4 in the cochlear nuclei of P7 mice 15 min after CX. Pretreatment with saline (sal) served as a vehicle control (23.19 ± 1.24%; t = 18.67; **p < 0.01) and did not differ from untreated (−) animals (26.60 ± 1.28; t = 20.74; **p < 0.01). Using paired t tests, means were compared with 0.0. B, Fluorescent photomicrographs of ApopTag-labeled AVCN neurons of P7 mice 24 h after CX. Images shown are AVCN on the side ipsilateral to CX. Pretreatment with 30 mg/kg (VIV 30) causes attenuated ApopTag labeling compared with untreated and saline-treated animals. Scale bar, 75 μm. C, Analysis of images (like those shown in B) comparing the number of apoptotic neurons on the side ipsilateral versus contralateral to CX. Pretreatment with VIV 30 (6.45 ± 0.40; n = 5; t = 6.131;***p < 0.001), VIV 20 (20 mg/kg; 10.80 ± 2.98; n = 7; t = 6.423; ***p < 0.001), or FK506 (4.31 ± 0.96; n = 4; t = 7.30; ***p < 0.001) significantly attenuated deafferentation-induced ApopTag labeling in P7 AVCN neurons 24 h after CX. Saline group does not significantly differ from untreated group. One-way ANOVA was used to compare means with untreated group.
Recently, the activation of NFATc4 has been linked to the induction of apoptotic pathways in the CNS after drug administration and ischemia (Jayanthi et al., 2005; Zhang et al., 2005). Because we have shown that CX induces NFATc4 activation in AVCN neurons at ages that are susceptible to CX-induced apoptosis, we asked whether or not CX-induced apoptosis is dependent on the activation of NFAT. Images of P7 CX AVCN neurons showed plentiful labeling with ApopTag in both untreated and saline-treated animals (Fig. 4B). In contrast, ApopTag labeling was significantly reduced in animals pretreated with 30 mg/kg 11R-VIVIT (Fig. 4B, VIV 30). Ratios of the number of ApopTag-labeled AVCN neurons on the deafferented side versus the intact side (CX/CTL) were significantly attenuated in mice pretreated with 20 mg/kg 11R-VIVIT (VIV 20), 30 mg/kg 11R-VIVIT (VIV 30), or FK506 (Table 1, Fig. 4C). Means were also compared with 1.0 with a paired t test to determine whether the CX-induced ApopTag labeling was statistically significant; values are ratios of CX/CTL, and 1.0 represents no change after CX. The results indicate that treatment with 30 mg/kg 11R-VIVIT, FK506, or 20 mg/kg 11R-VIVIT did not completely eliminate an increase of ApopTag labeling after CX. The data in Figure 4 suggest that NFAT plays a role in the CX-induced apoptosis of AVCN neurons in mice at P7, an age that lies within the critical period.
To verify that apoptosis of AVCN neurons after CX eventually leads to the death and loss of AVCN neurons, neuron number was examined and compared between the AVCN ipsilateral to surgery (CX) and contralateral to surgery (CTL) in P7 mice. After 96 h of CX, a time when neuron loss in the AVCN reaches a maximum in P7 mice (Mostafapour et al., 2000), images of cresyl violet-stained AVCN sections were acquired. The AVCN of P7 mice appeared to have more neurons on the side contralateral to CX (Fig. 5A) versus ipsilateral to CX (Fig. 5B). Mice pretreated with 11R-VIVIT showed less neuron loss between the AVCN contralateral to CX (Fig. 5C) versus ipsilateral to CX (Fig. 5D). Analysis of neuron number showed that pretreatment with the NFAT inhibitor 11R-VIVIT or FK506 significantly attenuates the percentage loss of AVCN neurons (Table 1, Fig. 5E). The means in Figure 5E were also compared with 0.0 using a paired t test to determine whether CX-induced neuron death was statically abolished after treatment with NFAT inhibitor. Pretreatment with 11R-VIVIT or FK506 resulted in a percentage loss of AVCN neurons that is significantly different from 0.0. The results in Figure 5 demonstrate that, at age P7, significant loss of AVCN neurons results from 96 h of CX and that the CX-induced death of AVCN neurons is partly mediated by NFAT.
Figure 5.

