

Abstract
In several brain regions, microglia actively promote neuronal apoptosis during development. However, molecular actors leading microglia to trigger death remain mostly unknown. Here, we show that, in the developing hippocampus, apoptotic neurons are contacted by microglia expressing both the integrin CD11b and the immunoreceptor DAP12. We demonstrate that developmental apoptosis decreases in mice deficient for CD11b or DAP12. In addition, function-blocking antibodies directed against CD11b decrease neuronal death when injected into wild-type neonates, but have no effect when injected into DAP12-deficient littermates. This demonstrates that DAP12 and CD11b act in converging pathways to induce neuronal death. Finally, we show that DAP12 and CD11b control the production of microglial superoxide ions, which kill the neurons. Thus, our data show that the process of developmental neuronal death triggered by microglia is similar to the elimination of pathogenic cells by the innate immune cells.
Keywords: microglia, developmental death, apoptosis, integrin, CD11b, DAP12
Introduction
Recent studies have shown that, aside their phagocytic activity, microglia are able to promote developmental neuronal apoptosis. The molecular mediators of microglial-induced cell death have been identified in several in vitro systems. In the developing retina, the neuronal death studied in eye explants results from the production of NGF by invading microglia (Frade and Barde, 1998). In spinal cord explants, microglial tumor necrosis factor α (TNFα) commits differentiating motoneurons to die unless they are rescued by muscle-derived trophic factors (Sedel et al., 2004). In developing cerebellar slices, microglia induce Purkinje cell apoptosis by releasing superoxide ions produced by a respiratory burst (Marín-Teva et al., 2004). However, the mechanisms and molecules controlling microglial ability to kill neurons remain mostly unknown.
Microglia are cells of the innate immune system, which is the front line of host defense against invading pathogens or pathological cells (Vivier and Malissen, 2005). We thus thought to investigate whether effector mechanisms used by peripheral innate immune cells such as macrophages and neutrophils are also active in microglia. Neutrophils induce the death of invading pathogens by the production of reactive oxygen species (ROS), which is triggered by the activation of the CD11a or CD11b subunits of β2-integrin (Nathan et al., 1989; Abram and Lowell, 2007). In addition, this integrin signaling requires DAP12 and Fc receptor γ (FcRγ), which are adaptors containing an immunoreceptor tyrosine-based activation motif (ITAM) acting through syk kinase pathway (Mócsai et al., 2006).
In the brain, CD11b integrin expression is restricted to microglia and is a hallmark of their activation (Block et al., 2007). CD11b has recently been implicated in the death of dopaminergic neurons on inflammation (Pei et al., 2007). DAP12 expression, which is not detected in adult brain, is restricted to developing microglia (Roumier et al., 2004). Mutation of DAP12 in humans induces a presenile dementia called Nasu-Hakola disease (Paloneva et al., 2000). Based on the role of CD11b and DAP12 in inducing cell death in the innate immune system, we have now characterized in vivo their involvement in the induction of developmental neuronal death in the hippocampus. Our results demonstrate that microglia use innate immunity mechanisms to induce the apoptosis of neurons committed to death during development.
Materials and Methods
Animals and surgery.
DAP12 mutated mice are the KΔY75 mice (here referred to as DAP12KI), in which the KARAP/DAP12 protein is expressed but bears a loss-of-function mutation (Tomasello et al., 2000). DAP12KI mice were backcrossed eight times with C57BL/6 mice. CD11b−/− mice (van Spriel et al., 2001) were kindly provided by Dr. J. H. Leusen (University Medical Center, Utrecht, The Netherlands) and were on a C57BL/6 genetic background. The day of birth was considered as postnatal day 0 (P0). P0 pups were cryoanesthetized on ice for 1 min. Two microliters of F4/80 or CD11b blocking antibody (0.5 mg/ml) mixed with fast green (0.1 mg/ml) were injected intracerebroventricularly into the left hemisphere using a glass capillary. The pups were then placed on a heating pad with their original nesting material for 5 min and returned to their mother for additional recovery. The mice were processed for immunohistochemistry 24 h after antibody injection. Brains were dissected after intracardiac perfusion with 4% paraformaldehyde and 2 h postfixation at 4°C. They were then cryoprotected overnight (o/n) at 4°C in PBS plus 20% sucrose before rapid freezing. The experimental procedures were approved by the Regional Animal Experimentation Ethics Committee (p3-2005-012; p3-2007-025).
Immunohistochemistry.
