

Abstract
Reduced activity of the mitochondrial respiratory chain and in particular of complex I is implicated not only in the etiology of Parkinson's disease but also in other forms of parkinsonism in which striatal neurodegeneration occurs, such as progressive supranuclear palsy.
The pesticide rotenone inhibits mitochondrial complex I and reproduces features of these basal ganglia neurological disorders in animal models. We have characterized the electrophysiological effects of rotenone in the striatum as well as potential neuroprotective strategies to counteract the detrimental effects of this neurotoxin. We found that rotenone causes a dose-dependent and irreversible loss of the corticostriatal field potential amplitude, which was related to the development of a membrane depolarization/inward current in striatal spiny neurons, coupled to an increased release of both excitatory amino acids and dopamine (DA).
In particular, we have investigated whether glutamate, DA, and GABA systems might represent possible targets for neuroprotection against rotenone-induced striatal neuronal dysfunction. Interestingly, whereas modulation of glutamatergic transmission was not neuroprotective, blockade of D2-like but not D1-like DA receptors significantly reduced the rotenone-induced effects via a GABA-mediated mechanism. In addition, because antiepileptic drugs (AEDs) modulate multiple transmitter systems, we have analyzed the possible neuroprotective effects of some AEDs against rotenone. We found that carbamazepine, unlike other tested AEDs, exerts a potent neuroprotective action against rotenone-induced striatal neuronal dysfunction. This neuroprotection was observed at therapeutically relevant concentrations requiring endogenous GABA. Differential targeting of GABAergic transmission may represent a possible therapeutic strategy against basal ganglia neurodegenerative disorders involving mitochondrial complex I dysfunction.
Keywords: antiepileptic drugs, dopamine, GABA, Parkinson's disease, rotenone, striatum
Introduction
Reduced activity of the mitochondrial respiratory chain and particularly complex I are implicated in the etiology of both Parkinson's disease (PD) and progressive supranuclear palsy (PSP), although these degenerative diseases differ substantially in their clinical features, distinctive pattern of neuronal cell loss, and pathological hallmarks. In PD, dopaminergic neurons in the substantia nigra (SN) degenerate. This degeneration is coupled to the formation of Lewy bodies, spherical proteinaceous structures containing the presynaptic protein α-synuclein (Spillantini et al., 1998; Braak and Braak, 2000).
Conversely, PSP is an atypical parkinsonian syndrome characterized by dorsorostral midbrain atrophy associated with severe neuronal loss in the substantia nigra, loss of striatal GABAergic projecting spiny neurons, and cerebral tau protein pathology (Daniel et al., 1995; Burn and Lees, 2002; Pearce, 2007).
Experimental studies have reported that generalized complex I inhibition by rotenone damages both nigral and striatal neurons, in a pattern reminiscent of PSP pathology (Ferrante et al., 1997; Höglinger et al., 2003, 2005, 2006; Centonze et al., 2006).
The inhibition of the complex I shifts the cell into a state of oxidative stress and triggers several bidirectional links with the excitotoxic death pathway that is believed to substantially contribute to neuronal death in both PD and PSP. In a cellular model of PD, rotenone-mediated neurotoxicity was primarily associated with increased oxidative stress and/or proteasomal dysfunction (Betarbet et al., 2000; Sherer et al., 2002, 2003a,b, 2007).
Glutamate-mediated excitotoxicity is believed to substantially contribute to neuronal death during degenerative processes, and evidence exists that, at least under certain circumstances, mitochondrial impairment may sensitize neurons to glutamate NMDA receptor-mediated excitotoxicity (Greene and Greenamyre, 1996; Luetjens et al., 2000; Calabresi et al., 2001). One of the possible explanations for the cell-type-specific vulnerability induced by rotenone in the basal ganglia is the dopamine (DA) dependence of the rotenone-induced neurodegeneration. In fact, it is possible that the high endogenous DA levels present in both the pars compacta of the substantia nigra and the striatum render these structures selectively prone to toxicity induced by mitochondrial complex inhibition.
Glutamate and DA signaling pathways could act synergistically to modulate mitochondrial dysfunction and striatal neurodegeneration (Di Filippo et al., 2006; Calabresi et al., 2007). This detrimental synergism could generate a downstream effect causing loss and/or dysfunction of striatal GABAergic spiny neurons.
The aim of the present study is to investigate the acute electrophysiological effects of the inhibition of mitochondrial complex I by rotenone and to analyze whether glutamate, DA, and GABA systems are possible targets for neuroprotection against rotenone-induced striatal neuronal dysfunction. Moreover, because antiepileptic drugs (AEDs) modulate multiple transmitter systems (Calabresi et al., 2003), we have analyzed the possible neuroprotective effects of some AEDs against rotenone-induced neurotoxicity.
Materials and Methods
Preparation and maintenance of slices for electrophysiological recordings.
All the experiments were conducted in conformity with the European Communities Council Directive of November 1986 (86/609/ECC). Corticostriatal coronal slices (thickness, 270 μm) and hippocampal slices (thickness, 400 μm) were cut from 1 to 2-month-old male Wistar rats (n = 120) (Charles River Laboratories) using a vibratome. Preparation and maintenance of rat corticostriatal slices have been described previously (Calabresi et al., 1997; Picconi et al., 2003). The hippocampus was extracted and cut in ice-cold artificial CSF (ACSF) with a vibratome into transverse slices, which were allowed to recover in oxygenated ACSF at 30°C for 30 min and then at room temperature for an additional 1–2 h before experimental recordings. A single slice was then transferred to a recording chamber and submerged in a continuously flowing Krebs' solution (34°C; 2.5–3 ml/min) bubbled with a 95% O2–5% CO2 gas mixture. The composition of the solution was as follows (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 10 glucose, and 25 NaHCO3. Drugs were bath applied by switching the solution to one containing known concentrations of drugs. Total replacement of the medium in the chamber occurred within 1 min.
Electrophysiology.
Intracellular recordings of striatal medium spiny neurons were obtained by using sharp microelectrodes pulled from borosilicate glass pipettes backfilled with 2 mol/L KCl (30–60 MΩ). An Axoclamp 2B amplifier (Molecular Devices) was connected in parallel to an oscilloscope (Hameg Instruments) to monitor the signal in “bridge” mode and to a personal computer for acquisition of the traces using a pClamp9 software (Molecular Devices). After the impalement of the neuron, a small amount of current (5–20 pA) might be injected via the recording electrode when necessary. Only neurons electrophysiologically identified as spiny neurons were considered for the experiments (Calabresi et al., 1997).
For patch-clamp recordings, neurons were visualized using differential interference contrast (Nomarski) and infrared microscopy (Olympus Europa). Whole-cell recordings (voltage clamp at −80 mV) were performed with borosilicate glass pipettes (4–7 MΩ) filled with intracellular solution containing with the following (in mm): 145 K+-gluconate, 0.1 CaCl2, 2 MgCl2, 0.1 EGTA, 10 HEPES, 0.3 GTP, and 2 Mg-ATP, adjusted to pH 7.3 with KOH. Signals were amplified with a Multiclamp 700B amplifier, recorded, and stored on a personal computer using pClamp10 (Molecular Devices). Whole-cell access resistance was 5–30 MΩ. The rotenone current–voltage relationship (I–V) was investigated by measuring ramp I–V values (1.5 s ramp −80 to +40 mV executed in the presence of 1 μm rotenone).
Electrodes for extracellular recordings were filled with 2 mol/L NaCl (15–20 MΩ). An Axoclamp 2B amplifier (Molecular Devices) was used for extracellular recordings. The field potential amplitude was defined as the average of the amplitude from the peak of the early positivity to the peak negativity and the amplitude from the peak negativity to peak late positivity (Costa et al., 2004, 2006).
A corticostriatal field potential was evoked every 10 s by means of a bipolar electrode connected to a stimulator unit (Grass Telefactor). The stimulating electrode was located in the cortical areas close to the recording electrode or in the white matter between the cortex and the striatum to activate corticostriatal fibers. The recording electrodes were invariably placed within the striatum. For hippocampal field potentials measurements, the stimulating electrode was inserted, under visual control, into the Schaffer collateral fibers, and the recording electrode was inserted into CA1 region. Quantitative data are expressed as a percentage of the field potential amplitudes with respect to the relative control amplitude values, the latter representing the mean of responses recorded during a stable period (15–20 min). All the putative neuroprotective drugs used in the study were applied 15–20 min before the onset of the bath application of rotenone. Off-line analysis was performed using Clampfit (Molecular Devices) and Microcal Origin software. ANOVA and Bonferroni's post hoc test were used for statistical analysis. Values given in the figures and text are mean ± SEM. The significance level was established at *p < 0.05, **p < 0.01, and ***p < 0.001.
