

Abstract
In mammals, the thalamus plays important roles for cortical processing, such as relay of sensory information and induction of rhythmical firing during sleep. In neurons of the avian cerebrum, in analogy with cortical up and down states, complex patterns of regular-spiking and dense-bursting modes are frequently observed during sleep. However, the roles of thalamic inputs for shaping these firing modes are largely unknown. A suspected key player is the avian thalamic nucleus uvaeformis (Uva). Uva is innervated by polysensory input, receives indirect cerebral feedback via the midbrain, and projects to the cerebrum via two distinct pathways. Using pharmacological manipulation, electrical stimulation, and extracellular recordings of Uva projection neurons, we study the involvement of Uva in zebra finches for the generation of spontaneous activity and auditory responses in premotor area HVC (used as a proper name) and the downstream robust nucleus of the arcopallium (RA). In awake and sleeping birds, we find that single Uva spikes suppress and spike bursts enhance spontaneous and auditory-evoked bursts in HVC and RA neurons. Strong burst suppression is mediated mainly via tonically firing HVC-projecting Uva neurons, whereas a fast burst drive is mediated indirectly via Uva neurons projecting to the nucleus interface of the nidopallium. Our results reveal that cerebral sleep-burst epochs and arousal-related burst suppression are both shaped by sophisticated polysynaptic thalamic mechanisms.
Keywords: sleep, modulation, arousal, glutamate, lidocaine, GABA, antidromic, orthodromic, acetylcholine, relay, reticular, lemniscus
Introduction
In mammals, the thalamus is a central relay of ascending sensory information and of corticocortical information (McCormick and Feeser, 1990; Guillery and Sherman, 2002). Mammalian thalamic neurons act in a “relay” mode of high regular spike rates in awake subjects and in a “burst” mode in sleeping subjects (Glenn and Steriade, 1982; Weyand et al., 2001; Swadlow et al., 2002); their afferents are divided into drivers (which for higher-order thalamic relays are of cortical origin) and modulators (with origin mostly in the brainstem and the reticular nucleus). Currently, almost nothing is known about the physiology of thalamic projection neurons in the avian brain.
In the songbird thalamus, there are several cerebrum-projecting nuclei involved in sensory processing and song control. For example, auditory input from the brainstem is relayed to the cerebrum mainly via the thalamic nucleus ovoidalis (Vates et al., 1996). Auditory input is also relayed via the thalamic uvaeform nucleus (Uva), although Uva appears not to have a critical function in eliciting auditory responses in its target areas, which are HVC and the nucleus interface of the nidopallium (NIf) (Fig. 1a) (Nottebohm et al., 1982; Coleman et al., 2007). Uva also receives somatosensory and visual inputs (Wild, 1994). As in mammalian thalamic relays, Uva receives information about ongoing motor instructions via respiratory and brainstem nuclei (Reinke and Wild, 1998) and is innervated by cholinergic fibers (Akutagawa and Konishi, 2005). One of Uva's behavior-related functions is to control the temporal patterning of learned songs (Williams and Vicario, 1993; Coleman and Vu, 2005).
Figure 1.
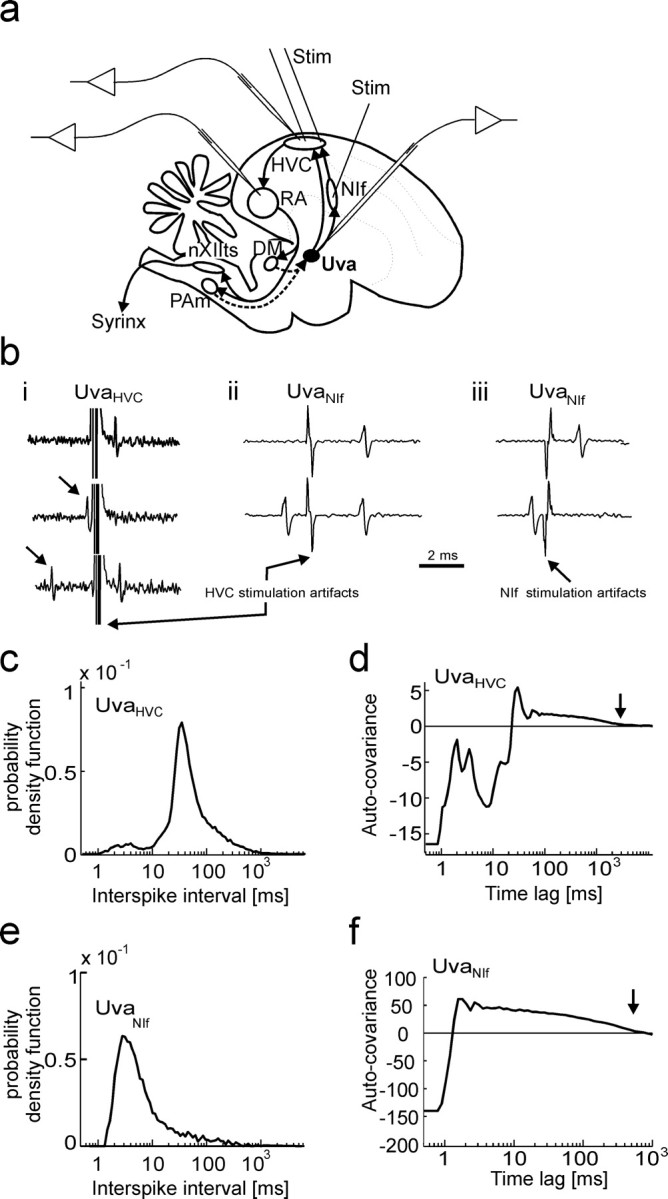
Identification of Uva neurons using antidromic stimulation in HVC. a, Schematic drawing of a song control pathway, showing the main premotor and motor areas and the experimental design. Uva projects to NIf and to HVC. HVC projects to RA, which in turn innervates neurons in DM in the midbrain and respiratory neurons in PAm in the medulla. These subpallial structures both project back to Uva, thus closing a respiratory–motor feedback loop (dashed arrows). We identified Uva neuron type by electrical stimulation in HVC and recorded from RA, HVC, NIf, and Uva neurons of the right hemisphere. Stim, Stimulation. b, Electrical stimulation in HVC elicits spike responses in Uva neurons. bi, UvaHVC neurons exhibit spike collisions in response to HVC stimulation (200 μA) at small time lags after spontaneous spikes (arrows), but not at large time lags. bii, biii, UvaNIf neurons respond to 60 μA HVC stimulation (bii) and to 30 μA NIf stimulation (biii), but spike collisions are only observed for NIf stimulation, not for HVC stimulation (bii, biii, bottom traces). c, During sleep, the average ISI pdf of n = 11 UvaHVC spike trains peaks at an interval of 30–40 ms. UvaHVC cells produce few bursts (ISIs smaller than 10 ms). d, The average auto-covariance function of n = 11 UvaHVC spike trains has a long tail up to a time lag of 2–3 s (arrow), indicating frequent epochs of increased firing rates. e, f, The average ISI probability density function (e) and the average auto-covariance function (f) of UvaNIf neurons reveal frequent burst ISIs and epochs of increased firing lasting several hundreds of milliseconds (arrow).
In many vocal control areas, such as NIf, HVC, and the downstream robust nucleus of the arcopallium (RA), there are auditory neurons that are selective to the bird's own song (Katz and Gurney, 1981; McCasland and Konishi, 1981; Coleman and Mooney, 2004; Theunissen et al., 2004). Auditory responsiveness and selectivity are strongly modulated by the behavioral state of birds, such as sleep, wakefulness, anesthesia, and arousal (Vicario and Yohay, 1993; Dave et al., 1998; Schmidt and Konishi, 1998; Cardin and Schmidt, 2003). For example, during sleep, neurons in HVC and RA respond to auditory stimulation, but responses can be gated off by arousal or by electrical stimulation of Uva, with a remarkably rapid transition on a millisecond time scale (Williams and Nottebohm, 1985; Williams, 1989; Nick and Konishi, 2001; Coleman et al., 2007). A similar gating also applies to spontaneously generated spike bursts that are frequently observed in HVC and RA neurons during sleep, but almost never in awake, nonsinging birds (Yu and Margoliash, 1996; Dave et al., 1998; Hahnloser et al., 2002). Both sleep bursts and auditory responses in HVC are driven from NIf (Cardin and Schmidt, 2004; Coleman and Mooney, 2004; Hahnloser and Fee, 2007). To date, little is known about the neural mechanisms by which Uva projection neurons influence and gate sleep-related activity in the cerebrum. In head-fixed zebra finches, we explore the involvement of Uva for the generation of spontaneous and auditory-evoked activity in NIf, HVC, and RA.
Materials and Methods
Our experimental system of head-fixed, sleeping birds and methods of spike train data analysis have been described previously (Hahnloser et al., 2006). All experiments were performed in accord with protocols approved by the Veterinary Office of the Canton of Zurich, Switzerland, and in accordance with the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, 1996).
Subjects.