Neuron loss in the AVCN is partly dependent on activation of NFAT. A–E, Cresyl violet-stained sections of AVCN at age P7 show that neuron number is greater in the intact AVCN (A) compared with deafferented AVCN that has undergone 96 h of CX (B). C, D, Pretreatment with 30 mg/kg 11R-VIVIT (VIV) results in no visible difference in neuron number between the intact AVCN (C) and the 96 h of CX AVCN (D). The boxed region in the top half of A–D is enlarged and shown in the bottom half of each panel. Scale bar: A, top, 140 μm; A, bottom, 35 μm. E, Analysis of images like those shown in A–D. The percentage neuron loss at age P7 is significantly attenuated compared with the untreated group (−) with VIV pretreatment (30 mg/kg, 11.08 ± 0.91; t = 9.42; ***p < 0.001) or FK506 (8.00 ± 1.41; t = 11.12; ***p < 0.001; means compared with − group with one-way ANOVA).
NFAT regulation of FasL
CaN-dependent activation of NFAT has been shown to result in upregulation of the death receptor ligand FasL (Holtz-Heppelmann et al., 1998; Jayanthi et al., 2005). FasL binds to its receptor Fas and triggers the Fas/FasL apoptotic death cascade (Suda and Nagata, 1994) that results in cleavage of DNA, which is detected by ApopTag–TUNEL. To determine whether CX-induced activation of NFATc4 and subsequent apoptosis of AVCN neurons is linked to the Fas/FasL death pathway, we examined the expression of FasL after CX at P7 when the cochlear nucleus neurons are susceptible to deafferentation-induced apoptosis. After CX for 1 h, the fold change of FasL mRNA expression in CX cochlear nuclei versus intact cochlear nuclei at P7 was measured with semiquantitative RT-PCR. In untreated mice, FasL mRNA expression was significantly increased after CX in both untreated and saline-treated animals. Pretreatment with 11R-VIVIT or FK506 eliminated the CX-induced increase of FasL mRNA expression (Fig. 6A). Figure 6B shows the fold change in expression of Fas and FasL after deafferentation, demonstrating that the effect of CX on FasL gene expression is specific to FasL and not gene expression in general. In addition to using GAPDH as a housekeeping gene, two additional commonly used housekeeping genes (S15 and RPL3) were used to confirm the validity of GAPDH as a control and eliminate the possibility of an artifact-induced effect on FasL expression. With use of the alternative housekeeping genes, FasL had a significant increase in its mean fold change of expression. Fas had no significant mean fold change after deafferentation. Similarly, FasL protein expression was measured after 6 h of CX at P7 with Western blot analysis as shown in Figure 6C. Lanes 1–4 are of the nondeafferented cochlear nucleus tissue lysate from four separate animals, and lanes 5–8 contain the corresponding cochlear nucleus tissue lysate that underwent 6 h of CX (Fig. 6C, lanes 1 and 5 are from the same animal, as well as 2,6, 3,7, and 4,8). The analyzed Western blot data are ratios of the level of FasL protein in CX/CTL and reflect a significant change in FasL protein expression after CX in both untreated and saline-treated animals (Fig. 6D). Blockade of NFAT activation by pretreatment with 30 mg/kg 11R-VIVIT abolished the CX-induced increase of FasL protein in vivo (Fig. 6D).
Figure 6.

Expression of FasL follows activation of NFATc4 and may play a role in deafferentation-induced NFAT-dependent apoptosis during a critical period of development. A, Data were obtained by semiquantitative RT-PCR. At P7, FasL mRNA expression is significantly increased 1 h after CX in normal (1.85 ± 0.22; n = 4; t = 3.81; *p < 0.05) and saline-treated (sal) (1.73 ± 0.15; n = 3; t = 4.93, *p < 0.05) animals. Pretreatment with 11R-VIVIT (VIV) (30 mg/kg) or FK506 attenuated the increase of FasL mRNA after CX. Means compared with 1.0 with paired t tests. B, Semiquantitative RT-PCR for both Fas and FasL demonstrating the specificity of CX-induced effects on gene transcription. The housekeeping genes S15 and RPL3 were used in addition to GAPDH to ensure that CX is not affecting the loading control. Fas had no significant mean fold change of expression. The mean fold change of FasL expression remained significant regardless of the housekeeping gene (GAPDH, 1.85 ± 0.22, t = 3.81, *p < 0.05; S15, 1.64 ± 0.12, t = 5.46, *p < 0.05; RPL3, 2.48 ± 0.22, t = 6.71, *p < 0.05). Means compared with 1.0 with paired t tests. C, FasL protein expression was measured after 6 h of CX at P7 with Western blot analysis. Western blot of nondeafferented cochlear nucleus tissue (lanes 1–4) and 6 h of CX cochlear nucleus tissue (lanes 5–8) are shown and demonstrate increased FasL protein in the deafferented samples. D, Analysis of Western blots of FasL protein show increased levels of FasL protein in untreated (fold change, 1.20 ± 0.07; n = 7; t = 11.0; ***p < 0.0001) and saline-treated (t = 4.9; *p < 0.05) animals after 6 h of CX. Pretreatment with VIV attenuated the increase in FasL protein. With paired t tests means were compared with 1.0, which represents no change of expression.
