

Abstract
cAMP regulates secretory processes through both PKA-independent and PKA-dependent signaling pathways. Their relative contributions to fast neurotransmission are unclear at present, although forskolin, which is generally believed to enhance intracellular cAMP levels by stimulation of adenylyl cyclase activity, was shown to increase vesicular release probability (p) and the number of releasable vesicles (N) in various neuronal preparations. Using low-frequency (0.2 Hz) electrophysiological recordings in the presence of the Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP (ESCA1), we find that Epac activation by this analog accounts on average for 38% of the forskolin-induced increase in evoked EPSC amplitudes and for 100% of the forskolin-induced increase in miniature EPSC (mEPSC) frequency in dissociated autaptic neuronal cultures from mouse hippocampus. From paired-pulse facilitation experiments, and considering the enhancement of mEPSC frequency, we conclude that ESCA1-induced Epac activity is presynaptic in origin and increases p. In addition, preapplication of ESCA1 augmented a subsequent enhancement of evoked EPSC amplitudes by phorbol ester (PDBu). This effect was maximal when ESCA1 application preceded the PDBu application by 3 min. Because the PDBu response was abolished after downregulation of intracellular PKC activity, we conclude that ESCA1-induced Epac activation leads to presynaptic changes involving Epac-to-PKC signaling.
Keywords: cAMP, Epac, vesicular release probability, excitatory autaptic neurons, PKC, glutamatergic synapse
Introduction
Greengard et al. (1970) showed that neuronal activity increases cAMP levels in superior cervical ganglia. At excitatory mammalian cerebral synapses, cAMP is known to enhance glutamate release from presynaptic terminals (Weisskopf et al., 1994; Leßmann and Heumann, 1997). Traditionally, this cAMP-mediated enhancement of neurotransmitter release was attributed to the direct activation of protein kinase A (PKA) (Castellucci et al., 1980; Chavez-Noriega and Stevens, 1994). PKA not only inhibits potassium channel function (Siegelbaum et al., 1982), but also regulates vesicular dynamics by phosphorylation of synapsins, SNAP-25, and voltage-dependent calcium channels (VDCCs) (Hell et al., 1995; Nagy et al., 2004; Menegon et al., 2006). PKA has been implicated in the expression of specific forms of long-term potentiation and long-term depression (Nguyen and Woo, 2003). However, basal neurotransmission and short-term plasticity were unaffected in neurons deficient for different PKA subunits (Brandon et al., 1997).
In 1998, two novel cAMP receptors, Epac1 and Epac2, were identified (de Rooij et al., 1998; Kawasaki et al., 1998). Since then, PKA-independent, Epac-mediated modulation of secretory processes was reported for insulin-secreting β-cells (Holz, 2004), melanotrophs (Sedej et al., 2005), the crayfish neuromuscular junction (Zhong and Zucker, 2005), the calyx of Held (Sakaba and Neher, 2003), and cortical neurons (Huang and Hsu, 2006). In pancreatic β-cells and melanotrophs, exocytosis is marked by two distinct components: the fast component is Epac dependent, whereas the slow component relies on PKA activity. Epac increases the size of the readily releasable pool (RRP) of secretory granules (Eliasson et al., 2003). At the calyx of Held synapse, the adenylyl cyclase agonist forskolin enhances both the number of releasable vesicles (N) and their release probability (p) (Kaneko and Takahashi, 2004). Huang and Hsu (2006) reported similar changes in p and N for cortical neurons. However, they conclude that Epac activity does not contribute to the observed forskolin effect.
Here, we were interested in the contribution of Epac to the forskolin-mediated facilitation of neurotransmitter release in cultured excitatory autaptic neurons. Application of the specific Epac agonist 8-pCPT-2′-O-Me-cAMP (ESCA1) [term adopted from Holz et al. (2008)] accounts on average for 38% of a forskolin-mediated increase in evoked EPSC amplitudes. Epac activation sufficiently explains the forskolin-induced enhancement of miniature EPSC (mEPSC) frequency, whereas no change of the sucrose-releasable pool of vesicles was observed. We conclude that Epac activity increases vesicular release probability (p).
Additionally, prior activation of Epac augmented a subsequent enhancement of evoked EPSC amplitudes by phorbol ester, presumably involving Epac-to-PKC signaling. Potentiation of evoked release by phorbol ester results from an increase in the vesicle number in the RRP (Waters and Smith, 2000).
Our results suggest that Epac either increases the mean vesicular release probability or, in view of the heterogeneity of vesicular release probability (Rosenmund et al., 1993; Moulder and Mennerick, 2005), modulates a shift between different populations of vesicles. Moreover, potentiation of evoked EPSCs and mEPSC frequency was dependent on the initial EPSC amplitude (EPSC0) of a neuron, and we relate this to the state of maturation of the culture.
Materials and Methods
Cell culture.
For electrophysiological experiments, individual murine and rat hippocampal neurons forming recurrent excitatory synapses (autapses) were grown in microisland cultures according to a modified protocol by Bekkers and Stevens (1991). To obtain astrocytes and hippocampal neurons, newborn rats and mice were decapitated according to the rules of the state and animal welfare committee. Neurons were isolated from the dentate gyrus (DG) and the hippocampal CA1 region and were seeded at low density on top of astrocyte microislands. Neuronal cultures were allowed to mature in Neurobasal A medium supplemented with 2% B27, 1% GlutaMax-I, 0.2% penicillin/streptomycin (at 5% CO2 and 95% humidity) for at least 8 d before electrophysiological recordings. Only islands containing single neurons were examined. For immunoblot analysis neuronal cultures were prepared from the dentate gyrus and the CA1 region of the hippocampal formation of postnatal day 0 (P0)/P1 animals (rat and mouse) and grown in collagen/poly-d-lysine-coated Petri dishes (10 cm diameter) in the absence of an astrocyte feeder layer. If not stated otherwise, cell culture solutions were purchased from Invitrogen.
Immunoblot analysis.
Before protein lysate preparation, the cultures were left untreated (mock pretreatment with external solution used for electrophysiological recordings containing 2 mm Ca2+/1 mm Mg2+) for control lysates or were pretreated for 2 min with ESCA1 (Biolog), forskolin (Sigma), or 6-Bnz-cAMP (Biolog). The duration of the drug treatment and the drug concentrations were chosen as indicated for the electrophysiological experiments. Protein lysates were generated on day in vitro 10–11 (DIV 10–11) under maintenance of a working temperature of 4°C. The neuronal cultures were washed with ice-cold Tris-buffered saline (TBS; 30 mm Trizma-Base, 140 mm NaCl, and 2.7 mm KCl) of pH 7.4–7.6 to stop the pretreatment of the cells and remove the drugs applied. The cells were mechanically detached in TBS containing 100 mm phenylmethylsulfonyl fluoride (PMSF) and subjected to centrifugation for 10 min at 600–800 × g and 4°C. The cell precipitate was lysed for 15 min on ice with a Tris-HCl-based buffer (50 mm, pH 8) containing NaCl (120 mm), EDTA (5 mm), and Nonidet P-40 Substitute (0.5%, v/v), and to which protease- and phosphatase-inhibitors like aprotinin (20 μg/ml), leupeptin (10 μg/ml), PMSF (100 μg/ml), NaF (50 mm), and Na3VO4 (200 μm) were added. Cytosolic protein extracts were obtained by centrifugation at 13,000 × g (4°C, 15 min). Protein content was determined by a standard Bradford assay. SDS-PAGE (Laemmli, 1970) was performed either on a 7% gel with each lane containing 8 μg of protein (Fig. 1A) or on a 7.5% gel with each lane containing 3 μg of protein (Fig. 1B1). For Western blot analysis, proteins were transferred to polyvinylidene fluoride membranes (Roche). We generally used TBS solution containing either 0.05% (v/v) (for blocking and antibody solutions) or 0.1% (v/v) (for wash steps) Tween 20 during the analysis of the Western blots. When monitoring phosphorylation levels of synapsin, we blocked or probed the membranes in the presence of 3.5% (w/v) bovine serum albumin (Fraction V; Sigma) instead of nonfat dry milk. Antigen–antibody complexes were visualized by species-specific horseradish-peroxidase-conjugated secondary antibodies and the enhanced chemiluminescence system (SuperSignal West Femto; Pierce). Signal intensities were quantified densitometrically (LumiImager, LumiAnalyst software; Boehringer Ingelheim). The antibodies (polyclonal) were applied in the following dilutions: phospho-synapsin (Ser9; Cell Signaling Technology), 1:1000; β-actin (I-19; Santa Cruz Biotechnology), 1:500; Epac1 (GeneTex), 1:500; Epac2 (M-18; Santa Cruz Biotechnology), 1:500.
Figure 1.