Zebra finches (Taeniopygia guttata) were obtained from commercial suppliers Qualipet (Dietlikon, Switzerland) and Animal Diffusion (Villarimboud, Switzerland) and our own breeding colony. Data were taken from a total of 40 adult (>90-d-old) zebra finches.
Surgery.
Birds were anesthetized with 1–3% isoflurane in oxygen, and small holes (∼200 μm) were made in the dura over HVC, RA, and Uva; wound margins were treated with xylocaine gel (2%; Astrazeneca, Zug, Switzerland). The animal was placed in a small foam restraint and subsequently moved to the recording apparatus without further anesthesia. Unlike in previous experiments, we did not administer melatonin to promote sleep.
Electrical stimulation and electrophysiology.
For antidromic identification of UvaHVC neurons, we inserted bipolar stimulation electrodes into HVC (Teflon-insulated 50-μm-diameter stainless steel wires spaced 0.5 mm apart) (Fig. 1a). Electrical stimulation was produced using an isolated stimulation unit (A.M.P.I., Jerusalem, Israel) delivering single monophasic 50–500 μA current pulses of 0.2 ms duration. Antidromic identification of UvaNIf neurons was performed in different birds using monopolar current pulses of 0.2 ms duration, delivered through 100 kΩ anodal tungsten electrodes placed in the centers of NIf and HVC (cathodal electrodes were placed on the surface of the brain). In 3 of 11 putative UvaNIf neurons, we did not observe spike responses to HVC stimulation even at very high stimulation amplitudes. We decided to include these cells in our analysis of UvaNIf neurons, because results were qualitatively similar for these cells.
Note that we also identified an additional class of putative Uva interneurons that responded to low-intensity stimulation in HVC and NIf but did not exhibit spike collisions in response to either NIf or HVC stimulation. We did not extensively record and analyze these cells, their firing behaviors were similar to those of UvaNIf cells. We also encountered Uva neurons with very low latency variability (<100 μs) but quite frequent absences of spike collisions (no collisions in response to ∼20% of stimuli). Because such lack of collisions could be attributable either to synaptically generated spike bursts in projection neurons or to indirect antidromic activation of nonprojection neurons, we took a conservative stance and discarded these cells from further analysis.
RA and HVC recordings were performed with sharp glass electrodes (5–15 MΩ, borosilicate, 1.0 mm outer diameter, 0.7 mm inner diameter) filled with 3 m KCl. HVC interneurons were identified based on their high spike rates compared with HVC projection neurons (Hahnloser et al., 2006). Uva recordings were performed either with similar glass electrodes or with tungsten metal electrodes (2–5 MΩ; Micro Probe, Gaithersburg, MD). Extracellular signals were bandpass filtered (0.3–13 kHz) and digitized to 16 bits precision at a sampling rate of 30 kHz on a Pentium-based PC running custom Labview software (National Instruments, Austin, TX).
Electrical Uva stimulation in Figure 10 was performed using a monopolar stimulation electrode placed in the center of Uva. Orthodromic response thresholds in HVC were typically 10 μA, and in NIf they were typically 5 μA.
Figure 10.
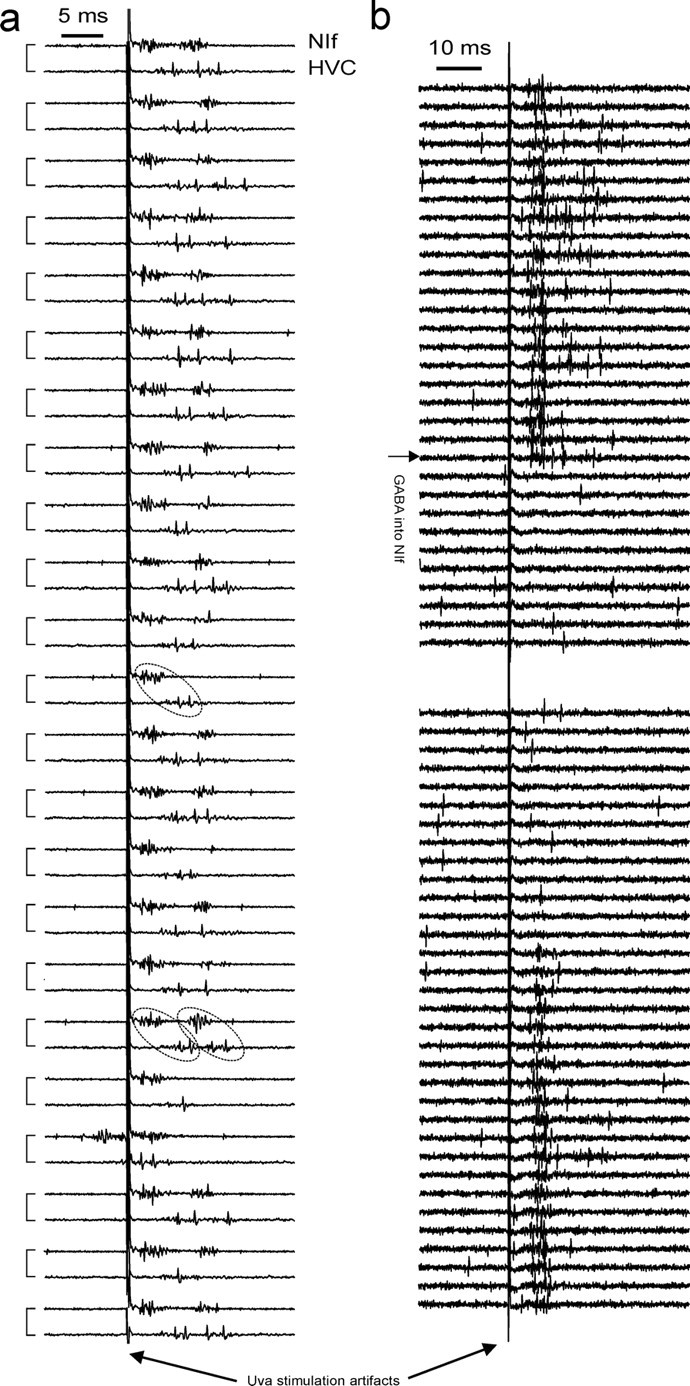
Uva drive of HVC is mediated via NIf. a, Uva stimulation (10 μA) leads to short-latency responses in NIf (top trace in paired traces) and long-latency responses in HVC (bottom trace in paired traces). Simultaneously recorded traces are linked in pairs on the left of the plot. HVC and NIf response strengths (rms of voltage traces) are correlated: single-burst responses (single dashed ellipse) and double-burst responses (two dashed ellipses) appear mostly in pairs. b, Inactivating NIf (55 nl of 250 mm GABA injection; horizontal arrow) completely suppresses orthodromic HVC responses to 70 μA Uva stimulation. Recovery is observed after several minutes.
Drug and dye injections.
Injections were made from pulled-glass pipettes (∼20 μm tip size) using a pressure injection system (Picospritzer II; Parker Hannifin, Fairfield, NJ). We injected saline (0.9% NaCl), GABA (250 mm; Sigma-Aldrich, Buchs, Switzerland), or lidocaine (4% dissolved in 0.9% NaCl; Sigma-Aldrich) at 20 psi in ∼8 ms steps. We controlled the injected volume by assessment of the meniscus displacement within the pipette. To inactivate Uva for prolonged periods as in Figure 8a, we injected ∼10 nl of GABA every 10 s. After Uva recording and injection experiments, a small dose of fluororuby (23 nl) or Alexa Fluor 488 (44 nl) was injected into Uva. In two birds, Alexa Fluor 488 was coinjected with either GABA or glutamate to assess drug leakage. Animals were killed by intramuscular injection of 20% Nembutal or by cranial dislocation. The brain was removed for histological examination of unstained slices to verify the locations of drug injection sites (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In none of the birds injected with glutamate was leakage of Alexa Fluor into the basal forebrain or the brainstem detectable.
Figure 8.