Cryostat sections (20 μm; Frigocut; Leica Microsystems) were incubated o/n at 4°C with primary antibodies (or 48 h for anti-DAP12 antibody) plus Triton X-100 (0.1%) plus gelatin (0.12%). Primary antibodies were as follows: polyclonal rabbit anti-activated caspase 3 (R&D Systems), monoclonal rat anti-F4/80 (Serotec), monoclonal rat anti-CD11b (M1/70; BD Biosciences), and polyclonal rabbit anti-DAP12 (Millipore Bioscience Research Reagents). Secondary antibodies were as follows: goat anti-rabbit Cy3-conjugated, goat anti-rabbit Cy5-conjugated (Jackson ImmunoResearch), and goat anti-rat Alexa 488-conjugated (Invitrogen).
Detection and neutralization of superoxide ions.
Production of 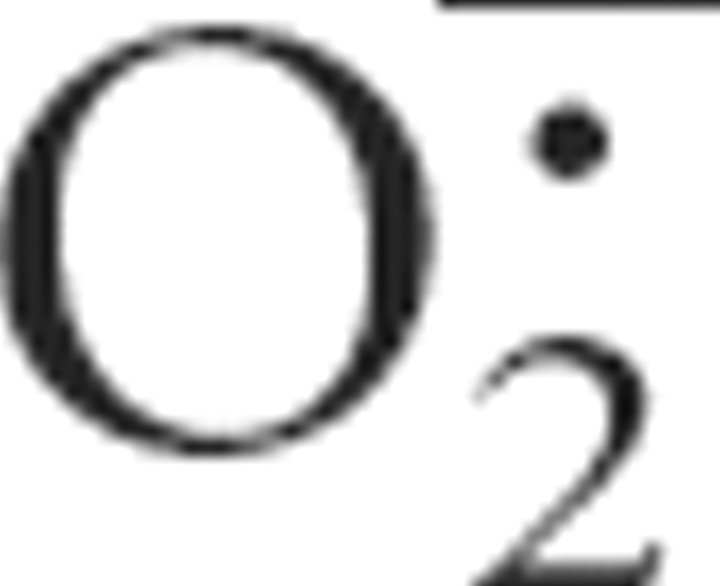 was visualized through its ability to specifically oxidize dihydroethidine (DHE) into ethidium (Bindokas et al., 1996), which binds to the DNA of the cells in which it is produced. Dihydroethidine (Invitrogen) was dissolved in dimethylsulfoxide (DMSO), and then diluted in sterile PBS and immediately injected subcutaneously into newborn C57BL/6 mice (1 mg/ml; 20 μl per animal; 10% DMSO final concentration). Neutralization of superoxide ion was achieved by subcutaneous injection of N-tert-butyl-phenylnitrone (PBN) (Sigma-Aldrich). PBN was dissolved in DMSO, and then diluted in sterile PBS and immediately injected subcutaneously into newborn C57BL/6 mice (10 mg/ml; 20 μl per animal; 10% DMSO final concentration).
was visualized through its ability to specifically oxidize dihydroethidine (DHE) into ethidium (Bindokas et al., 1996), which binds to the DNA of the cells in which it is produced. Dihydroethidine (Invitrogen) was dissolved in dimethylsulfoxide (DMSO), and then diluted in sterile PBS and immediately injected subcutaneously into newborn C57BL/6 mice (1 mg/ml; 20 μl per animal; 10% DMSO final concentration). Neutralization of superoxide ion was achieved by subcutaneous injection of N-tert-butyl-phenylnitrone (PBN) (Sigma-Aldrich). PBN was dissolved in DMSO, and then diluted in sterile PBS and immediately injected subcutaneously into newborn C57BL/6 mice (10 mg/ml; 20 μl per animal; 10% DMSO final concentration).
Quantifications.
For quantitative analysis, 20 μm slices were cut using a cryostat starting at a random point within the hippocampal commissure. Eight slices, 100 μm distant from each other, were then quantified. The same number of slices was obtained from wild-type (WT) and mutated hippocampus. Caspase-3-positive cells and ethidium-positive cells were quantified using an upright microscope (Leica DMR and a Princeton Scientific Instruments CCD1300 camera). Contacts between caspase-3-positive or ethidium-positive cells and microglia were quantified using confocal microscopes (microscope; Leica DM5000B; spinning disk CSU10; Yokogawa; camera, COOLSNAP HQ2; Photometrics). Stacks were acquired sequentially, and then overlaid. Contacts were assessed in each slice of the stack (every micrometer).
Statistical analysis.