Preparation of synaptosomes.
Two-month-old Wistar rats were housed at constant temperature (22 ± 1°C) and relative humidity (50%) under a regular light/dark schedule (lights on from 7:00 A.M. to 7:00 P.M.). Food and water were available ad libitum. The animals were killed by decapitation and striata were rapidly dissected at 0–4°C. Animal care and experimental procedures were conducted in compliance with the institutional guidelines and with international laws and policies (European Communities Council Directive of November 24, 1986, 86/609/EEC).
Rat striata were homogenized in 40 vol of 0.32 m sucrose buffered at pH 7.4 with phosphate (final concentration, 0.04 m). The homogenates were centrifuged at 1000 × g for 5 min to remove nuclei and cellular debris, and synaptosomal fractions were isolated from the supernatants by centrifugation at 12,000 × g for 20 min. The synaptosomal pellets were then resuspended in a standard physiological medium having the following composition (mm): 125 NaCl, 3 KCl, 1.2 MgSO4, 1.2 CaCl2, 1 NaH2PO4, 22 NaHCO3, and 10 glucose (aeration with 95% O2 and 5% CO2), pH 7.2–7.4.
Release experiments.
Rat striatal synaptosomes were incubated in the presence of [3H]d-aspartate ([3H]d-ASP) (final concentration, 80 nm) or in the presence of [3H]dopamine ([3H]DA) (final concentration, 50 nm) at 37°C, for 15 min, in a rotary water bath and in an atmosphere of 95% O2 and 5% CO2. After labeling, identical aliquots of the synaptosomal suspensions were layered on microporous filters at the bottom of parallel superfusion chambers (Raiteri et al., 1974) thermostated at 37°C. Synaptosomes were then superfused at 37°C with standard physiological solution (flow rate, 0.5 ml/min) aerated with 95% O2 and 5% CO2 for a total time of 57 min. After 36 min of superfusion to equilibrate the system, seven consecutive 3-min fractions were collected. Synaptosomes were exposed to rotenone starting at the end of the second fraction collected (t = 42 min) until the end of the superfusion. When used, transporter blockers dl-threo-β-benzyloxyaspartic acid (dl-TBOA) or 1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-hydroxy-3-phenylpropyl) piperazinyl decanoate (GBR12909) were introduced 11 min before rotenone. When indicated, at t = 20 min of superfusion, standard physiological solution was replaced with a solution from which Ca2+ ions were omitted. Appropriate controls were always run in parallel. Fractions collected and superfused synaptosomes were then counted for radioactivity.
The amount of radioactivity released into each superfusate fraction was expressed as a percentage of the total synaptosomal tritium content at the start of the fraction collected (fractional efflux). The amount of drug effects were expressed as percentage of increase over basal release and were evaluated as the ratio between the percentage of tritium released into each fraction after the first (b2–b7) and that in the first fraction collected (b1). These ratios were compared with the corresponding values obtained under control conditions (no rotenone added). Differences were analyzed by nonparametric Kruskal–Wallis test followed by Mann–Whitney U test. Data were considered significant at the level of p < 0.05.
Rats with 6-hydroxydopamine-induced striatal DA-denervation.
Nine deeply anesthetized rats were injected with 6-hydroxydopamine (6-OHDA) (8 μg/4 μl saline containing 0.1% ascorbic acid) into the medial forebrain bundle at a rate of 0.38 μl/min (Picconi et al., 2003). Seven rats were injected only with vehicle at the same coordinates. Fifteen days later, the rats were tested with 0.05 mg/kg subcutaneous apomorphine, and contralateral turns to the lesion were counted for 40 min. The rats who showed >200 contralateral turns were put in the group of the DA-denervated animals. Sham-operated animals did not show turning behavior. One and a half months after the lesion, the rats were used for electrophysiological experiments. The severity of the lesion was confirmed afterward by striatal and nigral tyrosine hydroxylase immunohistochemistry (Picconi et al., 2003).
Immunohistochemistry.
After decapitation, the caudal parts of the brain corresponding to the SN were kept in 4% paraformaldehyde for 1 d and finally stored in 25% phosphate-buffered (PB) sucrose for 24–48 h before cutting. Forty-micrometer-thick coronal sections were cut on a microtome (Microm) and collected in sequence in vials containing 1 ml of PB. Sections at the level of the SN were processed free floating for double immunofluorescence using antibodies against tyrosine hydroxylase (TH) and neuron-specific nuclear antigen (NeuN). Sections were incubated with a mixture of the following antibodies: mouse anti-NeuN (1:200; IgM; Millipore Bioscience Research Reagents) and rabbit anti-TH (1:500; Santa Cruz Biotechnology). After incubation with primary antibodies and subsequent washing with PB, sections were incubated in a mixture of secondary antibodies: cyanine 3 (Cy3)-conjugated donkey anti-mouse IgM and Cy2-conjugated donkey anti-rabbit IgG (1:100; Jackson ImmunoResearch). Sections were mounted in gelatin-coated slides, air dried, and coverslipped with Gel-Mount (Biomeda).
For each section, a picture of the SN was taken using the 10× magnification of a confocal microscope CLSM (LSM 510; Carl Zeiss).
Mice transgenic for truncated human α-synuclein (1–120).
Seven mice transgenic for truncated human α-synuclein (1–120), generated as reported previously (Tofaris et al., 2006), and six wild-type (WT) mice were used for electrophysiological recordings. In these transgenic mice (TG Syn 120), the expression of truncated human α-synuclein (1–120), driven by the tyrosine hydroxylase promoter on a mouse α-synuclein null background, leads to the formation of pathological inclusions in the substantia nigra and olfactory bulb and to the reduction in striatal DA levels. At the behavioral level, the transgenic mice show a progressive reduction in spontaneous locomotion (Tofaris et al., 2006).
Chemicals.
Powders were dissolved in water or DMSO and then stored at −20°C in aliquots. Each aliquot was only used the day of experiment and then discarded. Drugs were applied by dissolving them to the desired final concentration in the external Krebs' solution. l-2-Amino-5-phosphonovaleric acid (l-APV), dl-TBOA, 3-aminopropyl-cyclohexylmethyl phosphinic acid (CGP46381), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), GBR12909, memantine hydrochloride, picrotoxin, SCH23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride], l-sulpiride, and tetrodotoxin (TTX) were from Tocris Bioscience. Bicuculline, carbamazepine (CBZ), 6-OHDA, lamotrigine (LTG), MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate], 3-nitropropionic acid (3-NP), rotenone, and valproic acid (VPA) were from Sigma-Aldrich. Topiramate (TPM) was from Johnson & Johnson. [3H]d-Aspartate (specific activity, 38 Ci/mmol) and [3H]dopamine (specific activity, 38 Ci/mmol) were purchased from GE Healthcare.
Results
Action on rotenone on extracellularly recorded striatal field potentials
To test whether inhibition of mitochondrial complex I by rotenone was able to alter the electrical neuronal activity in a corticostriatal brain slice preparation, field potentials were recorded from the striatum after the activation of glutamatergic corticostriatal inputs. As shown in Figure 1A, rotenone induced a progressive reduction of the field potential amplitude. After 30–35 min of application of this toxin, the loss of the field potential amplitude was almost complete by using a dose of 1 μm. As shown in Figure 1A, the effect of rotenone was concentration dependent (at least n = 10 for each concentration). After the washout of this toxin, no recovery of the field potential was observed at all doses used (n = 10 for each concentration) (data not shown). This observation suggests that rotenone causes irreversible electrophysiological alterations in striatal neurons probably reflecting neuronal death (Centonze et al., 2006; Tozzi et al., 2007).
Figure 1.

The mitochondrial complex I inhibitor rotenone reduces the corticostriatal field potential and produces an irreversible membrane depolarization of striatal spiny neurons. A, Time course of the mean field potential amplitude measured in the presence of rotenone 0.1, 0.3, and 1 μm bath applied for 35 min. The traces represent a field potential recorded in control condition and at 20 and 30 min after the application of 1 μm rotenone. B, Representative trace showing the membrane depolarization of a spiny neuron in the presence of 1 μm rotenone and time course of the mean membrane potential amplitude of spiny neurons recorded in control condition and in the presence of 1 μm rotenone. C, Time course of the mean field potential amplitude recorded in the presence of 1, 30, and 100 μm rotenone, applied for 35 min in CA1 hippocampal slices. The traces represent a field potential recorded in control condition and at 20 and 30 min after the application of 100 μm rotenone. In Figures 1–9, the number of experiments represented in the graphs are indicated in text.