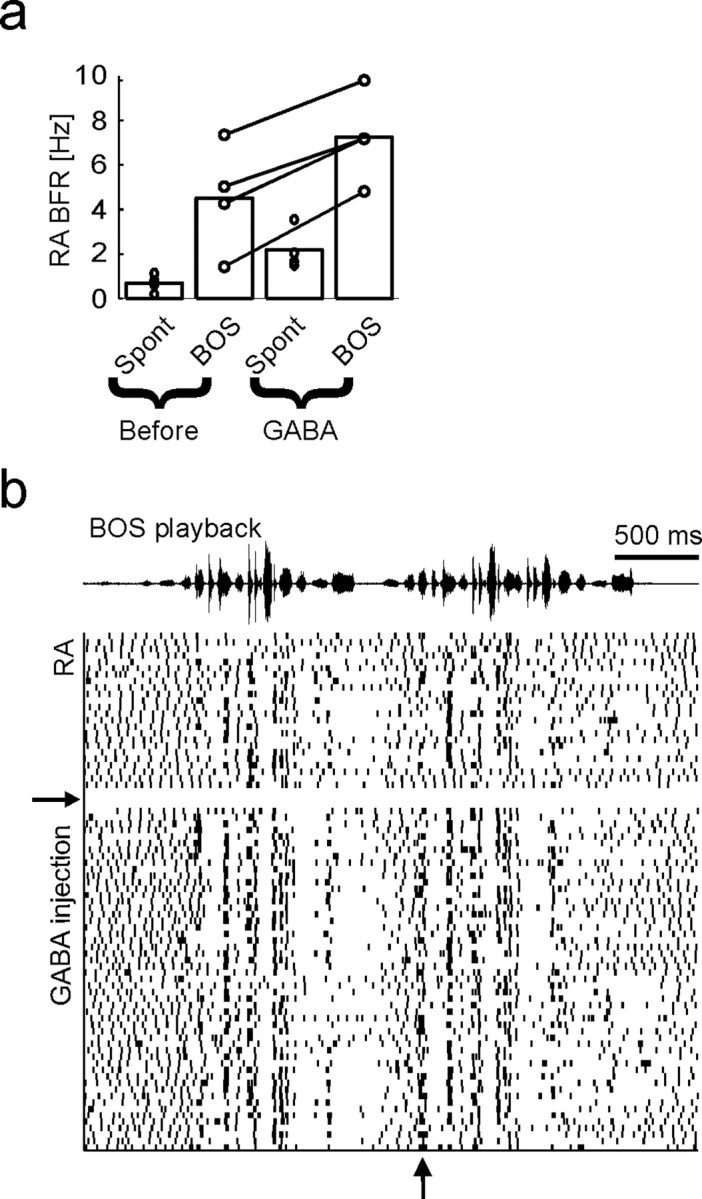
Uva inactivation enhances RA burst responses to BOS playback. a, Bar plot showing that RA burst firing rates in response to BOS playback increase during Uva inactivation (n = 4 steady GABA injections in 3 birds for a duration of 16 BOS stimuli each). RA neurons respond to BOS playback both before and during Uva inactivation (“Spont” bars indicate spontaneous RA BFRs measured during silent periods just before playback onsets). b, Raster plot of an RA spike train in response to BOS playback before and after GABA is steadily injected into Uva (GABA injection onset is indicated by the horizontal arrow). Uva inactivation slightly increases temporally locked RA burst responses (vertical arrow).
Statistics.
Unless specified, all statistical tests refer to the Wilcoxon rank sum test of equal medians. Statistical significance was defined by p < 0.01. For the analysis of spontaneous activity, we included all cells for which at least 200 s of data with the bird's eyes closed were recorded, and for the analysis of auditory responses, we included all cells exposed to at least 16 renditions of the bird's own song (BOS).
Instantaneous firing rate.
In the figures, we represented spike trains by the instantaneous firing rate (IFR) function, a continuous function defined by the inverse of the interspike interval surrounding time t.
Burst firing rate.
A burst spike train is formed by removing single spikes from a spike train (single spikes are defined as spikes that do not form an interspike interval smaller than 10 ms with either the preceding or the following spike). The average firing rate of the remaining spikes in a burst spike train is referred to as the burst firing rate (BFR) (see Figs. 4b, 7b, 8a).
Figure 4.

UvaHVC firing negatively correlates with RA bursting during BOS playback. a, Raster plots of simultaneously recorded UvaHVC and RA spike responses (tick marks) to BOS playback. UvaHVC spikes are not obviously locked to the stimulus, but RA burst spikes are. b, The burst firing rate in RA neurons to BOS playback is a monotonically decreasing function of the firing rate in UvaHVC neurons (mean ± SEM; n = 8 UvaHVC–RA pairs).
Figure 7.

Uva inactivation leads to burst-rate increases in RA neurons. a, IFR function of an RA neuron during sleep. During the time interval marked by the thick horizontal line, 115 nl of lidocaine is injected into Uva, leading to a transient increase in RA bursting. b, Bar plot summarizing the average increase in RA burst-firing rate in the 60 s interval after lidocaine injections (filled circles) and GABA injections (open circles), compared with the 60 s interval before the injections (n = 7 injections in 3 birds). The rightmost bar depicts the average burst-firing rate in the interval 400–460 s after the injections (recovery). c, The average ISI pdf of RA neurons before the injection (solid line) and during inactivation (dashed line). Both curves exhibit peaks at 5 and ∼5 ms (arrows), but of different relative amplitudes. Inset, ISI pdfs of RA burst spike trains reveal that burst shapes are unchanged during Uva inactivation (left peaks are identical), but interburst intervals are shortened: the peak associated with interburst intervals shifts from 1.2 to 0.7 s during Uva inactivation (tilted arrow).
Interspike interval probability density function.
To display the firing statistics of neurons, we computed the interspike interval (ISI) probability density function (pdf) h(τ) (τ stands for the ISI). Bin centers τi were chosen on a logarithmic scale (i = 1, …, 100); h(τi) is simply a normalized ISI histogram satisfying Σih(τi) = 1.
Cross-covariance function.
The (cross-) covariance function CAB(t) between two spike trains ρA and ρB (modeled as sums of δ functions) is a measure of relative spike density fluctuation. It is a function of the time lag t between spike pairs and is computed as
 |
where ρA and ρB are the mean firing rate of neurons A and B, and T is the duration of the recording. The first term on the right side of this equation is known as the cross-correlation function. When the spike train in just a single neuron A is inserted into this equation (ρA = ρB), the resulting function is the auto-covariance function. The average auto-covariance functions in UvaHVC and UvaNIf cells are displayed in Figure 1, d and f.
We smoothed covariance functions by convolution with a Gaussian window of SD of 20 ms in Figures 2e and 5ci, and of 4 ms in Figures 2c, 3, and 5cii. The smoothed covariance functions were downsampled by summation of covariance values over 1 ms bins. The significance of (positive or negative) peaks in covariance functions were assessed using 99% confidence thresholds that corresponded to three jackknife SDs (Thomson and Chave, 1991). The jackknife SDs were estimated by removing spikes in 20 s windows (Hahnloser et al., 2006). Only significant peaks are reported in the text.
Figure 2.

UvaHVC neuron spikes are negatively correlated with spikes in HVCI neurons and bursts in RA neurons. a, Extracellular record of an UvaHVC–HVCI pair recorded during sleep. b, IFR functions of the neurons in a. Periods of high UvaHVC firing (top, thick horizontal bars) are coincident with periods of reduced HVCI bursting (bottom). UvaHVC bursts >100 Hz have been truncated for better visibility of low firing rates. c, The cross-covariance function of this neuron pair (top) and the average covariance function of n = 7 UvaHVC–HVCI pairs in three birds (bottom) both exhibit a broad dip extending up to 3 s of UvaHVC spikes and sharp positive and negative peaks close to zero time lag (arrows). To not smear over the sharp peaks, almost no smoothing was applied (4-ms-wide Gaussian). d, e, Same as b and c, but for RA instead of HVCI neurons (n = 8 UvaHVC–RA pairs). RA single spikes have been removed for the computation of covariance functions (see Materials and Methods). Smoothing was performed with a 20-ms-wide Gaussian.
Figure 5.

UvaNIf neurons. a, Raw traces of a simultaneously recorded UvaNIf–HVCI pair. UvaNIf bursts frequently precede HVCI bursts. b, The IFR plots of the same neuron pair reveals dense UvaNIf bursting. ci, The covariance function of this neuron pair exhibits a negative dip that extends up to several seconds after UvaNIf spikes (top trace, 20 ms smoothing kernel). cii, The average covariance function of eight UvaNIf–HVCI pairs (4 ms kernel) reveals a very large positive peak at a time lag of 3 ms of UvaNIf burst spikes (solid line) and a flat and negative covariance behavior for UvaNIf single spikes (dashed line). Note the different time scales in ci and cii.
Figure 3.

UvaHVC bursts correlate with RA and HVCI bursts during sleep. a, IFR plots of a simultaneously recorded UvaHVC–HVCI pair (no clipping of UvaHVC firing rates). Inset, The raw signals of an UvaHVC burst (arrow) followed by an HVCI burst. b, The covariance functions of the same neuron pair for UvaHVC single spikes (dashed line) and UvaHVC burst spikes (solid line): UvaHVC bursts tend to precede HVCI spikes. c, The population UvaHVC–HVCI covariance function for UvaHVC single spikes (dashed line) has a negative dip at 0 ms (arrow), and for UvaHVC burst spikes (solid line) it has a peak at 10 ms (n = 7 neuron pairs). For better visibility, the burst-related covariance function has been stretched by a factor of four in the vertical direction (labeled “4×”). d, The population UvaHVC–RA covariance functions for RA burst spikes and UvaHVC single spikes (dashed line) and for burst spikes in both RA and UvaHVC neurons (solid line; the peak at 16 ms is weakly significant; p = 0.05; n = 9 neuron pairs). Note that the population covariance functions in c and d are based on concatenating all simultaneously recorded spike trains, because averaging over individual covariance functions as in Figure 2c did not work well (some UvaHVC cells did not produce enough burst spikes). e, Bar plot of the average number of HVCI spikes per 30 ms window as a function of the average number of (all) UvaHVC spikes fired in the preceding 30 ms windows. Error bars indicate SEM.