Quantifications were performed blindly to the phenotype or to the treatment on neonates from at least two different litters from different mothers. Quantitative data were analyzed by two-way ANOVA to compare WT and mutated or treated mice using Prism 3.0 computer software. Data are expressed as mean ± SEM. Values of p < 0.01 were considered statistically significant.
Results
Hippocampal apoptotic neurons contact microglia that express DAP12 and CD11b
Hippocampal developmental death takes place around birth (Knuesel et al., 2005). To compare this process with the elimination of pathogens after contact by the innate immune system, we first investigated the contact between microglia and apoptotic cells during this period. In the developing hippocampus, apoptotic cells were identified by their expression of activated caspase-3 (Fig. 1a) and their condensed or fragmented nuclei observed on 4′,6′-diamidino-2-phenylindole (DAPI) staining (Fig. 2f). At this stage, apoptosis mostly involves neurons (Ferrer et al., 1990a,b; Knuesel et al., 2005). Cells expressing activated caspase-3 were only found in the rostral part of the hippocampus, in the intermediate zone and the pyramidal cell layer of the subiculum (Fig. 1a) and to a lesser extent in the CA3 area (data not shown). Most apoptosis occurred during the early postnatal stages, between P0 and P1 (Fig. 1d). We examined the contacts between dying neurons and microglia characterized by their immunoreactivity against F4/80 (Fig. 1b,c). At P0, 51 ± 1% of the caspase-3-positive cells (208 cells from n = 3 animals) were in close contact with microglial processes (Fig. 1c). This proportion is comparable with that found in the cerebellum in which microglia promote neuronal death (Marín-Teva et al., 2004). This result favors the hypothesis of an active role of microglia in the neuronal apoptosis process. In immune cells, adhesive interactions leading to target cell death critically involve the immunoreceptor DAP12, and the β2-integrins CD11a and CD11b (Mócsai et al., 2006). As a first step toward the characterization of these proteins in developmental neuronal death induction, we characterized their expression in the developing hippocampus. No expression of CD11a was found (data not shown). In contrast, a subpopulation of microglia mostly localized in the subiculum and to a lesser extent in CA3 and in the fimbria expressed both CD11b and DAP12 (Fig. 2). Using confocal microscopy, we found that 96 ± 4% (310 cells from n = 3 animals) of the F4/80-positive microglia contacting activated-caspase-3-positive cells also expressed CD11b. In addition, as illustrated in Figure 2, d–f, all CD11b-positive microglia also expressed DAP12. Finally, we found that DAP12-expressing microglia also expressed TREM-2 (Fig. 2b,c), which is the DAP12-associated receptor (Bouchon et al., 2001) in microglia. Figure 2, d–f, shows that these microglia were contacting apoptotic cells characterized by their condensed DAPI staining. These results show that microglia that are in contact with apoptotic cells express CD11b, DAP12, and TREM-2.
Figure 1.

Apoptotic neurons in neonate hippocampus are in contact with microglia. Double labeling of P0 hippocampus with antibodies against activated caspase-3 (red) (a) and F4/80 (green) (b). The arrow and arrowhead indicate caspase-3-positive cells that are apposed or not with microglia, respectively. Scale bar, 100 μm. py, Pyramidal cell layer. c, Higher magnification of the subiculum. Activated caspase-3-positive cells are surrounded (arrow) or contacted (arrowhead) by microglial processes. Scale bar, 10 μm. d, Quantification of developmental death in the hippocampus between E17 and P3 in WT (white bar) or in DAP12KI (black bar). Shown are mean ± SEM. ***p < 0.0001; **p < 0.001.
Figure 2.

Microglia contacting apoptotic cells in the developing hippocampus express CD11b and DAP12 and TREM-2. a, Activated caspase-3-positive cells (red; arrowheads) are apposed to CD11b-positive microglia (green). py, Pyramidal cell layer. Scale bar, 50 μm. b, c, Double labeling of neonate hippocampus with anti-DAP12 antibody (b) and TREM-2 antibody (c). Scale bar, 20 μm. d–f, Triple labeling of P0 hippocampus with anti-DAP12 antibody (d), anti-CD11b antibody (e), and DAPI (f). The arrows indicate double-labeled microglia in contact with condensed nucleus. The arrowhead shows a condensed nucleus not in contact with labeled microglia. Scale bar, 50 μm.