Together, these data indicate that NFAT-mediated transcription and translation of FasL result after CX. Furthermore, this suggests that the Fas/FasL death pathway may play a role in NFAT-mediated apoptosis of CX AVCN neurons during the critical period when they are vulnerable to CX-induced death.
Discussion
The susceptibility of AVCN neurons to deafferentation-induced death occurs during a short span of time during development, referred to here and in former works as a critical period (Tierney et al., 1997; Mostafapour et al., 2000). Consistent with that reported by Mostafapour et al. (2000), we have shown that mice at age P7 are within this critical period, but, by age P14 and certainly P21, the critical period is over and the survival of the neurons is no longer dependent on afferent input from the eighth cranial nerve. It is not known what differentiates AVCN neurons during the critical period from those outside of the critical period. The transcription factor NFATc4 is expressed in AVCN neurons both during and after the critical period. However, its activation exclusively occurs after deafferentation during the critical period (P7) and not after (P14 and P21). We show that the activation/nuclear translocation of NFATc4 after deafferentation of AVCN neurons is rapid (by 15 min) and lasts for up to 45 min by immunohistochemical analysis. However, our Western blots of fractionated lysates show that NFATc4 remains in the nucleus for <30 min. This is likely attributable to reduced detection of NFATc4 in the AVCN used for Western blots because of the inclusion of additional cell types (i.e., glial cells, which do not express NFATc4), whereas only the AVCN neurons were considered for immunohistochemical analysis.
During the critical period of deafferentation-induced death of AVCN neurons, activation of NFATc4 coincides with susceptibility to apoptosis as well as loss of neurons. Furthermore, inhibition of NFAT indicates that apoptosis and death of AVCN neurons during the critical period is significantly, although not entirely, dependent on the activation of NFAT. Even so, these data indicate that the mechanism through which NFATc4 is activated during the critical period may characterize, at least in part, what defines their susceptibility to deafferentation-induced death. Because activation of NFATc4 is a calcium-mediated event, there may be differences in calcium signaling or buffering after deafferentation during the critical period versus outside of the critical period.
NFAT proteins are specialized for sensing and responding to dynamic changes in intracellular calcium concentration because of their ability to rapidly translocate to and from the nucleus via the opposing activities of CaN and kinases (Loh et al., 1996). As soon as 1 min after either a high but short or low but sustained increase in intracellular calcium concentration, NFAT can become activated and remain activated for several minutes after stimulation (Dolmetsch et al., 1997). Therefore, the nature of the deafferentation-induced calcium signal remains to be determined in terms of duration, magnitude, and origin. The time course of NFATc4 activation necessary for NFAT-mediated transcription in neurons observed here is similar to a previous result reporting stimulation-induced (depolarization with 90 mm K+) nuclear translocation of NFATc4 in hippocampal neurons by 15 min with a duration of at least 60 min (but <120 min) and subsequent NFATc4-dependent gene transcription (Graef et al., 1999). This is similar to our results in that we also show rapid activation of NFATc4 by 15 min after deafferentation. The duration of NFATc4 activation in our system is shorter (lasting 45 min rather than 60–120 min), but our observations were made in a unique population of cells, in vivo, and under a different stimulus paradigm. Although it may seem paradoxical that NFATc4 activation in hippocampal neurons is stimulus dependent and the activation of NFATc4 in AVCN neurons occurs after deafferentation, one must remember that, in addition to source, important parameters dictating the effect of a calcium signal include duration, magnitude, and physical location (Franks and Sejnowski, 2002), all of which may be similar or variable in the two experimental systems but obviously combine so as to result in a CaN/NFATc4 activating signal. In vivo deactivation of NFATc4 in AVCN neurons may be a more rapid process than that observed in cultured hippocampal neurons. Inhibition of NFAT resulted in inhibition of FasL transcript and protein expression, demonstrating that activation of NFATc4 for 45 min is sufficient to mediate the expression of FasL in AVCN neurons.