Action of rotenone on striatal spiny neurons intracellularly recorded in vitro
The effect of rotenone was also investigated by using intracellular recordings from 58 electrophysiologically identified principal spiny neurons. The main characteristics of these cells have been described in detail previously (Calabresi et al., 1997, 2001). These cells had high resting membrane potential (−84 ± 5 mV), low apparent input resistance (39 ± 9 MΩ), and action potentials of short duration (1.1 ± 0.3 ms) and high amplitude (102 ± 4 mV). These cells were silent at rest and showed tonic firing activity during a long-duration depolarizing pulse. At the concentration of 1 μm, rotenone induced a slow membrane depolarization starting after 5–10 min from the onset of the perfusion (n = 35) (Fig. 1B). The rotenone-induced depolarization reached a plateau after 30 min from the onset of the application, when the membrane potential of the neuron was approximately −20 mV (Fig. 1B). Also in this case, the electrophysiological effect induced by the toxin was irreversible after its washout (n = 5) (data not shown). It is worth noting that the rotenone-induced membrane depolarization was coupled to a reduction of the input resistance of the recorded cells (n = 7) (data not shown).
Action of rotenone on CA1 hippocampal field potentials
To better estimate the specificity of the toxic action of rotenone in striatum, we compared the effect of rotenone on corticostriatal field potentials with that one observed on CA1 hippocampal field potentials (1, 30, and 100 μm) (n = 7 for each concentration). The CA1 hippocampal area is, in fact, a brain region highly vulnerable to energy deprivation induced by either ischemia or oxygen and glucose deprivation (Sims and Pulsinelli, 1987; Calabresi et al., 2000). Field potentials were recorded in the CA1 region after the activation of collateral Schaffer inputs. Interestingly, as reported in Figure 1C, 1 μm rotenone produced, at 35 min, only a slight reduction of the hippocampal field potential amplitude (17 ± 4.8%), suggesting a considerably less pronounced sensitivity of this structure to rotenone compared with striatum. Accordingly, higher concentrations (30 and 100 μm) of rotenone were required to produced an almost complete loss of the hippocampal CA1 field potentials (see traces and time course in Fig. 1C).
Rotenone produces an inward current in striatal spiny neurons recorded by whole-cell patch-clamp configuration
To further characterize the response of striatal spiny neurons to rotenone, we performed whole-cell patch-clamp recordings from these cells. Also in these experimental configuration, spiny neurons exhibited tonic firing discharge evoked by depolarizing current pulses (Fig. 2A) and inward rectification detected by current–voltage plots (Fig. 2B,C). The mean holding current for these cells was +98.1 ± 8.9 pA (n = 12; Vhold = −80 mV). Bath application of 1 μm rotenone produced an inward current (Fig. 2D,E) whose time course mimicked the membrane depolarization observed in current-clamp experiments performed with sharp electrodes (Fig. 1B). The mean amplitude of the rotenone-induced inward current, measured at 30 min from the onset of the application, was +267 ± 27.8 pA (n = 9) (Fig. 2D,E). We also measured the reversal potential of this current by applying voltage ramps (from −80 to +40 mV) before and during the application of rotenone (25–30 min from the onset of the application). The reversal potential was −18 ± 5 mV (n = 5) (data not shown).
Figure 2.

Inhibition of mitochondrial complex I produces an inward current in striatal spiny neurons. A, B, I–V traces of a striatal spiny neuron recorded in whole-cell patch clamp in current-clamp (A) and voltage-clamp (B) mode. C, Current–voltage relationship of a pool of striatal spiny neurons obtained during hyperpolarizing and depolarizing steps of current. D, Time course of the mean inward current produced in the presence of 1 μm rotenone bath applied for 40 min. E, Inward current produced in the presence of 1 μm rotenone applied for 30 min in an whole-cell patch-clamp experiment.
Rotenone enhances glutamate and DA release from striatal synaptosomes by reverting DA and glutamate carrier activity
Both endogenous glutamate and DA might play a role in the rotenone-induced toxicity (Greene and Greenamyre, 1996; Di Filippo et al., 2006; Höglinger et al., 2006). Thus, we investigated whether rotenone was able to modulate the release of excitatory amino acids and DA.
Exposure of rat striatal synaptosomes to rotenone elicited a marked and prolonged release of [3H]d-ASP. The toxin caused the release of [3H]d-ASP in a concentration-dependent manner, being maximally active when added up to 0.1–1 μm (Fig. 3A) (n = 4 in triplicate; *p < 0.05). Two main mechanisms of exit usually account for neurotransmitter release from isolated nerve terminals: the Ca2+-dependent exocytosis and/or the reversal of neurotransmitter transporters. To investigate the mode of exit of [3H]d-ASP when synaptosomes were exposed to the neurotoxin, we evaluated on one hand the effects of Ca2+ omission from the external medium, and, on the other hand, the consequences of blockade of glutamate membrane transporter. We focused on the release of [3H]d-ASP evoked by 0.1 μm, because, at this concentration, the neurotoxin was found to be maximally active. The release of [3H]d-ASP evoked by 0.1 μm rotenone was significantly reduced by dl-TBOA (10 μm), a selective blocker of glutamate transporter (Fig. 3B) (n = 4 in triplicate; *p < 0.05). At the concentration applied, the carrier blocker was unable to affect, on its own, the spontaneous release of [3H]d-ASP (data not shown).
Figure 3.

Rotenone enhances [3H]d-aspartate and [3H]dopamine spontaneous release from rat striatal synaptosomes through calcium-dependent and transporter-mediated mechanisms. A, Concentration-dependent effects of rotenone on [3H]d-aspartate release. B, Effects of dl-TBOA or calcium deprivation of the rotenone-induced [3H]d-aspartate release. C, Concentration-dependent effects of rotenone on [3H]dopamine release. D, Effects of GBR12909 or calcium deprivation of the rotenone-induced [3H]dopamine release. Data represent mean ± SEM of four different experiments run in triplicate. *p < 0.05 versus controls; #p < 0.05 versus rotenone at 0.1 (B) or 1 μm (D). Horizontal axes in the graphs represent the ratio between the percentage of tritium released into each fraction after the first (b2–b7) and that in the first fraction collected (b1).
Also omission of Ca2+ ions from the superfusion medium was found to reduce significantly the release of [3H]d-ASP induced by 0.1 μm rotenone, although, in this case, the inhibition was less pronounced than that induced by blockade of glutamate transporter (Fig. 3B) (n = 4 in triplicate; *p < 0.05). Omission of external Ca2+ ions did not affect, on its own, the spontaneous release of tritium (data not shown).
Similarly, exposure of rat striatal synaptosomes to rotenone elicited a marked and prolonged release of [3H]DA. The toxin caused the release of [3H]DA in a concentration-dependent manner, the highest effect being observed at the concentration of 1 μm (Fig. 3C) (n = 4 in triplicate; *p < 0.05). Also in this case, the effect of rotenone (1 μm) was partially reduced by either GBR12909 (0.1 μm), a selective blocker of DA transporter, or Ca2+ ion deprivation (Fig. 3D) (n = 4 in triplicate; *p < 0.05), although to a different extent. Both GBR12909 or Ca2+ removal did not affect, on their own, the spontaneous release of [3H]DA (data not shown).
Targeting glutamate receptors fails to exert neuroprotective effects
Because the rotenone-induced release of excitatory amino acids was dependent on the reversal activity of the glutamate transporter, we tested whether the selective inhibition of the glutamate transporter by dl-TBOA was able to exert a neuroprotective effect on the irreversible loss of the field potential amplitude induced by this toxin. As shown in Figure 4A, 10 μm dl-TBOA did not significantly alter the irreversible electrophysiological effects induced by 1 μm rotenone (n = 6; p = 0.89) and did not change the time course of the field potential amplitude in the presence of 0.1 μm rotenone (n = 5; p = 0.94).
Figure 4.

Modulation of glutamatergic transmission does not affect the electrophysiological effect induced by inhibition of mitochondrial complex I, although it reduces the action of a complex II inhibitor. A, The glutamate transporter blocker dl-TBOA (10 μm), bath applied before and during the application of 0.1 and 1 μm rotenone, did not prevent the rotenone-induced reduction of the field potential amplitude. B, The preincubation with the NMDA receptor antagonists MK-801, memantine, and APV did not prevent the effect of rotenone. C, The NMDA and AMPA receptor antagonists APV and CNQX did not affect the rotenone-dependent membrane depolarization. D, The NMDA receptor antagonists MK-801 or memantine significantly reduced the electrophysiological effect of 3-NP, an inhibitor of mitochondrial complex II, on the field potential amplitude.