Developmental death in the hippocampus requires CD11b and DAP12
We next investigated the role of microglial CD11b and DAP12 in the process of neuronal death. We first analyzed the activated caspase-3-positive cells in a mouse line bearing a loss-of-function mutation in the DAP12 gene [mouse line KΔ75 (Tomasello et al., 2000) referred to here as DAP12KI mutant]. Apoptotic cells could be found in the same hippocampal regions in WT and in DAP12KI mutant neonates (Fig. 3a,b). We next quantified the caspase-3-positive cells in hippocampus from embryonic day 17 (E17) to P3. As illustrated in Figure 3b, and quantified in Figure 1d, apoptosis is significantly decreased in DAP12KI mutant compared with WT hippocampus at P0 (WT, 7.8 ± 0.5 cells/slice; DAP12KI, 2.9 ± 0.3 cells/slice; n = 5 mice; p < 0.0001) and P1 (WT, 7.0 ± 0.6 cells/slice; DAP12KI, 4.7 ± 0.3 cells/slice; n = 3 mice; p = 0.0002), when the level of developmental death reaches its maximum in WT animals. No difference was observed before or after these developmental stages, indicating that DAP12 is required at P0 and P1 for developmental apoptosis in the hippocampus. In addition, microglia could be detected in the parenchyma of DAP12KI mutant hippocampus (Fig. 3b, inset) showing that the decreased apoptotic cell number in mutant is not attributable to the absence of microglia. We next investigated whether the integrin CD11b was involved in the induction of developmental death. For this purpose, we analyzed cell death in early postnatal hippocampus of mice mutated in the gene encoding CD11b (CD11b−/− mice) (van Spriel et al., 2001). As illustrated in Figure 3c, fewer caspase-3-positive cells were found in CD11b−/− P0 hippocampus compared with WT mice (WT, 7.8 ± 0.5 cells/slice; CD11b−/−, 3.5 ± 0.5 cells/slice; n = 5 mice; p < 0.0001). As in DAP12KI, microglia were present in the hippocampus of CD11b−/− mutant neonates (Fig. 3c, inset). The above-described experiments demonstrate that microglial DAP12 and CD11b are both required for developmental death in the hippocampus.
Figure 3.

Decreased developmental apoptosis on DAP12 or CD11b inactivation. Anti-activated caspase-3 immunoreactivity in the hippocampus of neonates: WT (a); DAP12KI (b); CD11b−/− (c). The inset shows the codetection of F4/80 immunoreactivity in the subicular region. d–g, Codetection of activated caspase-3-positive cells (red; arrows) and microglia (green) in the subicular region of WT (d, e) or DAP12KI (f, g) P1 mice injected 24 h earlier with rat anti-CD11b function-blocking antibody (d, f) or rat anti-F4/80b antibody (e, g). The green staining shows the detection of rat antibodies present in the tissue by labeling the slices with an anti-rat antibody coupled to Alexa 488 fluorophore without previous incubation with primary antibodies. Scale bars, 100 μm.
Deficiency of DAP12 or CD11b leads to a comparable decrease in apoptosis, suggesting that both mutations could alter the same pathway. If DAP12 and CD11b promote developmental death through different pathways, then their deficiencies should be cumulative. To test putative additive effects of DAP12 and CD11b deficiencies, we injected function-blocking anti-CD11b antibodies into lateral ventricles of P0 DAP12KI mice, and assessed the effect on developmental apoptosis 24 h later, at P1. As illustrated in Figure 3, d–g, function-blocking CD11b antibodies induced a significant decrease (26 ± 3%; n = 7 mice; p < 0.0001) in the number of apoptotic neurons when injected into WT hippocampus compared with the injection of nonrelevant F4/80 antibody, which is raised in the same species (rat) and is of the same IgG2b isotype. In contrast, CD11b-blocking antibody had no effect when injected into DAP12KI neonates (Fig. 3f,g) (F4/80, 5.6 ± 0.9 cells/slice; CD11b, 5.0 ± 0.7; n = 4 mice for each condition; p = 0.9287). We verified that the injected anti-CD11b or anti-F4/80 antibodies had reached microglia by labeling the hippocampal slices from WT or mutated injected animals with secondary anti-rat antibody coupled to Alexa 488 fluorophore without previous incubation with primary antibodies. Figure 3, d–g, shows that microglia in the subiculum were labeled by this secondary antibody. This shows that injection has not prevented microglial invasion of hippocampal parenchyma and that CD11b is expressed by DAP12KI microglia. Thus, the lack of effect of CD11b-blocking antibody is not attributable to the lack of CD11b expression or to the absence of microglia. The above-described experiments demonstrate that microglial CD11b and DAP12 act in the same pathway to promote developmental death of neurons.