Genes transcribed by activated NFATc4 may lead to the eventual death of a subpopulation of deafferented AVCN neurons. Considering that FasL expression has been shown to be dependent on NFAT activation (Holtz-Heppelmann et al., 1998; Jayanthi et al., 2005) and that FasL results in activation of an apoptotic pathway (Suda and Nagata, 1994; Raoul et al., 1999; Raoul et al., 2002), FasL is a good candidate for involvement in the mechanism of deafferentation-induced AVCN neuronal death. Although it is interesting to note that cell death in deafferented avian cochlear nucleus does not depend on changes in gene transcription (Garden et al., 1995), it is difficult to make comparisons with the mechanisms of deafferentation-induced death in AVCN attributable to the incomplete understanding of the specific mechanisms underlying the death in either system. The presence and/or expression levels of NFATc4, Fas, and FasL are unknown in chick cochlear nucleus under normal conditions as well as after deafferentation. Additional studies are required to make a sound comparison between mechanisms of deafferentation-induced neuronal death in avian and murine cochlear nuclei.
FasL is a good candidate for involvement in the mechanism of deafferentation-induced AVCN neuronal death. Here, NFAT regulates the expression of FasL in AVCN neurons as indicated by the abolition of increased FasL expression by specific inhibition of NFAT activation. Additionally, apoptosis of deafferented AVCN neurons is shown to be partly dependent on activation of NFAT as suggested by the significant attenuation of apoptosis after inhibition of NFAT activation.
One mechanism by which upregulation of FasL can result in deafferentation-induced death of AVCN neurons is by inserting into the cell membrane and binding to its receptor Fas, which is expressed on the membranes of adjacent neurons, subsequently triggering apoptosis in the Fas-expressing neurons. This is a potential mechanism, because Fas is endogenously expressed in mammalian cochlear nucleus tissue (Guan et al., 2005), although its expression pattern in the cochlear nuclei of mice has not yet been characterized in detail. The level of expression of FasL may be the limiting factor that regulates the induction of apoptosis in AVCN neurons, which depends on activation of NFAT. Note that we hypothesize that the AVCN neurons with activated NFATc4 after deafferentation are not the neurons that die but the neurons that induce death in neighboring neurons that express Fas.
Previous research on the proto-oncogene bcl-2 supports our finding that NFAT-mediated expression of FasL after deafferentation is a likely contributor to the deafferentation-induced apoptosis of AVCN neurons because this hypothesis fits logically into previous studies that examine Bcl-2 not only in the context of the Fas/FasL death pathway but also in the context of activity-dependent survival of neurons in the auditory system. Bcl-2 is an antiapoptotic protein that significantly inhibits Fas/FasL-mediated cell death (Itoh et al., 1993). In addition, bcl-2 was also shown to inhibit apoptosis induced by NFAT-mediated expression of FasL (Srivastava et al., 1999). Interestingly, overexpression of bcl-2 eliminated neuron death of deafferented AVCN during the critical period (Mostafapour et al., 2002). This implies that bcl-2 may be inhibiting the upregulation of FasL expression that follows deafferentation of AVCN neurons that are beyond the critical period, ultimately preventing their death by suppressing the Fas/FasL apoptotic pathway. Similarly, Harris et al. (2005) found, using microarray analysis, that AVCN from mice within the critical period express higher levels of transcript for proapoptotic genes than AVCN from mice beyond the critical period and, conversely, AVCN from mice beyond the critical period express higher levels of transcript for prosurvival genes than AVCN from mice within the critical period. This indicates a developmental “switch” in AVCN from a complement of cellular mechanisms favoring induction of an apoptotic pathway to a complement of mechanisms that prevent or eliminate the apoptotic pathways. Our data are consistent with this and suggest that NFAT may be a molecular mechanism underlying, in part, this developmental switch.