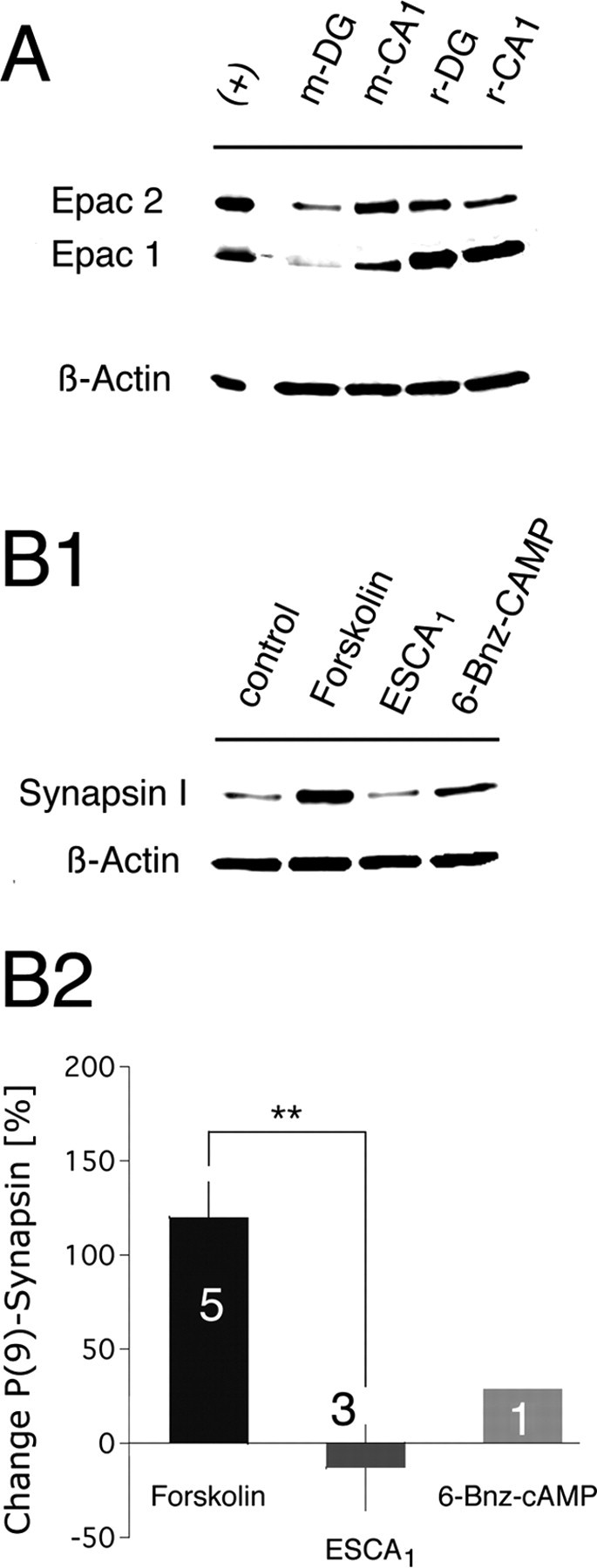
Expression of Epac in neuronal cell cultures. A, Cultured autaptic neurons express both Epac isoforms. Lanes, Rat cerebellum lysate, positive control (+), murine dentate gyrus neurons (m-DG), murine CA1 neurons (m-CA1), rat dentate gyrus neurons (r-DG), rat CA1 neurons (r-CA1). Protein bands were detected at the following molecular weights: 126 kDa for Epac2, 100 kDa for Epac1, and 43 kDa for β-actin. Epac1 experiments were performed in triplicate. B1, Test for the specificity of the Epac-activating cAMP analog, ESCA1. A Western blot analysis of cAMP- or cAMP-analog-mediated phosphorylation of synapsin I at its PKA phosphorylation site (Ser9) is shown. For drug treatment procedure, see Materials and Methods. β-Actin controls for equal protein loading. B2, Quantitative change in the phosphorylation level of the PKA site of synapsin I. To correct for nonspecific effects (unrelated to drug treatment), which might have been introduced by variability in protein loading, P-Ser9 synapsin I chemiluminescence intensities were first normalized to the intensities of the corresponding β-actin values. These values were further normalized to the control intensity value to determine the increase or decrease in P-Ser9 levels after drug treatment. Values are shown as mean ± SEM based on the number of different Western blot performances. Statistical significance was tested by an unpaired two-tailed Student's t test assuming unequal variances (see Materials and Methods). Numbers within histogram bars represent the number of measurements.
Electrophysiological recordings.
Evoked EPSCs were recorded from isolated cultured autaptic neurons from DIV 8 to DIV 15 at a holding potential of −70 mV with an EPC-9 patch-clamp amplifier (HEKA). Neurons were depolarized from −70 to 0 mV for 2 ms every 5 s (0.2 Hz). The recorded EPSC trace length was 1 s. Current traces were digitized at 20 kHz and filtered at 3 kHz. The extracellular solution (10 mm HEPES, 10 mm glucose, 120 mm NaCl, and 2.4 mm KCl, pH 7.5–7.6; 298–305 mOsm) contained 2 mm CaCl2 and 1 mm MgCl2. Fire-polished patch electrodes (GB150F-8P; Science Products) with resistances of 2.5–3.5 MΩ were filled with standard internal KCl solution (136 mm KCl, 17.8 mm HEPES, 12 mm phosphocreatine Na2, 4 mm ATP-Mg, 1 mm K-EGTA, 0.3 mm GTP-Na2, and 0.6 mm MgCl2, pH 7.4). The uncompensated series resistance did not exceed 4 MΩ during whole-cell recording of autaptic neurons.
Drug application.
Drug application was performed by a fast perfusion system with a solution exchange time ranging from 190 to 300 ms depending on the viscosity of the solution applied as previously described (Rosenmund et al., 1995). The flow rate from the flow pipes was 0.52 ± 0.03 ml/min (mean ± SD), corresponding to a rate of 0.07 m/s. In addition to the local perfusion with flow pipes, the whole bath (volume, 2 ml) was perfused at 0.8 ml/min to prevent accumulation of drugs. Adequate positioning and solution exchange was examined at the beginning and end of each experiment. After every neuronal recording, the bath chamber was cleaned with 70% ethanol and demineralized water to ensure that no drug-related cross-contamination occurred from one cell measurement to the next. The drugs used and their concentrations were as follows: 6-Bnz-cAMP (150 μm; Biolog), ESCA1 (50 μm; Biolog), forskolin (50 μm; Sigma), phorbol-12,13-dibutyrate (PDBu) (1 μm; Merck Biosciences), and tetrodotoxin (300 nm; Sigma). Most compounds were diluted into the external solution from concentrated dimethylsulfoxide (DMSO)-stock solutions. The final concentrations of DMSO were <0.1%.
Analysis.
Off-line analysis was performed with AxoGraph 4.5 (Molecular Devices), Excel (Microsoft), KaleidaGraph 3.0 (Synergy Software), and IgorPro (WaveMetrics) software. Before analysis with AxoGraph, EPSCs were offset corrected. The EPSC0 was used as a reference when comparing the effects under control and drug conditions. The EPSC0 specifies the steady-state EPSC amplitude measured before drug application under control condition (and equals the mean EPSC amplitude usually recorded from a given neuron over the first 2 min). The drug effect was described by the potentiation factor. This factor was calculated according to the following equation:
 |
The potentiation factor corresponds to a normalization of the data and is denoted by “EPSC Potentiation” in the presented figures. A potentiation factor of 1.0 means that the EPSC amplitudes remained unchanged after drug treatment. When time courses are shown, the equation presented above was modified to
 |
In this case, the applied solution represented either the control or the drug solution.
Paired-pulse facilitation was calculated as the amplitude of the second EPSC minus the first, divided by the first.
mEPSCs were detected using a sliding template algorithm (Clements and Bekkers, 1997). The mEPSC template (linear summation of two exponentials) length of 15 ms allowed detection of nonoverlapping mEPSCs. The template was automatically offset to account for fluctuations in the baseline, and the amplitude was scaled to fit the analysis window. Events were only accepted as mEPSCs when the calculated detection criterion exceeded the SD of the background noise at least 3.5-fold. To avoid false positive events, we considered only those mEPSCs that fitted to all of the following imposed criteria: amplitude interval [−100; −8] pA, rise time [0.24; 0.71] ms, and width [0.67; 4] ms. Events that fulfilled these criteria were then averaged to obtain a mean amplitude and charge per neuron. The total number of releasable vesicles was determined by a 4.5 s application of external saline solution made hypertonic by the addition of 500 mm sucrose. For every cell analyzed, the total number of releasable vesicles was defined as the charge during the transient burst of exocytotic activity that followed application of the hypertonic solution divided by the mean charge of the corresponding mEPSC (Rosenmund and Stevens, 1996). A pedestal defined by the late-sucrose response was subtracted from the total charge.
Generally, the measured data are reported as mean ± SEM, if they were averaged over the maximal EPSC0 range. If smaller, defined EPSC0 intervals were considered, the variability in the measured effects was of interest, and the data are presented as mean ± SD. The calculation of averages and errors was based on the total number of cells measured. Our analysis showed that the average data were largely independent on variabilities introduced by differences between cell cultures. The total number of cells and the number of cultures are given by N and n, respectively.