Decreased production of superoxide by DAP12 or CD11b mutated microglia
One of the main methods macrophages or microglia use to achieve cell death is the production of ROS (Théry et al., 1991; Block et al., 2007). We took advantage of the property of superoxide ions to specifically oxidize DHE to ethidium, which then binds to the DNA or RNA in the cells in which it has been produced (Bindokas et al., 1996). We first assessed the production of in neonate hippocampus by analyzing the localization of ethidium in P0 mice injected with DHE. Ethidium fluorescence was almost exclusively restricted to the subiculum (Fig. 4a) and CA3 regions (data not shown) in the rostral part of the hippocampus, which are the regions of high level of developmental apoptosis (Figs. 1, 3). In addition, 70 ± 6% of the ethidium-positive cells were F4/80-positive microglia (211 cells from n = 6 animals) (Fig. 4a,b). Up to 30 ± 4% (156 cells from n = 4 mice) of these superoxide-producing microglia were contacting apoptotic neurons (Fig. 4c). The remaining superoxide-producing microglia are most probably contacting neurons that have not yet triggered the apoptotic program and thus do not express activated caspase-3.
Figure 4.
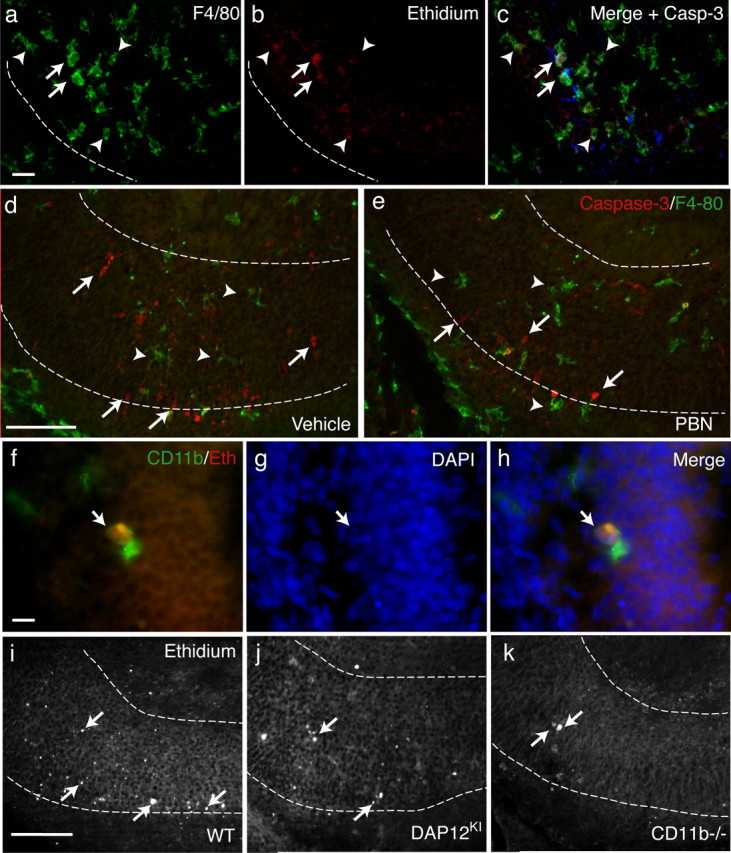
DAP12- and CD11b-dependent production of superoxide ions induces the developmental death of neurons. a–c, In the subiculum of WT neonate, triple detection of microglia by F4/80 immunoreactivity (a); cells producing superoxide ions revealed by the oxidation of DHE into red-fluorescent ethidium (b). c, Merge image with activated caspase-3 staining (blue). Some ethidium-producing microglia contact apoptotic cells (arrows) but most do not (arrowheads). Scale bar, 20 μm. d, e, Double detection of caspase-3 labeling (red) and F4/80 immunoreactivity (green) in hippocampus on systemic injection of vehicle (d) or free radical scavenger PBN (e). Scale bar, 50 μm. f, High magnification of a CD11b-positive microglia (green) that has accumulated ethidium (red). Scale bar, 10 μm. g, DAPI staining revealed condensed nucleus (arrow) of apoptotic cells. h, Merge image showing a contact between superoxide producing CD11b-positive microglia and the apoptotic nucleus. i–k, Oxidation of DHE in WT (i), DAP12KI (j), or CD11b−/− (k) P0 hippocampus. Scale bar, 50 μm. The arrows indicate ethidium-positive cells.