We further tested the possible role of glutamatergic neurotransmission on the rotenone-induced neuronal dysfunction by analyzing whether the application of the NMDA receptor antagonists could exert a neuroprotective action against the irreversible electrophysiological effects caused by this toxin (Centonze et al., 2006). As shown in Figure 4B, neither 30 μm MK-801, a potent noncompetitive antagonist of the NMDA receptor, nor 10 μm memantine, a low-affinity noncompetitive NMDA receptor antagonist, altered the rotenone-induced effects. Also, 50 μm APV, a competitive NMDA receptor antagonist, failed to prevent or to reduce the rotenone-induced effects (n = 5 for each drug; p = 0.85, p = 0.93, and p = 0.82, respectively). Accordingly, as reported in Figure 4C, both 50 μm APV and 10 μm CNQX, an AMPA receptor antagonist, did not alter the rotenone-induced membrane depolarization of striatal spiny neurons intracellularly recorded. Conversely, the NMDA receptor antagonists, at the same concentrations, were able to significantly reduce the irreversible reduction of the field potential amplitude caused by 3-NP, a mitochondrial toxin selectively blocking complex II (Calabresi et al., 2001; Saulle et al., 2004) (n = 6 for each drug; *p < 0.05 for both drugs) (Fig. 4D).
Neuroprotective effects by selectively targeting D2-like mediated dopaminergic transmission: the role of the GABA system
Because we have shown that exposure of rat striatal synaptosomes to rotenone elicited also a marked and prolonged release of [3H]DA, in a subsequent set of electrophysiological experiments, we analyzed the possibility that endogenous DA influences the electrophysiological changes induced by rotenone. Thus, we first preincubated the slices with 1 μm GBR12909 to modulate the level of endogenous DA by blockade of DA transporter. As shown in Figure 5A, this experimental procedure did not significantly affect the time course of the rotenone-induced electrophysiological changes (n = 6; p = 0.10). We then tested whether the selective antagonism of either D1-like DA receptors or D2-like receptors could influence the effects of the toxin. As shown in Figure 5A, the application of 10 μm SCH23390, an antagonist of D1-like receptors, did not affect the rotenone-induced alterations (n = 5; p = 0.98). Conversely, bath application of 10 μm l-sulpiride, an antagonist of D2-like receptors, significantly delayed the irreversible electrophysiological changes triggered by rotenone (n = 7; **p < 0.01) (Fig. 5B). To better address the effect of l-sulpiride in modulating the rotenone-mediated effect in striatum, we also performed recordings of striatal spiny neurons by using either intracellular recordings with sharp electrodes or whole-cell patch-clamp recordings. The D2-like receptor antagonist l-sulpiride (10 μm) significantly delayed the rotenone-induced membrane depolarization. We also investigated whether the effect of l-sulpiride could be influenced by glutamatergic transmission. As shown in Figure 5C, the effect of l-sulpiride was not different when applied together with the glutamate receptor antagonists APV (50 μm) plus CNQX (10 μm) (n = 5; p > 0.05). APV plus CNQX did not affect per se the rotenone-mediated membrane depolarization (Fig. 5C) (n = 4; p > 0.05).
Figure 5.

Blockade of D2-like but not D1-like DA receptors reduces the rotenone-induced effect via a GABA-mediated mechanism. A, Both the dopamine transporter blocker GBR12909 (1 μm) and the D1 receptor antagonist SCH23390 (10 μm), bath applied before and during the application of 1 μm rotenone, did not prevent the reduction of the mean field potential amplitude produced by the application of 1 μm rotenone. B, Conversely, the preincubation with the D2 receptor antagonist l-sulpiride (sulp; 10 μm) significantly reduced the rotenone-mediated reduction of the field potential amplitude. Note, however, that the concomitant application of SCH23390 reversed the effect exerted by l-sulpiride. C, The histogram summarizes cumulative data of membrane potential changes recorded 20 min after the onset of 1 μm rotenone showing that the action of l-sulpiride was not affected by the glutamate receptors antagonists APV and CNQX. D, Time courses of the rotenone-induced mean inward current in control conditions and in the presence of 10 μm l-sulpiride. E, The l-sulpiride-mediated effect against the rotenone-induced dysfunction was blocked by both the GABAA receptor antagonist bicuculline (bic; 3 μm) and the chloride channel blocker picrotoxin (PTX; 30 μm) in field potential experiments.
As shown in Figure 5D, also the rotenone-mediated inward current, measured in patch-clamp recordings, was significantly reduced when the slices were preincubated (15 min) with 10 μm l-sulpiride (n = 6; ***p < 0.001; sulp vs control), further supporting the role of endogenous DA in affecting the electrophysiological changes produced by rotenone in striatum. It has been reported that activation of D2 receptors in the striatum decreases GABA-mediated synaptic transmission (Centonze et al., 2003, 2004). Therefore, it is conceivable that the neuroprotective effect observed in the presence of l-sulpiride is attributable to the blockade of the D2-mediated depression of GABAergic striatal transmission. Accordingly, experimental findings support the hypothesis that a decrease of GABA-mediated transmission is highly detrimental in conditions of energy deprivation (Costa et al., 2004). In line with this hypothesis, the neuroprotective effect of l-sulpiride was reversed by a very low concentration of the GABAA receptor antagonist bicuculline (3 μm; n = 5; p = 0.91) (Fig. 5E) as well as by picrotoxin (30 μm; n = 5; p = 0.93) (Fig. 5E), a drug blocking the GABA-activated chloride ionophore, suggesting that endogenous GABA is implicated in the neuroprotective effects of l-sulpiride. None of the tested drugs affected per se the field potential amplitude.
Because it has been reported that D1-like and D2-like DA receptors exert an opposite influence on striatal GABA release (Harsing and Zigmond, 1997), we investigated whether the concomitant antagonism of D1-like receptor would counteract the neuroprotective effect exerted by l-sulpiride alone. Accordingly, we found that, in the presence of l-sulpiride plus SCH23390, rotenone produced effects similar to those observed under control conditions (n = 6; p = 0.82) (Fig. 5B).
Complete DA denervation induced by 6-OHDA does not affect the rotenone-induced electrophysiological effects
Because l-sulpiride was able to modulate the electrophysiological effect of rotenone suggesting a possible involvement of endogenous DA, we further analyzed the possible role of this neurotransmitter in the effects of rotenone by using the 6-OHDA experimental model of PD (Picconi et al., 2003). In fact, the unilateral nigral lesion by 6-OHDA induces a complete striatal denervation. In slices obtained from fully DA-denervated rats (n = 9), rotenone induced a progressive reduction of the field potential amplitude that was not significantly different from that observed in sham-operated rats (n = 7; p = 0.93) (Fig. 6A,B).
Figure 6.

The electrophysiological effects of rotenone are not affected in rats with complete striatal dopamine denervation by 6-OHDA and in mice transgenic for truncated human α-synuclein. A, Representative sections of TH (green) and NeuN (red) double immunostaining in rats with 6-OHDA-induced lesion of the substantia nigra pars compacta. On the left (ipsi) is shown the lesioned side, and on the right (contra) is shown the contralateral part. Scale bar, 100 μm. B, The graph shows that time course and the amplitude of the rotenone-induce electrophysiological effect were not different in DA-denervated and sham-operated rats. C, The effect of this toxin was similar in mice transgenic for truncated human α-synuclein (α-syn120 mice) and in wild-type mice.
Slices obtained from mice transgenic for truncated human α-synuclein show rotenone-induced electrophysiological responses similar to those obtained in WT mice
Mitochondrial impairment at complex I, oxidative stress, and α-synuclein aggregation have been implicated in PD pathogenesis, but how they are related to each other is unclear. Thus, it is possible that the toxic effects of rotenone could be modified by genetic manipulation of α-synuclein. Mice transgenic for truncated human α-synuclein (1–120) show pathological inclusions in the substantia nigra, partial but significant decrease in striatal DA levels, and progressive reduction in spontaneous locomotion (Tofaris et al., 2006). For these reasons, we tested whether in these transgenic mice (n = 7) the electrophysiological response to rotenone could differ from that seen in WT animals (n = 6). As shown in Figure 6C, the progressive loss of field potential amplitude induced by rotenone was similar in slices obtained from these two experimental groups (p = 0.91).