For normally distributed data, statistical analysis was performed using either the paired or unpaired two-tailed Student's t test with *p < 0.05, **p < 0.01, and ***p < 0.001. In the unpaired case, unequal variances were considered. Samples showing no normal distribution were statistically analyzed by the nonparametric Kolmogorov–Smirnov (KS) test.
We adopted the acronym ESCA (Epac-selective cAMP analog) from Holz et al. (2008) for the 2′-O-methyl-substituted cAMP analogs that activate the Epac protein family. ESCA1 specifically denotes 8-pCPT-2′-O-Me-cAMP.
Results
Cultured autaptic neurons express both Epac (1, 2) isoforms
Electrophysiological experiments performed at the calyx of Held have shown that the rate of vesicle recruitment is modulated by the intracellular level of cAMP and that this modulation involves Epac activity (Sakaba and Neher, 2003; Kaneko and Takahashi, 2004). In the murine and rat brain, both Epac isoforms were detected at the mRNA expression level (Kawasaki et al., 1998; Ozaki et al., 2000). Here we determined the protein expression of Epac1 and Epac2 in cultured neurons from the DG and the CA1 region of the hippocampal formation (CA1) of both murine and rat origin (see Materials and Methods). Western blot analysis of the corresponding neuronal protein lysates showed that cultured murine and rat hippocampal neurons express both Epac isoforms (Fig. 1A).
ESCA1 is a specific activator of Epac in autaptic neurons
At present, specificity of cAMP-induced Epac actions is mainly tested by ESCAs (Holz et al., 2008). ESCAs incorporate a 2′-O-methyl substitution on the ribose ring of cAMP. This modification impairs their ability to activate PKA, while leaving their ability to activate Epac unaffected, when used at submillimolar concentrations in vitro (Enserink et al., 2002; Christensen et al., 2003). At 50 μm, ESCA1 activates Epac maximally, whereas PKA function is barely affected (∼5–7%) (Christensen et al., 2003). By quantifying the amount of phosphorylation at the PKA phosphorylation site (Ser9) of the presynaptic protein marker synapsin I, we confirmed the Epac specificity of ESCA1 in neuronal cultures (Johnson et al., 1972; Menegon et al., 2006). Figure 1B1 shows the amount of synapsin I phosphorylation at Ser9 after treatment of neuronal DG cultures with either ESCA1 (50 μm), forskolin (50 μm), or 6-Bnz-cAMP (100 μm) (see Materials and Methods). Both forskolin and the PKA-activating cAMP analog 6-Bnz-cAMP, increased synapsin I phosphorylation at Ser9 (Fig. 1B1,B2). Forskolin enhanced synapsin I phosphorylation significantly by 120 ± 20% (mean ± SEM; N = 5), whereas 6-Bnz-cAMP enhanced the phosphorylation at Ser9 by 29% (N = 1) (Fig. 1B2). In contrast, application of ESCA1 (N = 3) did not increase the phosphorylation level of the synapsin I-PKA site compared with control condition.
Application of ESCA1 enhances neurotransmitter release
Analysis of neurotransmitter release from excitatory cultured autaptic neurons of the murine dentate gyrus revealed that external application of ESCA1 (50 μm) increased EPSC amplitudes during low stimulation frequency (0.2 Hz). On average, EPSC amplitudes were potentiated by 23 ± 3% (mean ± SEM; N = 60) (Fig. 2A). This action of ESCA1 was mimicked by structurally related ESCAs, which displayed different membrane permeabilities and were either resistant to enzymatic hydrolysis or showed higher affinity toward Epac (see supplemental material, available at www.jneurosci.org) (Fig. 1B). This indicates that neither stability nor the concentration of ESCA1 were limiting factors in our experiments.
Figure 2.

ESCA1 enhances neurotransmitter release at excitatory autaptic neurons from dentate gyrus. A, Average EPSC amplitude potentiation (mean ± SEM) by 50 μm ESCA1 (EPSC0 = 5.43 ± 0.51 nA, mean ± SEM) and 50 μm forskolin (EPSC0 = 4.95 ± 0.48 nA, mean ± SEM). Statistical significance was analyzed by using the paired t test. The statistical significance of the difference between the ESCA1 and the forskolin effect was tested by the KS test. B1, Average time course of EPSC amplitudes in the presence and absence of ESCA1 (mean ± SEM). ●, ESCA1; N = 27; n = 5 (EPSC0 = 7.01 ± 0.94 nA, mean ± SEM); □, average EPSC amplitude rundown under control condition; N = 17; n = 2 (EPSC0 = 9.2 ± 1.5 nA, mean ± SEM). The black bar represents the onset and duration of drug application. B2, Average time course of EPSC amplitudes in the presence and absence of forskolin (mean ± SEM). Markers were used in analogy to the top panel. N = 13; n = 1 (EPSC0 = 4.76 ± 0.81 nA, mean ± SEM). C, Analysis of the changes in the AP-related Na+- and K+-current peaks (mean ± SEM). AP-related sodium and potassium currents were observed before every EPSC. Top left, A typical waveform, indicating negative and positive peaks as values representative for Na+ currents and K+ currents. Peak current changes were analyzed for the control rundown condition (top right) and for both drug conditions (middle, ESCA1; bottom, forskolin). The changes both during the steady-state phase of drug application and 3 min after drug removal are shown. In the control rundown experiment, the time intervals for analysis were chosen to match those used in the drug experiments. Significance was tested by a paired t test. The numbers within the histogram bars are N values, representing the number of cells. For the control rundown condition, 13 cells were analyzed.
In a second set of experiments, application of forskolin significantly enhanced EPSC amplitudes on average by a factor of 60 ± 6% (mean ± SEM; N = 46) (Fig. 2A). Forskolin is generally believed to enhance intracellular cAMP levels by activation of adenylyl cyclases (Vincent and Brusciano, 2001; Willoughby and Cooper, 2006). Thus, Epac activation can explain on average ∼38% of the forskolin-induced increase in evoked EPSC amplitudes. A contribution of Epac activity to the observed forskolin response is further supported by the analysis of the washout phases of both the ESCA1 and the forskolin response (Fig. 2B1,B2). After ESCA1 application, the washout phase was fitted by a monoexponential function with a decay time constant of 13 ± 1 s (mean ± SD) (Fig. 2B1). In the case of forskolin application, the washout phase was best described by a biexponential function, where the fast decay time constant was 18 ± 3 s (mean ± SD), followed by a much slower decay with a time constant of 192 ± 23 s (Fig. 2B2). Therefore, the forskolin effect on EPSC amplitudes can be attributed to complex cAMP signaling. We attempted to demonstrate experimentally a possible role of HCN channels in the cAMP- or ESCA1-mediated responses (Zhong and Zucker, 2005), however without positive results (see supplemental material, available at www.jneurosci.org) (Fig. 2). Our electrophysiological recordings suggest that HCN channel function is absent from autaptic excitatory DG neurons. In contrast to rat dentate granule cells, which show a high HCN channel mRNA and protein expression level for all known isoforms (Monteggia et al., 2000; Notomi and Shigemoto, 2004), HCN isoforms are absent or barely detectable above background in murine dentate granule cells (Moosmang et al., 1999; Santoro et al., 2000). Overall, our data indicate that Epac activity contributes to the forskolin response in murine excitatory autaptic DG neurons, that it is involved in the fast mode of the forskolin response, and that it does not act via HCN channels.
Finally, we analyzed the action potential (AP)-related Na+ and K+ currents that preceded the autaptic EPSC (Fig. 2C, top left). Changes in these voltage-gated currents may affect our measurements of evoked EPSCs by two means: first, the overlap of the time course of these voltage-gated currents and the EPSC may contaminate measurements of the EPSC amplitude, and second, their modulation by drug application may change the shape of the action potential and thereby additionally modulate neurotransmitter release. To exclude a contamination of the measured EPSC by AP-related current traces, we applied CNQX to measure the postsynaptic AMPA receptor current as a difference current. The size of the EPSC amplitude was not affected by the shape of the preceding AP-related current traces (see supplemental material, available at www.jneurosci.org) (Fig. 1A). Forskolin-induced changes in the EPSC amplitude were confirmed. Still, ESCA1 and forskolin differently inhibited the AP-related currents (Fig. 2C). ESCA1 depressed only the Na+ current significantly, whereas forskolin reduced both the Na+ and the K+ currents. In contrast to the Na+ current, forskolin-mediated inhibition of the K+ current was reversible. Neither Na+ nor K+ currents showed a significant rundown during control EPSC amplitude recordings. These observations provide further evidence that ESCA1 specifically targets Epac at the concentration used. The inhibition of K+ current can be attributed to PKA activity (Siegelbaum et al., 1982).