Figure 7 is a schematic model of our hypothesis for CX-induced apoptosis of cochlear nucleus neurons during the developmental critical period for activity-dependent survival. This hypothesis is consistent with the data presented in the current study as well as in previous reports. CX leads to an increase in intracellular calcium concentration that binds to and activates CaM. Calcium/CaM then binds to CaN, leading to rapid dephosphorylation, activation, and nuclear translocation of NFATc4 as shown by this study (Figs. 2–4). Activated/nuclear NFAT then induces expression of the death receptor ligand FasL (Fig. 6), which contributes to the death of deafferented cochlear nucleus neurons by triggering the apoptotic death cascade (Figs. 1, 4, 5) in neurons that express, either endogenously or deafferentation-induced, the death receptor Fas, perhaps only during the critical period. This is shown as a calcium-mediated process because inhibition of NFAT activation specifically via the calcium-mediated pathway eliminates increased FasL expression and attenuates neuronal death in vivo. Also, upregulation of bcl-2 is able to prevent CX-induced death of AVCN neurons, and we hypothesize that this is attributable to its ability to inhibit Fas/FasL-mediated apoptosis (Itoh et al., 1993) and, more specifically, its ability to inhibit NFAT-mediated death through FasL (Srivastava et al., 1999). The events shown in Figure 7 that occur after deafferentation and up to the point of FasL expression have been shown in the current study or previously; the events beyond this that show the presence of Fas receptors on neighboring neurons and their activation-dependent apoptosis need to be further studied, as does the hypothesized role that Bcl-2 plays in this mechanism.
Figure 7.

A proposed model for CX-induced apoptosis of cochlear nucleus neurons during a developmental critical period. After CX, NFATc4 is activated by Ca/CaM/CaN and translocates to the nucleus in which it mediates transcription and translation of the death receptor ligand FasL. We hypothesize that upregulation of FasL expression results in apoptosis of adjacent neurons that express the death receptor Fas. Bcl-2 may play a prosurvival role in this mechanism by inhibiting initiation of apoptosis via Fas/FasL.
Classically, the role of NFAT is described in the context of lymphocyte function and its role in the immune system. Alternatively, this study provides an example of a role for NFAT in the plasticity of the nervous system, a role that is becoming more common (Groth and Mermelstein, 2003; Hernandez-Ochoa et al., 2007). In summary, we have shown that NFATc4 is activated after deafferentation and that this event leads to increased FasL expression and apoptosis of cochlear nucleus neurons. These studies present convincing evidence for the involvement of NFAT in the deafferentation-induced apoptosis of deafferented AVCN neurons, but the specific cellular pathways remain to be elucidated. Regardless, this research provides a novel mechanism through which deafferentation-induced death of AVCN neurons and possibly other types of sensory neurons may occur.
Footnotes
This work was supported by the National Institute on Deafness and Other Communication Disorders Grant DC05012 and The Minnesota Medical Foundation. We thank Dr. Paul Mermelstein for scientific discussion and insightful comments regarding this manuscript, Dr. Thomas Parks for critical reading of this manuscript, Dr. Xiao-Qing Tang for assistance with cochlea removal surgeries, Brittani Conway for help with immunohistochemical processing and analysis, and Alexis Gerber for assistance with neuronal counting.
References
- Aramburu et al., 1999.Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- Born and Rubel, 1985.Born DE, Rubel EW. Afferent influences on brain stem auditory nuclei of the chicken: neuron number and size following cochlea removal. J Comp Neurol. 1985;231:435–445. doi: 10.1002/cne.902310403. [DOI] [PubMed] [Google Scholar]
- Bradley et al., 2005.Bradley KC, Groth RD, Mermelstein PG. Immunolocalization of NFATc4 in the adult mouse brain. J Neurosci Res. 2005;82:762–770. doi: 10.1002/jnr.20695. [DOI] [PubMed] [Google Scholar]