Cross-covariance functions can have wide peaks or troughs resulting from slow comodulated firing in cell pairs. To subtract these comodulations, we computed the coherency function γAB(ω) between cells A and B in the frequency domain by Fourier transforming and normalizing the cross-covariance function (see also Kimpo et al., 2003):
 |
where CAA(t) and CBB(t) are the auto-covariance functions of the spike trains. The coherency functions were inspected for peaks in the time domain γAB(t) after inverse Fourier transformation of γAB(ω). Smoothing of coherency functions γAB(t) was done by convolution with a Gaussian of SD of 4 ms.
Results
Antidromic stimulation experiments reveal Uva neurons projecting to NIf and to HVC
Using antidromic stimulation in HVC and NIf, and single-unit recordings in Uva of head-fixed zebra finches (see Materials and Methods), we were able to distinguish two classes of Uva projection neurons. HVC-projecting Uva neurons (UvaHVC neurons) exhibited spike collisions when stimulated in HVC at small time lags after spontaneous spikes (Fig. 1bi). Putative NIf-projecting Uva neurons (UvaNIf neurons) exhibited spike collisions in response to low-amplitude NIf stimulation (Fig. 1biii). To eliminate a possible confound with UvaHVC neurons that tended to also be activated by NIf stimulation as a result of their axons passing in close proximity to NIf, we also performed HVC stimulation to identify UvaNIf neurons: UvaNIf neurons often responded to HVC stimulation but did not display spike collisions, consistent with orthodromic activation of UvaNIf neurons via UvaHVC axons (Fig. 1bii).
Additional analysis showed that near-threshold HVC stimulation at 1 Hz produced latencies to the first spike in UvaHVC neurons in the range 1.1–5.4 ms (mean, 2.5 ± 1.6 ms; n = 11 cells), with small latency variability (SD) in the range 16–70 μs (mean, 44 ± 19 μs; n = 11). UvaNIf neurons exhibited small spike latencies and small latency variability to NIf stimulation (latencies, 0.8–1.7 ms; variability, 13–60 μs; n = 11 UvaNIf neurons). Thus, antidromic activation is associated with small spike-latency variability. However, the converse was not always true, because in some neurons with small latency variability, we quite frequently observed absence of spike collisions (see also Materials and Methods). Therefore, all Uva cells were identified using the collision test.
During sleep, UvaHVC neurons produce mostly single spikes, whereas UvaNIf neurons frequently produce spike bursts
During sleep, UvaHVC neurons had an average firing rate of 22 Hz (n = 11); they produced ISIs in a typical 30–40 ms range, and infrequently they also produced bursts (ISIs smaller than 10 ms) (Fig. 1c). On average, UvaHVC neurons burst every 12 s with 2.8 spikes per burst and 306 Hz burst firing rate; in total, <1.5% of UvaHVC spikes were associated with bursts. Firing rates in UvaHVC neurons were quite irregular with frequent epochs of increased firing, as revealed by the average auto-covariance function (see Materials and Methods and Fig. 1d).
Similar to UVAHVC neurons, UvaNIf neurons had an average firing rate of 21 Hz (n = 8); however, UVANIf neurons burst more frequently with an average of 2.3 bursts per second, 3.0 spikes per burst, and 326 Hz burst firing rate. In total, ∼35% of UvaNIf spikes occurred in bursts. By assessment of ISI pdfs and auto-covariance functions, UvaNIf neurons produced relatively few single spikes and displayed shorter epochs of increased firing than did UvaHVC neurons (Fig. 1e,f).
In the following, to distinguish between influences of Uva single spikes and Uva burst spikes (the complement of single spikes), we frequently removed either single spikes or burst spikes from Uva spike trains before the analysis. Accordingly, in the following we distinguish between (regular) spike measures, single-spike measures (burst spikes removed), and burst-spike measures (single spikes removed). In all neuron types, our criterion for single spikes was that interspike interval pairs had to exceed 10 ms each. This definition is commensurate with our previous work and coincides nicely with the location of local minima in ISI pdfs and auto-covariance functions of UvaHVC neurons (Fig. 1c,d).
Tonic firing in UvaHVC neurons has suppressive effects on HVCI and RA neurons
We studied the relationships between Uva and HVC activity by recording from HVC interneurons (HVCI neurons), but not from HVC projection neurons, because the latter fire extremely sparsely during sleep. To explore the influence of Uva in shaping activity along the motor pathway, we also recorded from RA neurons, known to be strongly driven by activity in RA-projecting HVC neurons (Hahnloser et al., 2006). HVC is necessary for generation of RA spike bursts but not RA single spikes. To avoid corruption of our analysis by RA single spikes, we often removed single spikes from RA spike trains before the analysis.
HVCI neurons and RA neurons display strongly modulated firing patterns during sleep (Danóczy and Hahnloser, 2006; Hahnloser et al., 2006). In paired recordings with UvaHVC neurons, we found that occasional periods of suppressed bursting in HVCI and RA neurons were often associated with periods of high tonic firing rates in UvaHVC neurons (Fig. 2a,b,d). Covariance functions between spikes in UvaHVC and HVCI neurons, as well as between spikes in UvaHVC and burst spikes in RA neurons, both displayed a large and wide negative peak (dip) that extended up to several seconds after UvaHVC spikes: in seven of seven UvaHVC–HVCI pairs and in eight of nine UvaHVC–RA pairs (RA single spikes removed), the median covariance in the interval [0 s, 2 s] of UvaHVC spikes was significantly smaller than in the interval [−10 s, −8 s] (Fig. 2c,e). Thus, after UvaHVC spikes there was a robust and long-lasting suppression of HVCI firing. Also, in six of seven UvaHVC–HVCI pairs, a sharp peak a few tens of milliseconds before UvaHVC spikes (Fig. 2c, top arrow) emerged, suggesting that HVCI spikes drive spikes in UvaHVC cells after a few tens of milliseconds. Finally, in four of seven UvaHVC–HVCI pairs, there was a sharp dip ∼1 ms after UvaHVC spikes, indicating that UvaHVC cells might mediate fast inhibition onto HVC.
How can we reconcile the apparent coexistence of fast and slow inhibition mediated by UvaHVC spikes? Of course, the wide cross-covariance dips do not necessarily imply that single UvaHVC spikes mediate long-lasting inhibition, because important factors that contribute to the width of cross-covariance functions are the widths of associated auto-covariance functions (i.e., the auto-covariances of UvaHVC and HVCI spike trains). One method of subtracting effects of auto-covariance is to inspect coherency functions rather than cross-covariance functions (see Materials and Methods). Indeed, when we evaluated coherency functions in UvaHVC–HVCI pairs (data not shown), the wide dips disappeared, but the sharp peaks and dips close to zero time lag remained, reinforcing the view that Uva is indirectly excited by HVC spikes and feeds back fast and brief inhibition.
In RA neurons, no sharp dip was seen, but positive peaks were seen at −7 ms (on average), implying that UvaHVC spikes were often preceded first by HVCI spikes, then by RA bursts. Note that the covariance results for RA neurons depended strongly on removal of single RA spikes. In contrast, results remained qualitatively unchanged when single HVCI spikes were removed, presumably because most HVCI neurons have low tonic firing rates (Hahnloser et al., 2006).
UvaHVC and HVCI neurons often burst sequentially, with HVCI bursts following UVAHVC bursts (Fig. 3a). To explore possible excitatory effects of UvaHVC bursts, we tested whether the positive peaks in covariance functions in UvaHVC cells were associated with single UvaHVC spikes or with burst spikes. Interestingly, for both HVCI and RA neurons, when removing UvaHVC burst spikes, the positive covariance peaks remained at negative time lags. However, when we removed single UvaHVC spikes in five of six UvaHVC–HVCI pairs, the positive covariance peaks appeared at positive time lags (one UvaHVC cell did not produce bursts), suggesting an excitatory effect of UvaHVC bursts (Fig. 3 b–d). Furthermore, UvaHVC bursts appeared to have graded effects on HVCI bursts: the average number of HVCI spikes fired in 30 ms windows was an increasing function of the average number UvaHVC spikes fired in preceding 30 ms windows (Fig. 3e). This result was quite robust for HVCI and UvaHVC time windows ranging from 20 to 100 ms.
In summary, until now our results suggest that single UvaHVC spikes are inhibitory and driven by cerebral feedback via the brainstem (e.g., via the dorsomedial nucleus of the intercollicular complex and the nucleus paraambigualis) and that UvaHVC bursts are excitatory and drive cerebral bursts.