Potentiation induced by pharmacological agents depends on the initial EPSC amplitude, the type of neuronal preparation, and animal species
The magnitude of EPSC amplitude potentiation induced by short-term application of forskolin (2 min) or ESCA1 (1.2 min) was dependent on the EPSC0 (for definition, see Materials and Methods) (Fig. 3A). Regardless of the EPSC0 amplitude, ESCA1 invariably caused less potentiation than forskolin. The forskolin response was particularly pronounced for EPSC0 < 1 nA, a property not shared by the ESCA1-mediated activation of Epac. However, dependency on EPSC0 was not restricted to pharmacological agents modulating cAMP signaling. Similarly, an increase in EPSC amplitudes induced by PDBu, a phorbol ester known to presynaptically enhance neurotransmitter release (Malenka et al., 1986), was more pronounced for small EPSC0 than for larger ones (Fig. 3B). In Fig. 3C, we correlated the drug-induced EPSC amplitude potentiation with the age of the dentate gyrus culture (DIV) to account for a possible maturation aspect that may underlie the observed effect dependency on EPSC0. Overall, the EPSC potentiation effects were larger for younger (DIV 8–10) than for older (DIV 11–15) cell cultures (Fig. 3C1).
Figure 3.

The magnitude of the drug-induced EPSC amplitude potentiation can be correlated to the size of the EPSC0 amplitude. Electrophysiological recordings of excitatory DG neurons. A, Left, cAMP-related EPSC amplitude potentiation versus the EPSC0 amplitude (mean ± SD). ■, ESCA1 (50 μm); N = 60; n = 13; △, forskolin (50 μm); N = 46; n = 3. Individual data points were binned in classes of 1 nA for EPSC0 > 0.5 nA. A monoexponential fit is applied to the ESCA1 values. The forskolin data were fitted by a biexponential function. Right, Representative EPSC sample traces before and after ESCA1 or forskolin application for both small (a) and large (b) EPSC0, as marked by arrows in the left panel. AP-related currents were blanked out. B, Left, EPSC amplitude potentiation induced by PDBu (1 μm) versus the EPSC0 amplitude (mean ± SD). ○, Murine DG; N = 35; n = 7; ◇, murine CA1; N = 5; ♦, rat CA1; N = 16; n = 3. Data binning and layout of the figure are as in A. The PDBu data were fitted by a biexponential function. Right, The time course of the PDBu effect on EPSC amplitudes (mean ± SEM) depends on the hippocampal region used for the preparation of neuronal cultures. ○, Murine DG; N = 7; n = 1 (EPSC0 = 3.33 ± 0.51 nA, mean ± SEM); ♦, rat CA1; N = 6; n = 1 (EPSC0 = 3.37 ± 0.7 nA, mean ± SEM). C1, Drug-induced EPSC amplitude potentiation as a function of culture age (DIV; mean ± SEM). A, DIV 8–10; B, DIV 11–15. The numbers within the histogram bars are N values, representing the number of cells. Significance was tested by a KS test. C2, Dependency of the EPSC0 on the age of the culture (DIV; mean ± SD). ●, Mean control EPSC0 for a given culture age. The number in brackets indicates the number of cells measured for a given culture age. Gray open circles indicate individual EPSC0 data points measured at a certain culture age; white open squares, triangles, and circles indicate mean EPSC0 values of the cells from C1.
The age of the culture not only determined the amount of EPSC amplitude potentiation, it additionally influenced the mean EPSC0 amplitude. The increase in the mean EPSC0 amplitude with age could be described by a power law (Fig. 3C2). However, the variation in EPSC0 was large for any given age. To reduce this experimental variability, we chose the EPSC0 amplitude as our reference for further analysis of the drug-mediated EPSC amplitude potentiation.
Dependency on EPSC0 extended to properties beyond those of the evoked EPSCs. Under control condition, both mEPSC amplitudes and frequency increased in size with larger EPSC0 values (Fig. 4A). Mean frequency of quantal events covered a 22.6-fold range (from 0.18 ± 0.06 Hz at an EPSC0 of 0.4 nA to 4.06 ± 1.12 Hz at an EPSC0 of 12 nA, mean ± SEM) (Fig. 4C). Correlation of control mEPSC amplitudes and frequency with the age of the culture showed that both increased with age, being significantly larger for DIV 11–14 than for DIV 8–10. The average control mEPSC amplitude significantly (p < 0.01) increases from 44 ± 2 pA (mean ± SEM; N = 69) for DIV 8–10 to 52 ± 2 pA (mean ± SEM; N = 32) for DIV 11–14, whereas the average control mEPSC frequency significantly (p < 0.001) increases from 1 ± 0.02 Hz (mean ± SEM; N = 69) to 3.5 ± 0.6 Hz (mean ± SEM; N = 32). Similar effects of cell culture age on mEPSC frequency and amplitude were previously observed by Gottmann et al. (1994).
Figure 4.
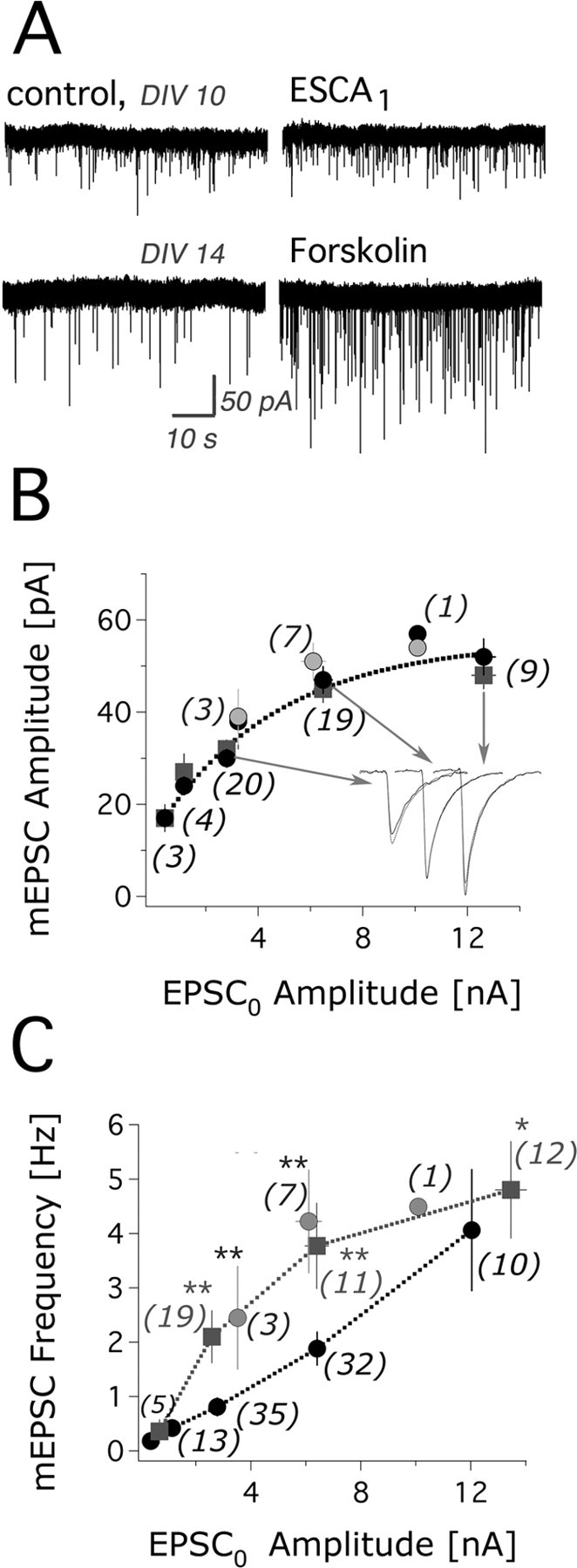
ESCA1 application enhances mEPSC frequency. For every individual cell, mEPSC events were recorded for at least 1 min from murine DG neurons in the absence or presence of cAMP-related pharmacological agents. To prevent AP-related calcium influx, sodium channels were blocked by coapplication of 300 nm TTX. A, Representative mEPSC sample traces for the control (DIV 10 and DIV 14) condition and the corresponding ESCA1 (50 μm) or forskolin (50 μm) treatment. B, mEPSC amplitude versus the EPSC0 amplitude. Black circles, Control; squares, ESCA1; N = 47; n = 3; gray circles, forskolin; N = 11; n = 2. Inset, Mean mEPSC sample traces are shown for control and ESCA1 treatment. Arrows indicate their corresponding data points in the curve. C, mEPSC frequency versus the EPSC0 amplitude. Significance was tested by a paired t test. Black circles, Control; N = 101; n = 9; squares, ESCA1; N = 47; n = 3; gray circles, forskolin (50 μm); N = 11; n = 2. In B and C, mEPSC data are pooled over the following EPSC0 intervals: from >0 to <1 nA, from 1 to <2 nA, from 2 to <4 nA, from 4 to <10 nA, and ≥10 nA. The values are presented as mean ± SEM. The number of cells in the EPSC0 classes is given in parentheses.