To demonstrate a direct role of microglial superoxide ions in the induction of apoptosis, we analyzed the effect of the free radical scavenger PBN, which efficiently traps ROS including (Carney et al., 1991). We administered PBN systemically via subcutaneous injection in wild-type neonates. Figure 4, d and e, shows that, 3 h after PBN injection, a significant decrease in neuronal death in P0 hippocampus was observed compared with the vehicle-treated controls (vehicle, 10.7 ± 1.2 cells/slice; PBN, 7.1 ± 0.6 cells/slice; p < 0.0001; n = 6 mice). Yet, microglia are still present in the parenchyma of the hippocampus after PBN treatment, as illustrated in Figure 4e. This demonstrates that, during hippocampus development, superoxide produced by microglia can induce neuronal death.
In neutrophils, the production of reactive oxygen radicals is integrin-dependent and requires DAP12 (Mócsai et al., 2006). In developing hippocampus, we found that all the ethidium-positive microglia expressed the CD11b integrin (Fig. 4f). To investigate whether ROS production is dependent on the CD11b/DAP12 pathway, we injected DAP12KI and CD11b−/− neonates with DHE, and quantified the number of ethidium-positive microglia. Figure 4, i–k, shows that, in DHE-injected DAP12KI or CD11b−/− mice, ethidium fluorescence was found in the same regions as in WT, but in a significantly lower number of microglia (WT, 11.0 ± 0.5 cells/slice; DAP12KI, 7.1 ± 0.5 cells/slice; n = 3 mice; p < 0.0001; CD11b−/−, 6.9 ± 0.6 cells/slice; p = 0.0026; n = 3 mice). We conclude that the production of the superoxide ions by microglia is DAP12-dependent. In DAP12KI hippocampus, among the remaining ethidium-positive microglia, the same proportion of cells was contacting dying neurons as in WT mice (DAP12KI, 34 ± 2%; 181 cells from n = 5 animals; p = 0.1724; CD11b−/−, 35 ± 4%; 138 cells from n = 3 animals; p = 0.4578). This shows that, when ROS has been produced by microglia, DAP12 and CD11b are no more required to achieve the death of neurons.
Discussion
In the cerebellum, developmental death of Purkinje cells is achieved through the production of superoxide ions by contacting microglia (Marín-Teva et al., 2004). However, the microglial proteins and mechanisms that trigger microglial induction of neuronal death remain unknown. We have now identified microglial factors involved in death induction in the developing hippocampus. Our data support a model in which developmental death of neurons in the hippocampus is triggered by microglial CD11b and DAP12 that cooperate to promote the production of superoxide ions on contact with neurons committed to die. TREM-2 is a DAP12-associated receptor (Bouchon et al., 2001) and is coexpressed with CD11b and DAP12 in microglia. TREM-2 may also be involved in this induction of developmental neuronal death. The fact that residual apoptosis is observed on DAP12 or CD11b deficiency or when superoxide ions are trapped indicates that, in developing hippocampus, these factors act synergistically with other yet-uncharacterized pathways to induce neuronal death. Microglia and macrophages produce a wide variety of factors including NGF, TNFα, IL-1β (interleukin-1β), NO (nitric oxide), or glutamate, which can trigger apoptosis in cultured neurons (Piani et al., 1991; Chao et al., 1992) (for review, see Bessis et al., 2007) or in explants (Frade and Barde, 1998; Sedel et al., 2004). Although some of these factors are involved in developmental death in the hippocampus (data not shown), their synergistic action with the DAP12/CD11b pathway remains to be demonstrated.
The developmental process that we have now described resembles the recognition and death induction of exogenous or altered cells by innate immunity cells (Mócsai et al., 2006) such as macrophages or neutrophils. Neutrophils fight against invading microorganisms by producing a CD11a and CD11b integrin-dependent respiratory burst that requires DAP12 signaling together with signaling through FcRγ, another ITAM-bearing molecule (Mócsai et al., 2004, 2006). In the developing hippocampus, we did not detect CD11a expression, but FcRγ (Loughlin et al., 1992) and several integrins are expressed by microglia (Milner and Campbell, 2003) and might also promote the developmental death of neurons. However, in both the immune and nervous systems, the molecular interactions between the ITAM-containing and the integrins remain to be understood.
In nervous tissue, dying cells are eliminated by microglia (Ferrer et al., 1990b). The decreased number of caspase-3-positive cells found in mutant context could be attributable to increased phagocytosis of dying neurons by DAP12- or CD11b-deficient microglia. This possibility is, however, unlikely because DAP12 mutation does not increase the phagocytic activity of cultured microglia (Takahashi et al., 2005). Furthermore, we show that, in the DAP12KI and in the CD11b−/− hippocampus, there are fewer ROS-producing microglia than in WT, but these microglia contact the same proportion of dying neurons than WT ROS-positive microglia. This indicates that the alterations of death induction induced by DAP12 deficiency occur upstream of microglial production of superoxide.