- Catsicas et al., 1992.Catsicas M, Pequignot Y, Clarke PG. Rapid onset of neuronal death induced by blockade of either axoplasmic transport or action potentials in afferent fibers during brain development. J Neurosci. 1992;12:4642–4650. doi: 10.1523/JNEUROSCI.12-12-04642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen et al., 1999.Chen RW, Saunders PA, Wei H, Li Z, Seth P, Chuang DM. Involvement of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and p53 in neuronal apoptosis: evidence that GAPDH is upregulated by p53. J Neurosci. 1999;19:9654–9662. doi: 10.1523/JNEUROSCI.19-21-09654.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree and Olson, 2002.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Dolmetsch et al., 1997.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- Franks and Sejnowski, 2002.Franks KM, Sejnowski TJ. Complexity of calcium signaling in synaptic spines. BioEssays. 2002;24:1130–1144. doi: 10.1002/bies.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden et al., 1995.Garden GA, Redeker-DeWulf V, Rubel EW. Afferent influences on brainstem auditory nuclei of the chicken: regulation of transcriptional activity following cochlea removal. J Comp Neurol. 1995;359:412–423. doi: 10.1002/cne.903590305. [DOI] [PubMed] [Google Scholar]
- Graef et al., 1999.Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Groth and Mermelstein, 2003.Groth RD, Mermelstein PG. Brain-derived neurotrophic factor activation of NFAT (nuclear factor of activated T-cells)-dependent transcription: a role for the transcription factor NFATc4 in neurotrophin-mediated gene expression. J Neurosci. 2003;23:8125–8134. doi: 10.1523/JNEUROSCI.23-22-08125.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan et al., 2005.Guan YG, Wang TH, Ni W, Li L, Lu YC, Gao ZY. Distribution of Fas and FasL in the central nervous system of adult rhesus (in Chinese) Sichuan Da Xue Xue Bao Yi Xue Ban. 2005;36:322–324. [PubMed] [Google Scholar]
- Gupta et al., 1985.Gupta RC, Khandelwal RL, Sulakhe PV. Isolation and characterization of calcineurin from bovine brain. Can J Physiol Pharmacol. 1985;63:1000–1006. doi: 10.1139/y85-165. [DOI] [PubMed] [Google Scholar]
- Harris et al., 2005.Harris JA, Hardie NA, Bermingham-McDonogh O, Rubel EW. Gene expression differences over a critical period of afferent-dependent neuron survival in the mouse auditory brainstem. J Comp Neurol. 2005;493:460–474. doi: 10.1002/cne.20776. [DOI] [PubMed] [Google Scholar]
- Hashisaki and Rubel, 1989.Hashisaki GT, Rubel EW. Effects of unilateral cochlea removal on anteroventral cochlear nucleus neurons in developing gerbils. J Comp Neurol. 1989;283:5–73. doi: 10.1002/cne.902830402. [DOI] [PubMed] [Google Scholar]
- Hernandez-Ochoa et al., 2007.Hernandez-Ochoa EO, Contreras M, Cseresnyes Z, Schneider MF. Ca2+ signal summation and NFATc1 nuclear translocation in sympathetic ganglion neurons during repetitive action potentials. Cell Calcium. 2007;41:559–571. doi: 10.1016/j.ceca.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho et al., 1995.Ho SN, Thomas DJ, Timmerman LA, Li X, Francke U, Crabtree GR. NFATc3, a lymphoid-specific NFATc family member that is calcium-regulated and exhibits distinct DNA binding specificity. J Biol Chem. 1995;270:19898–19907. doi: 10.1074/jbc.270.34.19898. [DOI] [PubMed] [Google Scholar]
- Holtz-Heppelmann et al., 1998.Holtz-Heppelmann CJ, Algeciras A, Badley AD, Paya CV. Transcriptional regulation of the human FasL promoter-enhancer region. J Biol Chem. 1998;273:4416–4423. doi: 10.1074/jbc.273.8.4416. [DOI] [PubMed] [Google Scholar]
- Itoh et al., 1991.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66:233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- Itoh et al., 1993.Itoh N, Tsujimoto Y, Nagata S. Effect of bcl-2 on Fas antigen-mediated cell death. J Immunol. 1993;151:621–627. [PubMed] [Google Scholar]
- Jain et al., 1993.Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T, Rao A. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature. 1993;365:352–355. doi: 10.1038/365352a0. [DOI] [PubMed] [Google Scholar]