In summary, our results suggest that a maturation aspect may underlie the EPSC0 amplitude and needs to be considered as a normalizing factor. Therefore, we will use the EPSC0 as a reference in all our analyses of drug-induced changes in evoked EPSCs. The EPSC0 reflects a certain initial functional state of a neuron, which is determined by the number of synapses, the quantal size, and the release probability of a vesicle.
Differences between species or neuronal preparations additionally influenced the drug-related enhancement of neurotransmitter release. Unlike in murine DG and CA1 neurons, ESCA1 application did not elicit an acute response in rat CA1 neurons (data not shown). In contrast, the PDBu response showed no significant species difference when comparing rat and murine CA1 neurons. However, the PDBu response was more pronounced for murine DG neurons than for murine/rat CA1 neurons (Fig. 3B). When considering an EPSC0 of 3.33 ± 0.51 nA (mean ± SEM), the PDBu-induced steady-state EPSC amplitude potentiation in rat CA1 neurons was only half the steady-state potentiation observed for murine DG neurons (Fig. 3B, right).
ESCA1-mediated Epac activation is presynaptic in origin and increases the release probability for a fusion event
Both forskolin and ESCA1 enhanced mEPSC frequency (Fig. 4A,C) without changing the mEPSC amplitude (Fig. 4B). mEPSC frequency was increased by up to fivefold during ESCA1 application for young cell culture ages (DIV 8–10) (see supplemental material, available at www.jneurosci.org) (Fig. 3A2). The average increase was by a factor of 2.9 ± 0.5 (mean ± SEM; N = 20). The ESCA1-induced enhancement of mEPSC frequency was prominent for the EPSC0 amplitude interval of [2; 10] nA. For EPSC0 amplitudes <0.5 nA, no ESCA1-mediated potentiation of mEPSC frequency was detectable.
The average potentiation of mEPSC frequency by application of forskolin (3.4 ± 0.4, mean ± SEM; N = 10) was not significantly different from the one induced by ESCA1 (2.7 ± 0.6, mean ± SEM; N = 15) when considering similar distributions of EPSC0 values and the same cell culture ages (DIV 11–14) for both drug treatments. Therefore, we conclude that Epac activation can account for 100% of the forskolin effect on mEPSC frequency. Because we did not observe a change of mEPSC amplitude after brief ESCA1 application, the observed ESCA1 response on spontaneous release is presynaptic in origin. Paired-pulse experiments further validated the presynaptic locus of the ESCA1 response: there was on average a 55% decrease in paired-pulse facilitation with ESCA1, consistent with an increased probability of release (see supplemental material, available at www.jneurosci.org) (Fig. 3B).
Next, we addressed the question of whether the number of vesicles that can be released with a brief pulse of hypertonic sucrose solution is modulated by ESCA1 (Fig. 5). Application of 500 mm sucrose solution maximally releases all vesicles in close proximity to the active zone (Rosenmund and Stevens, 1996). We calculated the number of vesicles released by sucrose under control condition and in the presence of ESCA1 (for details on the calculation of the vesicle numbers, see Materials and Methods). ESCA1 was applied for 72 s before sucrose stimulation. For any given EPSC0 value, we detected no significant difference between the number of released vesicles in the absence or presence of ESCA1 (Fig. 5B). This shows that the average number of sucrose-releasable vesicles is not changed during application of ESCA1. Given the finding that ESCA1 increases action potential-evoked EPSCs and that quantal size is unchanged after Epac activation, we conclude that the drug increases the mean release probability. In view of the heterogeneity in release probability (Moulder and Mennerick, 2005), this change can be either a general increase in p or else a shift between different populations of vesicles.
Figure 5.

The number of sucrose-releasable vesicles is not affected by ESCA1 application. A, Representative control sucrose sample traces (a–e) for selected EPSC0 amplitudes (see B). Arrowheads indicate onset and end of sucrose (500 mm) application. B, Linear dependency of vesicle numbers on the EPSC0 amplitude (mean ± SD). The vesicle number was calculated as described in Materials and Methods. ●, Control; N = 50; n = 6; □, ESCA1 (50 μm); N = 44; n = 3. Data points were binned for the following EPSC0 intervals: <0.5 nA, from 0.5 to 1 nA, from >1 to <4 nA, from 4 to <10 nA, from 10 to <15 nA, and >15 nA. The number of cells in the EPSC0 classes is given in parentheses. The arrows (a–e) indicate the EPSC0 values for the control sample traces shown in A.
cAMP-to-PKC signaling occurs after ESCA1-induced Epac activation
Next, we tested whether application of ESCA1 and of phorbol esters may affect similar signaling pathways. Phorbol esters were shown to enhance evoked and spontaneous neurotransmitter release in various neuronal preparations (Malenka et al., 1986; Parfitt and Madison, 1993; Chen and Roper, 2003; Searl and Silinsky, 2003). They modulate both the number of readily releasable vesicles (Stevens and Sullivan, 1998; Waters and Smith, 2000) and the vesicular release probability (Rhee et al., 2002; Basu et al., 2007). We stimulated neurons with ESCA1 before application of PDBu (for details, see Fig. 6), and subsequently recorded the PDBu-mediated changes in EPSC amplitudes at 0.2 Hz in the absence of ESCA1. We found a prominent interaction between the two stimuli.
Figure 6.

 |
We analyzed murine DG neurons, which so far proved to be susceptible to ESCA1 application. In addition, rat CA1 neurons were chosen to serve as a negative control for a putative interaction of the PDBu- and the ESCA1-activated pathways, because application of ESCA1 did not elicit an acute increase in EPSC amplitudes in this preparation (data not shown). These mechanistic differences in the ESCA1 response for mouse and rat neurons may result from differential protein expression ratios for both Epac isoforms. Cultured murine neurons show a higher Epac2-to-Epac1 protein expression ratio than cultured rat neurons.
We analyzed the changes of the PDBu response induced by prestimulation with ESCA1, and differentiated between different classes of EPSC0. Prestimulation with the cAMP analog increased a subsequent PDBu response in murine DG neurons, but not in rat CA1 neurons (Fig. 6B). The difference was detectable for all EPSC0 values <10 nA. Statistical analysis of the PDBu potentiation values for murine DG neurons showed that the ESCA1-induced enhancement of the PDBu response is significant (p < 0.0001). Thus, a modulation of the PDBu response occurs only for neurons, which also show an increase of neurotransmitter release during prestimulation with the Epac-specific cAMP analog (see above). Sample traces from murine DG neurons show the ESCA1-induced enhancement of the PDBu response (Fig. 6A). The amount of potentiation depended on the time interval between ESCA1 and PDBu application. The resulting time course of relative EPSC amplitude potentiation peaked at the interapplication interval (IAI) of 3 min (Fig. 6C), with the enhancement of the PDBu response developing slowly and decaying over several minutes. For murine CA1 neurons, the size of the PDBu response enhancement at an interapplication interval of 1 min was comparable with that observed for murine DG neurons (other IAIs were not analyzed). Rat CA1 neurons showed an ESCA1-induced PDBu response enhancement neither at an interapplication interval of 1 min nor at one of 2 min. These data demonstrate that for murine DG and CA1 neurons, ESCA1 application exhibits a slow mode of action in addition to its rapid enhancement of EPSC amplitude during low-frequency stimulation. Considering the fast washout of the ESCA1 response (τ ∼ 13 ± 1 s; mean ± SD) (Fig. 2B1), we conclude that the ESCA1-mediated enhancement of the PDBu response over a time scale of minutes presumably involves a complex signaling cascade either converging to or complementing PDBu-activated signaling pathways.
To define the role of the classical phorbol ester receptor, PKC, for the observed cAMP-to-PDBu effect, we inhibited the enzymatic kinase function in situ in the absence of ESCA1-mediated Epac stimulation. During electrophysiological recordings, preapplication of the PKC-antagonist bisindolylmaleimide I (Bis I; 1.2 μm) significantly (p < 0.0001) inhibited the PDBu-mediated potentiation of EPSC amplitudes on average by 60 ± 19% (mean ± SD; N = 31) (Fig. 7). In addition, it led to a long-lasting suppression of EPSCs (Fig. 7A) as reported by Rhee et al. (2002). Bis I does not inhibit the activity of Munc13, another putative presynaptic PDBu target crucial for neurotransmitter release, in vitro (Betz et al., 1998). Thus, the nonblocked PDBu response may be attributable to activation of Munc13. However, we cannot exclude that an incomplete kinase block may explain the remaining PDBu response as well in this experimental design. Moreover, application of Bis I reduced AP-related sodium and potassium currents by 12.4 ± 1.6% (mean ± SEM) and 14 ± 1.7% (mean ± SEM), respectively. Both current type changes are significant at the 1% level.
Figure 7.