Carbamazepine, but not other AEDs, exerts neuroprotective effects against rotenone-induced toxicity
AEDs target more molecular sites in the brain, altering voltage-operated ion channels and chemical transmission through the interaction with neurotransmitters receptors, their metabolism, and ion channels (Calabresi et al., 2003; Costa et al., 2004, 2006; Rogawski and Löscher, 2004). The interaction with these multiple sites decreases abnormal brain excitability and protects vulnerable neurons in conditions requiring a high energy demand such as neuronal hyperactivity as well as metabolic impairment (Calabresi et al., 2000, 2003). For this reason, we investigated the possible neuroprotective effects of various AEDs against the irreversible functional alterations caused by rotenone in striatal neurons. As reported in Figure 7, A and B, only 30 μm CBZ significantly reduced the electrophysiological changes caused by rotenone (n = 8; ***p < 0.001), whereas 10 μm LTG, 100 μm TPM, or 300 μm VPA did not exert significant neuroprotection (n = 8 for each drug; p > 0.05). The application of 30 μm CBZ for 15–20 min did not alter per se the amplitude of the field potential (data not shown). The neuroprotective effect of CBZ was concentration dependent, with an EC50 of 11.8 μm (Fig. 7C).
Figure 7.

The antiepileptic drug carbamazepine reduces in a dose-dependent manner the rotenone-induced reduction of the field potential amplitude. A, The rotenone-mediated reduction of the field potential amplitude was significantly reduced by the antiepileptic drug CBZ (30 μm), whereas LTG (10 μm), TPM (100 μm), and VPA (300 μm) did not exert any significant neuroprotection. B, Example traces of corticostriatal field potentials recorded in control condition and at different times from the onset of the application of rotenone alone (top traces) and in the presence of rotenone plus 30 μm CBZ (bottom traces). C, Dose–response curve of the effect of CBZ on the field potential amplitude measured after 60 min from the onset of the application of 1 μm rotenone.
To further analyze the CBZ-induced neuroprotection against the rotenone-induced neurotoxicity, we also performed intracellular recordings with sharp microelectrodes. Interestingly, the preincubation of the slices with 30 μm CBZ significantly delayed the onset of the rotenone-induced membrane depolarization (n = 5; *p < 0.05) (Fig. 8A,B). This concentration of CBZ did not alter per se the membrane potential and the input resistance of the recorded neurons (data not shown). We also investigated the action of CBZ by whole-cell patch-clamp recordings; accordingly, 30 μm CBZ significantly depressed the rotenone-mediated inward current (n = 6; ***p < 0.0001, CBZ vs control) (Fig. 8C).
Figure 8.
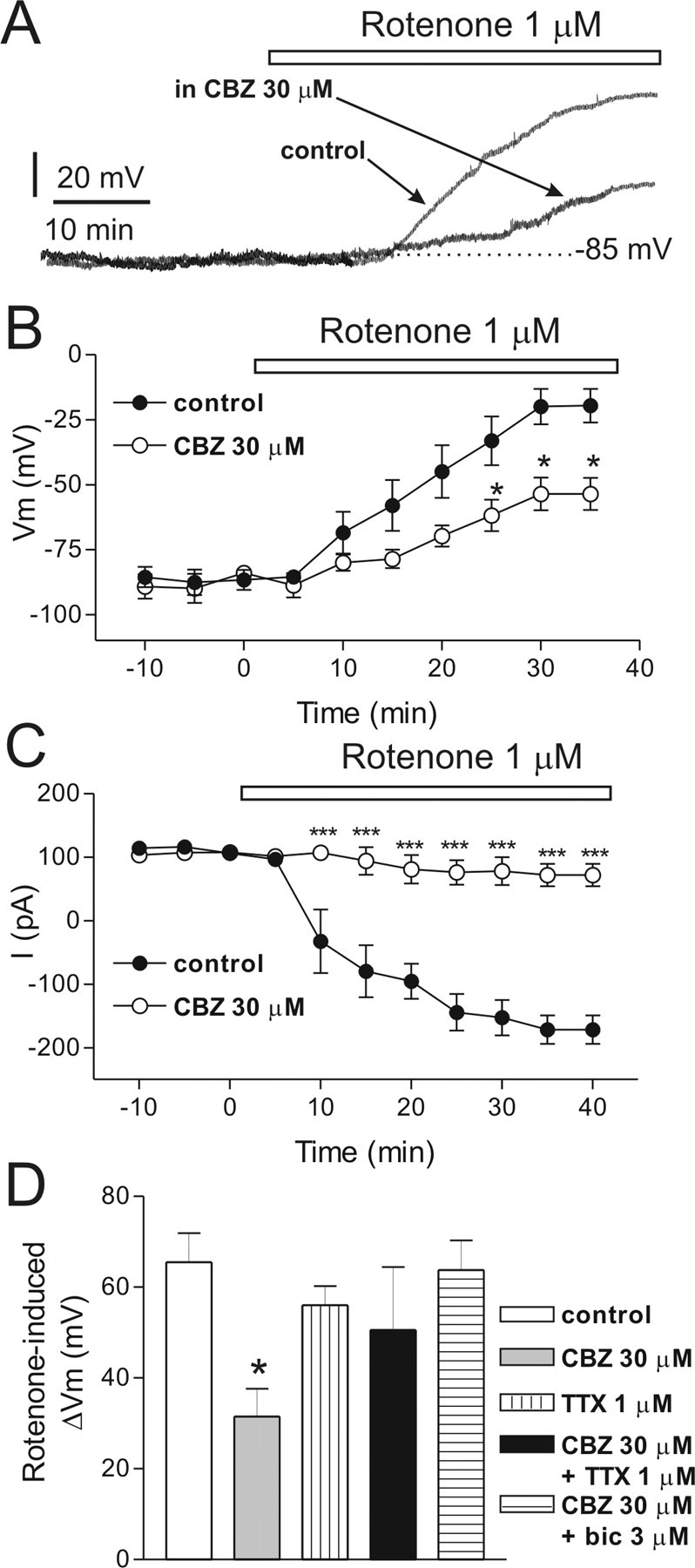
Carbamazepine reduces the rotenone-induced membrane depolarization/inward current of striatal spiny neurons via a TTX-sensitive and a GABA-mediated mechanism. A, Traces represent the membrane depolarizations of two single spiny neurons recorded either after the application of 1 μm rotenone alone (control) or in the presence of rotenone plus 30 μm CBZ. In both neurons, the resting membrane potential was −85 mV. Note that, in the presence of CBZ, the rotenone-induced depolarization was smaller. B, Cumulative data of the time courses of the rotenone-mediated membrane depolarization in control condition and in the presence of 30 μm CBZ. C, Time courses of the rotenone-induced mean inward current in control conditions and in the presence of 30 μm CBZ preincubated for 15 min in isolation and bath applied for 40 min together with rotenone. D, The histogram represents the membrane depolarization recorded 30 min after the onset of rotenone application in control condition and in various pharmacological conditions. bic, Bicuculline.
TTX does not affect per se rotenone-induced effects, but it blocks carbamazepine-induced neuroprotection
Modulation of gating of the brain Na+ channels accounts for the efficacy of several AEDs in epilepsy (Rogawski and Löscher, 2004). In particular, blockade of Na+ channels has been considered one of the major mechanisms implicated in the therapeutic action of CBZ. Thus, to explore the possibility that the neuroprotective effect exerted by CBZ against rotenone-induced electrophysiological alterations was mediated by an inhibitory action on Na+ channels, we analyzed whether the CBZ-mediated neuroprotection was mimicked by TTX, a selective Na+ channel blocker. Surprisingly, 1 μm TTX did not exert any neuroprotective effect per se with respect to control (n = 4; p = 0.99), although it fully abolished the neuroprotective effect of CBZ on rotenone-induced membrane depolarization (n = 5; *p < 0.05) (Fig. 8D). These latter findings suggest that the inhibition of Na+ channels is not directly implicated in the neuroprotective effects of CBZ, although this AED seems to exert its action against rotenone-induced toxicity via a mechanism requiring intact synaptic transmission. In fact, TTX does not only block postsynaptic Na+ channels, but it also inhibits synaptic transmission via a presynaptic mechanism of action.
Endogenous GABA is critically involved in the carbamazepine-mediated neuroprotection via GABAA but not GABAB receptors
Which is the endogenous neurotransmitter involved in the neuroprotective action of CBZ? To address this issue, we analyzed the possible involvement of endogenous GABA in the neuroprotective action of CBZ against rotenone effect. In particular, we tested whether a low concentration of bicuculline was able to block the neuroprotective action of CBZ. As shown in Figure 8C, 3 μm bicuculline fully abolished the effect of CBZ on the rotenone-induced membrane depolarization (n = 4; p = 0.99) suggesting also that the neuroprotective effect of this AED is critically dependent on the modulation of endogenous GABA. The same concentration of bicuculline did not alter per se the membrane potential and the input resistance of the recorded neurons (data not shown). Interestingly, the neuroprotective effect of CBZ was also blocked by 30 μm picrotoxin (n = 5; p = 0.90) (data not shown).