Spikes are responsive to playback of the bird's own song in few UvaHVC neurons
Multiunit recordings in anesthetized birds have revealed strong auditory responses in Uva to playback of BOS (Coleman et al., 2007), suggesting that such Uva responses may be relayed to the cerebrum also during sleep. To that end, we investigated auditory responses of identified Uva projection neurons in sleeping birds. Whereas RA cells burst in a more or less stereotypical manner in response to BOS playback, we found no obvious BOS-locked firing in many UvaHVC cells (Fig. 4a). In total, only two of eight UvaHVC cells (n = 4 birds) had significantly increased average firing rates during BOS playback compared with equally sized time windows before playback (p < 0.01). The number of BOS-responsive UvaHVC cells did not change when either UvaHVC single spikes or burst spikes were removed, suggesting that weak BOS responses are associated with both single spikes and spike bursts. Covariance functions between UvaHVC spikes and RA burst spikes elicited by BOS playback (data not shown) were similar to covariance functions in Figure 2e, suggesting that effects of Uva on its targets do not qualitatively change during sensory processing. Most interestingly, the average rate of RA burst spikes (the BFR) in response to BOS playback was a smooth and decreasing function of the number of UvaHVC spikes (Fig. 4b). These results indicate graded suppressive effects of UvaHVC spikes onto both spontaneous and auditory-evoked RA bursts.
Paired UvaNIf–HVCI recordings reveal an excitatory drive mediated by UvaNIf bursts
UvaNIf neurons tended to burst densely during sleep, with many UvaNIf spikes preceding HVCI spikes by a few milliseconds (Fig. 5a,b). UvaNIf–HVCI covariance functions displayed a negative dip that extended up to 2 s after UvaNIf spikes, and they peaked at positive time lags of UvaNIf spikes (7–9 ms; seven of eight UvaNIf–HVCI pairs; three birds). In all seven pairs, the average covariance peak persisted no matter whether all UvaNIf spikes were included or burst spikes only (Fig. 5ci,cii). In contrast, removal of UvaNIf burst spikes caused the covariance peak to disappear in all seven pairs, resulting in a flat and typically negative function (Fig. 5cii).
In summary, both types of Uva projection neurons displayed single spikes that led to reduced HVCI and RA bursting on a long time scale, and they displayed spike bursts that tended to precede HVCI and RA bursts by a few milliseconds. The major difference between the two types of Uva projection neurons was that UvaNIf neurons produced bursts ∼30 times more often than did UvaHVC neurons, implying that predominance of UvaNIf burst spikes appeared to strongly drive HVC bursts, whereas predominance of UvaHVC single spikes appeared to suppress HVC bursts.
In some UVANIf neurons, burst spikes are BOS responsive
We were able to record from seven UvaNIf neurons during BOS playback. In three of seven cases (n = 4 birds), the UvaNIf neuron significantly increased its firing rate during playback (p < 0.01), with an example shown in Figure 6. In Figure 6, also note that after approximately nine BOS playbacks, the UvaNIf neuron reduced its firing density, whereas the simultaneously recorded HVCI neurons increased its burst responses; presumably this comodulated firing was caused by the bird's arousal. The fraction of responsive UvaNIf neurons did not change when we evaluated BOS responsiveness for burst spikes only. However, only a single UvaNIf remained BOS responsive when UvaNIf burst spikes were removed. Similar to the spontaneous case, BOS-related UvaNIf–HVCI covariance functions (data not shown) peaked at positive time lags, suggesting that UvaNIf spikes may be involved not only in generation of HVC spontaneous sleep bursts but also in shaping HVC auditory responses.
Figure 6.
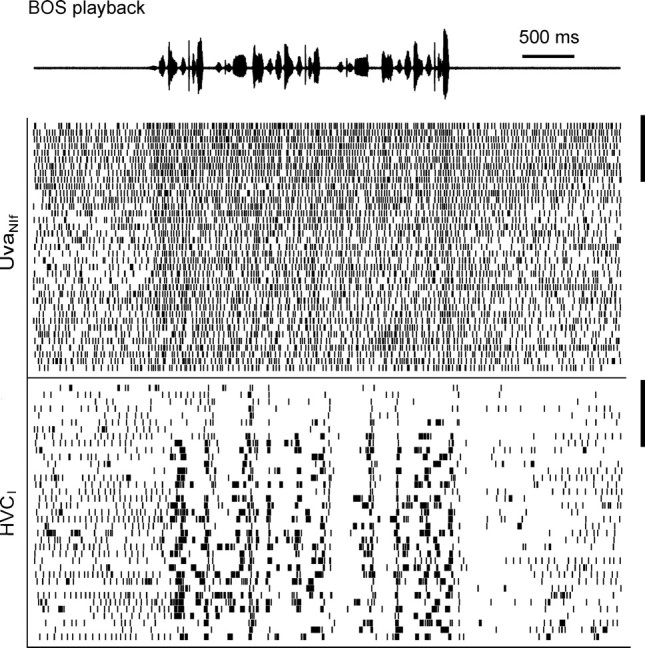
Spike raster plot of a UvaNIf–HVCI pair during BOS playback. The UvaNIf neuron (top raster) increases its firing during playback. During the first few song playbacks on top (delimited by the two vertical bars on the right), the HVCI neuron produces only very few bursts. At the same time, the UvaNIf cell displays higher tonic background firing (both before and during playback).
Pharmacological inactivation of Uva increases cerebral bursting
We explored whether the suppressive effects of UvaHVC single spikes were dominant over the excitatory effects of UvaNIf bursts by pharmacologically manipulating Uva activity. If UvaNIf bursts are more important for the generation of HVCI bursts, then HVCI bursting should diminish after Uva inactivation. If, however, UvaHVC single spikes are able to prevent HVCI neurons from bursting, then inactivating Uva should lead to increased HVCI burst firing rates. We reversibly silenced Uva by injecting the sodium-channel blocker lidocaine (4% lidocaine), or, to avoid accidental inactivation of fibers of passage, the inhibitory neurotransmitter GABA (250 mm GABA in 0.9% NaCl) into Uva of sleeping birds. Injected volumes were either 110 or 55 nl, corresponding to spheres of ∼300 and 240 μm radius, both larger than the presumed volume of Uva. No qualitative differences between GABA and lidocaine injections were seen: in either case, HVCI bursting (n = 3) and RA bursting (n = 7) transiently increased for a few minutes after Uva inactivation (Fig. 7a,b). We analyzed the RA data in more detail and found that many aspects of RA neuron spike trains were identical before and after the injections: normalized interspike-interval histograms exhibited two identically located peaks corresponding to a regular firing mode and a bursting mode (Fig. 7c). However, the relative heights of these peaks during Uva inactivation were such that RA neurons spent more time in the bursting mode when UvaHVC spikes were absent. Indeed, when we removed RA single spikes, we found that RA burst shapes were unchanged during Uva inactivation (as assessed by the density of small interspike intervals), except that RA bursts occurred more often during inactivation (as assessed by the left-shift of the ISI peak corresponding to interburst intervals) (Fig. 7c, inset).
BOS responses in RA neurons are enhanced by Uva inactivation
RA neurons significantly increased their burst firing rates in response to BOS playback, both before and during Uva inactivation (Fig. 8a). Despite this increase in playback-evoked responses, high temporal precision of individual RA bursts was maintained during Uva inactivation (Fig. 8b). Thus, Uva seems to have no major influence on the detailed timing of stimulus-evoked RA bursts. BOS-response enhancement was specific to Uva inactivation: Uva injections of vehicle (0.9% NaCl) and GABA injections 0.8 mm dorsal of Uva did not cause significant changes in RA bursting (n = 2 birds each). In two birds, GABA injections 0.8 mm anterior of Uva caused strong suppression of RA responses to BOS playback; histological examination revealed fluorescent staining of the lateral part of nucleus ovoidalis, suggesting that the decrease of RA auditory responses was caused by diminished auditory input, but not by Uva inactivation. These results show that the presumed UvaNIf drive of HVC activity during sleep and during BOS playback (Fig. 6) has little relevance for generation of HVC and RA burst responses.
Pharmacological activation of Uva leads to burst suppression and to shut-off
To test further whether UvaHVC spikes can cause suppression of sleep-related and auditory-evoked HVCI and RA bursts, we injected the excitatory neurotransmitter glutamate (in 0.9% NaCl) into Uva. When we pressure injected small volumes (23 nl) of 12 mm glutamate into Uva of sleeping birds, we observed no significant reduction in RA bursting (n = 3 injections in 3 birds; p > 0.01). At equal volume but higher concentration (23 nl of 50–200 mm glutamate), we observed robust suppression of auditory-evoked and spontaneous RA bursts (Fig. 9a). Suppression was transient, and bursting recovered within 20–40 s. On average, 50 mm glutamate injections into Uva significantly reduced spontaneous and evoked RA burst-spike rates from 2.1 to 0.2 Hz (20 s windows before and after injection) (Fig. 9b).
Figure 9.