Inhibition of PKC activity by bisindolylmaleimide I reduces the PDBu response. A, Time course of the EPSC amplitude potentiation of dentate gyrus autapses by 1 μm PDBu in the absence or presence of 1.2 μm Bis I during 0.2 Hz stimulation (mean ± SEM). Horizontal bars show the onset and the duration of drug application. ●, Reference PDBu response (−Bis I); N = 24; n = 6 (EPSC0 = 4.35 ± 0.51 nA, mean ± SEM); ◇, PDBu response in the presence of Bis I (+Bis I); N = 31; n = 3 (EPSC0 = 4.69 ± 0.49 nA, mean ± SEM). B, PDBu-mediated EPSC amplitude potentiation in the absence (−) or presence (+) of Bis I versus EPSC0 (mean ± SD). Individual data points were binned in classes of 1 nA for EPSC0 > 0.5 nA, and mean drug effects were calculated for each class. The continuous line is the fit to the values from Figure 3B (left); ▼, expected reference PDBu response calculated from the EPSC0 amplitudes of the cells used in the Bis I experiment and the curve fit of the reference PDBu “potentiation curve” (continuous line) (Fig. 3B, left); ♦, mean PDBu response measured in the presence of Bis I; ◇, individual data points measured in the presence of Bis I.
Given the partial action of the PKC-blocking drug, we thought to test the involvement of PKC by an alternative approach in which we downregulate intracellular PKC activity. It was previously shown that prolonged stimulation of cells with PDBu (1 μm) downregulates PKC activity in differentiated PC12 cells and sympathetic neurons (Matthies et al., 1987). This PKC downregulation results from proteasomal degradation of the enzyme (Lee et al., 1997). Therefore, we preincubated murine DG neurons for 16 h with 1 μm PDBu and tested for the PDBu-mediated effect on EPSC amplitudes after a 5 min wash. This wash time was considered to be sufficient for the removal of the preapplied PDBu, given a washout time constant for the acute PDBu response of τwashout(PDBu) = 35.7 ± 4.1 s (mean ± SD) (Fig. 3B, right). The PDBu-mediated effect on EPSC amplitudes was reduced for all EPSC0 classes (Fig. 8A). On average, the PDBu response was significantly (p < 0.0001) reduced by 93 ± 12% (mean ± SD; N = 19). In contrast, mean EPSC0 values were not significantly different between control measurements (1.88 ± 0.36 nA, mean ± SEM; N = 15) and those after PDBu pretreatment (1.87 ± 0.42 nA, mean ± SEM; N = 19). Similarly, mEPSC frequency was not changed when comparing control (0.29 ± 0.15 Hz, mean ± SD; N = 6) and PDBu-pretreated (0.31 ± 0.09 Hz, mean ± SD; N = 5) neurons for a given class of EPSC0 values (0.83 ± 0.48 nA) (Fig. 8C,D). From the occurrence of mEPSCs at normal rates, we conclude that long-term pretreatment with PDBu did not cause a major loss of Munc13 protein function, because Varoqueaux et al. (2002) presented evidence that neurons lacking Munc13 show neither evoked nor spontaneous release, despite forming a normal number of synapses with typical ultrastructural features. We conclude that PKC activity is required for the enhancement of EPSC amplitudes by PDBu.
Figure 8.

Downregulation of PKC activity by prolonged preincubation of neuronal cultures with PDBu abolishes the PDBu response. Dentate gyrus neurons were incubated either with PDBu (1 μm) or with DMSO (control) for 16 h before electrophysiological recordings. A, EPSC amplitude potentiation ± preincubation with PDBu for 16 h. Individual data points were binned in classes of 1 nA for EPSC0 >0.5 nA. ●, Mean PDBu response reference values from Figure 3B (left); N = 21; n = 6 (EPSC0 = 2.18 ± 0.28 nA, mean ± SEM); ▲, mean PDBu responses after preincubation with PDBu; ◇, individual PDBu responses after preincubation with PDBu. B, Average (ave.) potentiation of the EPSC amplitudes by PDBu in the presence or absence of preincubation with PDBu (mean ± SEM, significance tested by a KS test). The recordings for both conditions were not obtained from identical cells. Therefore, we generated three different control values for the mean PDBu effect. m, Potentiation values measured in the absence of PDBu preincubation (N = 21; n = 5). c, Potentiation values generated from the biexponential curve fit of the PDBu reference curve (Fig. 3B, left) using the individual EPSC0 amplitudes measured after preincubation (N = 19; n = 2). d, Potentiation values after preincubation with DMSO for 16 h (volume of application corresponded to the one used for pretreatment with PDBu; N = 11; n = 3). The average EPSC0 ± SEM (2.10 ± 0.59 nA) for the initial amplitude distribution used for d in B is indicated by a light gray bar in A. C, Top two traces, Sample traces for control and preincubation condition. Lowest traces, Superimposed traces of detected mEPSC events (24 events for control; 29 events for the preincubation condition). D, Dependency of the mEPSC frequency on the preincubation condition. ♦, Mean control frequency; N = 6 (EPSC0 = 0.83 ± 0.48 nA, mean ± SD); ◇, individual control data points; gray circle, mean frequency after 16 h preincubation with PDBu; N = 5 (EPSC0 = 0.81 ± 0.82 nA, mean ± SD); ○, individual preincubation data points.
Finally, we observed that potentiation of neurotransmitter release by acute ESCA1 application persisted after prolonged preincubation with PDBu, whereas a subsequent PDBu response was abolished (see supplemental material, available at www.jneurosci.org) (Fig. 4). Moreover, ESCA1 did not lead to a PDBu response after subsequent PDBu application in neurons pretreated with PDBu.
Discussion
In the present study, we investigated the contribution of the novel cAMP-target Epac to the cAMP-mediated enhancement of glutamate release from presynaptic terminals at excitatory hippocampal synapses during low stimulation frequency. First, we showed that ESCA1-mediated Epac activation modulates neurotransmitter release from murine DG neurons by increasing the mean vesicular release probability, either because of a general increase in p or because of a shift between different populations of vesicles. Second, we provided evidence that brief ESCA1 application leads to presynaptic changes persisting for several minutes. These persistent changes were attributed to interacting ESCA1- and PKC-signaling pathways.
ESCA1-activated Epac function complements PKA activity
Specific activation of Epac by ESCA1 presynaptically increased neurotransmission from cultured excitatory neurons of the murine dentate gyrus (Figs. 1B1,B2, 2). Previous studies examining the calyx of Held (Sakaba and Neher, 2003; Kaneko and Takahashi, 2004) and the crayfish neuromuscular junction (Zhong and Zucker, 2005) reported similar PKA-independent presynaptic effects. In cultured excitatory DG neurons, brief ESCA1 application enhances EPSC amplitudes on average by 23 ± 3% (mean ± SEM; N = 60). This effect explains on average 38% of a forskolin-induced increase in evoked EPSC amplitudes (60 ± 6%, mean ± SEM; N = 46). In slices of layer V cortical neurons, ESCA1 or forskolin potentiated EPSP slopes to a comparable extent (Huang and Hsu, 2006). In contrast to our findings and to results from the crayfish neuromuscular junction (Zhong and Zucker, 2005), Huang and Hsu (2006) concluded that Epac activity does not contribute to the forskolin-mediated increase of EPSP slopes.
In view of the modest cAMP-dependent potentiation of EPSC amplitudes, the total activity of Epac may well be underestimated. A putative subestimation of the ESCA1 response could not be related to hydrolysis or membrane permeability of the drug (see supplemental material, available at www.jneurosci.org) (Fig. 1B1,B2). However, we cannot exclude an increase in the intracellular cAMP level during 0.2 Hz stimulation or else a putative constitutive activation of endogenous Epac or PKA. In fact, experiments performed by Chevaleyre et al. (2007) suggest that both PKA-dependent and PKA-independent cAMP-mediated signaling pathways are involved in the maintenance of synaptic transmission during low-frequency stimulation.
Traditionally, PKA was believed to be the key player when modulating plasticity at central excitatory synapses. Considering its short-term actions, PKA not only inhibits potassium channel function (Siegelbaum et al., 1982), but additionally phosphorylates synapsins, SNAP-25, and voltage-dependent calcium channels (Hell et al., 1995; Nagy et al., 2004; Menegon et al., 2006). In our experiments, potassium current was indeed affected by forskolin but not by ESCA1-induced Epac activation.
Acute application of pharmacological agents modified neurotransmitter release from cultured autaptic excitatory neurons depending on the EPSC0 (Fig. 3A,B) and the age of the culture (Fig. 3C). Similarly, in slices of layer V cortical neurons, potentiation of neurotransmitter release by forskolin exhibited a dependency on the age of the animal, being more pronounced for younger than for older animal ages (Huang and Hsu, 2006). Our forskolin data suggest that notably for small EPSC0 and younger cell culture ages (DIV 8–10), cAMP-signaling pathways other than those induced by Epac activation have a high impact on neurotransmission (Fig. 3A,C1,C2). Both HCN channels and PKA may account for the larger forskolin response. However, our experimental results failed to demonstrate the contribution of HCN channels to hyperpolarization-activated currents (see supplemental material, available at www.jneurosci.org) (Fig. 2A1,A2), in agreement with the absence or very low expression of HCN channel isoforms at the mRNA level in murine dentate granule cells (Moosmang et al., 1999; Santoro et al., 2000). On the other hand, PKA activation by the specific cAMP analog 6-Bnz-cAMP increased EPSC amplitudes in excitatory murine DG neurons during 0.2 Hz stimulation (see supplemental material, available at www.jneurosci.org) (Fig. 2B).
