

Abstract
Tolerance to the analgesic effects of opioids occurs after their chronic administration, a pharmacological phenomenon that has been associated with the development of abnormal pain sensitivity such as hyperalgesia. In the present study, we investigated the role of TRPV1, which is crucial for the transduction of noxious chemical and thermal stimuli, in morphine tolerance and tolerance-associated thermal hyperalgesia. After chronic morphine treatment, a marked increase in TRPV1 immunoreactivity (IR) was detected in L4 dorsal root ganglion (DRG) neurons, spinal cord dorsal horn, and sciatic nerve. Real-time reverse transcription (RT)-PCR demonstrated that TRPV1 mRNA was upregulated in spinal cord and sciatic nerve but not in the DRG. Intrathecal pretreatment with SB366791 [N-(3-methoxyphenyl)-4-chlorocinnamide], a selective antagonist of TRPV1, attenuated both morphine tolerance and associated thermal hyperalgesia. Chronic morphine exposure induced increases in phosphorylation of mitogen-activated protein kinases (MAPKs), including p38 MAPK-IR, extracellular signal-regulated protein kinase (ERK)-IR, and c-Jun N-terminal kinase (JNK)-IR, in L4 DRG neurons. Intrathecal administration of the selective p38, ERK, or JNK inhibitors not only reduced morphine tolerance and associated thermal hyperalgesia but also suppressed the morphine-induced increase of TRPV1-IR in DRG neurons, spinal cord, and sciatic nerve and of mRNA levels in spinal cord and sciatic nerve. Together, we have identified a novel mechanism by which sustained morphine treatment results in tolerance and tolerance-associated thermal hyperalgesia, by regulating TRPV1 expression, in a MAPK-dependent manner. Thus, blocking TRPV1 might be a way to reduce morphine tolerance.
Keywords: morphine, TRPV1, MAPK, tolerance, immunohistochemistry, RT-PCR, Western blot
Introduction
Besides the known side effects, the clinical utility of opioid analgesics is often hampered by the development of tolerance that necessitates dose escalation regardless of disease progression. The development of opioid tolerance has also been associated with enhanced pain sensitivity such as hyperalgesia in both laboratory and clinical settings (Sjogren et al., 1993; Mao et al., 1994, 2002; Vanderah et al., 2000, 2001). There is emerging evidence suggesting that opioid tolerance and pathological pain may share common cellular mechanisms (Mao et al., 1995a; Mayer et al., 1999), and the abnormal pain elicited by opioids is a critical factor in the behavioral manifestation of opioid tolerance because the same manipulations that block abnormal pain also block opioid tolerance (King et al., 2005b; Ossipov et al., 2005). Despite considerable progress, the molecular and cellular mechanisms mediating morphine tolerance are not yet completely understood.
The TRPV1 receptor, which plays a critical role in the transduction of noxious chemical and thermal stimuli (Caterina et al., 1997, 2000), is a nonselective cation channel gated by noxious heat, protons, and capsaicin. TRPV1 is observed in primary sensory neurons and in both central and peripheral sensory terminals of neurons (Szallasi et al., 1995, 2006; Tominaga and Julius, 2000). It appears to be critical in the development of thermal and mechanical hyperalgesia under pathological conditions (Caterina et al., 2000; Levine and Alessandri-Haber, 2007; Ma and Quirion, 2007). TRPV1 can be modulated by numerous mediators, including growth factors, neurotransmitters, peptides or small proteins, lipids, chemokines, and cytokines (Ma and Quirion, 2007). Furthermore, it has been suggested that nerve growth factor leads to an increase in TRPV1 levels in dorsal root ganglion (DRG) neurons and inflamed skin through the activation of p38 mitogen-activated protein kinase (MAPK) (Ji et al., 2002) and that phosphatidylinositol 3-kinase induces heat hyperalgesia, possibly by regulating TRPV1 activity, in an extracellular signal-regulated protein kinase (ERK)-dependent manner (Zhuang et al., 2004). These studies imply that TRPV1 is not simply a thermoreceptor, but its activity is modulated by various molecules that act through distinct pathways.
MAPKs, consisting of p38, ERK, and c-Jun N-terminal kinase (JNK), are downstream to many kinases and are activated in primary sensory neurons, dorsal horn neurons, and spinal glial cells by nociceptive stimuli, growth factors, and inflammatory mediators, contributing to the induction and maintenance of sensitization via transcriptional, translational, and posttranslational regulation. Inhibition of MAPKs has been shown to attenuate inflammatory and neuropathic pain (Ji, 2004; Obata and Noguchi, 2004; Obata et al., 2004; Ma and Quirion, 2005; Ji et al., 2007). Data from in vitro and in vivo experiments suggest that the phosphorylation of MAPK and cAMP response element-binding protein (CREB) plays a role in the chronic morphine-induced increase in calcitonin gene-related peptide (CGRP) and substance P (SP) levels in DRG neurons, indicating that MAPK inhibitors might have the potential to block or reverse tolerance to morphine-induced antinociception (Ma et al., 2001). Intrathecal administration of SB203580 [4-(4-fluorophenyl)2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1Himidazole], a specific p38 inhibitor, significantly attenuated the tolerance to morphine analgesia (Cui et al., 2006). Furthermore, attenuation of spinal ERK phosphorylation by intrathecal administration of the MAPK kinase (MEK) inhibitor U0126 [1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene] or knockdown of spinal ERK by antisense oligonucleotides not only decreased the scores of morphine withdrawal but also attenuated withdrawal-induced allodynia (Cao et al., 2005).
Although TRPV1 has been implicated in thermal hyperalgesia under pathological conditions, it is not clear whether it plays a role in morphine tolerance and associated thermal hyperalgesia. Because morphine tolerance and pathological pain are supposed to share similar mechanisms (Mao et al., 1995a; Mayer et al., 1999), we tested the hypothesis that activation of TRPV1 contributes to morphine tolerance. Furthermore, we investigated the involvement of the MAPK signaling pathway in the activation of TRPV1 induced by chronic morphine exposure.
Materials and Methods
Animals.
Male Sprague Dawley rats (220–250 g; Charles River) were used in all experiments. Animals were housed on a 14/10 h light/dark cycle with standard rodent chow and water available ad libitum. All experiments were approved by the Bavarian state authorities and were in accordance with Ethical Guidelines for Investigators of Experimental Pain in Conscious Animals (Zimmermann, 1983).
Intrathecal catheterization and drug delivery.