Dose-dependent effects of glutamate injections into Uva. a, IFR response of an RA neuron to BOS playback during sleep. During the time interval marked by the thick horizontal line, 23 nl of 50 mm glutamate is injected into Uva, leading to a transient suppression of BOS-locked bursting. b, Bar plot summarizing the reduction in RA burst-firing rate in the 15 s interval after glutamate injections, compared with the 15 s interval before the injection. The connected circles depict data from different injections (n = 3 birds). c, Spontaneous sleep-related spiking is shut off in this RA neuron after injecting 110 nl of 100 mm glutamate into Uva. Spiking recovers within several seconds, but with visibly different statistics than before. The inset shows the shut-off event in which extracellular spike amplitudes dissapear in noise. d, Illustration of dose dependence of shut-off effect. In this HVCI neuron, BOS responses were transiently suppressed after 3 and 7 nl injections of 1 m glutamate into Uva. Approximately 1 min after a 14 nl injection, the HVCI neuron shut off (tilted arrow).
At higher concentration and injected volumes, RA and HVCI neurons often dramatically increased their spike frequency, and RA spike amplitudes gradually diminished until they disappeared in noise, similar to what in a different set of experiments has been termed “neuron shut-off” (Shea and Margoliash, 2003) (Fig. 9c,d). The time interval between injections and observations of the shut-off state ranged from several seconds up to ∼1 min. Shut-off seemed to be facilitated by arousal, because in one bird shut-off coincided with eye opening on two occasions. RA neuron spiking recovered from the shut-off state within 1–2 min (n = 3 neurons in 3 birds), but in two cases, firing patterns were qualitatively different after recovery (Fig. 9c), suggesting that some irreversible damage to Uva may have occurred. For this reason, we discarded all data recorded after the observation of shut-off events.
Electrical Uva stimulation activates HVC via NIf
Our pharmacological experiments show that Uva's overall inhibitory influence on HVC and RA bursts correlates most strongly with the firing behaviors in UvaHVC cells, not in UvaNIf cells. To explicitly address the presumed excitatory drive provided by UvaNIf bursts, we electrically stimulated Uva before and after NIf inactivation. Electrical stimulation of Uva is known to activate HVC neurons (Williams, 1989; Coleman et al., 2007), but the relative contributions of the direct and indirect Uva pathways for this activation remain unclear. We found that low-intensity Uva stimulation (5–20 μA monophasic current pulses of 0.2 ms duration) led to spike responses in HVC neurons with a mean latency of 6.0 ms (range, 4.9–6.7 ms; n = 6 HVC neurons in 3 birds). Low-intensity Uva stimulation also led to orthodromic spike responses in NIf (5–10 μA current pulses of 0.2 ms duration), with a shorter average spike latency of 2.1 ms (range, 1.3–2.8 ms; n = 6 NIf neurons in 2 birds). NIf and HVC multiunit responses to Uva stimulation were strongly correlated: the correlation coefficient of root-mean-square extracellular voltages in HVC and NIf in 0.5–15 ms windows after Uva stimulation ranged from r = 0.45 to 0.68 (p < 10−5; n = 5 recording sites in 3 birds) (Fig. 10a).
Injections of 55 nl of GABA (250 mm) into NIf led to complete suppression of HVC responses to high-intensity (70 μA) Uva stimulation (n = 7 injections in 3 birds) (Fig. 10b). Orthodromic spike responses in HVC remained suppressed even when Uva stimulation amplitude was increased to >10 times the threshold value (up to 300 μA). GABA injections (55 nl) 500 μm ventral of NIf did not lead to suppression of HVC responses (n = 2 birds). In one of two birds, GABA injections (55 nl) 500 μm dorsal of NIf led to partial suppression of HVC responses. We reexamined this case by smaller injections (11 nl) that led to suppression when injected into NIf but not when injected 500 μm dorsally. We infer from these results that electrical Uva stimulation activates HVC via NIf and that UvaNIf bursts are indeed part of a functional excitatory pathway from Uva to HVC. Furthermore, the complete absence of HVC responses during NIf inactivation provides evidence that positive correlations between UvaHVC and HVCI bursts are attributable to common direct and indirect input from UvaNIf neurons.
Uva firing is modulated by arousal
In most song-control brain areas, such as RA and HVC, neural activity is strongly gated depending on the behavioral state of birds, such as sleep, wakefulness, and anesthesia (Dave et al., 1998; Schmidt and Konishi, 1998; Cardin and Schmidt, 2004). Based on the negative correlations between UvaHVC spikes and RA bursts, we expected UvaHVC neurons to increase their firing when birds are woken from sleep. Indeed, when we aroused birds by brief air puffs [the awake state was assessed by open eyes and by lack of bursts in RA neurons (Hahnloser et al., 2006)], UvaHVC firing rates significantly increased from 12.8 to 27.3 Hz (burst firing rates in RA neurons significantly decreased from 2.9 to 0.02 Hz) (Fig. 11a,b). Interestingly, although arousal led to increases in average UvaHVC firing rates, average UvaHVC burst firing rates significantly decreased from 0.49 to 0.10 Hz (Fig. 11biii). Arousal led to significant increase of UvaNIf firing rates from 15 to 27 Hz (n = 8 UvaNIf neurons in 4 birds) (Fig. 11biv). Unlike in UvaHVC neurons, arousal led to increased UvaNIf burst firing rates in all eight UvaNIf neurons examined (data not shown). Assuming an excitatory drive mediated by UvaNIf bursts onto NIf, these findings agree with our previous findings of little or no behavioral-state dependence of NIf projection neuron activity (Hahnloser and Fee, 2007).
Figure 11.

UvaHVC and UvaNIf firing depends on the behavioral state. a, IFR plots of an UvaHVC–RA pair. When the bird is aroused by a light air puff (arrow), RA bursting stops, whereas UvaHVC tonic firing increases. b, Bar plots summarizing firing changes related to arousal. Firing rates and burst firing rates were measured in equal time windows before and after the air puffs. The durations of these windows were set by the time during which the awake state could be verified in terms of open eyes. bi–biv, BFRs in RA (bi) and UvaHVC (biii) neurons drop significantly, whereas IFRs in UVAHVC neurons (bii) and in UvaNIf neurons (biv) significantly increase.
Uva firing contributes to RA burst suppression associated with arousal
Both the wake–sleep dependence of UvaHVC firing and the suppressive action of UvaHVC spikes onto RA bursts suggest that UvaHVC spikes might be strongly implicated in mediating the behavioral-state dependence of bursting in RA. To test for this possibility, we aroused birds by air puffs after Uva inactivation. If Uva controls the behavioral-state dependence of RA activity, then RA bursting should be unaffected by arousal when Uva is silenced. If, instead, a common neuromodulatory input to Uva and RA leads to behaviorally mediated firing modulation in these areas, then RA bursting should be strongly suppressed, regardless of Uva activity. We tested Uva's involvement in RA burst suppression by delivering arousing air puffs after reversibly inactivating Uva by large lidocaine or GABA injections. The time interval between injection onset and air puff delivery was on the order of 40 s (range, 20–60 s), during which we verified the bird's sleep state by visual control of closed eyelids. We found that waking birds after inactivating Uva significantly reduced bursting in RA but did not completely abolish it (Fig. 12a,b). Thus, it appears that UvaHVC spiking is only a part of the input required for RA to generate burst suppression associated with arousal.
Figure 12.

Uva activity contributes to state-dependent firing modulation in RA. a, Arousing birds by air puffs after lidocaine or GABA injections leads to partial suppression of RA bursting. b, Bar plot summarizing the reduction in RA burst firing measured in 30 s windows before and after waking, both within 3 min of the injections (n = 6 injections in 3 birds; lidocaine injections shown by filled circles and GABA injections shown by open circles).
Discussion
Our experiments revealed that during sleep, single spikes in UvaHVC neurons mediate inhibition and bursts in UvaNIf neurons mediate excitation to their respective cerebral targets. In the following, we discuss the evidence and possible implications for this dichotomy of Uva firing behavior.
Uva single spikes seem to have a distinctive inhibitory role, because the dips in spike-covariance functions involving Uva neurons and RA or HVC neurons were absent for Uva burst spikes, and because no covariance peak was seen at a positive time lag of Uva single spikes. In contrast, Uva bursts seem to have an excitatory role, because covariance functions with RA and HVC neurons displayed highly significant peaks at positive time lags of Uva burst spikes. UvaNIf bursts seem to be very effective in driving NIf activity, because extremely low Uva stimulation was sufficient to trigger strong orthodromic NIf responses. With estimated latencies for the Uva–HVC drive of 7 ms (Fig. 5c), and of 5 ms for the NIf–HVC drive (Hahnloser and Fee, 2007), we estimate that UvaNIf bursts drive NIf activity in just 2 ms. Because even high-intensity Uva stimulation was not able to activate HVC neurons during NIf inactivation, we speculate that the tendency of HVCI spikes to follow UvaHVC bursts arises mainly from common direct and indirect input from UvaNIf neurons rather than from direct excitatory drive provided to HVC by UvaHVC bursts.