We also confirmed the critical role of GABAA receptors in the CBZ-mediated neuroprotective effects by a field potential analysis. In fact, 3 μm bicuculline fully blocked the neuroprotective effects of CBZ on field potential amplitude (n = 6; ***p < 0.001), but this GABAA receptor antagonist changed per se neither the field potential amplitude nor the rotenone-induced electrophysiological changes (Fig. 9B). Also in this case, the effect of CBZ was blocked by picrotoxin (30 μm) application (n = 6; p = 0.92) (Fig. 9B).
Figure 9.

The neuroprotective effect of carbamazepine against rotenone is mediated by GABAA but not GABAB receptors and occludes the action of l-sulpiride. A, A low concentration of the GABAA receptor antagonist bicuculline (bic) blocked the neuroprotective effect of CBZ. B, The histogram summarizes cumulative data recorded 30 min after the onset of 1 μm rotenone, showing that the neuroprotective action of CBZ was antagonized by bicuculline and picrotoxin (PTX) but not by the GABAB receptor antagonist CGP46381. Note that bicuculline, picrotoxin, and CGP46381, when applied alone, did not significantly affect the rotenone-induced effect. C, The histogram shows that coadministration of 30 μm CBZ plus 10 μm l-sulpiride did not produce an additive neuroprotective action compared with the effect achieved when a single drug was given in isolation, suggesting that CBZ and l-sulpiride share a common mechanism of action. Cumulative data from the different experimental condition were obtained 20 min after the onset of the application of 1 μm rotenone.
Because AEDs targeting GABAergic transmission can exert neuroprotective effects against energy deprivation by increasing endogenous GABA levels and via the activation of both GABAA and GABAB receptors (Costa et al., 2004), we tested whether the neuroprotective effect of CBZ also involved the latter. The application of 100 μm CGP46381 neither altered per se the effect of rotenone on the field potential amplitude (n = 4; p = 0.99) (Fig. 9B) nor affected the CBZ-mediated neuroprotective effects against rotenone (n = 4; ***p < 0.001) (Fig. 9B), suggesting that GABAB receptors are not required for the CBZ-mediated neuroprotection.
The neuroprotective effects against rotenone exerted by l-sulpiride and CBZ are mutually occlusive
Because the neuroprotective effects against rotenone exerted by either l-sulpiride or CBZ require endogenous GABA, these actions should be mutually occlusive. To investigate this hypothesis, we coapplied 10 μm l-sulpiride and 30 μm CBZ (n = 7) (Fig. 9C). As shown in Figure 9C, the neuroprotective effect obtained by coapplying these drugs was not increased compared with that one observed giving a single drug in isolation. Surprisingly, although there was no significant difference in the amplitude of neuroprotection by comparing the effect of l-sulpiride alone with that one obtained after the coapplication of the two compounds (p > 0.05), the neuroprotective effects of CBZ given alone was even reduced when this drug was coadministered with l-sulpiride (***p < 0.001). These occlusion experiments further support the hypothesis that l-sulpiride and CBZ share, at least in part, the mechanism underlying the neuroprotective action against striatal neuronal dysfunction induced by mitochondrial complex I inhibition.
Discussion
In PD, nigral neurons degenerate, whereas postsynaptic striatal neurons are spared (Braak and Braak, 2000). Conversely, in some atypical parkinsonian syndromes, such as PSP, mainly striatal neurons degenerate (Daniel et al., 1995; Burn and Lees, 2002). Reduced activity of complex I of the mitochondrial respiratory chain has been implicated in both these clinical conditions (Schapira et al., 1990; Dawson and Dawson, 2003; Höglinger et al., 2005, 2006; Di Filippo et al., 2006; Sherer et al., 2007). Exposure to the pesticide rotenone damages striatal spiny neurons by inhibiting mitochondrial complex I (Ferrante et al., 1997; Fleming et al., 2004). Thus, the electrophysiological characterization of the effects of rotenone in the striatum as well as the possible neuroprotective strategies resulting from this study might have profound clinical implications.
Rotenone causes a concentration-dependent and irreversible loss of the corticostriatal field potential amplitude correlated to the development of a membrane depolarization/inward current in striatal spiny neurons. The rotenone-induced inward current was coupled to an increased membrane conductance, and it reversed at approximately −18 mV. This reversal potential suggests the involvement of a nonselective cationic conductance (Calabresi et al., 1997, 1999, 2000; Centonze et al., 2001; Saulle et al., 2004), although a possible increase in a generic leak conductance may partly be associated.
Because both striatal and hippocampal CA1 neurons are highly vulnerable to ischemia and metabolic stress (Sims and Pulsinelli, 1987; Calabresi et al., 2000), we tested the selective vulnerability to rotenone by comparing its effects in these brain areas. The concentrations of rotenone required to produce an almost complete loss of the field potential amplitude in the CA1 hippocampal area were much higher than those necessary to produce similar effects in the striatum, suggesting that this mitochondrial toxin reveals cell-type-specific differences in sensitivity after inhibition of mitochondrial complex I.
A concentration of rotenone causing the electrophysiological effects in striatal slices was also able to increase the release of excitatory amino acids and DA from striatal synaptosomes. Ca2+-dependent exocytosis and the reversal of neurotransmitter transporters represent the two main mechanisms of exit usually implicated in release from nerve terminals. Accordingly, Ca2+ omission in the external medium significantly attenuated the rotenone-induced release of excitatory amino acids and DA from striatal synaptosomes suggesting the involvement of an exocytotic mechanism for both these transmitters. Nevertheless, the effect of rotenone on the release of excitatory amino acids was reduced even more potently by dl-TBOA, a selective blocker of glutamate transporter, indicating also the involvement of the reversal of neurotransmitter transporter. dl-TBOA, however, did not reduce the electrophysiological effects of rotenone, showing that the modulation of glutamate release does not seem to play a central role in the irreversible electrophysiological changes induced by this toxin in the striatum. The lack of neuroprotective effects of the modulation of excitatory transmission against rotenone-induced irreversible electrophysiological effects was further confirmed by using either competitive or noncompetitive NMDA receptor antagonists as well by blocking AMPA receptors.
The experiments dealing with the possible neuroprotective effects obtained by targeting the striatal DA system provided more puzzling but interesting results. Although GBR12909 significantly reduced the rotenone-induced release of DA, this selective blocker of the DA transporter was ineffective against the electrophysiological effects of this toxin. Moreover, the irreversible electrophysiological effects of rotenone on striatal neurons were not attenuated by the complete striatal DA denervation achieved by the 6-OHDA rat model (Picconi et al., 2003) or in mice transgenic for truncated human α-synuclein (1–120) showing a partial reduction of striatal DA (Tofaris et al., 2006). Surprisingly, in control rats, whereas the antagonism of D1-like DA receptors by SCH23390 did not cause significant electrophysiological effects, the application of l-sulpiride, a D2-like DA receptor antagonist, significantly attenuated the effect of this toxin on field potentials, membrane potentials, and inward currents.
What is the mechanism underlying this neuroprotective effect? Activation of D2-like DA receptors in the striatum exerts multiple actions (Bozzi and Borrelli, 2006). Thus, several mechanisms might be potentially involved in the protective effects of l-sulpiride against rotenone. The possibility that the glutamate and DA signaling pathways could synergistically be responsible of a mitochondrial dysfunction and impairment of the neuronal integrity has been explored by simultaneously blocking both the NMDA/AMPA glutamate and D2-like DA receptors. Our findings, however, did not suggest any contribution of glutamate in modulating the electrophysiological effects of l-sulpiride.
Thus, one of the likely putative mechanisms is the removal of the D2 receptor-mediated inhibition of striatal GABA release. The activation of D2 receptors in the striatum decreases GABA-mediated synaptic transmission (Centonze et al., 2003, 2004; An et al., 2004). Thus, it is conceivable that the effect of l-sulpiride is attributable to the removal of the D2-mediated depression of GABAergic striatal transmission. Accordingly, the effect of l-sulpiride against rotenone was reversed by a very low dose of the GABAA receptor antagonist bicuculline as well as by the chloride channel blocker picrotoxin. Interestingly, suppression of inhibitory GABAergic transmission is highly detrimental, particularly during energy deprivation, and the positive modulation of endogenous GABA can represent a potential neuroprotective strategy in clinical conditions requiring a high energetic demand (Costa et al., 2004).