In some regions of the developing hippocampus, such as the fimbria, no apoptotic cells are found in the vicinity of microglia expressing both DAP12 and CD11b. The expression of DAP12 and CD11b is thus not sufficient to trigger the death of neurons. This suggests that the neurons committed to die have to express a kill-me signal that activates the microglial pathway of death induction. The best candidates are the ligands of the DAP12/TREM-2 receptor (Bouchon et al., 2001), which are unknown, or ligands of CD11b, such as ICAM-1 (Diamond et al., 1990) or the C3 complement proteins (Beller et al., 1982). In the immune system, binding of the β2-integrins to the C3b or iC3b components of the complement is a signal of innate recognition of nonself (Savill et al., 2002). It has recently been proposed that, in the nervous system, complement proteins might be a tag for the elimination of unwanted synapses (Stevens et al., 2007). It cannot be excluded that these proteins also label neurons developmentally committed to die.
Our findings strongly suggest that the physiological mechanisms that we have described here in developing hippocampus might also be relevant for traumatic or degenerative alterations of neuronal tissues. In brain pathologies or injury, microglial activation, often characterized by increased CD11b expression, are consistently observed (Kreutzberg, 1996) and are concomitant with pathological elimination of neurons (Block et al., 2007). In some cases, such as in axotomy of motoneurons (Zujovic et al., 2005), or in ischemia (Hedtjärn et al., 2004), DAP12 reexpression has also been demonstrated. In addition, it is known that injury-activated microglia generate massive quantities or reactive oxygen species that are associated with pathological processes leading to apoptosis (Lewén et al., 2000). These data suggest that, after brain injuries or degenerative processes and as observed during development, a CD11b/DAP12-dependent ROS production could contribute to neuronal death. In animal models of Parkinson's disease, microglia produce superoxide ions responsible for the degeneration of dopaminergic neurons (Wu et al., 2003), associated with an involvement of CD11b (Pei et al., 2007). We anticipate that the description of the developmental microglia-induced neuronal death will allow novel approaches of pathological neuronal death associated with neuroinflammation.
Footnotes
This work was supported by grants from Agence Nationale de la Recherche–Maladies Rares, Fédération pour la Recherche sur le Cerveau, and Institut de Recherche sur la Moelle Épinière et l'Encephale. A.B. is an investigator of the Centre National de la Recherche Scientifique. We gratefully thank Jeanette Leusen for the gift of the CD11b mice; Michel Mallat for the DHE, helpful advice, and comments on this manuscript; and Drs. Serge Marty, Marion Wassef, and Clothilde Théry for critical reading of this manuscript.
References
- Abram CL, Lowell CA. Convergence of immunoreceptor and integrin signaling. Immunol Rev. 2007;218:29–44. doi: 10.1111/j.1600-065X.2007.00531.x. [DOI] [PubMed] [Google Scholar]
- Beller DI, Springer TA, Schreiber RD. Anti-Mac-1 selectively inhibits the mouse and human type three complement receptor. J Exp Med. 1982;156:1000–1009. doi: 10.1084/jem.156.4.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessis A, Béchade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2007;55:233–238. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bouchon A, Hernández-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194:1111–1122. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JM, Starke-Reed PE, Oliver CN, Landum RW, Cheng MS, Wu JF, Floyd RA. Reversal of age-related increase in brain protein oxidation, decrease in enzyme activity, and loss in temporal and spatial memory by chronic administration of the spin-trapping compound N-tert-butyl-alpha-phenylnitrone. Proc Natl Acad Sci U S A. 1991;88:3633–3636. doi: 10.1073/pnas.88.9.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, Hibbs ML, Springer TA. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18) J Cell Biol. 1990;111:3129–3139. doi: 10.1083/jcb.111.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Serrano T, Soriano E. Naturally occurring cell death in the subicular complex and hippocampus in the rat during development. Neurosci Res. 1990a;8:60–66. doi: 10.1016/0168-0102(90)90058-m. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Bernet E, Soriano E, del Rio T, Fonseca M. Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience. 1990b;39:451–458. doi: 10.1016/0306-4522(90)90281-8. [DOI] [PubMed] [Google Scholar]