The age of the animals not only influences the size of the drug-induced change in evoked release, but also determines mEPSC amplitude and frequency. Both of these quantities increase with the animal age (Yamashita et al., 2003). In autaptic hippocampal cultures, similar observations were made. Older cell cultures show larger mean mEPSC amplitudes and higher mean mEPSC frequencies (Gottmann et al., 1994). This property is shared by our control mEPSC recordings. Thus, developmental changes may likely determine the observed dependency of pharmacological effects on EPSC0 in neuronal cell cultures. To eliminate experimental heterogeneity resulting from developmental changes, we analyzed short-term pharmacological effects with respect to the initial state of a neuron. Moreover, this revealed features of second-messenger-initiated molecular interactions underlying the developmentally determined neuronal plasticity.
The ESCA1 response sufficiently explains the observed increase in mEPSC frequency after a forskolin-induced elevation of intracellular cAMP levels. Quantal size was affected neither by application of forskolin nor by ESCA1 (Fig. 4B). In a recent study, forskolin enhanced mIPSC frequency without affecting quantal size in the presence of PKA inhibition by cannabinoid receptor antagonist (Chevaleyre et al., 2007). This study, therefore, is compatible with our findings.
Although increasing AP-evoked neurotransmitter release, ESCA1-mediated Epac activation did not change the number of releasable vesicles as measured by the application of 500 mm sucrose solution (Rosenmund and Stevens, 1996) (Fig. 5). However, this result does not necessarily reflect a general increase in p. In the past years, several publications have provided evidence that kinetically distinct heterogeneous release components occur during presynaptic depolarization in diverse model systems, such as the neuromuscular junction, chromaffin cells, hippocampal neurons, and the calyx of Held (Rahamimoff and Yaari, 1973; Neher and Zucker, 1993; Goda and Stevens, 1994; Sakaba and Neher, 2001; Moulder and Mennerick, 2005). Therefore, the Epac-induced change in mean p also may represent a shift between different populations of vesicles.
ESCA1- and PDBu-induced signaling pathways interact
Both the number of readily releasable vesicles (Stevens and Sullivan, 1998; Waters and Smith, 2000) and the vesicular release probability (Rhee et al., 2002; Basu et al., 2007) are thought to be subject to modification by PDBu. We showed that ESCA1 application enhanced a subsequent PDBu effect on EPSC amplitudes (Fig. 6). The amount of enhancement depended on the time interval between ESCA1 and PDBu application, and developed slowly over several minutes. Synergistic interaction of the forskolin- and PDBu-mediated modulation of excitatory synaptic transmission was previously observed in granule cells of the human dentate gyrus: prior application of forskolin occludes the effects of PDBu on mEPSC frequency (Chen and Roper, 2003).
Downregulation of PKC activity by prolonged neuronal pretreatment with PDBu (Matthies et al., 1987; Popp et al., 2006) indicated that PKC activity is required for the PDBu response (Fig. 8). Recently, Wierda et al. (2007) showed that the PDBu response was abolished in autaptic Munc18-1-null mutant neurons expressing a PKC-insensitive Munc18-1 protein. Munc18-1 is believed to facilitate vesicle docking in a PKC phosphorylation-independent manner in chromaffin cells: its phosphorylation by PKC enhances vesicle pool replenishment after a depleting stimulation at a postdocking stage (Nili et al., 2006). However, at present, little is known about the contribution of PKC-mediated regulation of VDCCs and calcium influx (DeRiemer et al., 1985; Swartz, 1993) to the observed PDBu response. From our experimental design, it is unclear whether and to what extent the observed interaction of ESCA1- and PDBu-induced signaling pathways involves changes of VDCC function and calcium influx.
Because preapplication of the Epac-specific cAMP analog ESCA1 increases the PDBu response (Fig. 6), and because this response is dependent on PKC/Munc18-1 interaction (Wierda et al., 2007), we suggest that Epac activity may enhance the PKC/Munc18-1-mediated vesicle pool replenishment by directly stimulating PKC/Munc18-1 activity or indirectly by regulating Munc13 function and vesicle priming. Two mechanisms can be proposed for this action of Epac: plasma-membrane-associated Epac1 (DiPilato et al., 2004) targets PLCε via Rap2B (Evellin et al., 2002) generating diacylglycerol. This may lead to the activation and translocation of PKCε to the plasma membrane (Hucho et al., 2005) or else to the activation of Munc13 via its C1 domain. Moreover, Rap2B activates the Cdc25 domain function of PLCε generating the active GTP form of Rap1 (Wing et al., 2003). Alternatively, Epac2 was shown to interact N-terminally with the PDZ domain of the active zone protein RIM1 (Ozaki et al., 2000). The currently available data establish that RIM1 has a central function in the regulation of neurotransmitter release (Lonart, 2002). It organizes the docking and priming of vesicles for exocytosis, by regulating the recruitment of Munc13-1 to the active zone (Andrews-Zwilling et al., 2006), thereby enabling the formation of a Munc13/RIM/Rab3 tripartite complex (Dulubova et al., 2005). In pancreatic β-cells, cAMP fully rescues the priming defects caused by Munc13-1 deficiency via Epac and PKA signaling pathways and requires downstream Munc13-1/Rim2 interaction: both the RRP size and the refilling rate of a releasable pool of vesicles are partially restored after ESCA1-mediated Epac activation, whereas PKA completely restores the RRP size and partially rescues refilling kinetics (Kwan et al., 2007).
Footnotes
This work was performed in the framework of the European Union-integrated project “EUSynapse” (LSHM-CT-2005-019055) and was supported by a grant from the German Research Foundation (Deutsche Forschungsgemeinschaft) to E.N. and Christian Rosenmund (R01296/5-4). I.G. is a fellow of the Graduate Program 521 (GRK521) of the German Research Foundation. We thank Holger Taschenberger for helpful discussions throughout the work, and Frank Würriehausen and Frank Köhne for expert technical assistance. Holger Taschenberger and Christian Rosenmund critically reviewed this manuscript. We are grateful to Reinhard Jahn for suggesting to test for the Epac specificity of ESCA1 in autaptic neurons by quantifying the phosphorylation of synapsin at its PKA phosphorylation site after Epac stimulation. Finally, we acknowledge the help of Hans Dieter Schmitt, who shared with us the LumiImager and provided valuable comments on Western blotting.