- Jayanthi et al., 2005.Jayanthi S, Deng X, Ladenheim B, McCoy MT, Cluster A, Cai NS, Cadet JL. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc Natl Acad Sci USA. 2005;102:868–873. doi: 10.1073/pnas.0404990102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee et al., 1979.Klee CB, Crouch TH, Krinks MH. Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc Natl Acad Sci USA. 1979;76:6270–6273. doi: 10.1073/pnas.76.12.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo et al., 2003.Kondo E, Harashima A, Takabatake T, Takahashi H, Matsuo Y, Yoshino T, Orita K, Akagi T. NF-ATc2 induces apoptosis in Burkitt's lymphoma cells through signaling via the B cell antigen receptor. Eur J Immunol. 2003;33:1–11. doi: 10.1002/immu.200390000. [DOI] [PubMed] [Google Scholar]
- Latinis et al., 1997.Latinis KM, Carr LL, Peterson EJ, Norian LA, Eliason SL, Koretzky GA. Regulation of CD95 (Fas) ligand expression by TCR-mediated signaling events. J Immunol. 1997;158:4602–4611. [PubMed] [Google Scholar]
- Lipton, 1986.Lipton SA. Blockade of electrical activity promotes the death of mammalian retinal ganglion cells in culture. Proc Natl Acad Sci USA. 1986;83:9774–9778. doi: 10.1073/pnas.83.24.9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu et al., 1991.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Livak and Schmittgen, 2001.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loh et al., 1996.Loh C, Shaw KT, Carew J, Viola JP, Luo C, Perrino BA, Rao A. Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J Biol Chem. 1996;271:10884–10891. doi: 10.1074/jbc.271.18.10884. [DOI] [PubMed] [Google Scholar]
- Lu et al., 2007.Lu Y, Harris JA, Rubel EW. Development of spontaneous miniature EPSCs in mouse AVCN neurons during a critical period of afferent-dependent neuron survival. J Neurophysiol. 2007;97:635–646. doi: 10.1152/jn.00915.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo et al., 1996.Luo C, Burgeon E, Carew JA, McCaffrey PG, Badalian TM, Lane WS, Hogan PG, Rao A. Recombinant NFAT1 (NFATp) is regulated by calcineurin in T cells and mediates transcription of several cytokine genes. Mol Cell Biol. 1996;16:3955–3966. doi: 10.1128/mcb.16.7.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin et al., 1998.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafapour et al., 2000.Mostafapour SP, Cochran SL, Del Puerto NM, Rubel EW. Patterns of cell death in mouse anteroventral cochlear nucleus neurons after unilateral cochlea removal. J Comp Neurol. 2000;426:561–571. doi: 10.1002/1096-9861(20001030)426:4<561::aid-cne5>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Mostafapour et al., 2002.Mostafapour SP, Del Puerto NM, Rubel EW. bcl-2 overexpression eliminates deprivation-induced cell death of brainstem auditory neurons. J Neurosci. 2002;22:4670–4674. doi: 10.1523/JNEUROSCI.22-11-04670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi et al., 2004.Noguchi H, Matsushita M, Okitsu T, Moriwaki A, Tomizawa K, Kang S, Li ST, Kobayashi N, Matsumoto S, Tanaka K, Tanaka N, Matsui H. A new cell-permeable peptide allows successful allogeneic islet transplantation in mice. Nat Med. 2004;10:305–309. doi: 10.1038/nm994. [DOI] [PubMed] [Google Scholar]
- Northrop et al., 1994.Northrop JP, Ho SN, Chen L, Thomas DJ, Timmerman LA, Nolan GP, Admon A, Crabtree GR. NF-AT components define a family of transcription factors targeted in T-cell activation. Nature. 1994;369:497–502. doi: 10.1038/369497a0. [DOI] [PubMed] [Google Scholar]
- Oertel and Young, 2004.Oertel D, Young ED. What's a cerebellar circuit doing in the auditory system? Trends Neurosci. 2004;27:104–110. doi: 10.1016/j.tins.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Parks, 1999.Parks TN. Cochlear influences on development of the brainstem auditory system. In: Hyson RL, Johnson F, editors. The biology of early influences. New York: Academic; 1999. pp. 15–34. [Google Scholar]