A polyethylene PE10 catheter was implanted in the lumbar subarachnoid space during deep isoflurane anesthesia according to the method described previously (Yaksh and Rudy, 1976). Animals were allowed to recover for at least 7 d before experiments. Those rats exhibiting postoperative neurological deficits (e.g., paralysis) or poor grooming were killed. For the “control” and “morphine tolerance” groups, rats were intraperitoneally injected twice daily with normal saline (NS) or morphine sulfate (Merck) at 10 mg/kg body weight at 12 h intervals (8:00 A.M. and 8:00 P.M., days 1–8), and 10 μl of 25% DMSO as a vehicle was intrathecally injected, followed by 10 μl of NS flush, 30 min before morning NS or morphine injection. For the “inhibitor/antagonist plus morphine” groups, 10 μl of the inhibitors or antagonist, including the selective p38 inhibitor SB203580 (10 μg; Calbiochem), the selective MEK1/2 inhibitor U0126 (10 μg; Calbiochem), the selective TRPV1 antagonist SB366791 [N-(3-methoxyphenyl)-4-chlorocinnamide] (30 μg; Sigma), and the selective JNK inhibitor SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one) (50 μg; Calbiochem) dissolved in 25% DMSO (Sigma), except for SP600125 in 40% DMSO, were intrathecally injected, followed by a 10 μl NS flush 30 min before the morning morphine challenge on each testing day (days 1–8). For testing of the effect of the inhibitors or antagonist per se, rats were intrathecally injected with the inhibitors or antagonist 30 min before the morning NS injection for the next 8 d.
Behavioral testing.
To test morphine tolerance, 30 min after the morning intraperitoneal injection of NS or morphine from day 1 to day 8, sensitivity to noxious heat was assessed using the device of Hargreaves et al. (1988) purchased from Ugo Basile. A radiant heat source was focused on the plantar surface of the hindpaw, and the time from the initiation of the radiant heat until paw withdrawal [paw-withdrawal latency (PWL)] was measured automatically. Additionally, rats were tested for tolerance-associated thermal hyperalgesia on day 8 before the last morning NS or morphine challenge. A maximal cutoff of 25 s was used to prevent tissue damage. Left and right paws were randomly tested once; the mean withdrawal latency was calculated from both sides. The behavioral testing was done by an investigator blind to the treatment.
Immunohistochemistry.
The L4 DRGs, L4–L5 spinal cord, and the mid-sciatic nerve with a length of 1 cm were removed on day 8 after behavioral testing. The tissues were mounted in Tissue Tek OTC (Dialec) embedding compound, quickly frozen in liquid nitrogen, and stored at − 80°C. DRGs (30 μm), spinal cord (30 μm), and sciatic nerve (10 μm) were sectioned on a cryostat and thaw mounted onto Superfrost Plus slides (Langenbrinck). The following antibodies were used for DRG immunohistochemistry using the ABC method: polyclonal rabbit antibodies for phosphorylated p38 (p-p38) (1:100; Cell Signaling Technology), p-ERK1/2 (1:50; Cell Signaling Technology), p- stress-activated protein kinase (SAPK)/JNK (1:50; Cell Signaling Technology), and TRPV1 (1:500; Affinity BioReagents). DRG sections were air dried for 30 min, blocked for 30 min with 0.3% H2O2 diluted in methanol, and then incubated overnight at 4°C with primary antibodies as above. On the following day, they were incubated with biotinylated secondary antibodies (Vector Laboratories) for 30 min and with avidin–biotin solution (Vector Laboratories) for another 30 min. Specific antibody binding was visualized using a standard diaminobenzidine peroxidase method. Immunofluorescence was performed for single staining for TRPV1 (1:500, polyclonal rabbit anti-TRPV1; Affinity BioReagents) in spinal cord and sciatic nerve and for double staining for TRPV1 [1:500, polyclonal goat anti-TRPV1 (Santa Cruz Biotechnology); this antibody produced identical immunostaining in DRG to that of polyclonal rabbit anti-TRPV1 using single staining] with μ receptor (1:500, polyclonal rabbit anti-μ receptor; Gramsch Laboratories) in DRG. In brief, sections were air dried for 30 min, postfixed for 10 min in acetone at − 20°C, blocked with 10% BSA/PBS for 30 min, and incubated overnight at 4°C. On the following day, they were incubated for 2 h in a cyanine 3 (Cy3)-conjugated or Cy2-conjugated secondary antibody (1:100; Dianova).
Image analysis.
Three sections from each animal for each marker were randomly collected. After immunostaining, they were viewed and digitized with a Zeiss Axiophot 2 microscope and quantitatively analyzed using Image Pro Plus software for DRG sections (version 4.0; Media Cybernetics) and NIH ImageJ software (version 1.38x) for spinal cord and sciatic nerve sections. All sections were captured with the camera settings held constant. The examiner was unaware of the identity of the slides.
For DRG sections, neuronal cell bodies were identified by the typical morphology and the presence of a nucleus. The density threshold for positive immunoreactivity (IR) was determined by averaging two or three cell bodies in each section that were judged to be minimally positive. All neurons for which the mean density exceeded the threshold were counted as positive, and the positive cells were expressed as a percentage of total counted DRG neurons [% = (positive cells/total counted cells) × 100]. The area of the TRPV1-IR neuronal profiles was measured and displayed as size–frequency distributions. Percentages of size frequency were calculated for each DRG as follows: % = (positive cells within a size range/total neuronal cells) × 100. To distinguish cell-size-specific changes, we characterized the DRG neurons as small-sized (<600 μm2), medium-sized (600–1200 μm2), and large-sized (>1200 μm2) neurons, according to their cross-sectional area (Fukuoka et al., 2001).
For quantitative analysis of TRPV1-IR in spinal cord, the outline of the superficial dorsal horn was manually traced (encompassing laminas I–II) for each image, and then an appropriate threshold was set such that only specific TRPV1-IR was accurately represented and light nonspecific background labeling was not detected. The threshold was the same for all images. The density limited to threshold in the outlined area was measured for each section. A mean value from both sides was determined for each rat. The relative TRPV1-IR density was shown as the ratio of the density of the treated groups to the control group. A similar approach has been used previously (Garrison et al., 1994; Martin et al., 1999).
For quantitative analysis of TRPV1-IR in sciatic nerve, the procedures were the same with that of spinal cord except that the density was measured within a fixed box size of 190 × 190 μm (∼50% of the whole endoneural area) placed at the middle zone of the images. A similar approach has been used by others (Hartmann et al., 2004; Blesch and Tuszynski, 2007).
Real-time PCR.