DA can exert an excitatory influence on depolarization-induced GABA release within the striatum via D1-like receptors and an inhibitory influence via D2-like receptors, suggesting an opposite function of these two receptors on striatal GABA release (Harsing and Zigmond, 1997). In line with the hypothesis that the effect of l-sulpiride is mediated by a GABA-mediated mechanism, we found that the electrophysiological effect of this D2 receptor antagonist is blocked when it is coadministrated with the D1-like receptor antagonist SCH23390. One of the effects of D1 receptor activation is excitation of GABAergic striatal interneurons (Bracci et al., 2002), and this could explain why the action of l-sulpiride is blocked when it is coadministrated with a D1-like receptor antagonist. Decreased GABA release from such interneurons is expected under these conditions as a result of decreased D1 receptor excitatory influence.
AEDs represent a clinically available strategy to increase GABAergic transmission. Several AEDs, in fact, enhance GABA-mediated inhibition via different mechanisms (Rogawski and Löscher, 2004; Meldrum and Rogawski, 2007) and mediate neuroprotective effects (Calabresi et al., 2003; Costa et al., 2004, 2006). We tested several AEDs, whose action possibly involves the potentiation of GABAergic inhibition (Calabresi et al., 2003; Costa et al., 2004, 2006; Rogawski and Löscher, 2004; Meldrum and Rogawski, 2007), against the effects of rotenone. Surprisingly, among various tested AEDs, CBZ was the only drug able to exert a neuroprotective effect against rotenone. The CBZ-mediated neuroprotection was dose dependent, and its EC50 (11.8 μm) fell within the therapeutic range estimated for this AED in clinical studies (Morselli et al., 1977). CBZ not only prevented the rotenone-induced suppression of the field potential amplitude, but it also significantly reduced the membrane depolarization/inward current triggered by this mitochondrial toxin. The neuroprotective effect of CBZ was not mimicked by TTX, indicating that the inhibition of Na+ channels does not represent a major mechanism of neuroprotection against the rotenone-induced toxicity. TTX, however, was able to prevent the neuroprotective effect of CBZ against rotenone, suggesting that a TTX-sensitive synaptic mechanism is required for the CBZ-mediated neuroprotection.
In line with the results obtained for l-sulpiride, also the neuroprotective action of CBZ was blocked by bicuculline and picrotoxin but not by a GABAB antagonist, further supporting the hypothesis that neuroprotective mechanisms against rotenone-induced neuronal dysfunction mainly involve an enhancement of GABA-mediated inhibition via activation of GABAA receptors. Moreover, occlusion experiments using coapplication of both l-sulpiride and CBZ have not shown an additive effect of these two compounds, further supporting the hypothesis that they share the same mechanism of action.
An interaction between CBZ and the GABA-mediated inhibition has not been considered in the past as a major mechanism for the antiepileptic effect of this drug, and other AEDs have been classically related to the modulation of this transmitter system (Rogawski and Löscher, 2004; Meldrum and Rogawski, 2007). In fact, the antiepileptic mechanisms of CBZ have been related to the effects on voltage-gated sodium channels and on the release of glutamate and other transmitters (Okada et al., 1997, 1998, 2002; Kawata et al., 2001; Murakami et al., 2001) although the involvement of endogenous GABA has not been fully characterized. However, recent microdialysis experiments in the rat hippocampus have shown that therapeutically relevant concentration of CBZ increases basal GABA release without affecting basal glutamate release by modulating intracellular Ca2+ via the ryanodine receptor (Yoshida et al., 2007).
A previous interesting study has shown that nanomolar concentrations of rotenone cause suppression of single-pulse evoked DA release monitored by carbon-fiber microelectrodes in guinea pig striatal slices, hydrogen peroxide generation, and membrane depolarization in spiny neurons (Bao et al., 2005). In the present study, we had difficulty in producing consistent electrophysiological effects in “healthy” striatal spiny neurons using such a low concentrations of rotenone. Possible specie differences (rat and mouse in our study vs guinea pig) might critically influence the obtained.
In the present study, we have focused on the effects of rotenone on striatal spiny neurons. Interestingly, this toxin produces complex membrane responses in another striatal neuronal subtype such as the cholinergic interneuron (Bonsi et al., 2004). Application of rotenone in striatal cholinergic interneurons triggers an early membrane hyperpolarization followed by a late depolarizing response (Bonsi et al., 2004). The early hyperpolarization observed in cholinergic neurons, which is absent in spiny neurons, may represent an intrinsic membrane neuroprotective mechanism that might account for the differential neuronal vulnerability of these two striatal neuronal subtypes during metabolic stress (Calabresi et al., 1997, 2000; Centonze et al., 2001).
Future studies using animals chronically exposed to low doses of rotenone are required to confirm the hypothesis that both D2 receptor antagonism and treatment with CBZ exert a neuroprotective effect against inhibition of mitochondrial complex I via a GABAA-mediated mechanism.
Footnotes
This study was supported by Progetti Finalizzati e Strategici Ministero della Salute 2004, 2005, 2006 (P.C., B.P.) and Fondazione Cassa di Risparmio Perugia (P.C.). M.G.S. acknowledges the support of the United Kingdom Parkinson's Disease Society and Medical Research Council. We thank Cristiano Spaccatini for his excellent technical assistance.
References
- An JJ, Bae MH, Cho SR, Lee SH, Choi SH, Lee BH, Shin HS, Kim YN, Park KW, Borrelli E, Baik JH. Altered GABAergic neurotransmission in mice lacking dopamine D2 receptors. Mol Cell Neurosci. 2004;25:732–741. doi: 10.1016/j.mcn.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Bao L, Avshalumov MV, Rice ME. Partial mitochondrial inhibition causes striatal dopamine release suppression and medium spiny neuron depolarization via H2O2 elevation, not ATP depletion. J Neurosci. 2005;25:10029–10040. doi: 10.1523/JNEUROSCI.2652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Bonsi P, Calabresi P, De Persis C, Papa M, Centonze D, Bernardi G, Pisani A. Early ionic and membrane potential changes caused by the pesticide rotenone in striatal cholinergic interneurons. Exp Neurol. 2004;185:169–181. doi: 10.1016/j.expneurol.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Bozzi Y, Borrelli E. Dopamine in neurotoxicity and neuroprotection: what do D2 receptors have to do with it? Trends Neurosci. 2006;29:167–174. doi: 10.1016/j.tins.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Pathoanatomy of Parkinson's disease. J Neurol. 2000;247(Suppl 2):II3–II10. doi: 10.1007/PL00007758. [DOI] [PubMed] [Google Scholar]
- Bracci E, Centonze D, Bernardi G, Calabresi P. Dopamine excites fast-spiking interneurons in the striatum. J Neurophysiol. 2002;87:2190–2194. doi: 10.1152/jn.00754.2001. [DOI] [PubMed] [Google Scholar]
- Burn DJ, Lees AJ. Progressive supranuclear palsy: where are we now? Lancet Neurol. 2002;1:359–369. doi: 10.1016/s1474-4422(02)00161-8. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Ascone CM, Centonze D, Pisani A, Sancesario G, D'Angelo V, Bernardi G. Opposite membrane potential changes induced by glucose deprivation in striatal spiny neurons and in large aspiny interneurons. J Neurosci. 1997;17:1940–1949. doi: 10.1523/JNEUROSCI.17-06-01940.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Marfia GA, Centonze D, Pisani A, Bernardi G. Sodium influx plays a major role in the membrane depolarization induced by oxygen and glucose deprivation in rat striatal spiny neurons. Stroke. 1999;30:171–179. doi: 10.1161/01.str.30.1.171. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Bernardi G. Cellular factors controlling neuronal vulnerability in the brain: a lesson from the striatum. Neurology. 2000;55:1249–1255. doi: 10.1212/wnl.55.9.1249. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Gubellini P, Picconi B, Centonze D, Pisani A, Bonsi P, Greengard P, Hipskind RA, Borrelli E, Bernardi G. Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine. J Neurosci. 2001;21:5110–5120. doi: 10.1523/JNEUROSCI.21-14-05110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Cupini LM, Centonze D, Pisani F, Bernardi G. Antiepileptic drugs as a possible neuroprotective strategy in brain ischemia. Ann Neurol. 2003;53:693–702. doi: 10.1002/ana.10603. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–219. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Centonze D, Marfia GA, Pisani A, Picconi B, Giacomini P, Bernardi G, Calabresi P. Ionic mechanisms underlying differential vulnerability to ischemia in striatal neurons. Prog Neurobiol. 2001;63:687–696. doi: 10.1016/s0301-0082(00)00037-x. [DOI] [PubMed] [Google Scholar]