UvaHVC cells seem to be driven by input from HVC and RA, because small positive covariance peaks were seen at negative time lags of single UvaHVC spikes. Although the peak times may deviate by >10 ms from assumed polysynaptic spike propagation times, the possibility that Uva cells are driven by indirect HVC input has interesting implications from the viewpoint of feedback control. In engineered systems (such as in amplifiers), negative feedback has a stabilizing function. Thus, if excitatory RA bursts are transmitted to Uva indirectly via the dorsomedial nucleus (DM) and/or the nucleus paraambigualis (PAm), and these bursts induce increased tonic spike rates in UvaHVC neurons (as suggested by our covariance analysis), then the suppressive action of UvaHVC single spikes can help to stabilize HVC and RA burst rates via a negative feedback loop.
Single spikes in UvaHVC neurons seem to dampen cerebral bursting not only during unperturbed sleep but also during sensory processing. In our head-fixed sleeping bird preparation, many fewer Uva cells were activated in response to BOS playback than we could have expected from the number of BOS-responsive multiunit sites in Uva of anesthetized animals (Coleman et al., 2007). One reason for this discrepancy could be related to anesthesia, and another reason could be that the multiunit analysis emphasizes spike bursts, the latter of which are carriers of BOS responses in Uva projection neurons. Recordings of single units in anesthetized animals will hopefully provide more insights into this conundrum in the future.
We have shown that Uva participates in arousal-triggered suppression of RA bursting. However, large lidocaine injections into Uva did not prevent arousal-related RA burst suppression from occurring. Therefore, our study agrees with evidence that wake–sleep gating in RA involves gating sources in addition to Uva, such as noradrenergic neuromodulation (Cardin and Schmidt, 2004). Interestingly, arousal did not suppress bursting in UvaNIf cells, but to the contrary, bursting in these cells was increased by arousal. Although the incongruent behaviors in the two Uva cell types is puzzling, this latter finding agrees with our observation that in our head-fixed bird preparation, NIfHVC neurons also burst in the awake state (Hahnloser and Fee, 2007). The observation of arousal-triggered increase in UvaNIf bursting also suggests the view that in HVC, UvaHVC single-spike suppression dominates over UvaNIf spike-burst excitation when in conflict.
The synaptic mechanisms by which UvaHVC single spikes are able to mediate HVC burst suppression are largely unknown. Intracellular recordings have revealed that electrical Uva stimulation can induce either IPSPs or EPSPs in HVC neurons (Coleman et al., 2007), although the monosynaptic nature and neurotransmitter type associated with these potentials have not been established yet. Similarly, the burst generation mechanisms in Uva projection neurons are currently unknown, and it is conceivable that similar to projection neurons in a different thalamic nucleus, Uva bursts are generated by a hyperpolarization-dependent mechanism (Luo and Perkel, 1999). Analogies of Uva projection neurons can also be drawn with thalamic relay cells in the mammalian literature. Similar to UvaHVC cells, thalamic relay cells in mammals can fire in regular and burst modes associated with different levels of alertness and sleep phases (McCormick and Bal, 1997; Sherman, 2001; Weyand et al., 2001; Saper et al., 2005). Bursts in thalamic relay neurons are positively correlated with spikes in their target areas (e.g., cortical interneurons) (Swadlow and Gusev, 2001), similar to our observations in Uva projection neurons. Furthermore, thalamic firing patterns in mammals are strongly regulated by cholinergic input (Steriade, 2004). The dense innervations of Uva by cholinergic fibers (Akutagawa and Konishi, 2005) suggests that mechanistically, the waxing and waning of tonic spiking in UvaHVC cells might be influenced by the cholinergic system. Cholinergic neuromodulation might be coincident in both types of Uva projection neurons; at least, this could explain some of their common behavior, such as the wide negative dips in covariance functions (compare Figs. 2c, 5c).
All our pharmacological Uva manipulations induced effects most compatible with presumed action on UvaHVC cells, not UvaNIf cells (with the possible exception of the shut-off state elicited by large glutamate injections, which we find hard to interpret). For example, when Uva cells are silenced by GABA injections, effects of uninterrupted bursting in HVCI and RA neurons are in agreement with absence of UvaHVC single spikes, not UvaNIf bursts. Similarly, we found that small doses of glutamate suppress HVC bursting, in agreement with the notion that glutamate injections increase tonic firing in UvaHVC cells, not bursting in UvaNIf cells. Overall, these findings can help us to refine the current view of sleep-burst generation in cerebral song-control areas, discussed in the following.
NIf seems to be the single source of cerebral sleep bursts: on the one hand, it was previously shown that NIf input is necessary for HVC sleep-burst generation; on the other hand, we have shown here that HVC cells continue to burst when Uva is silenced, suggesting that NIf bursting can be entrained intrinsically without Uva input. What then is the relationship between Uva and NIf activity during sleep? In HVC-projecting NIf neurons, there is a lack of pronounced burst epochs during sleep (Hahnloser and Fee, 2007). Here we have observed a similar absence of long and pronounced burst epochs in UvaNIf neurons (Figs. 1f, 4b). Presumably, the role of UvaNIf bursts is to affect the timing of NIfHVC bursts but not the overall rate of bursting in NIf and downstream areas. In simple words, NIf seems to be the static motor that drives cerebral sleep bursts and Uva the dynamic break that frequently overrules the NIf drive. In a recent modeling study, such Uva function produced excellent results in explaining a large body of pairwise spike correlation data in HVC and RA (Weber and Hahnloser, 2007).
The role of Uva activity for song production remains largely unknown, but we imagine that UvaHVC and UvaNIf neurons could have important roles, as elaborated in the following.
Singing is a bilateral behavior associated with highly synchronized activity between the right and left HVCs (Vu et al., 1998; Schmidt, 2003; Schmidt et al., 2004). Interhemispheric HVC synchronization contrasts with the absence of bilateral connections between forebrain song-control areas (including Uva). Bilateral connections exist between DM and PAm (feeding into Uva) (Reinke and Wild, 1998; Ashmore et al., 2005). The idea that Uva is involved in the mediation of interhemispheric synchronization is consistent with lesion studies, because bilateral and unilateral lesions of Uva severely impair singing with song recovery only after unilateral lesions (Williams and Vicario, 1993; Coleman and Vu, 2005). However, bilateral NIf lesions do not severely impair singing (Cardin et al., 2005), which implies that if Uva is indeed a bilateral coordinator, then we expect UvaHVC cells to be the main players involved. That is, we can imagine that bilaterally coordinated UvaHVC activity constitute a trigger signal for singing behavior. For example, before singing, UvaHVC firing could suppress HVC activity. In combination with an excitatory NIf burst (triggered by UvaNIf neurons), these could constitute an effective triggering signal for HVC premotor bursts. At least, such an idea is consistent with a putative inhibitory role of UvaHVC spikes and with theoretical work that suggests that inhibition has excellent synchronization properties (Van Vreeswijk et al., 1994; Bush and Sejnowski, 1996).
Footnotes
The authors declare no competing financial interests.
This work was supported by a Schweizerischer Nationalfonds professorship grant (R.H.R.H.) and a Forschungskredit of the University of Zurich. We thank Kevan Martin for discussions on functions of the mammalian thalamus and for help with this manuscript. R.H.R.H., A.N., K.N., and C.Z.-H.W. jointly recorded the data, with relative contributions in the listed order (from high to low). C.Z.-H.W. wrote the stimulation and data acquisition programs. R.H.R.H. analyzed the data and wrote this manuscript.