In separate groups of rat treated with intrathecal 25% DMSO plus intraperitoneal NS (control group), intrathecal 25% DMSO plus intraperitoneal morphine (morphine group), and intrathecal MAPK inhibitors plus intraperitoneal morphine (inhibitor plus morphine groups), the L4 DRGs, L4–L5 spinal cord, and the mid sciatic nerve with a length of 1 cm were harvested on day 8 after the injections. Tissue homogenization and RNA isolation were performed as described previously (Chen and Sommer, 2007). In brief, total RNA from each tissue was prepared using the TRIzol reagent (Invitrogen) and was quantified spectrophotometrically. Tissues were homogenized using a Polytron homogenizer (Kinematica). Relative TRPV1 mRNA levels were quantified with reverse transcription (RT)-PCR using the fluorescent TaqMan technology. Total RNA (500 ng) was reverse transcribed (TaqMan Reverse Transcription Reagents; Applied Biosystems) using random hexamers. PCR primer and probe specific for rat TRPV1 (Applied Biosystems identification number Rn01460299_m1) and 18S rRNA were obtained as TaqMan Predeveloped Assay Reagents for gene expression (Applied Biosystems). The 18S rRNA was used as an endogenous control. PCR was performed with equal amounts of cDNA in the GeneAmp 7700 sequence detection system (Applied Biosystems) using TaqMan Universal PCR Master Mix (Applied Biosystems). Reactions (total volume, 50 μl) were incubated at 50°C for 2 min, at 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Water controls were included to ensure specificity. Each sample was measured in triplicate, and data points were examined for integrity by analysis of the amplification plot. The comparative cycle threshold Ct method was used for relative quantification of gene expression. The amount of TRPV1 mRNA, normalized to the endogenous control (18S rRNA) and relative to a calibrator, is given by 2−ΔΔCt, with Ct indicating the cycle number at which the fluorescence signal of the PCR product crosses an arbitrary threshold set within the exponential phase of the PCR, and ΔΔCt = [(Cttarget (unknown sample) − Ctend.control (unknown sample))] − [(Cttarget (calibrator sample) − Ctend. control (calibrator sample))].
Western blot.
In separate groups of rat treated with intrathecal 25% DMSO plus intraperitoneal NS (control group) and intrathecal 25% DMSO plus intraperitoneal morphine (morphine group), the L3–L5 DRGs, L4–L5 spinal cord, and the mid sciatic nerve with a length of 1 cm were harvested on day 8 after the injections. Tissues were homogenized in PBS containing 1% Triton X-100 and then resuspended in Laemmli's sample buffer as described previously (Laemmli, 1970). Equal amounts of protein (75 μg) were fractionated by 10% SDS-PAGE and electroblotted onto a nitrocellulose membrane. Unspecific binding sites were blocked by incubation with 0.05% Tween 20 containing 5% nonfat milk in PBS. Nitrocellulose was first incubated with TRPV1 antibody (1:1000, polyclonal rabbit anti-TRPV1; Affinity BioReagents) and, after stripping, with a mouse monoclonal anti-β-actin antibody (1:20,000; Sigma) for 12 h at 4°C. Primary antibody binding was detected using peroxidase-coupled anti-rabbit or anti-mouse secondary antibodies (1:3000; GE Healthcare) and the ECLTM Advance Western Blotting Detection kit (GE Healthcare) after washing three times for 10 min each with 0.05% Tween 20 in PBS. The specific bands were quantified by densitometric analysis using Image Pro Plus software.
Statistical analysis.
For statistical analysis, SPSS (version 10.0) was used. Results are presented as means ± SEM. The data were analyzed by Student's t test, one-way or two-way ANOVA, followed by Tukey's post hoc test to determine differences between groups. p < 0.05 was considered to be statistically significant.
Results
Development of morphine tolerance and associated thermal hyperalgesia
Injection of morphine produced robust analgesia to thermal stimuli on day 1 (see Fig. 5A) (p < 0.001). Although morphine consistently produced significant antinociception until day 8 when compared with control animals (see Fig. 5A) (p < 0.001), the effect of morphine gradually declined during chronic exposure from day 3 to day 8, indicating that the rats had developed morphine tolerance (see Fig. 5A). Compared with PWLs of control rats on day 8 before the last NS injection, PWLs were decreased in morphine-tolerant rats when tested on day 8 before the last morphine injection (see Fig. 5B) (p < 0.001), indicating thermal hyperalgesia in morphine-tolerant rats. No significant changes in PWLs were detected throughout the whole observation period in control rats (see Fig. 5A).
Figure 5.

Effects of intrathecal administration of the TRPV1 inhibitor SB366791 on morphine tolerance (A) and tolerance-associated thermal hyperalgesia (B) assessed by the PWL test. Although morphine consistently produced significant antinociception until day 8 (A; ***p < 0.001, Veh + Mor vs Veh + NS), the effect gradually declined during chronic exposure from day 3 to day 8, which was accompanied by thermal hyperalgesia on day 8 (B; ***p < 0.001, Veh + Mor vs Veh + NS). Rats in the morphine group pretreated with SB366791 displayed significantly longer PWLs from day 3 to day 8 (A; $$$p < 0.001 for SB366791 + Mor vs Veh + Mor, and ###p < 0.001 for SB366791 + Mor vs Veh + NS) and also had attenuated morphine-induced thermal hyperalgesia on day 8 (B; ###p < 0.001, SB366791 + Mor vs Veh + Mor). Neither Veh + NS nor SB366791 + NS treatment altered pain thresholds throughout the observation period. Two-way (A) and one-way (B) ANOVA, followed by Tukey's test were used for statistical analysis. n = 6 for each group.
TRPV1 activation in DRG, spinal cord, and sciatic nerve after chronic morphine treatment
TRPV1-IR was present in small- and medium-sized DRG neurons, in spinal laminas I and II inner and in sciatic nerve of control animals (Fig. 1A,C,E), consistent with previous studies (Caterina et al., 1997, 2000; Ji et al., 2002).
Figure 1.
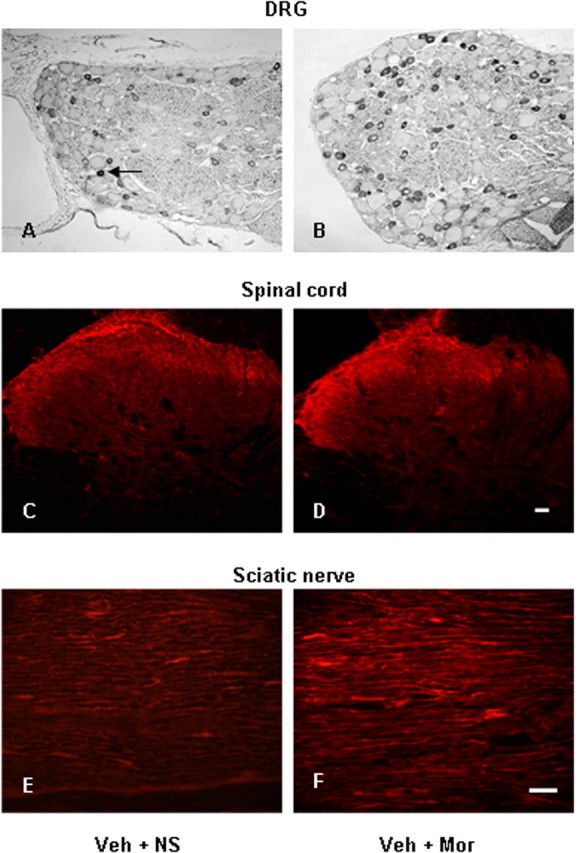
Photomicrographs of TRPV1-IR in DRG (A, B), spinal cord (C, D), and sciatic nerve (E, F) from control (A, C, E) and morphine (B, D, F) treated rats. A, Arrow indicates a TRPV1-IR DRG neuron. Scale bars: (in D) A–D, 20 μm; (in F) E, F, 40 μm. Veh, 25% DMSO; Mor, Morphine.