References
- Andrews-Zwilling YS, Kawabe H, Reim K, Varoqueaux F, Brose N. Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13–2. J Biol Chem. 2006;281:19720–19731. doi: 10.1074/jbc.M601421200. [DOI] [PubMed] [Google Scholar]
- Basu J, Betz A, Brose N, Rosenmund C. Munc13-1 C1 domain lowers the energy barrier for synaptic vesicle fusion. J Neurosci. 2007;27:1200–1210. doi: 10.1523/JNEUROSCI.4908-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Idzerda RL, McKnight GS. PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol. 1997;7:397–403. doi: 10.1016/s0959-4388(97)80069-4. [DOI] [PubMed] [Google Scholar]
- Castellucci VF, Kandel ER, Schwartz JH, Wilson FD, Nairn AC, Greengard P. Intracellular injection of the catalytic subunit of cyclic AMP-dependent protein kinase stimulates facilitation of transmitter release underlying behavioral sensitization in Aplysia. Proc Natl Acad Sci U S A. 1980;77:7492–7496. doi: 10.1073/pnas.77.12.7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-X, Roper SN. PKA and PKC enhance excitatory synaptic transmission in human dentate gyrus. J Neurophysiol. 2003;89:2482–2488. doi: 10.1152/jn.01031.2002. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Südhof TC, Purpura DP, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1α. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser H-G, Døskeland SO. cAMP analog mapping of Epac1 and cAMP kinase. J Biol Chem. 2003;278:35394–35402. doi: 10.1074/jbc.M302179200. [DOI] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRiemer SA, Strong JA, Albert KA, Greengard P, Kaczmarek LK. Enhamcement of calcium current in Aplysia neurons by phorbol ester and protein kinase C. Nature. 1985;313:313–316. doi: 10.1038/313313a0. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJT, Verheijen MHG, Cool RH, Nijman SMB, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, Südhof TC, Rizo J. A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity? EMBO J. 2005;24:2839–2850. doi: 10.1038/sj.emboj.7600753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson L, Ma X, Renström E, Barg S, Berggren P-O, Galvanovskis J, Gromada J, Jing X, Lundquist I, Salehi A, Sewing S, Rorsman P. SUR1 regulates PKA-independent cAMP-induced granule priming in mouse pancreatic B-cells. J Gen Physiol. 2003;121:181–197. doi: 10.1085/jgp.20028707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Evellin S, Nolte J, Tysack K, vom Dorp F, Thiel M, Oude Weernink PA, Jakobs KH, Webb EJ, Lomasney JW, Schmidt M. Stimulation of phospholipase C-ε by the M3 muscarinic acetylcholine receptor mediated by cyclic AMP and GTPase Rap2B. J Biol Chem. 2002;277:16805–16813. doi: 10.1074/jbc.M112024200. [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottmann K, Pfrieger FW, Lux HD. The formation of glutamatergic synapses in cultured central neurons: selective increase in miniature synaptic currents. Brain Res Dev Brain Res. 1994;81:77–88. doi: 10.1016/0165-3806(94)90070-1. [DOI] [PubMed] [Google Scholar]
- Greengard P, Kuo JF, Miyamoto E. Studies on the mechanism of action of cyclic AMP in nervous and other tissue. Adv Enzyme Regul. 1970;9:113–125. doi: 10.1016/s0065-2571(71)80040-7. [DOI] [PubMed] [Google Scholar]
- Hell JW, Yokoyama CT, Breeze LJ, Chavkin C, Catterall WA. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO J. 1995;14:3036–3044. doi: 10.1002/j.1460-2075.1995.tb07306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG. Epac: a new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic β-cell. Diabetes. 2004;53:5–13. doi: 10.2337/diabetes.53.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Chepurny OG, Schwede F. Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signal. 2008;20:10–20. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C-C, Hsu K-S. Presynaptic mechanism underlying cAMP-induced synaptic potentiation in medial prefrontal cortex pyramidal neurons. Mol Pharmacol. 2006;69:846–856. doi: 10.1124/mol.105.018093. [DOI] [PubMed] [Google Scholar]
- Hucho TB, Dina OA, Levine JD. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J Neurosci. 2005;25:6119–6126. doi: 10.1523/JNEUROSCI.0285-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EM, Ueda T, Maeno H, Greengard P. Adenosine 3′,5-monophosphate-dependent phosphorylation of a specific protein in synaptic membrane fractions from rat cerebrum. J Biol Chem. 1972;247:5650–5652. [PubMed] [Google Scholar]
- Kaneko M, Takahashi T. Presynaptic mechanism underlying cAMP-dependent synaptic potentiation. J Neurosci. 2004;24:5202–5208. doi: 10.1523/JNEUROSCI.0999-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan EP, Xie L, Sheu L, Ohtsuka T, Gaisano HY. Interaction between Munc13-1 and RIM is critical for glucagon-like peptide-1 mediated rescue of exocytosis defects in Munc13-1 deficient pancreatic beta-cells. Diabetes. 2007;56:2579–2588. doi: 10.2337/db06-1207. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lee H-W, Smith L, Pettit GR, Smith JB. Bryostatin 1 and phorbol ester down-modulate protein kinase C-α and -ε via the ubiquitin/proteasome pathway in human fibroblasts. Mol Pharmacol. 1997;51:439–447. [PubMed] [Google Scholar]
- Leßmann V, Heumann R. Cyclic AMP endogenously enhances synaptic strength of developing glutamatergic synapses in serum-free microcultures of rat hippocampal neurons. Brain Res. 1997;763:111–122. doi: 10.1016/s0006-8993(97)00406-x. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Glickman J, Donaldson JG, Robbins J, Kreis TE, Seamon KB, Sheetz MP, Klausner RD. Forskolin inhibits and reverses the effects of brefeldin A on Golgi morphology by a cAMP-independent mechanism. J Cell Biol. 1991;112:567–577. doi: 10.1083/jcb.112.4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G. RIM1: an edge for presynaptic plasticity. Trends Neurosci. 2002;25:329–332. doi: 10.1016/s0166-2236(02)02193-8. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol ester. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- Matthies HJG, Palfrey HC, Hirning LD, Miller RJ. Down regulation of protein kinase C in neuronal cells: effects on neurotransmitter release. J Neurosci. 1987;7:1198–1206. doi: 10.1523/JNEUROSCI.07-04-01198.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegon A, Bonanomi D, Albertinazzi C, Lotti F, Ferrari G, Kao H-T, Benfenati F, Baldelli P, Valtorta F. Protein kinase A-mediated synapsin I phosphorylation is a central modulator of Ca2+-dependent synaptic activity. J Neurosci. 2006;26:11670–11681. doi: 10.1523/JNEUROSCI.3321-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, Nestler EJ. Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Mol Brain Res. 2000;81:129–139. doi: 10.1016/s0169-328x(00)00155-8. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Biel M, Hofmann F, Ludwig A. Differential distribution of four hyperpolarization-activated cation channels in mouse brain. Biol Chem. 1999;380:975–980. doi: 10.1515/BC.1999.121. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Mennerick S. Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J Neurosci. 2005;25:3842–3850. doi: 10.1523/JNEUROSCI.5231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sørensen JB. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron. 2004;41:417–429. doi: 10.1016/s0896-6273(04)00038-8. [DOI] [PubMed] [Google Scholar]
- Neher E, Zucker RS. Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron. 1993;10:21–30. doi: 10.1016/0896-6273(93)90238-m. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Nili U, de Wit H, Gulyas-Kovacs A, Toonen RF, Sørensen JB, Verhage M, Ashery U. Munc18-1 phosphorylation by protein kinase C potentiates vesicle pool replenishment in bovine chromaffin cells. Neuroscience. 2006;143:487–500. doi: 10.1016/j.neuroscience.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- Parfitt KD, Madison DV. Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. J Physiol. 1993;471:245–268. doi: 10.1113/jphysiol.1993.sp019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp RL, Velasquez O, Bland J, Hughes P. Characterization of protein kinase C isoforms in primary cultured cerebellar granule cells. Brain Res. 2006;1083:70–84. doi: 10.1016/j.brainres.2006.01.110. [DOI] [PubMed] [Google Scholar]
- Rahamimoff R, Yaari Y. Delayed release of transmitter at the frog neuromuscular junction. J Physiol. 1973;228:241–257. doi: 10.1113/jphysiol.1973.sp010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee J-S, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Südhof TC, Takahashi M, Rosenmund C, Brose N. β phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not PKCs. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Feltz A, Westbrook GL. Synaptic NMDA receptor channels have a low open probability. J Neurosci. 1995;15:2788–2795. doi: 10.1523/JNEUROSCI.15-04-02788.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/s0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Direct modulation of synaptic vesicle priming by GABAB receptor activation at a glutamatergic synapse. Nature. 2003;424:775–778. doi: 10.1038/nature01859. [DOI] [PubMed] [Google Scholar]
- Santoro B, Chen S, Lüthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searl TJ, Silinsky EM. Phorbol esters and adenosine affect the readily releasable neurotransmitter pool by different mechanisms at amphibian motor nerve endings. J Physiol. 2003;553:445–456. doi: 10.1113/jphysiol.2003.051300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedej S, Rose T, Rupnik M. cAMP increases Ca2+-dependent exocytosis through both PKA and Epac2 in mouse melanotrophs from pituitary tissue slices. J Physiol. 2005;567:799–813. doi: 10.1113/jphysiol.2005.090381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegelbaum SA, Camardo JS, Kandel ER. Serotonin and cyclic AMP close single K+ channels in Aplysia sensory neurons. Nature. 1982;299:413–417. doi: 10.1038/299413a0. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee J-S, Brose N, Enk C, Reim K, Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U S A. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent P, Brusciano D. Cyclic AMP imaging in neurones in brain slice preparations. J Neurosci Methods. 2001;108:189–198. doi: 10.1016/s0165-0270(01)00393-4. [DOI] [PubMed] [Google Scholar]
- Waters J, Smith SJ. Phorbol esters potentiate evoked and spontaneous release by different presynaptic mechanisms. J Neurosci. 2000;20:7863–7870. doi: 10.1523/JNEUROSCI.20-21-07863.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Wierda KDB, Toonen RFG, de Wit H, Brussaard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54:275–290. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Cooper DM. Ca2+ stimulation of adenylyl cyclase generates dynamic oscillations in cyclic AMP. J Cell Sci. 2006;119:828–836. doi: 10.1242/jcs.02812. [DOI] [PubMed] [Google Scholar]
- Wing MR, Bourdon DM, Harden TK. PLC-ε: A shared effector protein in Ras-, Rho-, and Gαβγ-mediated signaling. Mol Interv. 2003;3:273–280. doi: 10.1124/mi.3.5.273. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ishikawa T, Takahashi T. Developmental increase in vesicular glutamate content does not cause saturation of AMPA receptors at the calyx of Held synapse. J Neurosci. 2003;23:3633–3638. doi: 10.1523/JNEUROSCI.23-09-03633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Zucker RS. cAMP acts on exchange protein activated by cAMP/cAMP-regulated guanine nucleotide exchange protein to regulate transmitter release at the crayfish neuromuscular junction. J Neurosci. 2005;25:208–214. doi: 10.1523/JNEUROSCI.3703-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]