References
- Akutagawa E, Konishi M. Connections of thalamic modulatory centers to the vocal control system of the zebra finch. Proc Natl Acad Sci USA. 2005;102:14086–14091. doi: 10.1073/pnas.0506774102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore RC, Wild JM, Schmidt MF. Brainstem and forebrain contributions to the generation of learned motor behaviors for song. J Neurosci. 2005;25:8543–8554. doi: 10.1523/JNEUROSCI.1668-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush P, Sejnowski T. Inhibition synchronizes sparsely connected cortical neurons within and between columns in realistic network models. J Comput Neurosci. 1996;3:91–110. doi: 10.1007/BF00160806. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Schmidt MF. Song system auditory responses are stable and highly tuned during sedation, rapidly modulated and unselective during wakefulness, and suppressed by arousal. J Neurophysiol. 2003;90:2884–2899. doi: 10.1152/jn.00391.2003. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Schmidt MF. Auditory responses in multiple sensorimotor song system nuclei are co-modulated by behavioral state. J Neurophysiol. 2004;91:2148–2163. doi: 10.1152/jn.00918.2003. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Raksin JN, Schmidt MF. Sensorimotor nucleus NIf is necessary for auditory processing but not vocal motor output in the avian song system. J Neurophysiol. 2005;93:2157–2166. doi: 10.1152/jn.01001.2004. [DOI] [PubMed] [Google Scholar]
- Coleman MJ, Mooney R. Synaptic transformations underlying highly selective auditory representations of learned birdsong. J Neurosci. 2004;24:7251–7265. doi: 10.1523/JNEUROSCI.0947-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MJ, Vu ET. Recovery of impaired songs following unilateral but not bilateral lesions of nucleus uvaeformis of adult zebra finches. J Neurobiol. 2005;63:70–89. doi: 10.1002/neu.20122. [DOI] [PubMed] [Google Scholar]
- Coleman MJ, Roy A, Wild JM, Mooney R. Thalamic gating of auditory responses in telencephalic song control nuclei. J Neurosci. 2007;27:10024–10036. doi: 10.1523/JNEUROSCI.2215-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danóczy M, Hahnloser RHR. Efficient estimation of hidden state dynamics from spike trains. In: Weiss Y, Schölkopf B, Platt J, editors. Advances in neural information processing systems, Vol 18; Proceedings of the 2005 conference; Cambridge, MA: MIT; 2006. pp. 227–234. [Google Scholar]
- Dave AS, Yu AC, Margoliash D. Behavioral state modulation of auditory activity in a vocal motor system. Science. 1998;282:2250–2254. doi: 10.1126/science.282.5397.2250. [DOI] [PubMed] [Google Scholar]
- Glenn LL, Steriade M. Discharge rate and excitability of cortically projecting intralaminar thalamic neurons during waking and sleep states. J Neurosci. 1982;2:1387–1404. doi: 10.1523/JNEUROSCI.02-10-01387.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW, Sherman SM. Thalamic relay functions and their role in corticocortical communication: generalizations from the visual system. Neuron. 2002;33:163–175. doi: 10.1016/s0896-6273(01)00582-7. [DOI] [PubMed] [Google Scholar]
- Hahnloser RHR, Fee MS. Sleep-related spike bursts in HVC are driven by the nucleus interface of the nidopallium. J Neurophysiol. 2007;97:423–435. doi: 10.1152/jn.00547.2006. [DOI] [PubMed] [Google Scholar]
- Hahnloser RHR, Kozhevnikov A, Fee MS. An ultrasparse code underlies the generation of neural sequences in a songbird. Nature. 2002;419:65–70. doi: 10.1038/nature00974. [DOI] [PubMed] [Google Scholar]
- Hahnloser RHR, Kozhevnikov AA, Fee MS. Sleep-related neural activity in a premotor and a basal-ganglia pathway of the songbird. J Neurophysiol. 2006;96:794–812. doi: 10.1152/jn.01064.2005. [DOI] [PubMed] [Google Scholar]
- Katz LC, Gurney ME. Auditory responses in the zebra finch's motor system for song. Brain Res. 1981;221:192–197. doi: 10.1016/0006-8993(81)91073-8. [DOI] [PubMed] [Google Scholar]
- Kimpo RR, Theunissen FE, Doupe AJ. Propagation of correlated activity through multiple stages of a neural circuit. J Neurosci. 2003;23:5750–5761. doi: 10.1523/JNEUROSCI.23-13-05750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Perkel DJ. A GABAergic, strongly inhibitory projection to a thalamic nucleus in the zebra finch song system. J Neurosci. 1999;19:6700–6711. doi: 10.1523/JNEUROSCI.19-15-06700.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCasland JS, Konishi M. Interaction between auditory and motor activities in an avian song control nucleus. Proc Natl Acad Sci USA. 1981;78:7815–7819. doi: 10.1073/pnas.78.12.7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Feeser HR. Functional implications of burst firing and single spike activity in lateral geniculate relay neurons. Neuroscience. 1990;39:103–113. doi: 10.1016/0306-4522(90)90225-s. [DOI] [PubMed] [Google Scholar]
- Nick TA, Konishi M. Dynamic control of auditory activity during sleep: correlation between song response and EEG. Proc Natl Acad Sci USA. 2001;98:14012–14016. doi: 10.1073/pnas.251525298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottebohm F, Kelley DB, Paton JA. Connections of vocal control nuclei in the canary telencephalon. J Comp Neurol. 1982;207:344–357. doi: 10.1002/cne.902070406. [DOI] [PubMed] [Google Scholar]
- Reinke H, Wild JM. Identification and connections of inspiratory premotor neurons in songbirds and budgerigar. J Comp Neurol. 1998;391:147–163. [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Schmidt MF. Pattern of interhemispheric synchronization in HVc during singing correlates with key transitions in the song pattern. J Neurophysiol. 2003;90:3931–3949. doi: 10.1152/jn.00003.2003. [DOI] [PubMed] [Google Scholar]
- Schmidt MF, Konishi M. Gating of auditory responses in the vocal control system of awake songbirds. Nat Neurosci. 1998;1:513–518. doi: 10.1038/2232. [DOI] [PubMed] [Google Scholar]
- Schmidt MF, Ashmore RC, Vu ET. Bilateral control and interhemispheric coordination in the avian song motor system. Ann NY Acad Sci. 2004;1016:171–186. doi: 10.1196/annals.1298.014. [DOI] [PubMed] [Google Scholar]
- Shea SD, Margoliash D. Basal forebrain cholinergic modulation of auditory activity in the zebra finch song system. Neuron. 2003;40:1213–1226. doi: 10.1016/s0896-6273(03)00723-2. [DOI] [PubMed] [Google Scholar]
- Sherman SM. Tonic and burst firing: dual modes of thalamocortical relay. Trends Neurosci. 2001;24:122–126. doi: 10.1016/s0166-2236(00)01714-8. [DOI] [PubMed] [Google Scholar]
- Steriade M. Acetylcholine systems and rhythmic activities during the waking–sleep cycle. Prog Brain Res. 2004;145:179–196. doi: 10.1016/S0079-6123(03)45013-9. [DOI] [PubMed] [Google Scholar]
- Swadlow HA, Gusev AG. The impact of ‘bursting’ thalamic impulses at a neocortical synapse. Nat Neurosci. 2001;4:402–408. doi: 10.1038/86054. [DOI] [PubMed] [Google Scholar]
- Swadlow HA, Gusev AG, Bezdudnaya T. Activation of a cortical column by a thalamocortical impulse. J Neurosci. 2002;22:7766–7773. doi: 10.1523/JNEUROSCI.22-17-07766.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen FE, Amin N, Shaevitz SS, Woolley SM, Fremouw T, Hauber ME. Song selectivity in the song system and in the auditory forebrain. Ann NY Acad Sci. 2004;1016:222–245. doi: 10.1196/annals.1298.023. [DOI] [PubMed] [Google Scholar]
- Thomson DJ, Chave AD, editors. Englewood Cliffs, NJ: Prentice Hall; 1991. Jackknifed error estimates for spectra, coherences, and transfer functions. [Google Scholar]
- Van Vreeswijk C, Abbott LF, Ermentrout GB. When inhibition not excitation synchronizes neural firing. J Comput Neurosci. 1994;1:313–321. doi: 10.1007/BF00961879. [DOI] [PubMed] [Google Scholar]
- Vates GE, Broome BM, Mello CV, Nottebohm F. Auditory pathways of caudal telencephalon and their relation to the song system of adult male zebra finches. J Comp Neurol. 1996;366:613–642. doi: 10.1002/(SICI)1096-9861(19960318)366:4<613::AID-CNE5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Vicario DS, Yohay KH. Song-selective auditory input to a forebrain vocal control nucleus in the zebra finch. J Neurobiol. 1993;24:488–505. doi: 10.1002/neu.480240407. [DOI] [PubMed] [Google Scholar]
- Vu ET, Schmidt MF, Mazurek ME. Interhemispheric coordination of premotor neural activity during singing in adult zebra finches. J Neurosci. 1998;18:9088–9098. doi: 10.1523/JNEUROSCI.18-21-09088.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber AP, Hahnloser RHR. Spike correlations in a songbird agree with a simple Markov population model. PLoS Comput Biol. 2007;3:e249. doi: 10.1371/journal.pcbi.0030249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand TG, Boudreaux M, Guido W. Burst and tonic response modes in thalamic neurons during sleep and wakefulness. J Neurophysiol. 2001;85:1107–1118. doi: 10.1152/jn.2001.85.3.1107. [DOI] [PubMed] [Google Scholar]
- Wild JM. Visual and somatosensory inputs to the avian song system via nucleus uvaeformis (Uva) and a comparison with the projections of a similar thalamic nucleus in a nonsongbird, Columba livia. J Comp Neurol. 1994;349:512–535. doi: 10.1002/cne.903490403. [DOI] [PubMed] [Google Scholar]
- Williams H. Multiple representations and auditory-motor interactions in the avian song system. Ann NY Acad Sci. 1989;563:148–164. [Google Scholar]
- Williams H, Nottebohm F. Auditory responses in avian vocal motor neurons: a motor theory for song perception in birds. Science. 1985;229:279–282. doi: 10.1126/science.4012321. [DOI] [PubMed] [Google Scholar]
- Williams H, Vicario DS. Temporal patterning of song production: participation of nucleus uvaeformis of the thalamus. J Neurobiol. 1993;24:903–912. doi: 10.1002/neu.480240704. [DOI] [PubMed] [Google Scholar]
- Yu AC, Margoliash D. Temporal hierarchical control of singing in birds. Science. 1996;273:1871–1875. doi: 10.1126/science.273.5283.1871. [DOI] [PubMed] [Google Scholar]