- Paxinos and Franklin, 2001.Paxinos G, Franklin KBJ. Ed 2. New York: Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Rao et al., 1997.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Raoul et al., 1999.Raoul C, Henderson CE, Pettmann B. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J Cell Biol. 1999;147:1049–1062. doi: 10.1083/jcb.147.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul et al., 2002.Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–1083. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Rubel et al., 2004.Rubel EW, Parks TN, Zirpel L. Assembling, connecting and maintaining the cochlear nucleus. In: Parks TN, Rubel EW, Fay RR, Popper A, editors. Plasticity of the auditory system. New York: Springer; 2004. pp. 8–48. Springer handbook of auditory research, Vol XXIII. [Google Scholar]
- Ruff and Leach, 1995.Ruff VA, Leach KL. Direct demonstration of NFATp dephosphorylation and nuclear localization in activated HT-2 cells using a specific NFATp polyclonal antibody. J Biol Chem. 1995;270:22602–22607. doi: 10.1074/jbc.270.38.22602. [DOI] [PubMed] [Google Scholar]
- Saunders et al., 1997.Saunders PA, Chalecka-Franaszek E, Chuang DM. Subcellular distribution of glyceraldehyde-3-phosphate dehydrogenase in cerebellar granule cells undergoing cytosine arabinoside-induced apoptosis. J Neurochem. 1997;69:1820–1828. doi: 10.1046/j.1471-4159.1997.69051820.x. [DOI] [PubMed] [Google Scholar]
- Simmons et al., 1981.Simmons PA, Rafols JA, Getchell TV. Ultrastructural changes in olfactory receptor neurons following olfactory nerve section. J Comp Neurol. 1981;197:237–257. doi: 10.1002/cne.901970206. [DOI] [PubMed] [Google Scholar]
- Srivastava et al., 1999.Srivastava RK, Sasaki CY, Hardwick JM, Longo DL. Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J Exp Med. 1999;190:253–265. doi: 10.1084/jem.190.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner et al., 1992.Steiner JP, Dawson TM, Fotuhi M, Glatt CE, Snowman AM, Cohen N, Snyder SH. High brain densities of the immunophilin FKBP colocalized with calcineurin. Nature. 1992;358:584–587. doi: 10.1038/358584a0. [DOI] [PubMed] [Google Scholar]
- Suda and Nagata, 1994.Suda T, Nagata S. Purification and characterization of the Fas-ligand that induces apoptosis. J Exp Med. 1994;179:873–879. doi: 10.1084/jem.179.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha and Stryker, 2005.Taha SA, Stryker MP. Molecular substrates of plasticity in the developing visual cortex. Prog Brain Res. 2005;147:103–114. doi: 10.1016/S0079-6123(04)47008-3. [DOI] [PubMed] [Google Scholar]
- Tierney et al., 1997.Tierney TS, Russell FA, Moore DR. Susceptibility of developing cochlear nucleus neurons to deafferentation-induced death abruptly ends just before the onset of hearing. J Comp Neurol. 1997;378:295–306. doi: 10.1002/(sici)1096-9861(19970210)378:2<295::aid-cne11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Trune, 1982.Trune DR. Influence of neonatal cochlear removal on the development of mouse cochlear nucleus. II. Dendritic morphometry of its neurons. J Comp Neurol. 1982;209:425–434. doi: 10.1002/cne.902090411. [DOI] [PubMed] [Google Scholar]
- Woischwill et al., 2005.Woischwill C, Karczewski P, Bartsch H, Luther HP, Kott M, Haase H, Morano I. Regulation of the human atrial myosin light chain 1 promoter by Ca2+-calmodulin-dependent signaling pathways. FASEB J. 2005;19:503–511. doi: 10.1096/fj.04-2201com. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2005.Zhang YJ, Mei HS, Wang C, Wang YL, Zhang YJ. Involvement of nuclear factor of activated T-cells (NFATc) in calcineurin-mediated ischemic brain damage in vivo. Yao Xue Xue Bao. 2005;40:299–305. [PubMed] [Google Scholar]
- Zirpel et al., 1995.Zirpel L, Lachica EA, Lippe WR. Deafferentation increases the intracellular calcium of cochlear nucleus neurons in the embryonic chick. J Neurophysiol. 1995;74:1355–1357. doi: 10.1152/jn.1995.74.3.1355. [DOI] [PubMed] [Google Scholar]
- Zirpel et al., 1998.Zirpel L, Lippe WR, Rubel EW. Activity-dependent regulation of [Ca2+]i in avian cochlear nucleus neurons: roles of protein kinases A and C and relation to cell death. J Neurophysiol. 1998;79:2288–2302. doi: 10.1152/jn.1998.79.5.2288. [DOI] [PubMed] [Google Scholar]
- Zirpel et al., 2000.Zirpel L, Janowiak MA, Veltri CA, Parks TN. AMPA receptor-mediated, calcium-dependent CREB phosphorylation in a subpopulation of auditory neurons surviving activity deprivation. J Neurosci. 2000;20:6267–6275. doi: 10.1523/JNEUROSCI.20-16-06267.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]