- Centonze D, Grande C, Usiello A, Gubellini P, Erbs E, Martin AB, Pisani A, Tognazzi N, Bernardi G, Moratalla R, Borrelli E, Calabresi P. Receptor subtypes involved in the presynaptic and postsynaptic actions of dopamine on striatal interneurons. J Neurosci. 2003;23:6245–6254. doi: 10.1523/JNEUROSCI.23-15-06245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Gubellini P, Usiello A, Rossi S, Tscherter A, Bracci E, Erbs E, Tognazzi N, Bernardi G, Pisani A, Calabresi P, Borrelli E. Differential contribution of dopamine D2S and D2L receptors in the modulation of glutamate and GABA transmission in the striatum. Neuroscience. 2004;129:157–166. doi: 10.1016/j.neuroscience.2004.07.043. [DOI] [PubMed] [Google Scholar]
- Centonze D, Prosperetti C, Barone I, Rossi S, Picconi B, Tscherter A, De Chiara V, Bernardi G, Calabresi P. NR2B-containing NMDA receptors promote the neurotoxic effects of 3-nitropropionic acid but not of rotenone in the striatum. Exp Neurol. 2006;202:470–479. doi: 10.1016/j.expneurol.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Costa C, Leone G, Saulle E, Pisani F, Bernardi G, Calabresi P. Coactivation of GABA(A) and GABA(B) receptor results in neuroprotection during in vitro ischemia. Stroke. 2004;35:596–600. doi: 10.1161/01.STR.0000113691.32026.06. [DOI] [PubMed] [Google Scholar]
- Costa C, Martella G, Picconi B, Prosperetti C, Pisani A, Di Filippo M, Pisani F, Bernardi G, Calabresi P. Multiple mechanisms underlying the neuroprotective effects of antiepileptic drugs against in vitro ischemia. Stroke. 2006;37:1319–1326. doi: 10.1161/01.STR.0000217303.22856.38. [DOI] [PubMed] [Google Scholar]
- Daniel SE, de Bruin VM, Lees AJ. The clinical and pathological spectrum of Steele–Richardson–Olszewski sindrome (progressive supranuclear palsy): a reappraisal. Brain. 1995;118:759–770. doi: 10.1093/brain/118.3.759. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Di Filippo M, Picconi B, Costa C, Bagetta V, Tantucci M, Parnetti L, Calabresi P. Pathways of neurodegeneration and experimental models of basal ganglia disorders: downstream effects of mitochondrial inhibition. Eur J Pharmacol. 2006;545:65–72. doi: 10.1016/j.ejphar.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Schulz JB, Kowall NW, Beal MF. Systemic administration of rotenone produces selective damage in the striatum and globus pallidus, but not in the substantia nigra. Brain Res. 1997;753:157–162. doi: 10.1016/s0006-8993(97)00008-5. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Zhu C, Fernagut PO, Mehta A, DiCarlo CD, Seaman RL, Chesselet MF. Behavioral and immunohistochemical effects of chronic intravenous and subcutaneous infusions of varying doses of rotenone. Exp Neurol. 2004;187:418–429. doi: 10.1016/j.expneurol.2004.01.023. [DOI] [PubMed] [Google Scholar]
- Greene JG, Greenamyre JT. Bioenergetics and glutamate excitotoxicity. Prog Neurobiol. 1996;48:613–634. doi: 10.1016/0301-0082(96)00006-8. [DOI] [PubMed] [Google Scholar]
- Harsing LG, Jr, Zigmond MJ. Influence of dopamine on GABA release in striatum: evidence for D1–D2 interactions and non-synaptic influences. Neuroscience. 1997;77:419–429. doi: 10.1016/s0306-4522(96)00475-7. [DOI] [PubMed] [Google Scholar]
- Höglinger GU, Féger J, Prigent A, Michel PP, Parain K, Champy P, Ruberg M, Oertel WH, Hirsch EC. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J Neurochem. 2003;84:491–502. doi: 10.1046/j.1471-4159.2003.01533.x. [DOI] [PubMed] [Google Scholar]
- Höglinger GU, Lannuzel A, Khondiker ME, Michel PP, Duyckaerts C, Féger J, Champy P, Prigent A, Medja F, Lombes A, Oertel WH, Ruberg M, Hirsch EC. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem. 2005;95:930–939. doi: 10.1111/j.1471-4159.2005.03493.x. [DOI] [PubMed] [Google Scholar]
- Höglinger GU, Oertel WH, Hirsch EC. The rotenone model of parkinsonism: the five years inspection. J Neural Transm. 2006;70:269–272. doi: 10.1007/978-3-211-45295-0_41. [DOI] [PubMed] [Google Scholar]
- Kawata Y, Okada M, Murakami T, Kamata A, Zhu G, Kaneko S. Pharmacological discrimination between effects of carbamazepine on hippocampal basal, Ca2+- and K+-evoked serotonin release. Br J Pharmacol. 2001;133:557–567. doi: 10.1038/sj.bjp.0704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetjens CM, Bui NT, Sengpiel B, Munstermann G, Poppe M, Krohn AJ, Bauerbach E, Krieglstein J, Prehn JH. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;200:5715–5723. doi: 10.1523/JNEUROSCI.20-15-05715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18–61. doi: 10.1016/j.nurt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli PL, Baruzzi A, Gerna M, Bossi L, Porta M. Carbamazepine and carbamazepine-10, 11-epoxide concentrations in human brain. Br J Clin Pharmacol. 1977;4:535–540. doi: 10.1111/j.1365-2125.1977.tb00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Okada M, Kawata Y, Zhu G, Kamata A, Kaneko S. Determination of effects of antiepileptic drugs on SNAREs-mediated hippocampal monoamine release using in vivo microdialysis. Br J Pharmacol. 2001;134:507–520. doi: 10.1038/sj.bjp.0704285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Kiryu K, Kawata Y, Mizuno K, Wada K, Tasaki H, Kaneko S. Determination of the effects of caffeine and carbamazepine on striatal dopamine release by in vivo microdialysis. Eur J Pharmacol. 1997;321:181–188. doi: 10.1016/s0014-2999(96)00938-7. [DOI] [PubMed] [Google Scholar]
- Okada M, Kawata Y, Mizuno K, Wada K, Kondo T, Kaneko S. Interaction between Ca2+, K+, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. Br J Pharmacol. 1998;124:1277–1285. doi: 10.1038/sj.bjp.0701941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Zhu G, Yoshida S, Kanai K, Hirose S, Kaneko S. Exocytosis mechanism as a new targeting site for mechanisms of action of antiepileptic drugs. Life Sci. 2002;72:465–473. doi: 10.1016/s0024-3205(02)02283-x. [DOI] [PubMed] [Google Scholar]
- Pearce JM. Progressive supranuclear palsy (Steele–Richardson–Olszewski syndrome): a short historical review. Neurologist. 2007;13:302–304. doi: 10.1097/01.nrl.0000254743.69160.b3. [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in l-DOPA-induced dyskinesia. Nat Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Raiteri M, Angelini F, Levi G. A simple apparatus for studying the release of neurotransmitters from synaptosomes. Eur J Pharmacol. 1974;25:411–414. doi: 10.1016/0014-2999(74)90272-6. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- Saulle E, Gubellini P, Picconi B, Centonze D, Tropepi D, Pisani A, Morari M, Marti M, Rossi L, Papa M, Bernardi G, Calabresi P. Neuronal vulnerability following inhibition of mitochondrial complex II: a possible ionic mechanism for Huntington's disease. Mol Cell Neurosci. 2004;25:9–20. doi: 10.1016/j.mcn.2003.09.013. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT. An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J Neurosci. 2002;22:7006–7015. doi: 10.1523/JNEUROSCI.22-16-07006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci. 2003a;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003b;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW, Greenamyre JT. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson's disease. J Neurochem. 2007;100:1469–1479. doi: 10.1111/j.1471-4159.2006.04333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR, Pulsinelli WA. Altered mitochondrial respiration in selectively vulnerable brain subregions following transient forebrain ischemia in the rat. J Neurochem. 1987;49:1367–1374. doi: 10.1111/j.1471-4159.1987.tb01001.x. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett. 1998;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Garcia Reitböck P, Humby T, Lambourne SL, O'Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, Spillantini MG. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human α-synuclein(1–120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi A, Costa C, Di Filippo M, Tantucci M, Siliquini S, Belcastro V, Parnetti L, Picconi B, Calabresi P. Memantine reduces neuronal dysfunctions triggered by in vitro ischemia and 3-nitropropionic acid. Exp Neurol. 2007;207:218–226. doi: 10.1016/j.expneurol.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Okada M, Zhu G, Kaneko S. Carbamazepine prevents breakdown of neurotransmitter release induced by hyperactivation of ryanodine receptor. Neuropharmacology. 2007;52:1538–1546. doi: 10.1016/j.neuropharm.2007.02.009. [DOI] [PubMed] [Google Scholar]