To test whether morphine tolerance and associated thermal hyperalgesia were mediated by regulating TRPV1, we first examined TRPV1 expression in the DRG of morphine-tolerant rats. Using immunohistochemistry, 30% of all DRG neurons of control rats displayed IR for TRPV1 (Figs. 1A, 2A). After 8 d of morphine treatment, a significant increase of TRPV1-IR (44%) was observed (Figs. 1B, 2A) (p < 0.001). The percentage of TRPV1-IR was increased in both small- and medium-sized neurons (<600 and 600–1200 μm2, respectively; p < 0.001) (Table 1, Fig. 1A,B). RT-PCR revealed no significant change of TRPV1 mRNA levels in the DRG after 8 d of morphine treatment (Fig. 2B).
Figure 2.

Chronic morphine increases TRPV1-IR in DRG, spinal cord, and sciatic nerve and mRNA levels in spinal cord and sciatic nerve. A, C, E, Immunohistochemistry showed an increase of TRPV1-IR in DRG neurons (A) and an enhanced density of TRPV1-IR in the spinal superficial dorsal horn (C) and in sciatic nerve (E) of chronic morphine-treated rats (***p < 0.001, Veh + Mor vs Veh + NS). B, D, F, RT-PCR revealed no significant change in TRPV1 mRNA levels in the DRGs (B) but increased TRPV1 mRNA levels in both spinal cord and sciatic nerve (D, F; ***p < 0.001, Veh + Mor vs Veh + NS) of morphine-tolerant rats. Student's t test was used for statistical analysis. n = 6 for each group.
Table 1.
Distribution of TRPV1-IR in small-, medium-, and large-sized DRG neurons
| DRG neurons | Veh + NS [% = (TRPV1-IR/total DRG neurons) × 100] | Veh + Mor [% = (TRPV1-IR/total DRG neurons) × 100] | p value |
|---|---|---|---|
| Small size (<600 μm2) | 28.9 ± 0.9 | 40.7 ± 1.7*** | p < 0.001 |
| Medium size (600–1200 μm2) | 0.97 ± 0.3 | 3.6 ± 0.5*** | p < 0.001 |
| Large size (>1200 μm2) | 0 | 0.09 ± 0.04 | p > 0.05 |
| Total | 29.8 ± 2.3 | 44.3 ± 2.6*** | p < 0.001 |
Size–frequency analysis illustrates that TRPV1-IR is predominantly localized in small-sized DRG neurons of control and morphine-tolerant rats. Chronic morphine treatment significantly increased the expression of TRPV1 in both small- and medium-sized neurons (<600 and 600–1200 μ m2, respectively; ***p < 0.001, Veh + Mor vs Veh + NS). Student's t test was used for statistical analysis. n = 6 for each group. Veh, Vehicle; Mor, morphine.
The density of TRPV1-IR in the superficial dorsal horn (Figs. 1D, 2C) (1.6-fold increase; p < 0.001) and in the sciatic nerve (Figs. 1F, 2E) (1.8-fold increase; p < 0.001) was substantially increased after 8 d of morphine treatment relative to control animals. TRPV1 mRNA levels in the spinal cord (Fig. 2D) (2.7-fold increase; p < 0.001) and sciatic nerve (Fig. 2F) (3.9-fold increase; p < 0.001) were increased in morphine-tolerant rats.
To assess total protein levels of TRPV1, we performed semiquantitative Western blot analysis of the DRG, spinal cord, and sciatic nerve. No significant difference in total protein levels could be detected in tissues from DRG, spinal cord, and sciatic nerve between morphine-tolerant and control rats (Fig. 3).
Figure 3.

Western blot analysis of TRPV1 in DRG, spinal cord, and sciatic nerve. A, Example of Western blot with TRPV1 antibody showing bands at 95 kDa in spinal cord of morphine-tolerant and control rats. B–D, Semiquantitative density measuring revealed no significant difference in spinal cord (B), DRG (C), and sciatic nerve (D) between morphine-tolerant and control rats (Veh + Mor vs Veh + NS). β-Actin served as loading control. Student's t test was used for statistical analysis. n = 6 for each group.
Colocalization of μ receptor with TRPV1 in DRG neurons
Double immunostaining was performed to determine the colocalization of μ receptor and TRPV1. Some small-sized DRG neurons contained IR for both μ receptor and TRPV1 in control and morphine-tolerant animals (Fig. 4C,F). Coexpression was seen in 8.8 and 17% of all neurons (percentage of expression relates to total number of DRG neurons; Student's t test, p < 0.001 between groups), in 31 and 37% of TRPV1-immunoreactive neurons (p > 0.05), and in 44 and 59% of μ receptor-immunoreactive neurons (p < 0.001) of control and morphine-tolerant rats, respectively.
Figure 4.

Double immunostaining of μ receptor (A, D) and TRPV1 (B, E) shows the colocalization in DRG neurons from control (A–C) and chronic morphine-treated (D–F) rats as indicated in yellow when both images are merged. Arrows in C and F indicate the DRG neurons immunoreactive for both TRPV1 and μ receptor. Scale bar (in F): A–F, 20 μm.
Blockade of TRPV1 attenuates morphine tolerance and associated thermal hyperalgesia
Based on these observations, we then designed an experiment to ascertain whether alteration of TRPV1 by morphine might be involved in morphine tolerance and associated thermal hyperalgesia. Once daily intrathecal injection of SB366791, a selective TRPV1 antagonist (Gunthorpe et al., 2004; Lappin et al., 2006; Xu et al., 2007), produced no effect on the basal thermal pain thresholds in rats receiving NS throughout the whole observation period (Fig. 5, SB366791 + NS group). There was no significant difference in morphine-induced analgesia on day 1 between rats pretreated with vehicle and rats pretreated with SB366791. From day 3 to day 8 after morphine, however, rats pretreated with vehicle showed decreased PWLs, whereas the SB366791 pretreated ones displayed significantly longer PWLs (Fig. 5A) (p < 0.001). Furthermore, the combined administration of SB366791 and morphine also effectively reduced the tolerance-associated thermal hyperalgesia when tested on day 8 (Fig. 5B) (p < 0.001).
Inhibition of the phosphorylation of MAPK reduces morphine tolerance and associated thermal hyperalgesia
To determine the functional roles of phosphorylation of MAPK in morphine tolerance and associated thermal hyperalgesia, the activation of p38, ERK, and JNK in DRG neurons was investigated. After 8 d of morphine administration, the p-p38-, p-ERK-, and p-JNK-IR in the DRG neurons was increased compared with the control animals (Fig. 6) (from 10.2, 4.6, and 3.6% to 27.6, 11.5, and 11.7%, respectively; p < 0.001), similar to the effect of chronic morphine on phosphorylation of MAPK in cultured DRG neurons (Ma et al., 2001). In separate groups of rats, the selective p38, ERK, or JNK inhibitors, SB203580, U0126, or SP600125, respectively, were intrathecally injected once daily 30 min before each morning NS or morphine challenge. We found that these treatments did not significantly affect the basal levels of phosphorylation of MAPK in DRG neurons in rats receiving NS (Fig. 6) but inhibited the increase of p-p38-, p-ERK-, and p-JNK-IR in DRG neurons induced by chronic morphine (from 27.6, 11.5, and 11.7% to 18.55, 3.4, and 6.36%, respectively; p < 0.001) (Fig. 6). Administration of the inhibitors SB203580, U0126, or SP600125 produced no significant changes in basal pain sensitivity in rats receiving NS throughout the observation period (Fig. 7); however, all of them significantly reduced both morphine tolerance (Fig. 7A,C,E) (p < 0.001) and associated thermal hyperalgesia (Fig. 7B,D,F) (p < 0.001).