- Frade JM, Barde YA. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron. 1998;20:35–41. doi: 10.1016/s0896-6273(00)80432-8. [DOI] [PubMed] [Google Scholar]
- Hedtjärn M, Mallard C, Hagberg H. Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 2004;24:1333–1351. doi: 10.1097/01.WCB.0000141559.17620.36. [DOI] [PubMed] [Google Scholar]
- Knuesel I, Elliott A, Chen HJ, Mansuy IM, Kennedy MB. A role for synGAP in regulating neuronal apoptosis. Eur J Neurosci. 2005;21:611–621. doi: 10.1111/j.1460-9568.2005.03908.x. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lewén A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- Loughlin AJ, Woodroofe MN, Cuzner ML. Regulation of Fc receptor and major histocompatibility complex antigen expression on isolated rat microglia by tumour necrosis factor, interleukin-1 and lipopolysaccharide: effects on interferon-gamma induced activation. Immunology. 1992;75:170–175. [Google Scholar]
- Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41:535–547. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- Milner R, Campbell IL. The extracellular matrix and cytokines regulate microglial integrin expression and activation. J Immunol. 2003;170:3850–3858. doi: 10.4049/jimmunol.170.7.3850. [DOI] [PubMed] [Google Scholar]
- Mócsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, Majumdar S, Lanier LL, Lowell CA, Nakamura MC. The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci U S A. 2004;101:6158–6163. doi: 10.1073/pnas.0401602101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mócsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, Srimal S, Farber C, Sanchez E, Kabbash L, Asch A, Gailit J, Wright SD. Cytokine-induced respiratory burst of human neutrophils: dependence on extracellular matrix proteins and CD11/CD18 integrins. J Cell Biol. 1989;109:1341–1349. doi: 10.1083/jcb.109.3.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloneva J, Kestilä M, Wu J, Salminen A, Böhling T, Ruotsalainen V, Hakola P, Bakker AB, Phillips JH, Pekkarinen P, Lanier LL, Timonen T, Peltonen L. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet. 2000;25:357–361. doi: 10.1038/77153. [DOI] [PubMed] [Google Scholar]
- Pei Z, Pang H, Qian L, Yang S, Wang T, Zhang W, Wu X, Dallas S, Wilson B, Reece JM, Miller DS, Hong JS, Block ML. MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. Glia. 2007;55:1362–1373. doi: 10.1002/glia.20545. [DOI] [PubMed] [Google Scholar]
- Piani D, Frei K, Do KQ, Cuénod M, Fontana A. Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett. 1991;133:159–162. doi: 10.1016/0304-3940(91)90559-c. [DOI] [PubMed] [Google Scholar]
- Roumier A, Béchade C, Poncer JC, Smalla KH, Tomasello E, Vivier E, Gundelfinger ED, Triller A, Bessis A. Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J Neurosci. 2004;24:11421–11428. doi: 10.1523/JNEUROSCI.2251-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- Sedel F, Béchade C, Vyas S, Triller A. Macrophage-derived tumor necrosis factor α, an early developmental signal for motoneuron death. J Neurosci. 2004;24:2236–2246. doi: 10.1523/JNEUROSCI.4464-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–2278. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry C, Chamak B, Mallat M. Cytotoxic effect of brain macrophages on developing. Eur J Neurosci. 1991;3:1155–1164. doi: 10.1111/j.1460-9568.1991.tb00050.x. [DOI] [PubMed] [Google Scholar]
- Tomasello E, Desmoulins PO, Chemin K, Guia S, Cremer H, Ortaldo J, Love P, Kaiserlian D, Vivier E. Combined natural killer cell and dendritic cell functional deficiency in KARAP/DAP12 loss-of-function mutant mice. Immunity. 2000;13:355–364. doi: 10.1016/s1074-7613(00)00035-2. [DOI] [PubMed] [Google Scholar]
- van Spriel AB, Leusen JH, van Egmond M, Dijkman HB, Assmann KJ, Mayadas TN, van de Winkel JG. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood. 2001;97:2478–2486. doi: 10.1182/blood.v97.8.2478. [DOI] [PubMed] [Google Scholar]
- Vivier E, Malissen B. Innate and adaptive immunity: specificities and signaling hierarchies revisited. Nat Immunol. 2005;6:17–21. doi: 10.1038/ni1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zujovic V, Luo D, Baker HV, Lopez MC, Miller KR, Streit WJ, Harrison JK. The facial motor nucleus transcriptional program in response to peripheral nerve injury identifies Hn1 as a regeneration-associated gene. J Neurosci Res. 2005;82:581–591. doi: 10.1002/jnr.20676. [DOI] [PubMed] [Google Scholar]