Figure 6.

Photomicrographs (A–D, F–I, K–N) and quantification (E, J, O) showing p-p38-IR (A–E), p-ERK-IR (F–J), and p-JNK-IR (K–O) in DRG neurons of the control, morphine, and the selective MAPK inhibitor pretreated control and morphine groups. Percentages of p-p38-, p-ERK-, and p-JNK-IR were increased after chronic morphine exposure (B, G, L and E, J, O; ***p < 0.001, Veh + Mor vs Veh + NS); this increase was reduced by treatment with the selective MAPK inhibitors (C, H, M and E, J, O; ###p < 0.001 for SB203580 + Mor, U0126 + Mor, or SP600125 + Mor vs Veh + Mor). No significant difference in the phosphorylation of MAPK was detected between control and the selective MAPK inhibitor pretreated control groups (SB203580 + NS, U0126 + NS, or SP600125 + NS vs Veh + NS). One-way ANOVA, followed by Tukey's test was used for statistical analysis. n = 6 for each group. Arrows in B, G, and L indicate p-p38-, p-ERK-, or p-JNK-IR DRG neurons. Scale bar (in N): A–D, F–I, K–N, 40 μm.
Figure 7.

Effects of repeated administration of the selective MAPK inhibitors on morphine tolerance (A, C, E) and associated thermal hyperalgesia (B, D, F) assessed by the PWL test. Although morphine consistently produced significant antinociception until day 8 (A, C, E; ***p < 0.001, Veh + Mor vs Veh + NS), the effect gradually declined during chronic exposure from day 3 to day 8, which was accompanied by thermal hyperalgesia on day 8 (B, D, F; ***p < 0.001, Veh + Mor vs Veh + NS). Pretreatment with the selective MAPK inhibitors, SB203580, U0126, or SP600125, produced significantly longer PWLs from day 3 to day 8 (A, C, E; $$$p < 0.001 for SB203580 + Mor, U0126 + Mor, or SP600125 + Mor vs Veh + Mor; ###p < 0.001 for SB203580 + Mor, U0126 + Mor, or SP600125 + Mor vs Veh + NS) and attenuated chronic morphine-induced thermal hyperalgesia on day 8 (B, D, F; ###p < 0.001, SB203580 + Mor, U0126 + Mor, or SP600125 + Mor vs Veh + Mor). None of the inhibitors altered pain thresholds in rats receiving NS throughout the observation period. Two-way (A, C, E) and one-way (B, D, F) ANOVA, followed by Tukey's test were used for statistical analysis. n = 6 for each group.
Inhibition of the phosphorylation of MAPK reduces the increase of TRPV1 induced by chronic morphine
To examine whether the activation of MAPK has a role in the increase of TRPV1 induced by chronic morphine, the IR and mRNA for TRPV1 in the DRGs, spinal cord, and sciatic nerve were compared in the control, morphine, and MAPK inhibitor pretreatment groups. None of the selective MAPK inhibitors by themselves influenced the IR and mRNA levels for TRPV1 (data not shown). However, all of them not only suppressed the chronic morphine-induced elevation of TRPV1-IR in DRG neurons (Figs. 8A–D, 9A) (from 44 to 34.4, 34.0, or 36.7% for the p38, ERK, or JNK inhibitor pretreatment groups, respectively; p < 0.001), spinal cord (Figs. 8E–H, 9C) (from 1.6-fold to 1.2-, 1.3-, or 1.1-fold for the p38, ERK, or JNK inhibitor pretreatment groups, respectively; p < 0.01 and p < 0.001), and sciatic nerve (Figs. 8I–L, 9E) (from 1.8-fold to 1.2-, 1.2-, or 1.4-fold for the p38, ERK, or JNK inhibitor pretreatment groups, respectively; p < 0.01 and p < 0.001) but also greatly reduced the increase of TRPV1 mRNA levels in spinal cord (Fig. 9D) (from 2.7-fold to 1.0-, 0.98-, or 0.89-fold for the p38, ERK, or JNK inhibitor pretreatment groups, respectively, p < 0.001) and sciatic nerve (Fig. 9F) (from 3.9-fold to 1.5-, 1.5-, or 2.1-fold for the p38, ERK, or JNK inhibitor pretreatment groups, respectively, p < 0.05 and p < 0.001). None of them altered TRPV1 mRNA levels in DRGs (Fig. 9B).
Figure 8.

Photomicrographs of TRPV1-IR in DRG (A–D), spinal cord (E–H), and sciatic nerve (I–L) from rats treated with morphine (A, E, I) and additional pretreatment with SB203580 (B, F, J), U0126 (C, G, K), or SP600125 (D, H, L). Arrow in A indicates a TRPV1-IR DRG neuron. Scale bars: (in H) A–H, 20 μm; (in L) I–L, 40 μm.
Figure 9.

Chronic morphine increases TRPV1-IR in DRG, spinal cord, and sciatic nerve and mRNA levels in spinal cord and sciatic nerve via the MAPK signaling pathway. Chronic morphine-induced increase of TRPV1-IR in DRG neurons (A) and enhanced density of TRPV-IR in the spinal superficial dorsal horn (C) and sciatic nerve (E) was reduced by pretreatment with the selective MAPK inhibitors (*p < 0.05, **p < 0.01, and ***p < 0.001 for SB203580 + Mor, U0126 + Mor, or SP600125 + Mor vs Veh + Mor). Pretreatment with the selective MAPK inhibitors did not produce significant effects on TRPV1 mRNA levels in the DRGs (B) but greatly reduced the increase of TRPV1 mRNA levels in both spinal cord and sciatic nerve (D, F; *p < 0.05 and ***p < 0.001 for SB203580 + Mor, U0126 + Mor, or SP600125 + Mor vs Veh + Mor) of morphine-tolerant rats. One-way ANOVA, followed by Tukey's test was used for statistical analysis. n = 6 for each group.
Discussion
In the present study, we demonstrated that TRPV1 was increased in the DRGs, spinal cord, and sciatic nerve of rats after chronic morphine treatment. Antagonism of TRPV1 by the selective TRPV1 antagonist SB366791 suppressed morphine tolerance and associated thermal hyperalgesia. Furthermore, inhibition of p38, ERK, and JNK activation by the selective MAPK inhibitors not only reduced morphine tolerance and associated thermal hyperalgesia but also reduced the morphine-induced increase of TRPV-IR in the DRGs, spinal cord, and sciatic nerve and of mRNA levels in spinal cord and sciatic nerve. These results for the first time suggest that the activation of TRPV1, which is induced by chronic morphine application via MAPK signaling pathways, contributes to morphine tolerance and associated thermal hyperalgesia.
It has been suggested that tissue injury- or inflammation-induced hyperalgesia and morphine tolerance may share common cellular mechanisms (Mao et al., 1995a; Mayer et al., 1999). Previous studies indicated that TRPV1 can be activated by tissue injury or inflammation and is also modulated by numerous mediators, including growth factors, neurotransmitters, peptides or small proteins, lipids, chemokines, and cytokines (Ma and Quirion, 2007). We found that chronic morphine exposure could increase TRPV1-IR in primary afferent neurons, as well as in spinal cord and sciatic nerve. In control rats, TRPV1-IR is predominantly in small-sized DRG neurons that are considered to be polymodal nociceptors with unmyelinated C-fibers. After chronic morphine treatment, the percentage of TRPV1-IR in small-sized DRG neurons was increased. Furthermore, there was an increased TRPV1-IR in medium-sized neurons with myelinated Aδ fibers, which are important in the development of hyperalgesia (Amaya et al., 2003). These findings imply a potential role of TRPV1 in morphine tolerance and associated thermal hyperalgesia. Previous RT-PCR studies did not show an increase in TRPV1 mRNA levels in the DRG after carrageenan- or complete Freund's adjuvant-induced inflammation (Tohda et al., 2001; Ji et al., 2002, Endres-Becker et al., 2007). We also did not detect the increase in TRPV1 mRNA levels in the DRG of morphine-tolerant animals. It is possible that chronic morphine enhances TRPV1 protein expression at the translational or posttranslational level in DRG neurons. In contrast, TRPV1 mRNA was increased in both spinal cord and sciatic nerve after chronic morphine treatment. It has been suggested that acute inflammation induced by carrageenan may induce bidirectional axonal transport of TRPV1 mRNA along primary afferents, and the mRNA may be involved in the biosynthesis of TRPV1 protein in the afferent terminals (Tohda et al., 2001). It is possible that chronic morphine might also increase the bidirectional axonal transport of TRPV1 mRNA along primary afferents and thus increase the level of TRPV1 in spinal cord and sciatic nerve. Additionally, the increased TRPV1-IR in spinal cord and sciatic nerve in the present study might also derive from protein synthesized in the DRG neurons, because TRPV1 protein can be transported to both the central and peripheral terminals of the primary afferent neurons (Szallasi et al., 1995; Guo et al., 1999; Ji et al., 2002). Considering that TRPV1-IR was almost abolished in rats that underwent dorsal rhizotomy (Guo et al., 1999; Valtschanoff et al., 2001) and no TRPV1 mRNA-positive cells could be detected in the dorsal horn in control rats (Tohda et al., 2001), we think that the increase of TRPV1-IR in spinal cord mainly derives from axonal transport along primary afferents, as we hypothesized above. However, Doly et al., (2004) found that 7% of the TRPV1-IR in spinal laminas I and II is localized in astrocytes. We cannot completely rule out that chronic morphine treatment might also directly increase TRPV1-IR in cells of nonprimary afferent origin. Western blot semiquantitative analysis did not show significant differences in total TRPV1 protein in the investigated tissues, which is most likely attributable to lack of sensitivity. Given that the differences seen with immunohistochemistry were ∼1.6 times, Western blot may not be sensitive enough to pick up this magnitude of difference. Another reason may be that the protein is denatured by the detergent, leading to conformational changes of the epitopes of the antibody binding sites (Burnette, 1981). Double-immunofluorescence labeling showed the colocalization of TRPV1 and μ receptor in DRG neurons of control and morphine-tolerant rats, suggesting that chronic morphine treatment may directly or indirectly activate TRPV1 at the subcellular level. Considering that TRPV1 plays an important role in thermal transduction, our results suggest that the activation of TRPV1 may have some intrinsic activity in morphine tolerance and associated thermal hyperalgesia. Our following results confirmed this point.
Intrathecal pretreatment with SB366791, a selective TRPV1 antagonist (Gunthorpe et al., 2004; Lappin et al., 2006; Xu et al., 2007), reduced morphine tolerance and associated thermal hyperalgesia without affecting the basal pain threshold of the control animals, suggesting that this receptor is involved in the effects induced by chronic morphine. Interestingly, acute morphine treatment inhibited, via a cAMP/PKA-dependent pathway, capsaicin-induced TRPV1 activity and thermal hyperalgesia (Endres-Becker et al., 2007). These results suggest that the activity of TRPV1 plays a differential role in acute morphine analgesia and in chronic morphine-induced tolerance. The mechanisms by which antagonism of TRPV1 attenuates morphine tolerance and associated thermal hyperalgesia are still unknown. TRPV1 is regarded as a polymodal molecular integrator in thermal nociception. It is conceivable that the morphine-tolerant rats with overexpression of TRPV1 are more sensitive to thermal stimuli compared with control animals. However, the contribution of TRPV1 goes beyond its role as a thermoreceptor. It has been shown that TRPV1-positive primary afferents are glutamatergic. Indeed, there is evidence that TRPV1 activation induces glutamate release (Li et al., 2004), and the TRPV1 antagonist SB366791 can inhibit glutamatergic synaptic transmission in rat spinal dorsal horn after peripheral inflammation (Lappin et al., 2006). Additionally, activation of TRPV1 evokes release of the peptide neurotransmitters, such as CGRP and SP, from central and peripheral endings of neurons, associated with nociceptive transmission and a neurogenic inflammatory response (Otsuka and Yoshioka, 1993; Geppetti and Holzer, 1996; Schicho et al., 2005). Interestingly, sustained morphine exposure enhances capsaicin-evoked release of the spinal CGRP (Gardell et al., 2002). Given that these neurotransmitters are involved in morphine tolerance (Mao et al., 1994, 1995b; Dunbar and Yaksh, 1996; Menard et al., 1996; Ma et al., 2000; King et al., 2005a,b; Ossipov et al., 2005; Vera-Portocarrero et al., 2007), we propose that chronic morphine activates TRPV1, which, at least partly, through modulation of neurotransmitters such as glutamate, CGRP, and SP, contributes to the tolerance and associated thermal hyperalgesia.
MAPKs transduce a broad range of extracellular stimuli into diverse intracellular responses by producing changes in transcription as well as by posttranslational and translational modification of target proteins. A recent set of studies suggested that MAPK are involved in neuronal plasticity, including long-term potentiation, learning and memory, and pain hypersensitivity (Ji, 2004; Thomas and Huganir, 2004; Ji et al., 2007). Here we found that chronic morphine induced an increase in the phosphorylation of p38, ERK, and JNK in DRG neurons, similar to the effect of chronic morphine on phosphorylation of MAPKs in cultured DRG neurons (Ma et al., 2001). Inhibition of the p38, ERK, and JNK phosphorylation by intrathecal injection of the selective MAPK inhibitors inhibited the increase of the MAPK phosphorylation in DRG neurons. Our findings in the selective p38 inhibitor experiments are in accordance with previous data showing that inhibition of p38-phosphorylation attenuated morphine tolerance (Cui et al., 2006). We have extended these findings and have shown that intrathecal pretreatment with the selective p38, ERK, or JNK inhibitors not only decreased morphine tolerance but also reduced the tolerance-associated thermal hyperalgesia. Activation of MAPK can regulate a variety of gene products, such as c-fos, brain-derived neurotrophic factor, neurokinin-1, and CGRP, through phosphorylation of CREB, which has been shown to be activated by chronic morphine treatment (Lonze and Ginty, 2002; McClung and Nestler, 2003). Previous studies demonstrated that TRPV1 expression could result from the p38 and ERK signaling pathway (Ji et al., 2002; Bron et al., 2003; Obata et al., 2004; Zhuang et al., 2004) and that long-lasting JNK/SAPK activation and c-Jun expression may participate in gene regulation (Kenney and Kocsis, 1998; Hou et al., 2003) in DRG neurons. In the present study, intrathecal application of the MAPK inhibitors suppressed the increase of TRPV1-IR in the DRGs, spinal cord, and sciatic nerve and of mRNA levels in spinal cord and sciatic nerve, induced by chronic morphine. Intrathecal administration of the inhibitors targeted to reach the DRG neurons (Ji et al., 2002) inhibited the phosphorylation of MAPK, which in turn decreased the TRPV1-IR in DRG neurons in morphine-tolerant rats. The increase of TRPV1-IR in spinal cord and sciatic nerve was suppressed, possibly attributable to the reduction in TRPV1 protein transported from primary afferent neurons to both the central and peripheral terminals (Szallasi et al., 1995; Guo et al., 1999; Ji et al., 2002). In addition, we cannot rule out the possibility that inhibition of the MAPK activation may have reduced the increase of TRPV1-IR in spinal cord and sciatic nerve by decreasing the bidirectional axonal transport of TRPV1 mRNA along primary afferents (Tohda et al., 2001), which may subsequently lead to reduced synthesis of TRPV1 in spinal cord and sciatic nerve. Our data imply, therefore, that the activation of MAPK in DRG neurons induced by chronic morphine participates in morphine tolerance and associated hyperalgesia by regulating the downstream target TRPV1. In other words, the MAPK signaling pathway contains the upstream regulators of TRPV1, which contribute to the tolerance of morphine and associated thermal hyperalgesia.
In summary, our data suggest that the activation of TRPV1 in the CNS and PNS is involved in the tolerance to morphine thermal analgesia and its modulatory effects involve the activation of upstream MAPK. TPRV1 and MAPK may therefore be potential targets for reducing morphine tolerance.
Footnotes
This work was supported by research funds from the University of Würzburg. We thank Barbara Dekant, Lydia Biko, Susanne Hellmig, and Barbara Gado for technical assistance and K. V. Toyka for continuous support.
References
- Amaya F, Oh-hashi K, Naruse Y, Iijima N, Ueda M, Shimosato G, Tominaga M, Tanaka Y, Tanaka M. Local inflammation increases vanilloid receptor 1 expression within distinct subgroups of DRG neurons. Brain Res. 2003;963:190–196. doi: 10.1016/s0006-8993(02)03972-0. [DOI] [PubMed] [Google Scholar]
- Blesch A, Tuszynski MH. Transient growth factor delivery sustains regenerated axons after spinal cord injury. J Neurosci. 2007;27:10535–10545. doi: 10.1523/JNEUROSCI.1903-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron R, Klesse LJ, Shah K, Parada LF, Winter J. Activation of Ras is necessary and sufficient for upregulation of vanilloid receptor type 1 in sensory neurons by neurotrophic factors. Mol Cell Neurosci. 2003;22:118–132. doi: 10.1016/s1044-7431(02)00022-2. [DOI] [PubMed] [Google Scholar]
- Burnette WM. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein. Anal Biochem. 1981;112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- Cao JL, He JH, Ding HL, Zeng YM. Activation of the spinal ERK signaling pathway contributes naloxone-precipitated withdrawal in morphine-dependent rats. Pain. 2005;118:336–349. doi: 10.1016/j.pain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sommer C. Activation of the nociceptin opioid system in rat sensory neurons produces antinociceptive effects in inflammatory pain: involvement of inflammatory mediators. J Neurosci Res. 2007;85:1478–1488. doi: 10.1002/jnr.21272. [DOI] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Doly S, Fischer J, Salio C, Conrath M. The vanilloid receptor-1 is expressed in rat spinal dorsal horn astrocytes. Neurosci Lett. 2004;357:123–126. doi: 10.1016/j.neulet.2003.12.051. [DOI] [PubMed] [Google Scholar]
- Dunbar S, Yaksh TL. Concurrent spinal infusion of MK801 blocks spinal tolerance and dependence induced by chronic intrathecal morphine in the rat. Anesthesiology. 1996;84:1177–1188. doi: 10.1097/00000542-199605000-00020. [DOI] [PubMed] [Google Scholar]
- Endres-Becker J, Heppenstall PA, Mousa SA, Labuz D, Oksche A, Schäfer M, Stein C, Zollner C. Mu-opioid receptor activation modulates transient receptor potential vanilloid 1 (TRPV1) currents in sensory neurons in a model of inflammatory pain. Mol Pharmacol. 2007;71:12–18. doi: 10.1124/mol.106.026740. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP, Jr, Lai J, Porreca F. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci. 2002;22:6747–6755. doi: 10.1523/JNEUROSCI.22-15-06747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Carlton SM. GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp Neurol. 1994;129:237–243. doi: 10.1006/exnr.1994.1165. [DOI] [PubMed] [Google Scholar]
- Geppetti P, Holzer P. Boca Raton, FL: CRC; 1996. Neurogenic inflammation. [Google Scholar]
- Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM, Luis Hannan S, Lappin SC, Egerton J, Smith GD, Worby A, Howett L, Owen D, Nasir S, Davies CH, Thompson M, Wyman PA, Randall AD, Davis JB. Identification and characterisation of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology. 2004;46:133–149. doi: 10.1016/s0028-3908(03)00305-8. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11:946–958. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hartmann B, Ahmadi S, Heppenstall PA, Lewin GR, Schott C, Borchardt T, Seeburg PH, Zeilhofer HU, Sprengel R, Kuner R. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron. 2004;44:637–650. doi: 10.1016/j.neuron.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Hou L, Li W, Wang X. Mechanism of interleukin-1 beta-induced calcitonin gene-related peptide production from dorsal root ganglion neurons of neonatal rats. J Neurosci Res. 2003;73:188–197. doi: 10.1002/jnr.10651. [DOI] [PubMed] [Google Scholar]
- Ji RR. Mitogen-activated protein kinases as potential targets for pain killers. Curr Opin Investig Drugs. 2004;5:71–75. [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Zhang YQ. Protein kinases as potential targets for the treatment of pathological pain. Handb Exp Pharmacol. 2007:359–389. doi: 10.1007/978-3-540-33823-9_13. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Kocsis JD. Peripheral axotomy induces long-term c-Jun amino-terminal kinase-1 activation and activator protein-1 binding activity by c-Jun and junD in adult rat dorsal root ganglia in vivo. J Neurosci. 1998;18:1318–1328. doi: 10.1523/JNEUROSCI.18-04-01318.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Gardell LR, Wang R, Vardanyan A, Ossipov MH, Malan TP, Jr, Vanderah TW, Hunt SP, Hruby VJ, Lai J, Porreca F. Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain. 2005a;116:276–288. doi: 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005b;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lappin SC, Randall AD, Gunthorpe MJ, Morisset V. TRPV1 antagonist, SB-366791, inhibits glutamatergic synaptic transmission in rat spinal dorsal horn following peripheral inflammation. Eur J Pharmacol. 2006;540:73–81. doi: 10.1016/j.ejphar.2006.04.046. [DOI] [PubMed] [Google Scholar]
- Levine JD, Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochim Biophys Acta. 2007;1772:989–1003. doi: 10.1016/j.bbadis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL. VR1 receptor activation induces glutamate release and postsynaptic firing in the paraventricular nucleus. J Neurophysiol. 2004;92:1807–1816. doi: 10.1152/jn.00171.2004. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R. The ERK/MAPK pathway, as a target for the treatment of neuropathic pain. Expert Opin Ther Targets. 2005;9:699–713. doi: 10.1517/14728222.9.4.699. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R. Inflammatory mediators modulating the transient receptor potential vanilloid 1 receptor: therapeutic targets to treat inflammatory and neuropathic pain. Expert Opin Ther Targets. 2007;11:307–320. doi: 10.1517/14728222.11.3.307. [DOI] [PubMed] [Google Scholar]
- Ma W, Zheng WH, Kar S, Quirion R. Morphine treatment induced calcitonin gene-related peptide and substance P increases in cultured dorsal root ganglion neurons. Neuroscience. 2000;99:529–539. doi: 10.1016/s0306-4522(00)00226-8. [DOI] [PubMed] [Google Scholar]
- Ma W, Zheng WH, Powell K, Jhamandas K, Quirion R. Chronic morphine exposure increases the phosphorylation of MAP kinases and the transcription factor CREB in dorsal root ganglion neurons: an in vitro and in vivo study. Eur J Neurosci. 2001;14:1091–1104. doi: 10.1046/j.0953-816x.2001.01731.x. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J Neurosci. 1994;14:2301–2312. doi: 10.1523/JNEUROSCI.14-04-02301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and opiate tolerance: a current view of their possible interactions. Pain. 1995a;62:259–274. doi: 10.1016/0304-3959(95)00073-2. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Phillips LL, Lu J, Mayer DJ. Increases in protein kinase C gamma immunoreactivity in the spinal cord of rats associated with tolerance to the analgesic effects of morphine. Brain Res. 1995b;677:257–267. doi: 10.1016/0006-8993(95)00161-i. [DOI] [PubMed] [Google Scholar]
- Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci. 2002;22:8312–8323. doi: 10.1523/JNEUROSCI.22-18-08312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WJ, Liu H, Wang H, Malmberg AB, Basbaum AI. Inflammation-induced up-regulation of protein kinase Cgamma immunoreactivity in rat spinal cord correlates with enhanced nociceptive processing. Neuroscience. 1999;88:1267–1274. doi: 10.1016/s0306-4522(98)00314-5. [DOI] [PubMed] [Google Scholar]
- Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci USA. 1999;96:7731–7736. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- Menard DP, van Rossum D, Kar S, St. Pierre S, Sutak M, Jhamandas K, Quirion R. A calcitonin gene-related peptide receptor antagonist prevents the development of tolerance to spinal morphine analgesia. J Neurosci. 1996;16:2342–2351. doi: 10.1523/JNEUROSCI.16-07-02342.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004;74:2643–2653. doi: 10.1016/j.lfs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- Otsuka M, Yoshioka K. Neurotransmitter functions of mammalian tachykinins. Physiol Rev. 1993;73:229–308. doi: 10.1152/physrev.1993.73.2.229. [DOI] [PubMed] [Google Scholar]
- Schicho R, Donnerer J, Liebmann I, Lippe IT. Nociceptive transmitter release in the dorsal spinal cord by capsaicin-sensitive fibers after noxious gastric stimulation. Brain Res. 2005;1039:108–115. doi: 10.1016/j.brainres.2005.01.050. [DOI] [PubMed] [Google Scholar]
- Sjogren P, Jonsson T, Jensen NH, Drenck NE, Jensen TS. Hyperalgesia and myoclonus in terminal cancer patients treated with continuous intravenous morphine. Pain. 1993;55:93–97. doi: 10.1016/0304-3959(93)90188-U. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Nilsson S, Farkas-Szallasi T, Blumberg PM, Hökfelt T, Lundberg JM. Vanilloid (capsaicin) receptors in the rat: distribution in the brain, regional differences in the spinal cord, axonal transport to the periphery, and depletion by systemic vanilloid treatment. Brain Res. 1995;703:175–183. doi: 10.1016/0006-8993(95)01094-7. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Cruz F, Geppetti P. TRPV1: a therapeutic target for novel analgesic drugs? Trends Mol Med. 2006;12:545–554. doi: 10.1016/j.molmed.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tohda C, Sasaki M, Konemura T, Sasamura T, Itoh M, Kuraishi Y. Axonal transport of VR1 capsaicin receptor mRNA in primary afferents and its participation in inflammation-induced increase in capsaicin sensitivity. J Neurochem. 2001;76:1628–1635. doi: 10.1046/j.1471-4159.2001.00193.x. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Julius D. Capsaicin receptor in the pain pathway. Jpn J Pharmacol 2000. 2000;83:20–24. doi: 10.1254/jjp.83.20. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Rustioni A, Guo A, Hwang SJ. Vanilloid receptor VR1 is both presynaptic and postsynaptic in the superficial laminae of the rat dorsal horn. J Comp Neurol. 2001;436:225–235. [PubMed] [Google Scholar]
- Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Malan TP, Ossipov MH, Lai J, Porreca F. Dynorphin promotes abnormal pain and spinal cord opioid antinociceptive tolerance. J Neurosci. 2000;20:7074–7079. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. 2001;21:279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Zhang ET, King T, Ossipov MH, Vanderah TW, Lai J, Porreca F. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain. 2007;129:35–45. doi: 10.1016/j.pain.2006.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GY, Winston JH, Shenoy M, Yin H, Pendyala S, Pasricha PJ. Transient receptor potential vanilloid 1 mediates hyperalgesia and is up-regulated in rats with chronic pancreatitis. Gastroenterology. 2007;133:1282–1292. doi: 10.1053/j.gastro.2007.06.015. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Analgesia mediated by a direct spinal action of narcotics. Science. 1976;192:1357–1358. doi: 10.1126/science.1273597. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigators of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]